Chitosan for Gene Delivery and Orthopedic Tissue Engineering Applications

Abstract
1. Introduction
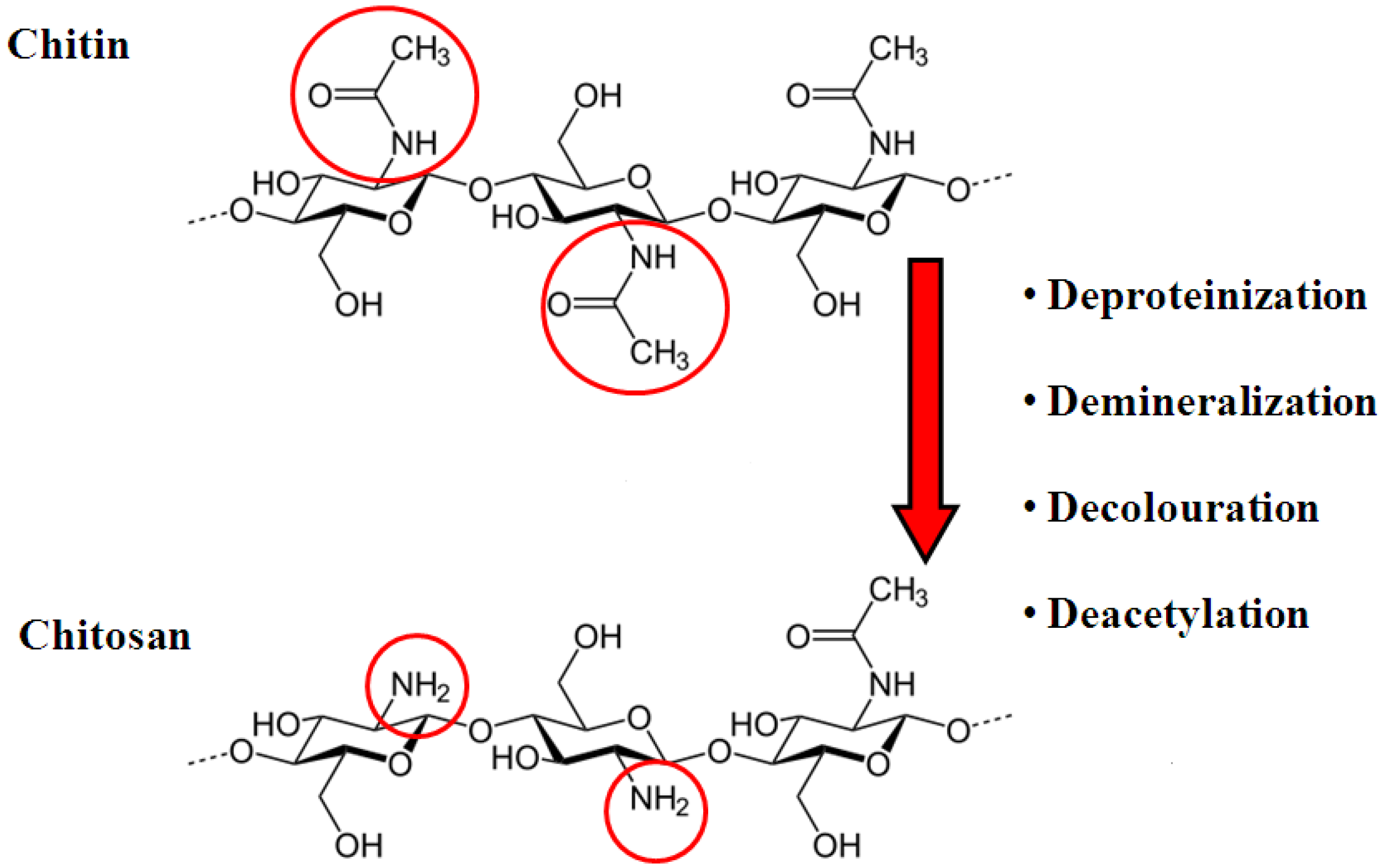
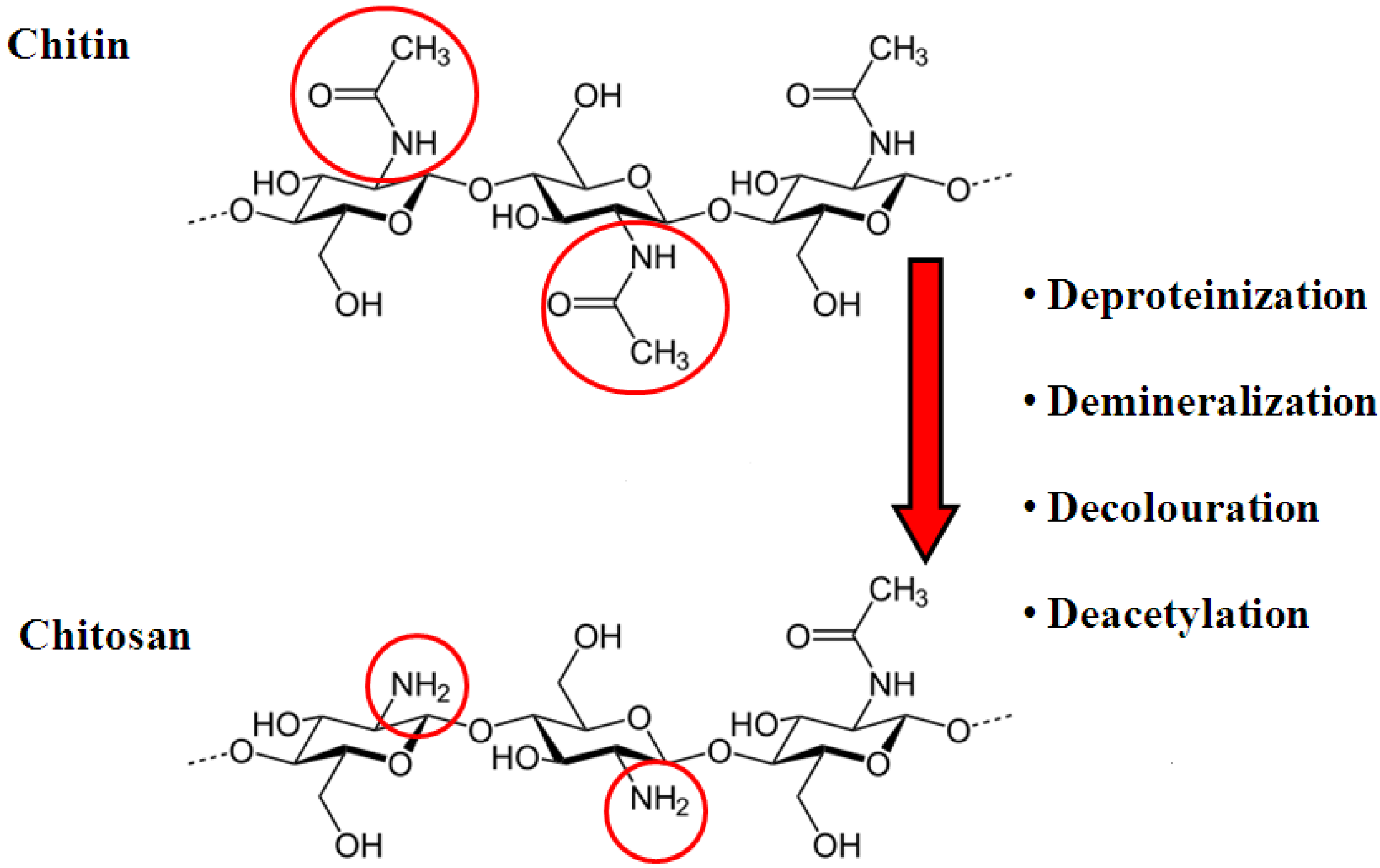
2. Chitosan
3. Gene Therapy
3.1. Viral Vectors
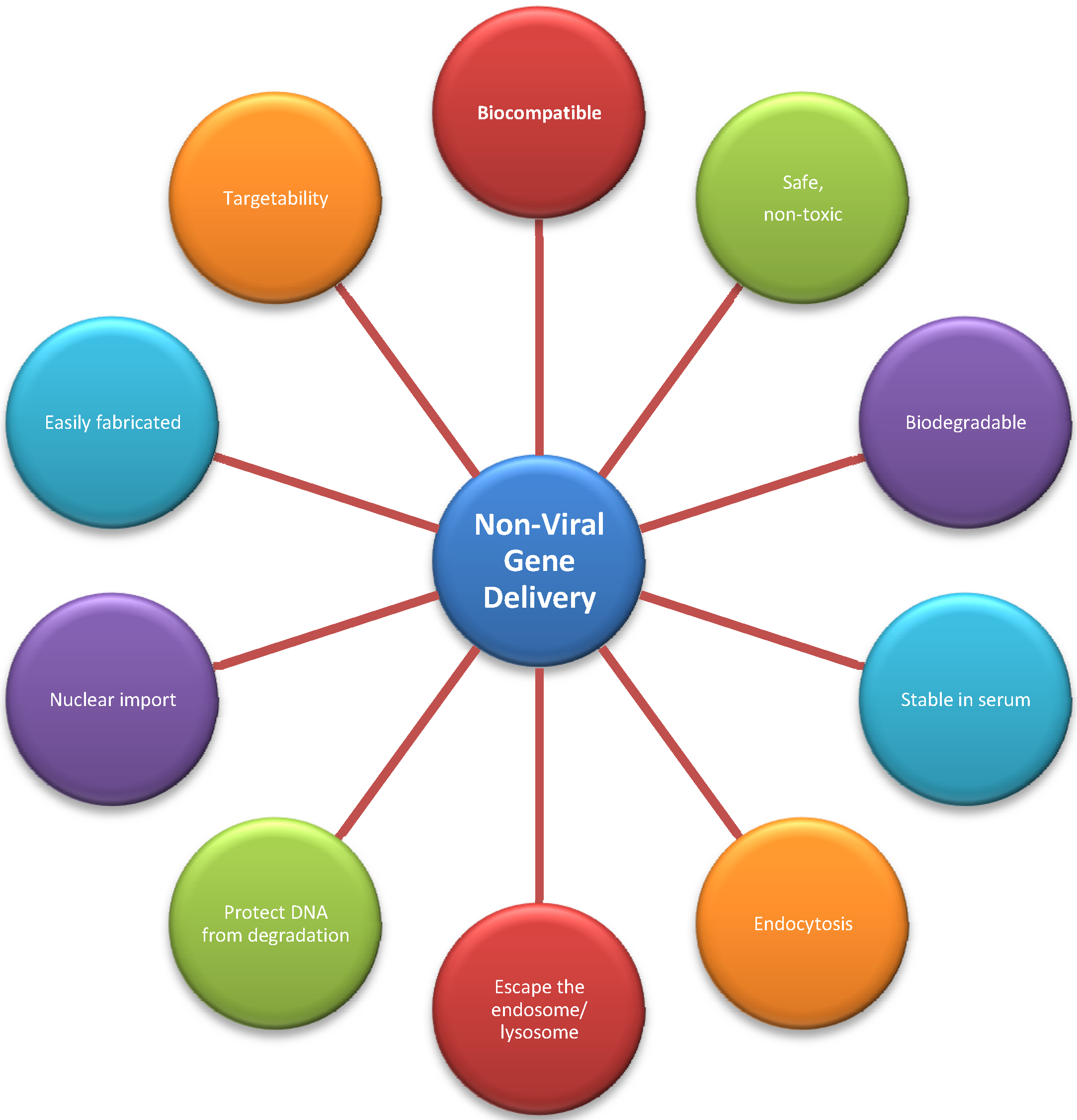
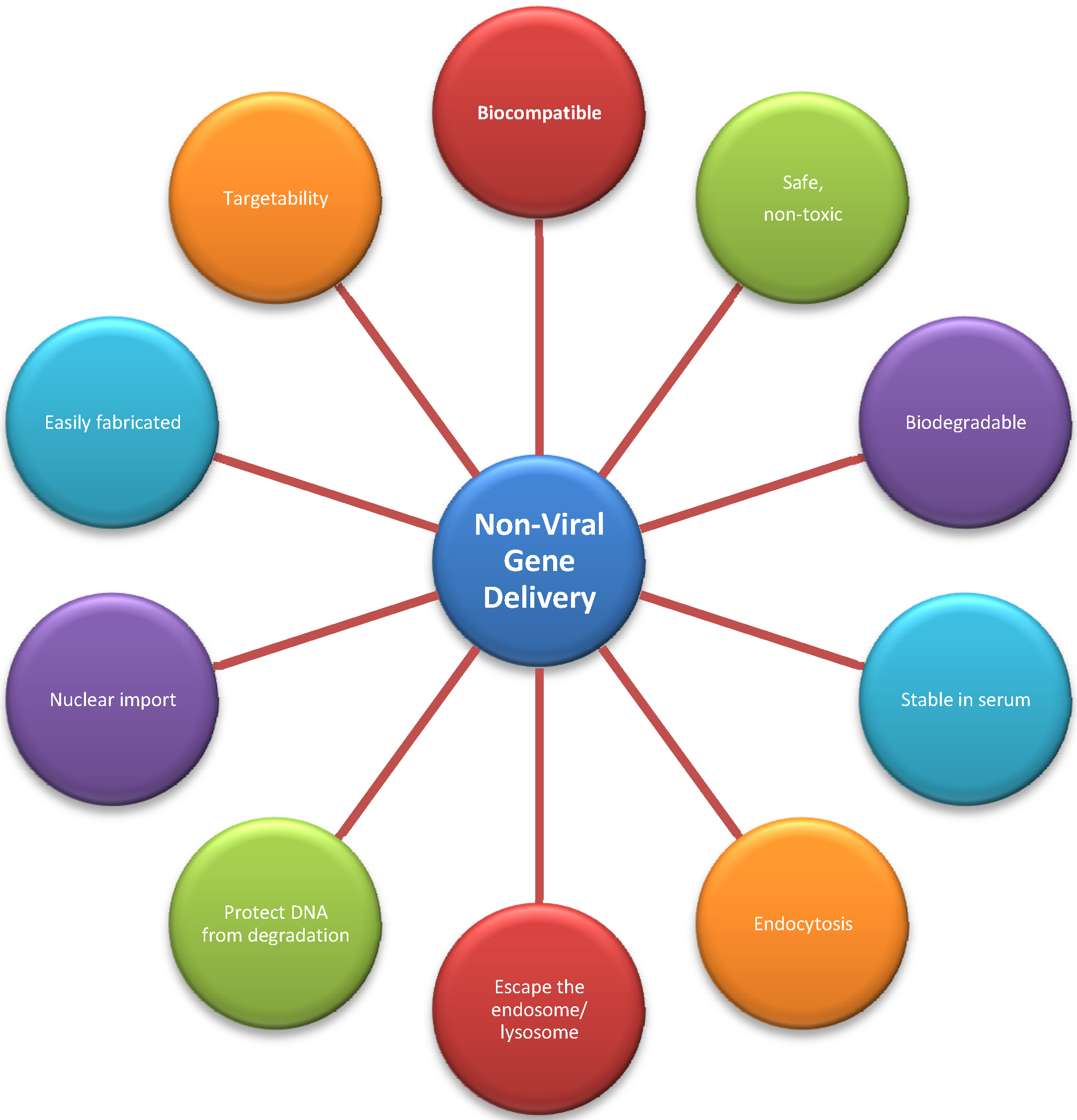
3.2. Non-Viral Vectors
4. Chitosan and Gene Therapy
4.1. Methods of Preparation of Chitosan-Nucleic Acid Complexes
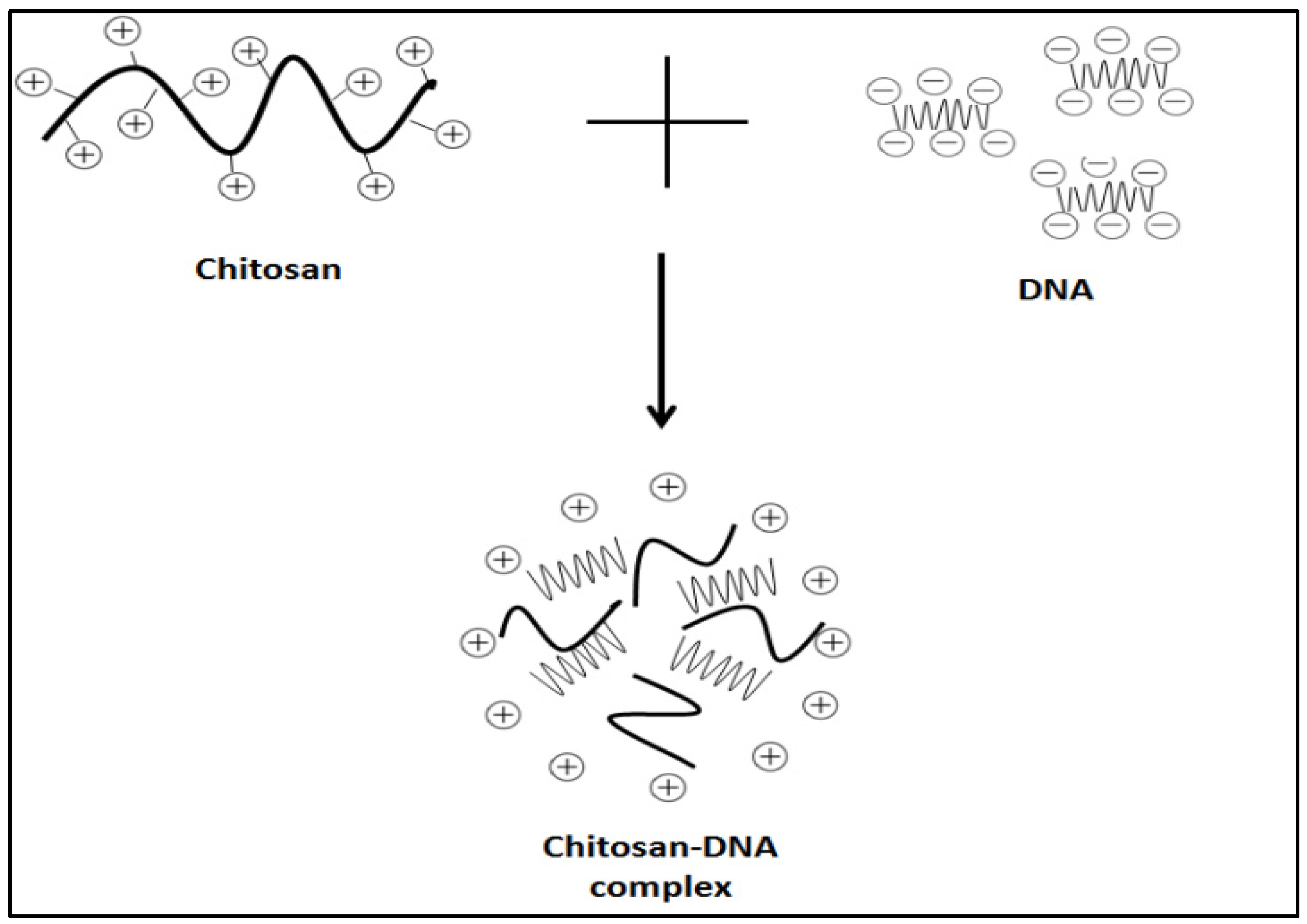
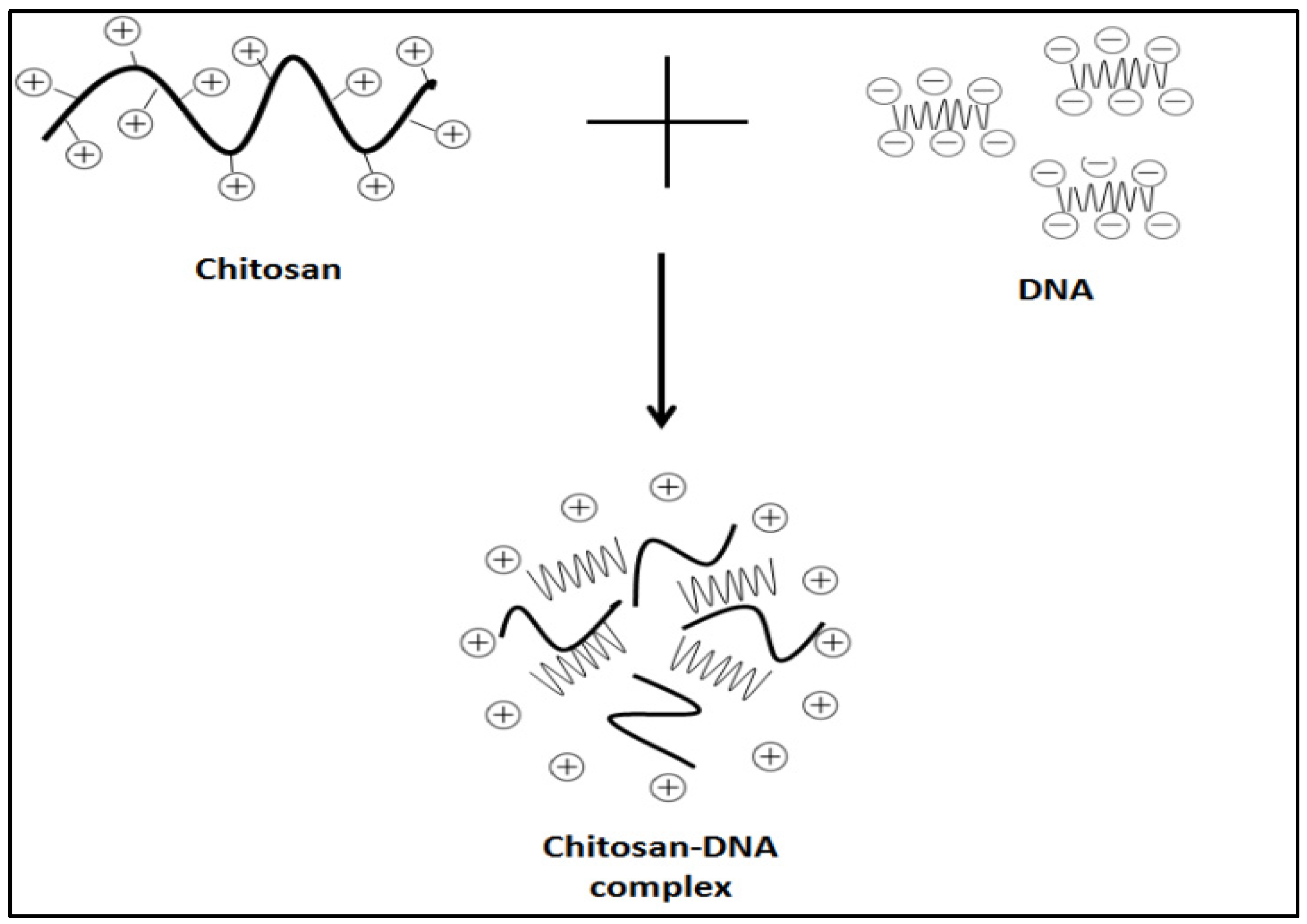
4.1.1. Electrostatic Interaction
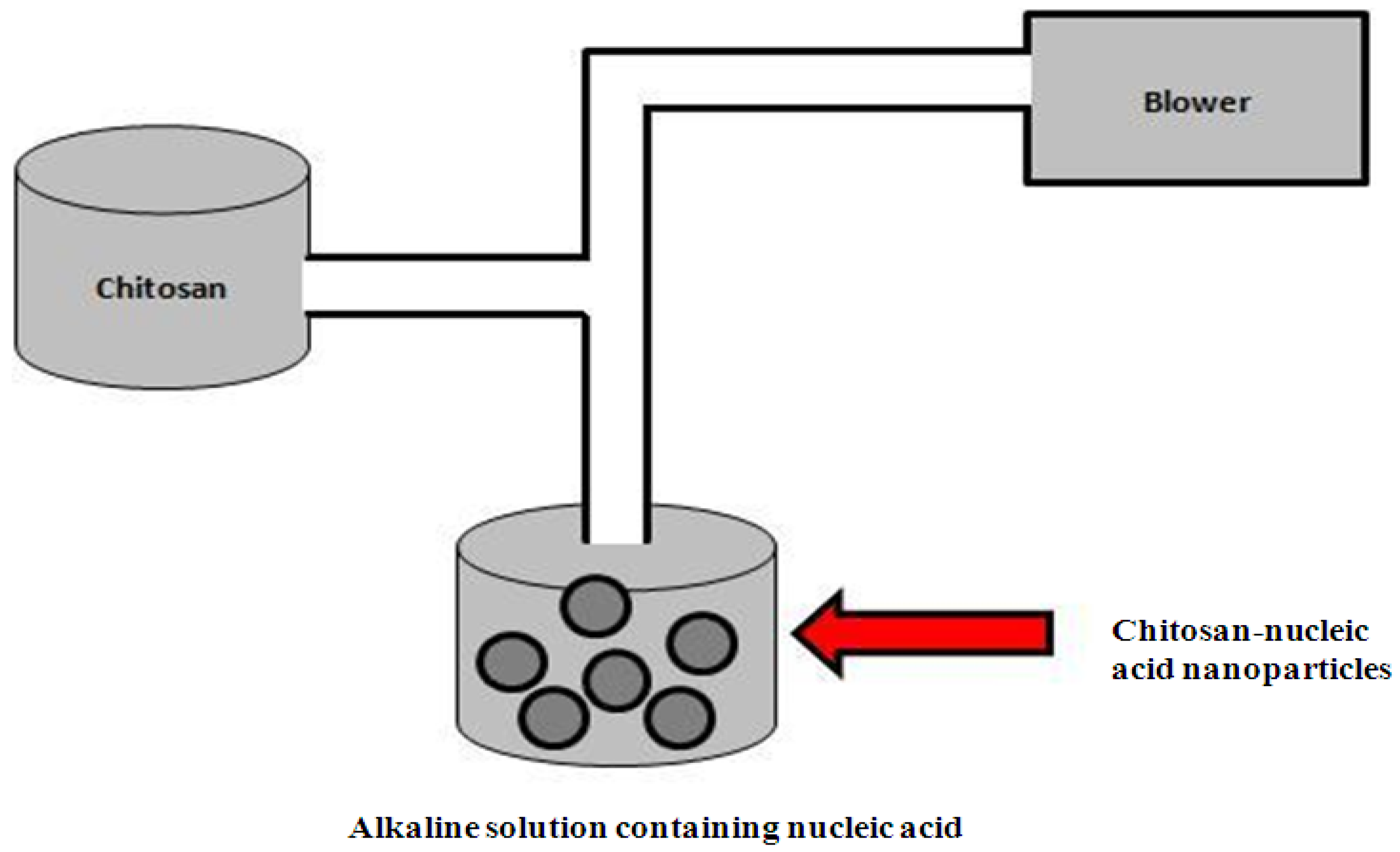
4.1.2. Ionic Gelation
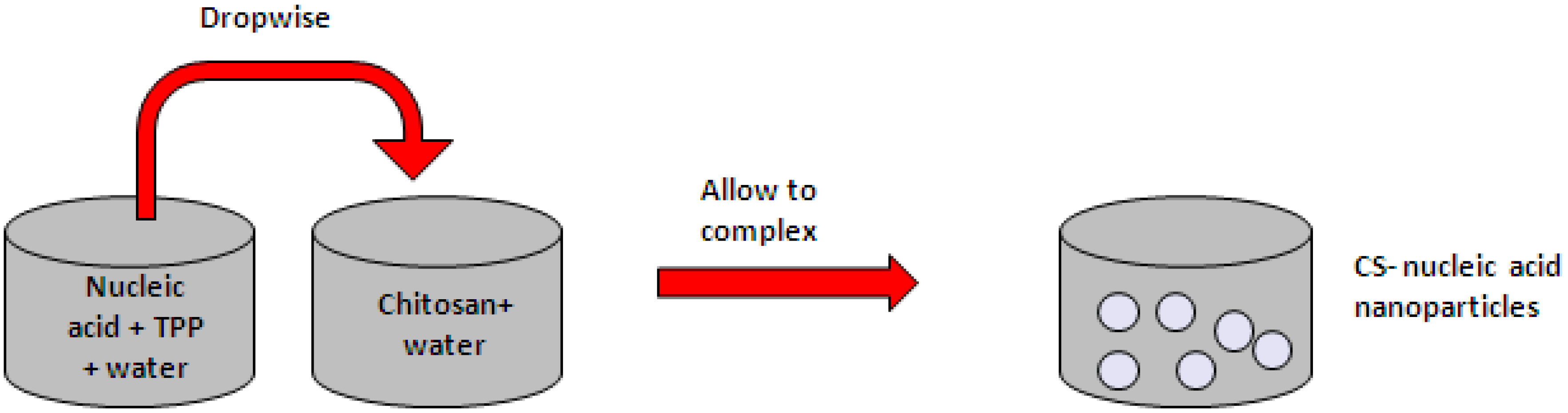
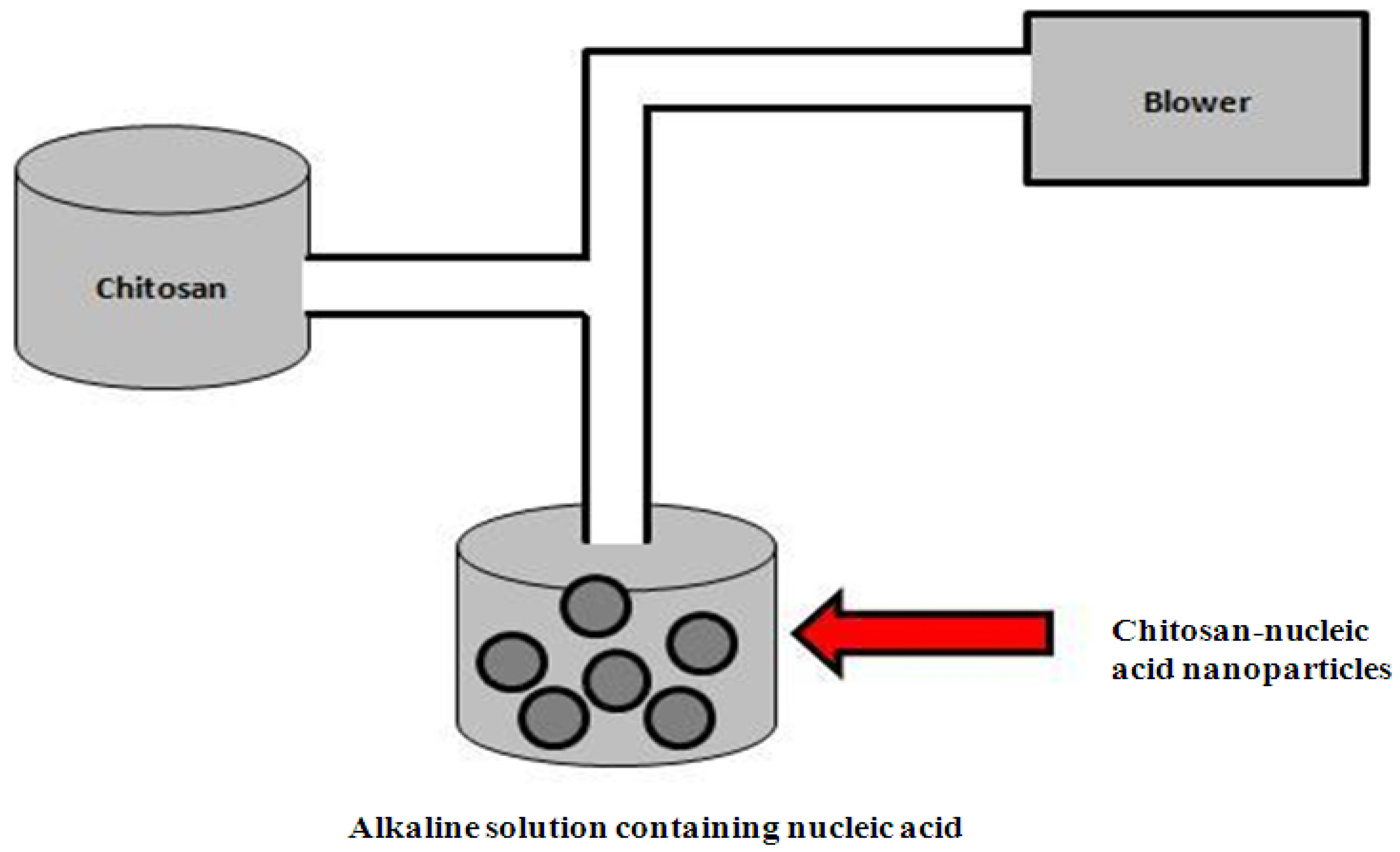
4.1.3. Complex Coacervation
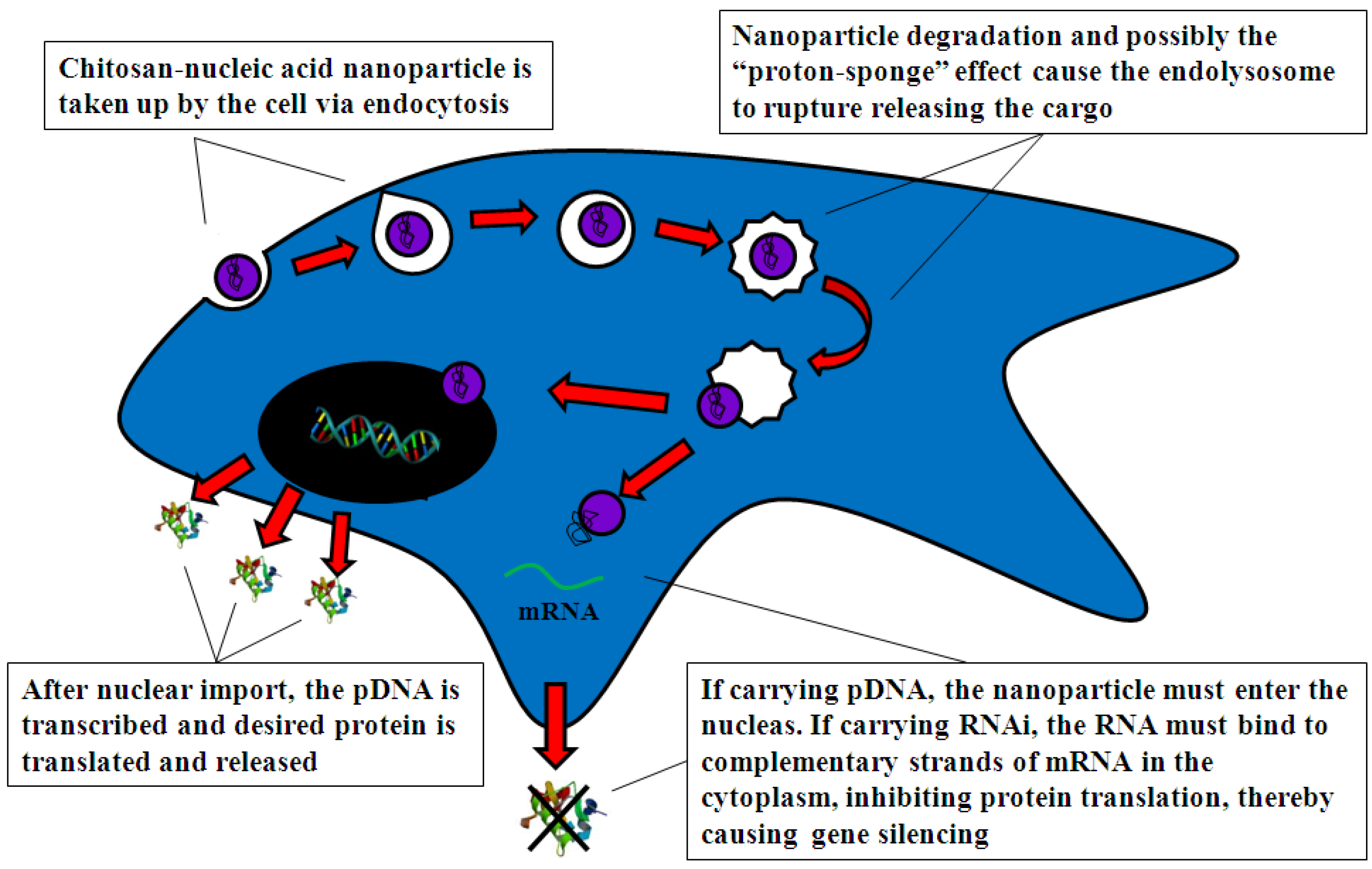
4.2. Mechanism of Chitosan-Nucleic Acid Complex Transfection
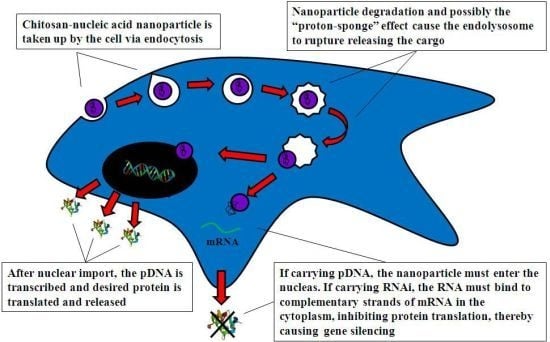
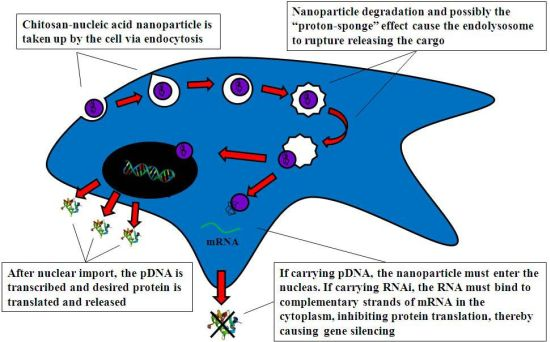
4.2.1. Cell Uptake
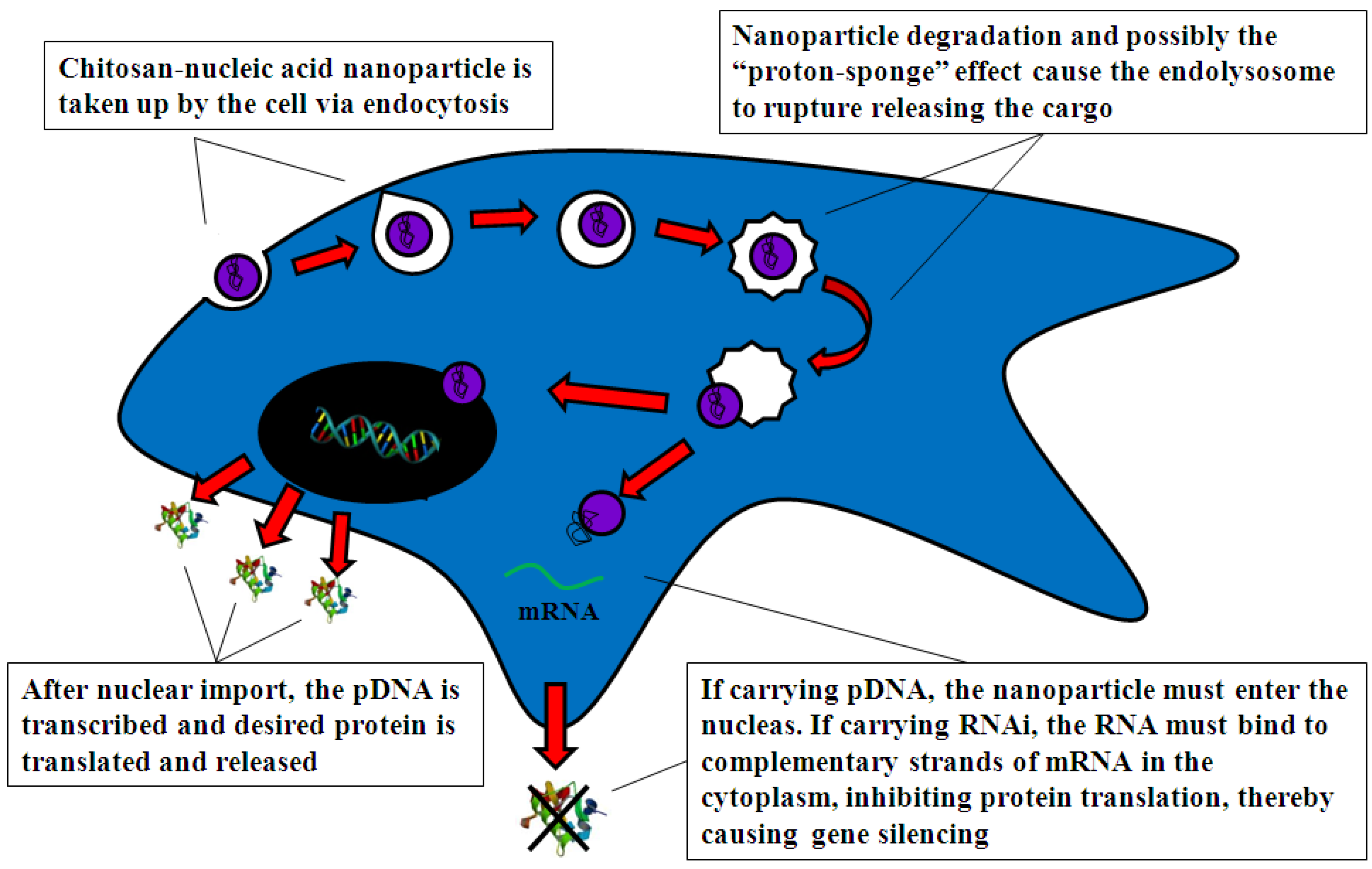
4.2.2. Intracellular Trafficking
4.2.3. Nuclear Import
4.3. Factors Affecting Chitosan-Nucleic Acid Transfection Efficiency
4.3.1. Molecular Weight
4.3.2. Degree of Deacetylation
4.3.3. N/P Ratio
4.3.4. Nucleic Acid Concentration
4.3.5. Nucleic Acid Dose
4.3.6. pH of Transfection Medium
4.3.7. Serum Content
4.3.8. Stability of Chitosan-Nucleic Acid Complexes
4.3.9. Toxicity of Chitosan Vectors
4.3.10. Modifications to Chitosan to Increase Transfection Efficiency
4.4. Cell Types Transfected By Chitosan Vectors in Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chitosan-pDNA complexes Cell Lines | |||||||
|---|---|---|---|---|---|---|---|
| Cell type | Origin | Mw | DNA content | +/− serum | pH of media | Transfection efficiency | Ref. |
| HEK293 | Murine | 390 kDa | 0.1–5 µg/well | + | 7.4 | 15%–20% | [73] |
| 390 kDa | 0.1–5 µg/well | + | 7.4 | 1%–18% (DNA dose dependent) | [15] | ||
| 160 kDa | 0.33 µg/well | - | 7.4 | 25% | [20] | ||
| 150, 400, 600 kDa | 5–10 µg/well | + | 7.4 | 104pg βgal/mg protein | [48] | ||
| 10, 40, 80, 150 kDa | 2.5 µg/well | + | 6.5 & 7.1 | 0%–40% (Mw, DD, N/P, pH dependent) | [104] | ||
| 113 kDa | 1 µg/well | + | 7.4 | 25% | [72] | ||
| 4.7, 8, 11.6, 16.4, 24.8, 32.9, 146 kD | 0.33 µg/well | - | 7 | 5%–60% (Mw, N/P dependent) | [66] | ||
| A549 | Human | 52 kDa, | 10 µg/well | + | 6.9 | 10 × 104 RLU | [111] |
| COS-1 | Simian | 7, 24, 32, 49, 74, 86, 92, 102, 230 and 540 kDa | 10 µg/well | + and − | 7.4 | + serum: 1 × 106 RLU/mg protein (102 kDa) | [94] |
| −serum: 7.5 × 105 RLU/mg protein (540 kDa) | |||||||
| HeLa | Human | (1) 52 kDa | 10 µg/well | + | 6.9 | [111] | |
| (2) 70 kDa | 6 µg/well | + | 7.4 | 106–108 RLU/mg protein | [87] | ||
| (3) 390 kDa | 0.1–5 µg/well | + | 7.4 | No transfection | [73] | ||
| Primary cells | |||||||
| MG63 | Human | 150, 400, 600 kDa | 5–10 µg/well | + | 7.4 | No transfection | [48] |
| MSCs | Human | 150, 400, 600 kDa | 5–10 µg/well | + | 7.4 | No transfection | [48] |
| Chitosan-RNAi ComplexesCell Lines | |||||||
|---|---|---|---|---|---|---|---|
| Cell type | Origin | Mw | RNAi content | +/− serum | pH | Gene Silencing Efficiency | Ref. |
| CHO K1 | Hamster | Chitosan hydrochloride 110 and 270 kDa; Chitosan Glutamate 160 and 470 kDa. | 4 pmol/well (96 well plate) | + | 7.4 | Up to 82% with 470 kDa formulation | [68] |
| HEK 293 | Human | Chitosan hydrochloride 110 and 270 kDa Chitosan Glutamate 160 and 470 kDa | 4 pmol/well (96 well plate) | + | 7.4 | Up to 44% with 470 kDa formulation | [68] |
| H1299 | Human | (1) 8.9–173 kDa (2) 114 kDa (3) 44–143 kDa | 50 nmol/well (24 well plate); 50 nmol/well (24 well plate); 37.5, 75 and 150 nmol/well(96 well plate). | - + and − + | 7.4 7.4 7.4 | Up to 80% at high Mw 77.9% Up to 80% | [100] [112] [113] |
| HepG2 | Human | 11.8 kDa | 10 pmol/well (96 well plate); 60 pmol/well (24 well plate). | + | 6.5 | 55% | [114] |
| LS174T | Human | 11.8–110.9 kDa | 10 pmol/well (96 well plate); 60 pmol/well (24 well plate). | + | 6.5 | 80% at low Mw | [114] |
| Primary cells | |||||||
| Peritoneal Macrophages | Human | 114 kDa | 50, 100, 200 nmol/well (24 well plate) | - | 7.4 | 86.9% | [112] |
4.5. Chitosan Vectors in Vivo
- (1)
- Ex vivo transfection refers to the transfection of cellsin vitro before applying them to the defect site or seeding them onto a scaffold and implanting that at the defect site. This method can involve extensive cell culture thus increasing the risk of cell contamination [120].
- (2)
- Gene therapy complexes can be injected in vivo into the systemic circulation or implanted directly into a defect site. The theory is that endogenous cells can endocytose the complexes and become transfected. However, there is a risk of rapid excretion and off-target transfection [121].
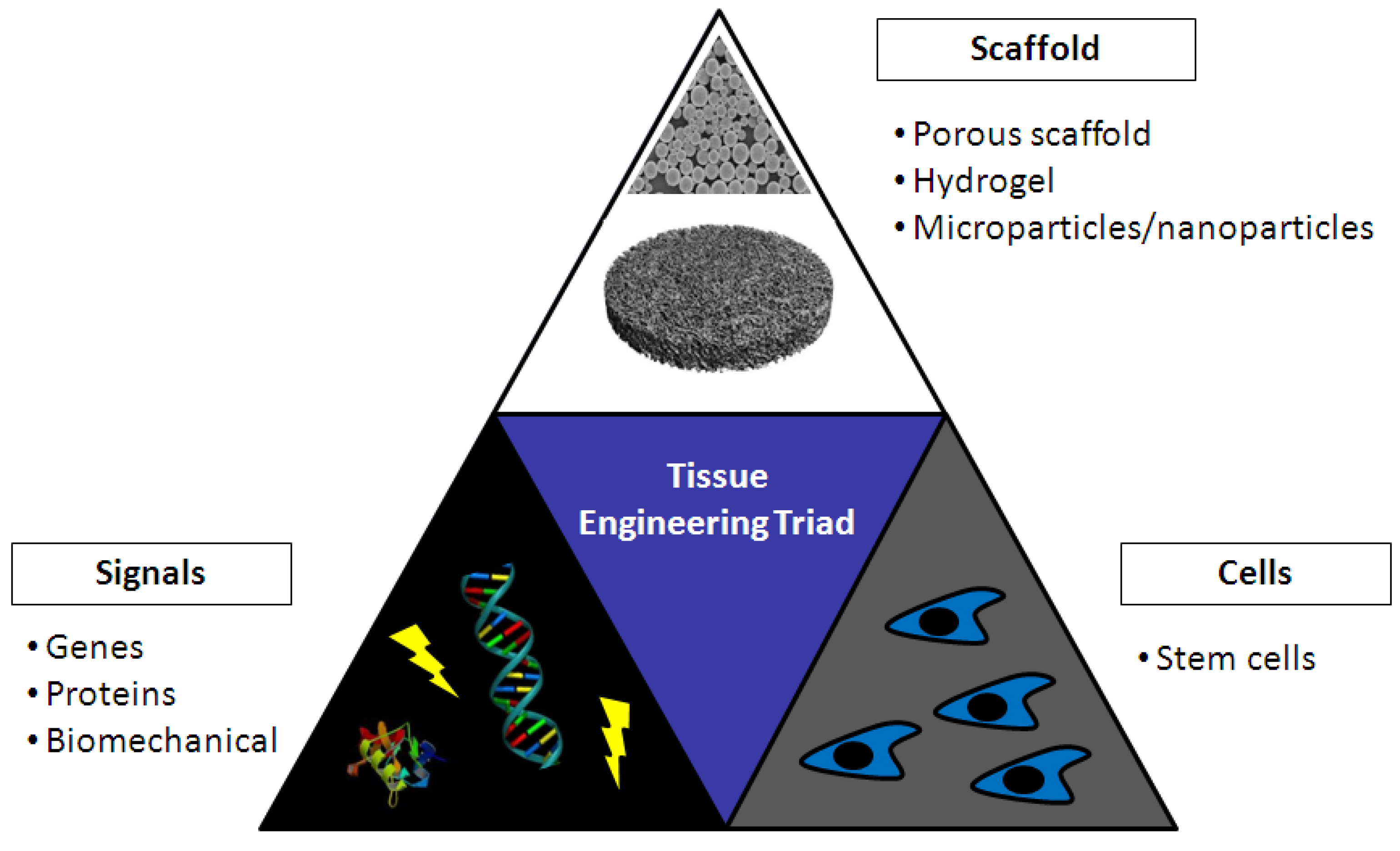
5. Tissue Engineering
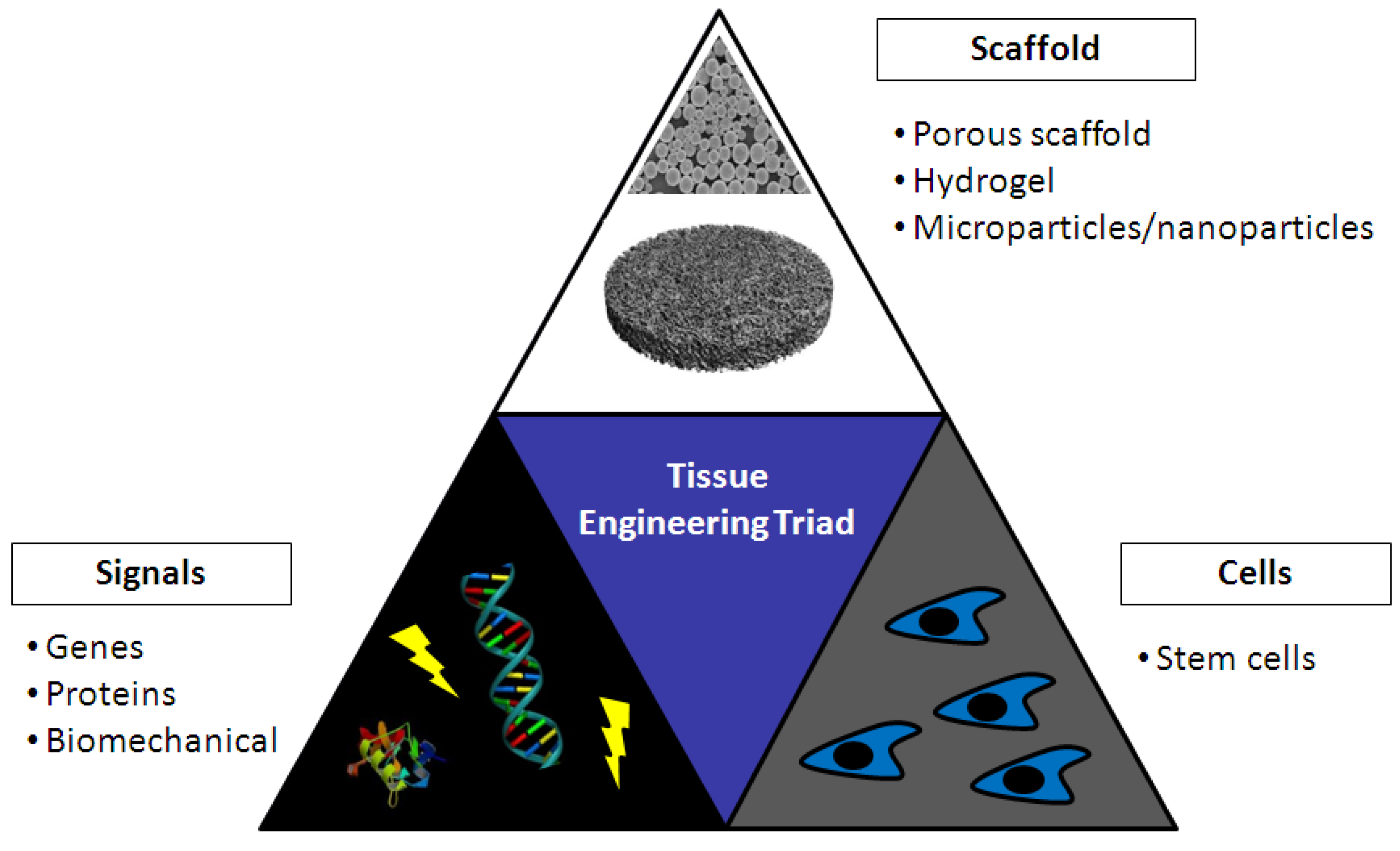
5.1. Chitosan in Bone Tissue Engineering
5.2. Chitosan in Cartilage Tissue Engineering
6. Gene Activated Matrices (GAMs)
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Friedmann, T.; Roblin, R. Gene therapy for human genetic disease. Science 1972, 175, 949–955. [Google Scholar]
- Alton, E. Progress and Prospects: Gene therapy clinical trials (Part 1). Gene Ther. 2007, 14, 1439–1447. [Google Scholar]
- Alton, E. Progress and Prospects: Gene therapy clinical trials (Part 2). Gene Ther. 2007, 14, 1555–1563. [Google Scholar]
- Hacein-Bey-Abina, S.; le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; de Villartay, J.P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S.; et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002, 346, 1185–1193. [Google Scholar]
- Edelstein, M.L.; Abedi, M.R.; Wixon, J. Gene therapy clinical trials worldwide to 2007 — An update. J. Gene Med. 2007, 9, 833–842. [Google Scholar]
- Malafaya, P.B.; Silva, G.A.; Reis, R.L. Natural-origin polymers as carriers and scaffolds for biomolecules and cell delivery in tissue engineering applications. Adv. Drug Deliv. Rev. 2007, 59, 207–233. [Google Scholar]
- Miyazaki, M.; Tsumara, H.; Wang, J.; Alanay, A. An update on bone substitutes for spinal fusion. Eur. Spine J. 2009, 18, 783–799. [Google Scholar]
- Lee, J.E.; Kim, K.E.; Kwon, I.C.; Ahn, H.J.; Lee, S.; Cho, H.; Kim, H.J.; Seong, S.C.; Lee, M.C. Effects of the controlled-released TGF-b1 from chitosan microspheres on chondrocytes cultured in a collagen/chitosan/glycosaminoglycan scaffold. Biomaterials 2004, 25, 4163–4173. [Google Scholar]
- Carragee, E.J.; Hurwitz, E.L.; Weiner, B.K. A critical review of recombinant human bone morphogenetic protein-2 trials in spinal surgery: Emerging safety concerns and lessons learned. Spine J. 2011, 11, 471–491. [Google Scholar]
- Jeon, O.; Jin, S.; Seok, H.; Bhang, S.; Kang, S.; Ae, M.; Jong, H.; Kim, B.S. Long-term delivery enhances in vivo osteogenic efficacy of bone morphogenetic protein-2 compared to short-term delivery. Biochem. Biophys. Res. Commun. 2008, 369, 774–780. [Google Scholar]
- Kumar, M.N.; Muzzarelli, R.A.; Muzzarelli, C.; Sashiwa, H.; Domb, A.J. Chitosan chemistry and pharmaceutical perspectives. Chem. Rev. 2004, 104, 6017–6084. [Google Scholar]
- Jeuniaux, C.; Voss-Foucart, M.F. Chitin biomass and production in the marine environment. Biochem. Syst. Ecol. 1991, 19, 347–356. [Google Scholar]
- Tran, D.L.; Pham, G.D.; Nguyen, X.P.; Vu, D.H.; Nguyen, N.T.; Tran, V.H.; Mai, T.T.T.; Nguyen, H.B.; Le, Q.D.; Nguyen, T.N.; et al. Some biomedical applications of chitosan-based hybrid nanomaterials. Adv. Nat. Sci.: Nanosci. Nanotechnol. 2011, 2. [Google Scholar] [CrossRef]
- Shi, C.; Zhu, Y.; Ran, X.; Wang, M.; Su, Y.; Cheng, T. Therapeutic potential of chitosan and its derivatives. J. Surg. Res. 2006, 133, 185–192. [Google Scholar]
- Mao, H.Q.; Roy, K.; Troung, V.L.; Janes, K.A.; Lin, K.Y.; Wang, Y.; August, J.T.; Leong, K.W. Chitosan-DNA nanoparticles as gene carriers: Synthesis, characterization and transfection efficiency. J. Control. Release 2001, 70, 399–421. [Google Scholar]
- Chandy, T.; Sharma, C.P. Chitosan-as a biomaterial. Artif. Cells Nanomedicine Biotechnol. 1990, 18, 1–24. [Google Scholar]
- Illum, L. Chitosan and its use as a pharmaceutical excipient. Pharm. Res. 1998, 15, 1326–1331. [Google Scholar] [CrossRef]
- Kean, T.; Thanou, M. Biodegradation, biodistribution and toxicity of chitosan. Adv. Drug Deliv. Rev. 2010, 62, 3–11. [Google Scholar] [CrossRef]
- Madihally, S.V.; Matthew, H.W.T. Porous chitosan scaffolds for tissue engineering. Biomaterials 1999, 20, 1133–1142. [Google Scholar]
- Köpping-Höggård, M.; Tubulekas, I.; Guan, H.; Edwards, K.; Nilsson, M.; Varum, K.; Artursson, P. Chitosan as a nonviral gene delivery system. Structure-property relationships and characteristics compared with polyethylenimine in vitro and after lung administration in vivo. Gene Ther. 2001, 8, 1108–1121. [Google Scholar]
- Chung, M.J.; Park, J.K.; Park, Y.I. Anti-inflammatory effects of low-molecular weight chitosan oligosaccharides in IgE-antigen complex-stimulated RBL-2H3 cells and asthma model mice. Int. Immunopharmacol. 2012, 12, 453–459. [Google Scholar]
- Köpping-Höggård, M.; Mel’nikova, Y.S.; Vårum, K.M.; Lindman, B.; Artursson, P. Relationship between the physical shape and the efficiency of oligomeric chitosan as a gene delivery system in vitro and in vivo. J. Gene Med. 2003, 5, 130–141. [Google Scholar]
- Roy, K.; Mao, H.Q.; Huang, S.K.; Leong, K.W. Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat. Med. 1999, 6, 387–391. [Google Scholar]
- Bonadio, J.; Smiley, E.; Patil, P.; Goldstein, S. Localized, direct plasmid gene delivery in vivo: Prolonged therapy results in reproducible tissue regeneration. Nat. Med. 1999, 6, 753–759. [Google Scholar]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 23–36. [Google Scholar] [CrossRef]
- Guo, P.; Coban, O.; Snead, N.M.; Trebley, J.; Hoeprich, S.; Guo, S.; Shu, Y. Engineering RNA for targeted siRNA delivery and medical application. Adv. Drug Deliv. Rev. 2010, 62, 650–666. [Google Scholar] [CrossRef]
- Van Rooij, E. The art of microrna research. Circ. Res. 2011, 108, 219–234. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function genomics: The miRNA genes. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Zhang, X.; Godbey, W.T. Viral vectors for gene delivery in tissue engineering. Adv. Drug Deliv. Rev. 2006, 58, 515–534. [Google Scholar] [CrossRef]
- Parker, A.L.; Newman, C.; Briggs, S.; Seymour, L.; Sheridan, P.J. Nonviral gene delivery: Techniques and implications for molecular medicine. Expert Rev. Mol. Med. 2003, 5, 1–15. [Google Scholar]
- Davis, M.E. Non-viral gene delivery systems. Curr. Opin. Biotechnol. 2002, 13, 128–131. [Google Scholar] [CrossRef]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar]
- Patil, S.D.; Rhodes, D.G.; Burgess, D.J. DNA-based therapeutics and DNA delivery systems: A comprehensive review. AAPS J. 2005, 7, E61–E77. [Google Scholar] [CrossRef]
- Walther, W.; Stein, U. Viral vectors for gene transfer a review of their use in the treatment of human diseases. Drugs 2000, 60, 249–271. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious. Nature 2001, 7, 33–40. [Google Scholar] [CrossRef]
- Hacein-bey-abina, S.; von Kalle, C.; Schmidt, M.; le Deist, F.; Wulffraat, N.; McIntyre, E.; Radfors, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for x-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef]
- Atkinson, H.; Chalmers, R. Delivering the goods: Viral and non-viral gene therapy systems and the inherent limits on cargo DNA and internal sequences. Genetica 2010, 138, 485–498. [Google Scholar] [CrossRef]
- Partridge, K.A.; Oreffo, R.O.C. Gene delivery in bone tissue engineering: Progress and prospects using viral and nonviral strategies. Tissue Eng. 2004, 10, 295–307. [Google Scholar] [CrossRef]
- Lundstrom, K.; Boulikas, T. Viral and non-viral vectors in gene therapy: Technology development and clinical trials. Technol. Cancer Res. Treat. 2003, 2, 471–485. [Google Scholar]
- Djurovic, S.; Iversen, N.; Jeansson, S.; Hoover, F.; Christensen, G. Comparison of nonviral transfection and adeno-associated viral transduction on cardiomyocytes. Mol. Biotechnol. 2004, 28, 21–31. [Google Scholar] [CrossRef]
- Kay, M.A. State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 2011, 12, 316–328. [Google Scholar] [CrossRef]
- Regnier, V.; Tahiri, A.; Andre, N.; Lemaitre, M.; Doan, T.L.; Preat, V. Electroporation-mediated delivery of 3'-protected phosphodiester oligodeoxynucleotides to the skin. J. Control. Release 2000, 67, 337–346. [Google Scholar] [CrossRef]
- Akaneya, Y.; Jiang, B.; Tsumoto, T. RNAi-induced gene silencing by local electroporation in targeting brain region. J. Neurophysiol. 2005, 93, 594–602. [Google Scholar] [CrossRef]
- McAllister, D.V.; Allen, M.G.; Prausnitz, M.R. Microfabricated microneedles for gene and drug delivery. Annu. Rev. Biomed. Eng. 2000, 2, 289–313. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadeiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Davis, H.L.; Whalen, R.G.; Demeneix, B.A. Direct gene transfer into skeletal muscle in vivo: Factors affecting efficiency of transfer and stability of expression. Hum. Gene Ther. 1993, 159, 151–159. [Google Scholar]
- Corsi, K.; Chellat, F.; Fernandes, J.C. Mesenchymal stem cells, MG63 and HEK293 transfection using chitosan-DNA nanoparticles. Biomaterials 2003, 24, 1255–1264. [Google Scholar]
- Anderson, D.G.; Akinc, A.; Hossain, N.; Langer, R. Structure/property studies of polymeric gene delivery using a library of poly(β-amino esters). Mol. Ther. 2005, 11, 426–434. [Google Scholar]
- Mintzer, M.A.; Simanek, E.E. Nonviral vectors for gene delivery. Chem. Rev. 2009, 109, 259–302. [Google Scholar] [CrossRef]
- Wu, G.Y.; Wu, C.H. Receptor-mediated in vitro gene transformation by a soluble DNA carrier system. J. Biol. Chem. 1987, 262, 4429–4432. [Google Scholar]
- Wu, G.Y.; Wu, C.H. Receptor-mediated gene delivery and expression in vivo. J. Biol. Chem. 1988, 263, 14621–14624. [Google Scholar]
- Wolfert, M.A.; Dash, P.R.; Nazarova, O.; Oupicky, D.; Seymour, L.W.; Smart, S.; Strohalm, J.; Ulbricj, K. Polyelectrolyte vectors for gene delivery: Influence of cationic polymer on biophysical properties of complexes formed with DNA. Bioconjug. Chem. 1999, 10, 993–1004. [Google Scholar]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar]
- Boussif, O.; Lezoualc, F.; Zanta, M.A.; Mergny, M.D.; Schermant, D.; Demeneixt, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar]
- Godbey, W.T.; Wu, K.K.; Mikos, A.G. Poly(ethylenimine ) and its role in gene delivery. J. Control. Release 1999, 60, 149–160. [Google Scholar] [CrossRef]
- Akinc, A.; Thomas, M.; Klibanov, A.M.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005, 7, 657–663. [Google Scholar]
- Tierney, E.G.; Duffy, G.P.; Hibbitts, A.J.; Cryan, S.A.; O'Brien, F.J. The development of non-viral gene-activated matrices for bone regeneration using polyethyleneimine (PEI) and collagen-based scaffolds. J. Control. Release 2012, 158, 304–311. [Google Scholar] [CrossRef]
- Behr, J.P. The proton sponge: A trick to enter cells the viruses did not exploit. Chimia 1997, 2, 34–36. [Google Scholar]
- Regnström, K.; Ragnarsson, E.G.E.; Fryknäs, M.; Köping-Höggård, M.; Artursson, P. Gene expression profiles in mouse lung tissue after administration of two cationic polymers used for nonviral gene delivery. Pharm. Res. 2006, 23, 475–482. [Google Scholar] [CrossRef]
- Dennig, J.; Duncan, E. Gene transfer into eukaryotic cells using activated polyamidoamine dendrimers. Rev. Mol. Biotechnol. 2002, 90, 339–347. [Google Scholar] [CrossRef]
- Gebhart, C.L.; Kabanov, A.V. Evaluation of polyplexes as gene transfer agents. J. Control. Release 2001, 73, 401–416. [Google Scholar] [CrossRef]
- Shakhbazau, A.; Isayenka, I.; Kartel, N.; Goncharova, N.; Seviaryn, I.; Kosmacheva, S.; Potapnev, M.; Shcharbin, D.; Bryszewska, M. Transfection efficiencies of PAMAM dendrimers correlate inversely with their hydrophobicity. Int. J. Pharm. 2010, 383, 228–235. [Google Scholar] [CrossRef]
- Liu, H.; Slamovich, E.B.; Webster, T.J. Less harmful acidic degradation of poly (lactic-co-glycolic acid) bone tissue engineering scaffolds through titania nanoparticle addition. Int. J. Nanomed. 2006, 1, 541–545. [Google Scholar] [CrossRef]
- Mumper, R.J.; Wang, J.; Claspell, J.M.; Rolland, A.P. Novel polymeric condensing carriers for gene delivery. Proc. Int. Symp. Control. Release Bioact. Mater. 1995, 22, 178–179. [Google Scholar]
- Strand, S.P.; Lelu, S.; Reitan, N.K.; Lange, C.D.; Artursson, P.; Vårum, K.M. Molecular design of chitosan gene delivery systems with an optimized balance between polyplex stability and polyplex unpacking. Biomaterials 2010, 31, 975–987. [Google Scholar]
- Mao, S.; Sun, W.; Kissel, T. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 2010, 62, 12–27. [Google Scholar] [CrossRef]
- Katas, H.; Alpar, O. Development and characterisation of chitosan nanoparticles for siRNA delivery. J. Control. Release 2006, 115, 216–225. [Google Scholar] [CrossRef]
- Strand, S.P.; Danielsen, S.; Christensen, B.E.; Vårum, K.M. Influence of chitosan structure on the formation and stability of DNA-chitosan polyelectrolyte complexes. Biomacromolecules 2005, 6, 3357–3366. [Google Scholar] [CrossRef]
- Calvo, P.; Remunan-Lopez, C.; Vila-Jato, J.L.; Alonso, M.J. Novel hydrophilic chitosan–polyethylene oxide nanoparticles as protein carriers. J. Appl. Polym. Sci. 1997, 63, 125–132. [Google Scholar] [CrossRef]
- Gan, Q.; Wang, T.; Cochrane, C.; Mccarron, P. Modulation of surface charge, particle size and morphological properties of chitosan–TPP nanoparticles intended for gene delivery. Colloid. Surface. B 2005, 44, 65–73. [Google Scholar] [CrossRef]
- Csaba, N.; Köpping-Höggård, M.; Alonso, M.J. Ionically crosslinked chitosan/tripolyphosphate nanoparticles for oligonucleotide and plasmid DNA delivery. Int. J. Pharm. 2009, 382, 205–214. [Google Scholar] [CrossRef]
- Leong, K.W.; Roy, K.; Walsh, S.M.; August, J.T. DNA-polycation nanospheres as non-viral gene delivery vehicles. J. Control. Release 1998, 53, 183–193. [Google Scholar] [CrossRef]
- Polk, A.; Amsden, B.; Yao, K.D.; Peng, T.; Goosen, M.F.A. Controlled release of albumin from chitosan—alginate microcapsules. J. Pharm. Sci. 1994, 83, 178–185. [Google Scholar] [CrossRef]
- Liu, L.S.; Liu, S.Q.; Ng, S.Y.; Froix, M.; Ohno, T.; Heller, J. Controlled release of interleukin-2 for tumour immunotherapy using alginate/chitosan porous microspheres. J. Control. Release 1997, 43, 65–74. [Google Scholar] [CrossRef]
- Kawashima, Y.; Handa, T.; Kasai, A.; Takenaka, H.; Lin, S.Y.; Ando, Y. Novel method for the preparation of controlled-release theophylline granules coated with a polyelectrolyte complex of sodium polyphosphate-chitosan. J. Pharm. Sci. 1985, 74, 264–268. [Google Scholar] [CrossRef]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef]
- Fernandez-Urrusano, R.; Calvo, P.; Remunan-Lopez, C.; Vila-Jato, J.L.; Alonso, M.J. Enhancement of nasal absorption of insulin using chitosan nanoparticles. Pharm. Res. 1999, 16, 1576–1581. [Google Scholar] [CrossRef]
- Pan, Y.; Li, Y.J.; Zhao, H.Y.; Zheng, J.M.; Xu, H. Bioadhesive polysaccharide in protein delivery system: Chitosan nanoparticles improve the intestinal absorption of insulin in vivo. Int. J. Pharm. 2002, 249, 139–147. [Google Scholar] [CrossRef]
- Xu, Y.; Du, Y. Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles. Int. J. Pharm. 2003, 250, 215–226. [Google Scholar]
- Ko, J.A.; Park, H.J.; Hwang, S.J.; Park, J.B.; Lee, J.S. Preparation and characterization of chitosan microparticles intended for controlled drug delivery. Int. J. Pharm. 2002, 249, 165–174. [Google Scholar] [CrossRef]
- Thibault, M.; Nimesh, S.; Lavertu, M.; Buschmann, M.D. Intracellular trafficking and decondensation kinetics of chitosan-pDNA polyplexes. Mol. Ther. 2009, 18, 1787–1795. [Google Scholar]
- Dautry-Varsat, A. Receptor-mediated endocytosis: The intracellular journey of transferrin and its receptor. Biochimie 1986, 68, 375–381. [Google Scholar] [CrossRef]
- Deshpande, D.; Toledo-Velasquez, D.; Wang, L.Y.; Malanga, C.J.; Ma, J.K.; Rojanasakul, Y. Receptor-mediated peptide delivery in pulmonary epithelial monolayers. Pharm. Res. 1994, 11, 1121–1126. [Google Scholar] [CrossRef]
- Henry, L.J.; Xia, D.I.; Wilke, M.E.; Deisenhofer, J.; Gerard, R.D. Characterization of the Knob domain of the adenovirus type fiber protein expressed in Escherichia coli. J. Virol. 1994, 68, 5239–5246. [Google Scholar]
- Hashimoto, M.; Morimoto, M.; Saimoto, H.; Shigemasa, Y.; Sato, T. Lactosylated chitosan for DNA delivery into hepatocytes: The effect of lactosylation on the physicochemical properties and intracellular trafficking of pDNA/chitosan complexes. Bioconjug. Chem. 2006, 17, 309–316. [Google Scholar]
- Erbacher, P.; Zou, S.; Bettinger, T.; Steffan, A.M.; Remy, J.S. Chitosan-based vector/DNA complexes for gene delivery: Biophysical characteristics and transfection ability. Pharm. Res. 1998, 15, 1332–1339. [Google Scholar] [CrossRef]
- Park, Y.K.; Park, Y.H.; Shin, B.A.; Choi, E.S.; Park, Y.R.; Akaike, T.; Cho, C.S. Galactosylated chitosan-graft-dextran as hepatocyte-targeting DNA carrier. J. Control. Release 2000, 69, 97–108. [Google Scholar] [CrossRef]
- Ishii, T.; Okahata, Y.; Sato, T. Mechanism of cell transfection with plasmid/chitosan complexes. Biochim. Biophys. Acta 2001, 1514, 51–64. [Google Scholar] [CrossRef]
- Nelson, N. Structure and pharmacology of the proton-ATPases. Trends Pharmacol. Sci. 1991, 12, 71–75. [Google Scholar] [CrossRef]
- Kichler, A.; Leborgne, C.; Coeytaux, E.; Danos, O. Polyethylenimine-mediated gene delivery: A mechanistic study. J. Gene Med. 2001, 3, 135–144. [Google Scholar] [CrossRef]
- Moreira, C.; Oliveira, H.; Pires, L.R.; Simo, S.; Barbosa, M.A.; Pe, A.P. Improving chitosan-mediated gene transfer by the introduction of intracellular buffering moieties into the chitosan backbone. Acta Biomater. 2009, 5, 2995–3006. [Google Scholar]
- Chang, K.; Higuchi, Y.; Kawakami, S.; Yamashita, F.; Hashida, M. Efficient gene transfection by histidine-modified chitosan through enhancement of endosomal escape. Bioconjug. Chem. 2010, 21, 1087–1095. [Google Scholar]
- Maclaughlin, F.C.; Mumper, R.J.; Wang, J.; Tagliaferri, J.M.; Gill, I.; Hinchcliffe, M.; Rolland, A.P. Chitosan and depolymerized chitosan oligomers as condensing carriers for in vivo plasmid delivery. J. Control. Release 1998, 56, 259–272. [Google Scholar] [CrossRef]
- Huang, M.; Khor, E.; Lim, L.Y. Uptake and cytotoxicity of chitosan molecules and nanoparticles: Effects of molecular weight and degree of deacetylation. Pharm. Res. 2004, 21, 344–353. [Google Scholar] [CrossRef]
- Kiang, T.; Wen, J.; Lim, H.W.; Leong, K.W. The effect of the degree of chitosan deacetylation on the efficiency of gene transfection. Biomaterials 2004, 25, 5293–5301. [Google Scholar] [CrossRef]
- Liu, W.; Sun, S.; Cao, Z.; Zhang, X.; Yao, K.; Lu, W.W.; Luk, K.D. An investigation on the physicochemical properties of chitosan/DNA polyelectrolyte complexes. Biomaterials 2005, 26, 2705–2711. [Google Scholar]
- Huang, M.; Fong, C.W.; Khor, E.; Lim, L.Y. Transfection efficiency of chitosan vectors: Effect of polymer molecular weight and degree of deacetylation. J. Control. Release 2005, 106, 391–406. [Google Scholar] [CrossRef]
- Köpping-Höggård, M.K.; Vårum, K.M.; Issa, M.; Danielsen, S.; Christensen, B.E.; Stokke, B.T.; Artursson, P. Improved chitosan-mediated gene delivery based on easily dissociated chitosan polyplexes of highly defined chitosan oligomers. Gene Ther. 2004, 11, 1441–1452. [Google Scholar] [CrossRef]
- Liu, X.; Howard, K.A.; Dong, M.; Andersen, M.Ø.; Rahbek, L.; Johnsen, M.G.; Hansen, O.C.; Besenbacher, F.; Kjems, J. The influence of polymeric properties on chitosan/siRNA nanoparticle formulation and gene silencing. Biomaterials 2007, 28, 1280–1288. [Google Scholar]
- Aiba, S. Studies on chitosan: 2. Solution stability and reactivity of partially N-acetylated chitosan derivatives in aqueous media. Int. J. Biol. Macromol. 1989, 11, 249–252. [Google Scholar] [CrossRef]
- Pangburn, S.; Trescony, P.; Heller, J. Lysozyme degradation of partially deacetylated chitin, its films and hydrogels. Biomaterials 1982, 3, 105–108. [Google Scholar] [CrossRef]
- Kamiyama, K.; Onishi, H.; Machida, Y. Biodisposition characteristics of N-succinyl-chitosan and glycol-chitosan in normal and tumor-bearing mice. Biol. Pharm. Bull. 1999, 22, 179–186. [Google Scholar] [CrossRef]
- Lavertu, M.; Methot, S.; Tran-Khanh, N.; Buschmann, M.D. High efficiency gene transfer using chitosan/DNA nanoparticles with specific combinations of molecular weight and degree of deacetylation. Biomaterials 2006, 27, 4815–4824. [Google Scholar]
- Nafee, N.; Taetz, S.; Schneider, M.; Schaefer, U.F.; Lehr, C.M. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: Effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomedicine 2007, 3, 173–183. [Google Scholar] [CrossRef]
- Smedt, S.C.D.; Demeester, J.; Hennink, W.E. Cationic polymer based gene delivery systems. Pharm. Res. 2000, 17, 113–126. [Google Scholar] [CrossRef]
- Romøren, K.; Pedersen, S.; Smistad, G.; Evensen, Ø.; Thu, B.J. The influence of formulation variables on in vitro transfection efficiency and physicochemical properties of chitosan-based polyplexes. Int. J. Pharm. 2003, 261, 115–127. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, S.; Wu, F.; Mao, Z.; Yu, C. Transfection of primary chondrocytes using chitosan-pEGFP nanoparticles. J. Control. Release 2006, 112, 223–228. [Google Scholar] [CrossRef]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef]
- Borel, F.; Logtenstein, R.V.; Koornneef, A.; Maczuga, P.; Ritsema, T.; Petry, H.; Deventer, S.J.; Jansen, P.L.; Konstantinova, P. In vivo knock-down of multidrug resistance transporters ABCC1 and ABCC2 by AAV-delivered shRNAs and by artificial miRNAs. J. RNAi Gene Silencing 2011, 7, 434–442. [Google Scholar]
- Sato, T.; Ishii, T.; Okahata, Y. In vitro gene delivery mediated by chitosan. Effect of pH, serum, and molecular mass of chitosan on the transfection efficiency. Biomaterials 2001, 22, 2075–2080. [Google Scholar] [CrossRef]
- Howard, K.A.; Rahbek, U.L.; Liu, X.; Damgaard, C.K.; Glud, S.Z.; Andersen, M.; Hovgaard, M.B.; Schmitz, A.; Nyengaard, J.R.; Besenbacher, F.; et al. RNA Interference in vitro and in vivo using a chitosan/siRNA nanoparticle system. Mol. Ther. 2006, 14, 476–484. [Google Scholar] [CrossRef]
- Holzerny, P.; Ajdini, B.; Heusermann, W.; Bruno, K.; Schuleit, M.; Meinel, L.; Keller, M. Biophysical properties of chitosan/siRNA polyplexes: Profiling the polymer/siRNA interactions and bioactivity. J. Control. Release 2012, 157, 297–304. [Google Scholar] [CrossRef]
- Alameh, M.; Dejesus, D.; Jean, M.; Darras, V.; Thibault, M.; Lavertu, M.; Buschmann, M.D.; Merzouki, A. Low molecular weight chitosan nanoparticulate system at low N:P ratio for nontoxic polynucleotide delivery. Int. J. Nanomed. 2012, 7, 1399–1414. [Google Scholar] [Green Version]
- Chen, M.; Gao, S.; Dong, M.; Song, J.; Yang, C.; Howard, K.A.; Kjems, J.; Besenbacher, F. Chitosan/siRNA nanoparticles encapsulated in PLGA nanofibers for siRNA delivery. ACS Nano 2012, 6, 4835–4844. [Google Scholar]
- Mourya, V.K.; Indamar, N.N. Trimethyl chitosan and its applications in drug delivery. J. Mater. Sci. Mater. Med. 2009, 20, 1057–1079. [Google Scholar] [CrossRef]
- Thanou, M.M.; Kotze, A.F.; Scharringhausen, T.; Leuben, H.L.; de Boer, A.G.; Verhoef, J.C.; Junginger, H.E. Effect of degree of quaternization of N-trimethyl chitosan chloride for enhanced transport of hydrophilic compounds across intestinal Caco-2 cell monolayers. J. Control. Release 2000, 64, 15–25. [Google Scholar] [CrossRef]
- Thanou, M.; Florea, B.I.; Geldof, M.; Junginger, H.E.; Borchard, G. Quaternized chitosan oligomers as novel gene delivery vectors in epithelial cell lines. Biomaterials 2002, 23, 153–159. [Google Scholar]
- Endo, M.; Kuroda, S.; Kondo, H.; Maruoka, Y.; Ohya, K.; Kasugai, S. bone regeneration by modified gene-activated matrix: Effectiveness in segmental tibial defects in rats. Tissue Eng. 2006, 12, 15–20. [Google Scholar]
- Evans, C.H. Gene therapy for bone healing. Expert Rev. Mol. Med. 2010, 12, 1–17. [Google Scholar] [CrossRef]
- Pelled, G.; Ben-Arav, A.; Hock, C.; Reynolds, D.G.; Yazici, C.; Zilberman, Y.; Zulma, G.; Awad, H.; Gazit, D.; Schwarz, E.M. Direct gene therapy for bone regeneration: Gene delivery, animal models , and outcome measures. Tissue Eng. Part B Rev. 2010, 16, 13–20. [Google Scholar]
- O’Rorke, S.; Keeney, M.; Pandit, A. Progress in polymer science non-viral polyplexes: Scaffold mediated delivery for gene therapy. Prog. Polym. Sci. 2010, 35, 441–458. [Google Scholar] [CrossRef]
- Csaba, N.; Garcia-Fuentes, M.; Alonso, M.J. The performance of nanocarriers for transmucosal drug delivery. Expert Opin. Drug Deliv. 2006, 3, 463–478. [Google Scholar]
- Puras, G.; Zarate, J.; Aceves, M.; Murua, A.; Díaz, A.R.; Avilés-Triguero, M.; Fernandez, E.; Pedraz, J.L. Low molecular weight oligochitosans for non-viral retinal gene therapy. Eur. J. Pharm. Biopharm. 2013, 83, 131–140. [Google Scholar] [CrossRef]
- Howard, K.A.; Paludan, S.R.; Behlke, M.A.; Besenbacher, F.; Deleuran, B.; Kjems, J. Chitosan/siRNA nanoparticle-mediated TNF-α knockdown in peritoneal macrophages for anti-inflammatory treatment in a murine arthritis model. Mol. Ther. 2008, 17, 162–168. [Google Scholar]
- Fröhlich, M.; Grayson, W.L.; Wan, L.Q.; Marolt, D.; Drobnic, M.; Vunjak-Novakovic, G. Tissue engineered bone grafts: Biological requirements, tissue culture and clinical relavence. Curr. Stem Cell Res. Ther. 2009, 3, 254–264. [Google Scholar]
- Rauh, J.; Milan, F.; Gu, K.P.; Stiehler, M. Bioreactor Systems for bone tissue engineering. Tissue Eng. Part B Rev. 2011, 17, 263–280. [Google Scholar] [CrossRef]
- Richardson, T.P.; Peters, M.C.; Ennett, A.B.; Mooney, D.J. Polymeric system for dual growth factor delivery. Nature 2001, 19, 1029–1034. [Google Scholar] [CrossRef]
- Curtin, C.M.; Cunniffe, G.M.; Lyons, F.G.; Bessho, K.; Dickson, G.R.; Duffy, G.P.; O’Brien, F.J. Innovative collagen nano-hydroxyapatite scaffolds offer a highly efficient non-viral gene delivery platform for stem cell-mediated bone formation. Adv. Mater. 2012, 24, 749–754. [Google Scholar]
- O’Brien, F.J. Biomaterial & scaffolds for tissue engineering. Mater. Today 2011, 14, 88–95. [Google Scholar] [CrossRef]
- Alvarez, K.; Nakajima, H. Metallic scaffolds for bone regeneration. Materials 2009, 2, 790–832. [Google Scholar] [CrossRef]
- Mano, J.F.; Silva, G.A.; Azevedo, H.S.; Malafaya, P.B.; Sousa, R.A.; Silva, S.S.; Boesel, L.F.; Oliveira, J.M.; Santos, T.C.; Marques, A.P.; et al. Natural origin biodegradable systems in tissue engineering and regenerative medicine: Present status and some moving trends. J. R. Soc. Interface 2007, 4, 999–1030. [Google Scholar] [CrossRef]
- Meinel, L.; Kaplan, D.L. Silk constructs for delivery of musculoskeletal therapeutics. Adv. Drug Deliv. Rev. 2012, 64, 1111–1122. [Google Scholar] [CrossRef]
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar] [CrossRef]
- Navarro, M.; Planell, J.A. Scaffolds for bone regeneration. Eur. Musculoskelet. Rev. 2011, 6, 1–5. [Google Scholar]
- Davis, H.E.; Leach, J.K. Designing bioactive delivery systems for tissue regeneration. Ann. Biomed. Eng. 2011, 39, 1–13. [Google Scholar] [CrossRef]
- Ladewig, K. Drug delivery in soft tissue engineering. Expert Opin. Drug Deliv. 2011, 8, 1175–1188. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tabata, Y. Tissue engineering by modulated gene delivery B. Adv. Drug Deliv. Rev. 2006, 58, 535–554. [Google Scholar] [CrossRef]
- Costa-Pinto, A.R.; Reis, R.L.; Neves, N.M. Scaffolds based bone tissue engineering: the role of chitosan. Tissue Eng. Part B Rev. 2011, 17, 331–347. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Rodrigues, M.T.; Silva, S.S.; Malafaya, P.B.; Gomes, M.E.; Viegas, C.A.; Dias, I.R.; Azevedo, J.T.; Mano, J.F.; Reis, R.L. Novel hydroxyapatite/chitosan bilayered scaffold for osteochondral tissue-engineering applications: Scaffold design and its performance when seeded with goat bone marrow stromal cells. Biomaterials 2006, 27, 6123–6137. [Google Scholar] [CrossRef]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef]
- Lienemann, P.S.; Lutolf, M.P.; Ehrbar, M. Biomimetic hydrogels for controlled biomolecule delivery to augment bone regeneration. Adv. Drug Deliv. Rev. 2012, 64, 1078–1089. [Google Scholar] [CrossRef]
- Gutowska, A.; Jeong, B.; Jasionowski, M. Injectable gels for tissue engineering. Anat. Rec. 2001, 349, 342–349. [Google Scholar] [CrossRef]
- Hastings, C.L.; Kelly, H.M.; Murphy, M.J.; Barry, F.P.; O’Brien, F.J.O.; Duffy, G.P. Development of a thermoresponsive chitosan gel combined with human mesenchymal stem cells and desferrioxamine as a multimodal pro-angiogenic therapeutic for the treatment of critical limb ischaemia. J. Control. Release 2012, 161, 73–80. [Google Scholar] [CrossRef]
- Adewuyi, S.; Kareem, K.T.; Atayese, A.O.; Amolegbe, S.A.; Akinremi, C.A. Chitosan-cobalt (II) and nickel (II) chelates as antibacterial agents. Int. J. Biol. Macromol. 2011, 48, 301–303. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, Y.; Fan, W.; Han, P.; Chang, J.; Yuen, J.; Zhang, M. Hypoxia-mimicking mesoporous bioactive glass scaffolds with controllable cobalt ion release for bone tissue engineering. Biomaterials 2012, 33, 2076–2085. [Google Scholar]
- Gleeson, J.P.; Plunkett, N.; O’Brien, F.J. Addition of hydroxyapatite improves stiffness, interconnectivity and osteogenic potential of a highly porous collagen-based scaffold for bone tissue regeneration. Eur. Cell. Mater. 2010, 20, 218–230. [Google Scholar]
- Cunniffe, G.M.; Dickson, G.R.; Partap, S.; Stanton, K.T.; O’Brien, F.J. Development and characterisation of a collagen nano-hydroxyapatite composite scaffold for bone tissue engineering. J. Mater. Sci. Mater. Med. 2010, 21, 2293–2298. [Google Scholar] [CrossRef]
- Finkemeier, C.G. Bone-grafting and bone-graft substitutes. J. Bone Joint Surg. 2002, 84A, 454–463. [Google Scholar]
- Giannoudis, P.V.; Dinopoulos, H.; Tsiridis, E. Bone substitutes: An update. Injury 2005, 36S, 20–27. [Google Scholar] [CrossRef]
- Tomford, W.W. Transmission Concepts through transplantation of musculoskeletal allografts. J. Bone Joint Surg. 1995, 77, 1742–1754. [Google Scholar]
- Heinemann, C.; Heinemann, S.; Bernhardt, A.; Worch, H.; Hanke, T. Novel textile chitosan scaffolds promote spreading, proliferation, and differentiation of osteoblasts. Biomacromolecules 2008, 9, 2913–2920. [Google Scholar] [CrossRef]
- Mathews, S.; Gupta, P.K.; Bhonde, R.; Totey, S. Chitosan enhances mineralization during osteoblast differentiation of human bone marrow-derived mesenchymal stem cells , by upregulating the associated genes. Cell Prolif. 2011, 44, 537–549. [Google Scholar] [CrossRef]
- Okamoto, Y.; Yano, R.; Miyatake, K.; Tomohiro, I.; Shigemasa, Y.; Minami, S. Effects of chitin and chitosan on blood coagulation. Carbohydr. Polym. 2003, 53, 337–342. [Google Scholar] [CrossRef]
- Park, T.G.; Hoon, J.; Wan, S. Current status of polymeric gene delivery systems. Adv. Drug Deliv. Rev. 2006, 58, 467–486. [Google Scholar] [CrossRef]
- No, H.K.; Park, N.Y.; Lee, S.H.; Meyers, S.P. Antibacterial activity of chitosans and chitosan oligomers with different molecular weights. Int. J. Food Microbiol. 2002, 74, 65–72. [Google Scholar] [CrossRef]
- Holmes, B.; Castro, N.J.; Zhang, L.G.; Zussman, E. Electrospun fibrous scaffolds for bone and cartilage tissue generation: Recent progress and future developments. Tissue Eng. Part B Rev. 2012, 18, 478–486. [Google Scholar]
- Geng, X.; Kwon, O; Jinho, J. Electrospinning of chitosan dissolved in concentrated acetic acid solution. Biomaterials 2005, 26, 5427–5432. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A.; Baldassarre, V.; Conti, F.; Ferrara, P.; Biagini, G.; Gazzanelli, G.; Vasi, V. Biological activity of chitosan: Ultrastructural study. Biomaterials 1988, 9, 247–252. [Google Scholar]
- Muzzarelli, R.A.A.; Zucchini, C.; Ilari, P.; Pugnaloni, A.; Belmonte, M.M.; Biagini, G.; Castaldini, C. Osteoconductive properties of methylpyrrolidinone chitosan in an animal model. Biomaterials 1993, 14, 925–929. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A.; Mattioli-Belmonte, M.; Tietz, C.; Biagini, R.; Ferioli, G.; Brunelli, M.A.; Fini, M.; Giardino, R.; Ilari, P.; Boagini, G. Stimulatory effect on bone formation exerted by a modified chitosan. Biomaterials 1994, 15, 1075–1081. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A.; Biagini, G.; Bellardini, M.; Simonelli, L.; Castaldini, C.; Fratto, G. Osteoconduction exerted by methylpyrrolidinone chitosan used in dental surgery. Biomaterials 1993, 14, 39–43. [Google Scholar]
- Yin, Y.; Ye, F.; Cui, J.; Zhang, F.; Li, X.; Yao, K. Preparation and characterization of macroporous chitosan-gelatin/beta-tricalcium phosphate composite scaffolds for bone tissue engineering. J. Biomed. Mater. Res. A 2003, 67A, 844–855. [Google Scholar] [CrossRef]
- Chesnutt, B.M.; Yuan, Y.; Buddington, K.; Haggard, W.O.; Bumgardner, J.D. Composite chitosan/nano-hydroxyapatite scaffolds induce osteocalcin production by osteoblasts in vitro and support bone formation in vivo. Tissue Eng. Part A 2009, 15, 1–29. [Google Scholar]
- Stephen, S.J.; Tholpady, S.S.; Gross, B.; Petrie-Aronin, C.E.; Botchway, E.A.; Nair, L.S.; Ogle, R.C.; Park, S.S. Injectable tissue-engineered bone repair of a rat calvarial defect. Laryngoscope 2010, 120, 895–901. [Google Scholar]
- Lee, J.; Nam, S.; Im, S.; Park, Y.; Lee, Y.; Seol, Y.; Chung, C.; Lee, S. Enhanced bone formation by controlled growth factor delivery from chitosan-based biomaterials. J. Control. Release 2002, 78, 187–197. [Google Scholar] [CrossRef]
- De la Riva, B.; Sánchez, E.; Hernández, A.; Reyes, R.; Tamimi, F.; López-Cabarcos, E.; Delgado, A.; Evora, C. Local controlled release of VEGF and PDGF from a combined brushite-chitosan system enhances bone regeneration. J. Control. Release 2010, 143, 45–52. [Google Scholar] [CrossRef]
- Roberts, S.; Genever, P.; McCaskie, A.; de Bari, C. Prospects of stem cell therapy in osteoarthritis. Regen. Med. 2011, 6, 351–366. [Google Scholar] [CrossRef]
- De Bari, C.; Kurth, T.B.; Augello, A. Mesenchymal stem cells from development to postnatal joint homeostasis, aging, and disease. Birth Defects Res. C Embryo Today 2010, 271, 257–271. [Google Scholar] [CrossRef]
- Hurst, J.M.; Steadman, J.R.; O’Brien, L.; Rodkey, W.G.; Briggs, K.K. Rehabilitation following microfracture for chondral injury in the knee. Clin. Sports Med. 2010, 29, 257–265. [Google Scholar] [CrossRef]
- Altman, R.D. Early management of osteoarthritis. Am. J. Manag. Care 2010, 16, S41–S47. [Google Scholar]
- Brittberg, M.; Lindarl, A.; Nilsson, A.; Ohlsson, C.; Isakkson, O.; Peterson, L. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplanatation. N. Engl. J. Med. 1994, 331, 889–895. [Google Scholar] [CrossRef]
- Pacifici, M.; Koyama, E.; Iwamoto, M. Mechanisms of synovial joint and articular cartilage formation: Recent advances, but many lingering mysteries. Birth Defects Res. C Embryo Today 2005, 248, 237–248. [Google Scholar] [CrossRef]
- Kosher, R.A.; Lash, J.W.; Minor, R.R. Environmental enhancement of in vitro chondrogenesis: IV. Stimulation of somite chondrogenesis by exogenous chondromucoprotein. Dev. Biol. 1973, 35, 210–220. [Google Scholar]
- Lindahl, U.; Hook, M. Glycosaminoglycans and their binding to biological macromolecules. Annu. Rev. Biochem. 1978, 47, 385–417. [Google Scholar] [CrossRef]
- Peluso, G.; Petillo, O.; Ranieri, M.; Santin, M.; Ambrosic, L.; Calabró, D.; Avallone, B.; Balsamo, G. Chitosan-mediated stimulation of macrophage function. Biomaterials 1994, 15, 1215–1220. [Google Scholar] [CrossRef]
- Usami, Y.; Okamoto, Y.; Minami, S.; Matsuhashi, A.; Kumazawa, N.H.; Tanioka, S.; Shigemasa, Y. Chitin and chitosan induce migration of bovine polymorphonuclear cells. J. Vet. Med. Sci. 1994, 56, 761–762. [Google Scholar] [CrossRef]
- Francis Suh, J.K.; Matthew, H.W.T. Application of chitosan-based polysaccharide biomaterials in cartilage tissue engineering: A review. Biomaterials 2000, 21, 2589–2598. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Lu, J.X.; Prudhommeaux, F.; Meunier, A.; Sedel, L. Effects of chitosan on rat knee cartilages. Biomaterials 1999, 20, 1937–1944. [Google Scholar] [CrossRef]
- Mattioli-Belmonte, M.; Gigante, A.; Muzzarelli, R.A.; Politano, R.; De Benedittis, A.; Specchia, N.; Buffa, A.; Biagini, G.; Greco, F. N,N-dicarboxymethyl chitosan as delivery agent for bone morphogenetic protein in the repair of articular cartilage. Med. Biol. Eng. Comp. 1999, 37, 130–134. [Google Scholar] [CrossRef]
- Chen, H.; Frankenburg, E.P.; Goldstein, S.A.; McCauley, L.K. Combination of local and systemic parathyroid hormone enhances bone regeneration. Clin. Orthop. Relat. Res. 2003, 416, 291–302. [Google Scholar] [CrossRef]
- Geiger, F.; Bertram, H.; Berger, I.; Lorenz, H.; Wall, O.; Eckhardt, C.; Simank, H.G. Vascular Endothelial growth factor gene-activated matrix (VEGF 165-GAM) enhances osteogenesis and angiogenesis in large segmental bone defects. J. Bone Miner. Res. 2005, 20, 2028–2035. [Google Scholar] [CrossRef]
- Beniash, E. Biominerals-hierarchical nanocomposites: The example of bone. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 47–69. [Google Scholar] [CrossRef]
- Keeney, M.; van den Beucken, J.J.P.; van der Kraan, P.M.; Jansen, J.A.; Pandit, A. The ability of a collagen/calcium phosphate scaffold to act as its own vector for gene delivery and to promote bone formation via transfection with VEGF165. Biomaterials 2010, 31, 2893–2902. [Google Scholar] [CrossRef]
- Huang, Y.C.; Simmons, C.; Kaigler, D.; Rice, K.G.; Mooney, D.J. Bone regeneration in a rat cranial defect with delivery of PEI-condensed plasmid DNA encoding for bone. Gene Ther. 2005, 12, 418–426. [Google Scholar] [CrossRef]
- Tierney, E.G.; McSorley, K.; Hastings, C.L.; Cryan, S.A.; O'Brien, T.; Murphy, M.J.; Barry, F.P.; O'Brien, F.J.; Duffy, G.P. High levels of ephrinB2 over-expression increases the osteogenic differentiation of human mesenchymal stem cells and promotes enhanced cell mediated mineralisation in a polyethyleneimine-ephrinB2 gene-activated matrix. J. Control. Release 2013, 165, 173–182. [Google Scholar] [CrossRef]
- Peng, L.; Cheng, X.; Zhuo, R.; Lan, J.; Wang, Y.; Shi, B.; Siqun, L. Novel gene-activated matrix with embedded chitosan/plasmid DNA nanoparticles encoding PDGF for periodontal tissue engineering. J. Biomed. Mater. Res. A 2009, 90, 564–576. [Google Scholar]
- Monaghan, M.; Pandit, A. RNA interference therapy via functionalized scaffolds. Adv. Drug Deliv. Rev. 2011, 63, 197–208. [Google Scholar] [CrossRef]
- Raftery, R.; Tierney, E.G.; Curtin, C.M.; Cryan, S.A.; O’Brien, F.J. Utilizing chitosan nanoparticles for the production of gene-activated matrices for bone regeneration. J. Tissue Eng. Regen. Med. 2012, 6, 291–292. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Raftery, R.; O'Brien, F.J.; Cryan, S.-A. Chitosan for Gene Delivery and Orthopedic Tissue Engineering Applications. Molecules 2013, 18, 5611-5647. https://doi.org/10.3390/molecules18055611
Raftery R, O'Brien FJ, Cryan S-A. Chitosan for Gene Delivery and Orthopedic Tissue Engineering Applications. Molecules. 2013; 18(5):5611-5647. https://doi.org/10.3390/molecules18055611
Chicago/Turabian StyleRaftery, Rosanne, Fergal J. O'Brien, and Sally-Ann Cryan. 2013. "Chitosan for Gene Delivery and Orthopedic Tissue Engineering Applications" Molecules 18, no. 5: 5611-5647. https://doi.org/10.3390/molecules18055611
APA StyleRaftery, R., O'Brien, F. J., & Cryan, S.-A. (2013). Chitosan for Gene Delivery and Orthopedic Tissue Engineering Applications. Molecules, 18(5), 5611-5647. https://doi.org/10.3390/molecules18055611