2005
In 2005, the synthesis of three fluorine-18- and seven carbon-11-labeled COX-2 inhibitors as well as one carbon-11-labeled COX-1 inhibitor was reported. Three of these compounds were investigated
in vivo. Wuest
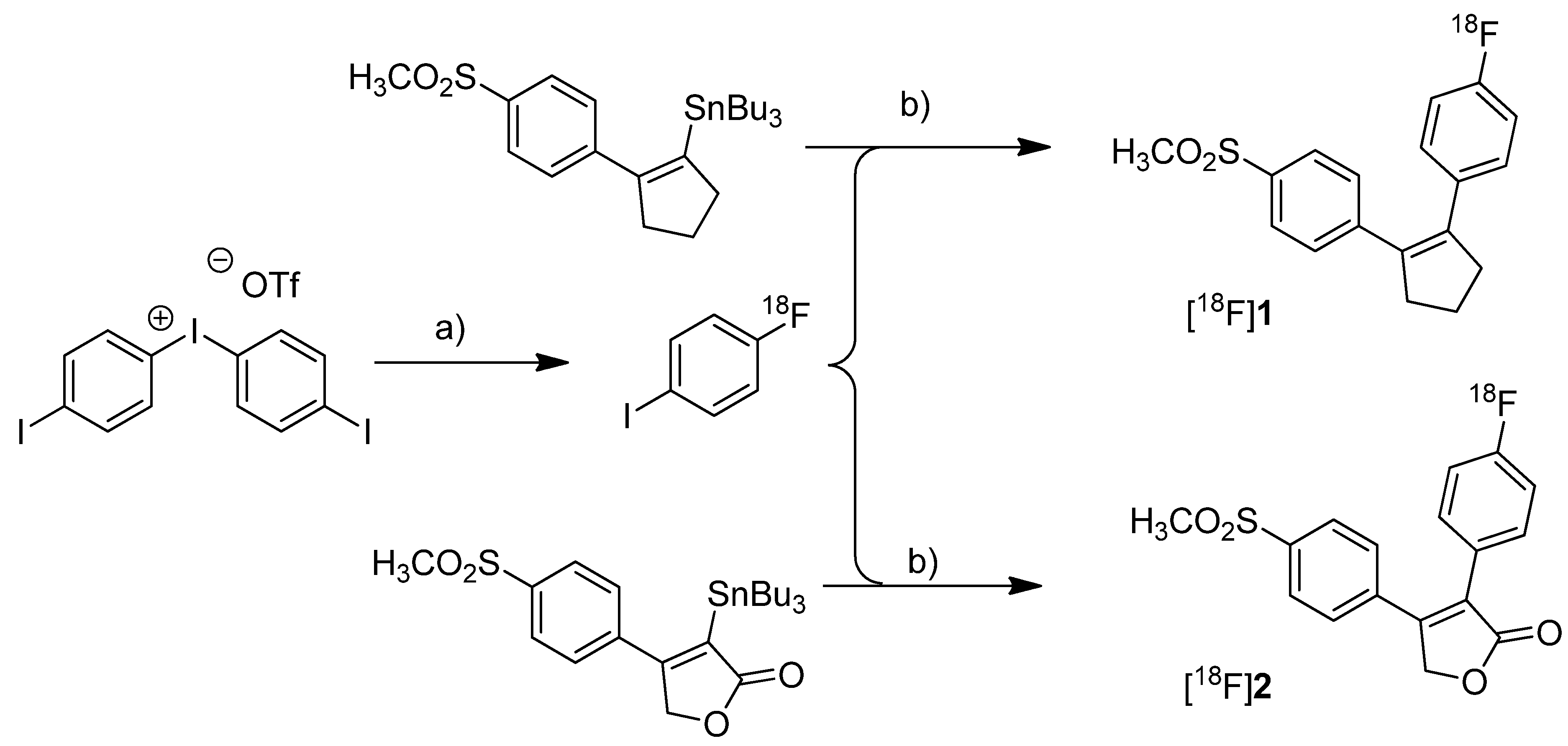
et al. presented the radiosynthesis of two COX-2 inhibitors, 1-[
18F]fluoro-4-(2-(4-methylsulfonyl)phenyl)cyclopent-1-enyl)benzene ([
18F]
1,
Scheme 1) and 3-(4-[
18F]fluorophenyl)-4-(4-(methylsulfonyl)phenyl)-5
H-furan-2-one ([
18F]
2,
Scheme 1), possessing a cyclopentene and a 2-(5
H)furanone ring as carbocyclic core [
41]. Compound
2 is a fluoro derivative of rofecoxib.
Scheme 1.
Synthesis of [18F]1 and [18F]2.
Scheme 1.
Synthesis of [18F]1 and [18F]2.
Reagents and conditions: (a) [18F]KF, K222, K2CO3, DMF, 120 °C; (b) different conditions, optimized: Pd2(dba)3/P(o-tolyl)3/CuI, DMF-toluene, 65 °C, 20 min.
Synthesis: The radiolabeling procedure was performed via Stille reaction. In detail, the tributyl stannyl precursor was reacted with 4-[18F]fluoroiodobenzene which was obtained from an iodonium salt by reaction with [18F]fluoride. Using Pd2(dba)3/P(o-tolyl)3/CuI as catalyst system in DMF/toluene at 65 °C for 20 min, the Stille cross-coupling gave compound [18F]1 and [18F]2 in an optimized radiochemical yield of 68% and 93% as determined by HPLC, respectively. In vitro/In vivo: Further information about in vitro or in vivo evaluation of the radiotracers was not given.
Toyokuni
et al. presented the synthesis of 4-(5-[
18F]fluoromethyl-3-phenylisoxazol-4-yl)-benzenesulfonamide ([
18F]
3,
Scheme 2), a [
18F]fluorinated analog of valdecoxib, and the results of a preliminary evaluation by PET [
33].
Scheme 2.
Synthesis of [18F]3.
Scheme 2.
Synthesis of [18F]3.
Reagents and conditions: (a) [18F]KF,K222, K2CO3, MeCN, 90 °C, 5 min; (b) 40% TCA in MeCN, 90 °C, 5 min.
Synthesis: The radiosynthesis started from a 4,4'-dimethoxytrityl-sulfonamide protected O-tosylated precursor and was performed by [18F]fluoride-for-tosylate exchange followed by acidic deprotection and purification by preparative HPLC. This gave chemically and radiochemically pure compound [18F]3 in 40% decay corrected radiochemical yield within 120 min after end of bombardment with a specific activity of about 74 GBq/µmol. In vitro: In vitro experiments for this tracer were not reported. In vivo: Instead, a preliminary small animal PET study using a normal mouse as well as a PET study using a vervet monkey was performed. The monkey whole-body biodistribution data allowed a dosimetry calculation for a 70 kg adult and an injection of 370 MBq [18F]3. This revealed that the maximum absorbed dose found in the urinary bladder would be below the recommended dose limitation ratio. Finally, further detailed evaluation of the radiotracer was hampered due to substantial de[18F]fluorination which was observed in mouse as well as in monkey by fluorine-18-activity enrichment in the skeleton after 15 min and 45 min, respectively.
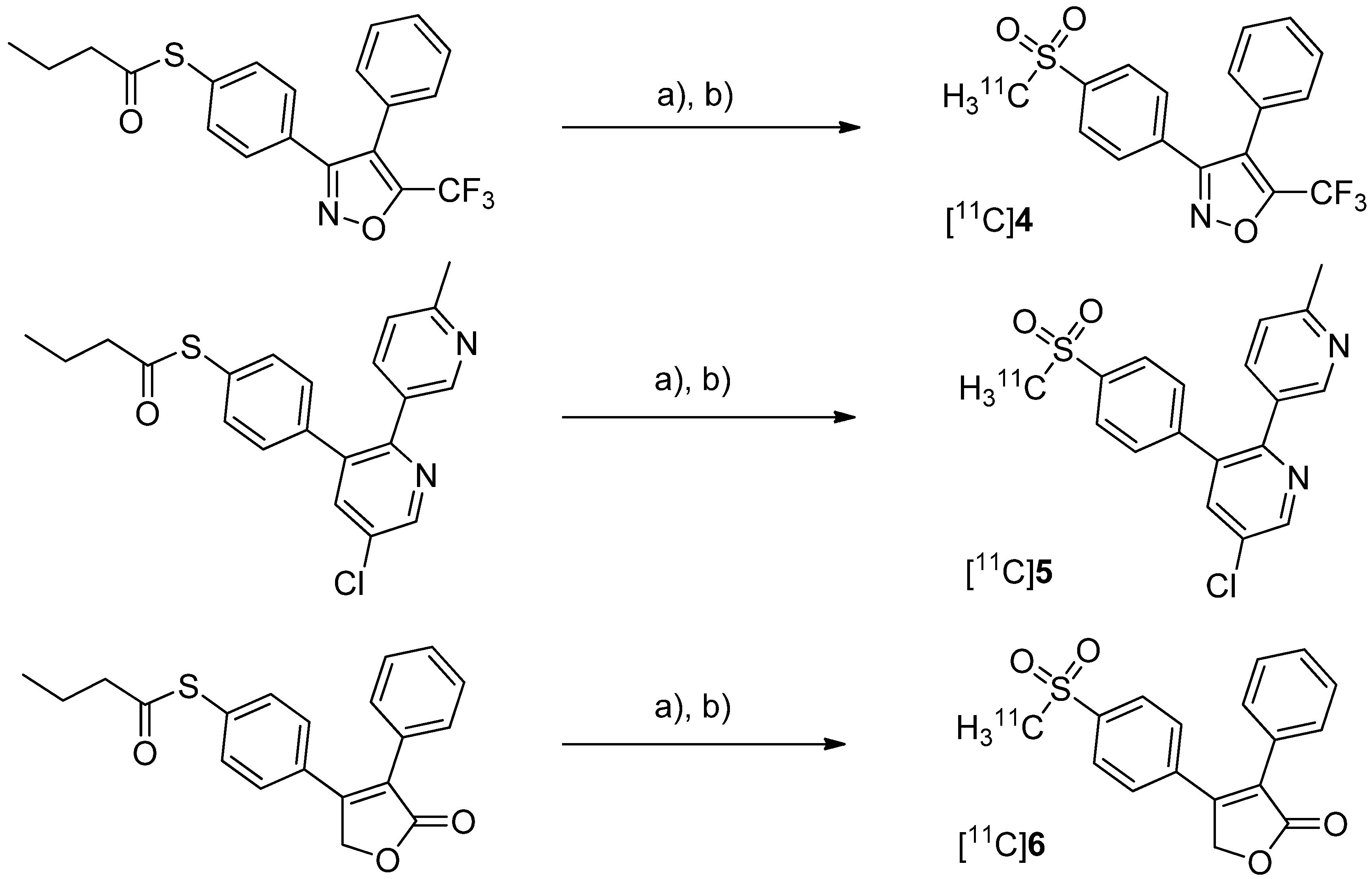
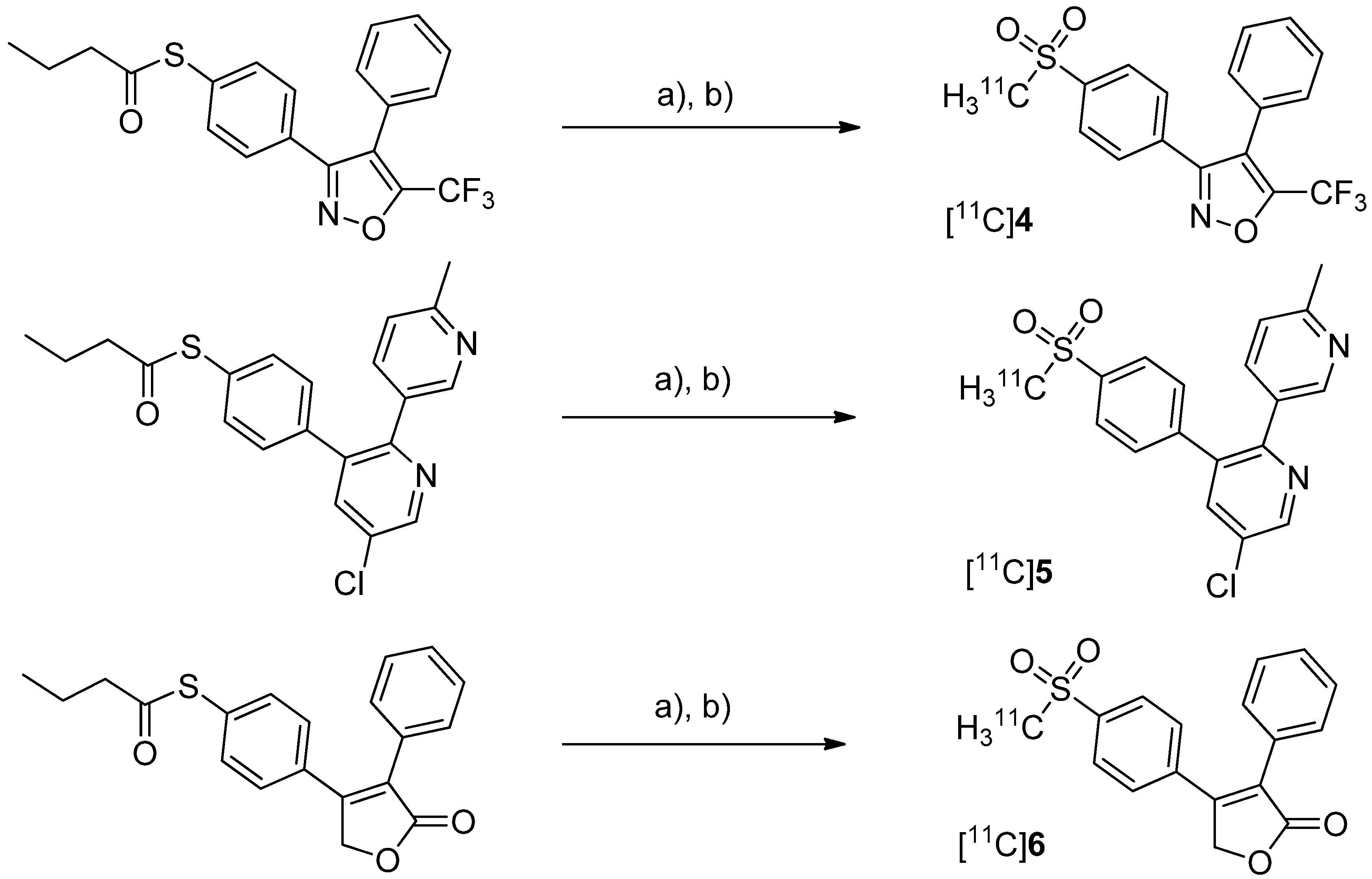
After having reported the synthesis of 3-(4-([
11C]methylsulfonyl)phenyl)-4-phenyl-5-(trifluoromethyl)isoxazole ([
11C]
4,
Scheme 3) [
47] and the synthesis of the celecoxib analog 4-(5-(4-[
11C]methylphenyl)-3-(trifluoromethyl)-1
H-pyrazol-1-yl)benzenesulfonamide ([
11C]
7,
Scheme 4) [
60] in conference contributions before, a detailed description of both radiotracers followed in 2005.
Scheme 3.
Synthesis of [11C]4–6.
Scheme 3.
Synthesis of [11C]4–6.
Reagents and conditions: (a) [11C]CH3I, TBAOH for [11C]4 and [11C]5 pyrrolidine for [11C]6, THF, r.t., 5 min; (b) Oxone®, MeOH/H2O (1:1), 70 °C and 2 min for [11C]4 and [11C]6, 75 °C and 3 min for [11C]5.
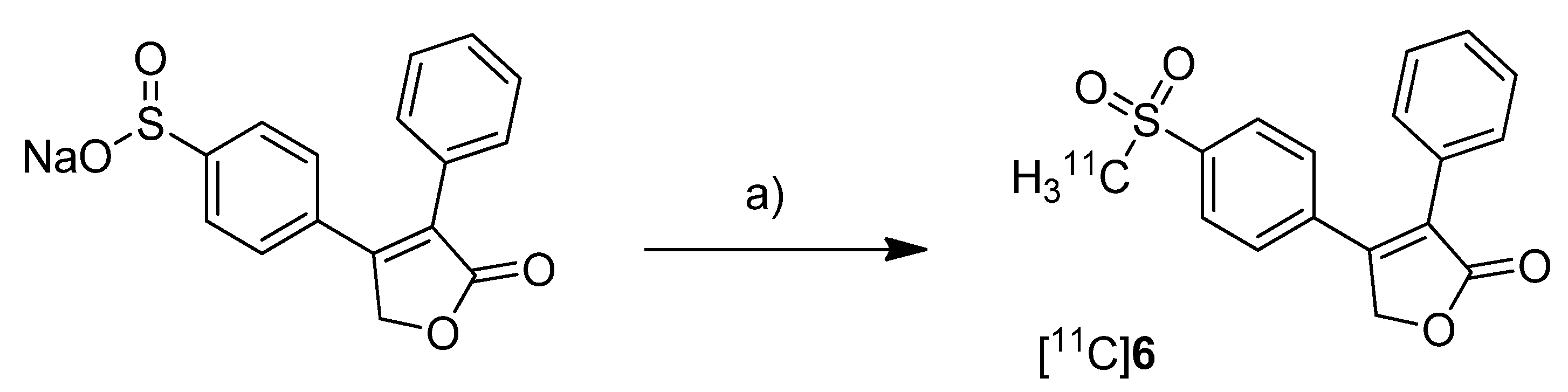
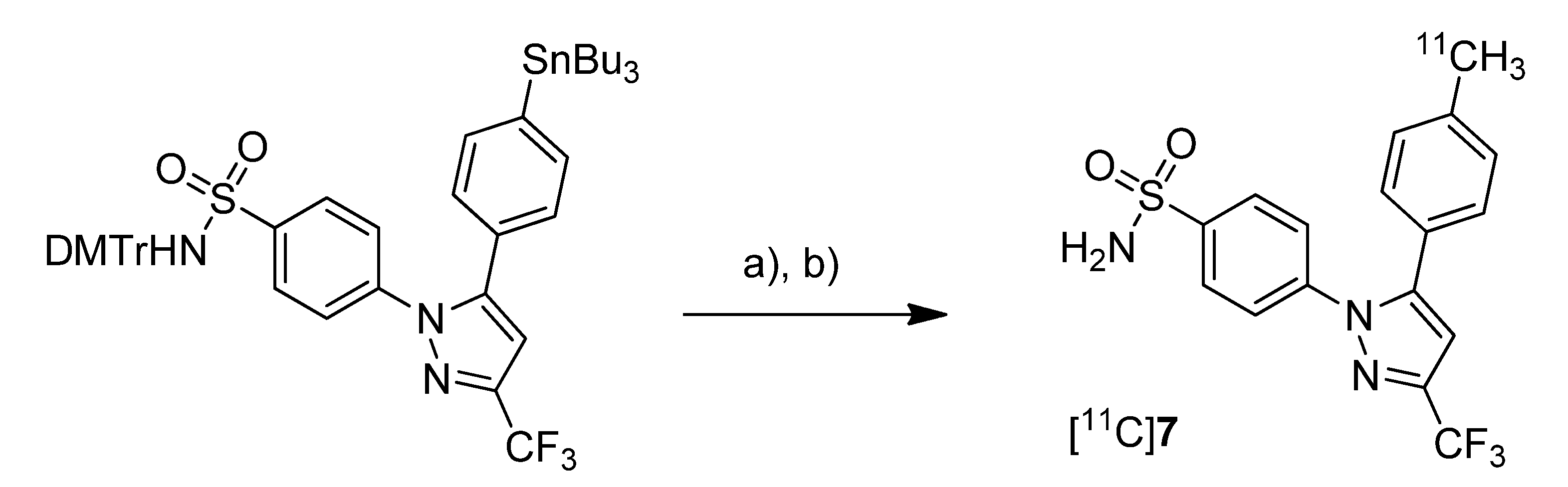
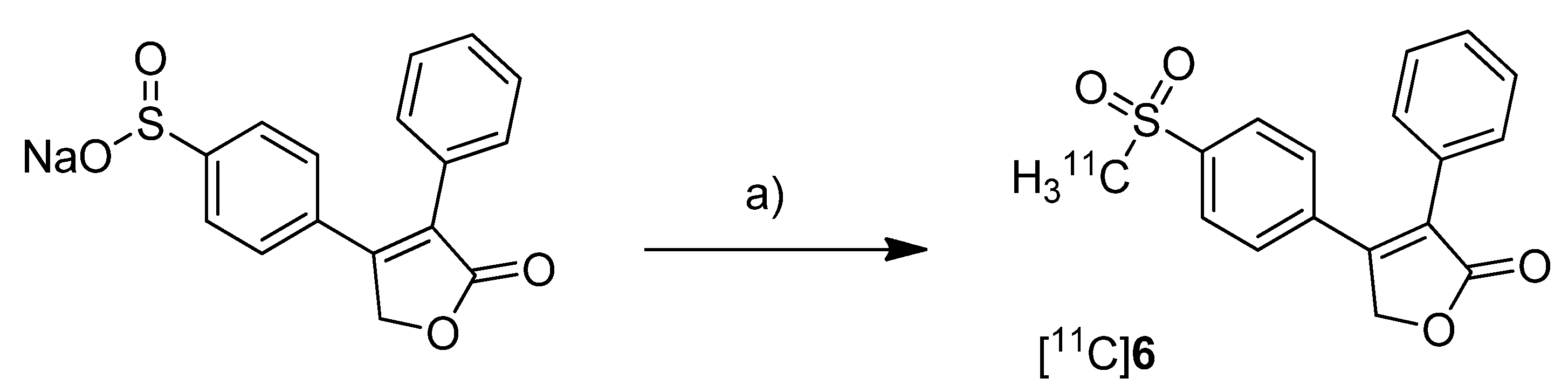
Majo
et al. presented the synthesis of three carbon-11-labeled COX-2 inhibitors thus exemplifying a new strategy for the synthesis of aryl [
11C]methylsulfones [
42]. 5-chloro-6'-methyl-3-(4-([
11C]methylsulfonyl)phenyl)-2,3'-bipyridine ([
11C]
5,
Scheme 3) and 4-(4-([
11C]methylsulfonyl)-phenyl)-3-phenylfuran-2(5
H)-one ([
11C]
6,
Scheme 3) are the isotopically labeled analogs of eterocoxib and rofecoxib.
Synthesis: The synthesis of all three compounds started with a thiobutyrate ester precursor which was converted into the radiolabeled COX-2 inhibitor by unraveling the free thiol under basic conditions followed by
S-[
11C]methylation and final oxidation of the thiomethyl ether to the methylsulfonyl moiety. In case of compound [
11C]
4 and [
11C]
5, tetrabutylammonium hydroxide in THF was used for the cleavage of the thiobutyrate ester. This failed for the synthesis of [
11C]
6 due to concomitant cleavage of the γ-butyrolactone ring. Therefore, in case of compound [
11C]
6 an excess of pyrrolidine in DMF was successfully used instead. This strategy provided compounds [
11C]
4, [
11C]
5 and [
11C]
6 after semi-preparative HPLC in a total synthesis time of 30 min and a decay corrected radiochemical yield of 37%, 28%, and 20% (based on [
11C]methyl iodide), respectively. The specific activity of [
11C]
4 was determined to be 74 ± 9.25 GBq/µmol.
In vitro/In vivo: Unfortunately, no further
in vitro or
in vivo evaluations have been presented in this work. [
11C]
6 was synthesized and evaluated by de Vries
et al. in 2008 [
29].
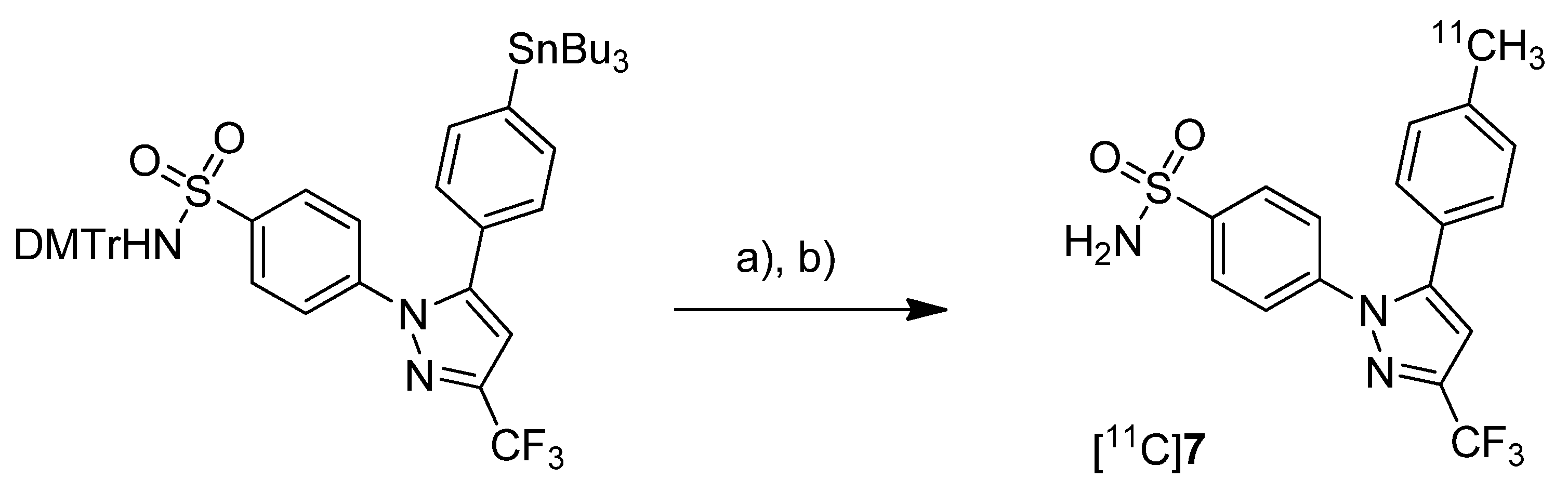
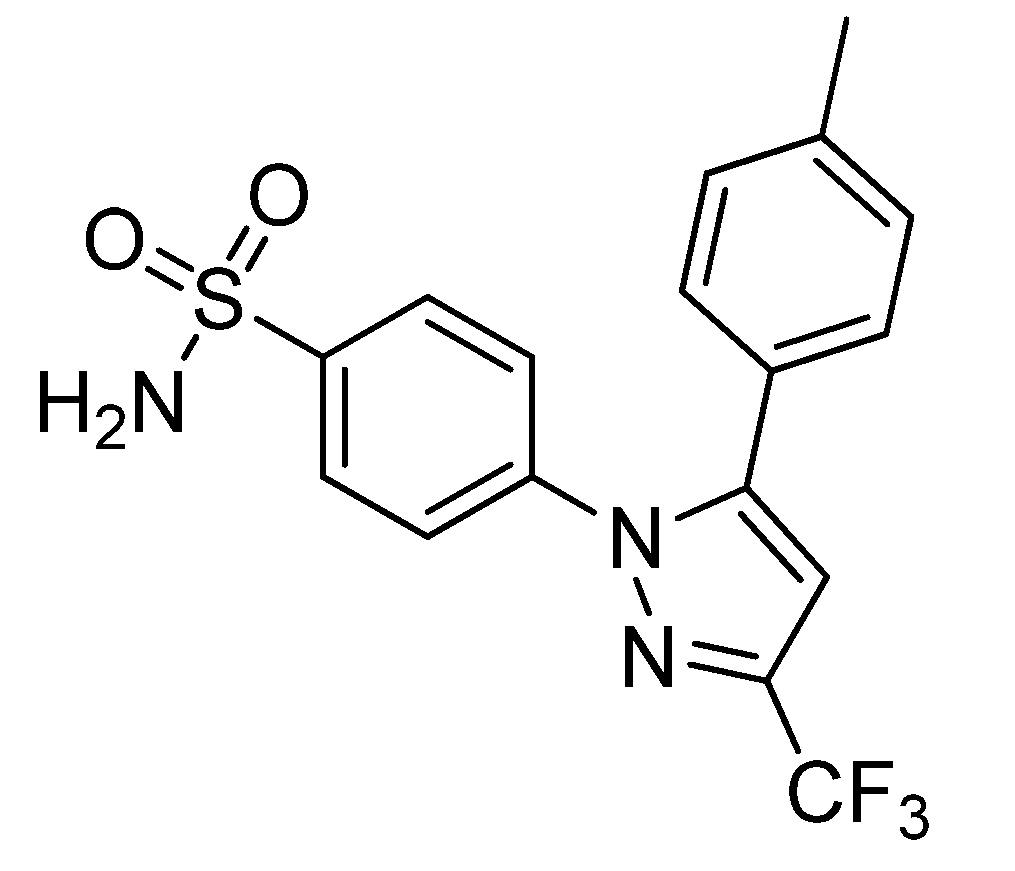
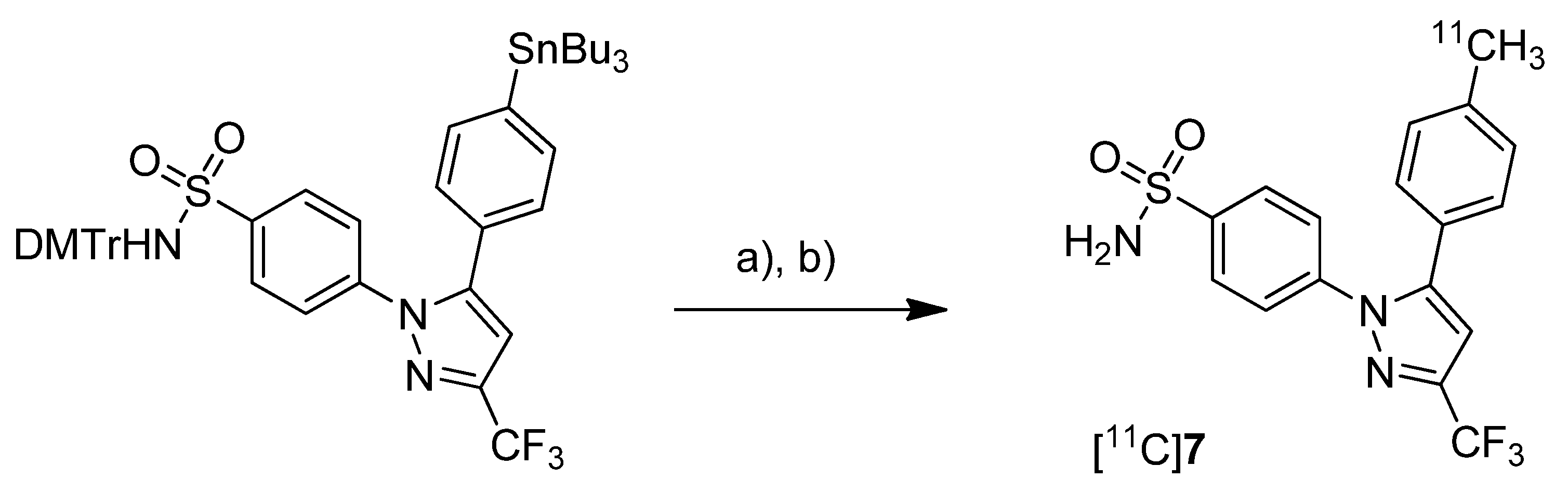
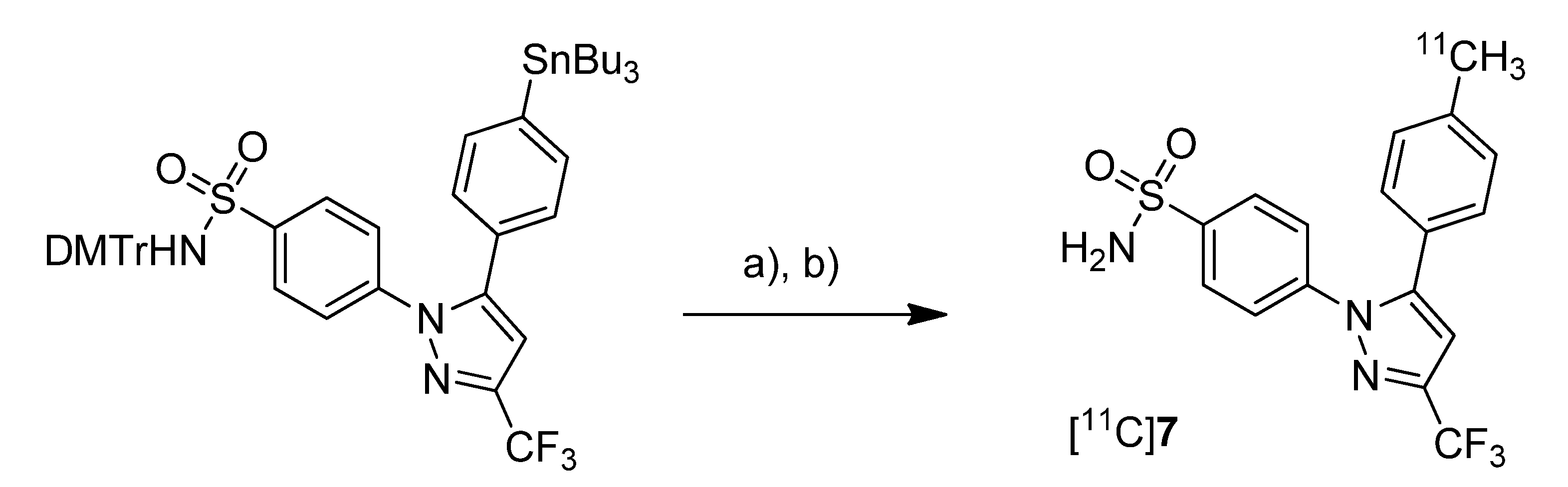
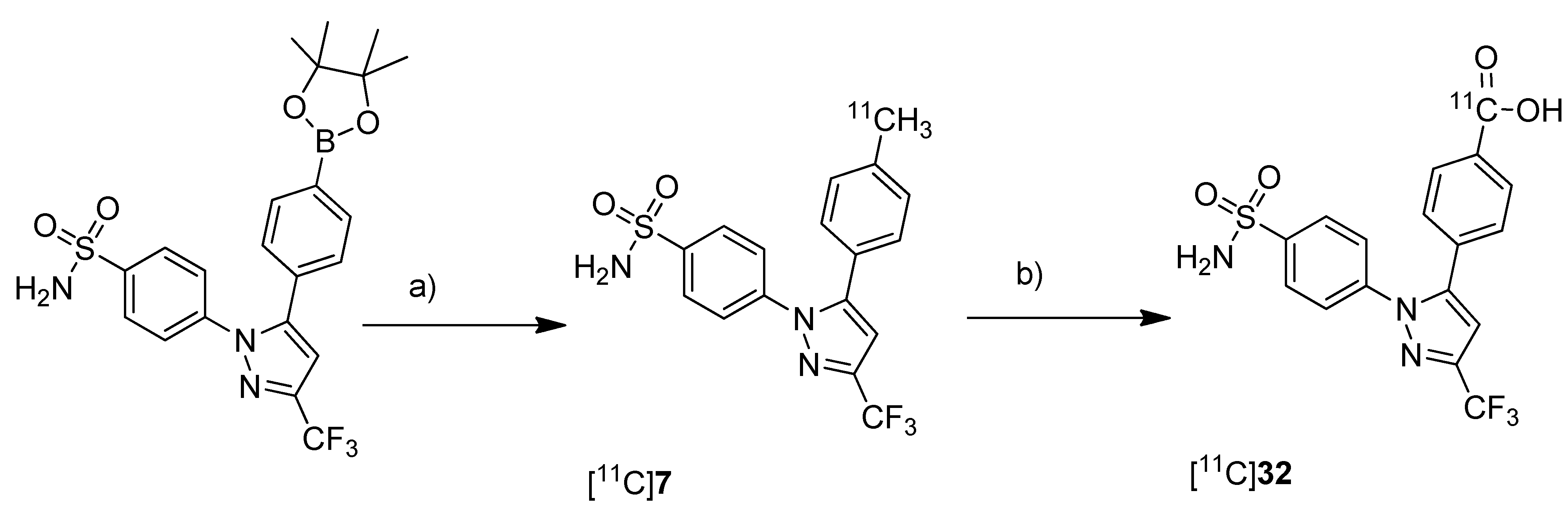
Prabhakaran
et al. reported the synthesis of 4-(5-(4-[
11C]methylphenyl)-3-(trifluoromethyl)-1
H-pyrazol-1-yl)benzenesulfonamide ([
11C]
7,
Scheme 4), the isotopically labeled carbon-11 analog of celecoxib [
61].
Scheme 4.
Synthesis of [11C]7.
Scheme 4.
Synthesis of [11C]7.
Reagents and conditions: (a) Pd2(dba)3, P(o-tolyl)3, [11C]CH3I, DMF, 135 °C, 4 min; (b) 20% TFA in DCM, 60 °C, 5 min.
Synthesis: The synthesis towards the [11C]methyl substituted 4-tolyl moiety of [11C]7 started from a stannylated and DMT-sulfonamide protected precursor by C-[11C]methylation and final acidic deprotection. The initial labeling reaction was achieved by trapping [11C]methyl iodide in an argon purged Pd2(dba)3/P(o-tolyl)3 solution of DMF, addition of the precursor and subsequent reaction at 135 °C for 4 min. The following deprotection with 20% trifluoroacetic acid (TFA) in dichloromethane and RP-HPLC purification gave compound [11C]7 in 8 ± 2% (n = 6) decay corrected radiochemical yield (based on [11C]methyl iodide) with a specific activity of 39.96 ± 6.66 GBq/µmol and a radiochemical purity >99%. For transmetallation of the tin precursor it was found to be essential to use reagents of high purity. The authors also pointed to the need to detect impurities of tin and palladium which were estimated to be less than 39 and 31 ppb using atomic absorption spectroscopy. In vitro/In vivo: Also in this work further in vitro or in vivo evaluation was not presented.
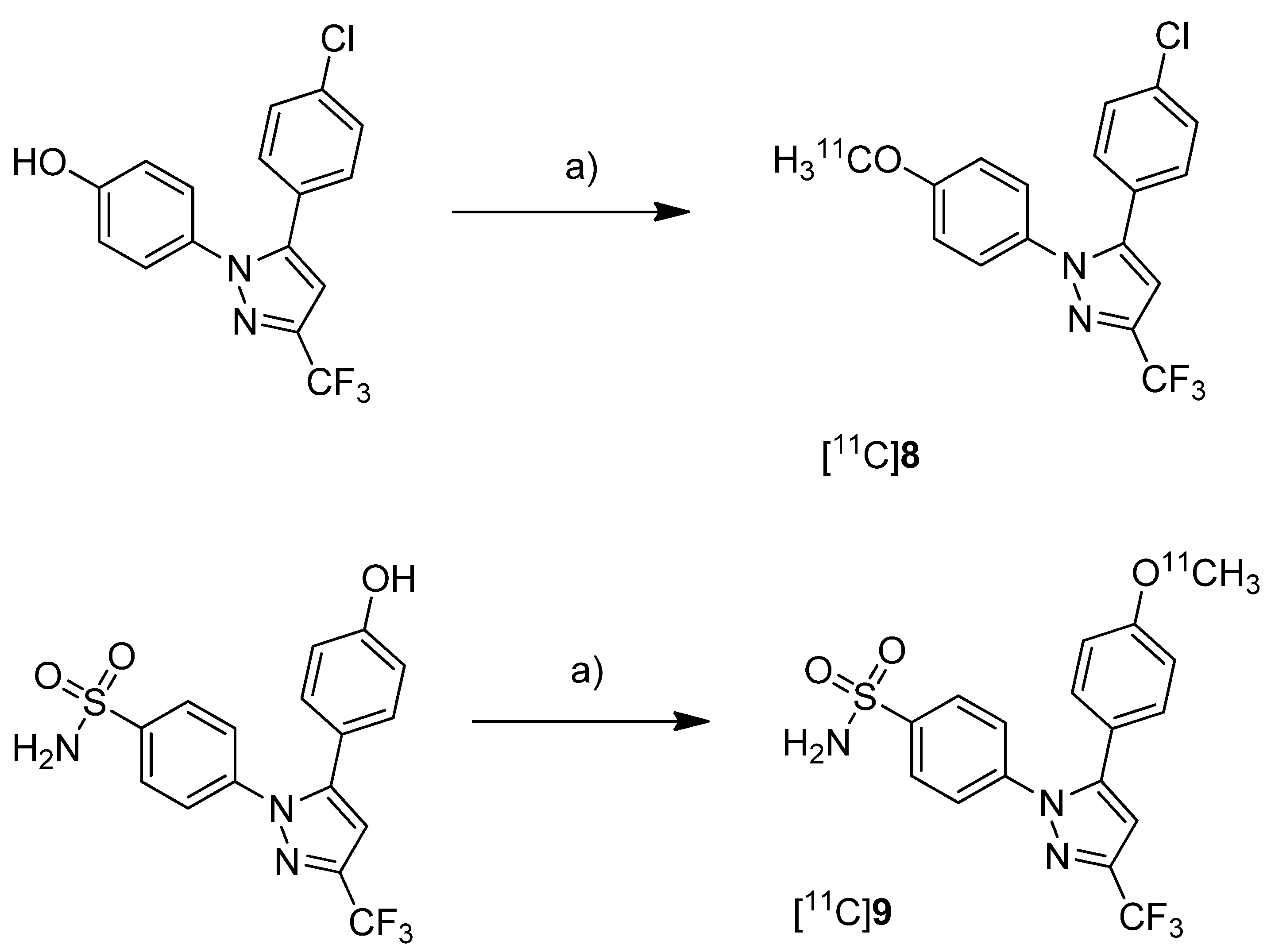
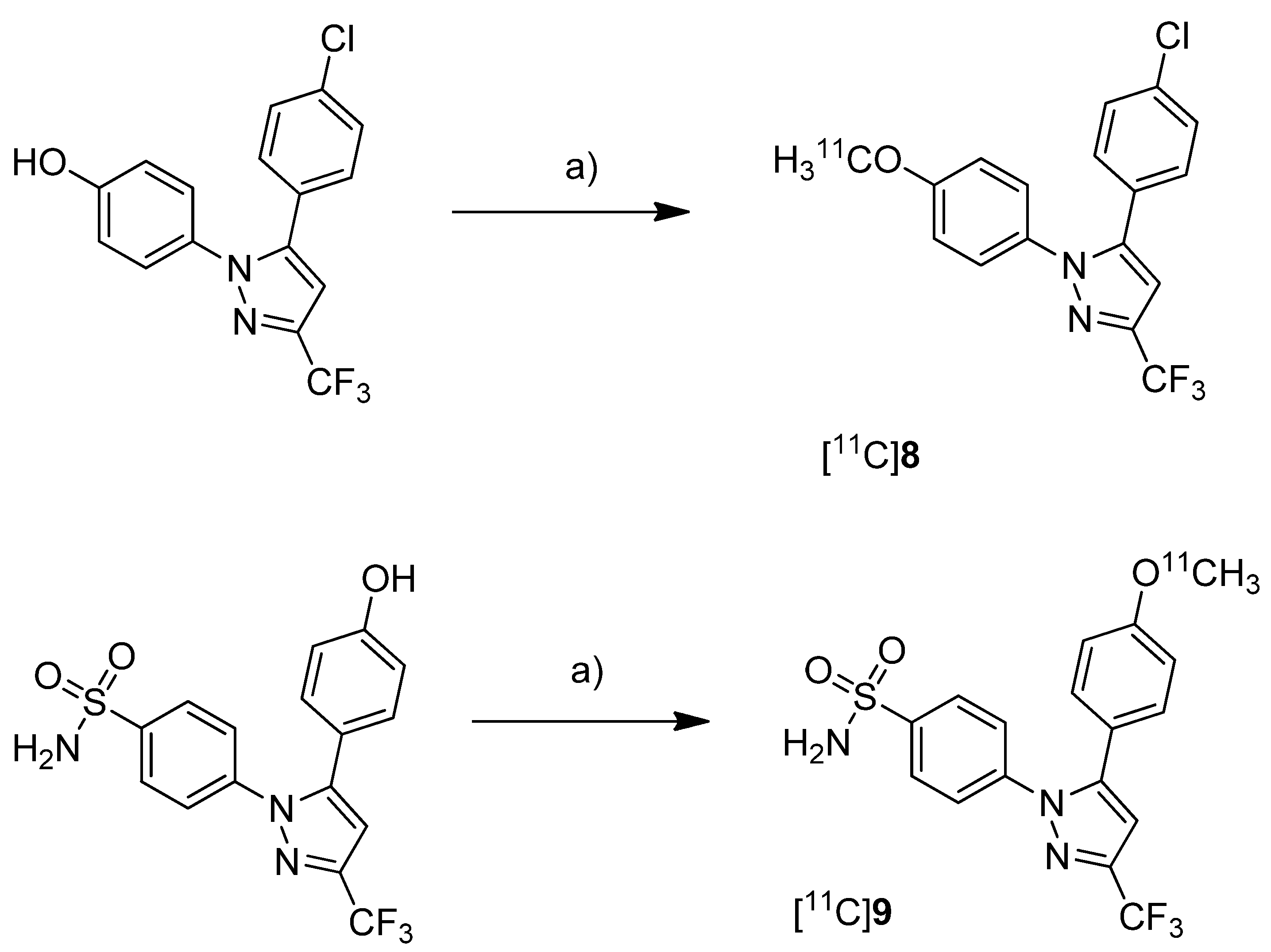
Fujisaki
et al. reported the radiosynthesis of 5-(4-chlorophenyl)-1-(4-[
11C]methoxyphenyl)-3-(trifluoromethyl)-1
H-pyrazole ([
11C]
8,
Scheme 5), a COX-1 selective inhibitor (IC
50(COX-1) = 9 nM; IC
50(COX-2) = 6.3 µM [
62]), and 4-(5-(4-[
11C]methoxyphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzenesulfonamide ([
11C]
9,
Scheme 5) a [
11C]methoxy analog of celecoxib (IC
50(COX-1) = 2.6 µM; IC
50(COX-2) = 8 nM [
62]) [
51]. Both compounds were evaluated
in vivo and
ex vivo. Synthesis: The radiolabeling procedure was similar for both compounds and started each time from the phenolic hydroxy precursor. The tracers were synthesized by
O-[
11C]methylation and the reaction conditions were optimized using [
11C]methyl iodide as well as [
11C]methyl triflate. The best results were obtained by using [
11C]methyl triflate and an equimolar amount of base with respect to the hydroxy precursor in acetone. This protocol yielded [
11C]
8 and [
11C]
9 in 74.5% and 46.6% decay corrected radiochemical yield (based on [
11C]methyl triflate) with a specific activity of 71–125 GBq/µmol and 27–61 GBq/µmol, respectively, and radiochemical purity >99% after 20 min total synthesis time. The log
P7.4 was determined to be 3.46 for [
11C]
8 and 3.06 for [
11C]
9.
In vitro:
In vitro experiments for these tracers were not reported.
In vivo/
Ex vivo: For
in vivo experiments male Donryu rats were used bearing subcutaneously an AH109A ascetic hepatoma cell xenograft tumor. COX-2 expression of the AH109A tumor cells was confirmed by Western blot analysis.
In vivo experiments revealed for [
11C]
8 and [
11C]
9 a high extend of non-specific binding. The COX-2 selective inhibitor [
11C]
9 showed high levels of radioactivity in the blood, a low uptake into the AH109A tumor and the brain and a much slower blood clearance in comparison to [
11C]
8. Metabolite analysis revealed that >93% of each compound remained unchanged in the brain and in the AH109A tissue after 30 min. The blocking studies by coinjection of [
11C]
9 with 8 or indomethacin as well as carrier-loading did not lower the uptake in any tissues except spleen and blood in case of carrier loading. In contrast, in few tissues radioactivity levels even increased. Although
ex vivo autoradiograms of rat brain showed regional differences in tracer distributions similar to the expression pattern of cyclooxygenases a selective blocking could not be shown.
Scheme 5.
Synthesis of [11C]8 and [11C]9.
Scheme 5.
Synthesis of [11C]8 and [11C]9.
Reagents and conditions: (a) different conditions, optimized: [11C]CH3OTf, aq. NaOH, acetone, r.t., 2 min.
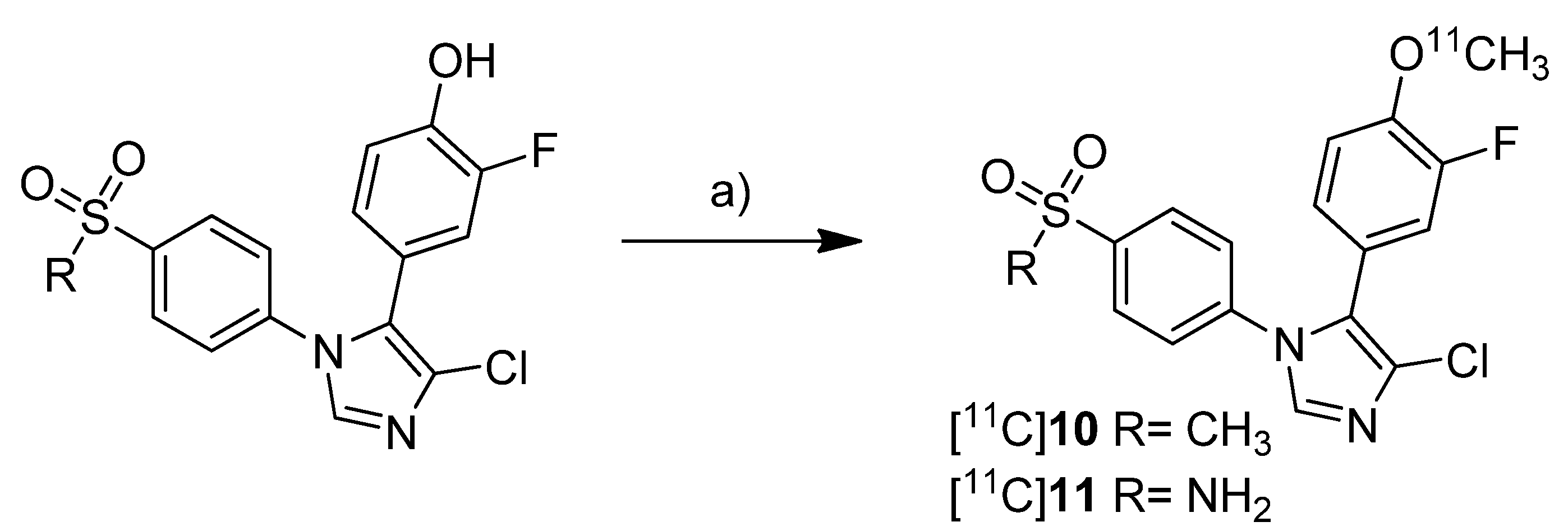
Together with the synthesis of [
11C]
8–
9, Kawamura
et al. presented the synthesis of [
11C]
10-11 (
Scheme 6) as part of a conference contribution [
63]. 4-Chloro-5-(3-fluoro-4-methoxyphenyl)-1-(4-(methylsulfonyl)phenyl)-1
H-imidazole (
10) and 4-(4-chloro-5-(3-fluoro-4-methoxyphenyl)-1
H-imidazol-1-yl)benzenesulfonamide (
11) were described as selective COX-2 inhibitors with IC
50(COX-2) of 4 nM and IC
50(COX-1) >10 µM for
10 and 5 nM and 3.3 µM for
11, respectively [
64].
Scheme 6.
Synthesis of [11C]10–11.
Scheme 6.
Synthesis of [11C]10–11.
Reagents and conditions: (a) different conditions, optimized: [11C]CH3OTf, aq. NaOH, acetone, r.t. or 120 °C, 1 min.
The reported reaction conditions [
51] were used that gave [
11C]
10 in 64.8–78.2% (n = 4) and [
11C]
11 in 66.1–74.1% (n = 4) decay corrected radiochemical yield (based on [
11C]methyl triflate).
2006
In 2006, the synthesis of one fluorine-18-labeled and three carbon-11-labeled COX-2 inhibitors was reported whereby the latter were investigated in vivo.
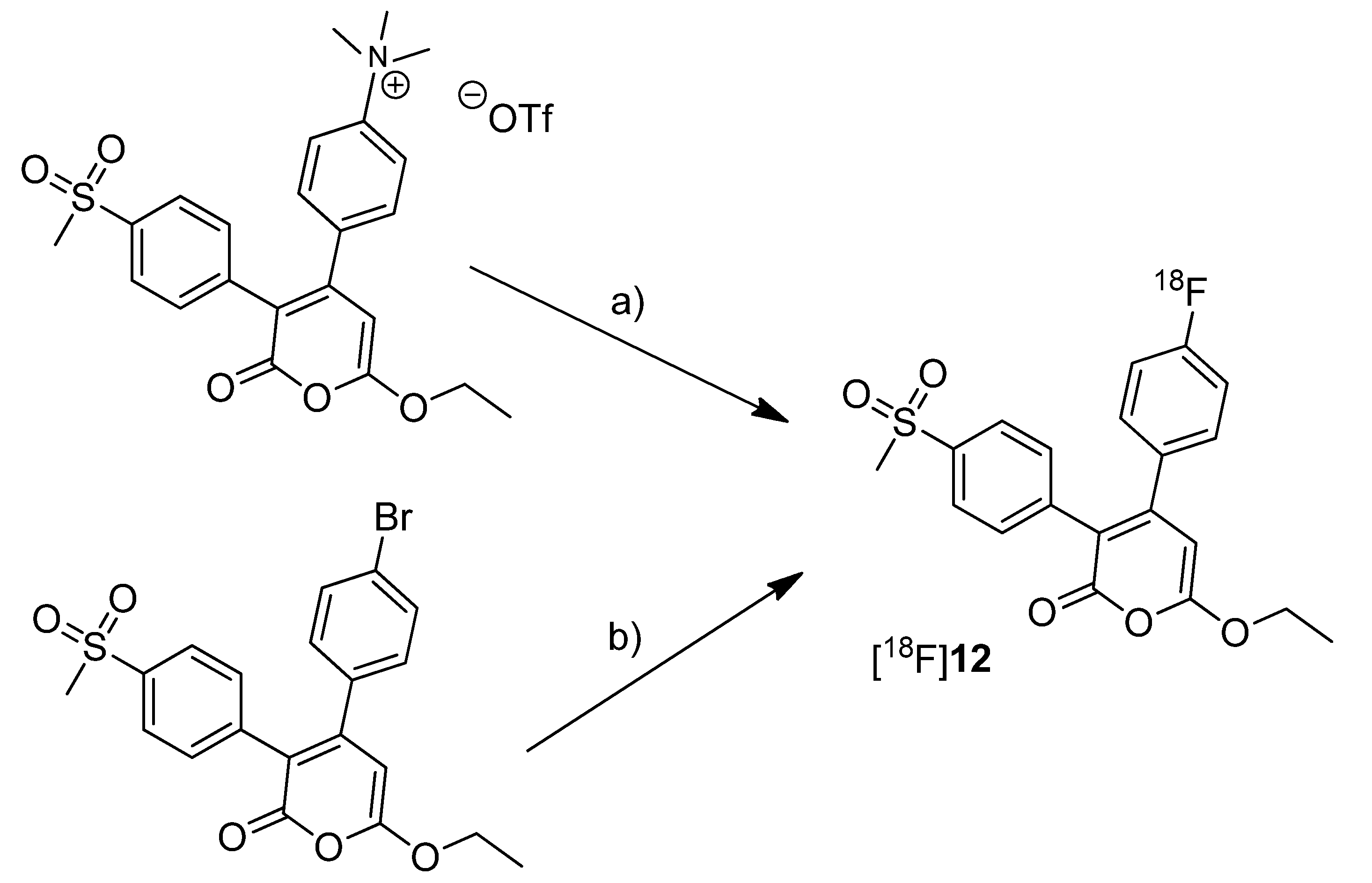
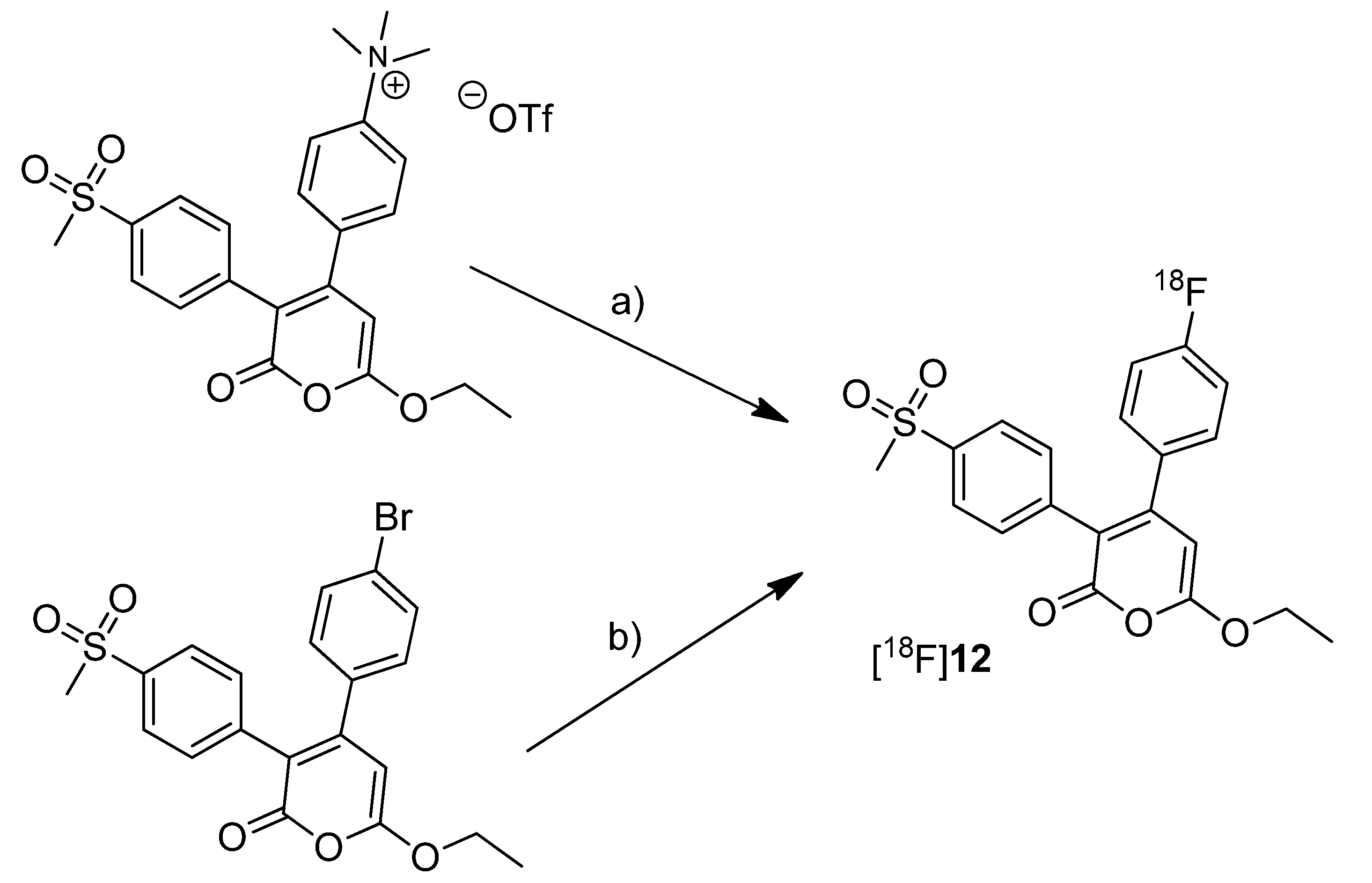
Tian and Lee reported the synthesis of the fluorine-18-labeled COX-2 inhibitor 6-ethoxy-4-(4-[
18F]fluorophenyl)-3-(4-(methylsulfonyl)phenyl)-2
H-pyran-2-one ([
18F]
12,
Scheme 7) (IC
50(COX-2) = 0.1 µM; SI = IC
50(COX-2)/IC
50(COX-1) = 2880 [
65]) [
28].
Scheme 7.
Synthesis of [18F]12.
Scheme 7.
Synthesis of [18F]12.
Reagents and conditions: (a) [18F]KF,K222, K2CO3, MeCN, 130 °C, 20 min; (b) [18F]KF,K222, K2CO3, DMSO, 160 °C, 30 min.
Synthesis: The radiosynthesis was performed starting from the trimethylammonium phenyl precursor as well as the bromo phenyl precursor by nucleophilic substitution with [18F]fluoride. For the trimethylammonium precursor, the optimized labeling condition was determined to be 130 °C in acetonitrile followed by semi-preparative HPLC that yielded [18F]12 in 14.6 ± 3.3% decay corrected yield with a specific activity of 18.02 ± 3.15 GBq/µmol after 60–70 min total synthesis time. Radiofluorination of the bromo phenyl precursor was performed at 160 °C in DMSO but this method gave the radiotracer only in 4% radiochemical yield and with low specific activity due to troublesome separation of the brominated precursor from the [18F]fluorinated product by semi-preparative HPLC. In vitro/In vivo: The in vitro or in vivo evaluation of [18F]12 was not presented.
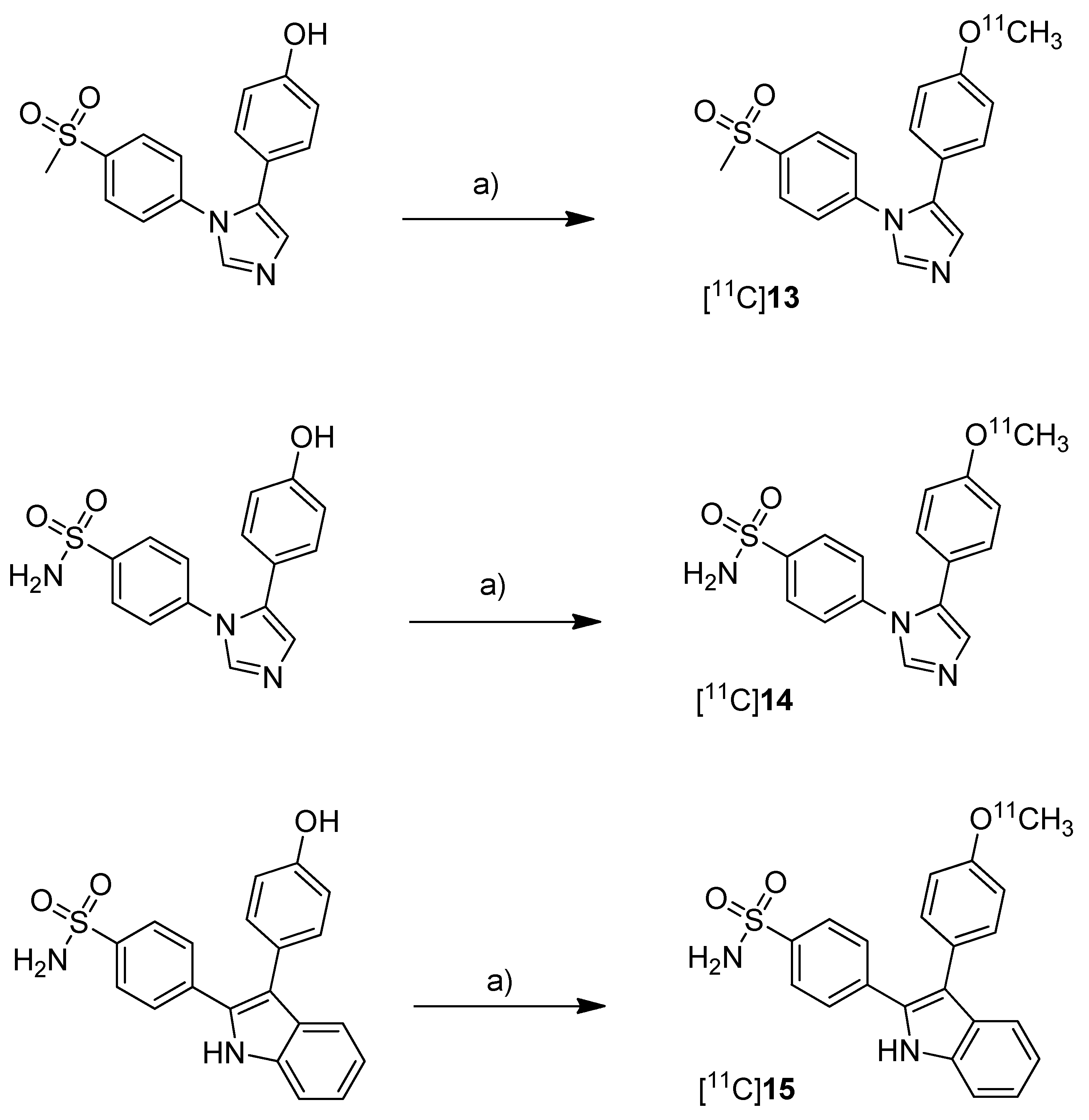
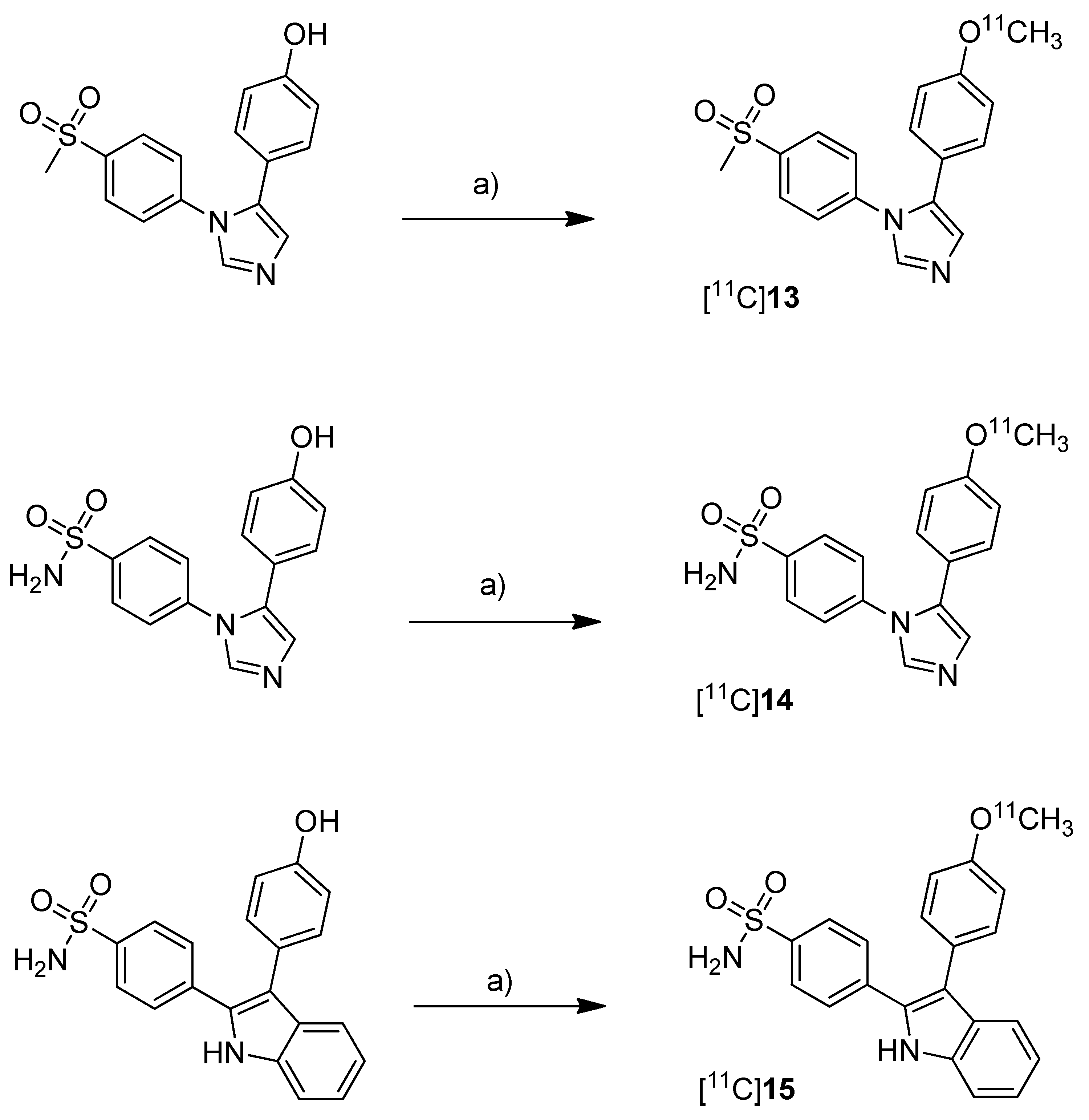
One year after the synthesis of [
11C]
9, Tanaka and coworkers published their results regarding the synthesis of three COX-2 inhibitors labeled with carbon-11 and the
in vitro,
in vivo and
ex vivo evaluation [
43]. Non-radioactive analogs were previously reported to have high affinity and to be highly selective towards COX-2 with IC
50(COX-2) and IC
50(COX-1) of 4 nM and > 10 µM for 5-(4-methoxyphenyl)-1-(4-(methylsulfonyl)phenyl)-1
H-imidazole (
13,
Scheme 8) [
64], 5 nM and > 3.3 µM for 4-(5-(4-methoxyphenyl)-1
H-imidazol-1-yl)benzenesulfonamide (
14,
Scheme 8) [
64] and 0.006 nM and > 10 µM for 4-(3-(4-methoxyphenyl)-1
H-indol-2-yl)benzenesulfonamide (
15,
Scheme 8) [
66].
Scheme 8.
Synthesis of [11C]13–15.
Scheme 8.
Synthesis of [11C]13–15.
Reagents and conditions: (a) different conditions: optimized: [11C]CH3OTf, aq. NaOH, acetone, r.t., 1 min.
Synthesis: The radiolabeling procedure started for all three compounds from the phenolic hydroxy precursor by O-[
11C]methylation. Using [
11C]methyl triflate in acetone with an equimolar amount of NaOH as the base and subsequent purification with semi-preparative HPLC gave [
11C]
13, [
11C]
14 and [
11C]
15 in decay corrected radiochemical yields (based on [
11C]methyl triflate) of 75.1% (n = 1),66.1–74.1% (n = 3) and 17.5–25.1% (n = 3), respectively, with a specific activity of 36–264 GBq/µmol within 20 min total preparation time. The log P
7.4 values were found to be between 1.89 and 2.33.
In vivo/Ex vivo: AH109A tumor bearing mice as described by Fujisaki et al. [
51] were used for the
in vivo experiments. The tissue distribution studies revealed that all three compounds showed the highest uptake in the liver. Some differences of the single radiotracers in blood uptake and organ distribution were found but there was no COX-specific binding observed in COX-2 expressing tissues such as brain, kidney, heart and lung, which was confirmed by blocking experiments. Also the tumor uptake was low showing no COX-specific blocking effects. Regional differences in brain uptake of [
11C]
13 were not further investigated. In contrast to compound [
11C]
13 and [
11C]
14 which were detected more or less unchanged in plasma and AH109A tumor tissue with more than 90% after 30 min, [
11C]
15 was found to be relatively unstable. 54% and 80% of original [
11C]
15 was detected after 30 min in plasma and in the AH109A tumor, respectively. Interestingly, [
11C]
14 and [
11C]
15 showed only very low uptake in the brain although lipophilicity would allow crossing the blood-brain barrier as discussed by the authors.
In vitro: For this, there were performed transcellular transport studies using human cells overexpressing the multidrug resistance protein 1 or permeability glycoprotein 1 (P-gp) and the P-gp inhibitor cyclosporine A (CsA). Although compound [
11C]
15 was shown to be an
in vitro substrate of P-gp which is responsible for the efflux of different drugs out of the brain, inhibition of P-gp
in vivo did not alter the brain uptake, a behavior that still needs to be elucidated.
2008
In 2008, the synthesis and
in vivo evaluation of further three carbon-11-labeled COX-2 inhibitors was described. Five years after the synthesis of [
18F]DuP-697, de Vries
et al. presented a new synthesis of [
11C]
6 (
Scheme 10) the isotopically labeled analog of rofecoxib, and its
in vivo evaluation because this was not performed yet [
29]. Because rofecoxib was shown to penetrate the brain resulting in high brain concentrations compared to other COXIBs, [
11C]
6 was initially evaluated as a brain tracer for COX-2 expression.
Scheme 10.
Synthesis of [11C]6.
Scheme 10.
Synthesis of [11C]6.
Reagents and conditions: (a) [11C]CH3I, DMF, 90 °C, 4 min.
Synthesis: The radiosynthesis of [
11C]
6 was performed starting from a different precursor than Majo
et al. [
42], the sulfinate, by reaction with [
11C]methyl iodide in DMF at 90 °C for 4 min and subsequent semi-preparative HPLC purification. This yielded [
11C]
6 in > 99% radiochemical purity within 40 min in 57 ± 9% (n = 23) decay corrected radiochemical yield (based on [
11C]methyl iodide) and a specific activity of 14 ± 8 GBq/µmol.
In vivo/
Ex vivo: Male Wistar rats were used for the evaluation of [
11C]
6. The distribution in the brain was determined with and without blocking by the COX-2 selective inhibitor
N-[2-(cyclohexyloxy)-4-nitrophenyl]methanesulfonamide (NS398). Interestingly, [
11C]
6 appeared to bind specifically to brain regions which were known to possess highest expression levels of COX-2. Furthermore, heterogeneous uptake in the brain was not observed after NS398 administration. The brain distribution of [
11C]
6 was measured also in herpes simplex virus infected animals which show intense inflammatory responses linked with high expression of COX-2 in the brain. This revealed a moderate but not significant increase of tracer uptake compared to healthy control animals. The rather homogenous uptake was confirmed by small animal PET imaging in parallel studies with [
11C]PK11195, a widely used PET tracer which targets microglia activation during neuroinflammation [
45], and corresponded to activated microglia. The uptake of [
11C]
6 in inflamed tissue was also investigated by a sterile inflammation model using turpentine injection into the muscle to induce inflammatory response. Here, also no enhanced tracer uptake into inflamed tissue was found and a blocking of uptake was only observed by using the nonselective COX inhibitor indomethacin but not by NS398. So finally, [
11C]
6 was not able to detect unambiguously the COX-2 expression in the tested inflammation models.
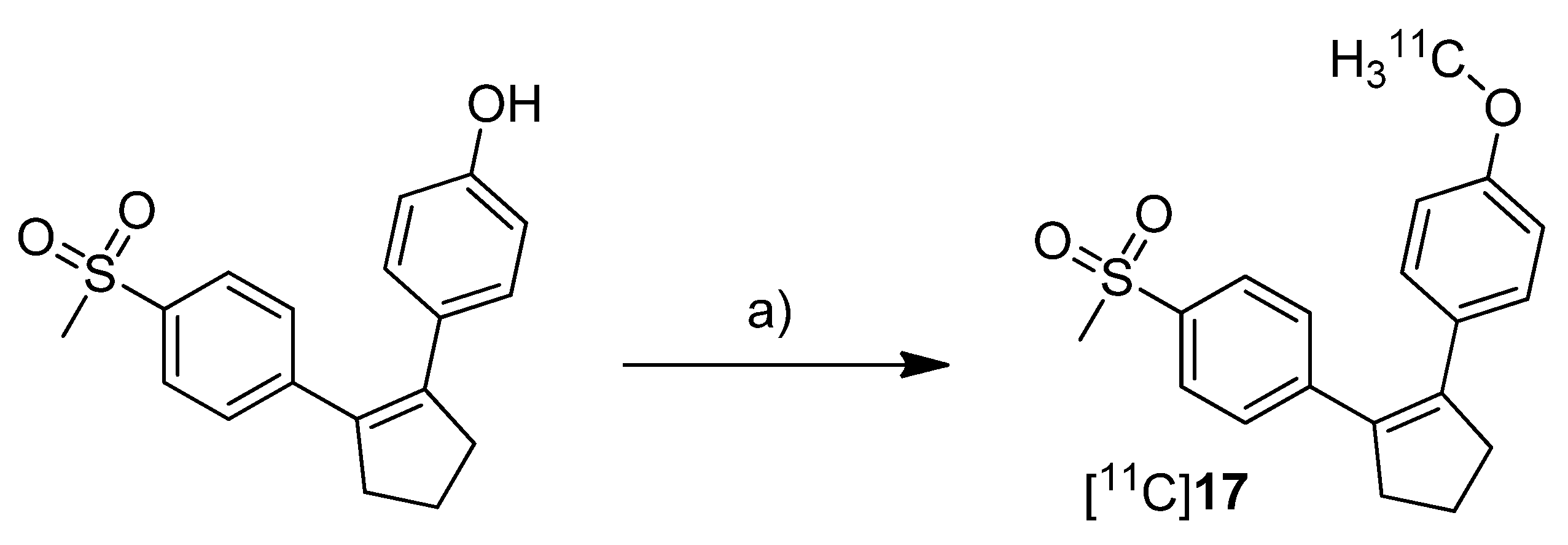
Three years after the synthesis of [
18F]
1 and [
18F]
2, Wuest
et al. presented the synthesis and the
in vitro and
in vivo evaluation of 1-[
11C]methoxy-4-(2-(4-(methylsulfonyl)phenyl)cyclopent-1-en-1-yl)benzene ([
11C]
17,
Scheme 11), a COX-2 selective inhibitor with an IC
50 value of 0.005 µM and 9.9 µM for COX-2 and COX-1, respectively [
30].
Scheme 11.
Synthesis of [11C]17.
Scheme 11.
Synthesis of [11C]17.
Reagents and conditions: (a) [11C]CH3I, NaOH, DMF, 60 °C, 3 min.
Synthesis: After [11C]methyl iodide was trapped in a mixture of DMF and 5 M NaOH at −20 °C the appropriate hydroxy precursor was radiolabeled by O-[11C]methylation at 60 °C for 3 min, purified by semi-preparative HPLC and formulated for the following experiments. In this way, [11C]17 was synthesized in 19% decay corrected radiochemical yield (based on [11C]carbon dioxide) with a specific activity of 20–25 GBq/µmol and a radiochemical purity > 95% within 35 min. In vitro: At first, [11C]17 was investigated in cell uptake studies with cell lines having a low baseline expression pattern of COX-2, namely unstimulated human (THP-1) and murine (RAW 264.7) monocytes/macrophages and quiescent murine fibroblasts (NIH 3T3). In a second experimental block cell lines with upregulated COX-2 expression were used, as like as human tumor cell lines (squamous cell carcinoma (FaDu) and colorectal adenocarcinoma (HT29)), mouse preadipocytes (3T3 L1) and 12-O-tetradecanoylphorbol-13-acetate (TPA)-stimulated THP-1 and RAW 264.7 cells. In these studies, [11C]17 showed specific uptake in COX-2 expressing cells which could also be blocked by pre-incubation with the non-radioactive reference. In vivo: Biodistribution studies in Wistar rats revealed a high uptake of [11C]17 in the liver, pancreas and white adipose tissue together with high initial brain uptake and elimination by hepatobiliary route. The finding that [11C]17 penetrates the BBB and accumulated in the adipose tissue was found to be consistent with the high calculated lipophilicity of the tracer of log P = 4.2 (ACDLab). Metabolite analysis revealed a fast metabolization to a single more polar compound so that after 60 min only 2% and 18% of intact [11C]17 were found in mouse and rat plasma, respectively. Compound [11C]17 was furthermore tested in nude mice bearing HT-29 xenograft tumors. Although an uptake of [11C]17 in the tumor tissue was clearly visible, this could not be blocked by pre-injection with the non-radioactive reference, pointing to a more non-specific uptake.
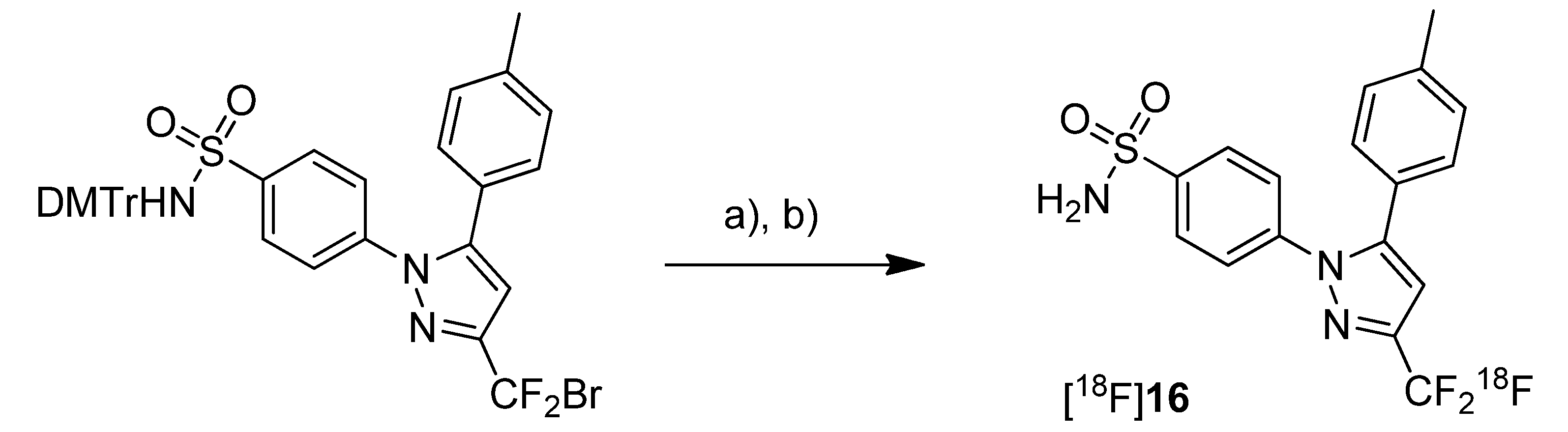
Three years after the synthesis of [
11C]celecoxib ([
11C]
7) by Prabhakaran
et al. [
61], de Vries
et al. presented their own studies with [
11C]
7 (
Scheme 12) to determine whether celecoxib affects the P-gp-mediated drug efflux [
67]. Within this work, the brain distribution of [
11C]
7 was determined by
ex vivo analysis and microPET imaging.
Scheme 12.
Synthesis of [11C]7.
Scheme 12.
Synthesis of [11C]7.
Reagents and conditions: (a) Pd2(dba)3, P(o-tolyl)3, [11C]CH3I, DMF, 130 °C, 5 min; (b) 200 µL TFA in 0.3 mL DMF, 130 °C, 5 min.
Synthesis: The synthesis of [
11C]
7 was based on the previously described protocol [
61] with several modifications including higher concentrations of the reactants and a modified work up after the deprotection step. In this way, the radiotracer was synthesized in 10–15% radiochemical yield with an average specific activity of 19 GBq/µmol.
In vivo/Ex vivo: To determine whether celecoxib is a P-gp substrate, Male Wistar rats were treated with 50 mg/kg CsA, a Pgp-inhibitor, and the brain uptake of [
11C]
7 was measured whereby untreated controls served as references.
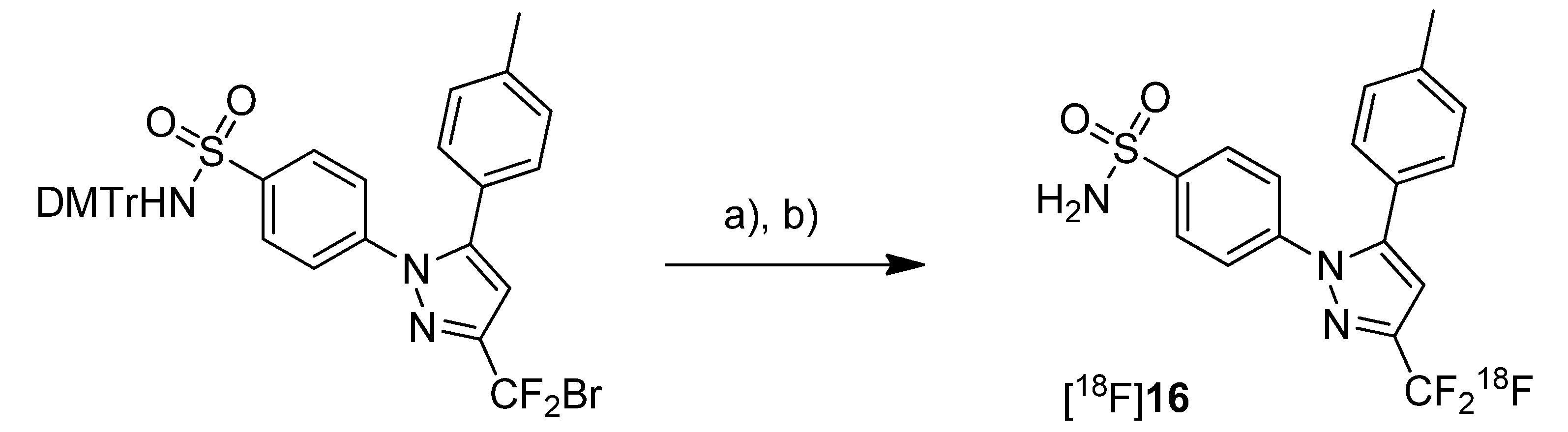
Ex vivo biodistribution studies revealed that the highest accumulation of [
11C]
7 was found in blood cells, liver, spleen, pancreas and lung as well. Generally, the uptake in these organs was higher than in plasma indicating a good distribution of the radiotracer. A relatively high uptake into the brain demonstrated that [
11C]
7 is able to penetrate the blood brain barrier (BBB). It was pointed out that this is in accordance with findings from Prabhakaran
et al. [
44] who showed that [
18F]
16, the fluorine-18 analog, has entered the brain. However, the radioactivity within the brain was homogenously distributed and did not reflect the distribution of COX-2 probably due to nonspecific binding. Further tests with inhibition by CsA revealed that celecoxib is a poor or even no substrate of P-gp. It was also shown that celecoxib is a poor inhibitor of P-gp in the BBB.
2010
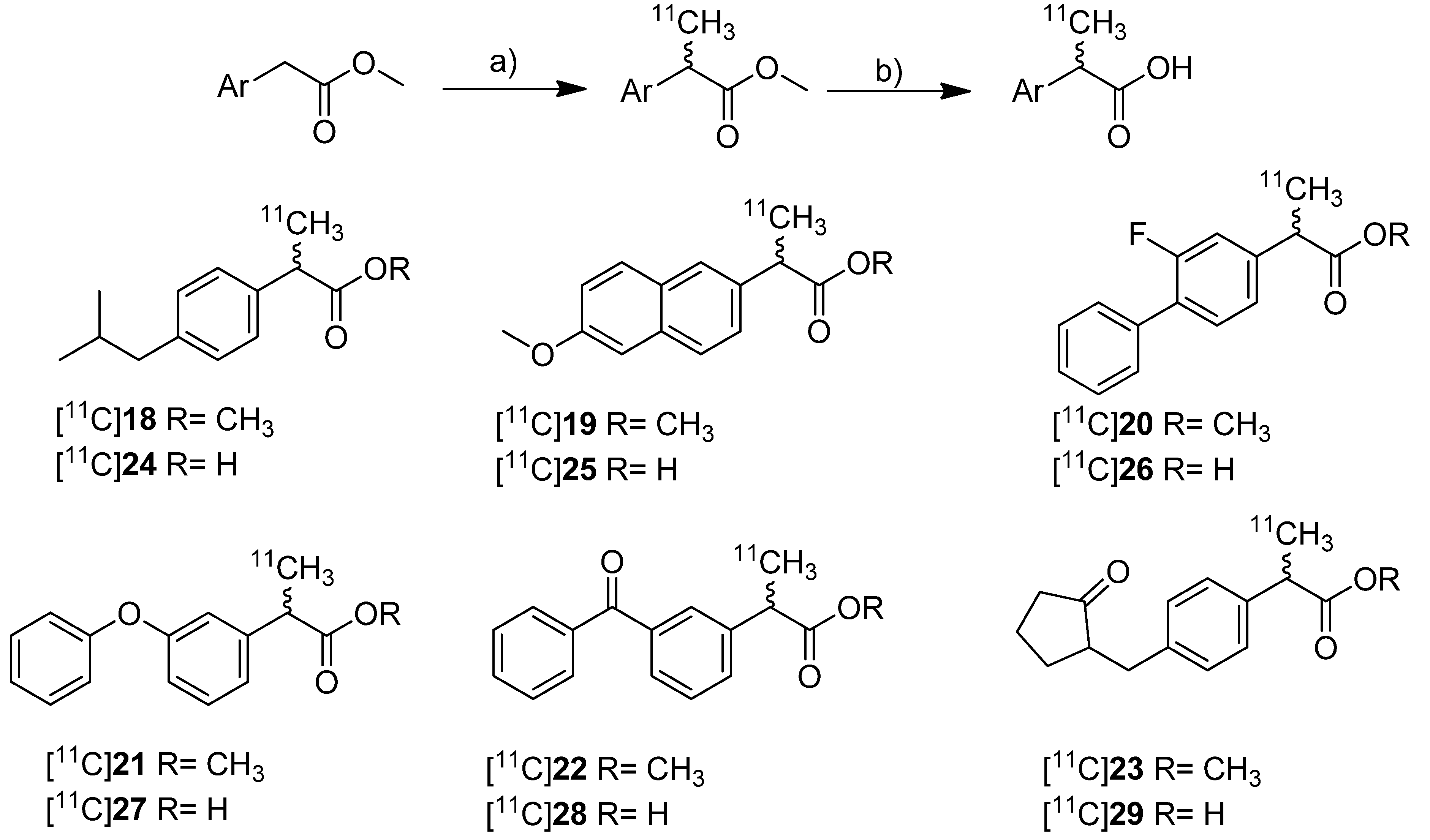
In 2010, synthesis and
in vivo evaluation of six carbon-11-labeled NSAIDs and their appropriate prodrugs was reported. Takashima-Hirano
et al. presented a general method to synthesize carbon-11-labeled 2-arylpropionic acids as unselective COX inhibitors and its esters as appropriate prodrugs [
45]. In this work, a set of carbon-11-labeled analogs of ibuprofen ([
11C]
24), naproxen ([
11C]
25), flurbiprofen ([
11C]
26), fenoprofen ([
11C]
27), ketoprofen ([
11C]
28) and loxoprofen ([
11C]
29) was synthesized (
Scheme 13) which shared this common structural motif of NSAIDs. The COX-inhibitory potency of the carboxylic acid derivatives was already described and exhibits no selectivity or rather selectivity towards COX-1 [
69,
70].
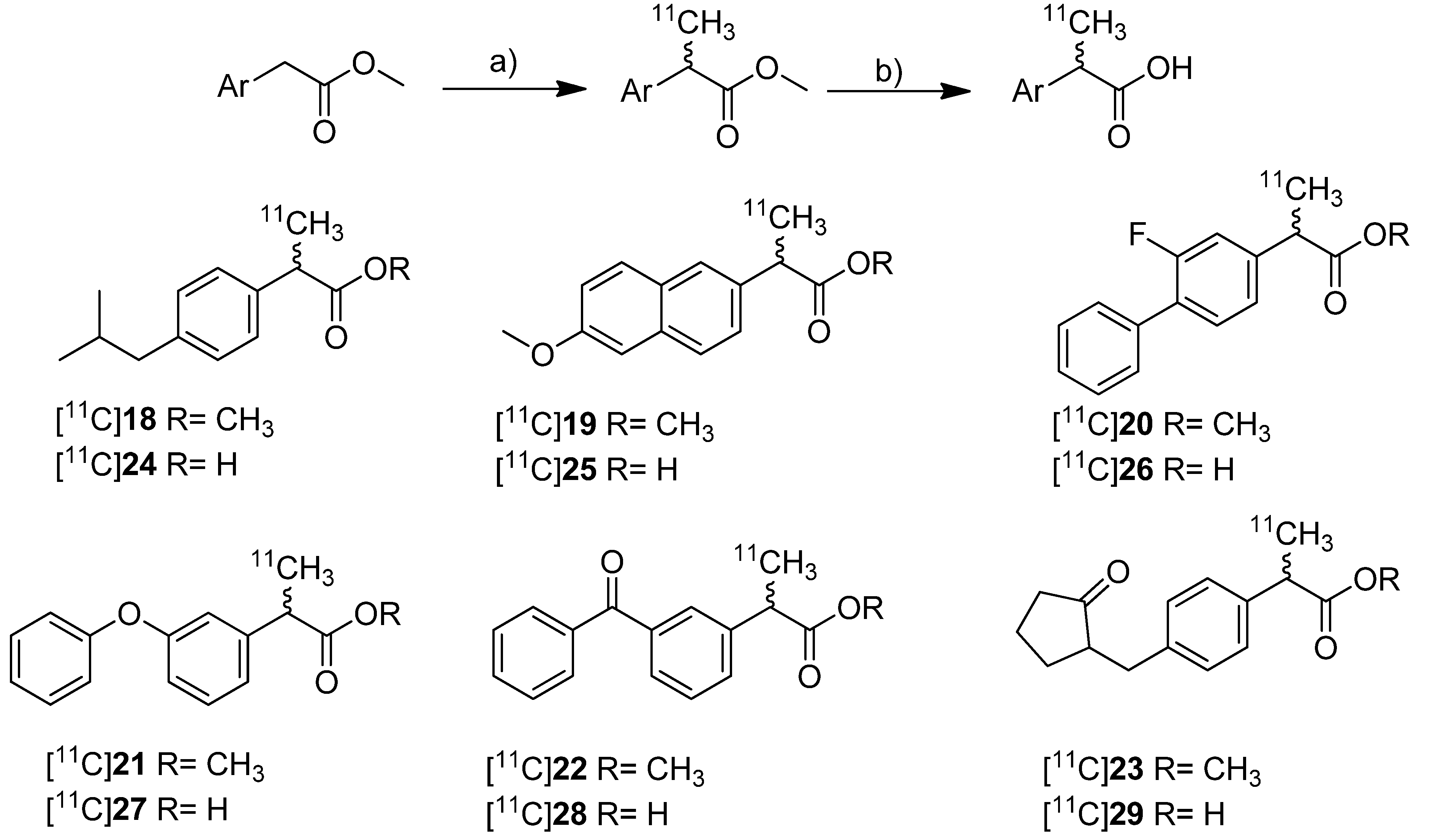
Scheme 13.
Synthesis of [11C]18–29.
Scheme 13.
Synthesis of [11C]18–29.
Reagents and conditions: (a) [11C]CH3I, NaH, DMF, 30 °C, 2 min; (b) for [11C]24–29: 2 M NaOH, DMF, 50 °C, 1 min.
Synthesis: The synthesis of [11C]18–23 was performed by C-[11C]methylation of the corresponding enolates which were generated in situ by deprotonation of the benzylic position with NaH and subsequent reaction with [11C]methyl iodide in DMF at 30 °C. This gave the methyl propionic acid esters [11C]18–23 after semi-preparative HPLC in 26–72% decay corrected radiochemical yield (based on [11C]methyl iodide) with specific activities between 20 ± 9 GBq/µmol for [11C]23 and 47 ± 11 GBq/µmol for [11C]22 within 28–41 min total synthesis time. Compounds [11C]18, [11C]19, [11C]22, and [11C]23 were sensitive towards radiolysis but this tendency could be prevented by addition of ascorbic acid and a radiosynthesis with an activity level less than 15 GBq [11C]methyl iodide. The carbon-11-labeled carboxylic acids [11C]24–29 were synthesized in an one pot reaction by hydrolysis of the esters [11C]18–23 after C-[11C]methylation by addition of 2 M NaOH solution and subsequent heating to 50 °C for 1 min. Purification by semi-preparative HPLC gave [11C]24–29 in 29–72% decay corrected radiochemical yield (based on [11C]methyl iodide) with specific activity between 20 ± 5 GBq/µmol for [11C]27 and 31 ± 12 GBq/µmol for [11C]23 within 28–42 min total synthesis time. In contrast to the esters, the carboxylic acids were found to be stable towards radiolysis. In vivo: [11C]22 and [11C]28 were evaluated in male Sprague-Dawley rats. Small animal PET studies of rats with lipopolysaccharide (LPS)-induced inflammation in the left striatum revealed a high accumulation in the LPS injection side and surrounding tissue after administration of [11C]22. The authors found that [11C]22 showed a similar uptake compared to [11C]PK11195 but with a better ratio regarding inflammatory region to background. In contrast to [11C]22, [11C]28 showed only low uptake into the brain. Metabolite analysis of [11C]22 in blood of normal rats revealed a fast hydrolysis to the biologically active form [11C]28 and a lower hydrolysis rate in brain. However, after uptake of the ester in the brain still more than 90% was hydrolyzed within 5 min into the acid derivative [11C]28 what was expected by the authors to be fast enough for detection of [11C]28 in the brain. After that time point, significant differences between inflamed region and background were observed. Further PET studies with [11C]18–23 showed that all compounds with exception of [11C]18 were accumulated in the LPS induced-inflammatory region of the brain with the highest accumulation of [11C]22 and best ratio of inflamed tissue to contralaterally lesioned area for [11C]19 and [11C]23. Blocking studies with [11C]22 were performed by simultaneous injection of the non-radioactive reference and revealed a significantly reduced accumulation of radioactivity in the LPS-induced inflammatory region.
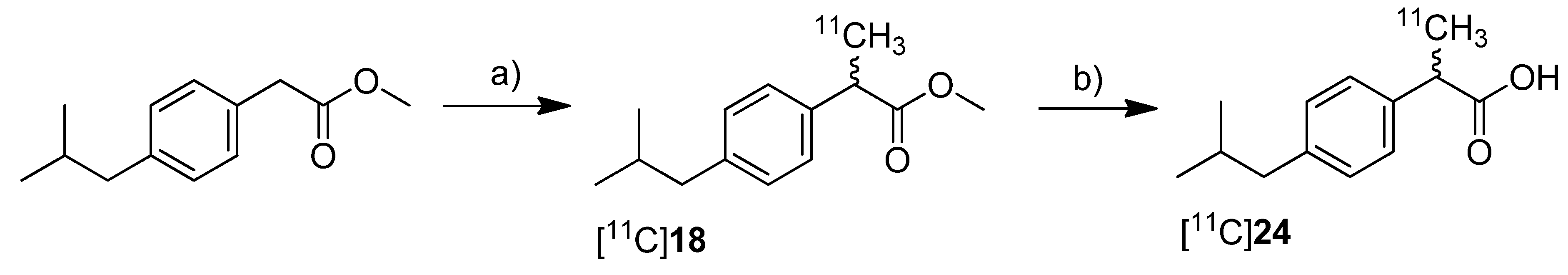
Kato
et al. presented the synthesis of [
11C]ibuprofen ([
11C]
24) as an example for the developed TBAF promoted synthesis of 2-arylpropionic acids [
71] (
Scheme 14). As valuable aspect in comparison to the procedure developed by Takashima-Hirano
et al. [
45], the authors point out that the use of TBAF instead of NaH allows performing the automated labeling reaction under homogenous conditions.
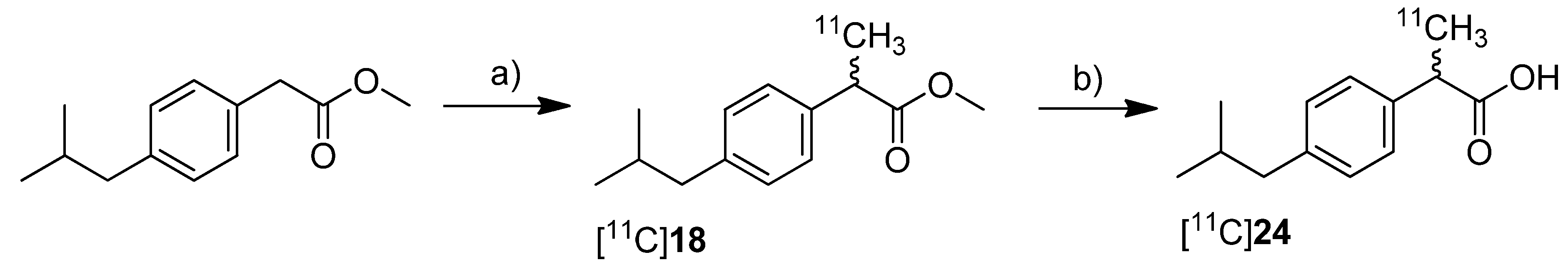
Synthesis: Starting from the appropriate phenyl acetic acid ester precursor and deprotonation of the benzylic position by TBAF in THF at room temperature, [
11C]
18 was synthesized by
C-[
11C]methylation with [
11C]methyl iodide. This yielded [
11C]
18 in 89.3 ± 3.3% decay corrected radiochemical yield determined by HPLC in the first step. As reported by Takashima-Hirano
et al. [
45] the authors observed as well significant radiolysis of [
11C]
18 when the synthesis started with > 15 GBq of [
11C]carbon dioxide. The radiosynthesis of [
11C]
24 was performed in an one pot reaction by
C-[
11C]methylation followed by addition of 1 M NaOH solution and methanol and subsequent alkaline hydrolysis at 80 °C for 3 min. In this way, [
11C]
24 was synthesized in 77.3 ± 4.9% decay corrected radiochemical yield determined by HPLC in two steps. Starting from 11.1 GBq of [
11C]carbon dioxide, the remote-controlled procedure gave > 1 GBq of [
11C]
24 after semi-preparative HPLC within 30 min total synthesis time.
In vitro/in vivo: Further information about
in vivo evaluation of the enantiomerically pure radiotracer was presented one year later by Kikuchi
et al. [
72].
Scheme 14.
Synthesis of [11C]18 and [11C]24.
Scheme 14.
Synthesis of [11C]18 and [11C]24.
Reagents and conditions: (a) [11C]CH3I, TBAF, THF, r.t. 30 s; (b) 1 M NaOH/MeOH 1:1, 80 °C, 3 min.
2011
In 2011, the synthesis of two fluorine-18-labeled and fifteen carbon-11-labeled COX-2 inhibitors and, for a part of them, the in vitro/in vivo evaluation as well the in vivo evaluation of the carbon-11-labeled prodrug of ketoprofen and carbon-11-labeled (R)- and (S)-ibuprofen was presented.
Scheme 15.
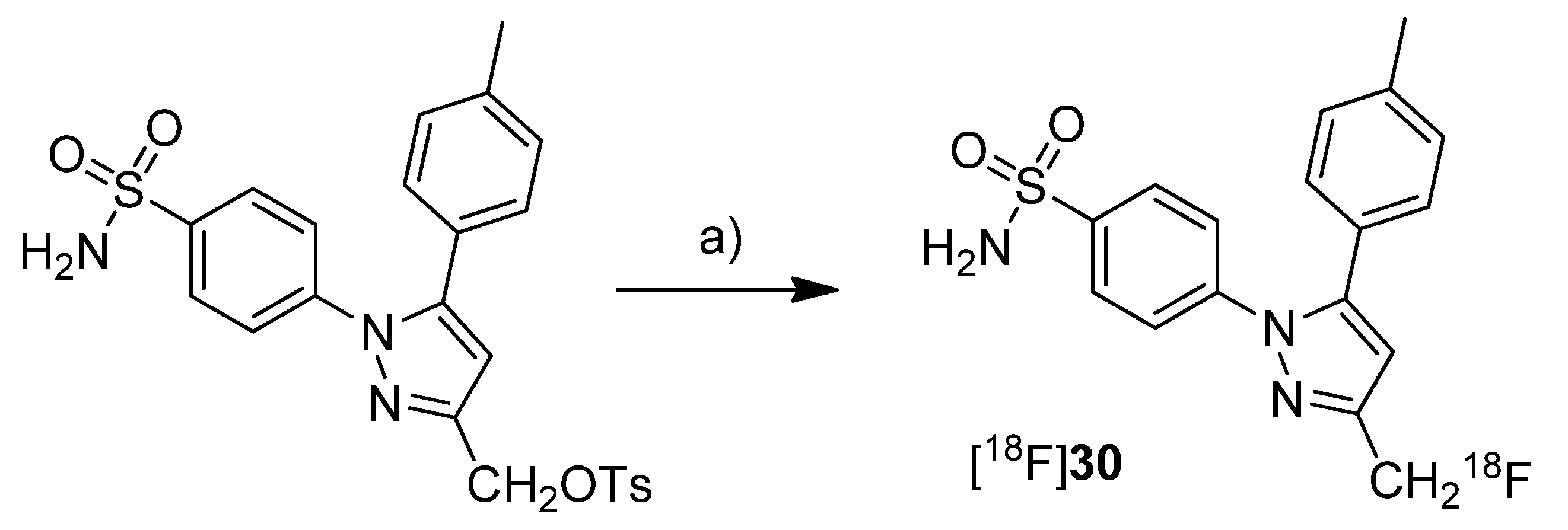
Synthesis of [18F]30.
Scheme 15.
Synthesis of [18F]30.
Reagents and conditions: (a) [18F]KF, K222, DMSO, microwave, 165 °C, 3 min.
Uddin
et al. presented the synthesis and
in vivo evaluation of 4-(3-([
18F]fluoromethyl)-5-(
p-tolyl)-1
H-pyrazol-1-yl)benzenesulfonamide ([
18F]
30,
Scheme 15), a fluorine-18-labeled celecoxib derivative [
48]. This compound was selected from a set of synthesized celecoxib and indomethacin derivatives due to its high and selective COX-2 inhibition potency. The non-radioactive compound 30 revealed
in vitro an IC
50 value of 0.16 µM against purified human COX-2, 0.08 µM against intact COX-2 expressing RAW 264.7 cells and > 4 µM against ovine COX-1.
Synthesis: [
18F]
30 was synthesized by fluorine-18-for-tosylate exchange in a microwave assisted reaction. For this, the tosylate precursor in DMSO was added to the dried [
18F]KF-kryptofix complex followed by heating to 165 °C and sequential additional heating by microwaves. This gave [
18F]
30 after semi-preparative HPLC in 25% radiochemical yield and in a radiochemical purity > 99%.
In vivo: For the evaluation of [
18F]
30, male Sprague-Dawley rats with a carrageenan-induced inflammation in the rear right paw were used. 1.5 h after injection of [
18F]
30, the animals were studied by small animal PET. The uptake in the inflamed paw was higher compared to the contralateral, non-inflamed paw and this enhanced uptake could be blocked by administration of celecoxib immediately prior to [
18F]
30 injection. These results were confirmed by post mortem analysis which revealed a 1.53 ± 0.5-fold (n = 5) higher amount of radioactivity in the inflamed compared to the non-inflamed paw. The specificity for COX-2 was examined by intraperitoneal (i.p.) administration of [
18F]
30 to wild-type and COX-2 null mice. In COX-2 null mice no statistical significant differences were observed between [
18F]
30 uptake into the carrageenan-induced inflamed paw compared to the non-inflamed paw. In contrast, wild-type mice showed a significantly higher uptake in the inflamed tissue. The stability of the tracer was tested by i.p. injection of the non-radioactive reference 30. The amount of defluorinated metabolite after 2 hours was detected by LC-ESI-MS in extracts of the pad tissue after euthanasia what revealed 8.8 ± 5% (n = 3) of defluorinated metabolite. [
18F]
30 was furthermore tested in tumor xenograft bearing female nude mice. For this, COX-2 negative human colorectal carcinoma (HCT116) and COX-2-expressing human head and neck squamous cell carcinoma (1483 HNSCC) tumor xenografts were allowed to grow in the right and left hip of the mice, respectively. Small animal PET studies revealed a significant uptake of [
18F]
30 in the COX-2 expressing tumor which was approximately 3-fold higher compared to the uptake in COX-2 null tumor tissue as determined by
ex vivo distribution analysis. Furthermore, the specific uptake of [
18F]
30 was demonstrated by i.p. administration of celecoxib and a vehicle immediately prior to radiotracer injection. In comparison, the tumor-to-muscle ratio was decreased from 2.94 ± 0.36 (n = 5) to 1.01 ± 0.16 (n = 4) when celecoxib was administrated instead of the vehicle. With this work, Uddin
et al. furnished proof for the first time that COX-2 can be visualized selectively and specific with a fluorine-18-labeled COX-2 inhibitor by means of PET.Al-Hourani
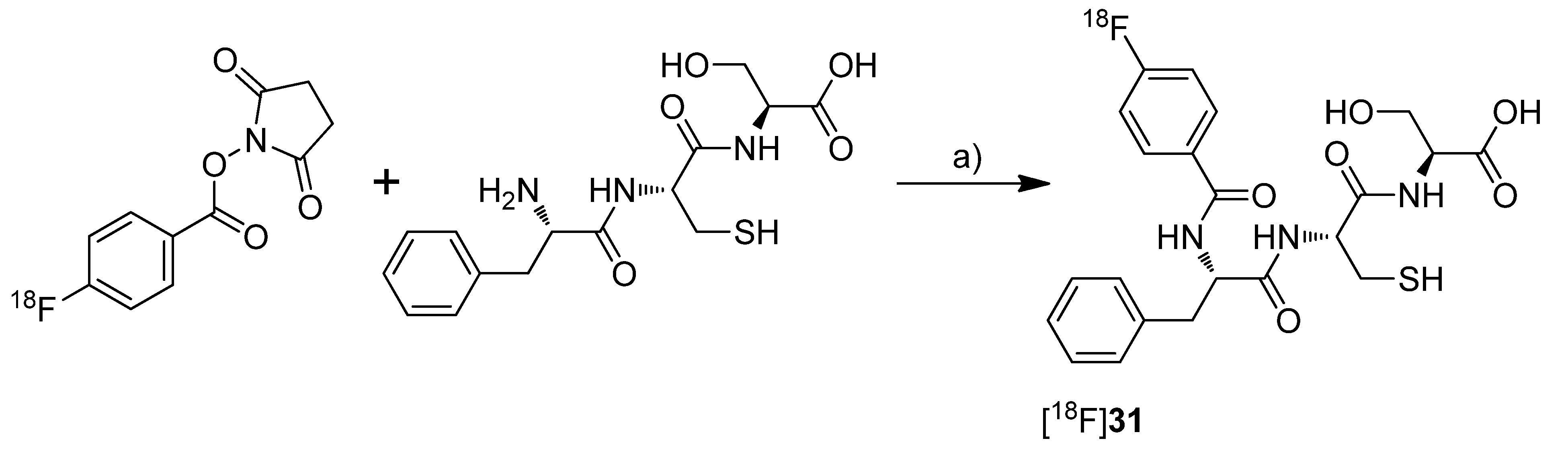
et al. presented in a poster presentation the synthesis and
in vitro results of the fluorine-18-labeled tripeptide
N-(4-[
18F]Fluorobenzoyl)-phenylalanyl-cysteyl-serine ([
18F]
31,
Scheme 16) possessing an IC
50 for COX-2 of 13 µM and for COX-1 of 27 µM [
73]. The synthesis and evaluation of this and other but less potent non-radiolabeled fluorobenzoylated di- and tripeptide derivatives was published recently [
74].
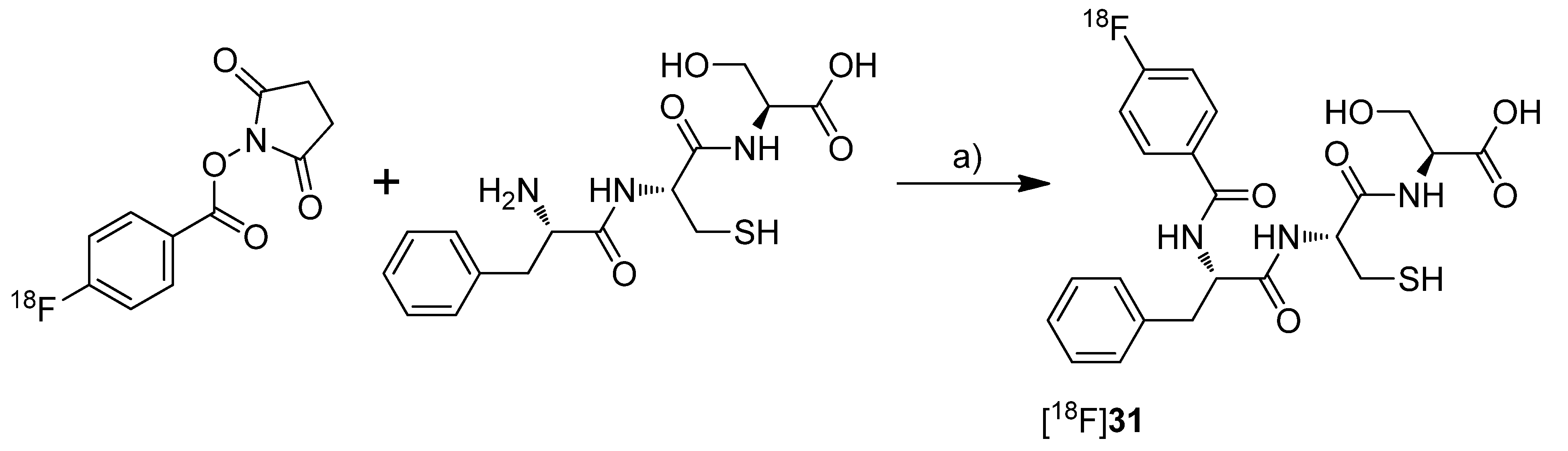
Synthesis: [
18F]
31 was synthesized by labeling the tripeptide precursor with
N-succinimidyl 4-[
18F]fluorobenzoate ([
18F]SFB) and subsequent semi-preparative HPLC in 5–8% radiochemical yield (based on [
18F]SFB) with a radiochemical purity exceeding 95% within 70 min.
In vitro: [
18F]
31 uptake was tested in COX-2 expressing Ward colon tumor cells but this revealed only low uptake which could not be blocked by celecoxib.
Scheme 16.
Synthesis of [18F]31.
Scheme 16.
Synthesis of [18F]31.
Reagents and conditions: (a) Borate buffer (pH = 8.3), 60 °C, 30 min.
Takashima-Hirano
et al. presented the synthesis and
in vivo evaluation of carbon-11-labeled celecoxib [
11C]
7 (
Scheme 7) and its major metabolite 4-(1-(4-sulfamoylphenyl)-3-(trifluoromethyl)-1
H-pyrazol-5-yl)benzoic acid [
11C]
32 (
Scheme 17) with the aim to examine the function of drug transporters for hepatobiliary excretion [
75].
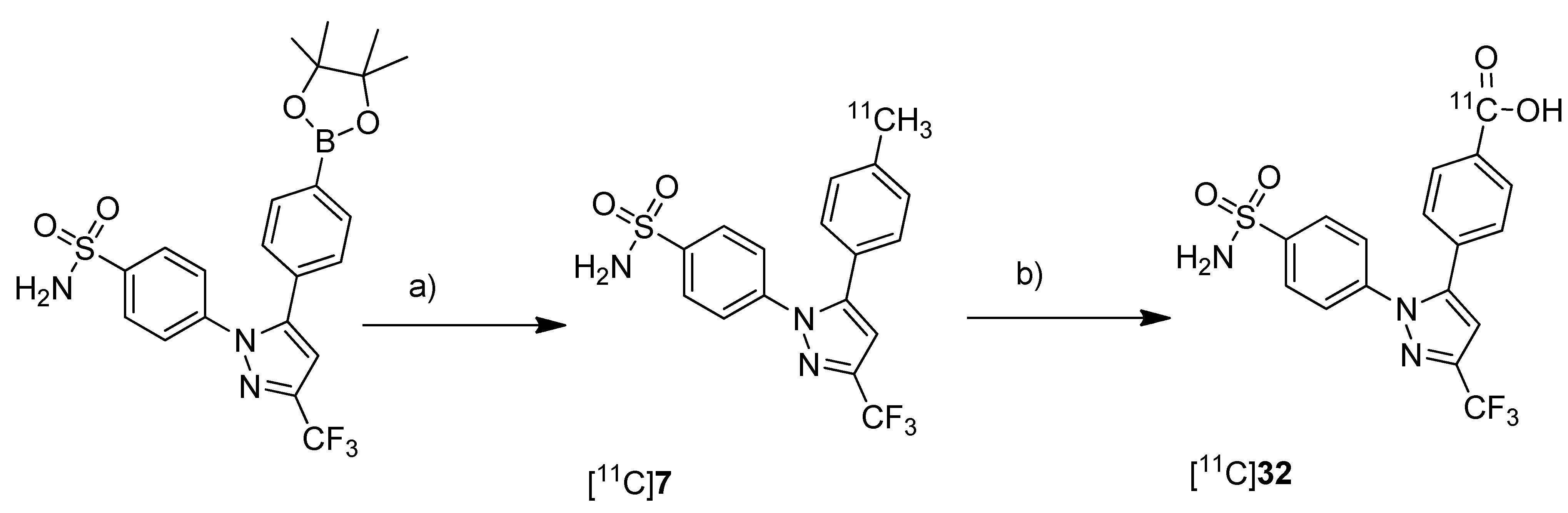
Synthesis: [
11C]
7 was synthesized in an one step reaction starting from an organoboron precursor by reaction with [
11C]methyl iodide in the presence of Pd
2(dba)
3, P(
o-tolyl)
3 and K
2CO
3 in DMF for 5 min at 130 °C. This yielded compound [
11C]
7 in 63 ± 23% (n = 7) decay corrected radiochemical yield with a specific activity of 84 ± 23 GBq/µmol within 30 min synthesis time from end of bombardment. [
11C]
32 was synthesized by subsequent oxidation of [
11C]
7 with KMnO
4 in 0.2 M NaOH under microwave irradiation. In this way, [
11C]
32 was synthesized in 55 ± 9% (n = 3) decay corrected radiochemical yield (based on [
11C]
7) with a specific activity of 39 ± 4 GBq/µmol after 50 min synthesis time from end of bombardement.
In vivo/Ex vivo: Both radiotracers have been evaluated with focus on their hepatobiliary excretion using male Sprague-Dawley rats. After initial uptake in the liver, [
11C]
7 moved to the intestine. Metabolite analysis revealed that [
11C]
7 was found in blood and liver with only small amounts of metabolites but that it was completely degraded to the carboxylic acid metabolite [
11C]
32 in the bile. When [
11C]
32 was injected, no further metabolites were detected and radioactivity was rapidly excreted by hepatobiliary and renal routes.
Scheme 17.
Synthesis of [11C]7 and [11C]32.
Scheme 17.
Synthesis of [11C]7 and [11C]32.
Reagents and conditions: (a) [11C]CH3I, Pd2(dba)3, P(o-tolyl)3, 65 °C, DMF, 4 min; (b) KMnO4, 0.2 M NaOH, microwave, 140 °C, 5 min.
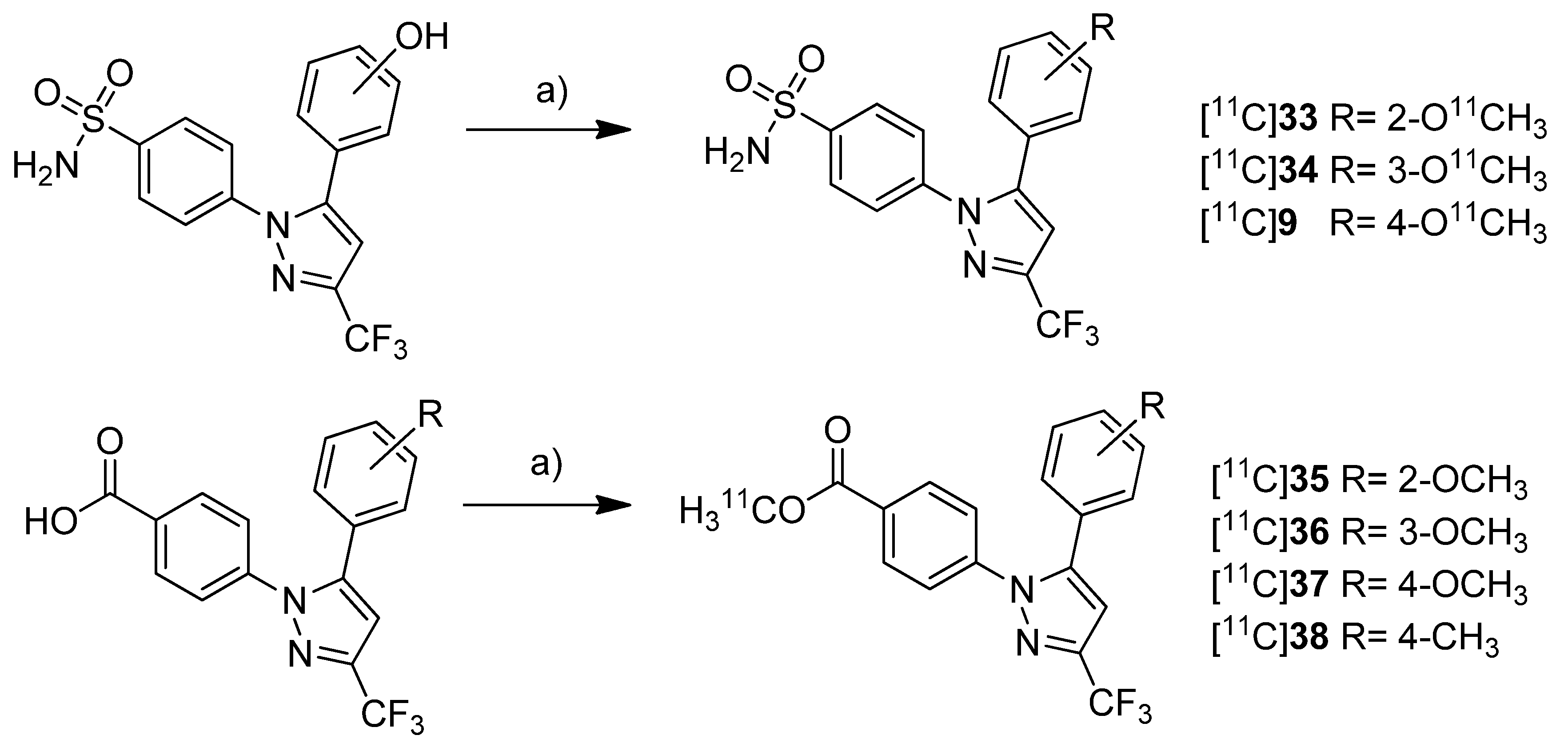
Gao
et al. presented the synthesis of one known ([
11C]
9) and six novel ([
11C]
33–
38) COX-2 inhibitors labeled with carbon-11 and the
in vitro evaluation of the corresponding non-radioactive reference compounds [
46] (
Scheme 18). The IC
50 values for COX-2 of the non-radioactive analogs 4-(5-(2-methoxyphenyl)-3-(trifluoromethyl)-1
H-pyrazol-1-yl)benzenesulfonamide (
33) and 4-(5-(4-methoxyphenyl)-3-(trifluoromethyl)-1
H-pyrazol-1-yl)benzenesulfonamide (
9) were reported earlier to be 290 nM and 9 nM [
62].
Scheme 18.
Synthesis of [11C]9 and [11C]33–38.
Scheme 18.
Synthesis of [11C]9 and [11C]33–38.
Reagents and conditions: (a) [11C]CH3OTf, 2 M NaOH, MeCN, 80 °C, 3 min.
The non-radioactive references were tested in a cell proliferation assay with a MDA-MB-435 tumor cell line. A statistical significant difference for the inhibition of proliferation at different concentration was found for compounds 35 and 37. 35 and 37 showed inhibition of cell proliferation in a range of 160–320 nM and at 160 nM, respectively. All other compounds did not significantly inhibit cell proliferation ranging between 20 and 1280 nM. Synthesis: All compounds were radiolabeled by O-[11C]methylation of the corresponding phenolic hydroxy or carboxylic acid precursor. In brief, [11C]methyl triflate was prepared as methylating agent from [11C]carbon dioxide and reacted with the precursor in acetonitrile basified with 2 N NaOH. This yielded the O-[11C]methylated ethers and esters after purification by solid phase extraction within 23 min in decay corrected radiochemical yields of 52 ± 2% (n = 5) and 57 ± 3% (n = 5) (based on [11C]carbon dioxide), respectively. The specific activity was determined to be 277.5 ± 92.5 GBq/µmol (n = 5) and the radiochemical purity was > 99%. In vitro/In vivo: No further in vitro or in vivo evaluation was presented.
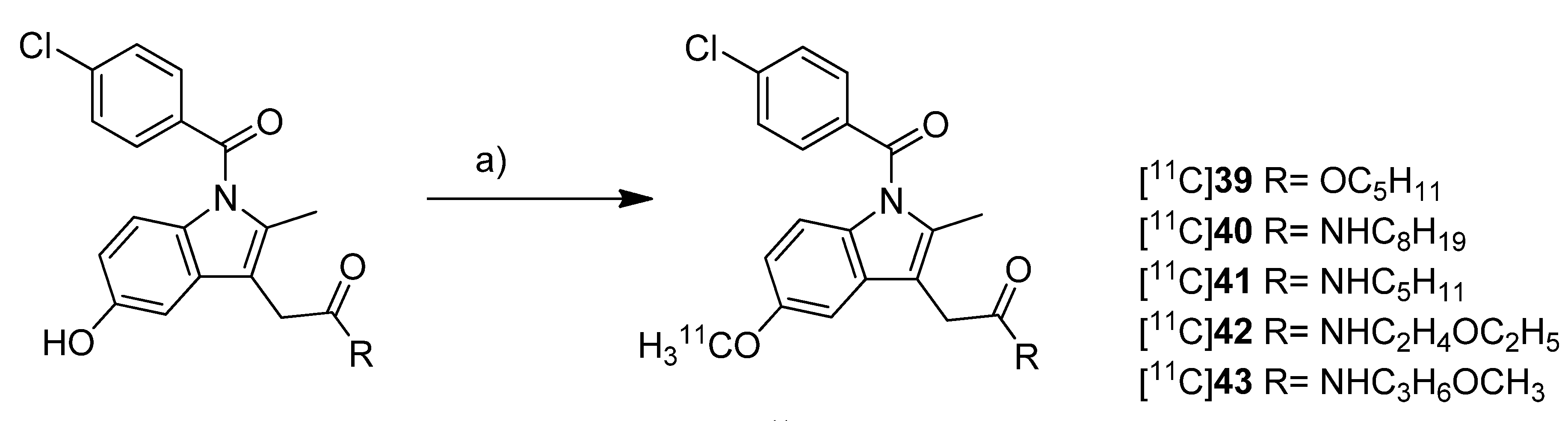
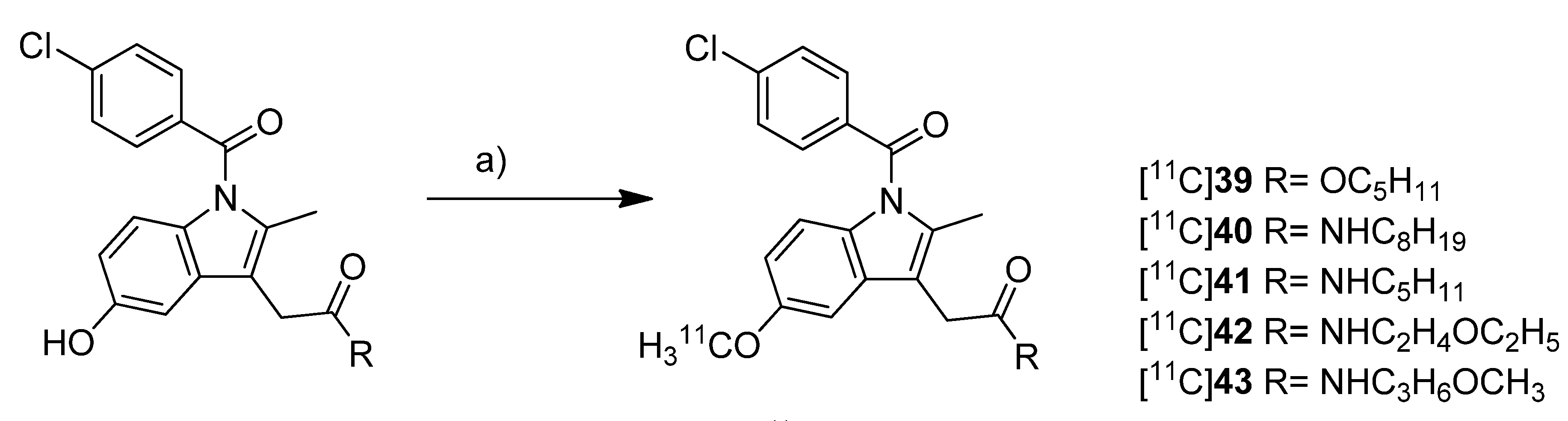
Scheme 19.
Synthesis of [11C]39–43.
Scheme 19.
Synthesis of [11C]39–43.
Reagents and conditions: (a) [11C]CH3OTf, NaOH, acetone, r.t.
Yamamoto
et al. presented five years after the synthesis of [
11C]
13–
15 the synthesis of five carbon-11-labeled indomethacin derivatives ([
11C]
39–
43,
Scheme 19) as well as their
in vitro and
in vivo evaluation [
35]. The non-radioactive analogs pentyl 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1
H-indol-3-yl)acetate (
39) and 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1
H-indol-3-yl)-
N-octyl-acetamide (
40) have been described earlier as potent COX-2 inhibitors [
76] and served as lead structure for the development of the presented compounds. The non-radioactive compounds were tested in an
in vitro COX assay which revealed that compound 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1
H-indol-3-yl)-
N-pentylacetamide (
41) had the highest affinity and selectivity for COX-2 with an IC
50 value for COX-2 of 0.039 µM and an IC
50 value for COX-1 of more than 100 µM. The other compounds were selective COX-2 inhibitors, too, but with IC
50 values for COX-2 in the range of 0.2–2.3 µM.
Synthesis: All compounds were radiolabeled by
O-[
11C]methylation from their corresponding desmethyl precursor. This was realized in detail by reaction with [
11C]methyl triflate in acetone in the presence of NaOH and purification with semi-preparative HPLC. That protocol gave the desired radiotracer in a decay corrected radiochemical yield (based on [
11C]methyl triflate) in the range of 55 ± 12% in case of [
11C]
40 to 71 ± 5% (n = 5) in case of [
11C]
42 within 21-27 min total synthesis time and a radiochemical purity of 93-97%. From [
11C]
39 to 2-(1-(4-chlorobenzoyl)-5-[
11C]methoxy-2-methyl-1
H-indol-3-yl)-
N-(3-methoxypropyl)acetamide ([
11C]
43)
, specific activity increased in the range of 22-331 GBq/µmol and lipophilicity determined as log
P7.4 decreased from 3.94 to 1.98.
In vivo/Ex vivo: [
11C]
39–
43 were evaluated
in vivo in male ddY mice. Compounds [
11C]
40–
43 showed a relatively fast blood clearance with a half-life of 1.3–3.8 min in contrast to compound [
11C]
39 with a half-life of 12.3 min. The radioactivity initially accumulated for all radiotracers in the liver and a peak concentration was observed in the small intestine reflecting predominant hepatobiliary clearance of the tracers or their metabolites, respectively. The brain uptake of all compounds was low and inversely related with the lipophilicity what was expected due to the increased plasma protein binding. Metabolite analysis revealed the low stability of the radiotracers
in vivo due to hydrolysis of the ester and amide functionality and further catabolic reactions. In case of [
11C]
39 no intact parent compound was found in plasma after 30 min and for compounds [
11C]
40–
43 the amount was 14–32%. In the brain only 9–26% of intact parent compound was detected as well. Blocking studies with either celecoxib, NS-398 or unlabeled compound did not inhibit brain uptake and did not influence brain-to-blood ratio what indicates non-specific binding of the tracers
in vivo except for [
11C]
41 what showed a small fraction of displaceable binding. To find out whether P-gp-mediated drug efflux played a role for the low brain uptake, all radiotracers were evaluated after pretreatment with CsA, a P-gp inhibitor. After pretreatment, a slightly increased uptake in the brain and increased brain-to-blood ratio was observed indicating a possible influence of P-gp in the transport of the radiotracer or an altered cytochrome P450-mediated metabolism caused by CsA.
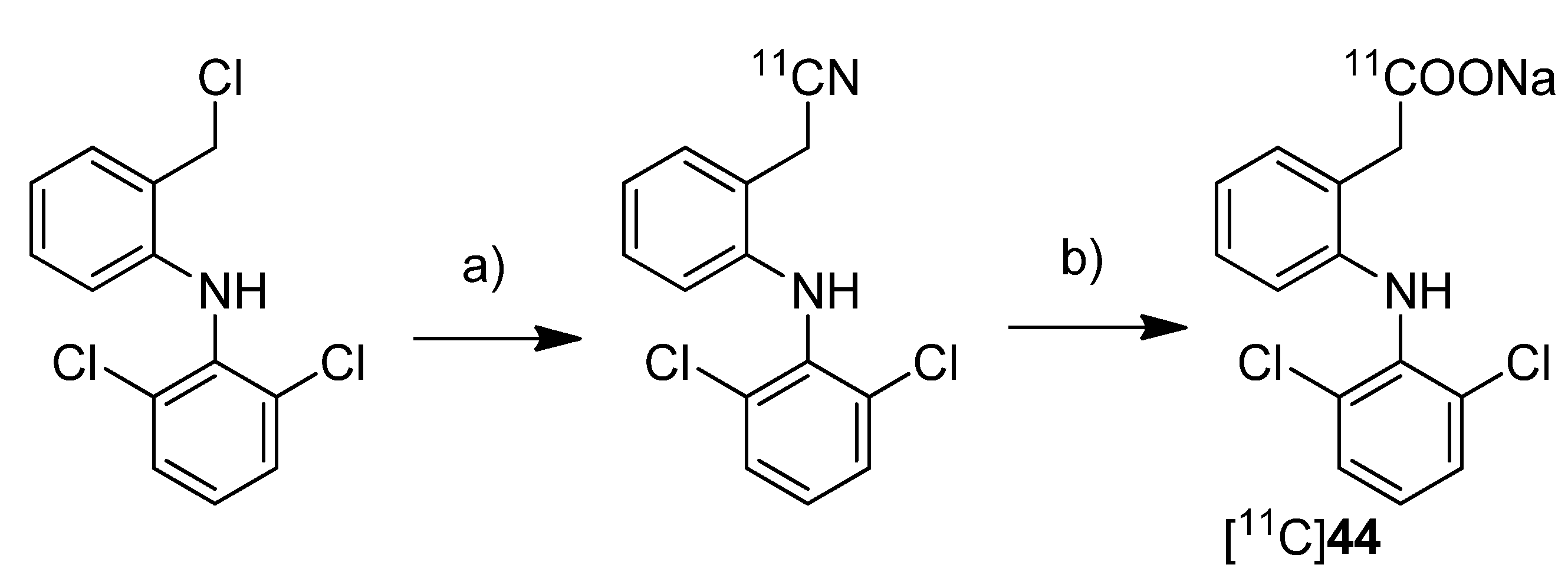
To investigate the transdermal penetration of a topical applied drug by means of PET, Petroni
et al. synthesized carbon-11-labeled diclofenac ([
11C]
44,
Scheme 20) and evaluated the tracer by means of PET after topical formulation [
40].
Scheme 20.
Synthesis of [11C]44.
Scheme 20.
Synthesis of [11C]44.
Reagents and conditions: (a) [11C]NaCN, DMSO, 110 °C, 10 min; (b) 10 M NaOH, 30% H2O2, DMSO/H2O, 135 °C, 10 min.
Synthesis: For the synthesis sodium [11C]cyanide was used which was produced starting from [11C]carbon dioxide by reduction to [11C]methane and subsequent reaction on platinum in the presence of anhydrous ammonia. By reacting the precursor 2-[(2,6-dichlorophenyl)amino]benzyl chloride with sodium [11C]cyanide in DMSO at 110 °C, the corresponding nitrile was formed in 84% radiochemical yield as determined by HPLC. Subsequently the hydrolysis was performed by addition of 1 M NaOH and H2O2 and heating to 135 °C. This gave [11C]44 in 43 ± 4% (n = 20) radiochemical yield as determined by HPLC. After preparative HPLC, [11C]44 was obtained in 97 ± 4% (n = 20) radiochemical purity and formulated for topical application with a non-pressurized sprayer. For this, the dried product was mixed with the diclofenac preparation of the manufacturer. The preliminary evaluation of the transdermal penetration of [11C]44 by means of PET showed that [11C]44 rapidly penetrated the keratinized layer of the skin and, hence, showed that PET is a valuable tool to explore transdermal absorption of active ingredients in topical formulations.
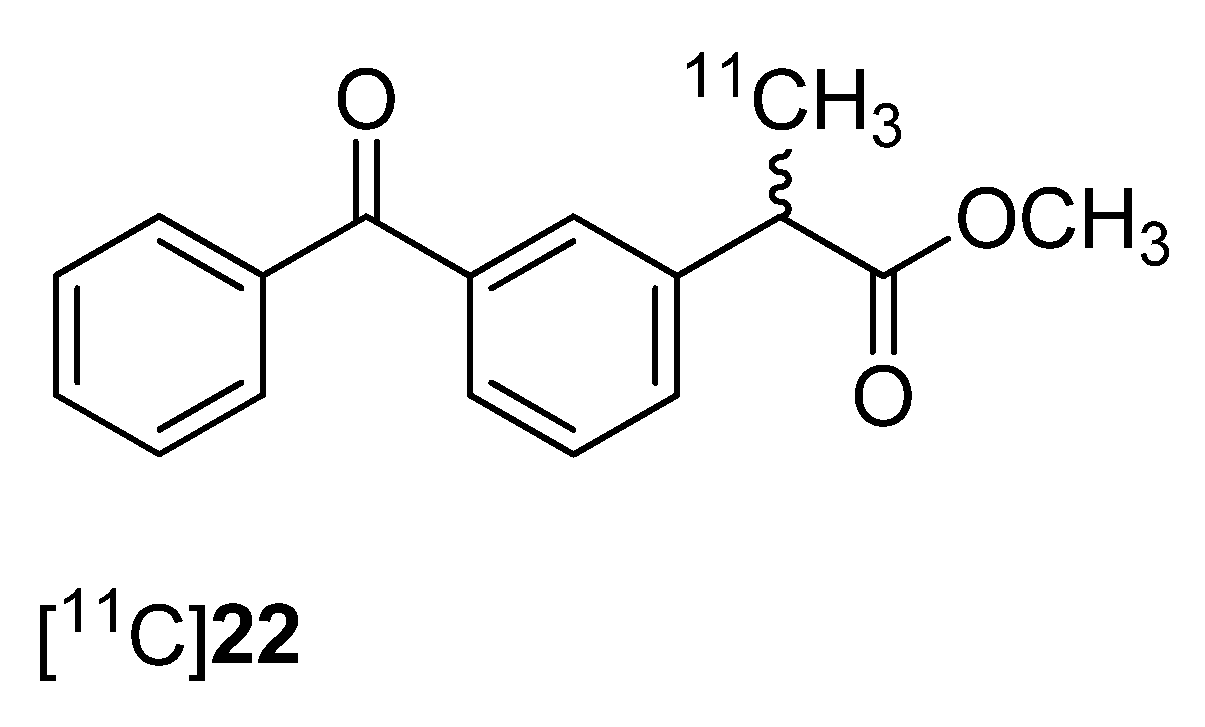
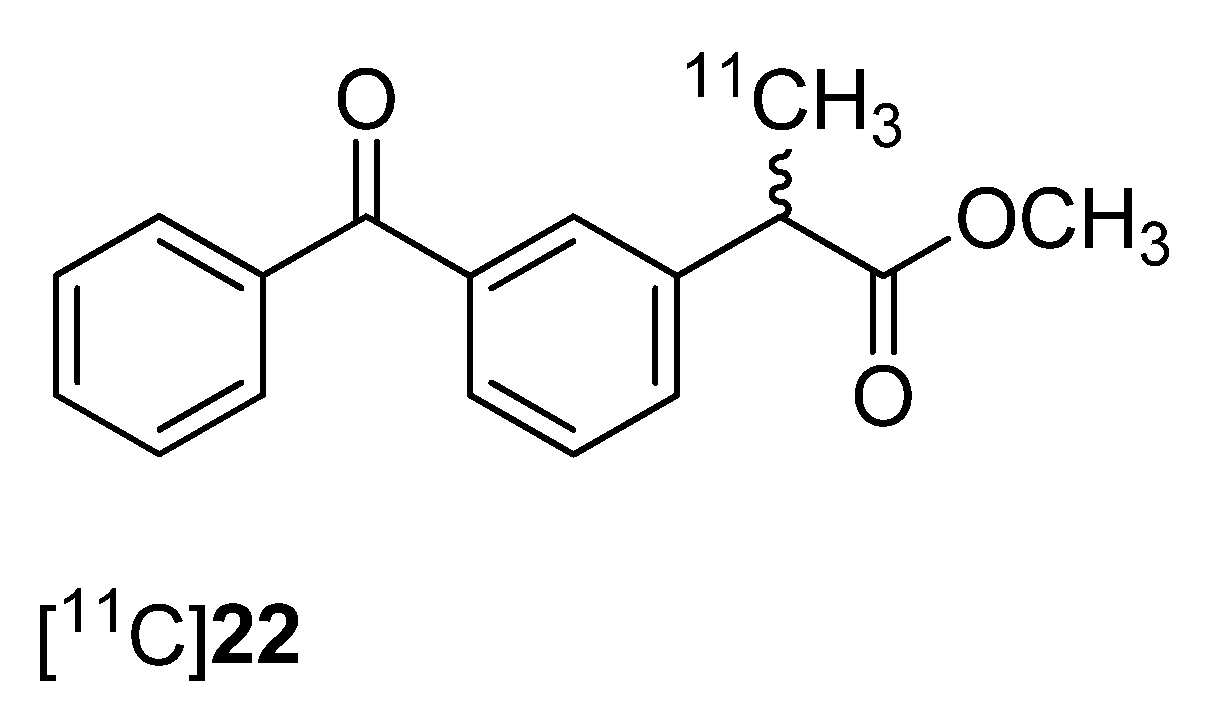
One year after Takashima-Hirano
et al. presented the synthesis of [
11C]ketoprofen methyl ester ([
11C]
22,
Figure 1) [
45], Shukuri
et al. published the results of further biological evaluations of this compound [
36].
Synthesis: [
11C]
22 was synthesized as previously reported [
45].
In vivo/
Ex vivo: The selectivity of [
11C]
22 was investigated with male mice homozygous deficient for COX-1 (COX-1
−/−) and COX-2 (COX-2
−/−) and their wild-type littermate controls.
Ex vivo autoradiography revealed a high accumulation in the brain of wild-type mice and COX-2
−/− mice and a significantly decreased uptake in COX-1
−/− mice showing the COX-1 selectivity of [
11C]
22 in the brain. Further studies were performed with male Sprague-Dawley rats with LPS-induced or quinoline acid-induced neuroinflammation. PET studies revealed that [
11C]
22 showed significant time dependent differences in the uptake to the inflamed regions of the brain which reached its maximum when the tracer was administrated 6 h after LPS injection. The appearance of activated microglia and macrophages was correlated to [
11C]
22 uptake and the expression of COX-1 but not COX-2 in these cells what was shown by immunostaining using IgG antibodies for COX-2 and COX-1. Similar results were obtained regarding the uptake of [
11C]
22 in the inflammatory regions as well as correlation to microglia and macrophage appearance when quinoline acid was used to induce excitotoxic neurodegeneration in the brain. The authors concluded that these results support the crucial proinflammatory role of COX-1 in neuroinflammatory processes.
Figure 1.
Structure of [11C]22.
Figure 1.
Structure of [11C]22.
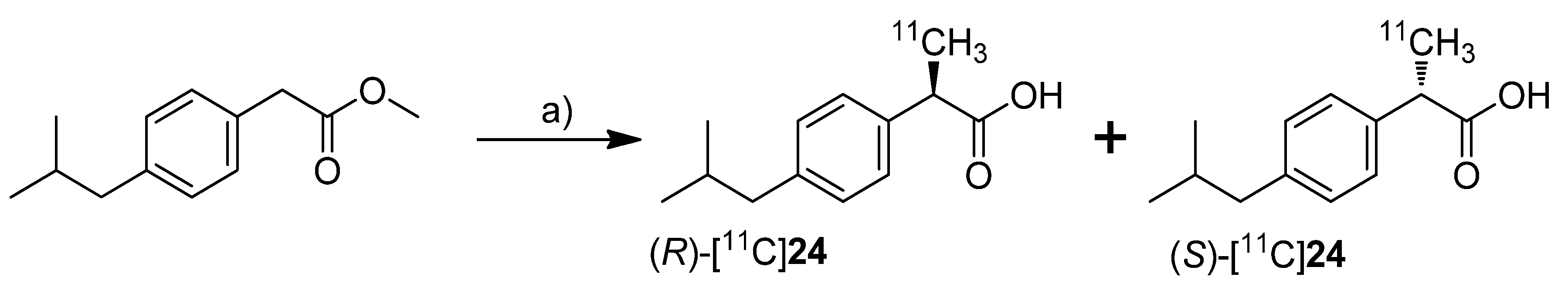
One year after Kato
et al. [
71] and Takashima-Hirano
et al . [
45] have published the synthesis of [
11C]
24, Kukushi
et al. presented the DMSO assisted synthesis, chiral separation and preliminary
in vivo evaluation of the (
R)- and (
S)-enantiomers of [
11C]
24 [
72] (
Scheme 21).
Scheme 21.
Synthesis of (R)-[11C]24 and (S)-[11C]24.
Scheme 21.
Synthesis of (R)-[11C]24 and (S)-[11C]24.
Reagents and conditions: (a) i: [11C]CH3I, TBAF, DMSO, r.t. 30 s; ii: 1 M NaOH, 60 °C, 2 min; iii: chiral semi-preparative HPLC.
Synthesis: The synthesis procedure was optimized regarding the solvent to suppress the radiolysis of the tracer. The improved procedure using lower amounts of the precursor and DMSO as radical scavenging solvent instead of THF gave [11C]18 in 93 ± 1%, and [11C]24 in 92 ± 3% (in two steps) decay corrected radiochemical yield as determined by HPLC. The chiral semi-preparative HPLC was conducted subsequently to the ester hydrolysis using a gradient of acetonitrile/phosphate buffer on a RP-CHIRALPAC OJ-RH column (DAICEL). In this way, in one synthesis more than 740 MBq of each enantiomer (R)-[11C]24 and (S)-[11C]24 was isolated (>3% decay uncorrected radiochemical yield based on [11C]carbon dioxide) with an specific activity of 66–289 GBq/µmol and radiochemical purity > 99%. The enantiomeric purity was > 95% and 90% for the (R)- and (S)-isomer, respectively. In vivo: (R)-[11C]24 and (S)-[11C]24 were tested in male mice using an anti-collagen induced arthritis model. In comparison to untreated mice, the accumulation of both enantiomers was more than twofold increased in arthritic joints but no differences were observed between the two isomers. For that and the fact that only the (S)-enantiomer exhibits COX-inhibition activity, the authors concluded that the accumulation mechanism of [11C]24 in the arthritic joint was independent of COX overexpression.
2012
In 2012, the synthesis of five new fluorine-18-labeled COX-2 inhibitors was presented, two of them with in vivo evaluation.
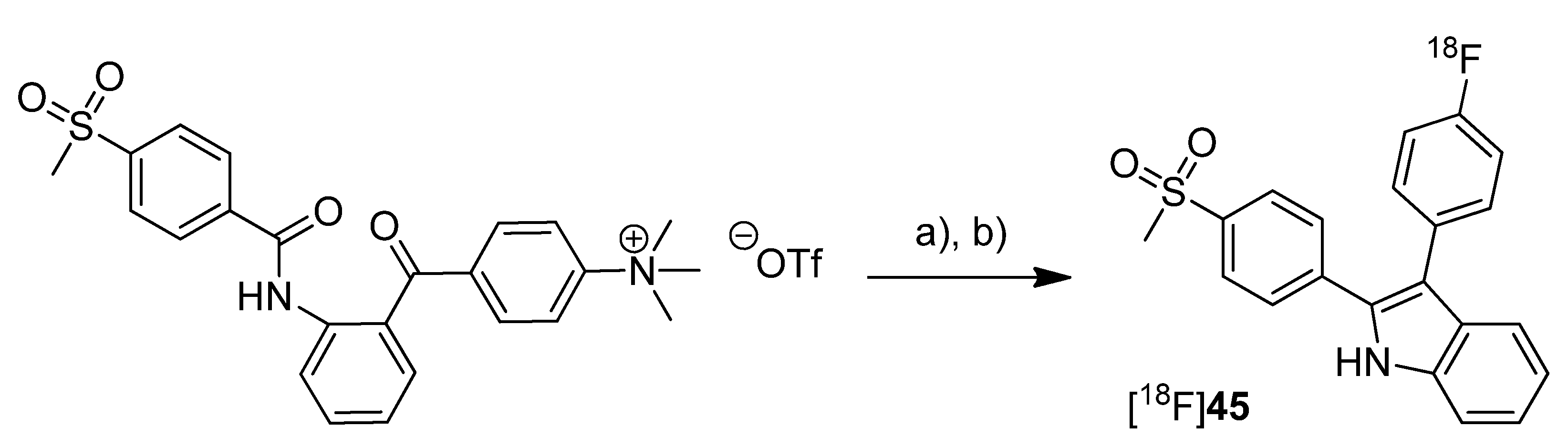
In 2012, our group published the automated synthesis and
in vitro and
in vivo evaluation of 3-(4-[
18F]fluorophenyl)-2-(4-(methylsulfonyl)phenyl)-1
H-indole ([
18F]
45) whose radiosynthesis via McMurry cyclization as a new radiolabeling approach has been presented as conference contribution [
77] one year earlier [
31] (
Scheme 22). The non-radioactive reference
45 was previously described as highly potent and selective COX-2 inhibitor with an IC
50 of 0.02 nM for COX-2 and >10 µM for COX-1 [
66]. By a fluorescence-based assay with the purified enzymes, the IC
50 value of
45 was determined to be 1.2 µM and 6.6 µM for COX-2 and COX-1, respectively. Furthermore, the COX-inhibition potency was verified by an LC-MS/MS based method determining the prostanoid levels in tumor cells and the inhibition of prostanoid synthesis after incubation with the non-radioactive reference compound.
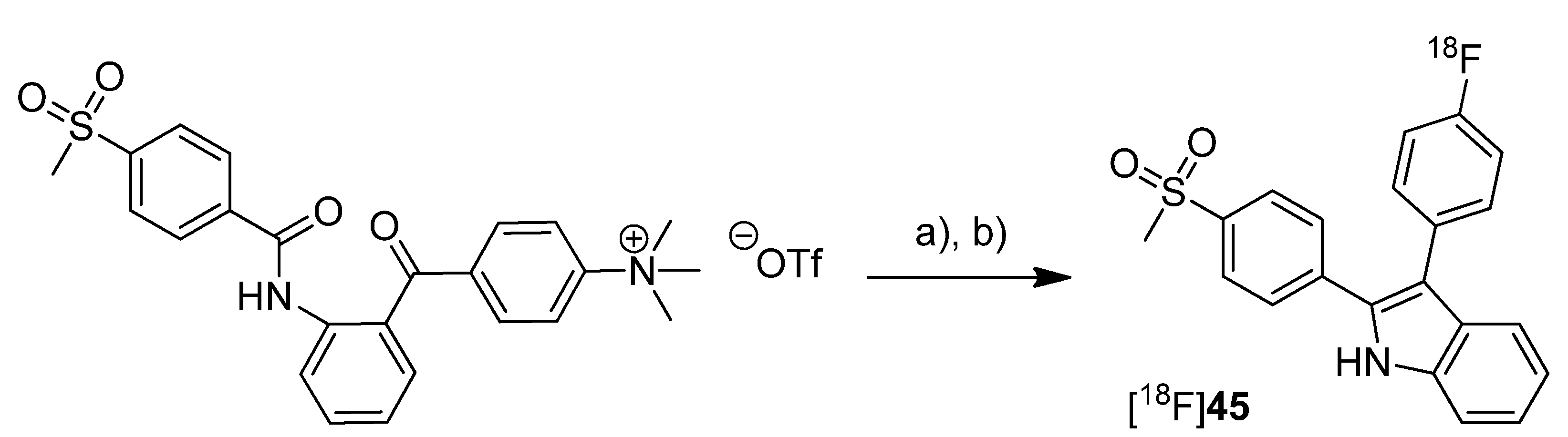
Scheme 22.
Synthesis of [18F]45.
Scheme 22.
Synthesis of [18F]45.
Reagents and conditions: (a) [18F]KF,K222, MeCN, 110 °C, 15 min; (b) TiCl4, Zn, THF, 90 °C, 15 min.
Synthesis: [18F]45 was synthesized in a two-step procedure which involved the labeling of the activated precursor molecule via [18F]fluorine-for-trimethylammonium exchange and was followed by the McMurry ring closure reaction. Different reaction conditions were tested and an optimized protocol for an automated radiosynthesis was presented. Reacting the dried [18F]fluoride-kryptofix complex with the precursor in acetonitrile at 110 °C for 15 min, removal of the solvent and dissolving in THF, and final cyclization in the presence of TiCl4 and Zn at 90 °C for 15 min formed [18F]45. A further solvent exchange to acetonitrile/water, filtration, semi-preparative HPLC and formulation gave [18F]45 in 10% decay corrected radiochemical yield with a specific activity of 74–91 GBq/µmol within 80 min total synthesis time. The log Doct7.4 of [18F]45 was determined to be 1.2 ± 0.2. In vitro: Cell uptake studies of [18F]45 were performed with several cell lines and models which were characterized regarding their COX expression pattern by Western blot analysis. As cell lines with low baseline COX-2 expression served unstimulated human monocytes (THP-1) and the melanoma cell line A375. TPA-stimulated human monocytes (THP-1) and a number of human tumor cell lines—FaDu, HT-29, and A2058 (melanoma)—were used as cell lines with upregulated COX-2 expression. The uptake studies were performed with monolayer cultures and, for selected cell lines, in suspension cultures (unstimulated THP-1). Furthermore, cell uptake experiments were performed in multicellular tumor spheroids with a diameter of 300 µm and 600 µm (HT-29). The uptake was consistent with the COX-1/COX-2 selectivity of [18F]45 and could be blocked by preincubation with the nonradioactive reference. Furthermore, hypoxia-induced COX-2 expression in large-sized spheroids was characterized by increased and specific uptake of [18F]45 and the hypoxia-specific radiotracer [18F]FMISO as well. The possible interaction with the P-gp efflux pump was examined in the presence of the P-gp inhibitor CsA and the calcium blocker verapamil but did not induce any significant effect on the tracer uptake. in vitro stability of [18F]45 was found to be high in rat whole blood and plasma with appearance of less than 5% of a more polar metabolite after 2 h. In vivo: The in vivo stability of [18F]45 was examined in male Wistar-Unilever rats by analysis of arterial blood samples revealing that after 1 h at least 75% of original compound was intact and two more polar metabolites with equal amount were formed. To evaluate the capacity of [18F]45 to detect COX-2 expressing tumors, the tracer was investigated by small animal PET studies in HT-29 tumor-bearing nude mice. Unfortunately, no substantial accumulation of the radiotracer was observed in the tumor tissue, instead the main activity was eliminated by the hepatobiliary route after fast accumulation in the liver. A substantial amount of activity was found in the harderian glands and brown adipose tissue.
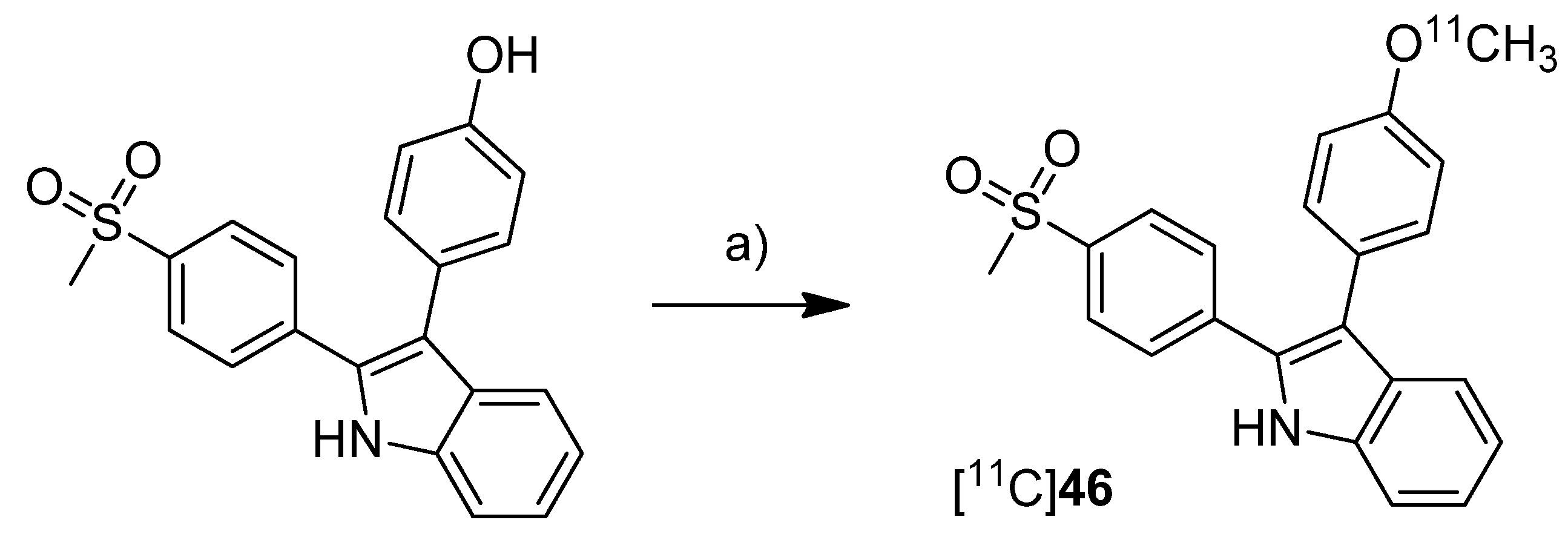
In 2012, we have summarized the results regarding the radiosynthesis and cell uptake studies of [
18F]
45 and another carbon-11-labeled COX-2 inhibitor, 3-(4-[
11C]methoxyphenyl)-2-(4-(methylsulfonyl)phenyl)-1
H-indole [
11C]
46, as a conference contribution [
37] (
Scheme 23).
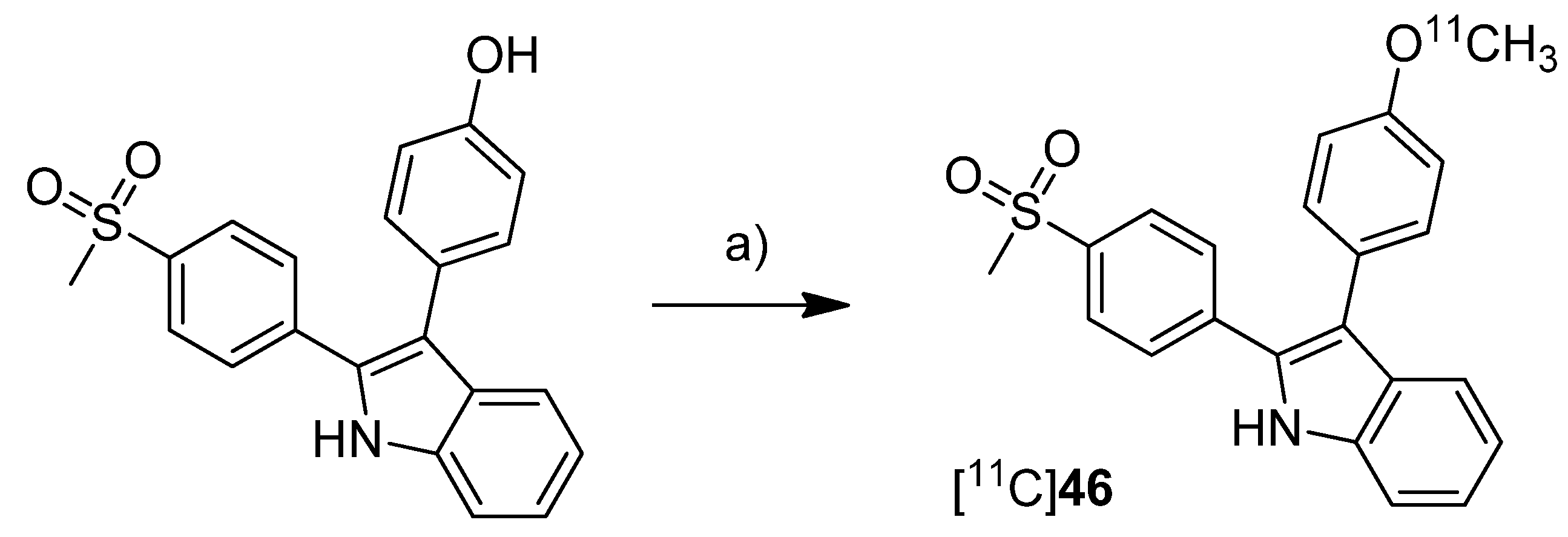
Synthesis: [
11C]
46 was synthesized by
O-[
11C]methylation from the corresponding desmethyl precursor with [
11C]methyl iodide in 23% decay corrected radiochemical yield with an specific activity of 79–89 GBq/µmol and 99% radiochemical purity.
In vitro: Comparing cell uptake studies were performed with [
11C]
45 and [
18F]
46 using the previously reported [
31] cell lines and models. [
11C]
46 showed like [
18F]
45 substantially higher uptake in COX-2 upregulated cells.
Scheme 23.
Synthesis of [11C]46.
Scheme 23.
Synthesis of [11C]46.
Reagents and conditions: (a) [11C]CH3I, 0.1 M NaOH, acetone, 80 °C, 2 min.
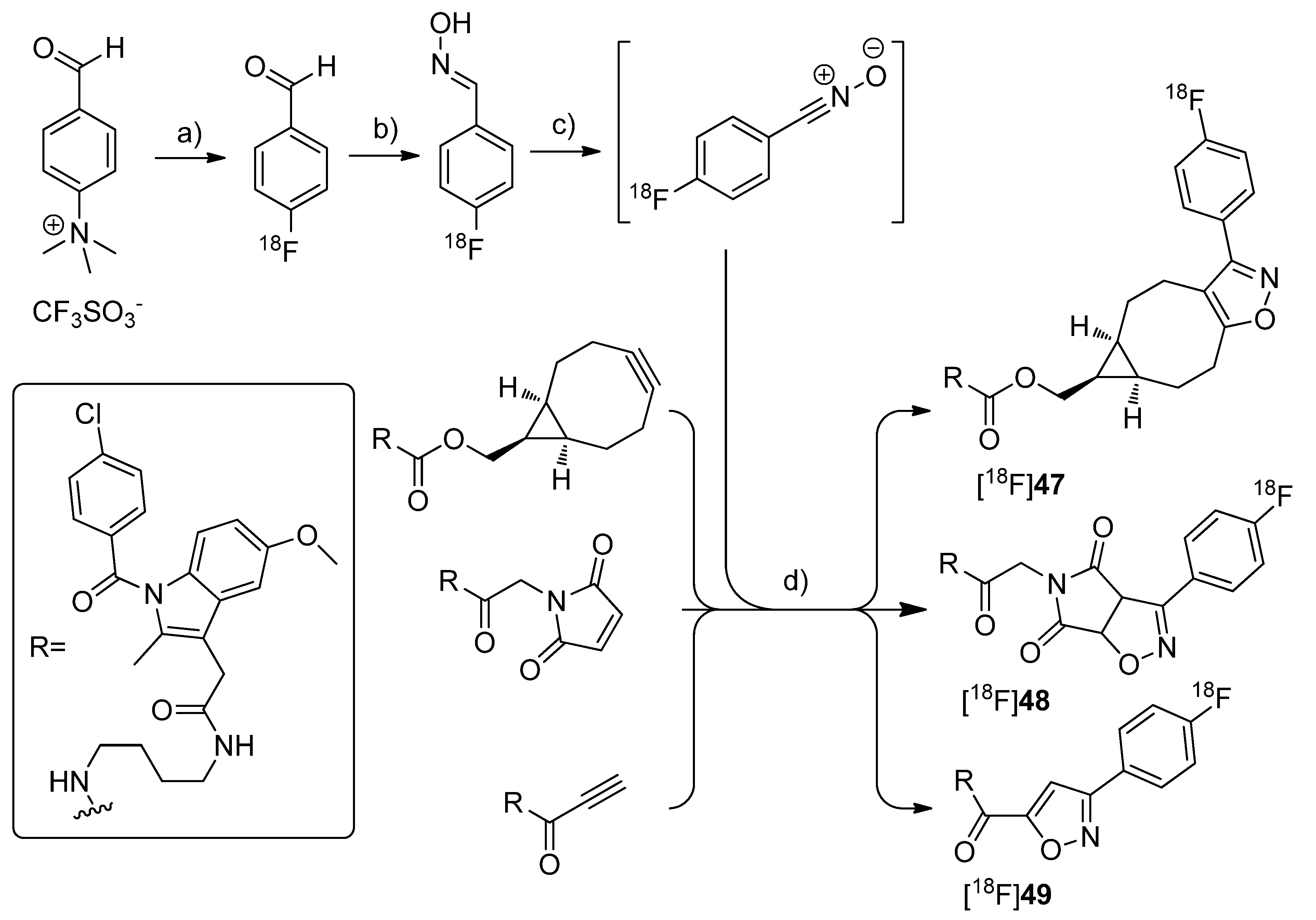
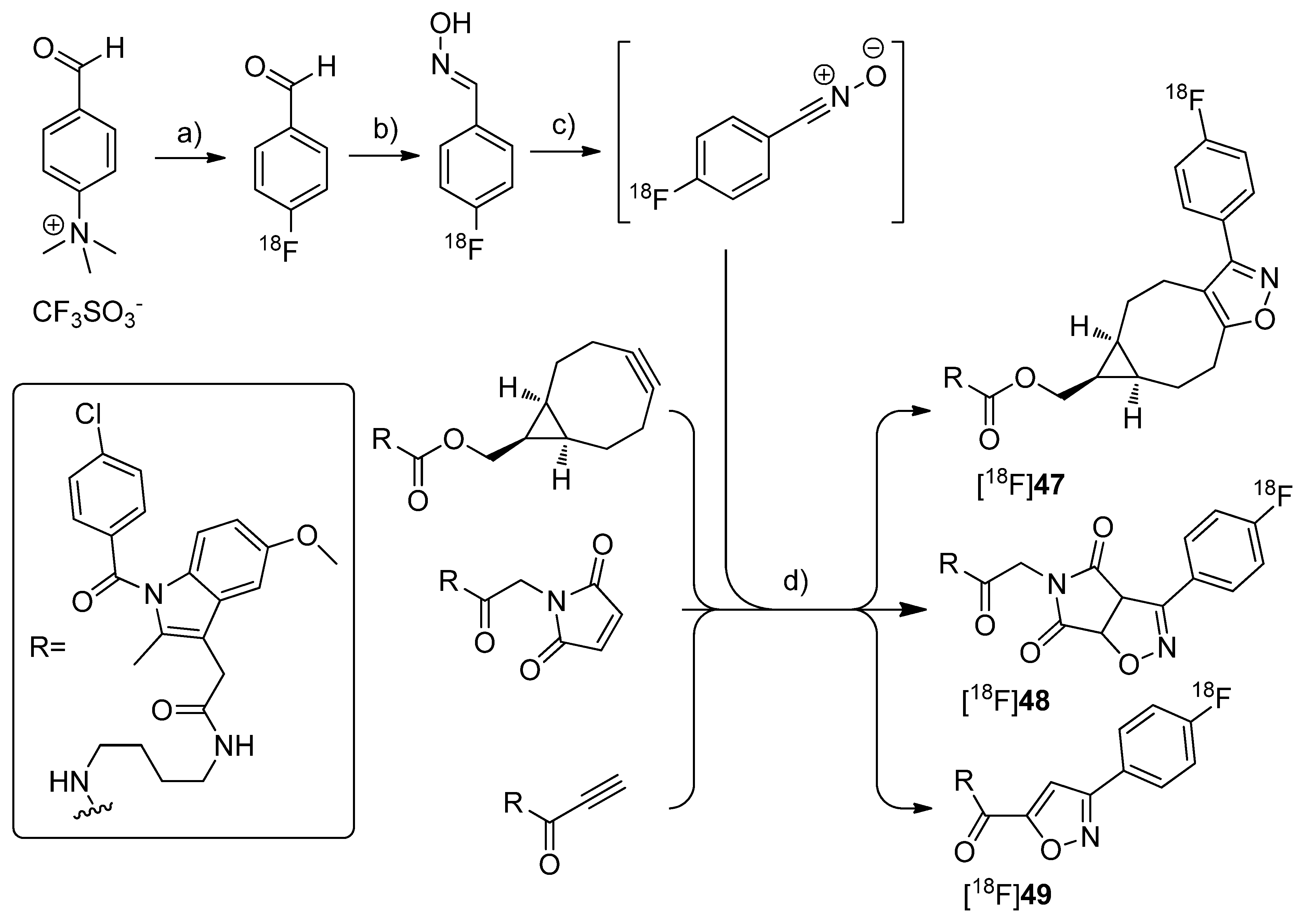
By demonstrating the potential of a new developed fluorine-18-labeling method, the metal-free [3+2]-nitrile oxide/alkyne cycloaddition, Zlatopolskiy
et al. synthesized three new isoxazole analogs of indomethacin esters [
18F]
47–
49 as potential COX-2 specific tracers [
78] (
Scheme 24). Similar fluorescent-dye labeled derivatives of indomethacin have been described by Uddin
et al. as the first
in vivo fluorescent optical imaging agents for COX-2 [
79].
Scheme 24.
Synthesis of [18F]47–49.
Scheme 24.
Synthesis of [18F]47–49.
Reagents and conditions: (a) [18F]KF,K222, DMSO, 85 °C, 10 min; (b) NH2OH·HCl, 1 M NaOH, MeOH, 40 °C, 10 min; (c) PIFA in MeOH for [18F]47 and [18F]48, BAIB in DMF for [18F]49, 5–10 s; (d) MeOH, r.t. for [18F]47, 40 °C for [18F]48, 60 °C for [18F]49, 10 min.
Synthesis: [18F]47–49 were synthesized in a four step procedure which involved the [3+2]-nitrile oxide/alkyne cycloaddition as final key step. 4-[18F]fluorobenzonitrile oxide was synthesized starting from [18F]fluoride and the trimethylammonium benzaldehyde precursor to yield [18F]fluorobenzaldehyde in the first step in 30-50% radiochemical yield within 50 min synthesis time. Afterwards, [18F]fluorobenzaldehyde was converted into the respective aldoxime by reaction with hydroxylamine hydrochloride and NaOH in methanol at 40 °C for 10 min with 92.1 ± 2.4% radiochemical yield (n = 23). To the crude 4-[18F]fluorobenzaldoxime (radiochemical purity > 90%) was then added phenyl iodine bis(trifluoroacetate) (PIFA) in case of [18F]47–48 and [bis(acetoxy)iodo]benzene (BAIB) in case of [18F]49 to generate in situ 4-[18F]fluorobenzonitrile oxide within 5–10 s. The in situ generated nitrile oxide was allowed to react with the appropriate labeling precursor for 10 min at room temperature, 40 °C and 60 °C to yield [18F]47 in 81.4 ± 2.0%, [18F]48 in 54.8 ± 1.3% and [18F]49 in 35.3 ± 4.7% radiochemical yield (n = 3) after preparative HPLC, respectively. Finally, [18F]47–49 were formulated in 20% ethanol for further biological experiments. In vitro/In vivo: A biological study is in progress but results are not published yet.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
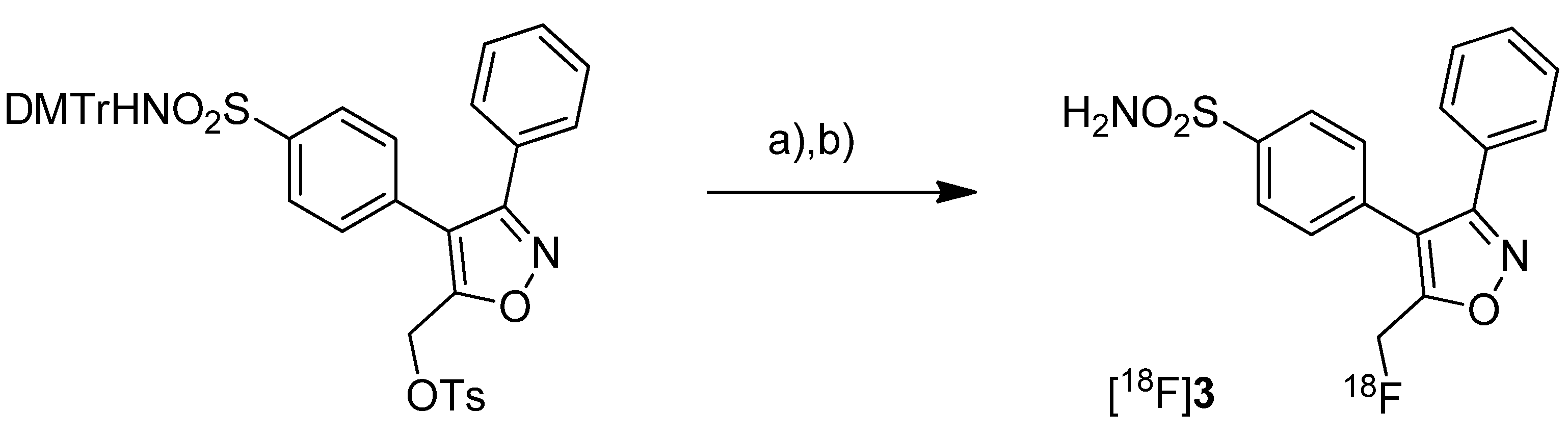{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}