Application of the Suzuki-Miyaura Reaction in the Synthesis of Flavonoids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
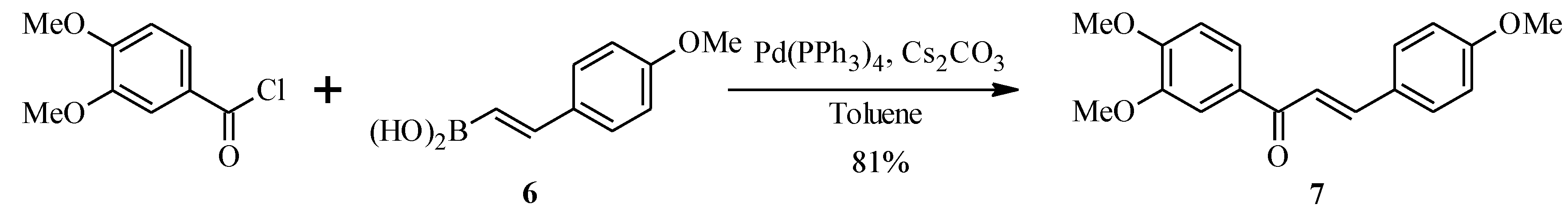{kind=link}
{kind=link}
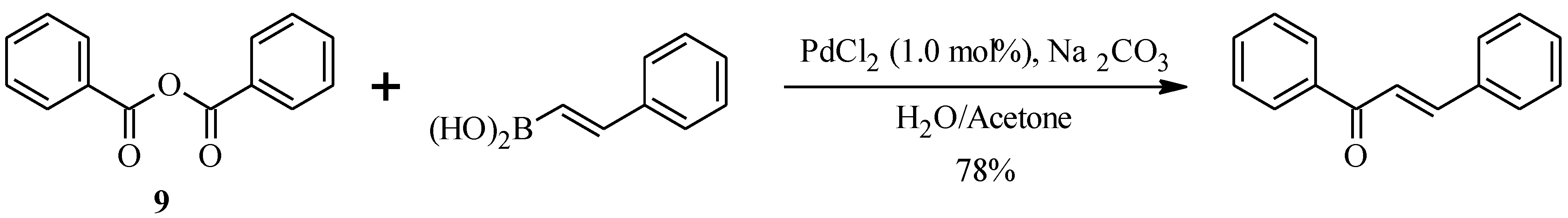{kind=link}
{kind=link}
{kind=link}
{kind=link}
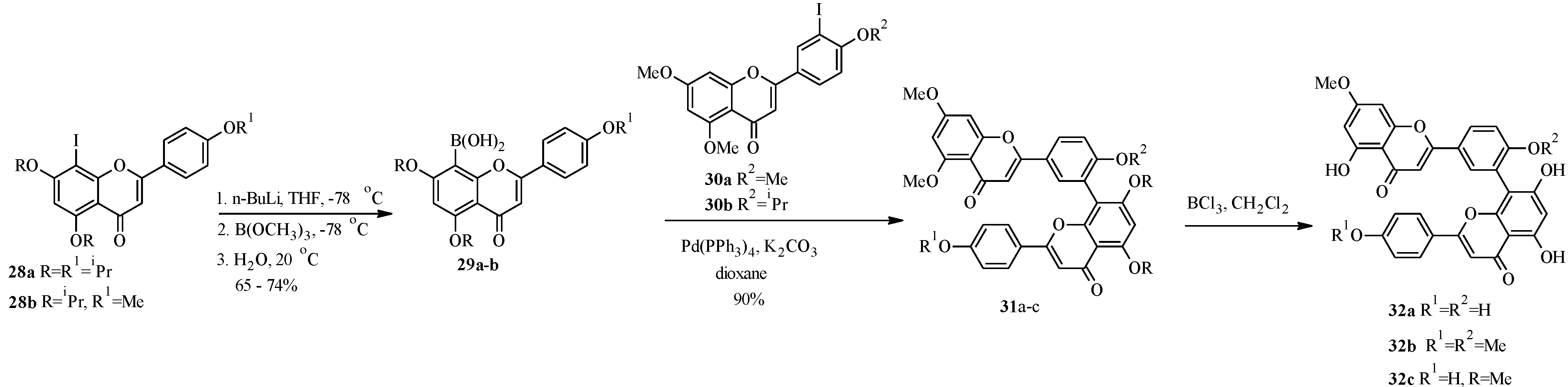{kind=link}
{kind=link}
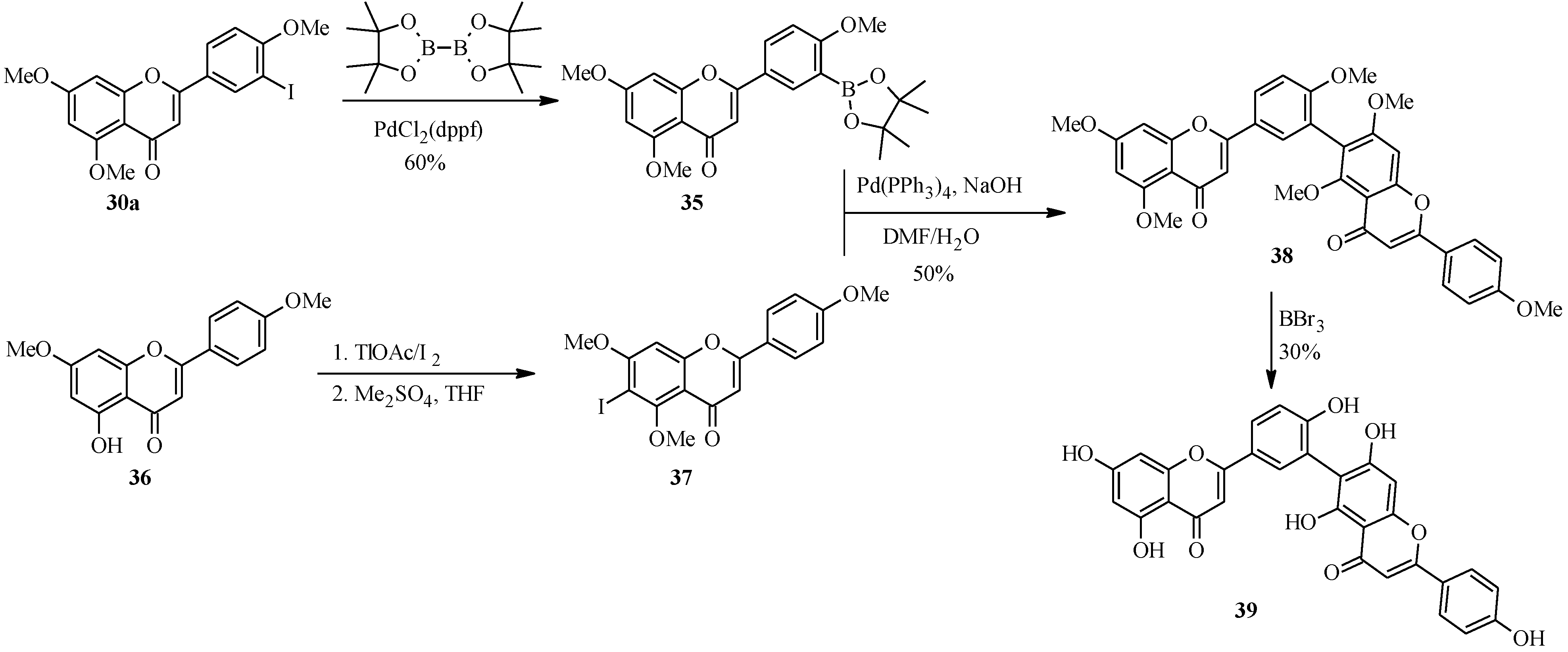{kind=link}
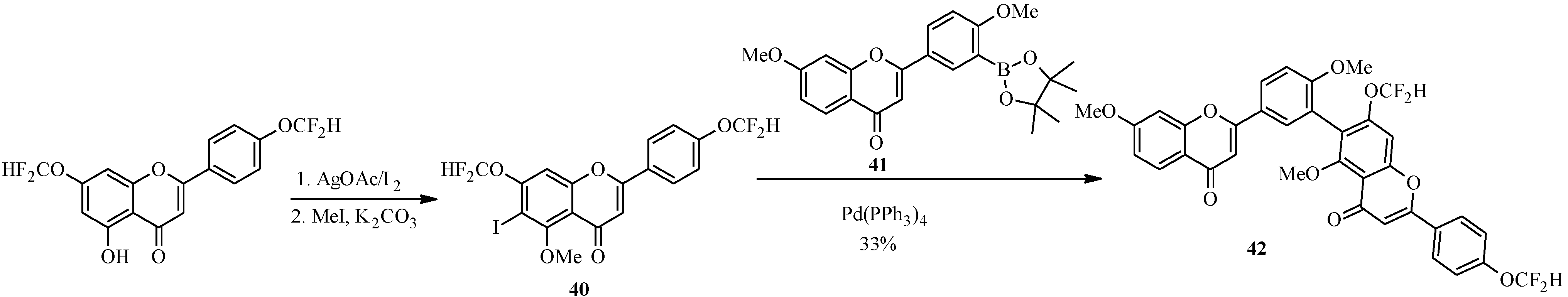{kind=link}
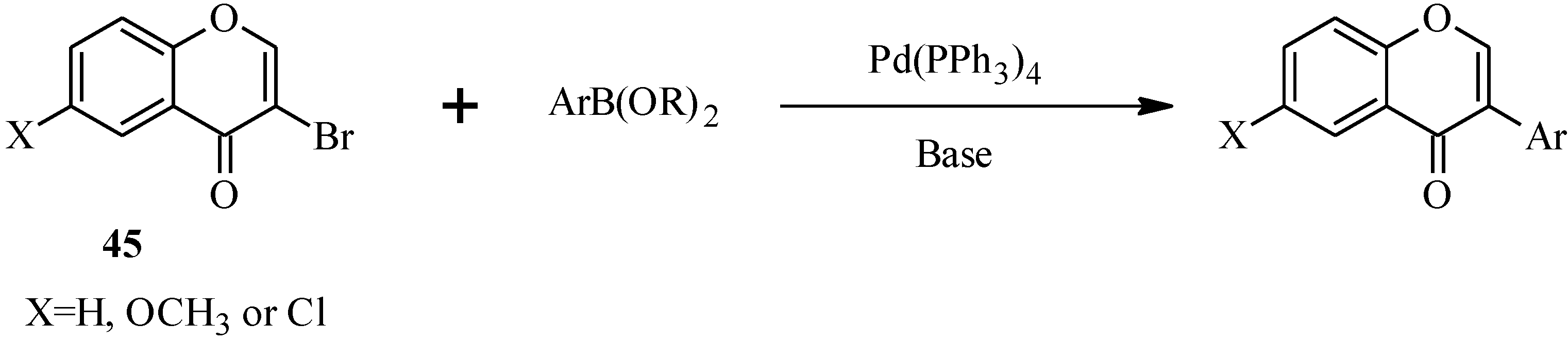{kind=link}
{kind=link}
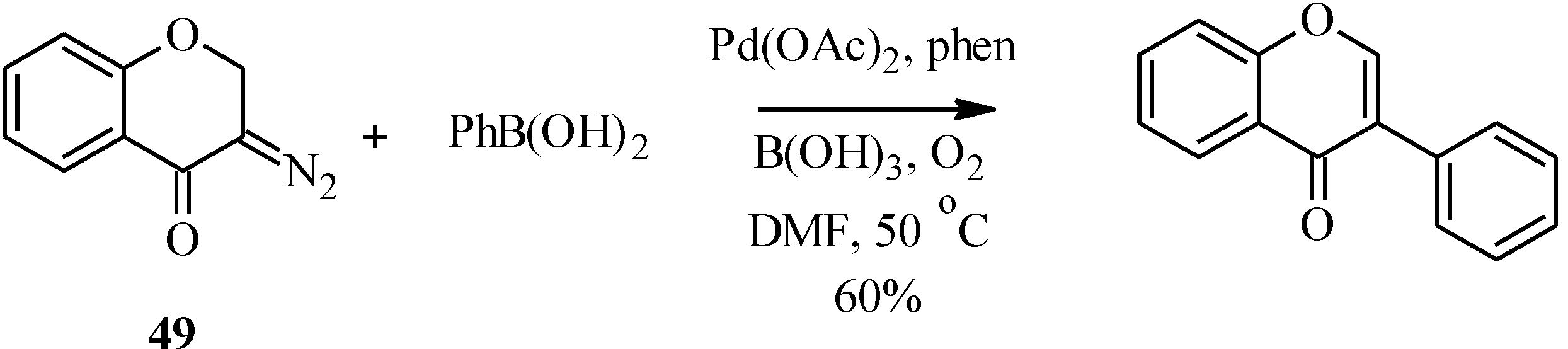{kind=link}
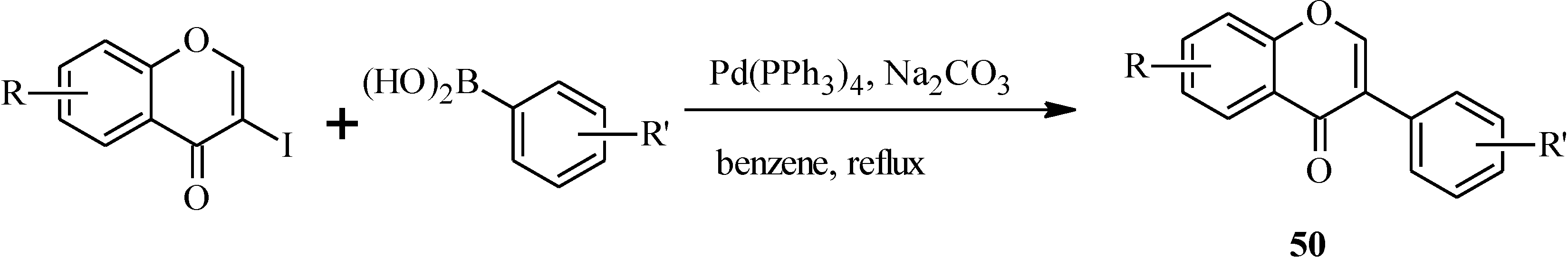{kind=link}
{kind=link}
{kind=link}
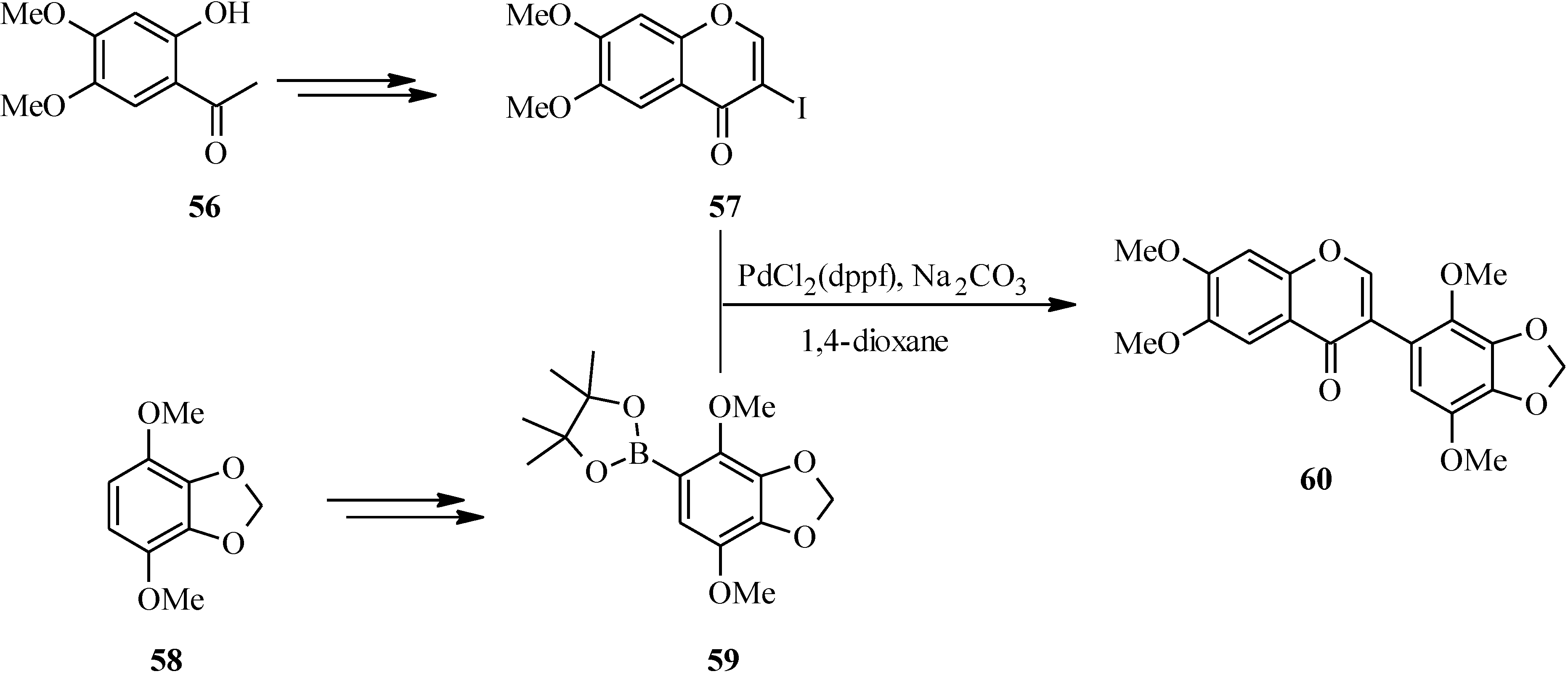{kind=link}
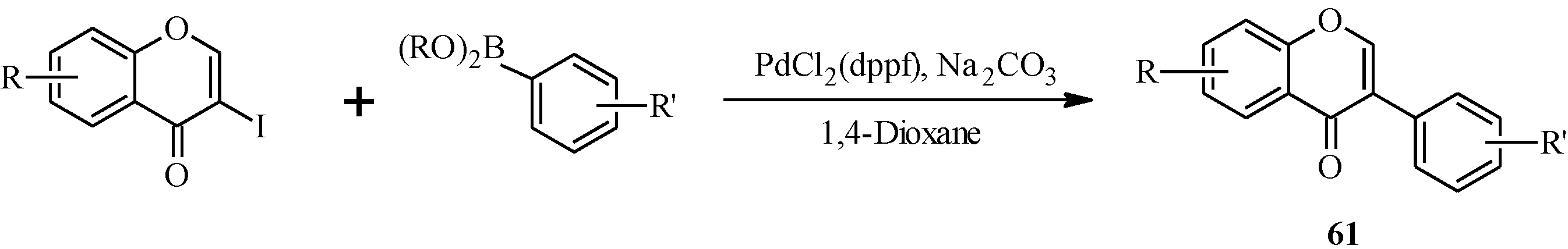{kind=link}
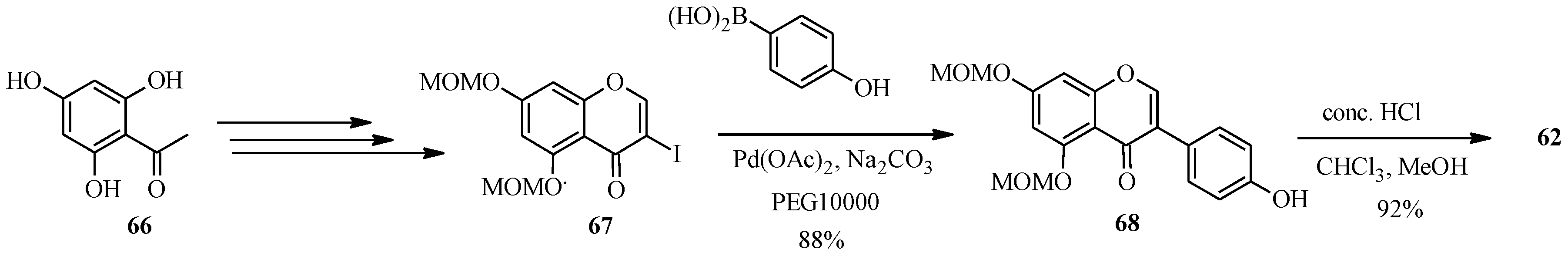{kind=link}
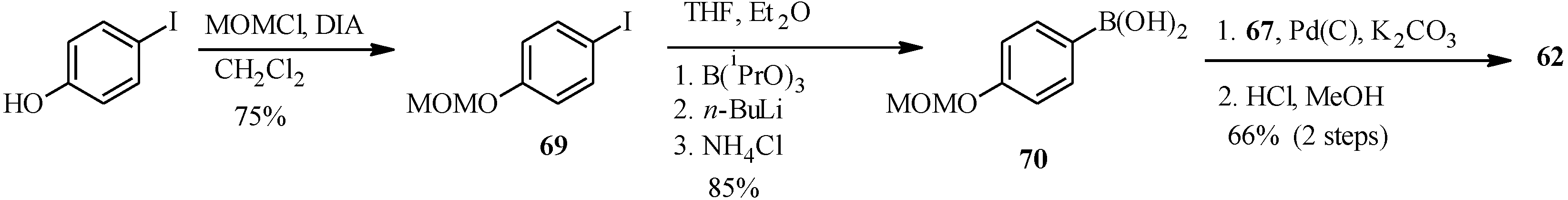{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
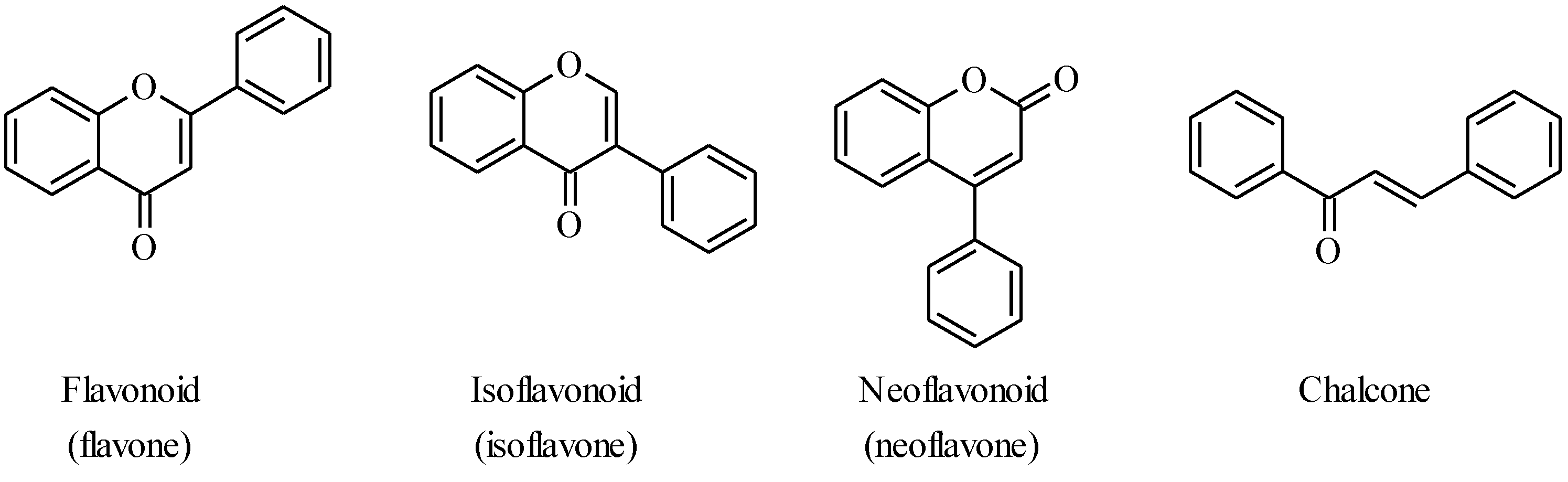
:1. Introduction
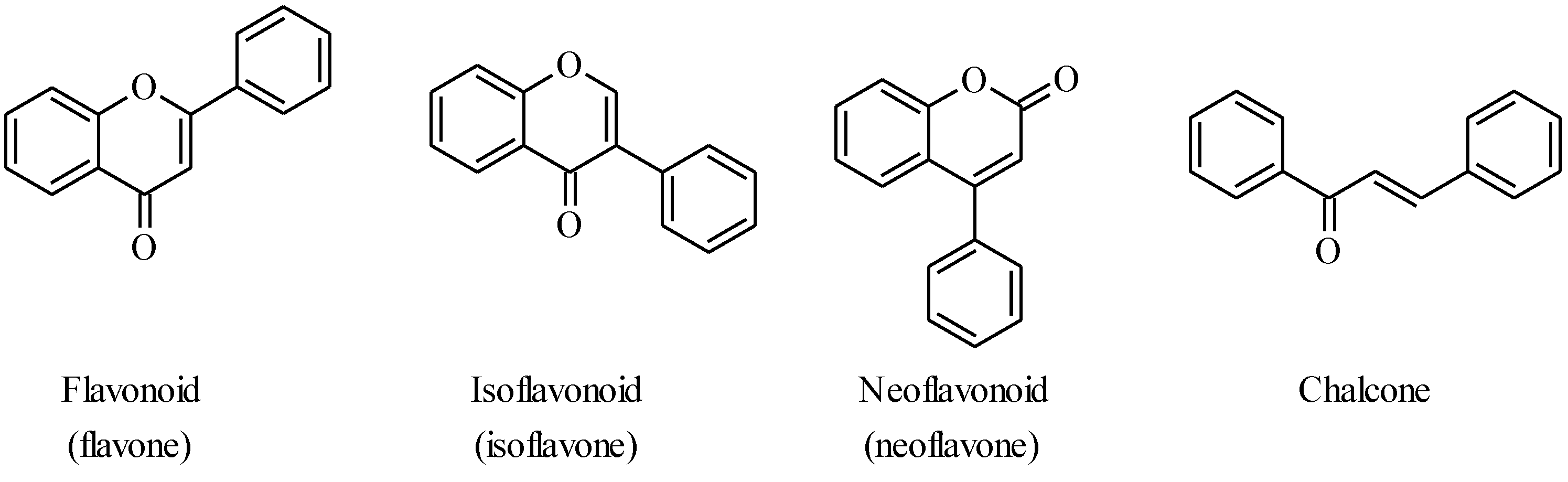
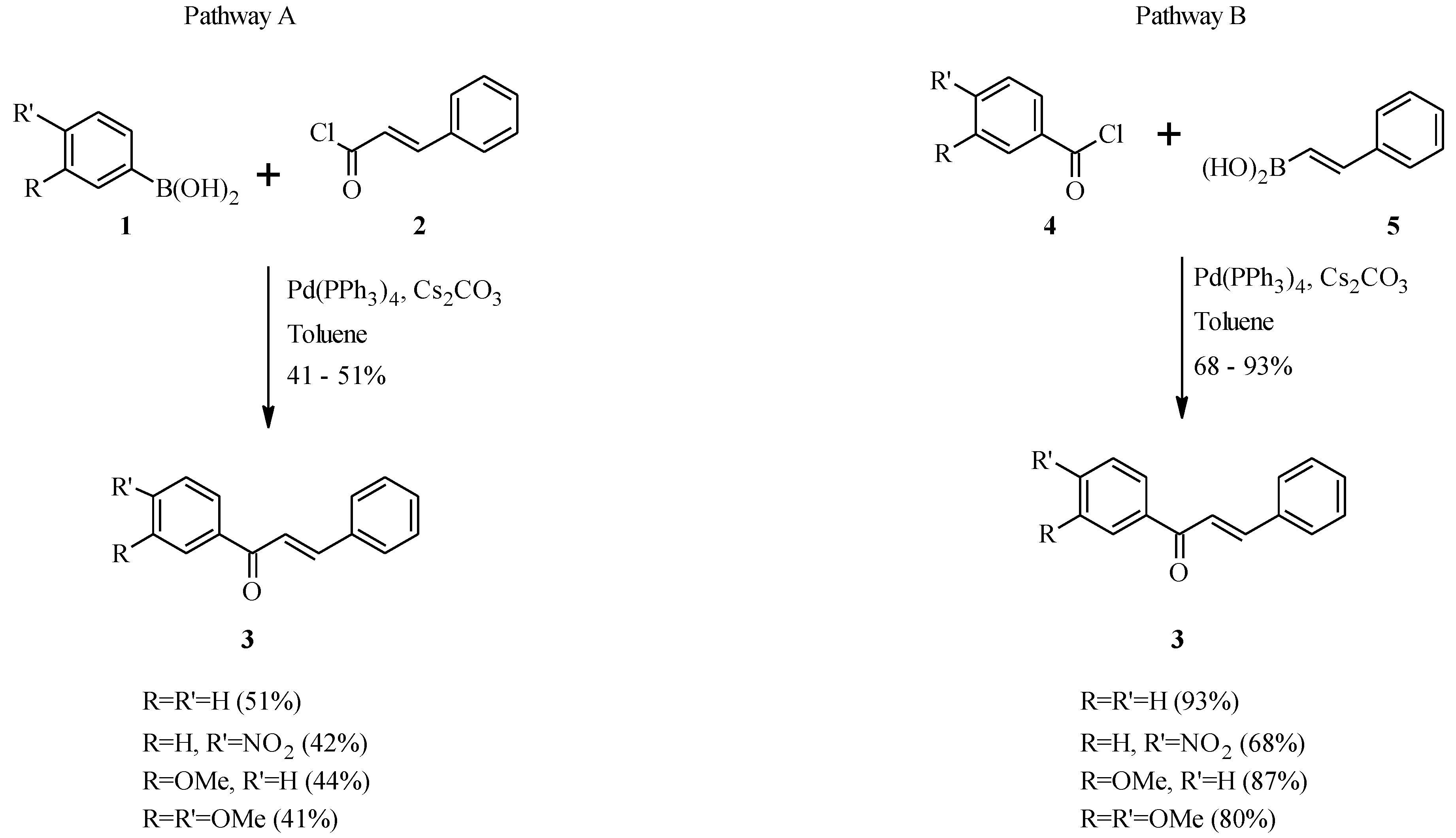
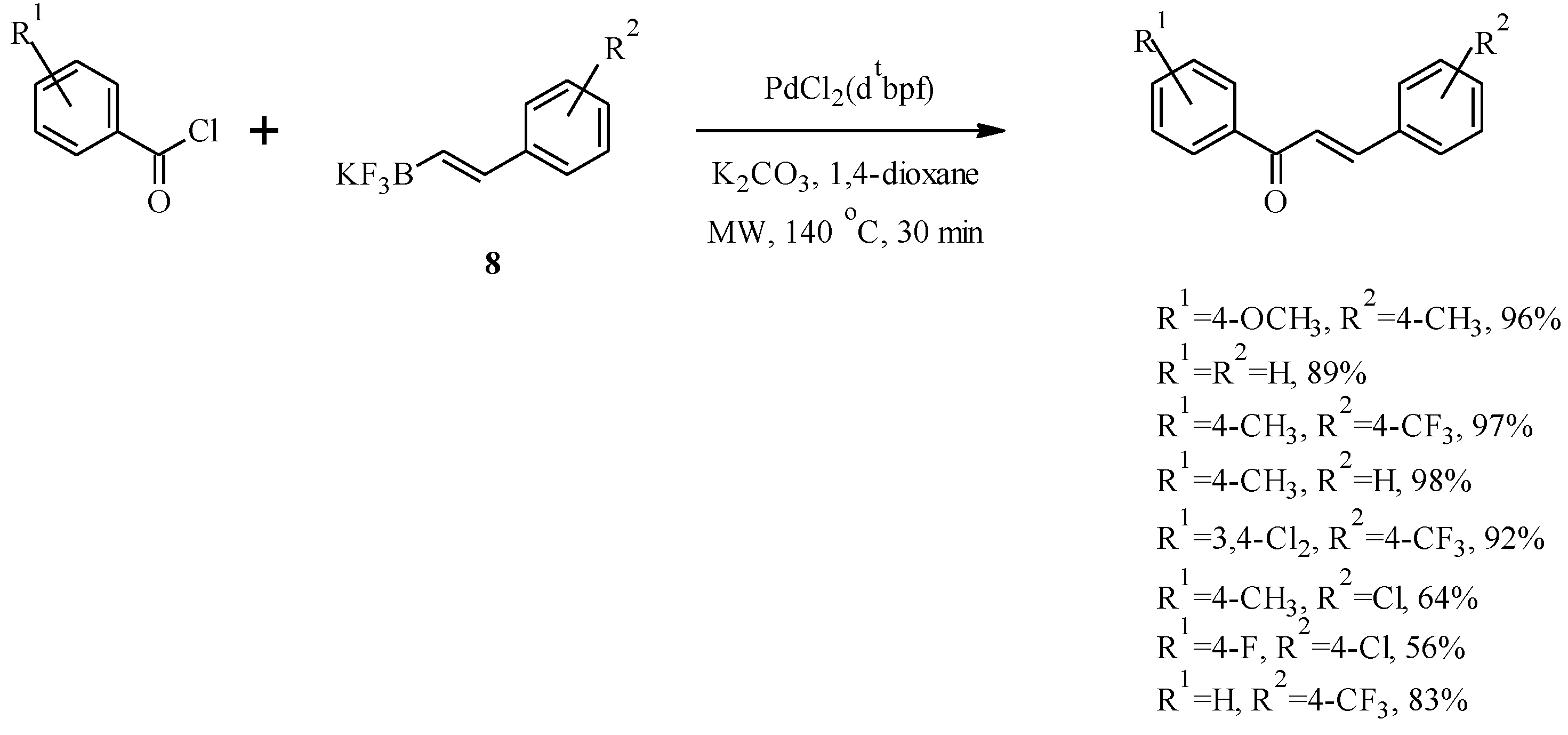
2. Chalcones
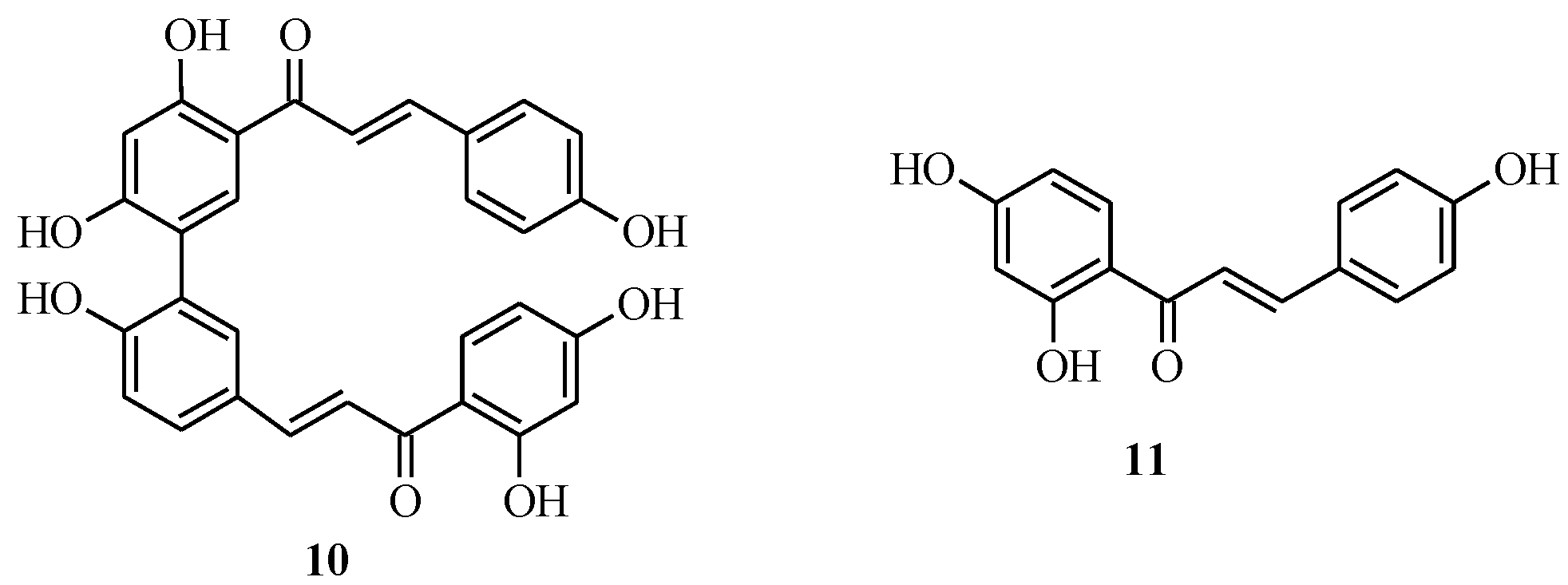
2.1. Chalcone Monomers
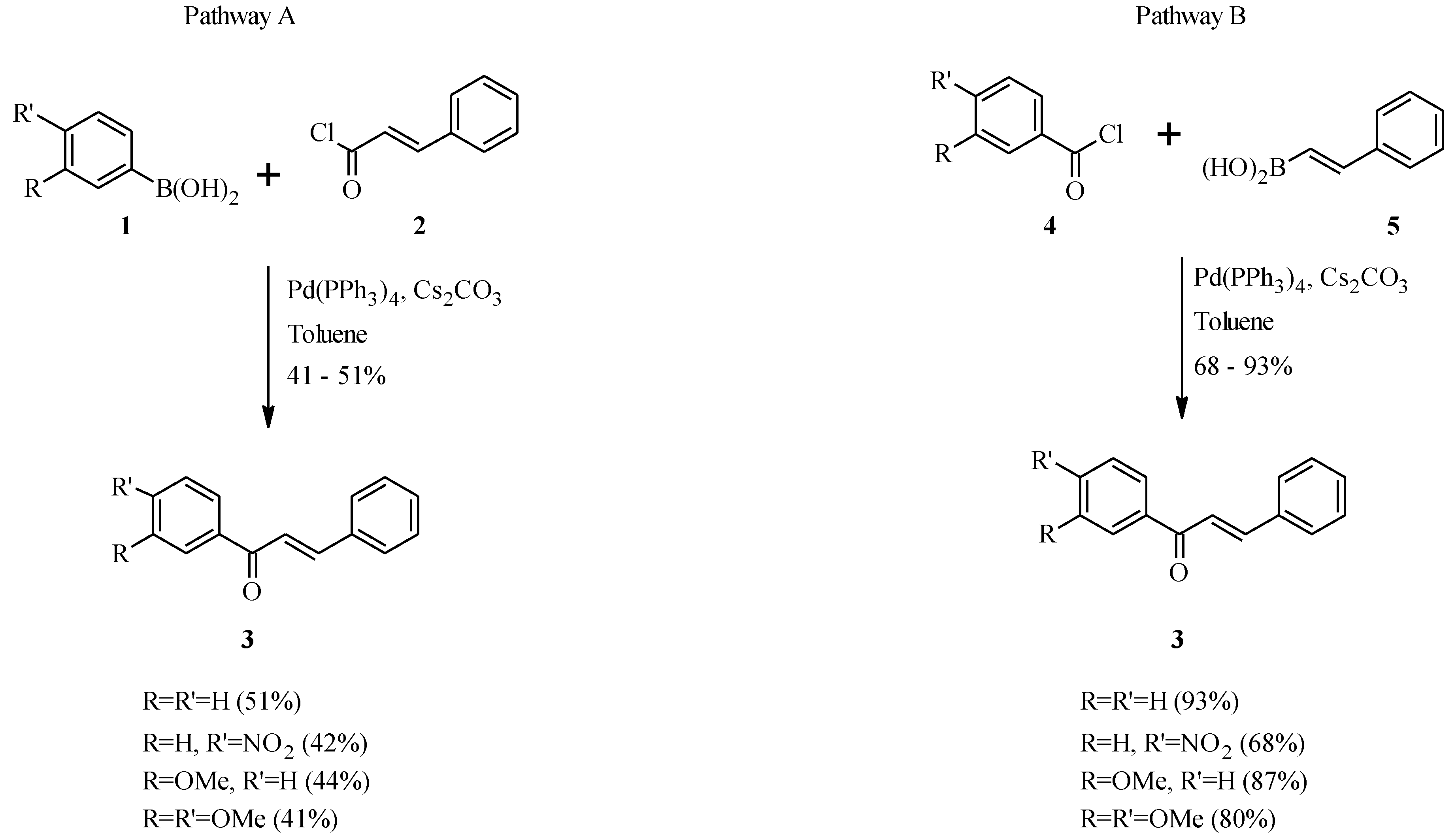
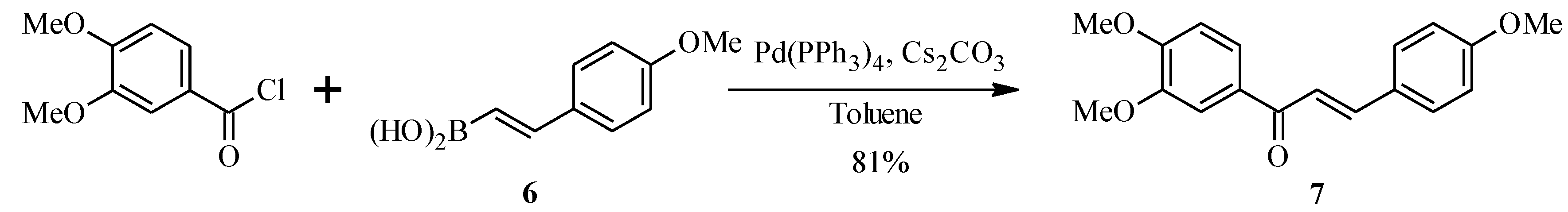

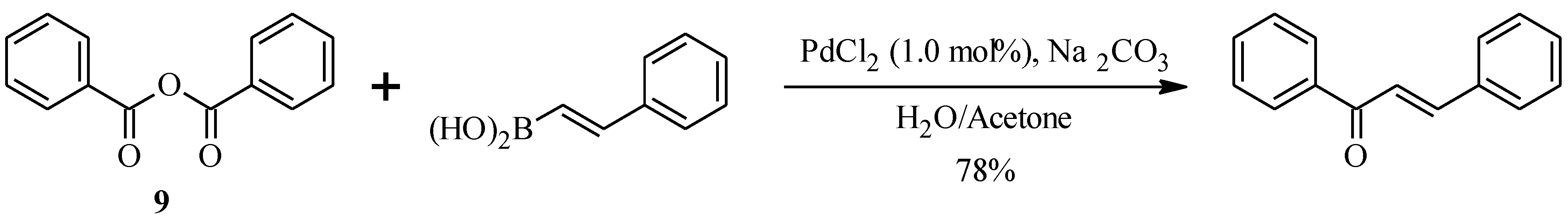
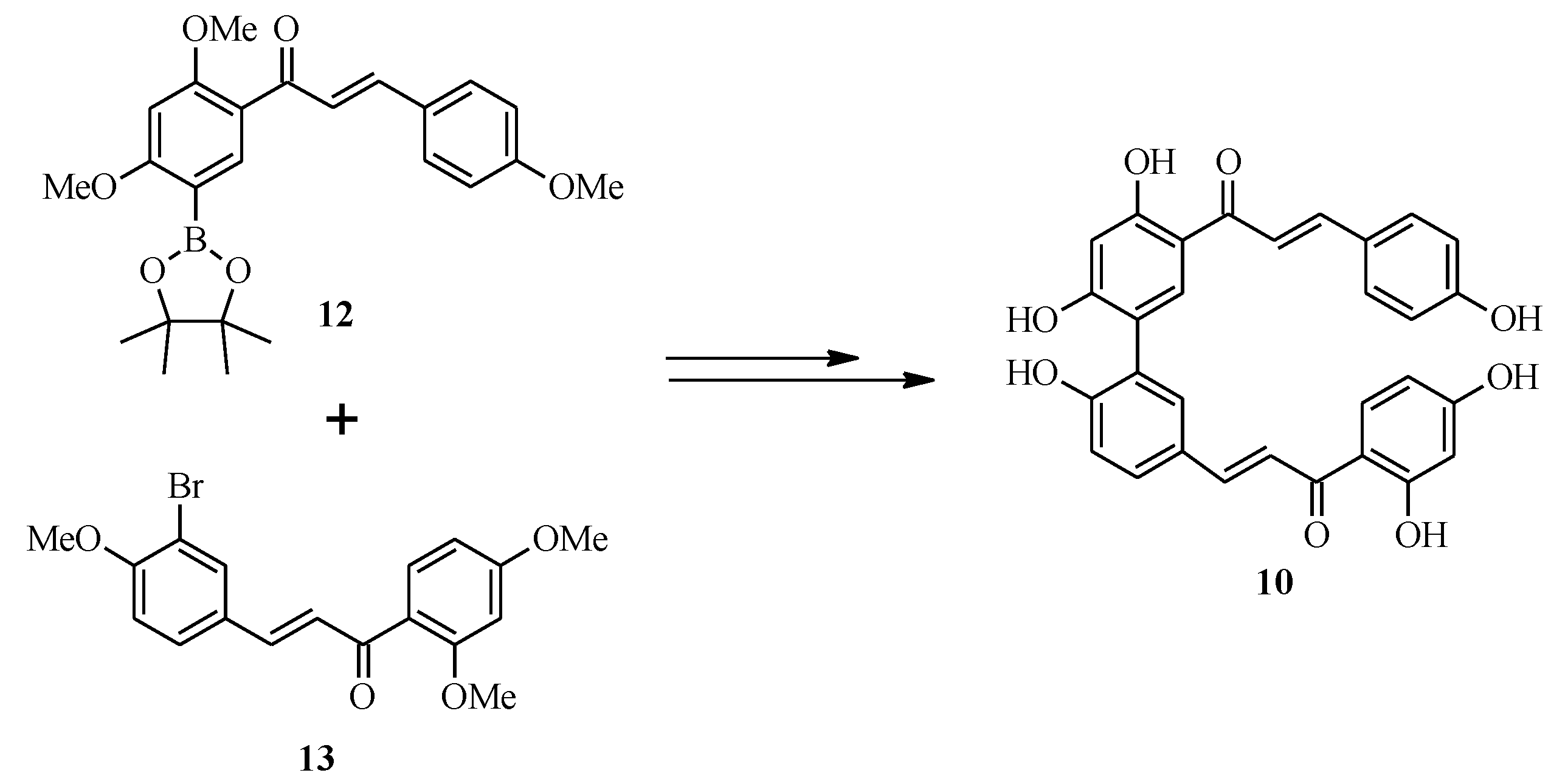
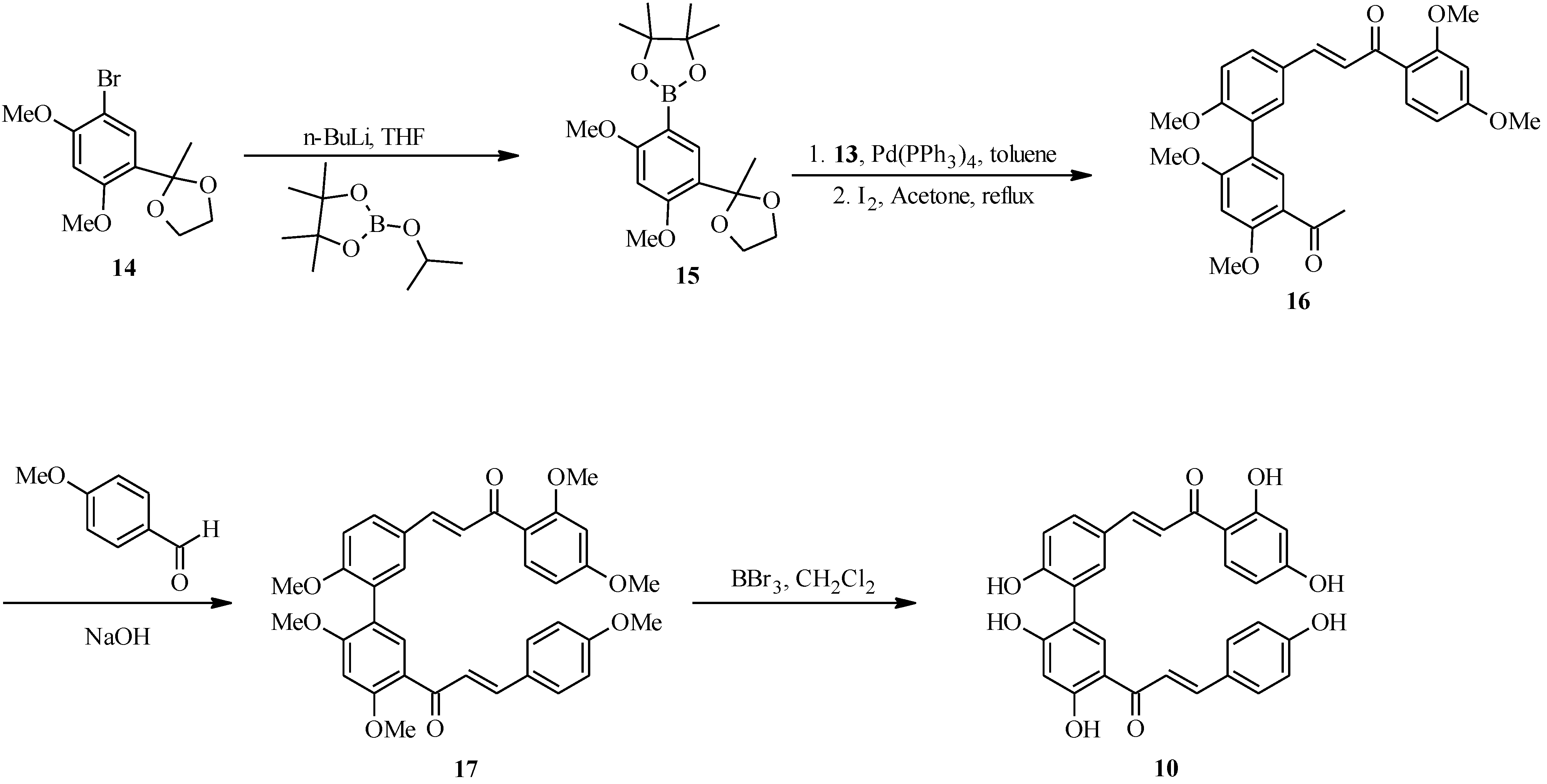
2.2. Bichalcones
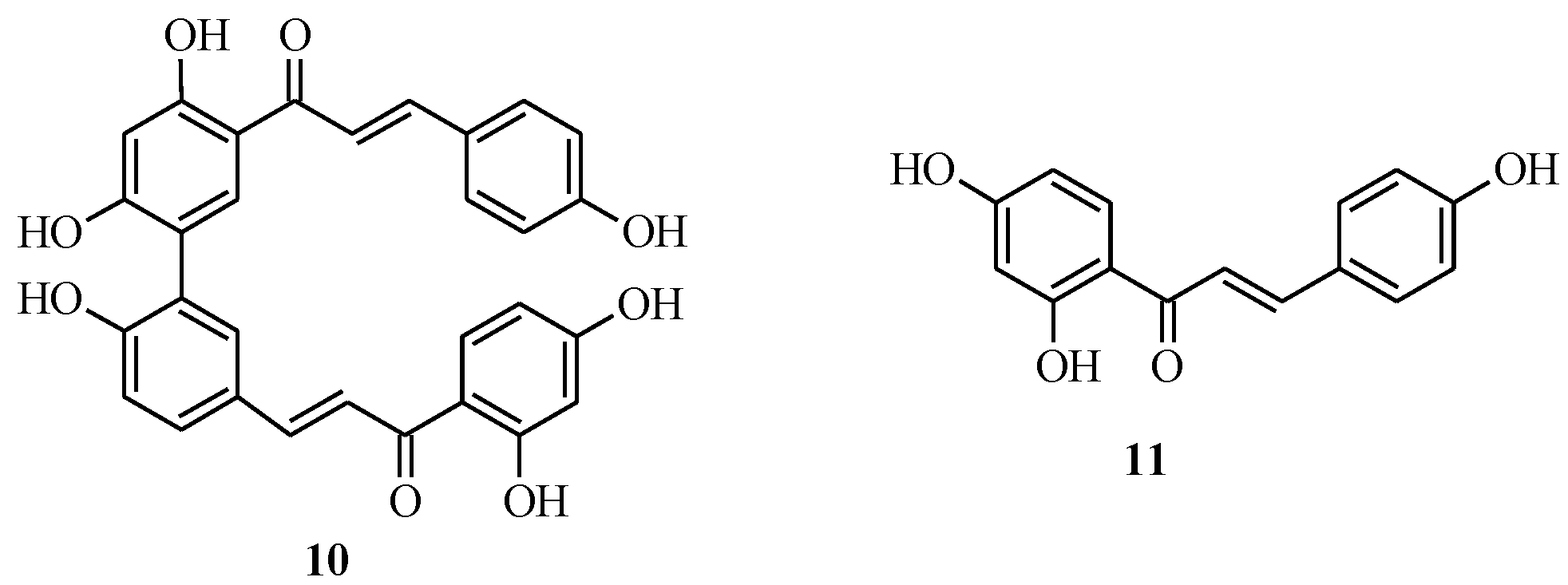
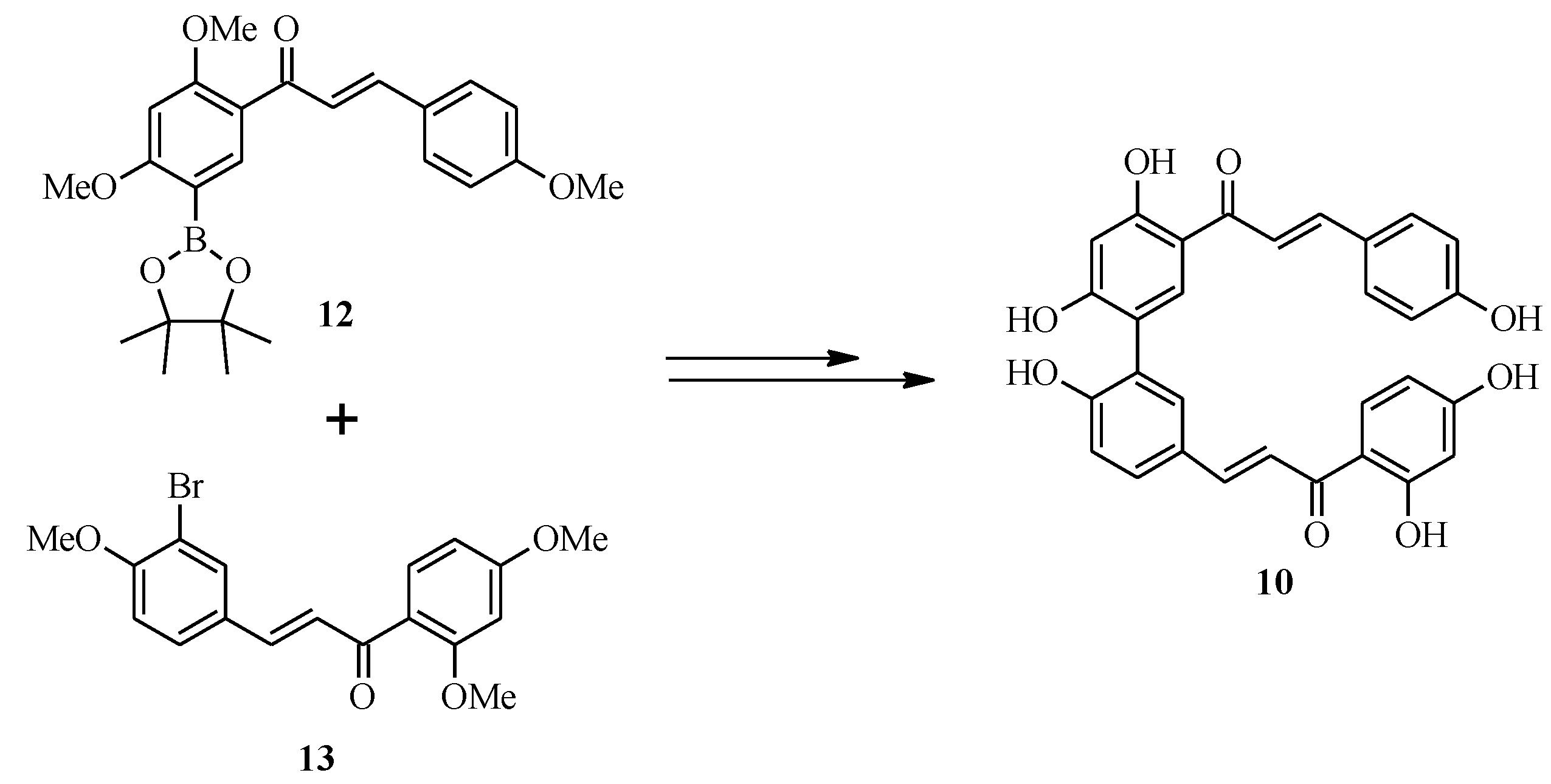
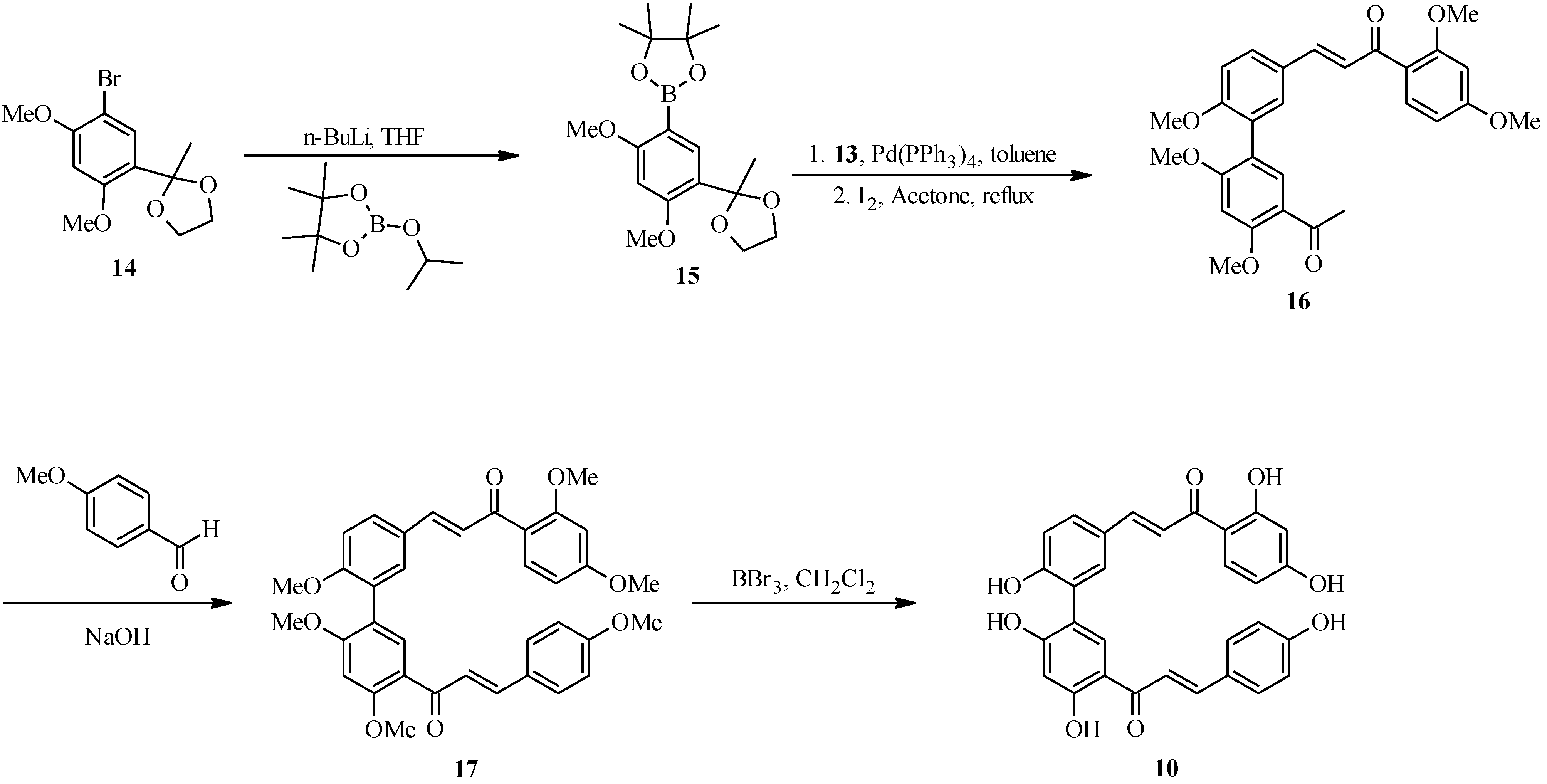
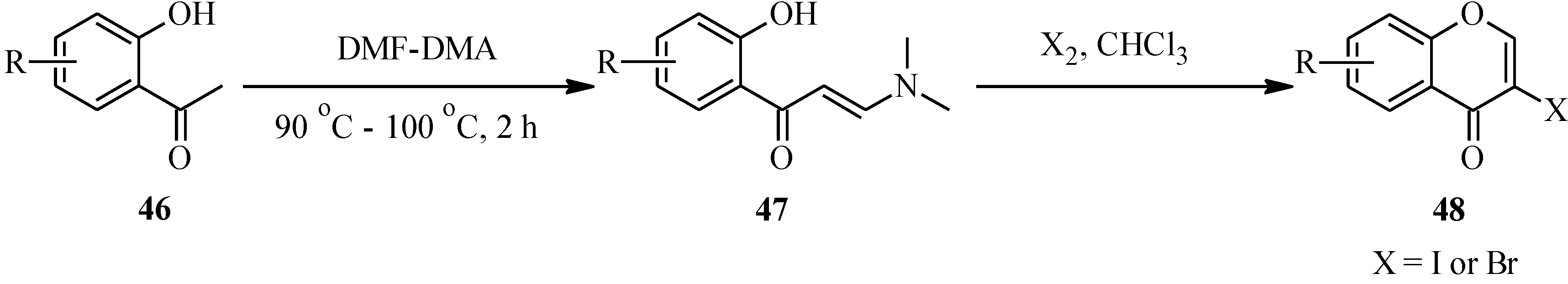
3. Flavones
3.1. Flavone Monomers
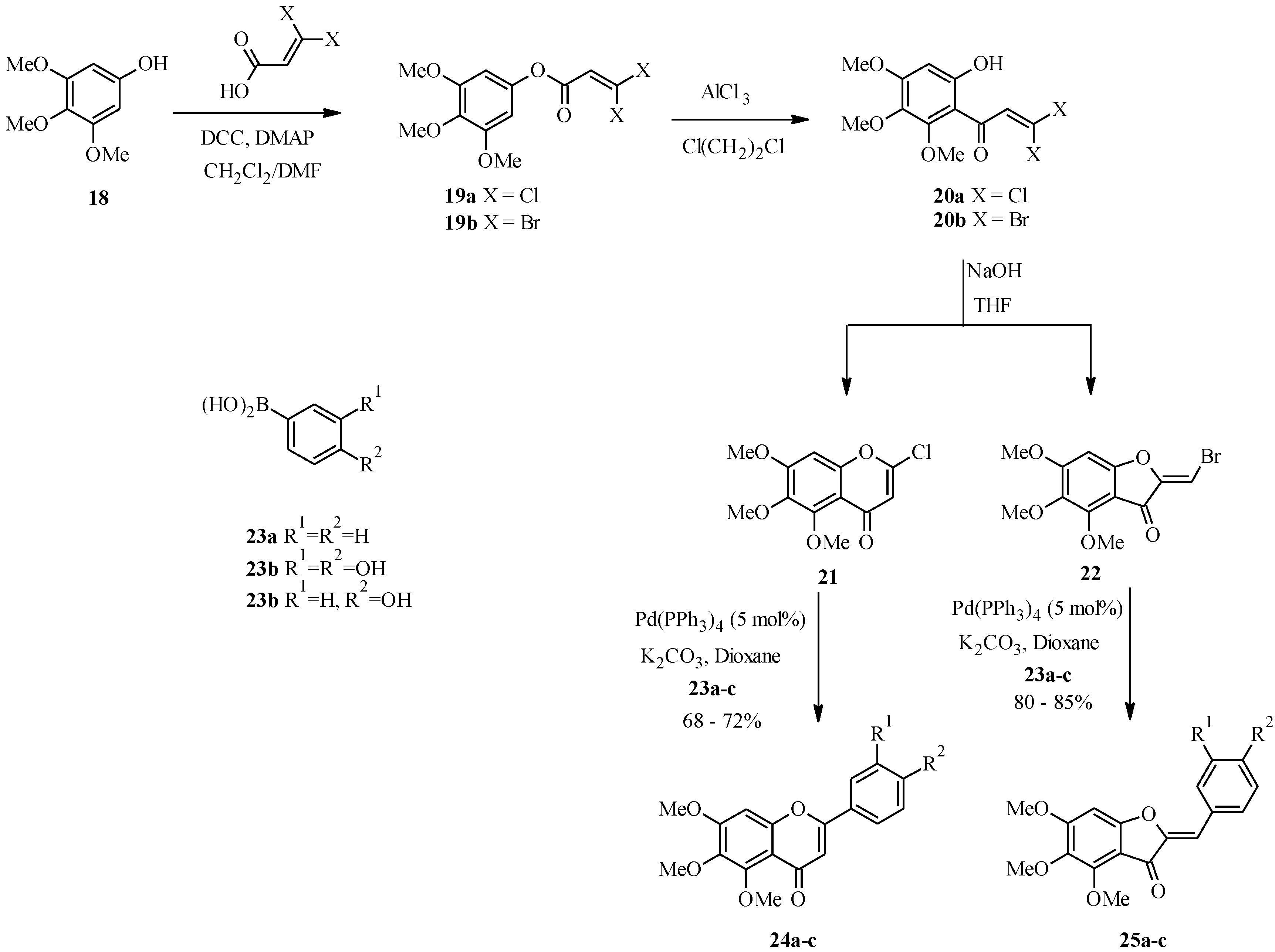
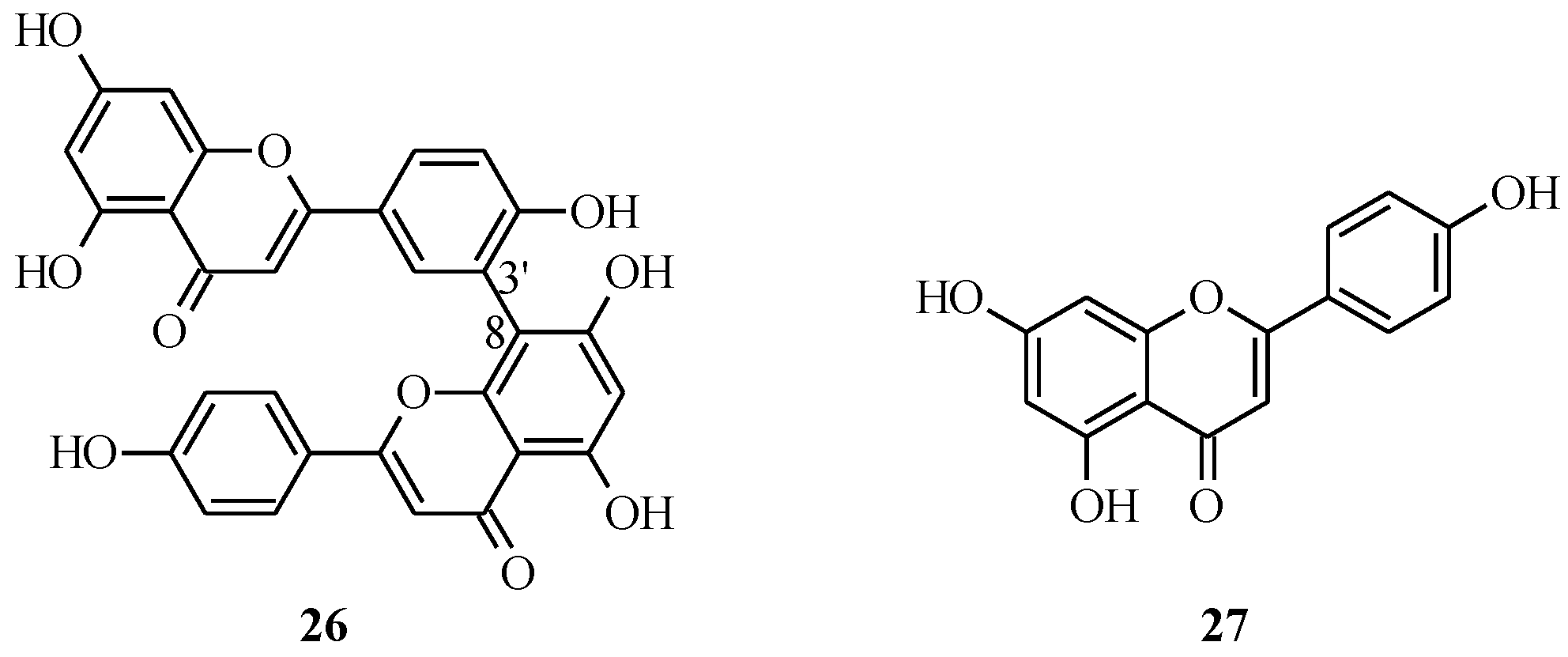
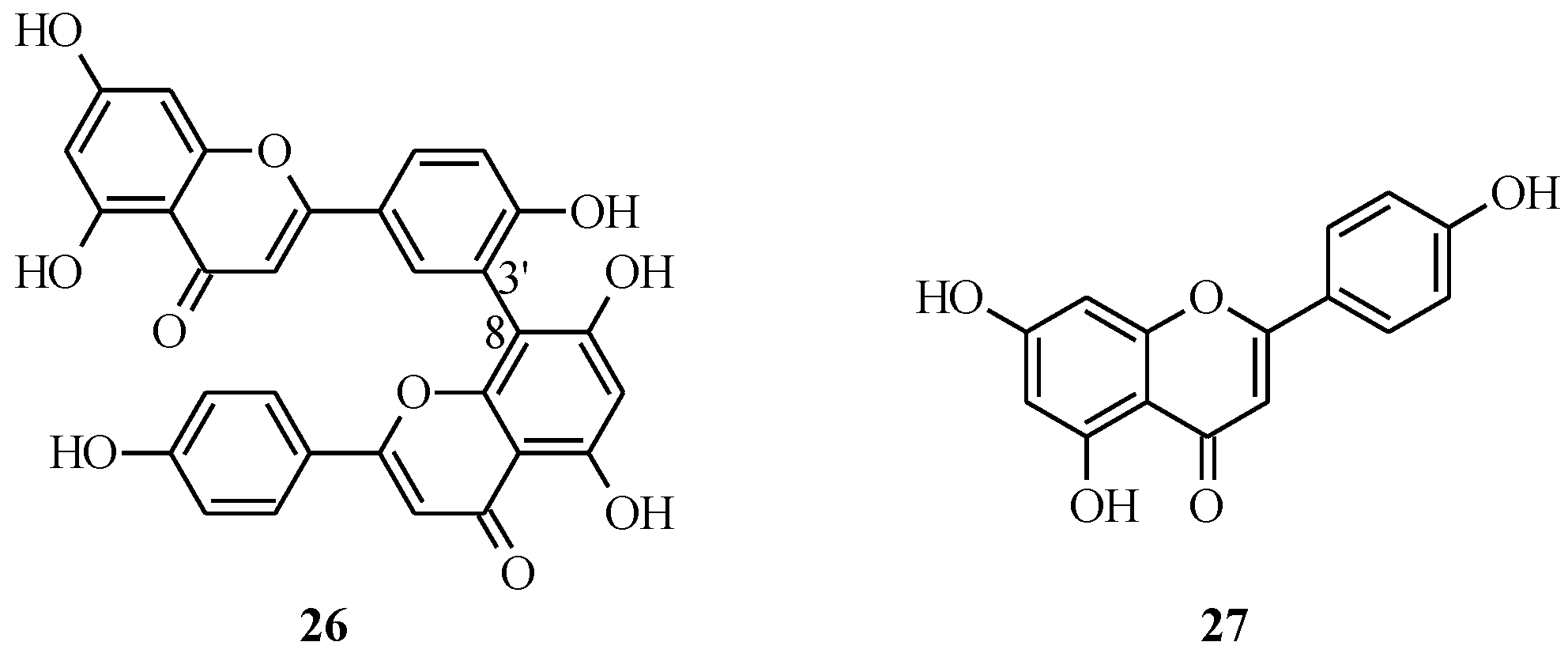
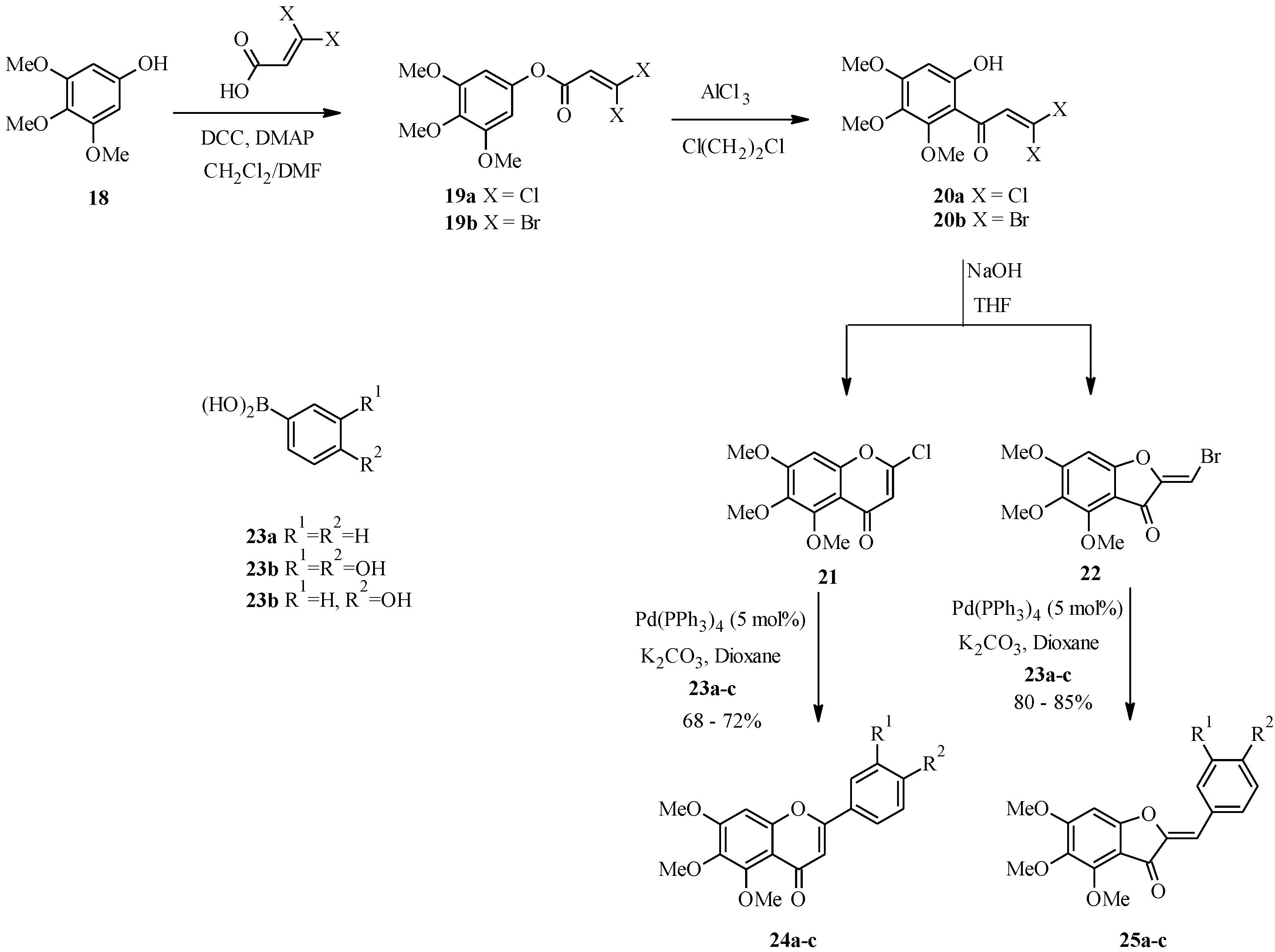
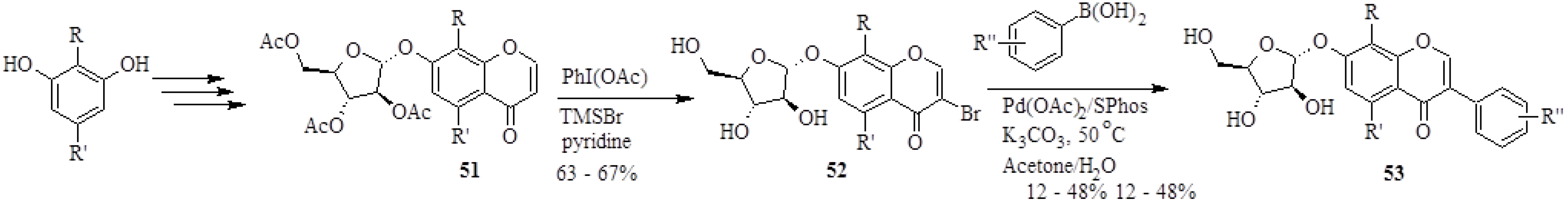
3.2. Biflavones
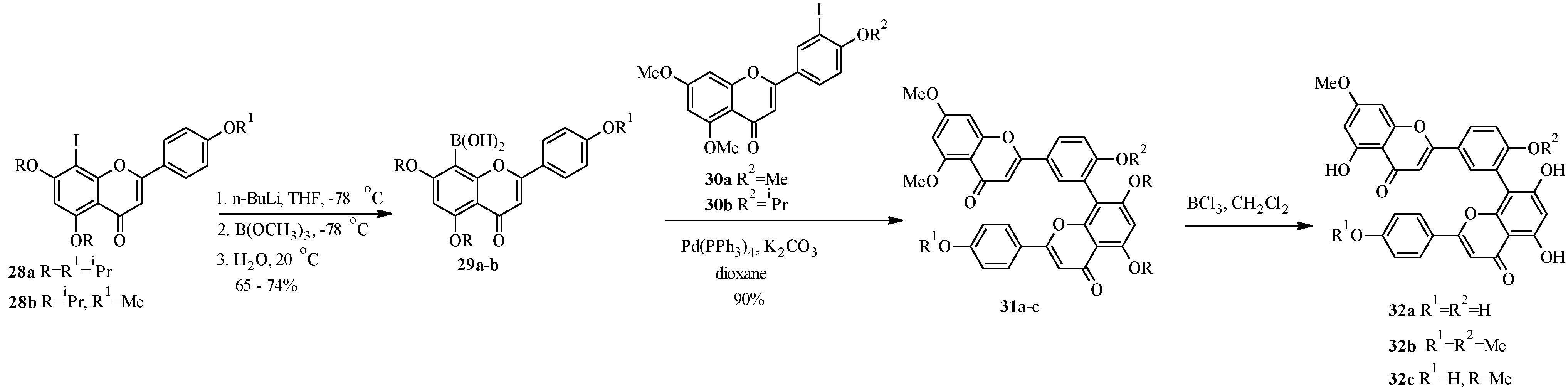
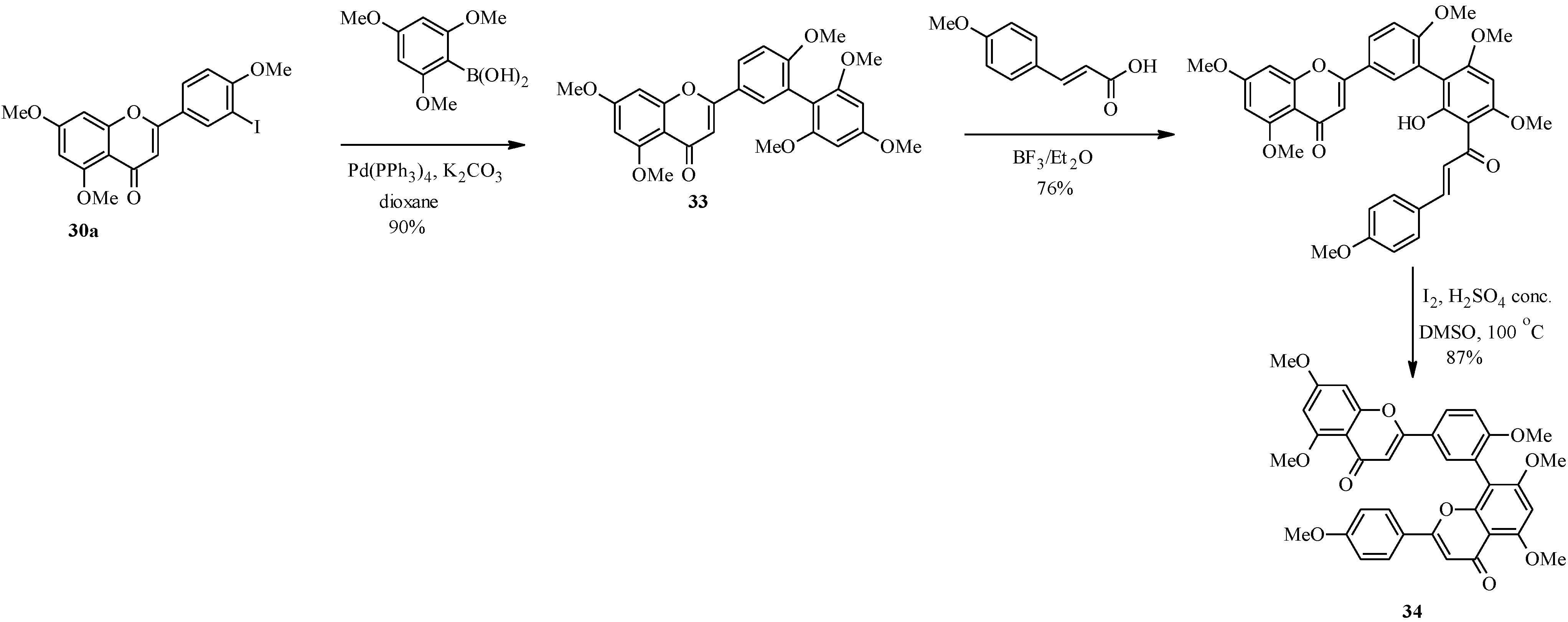

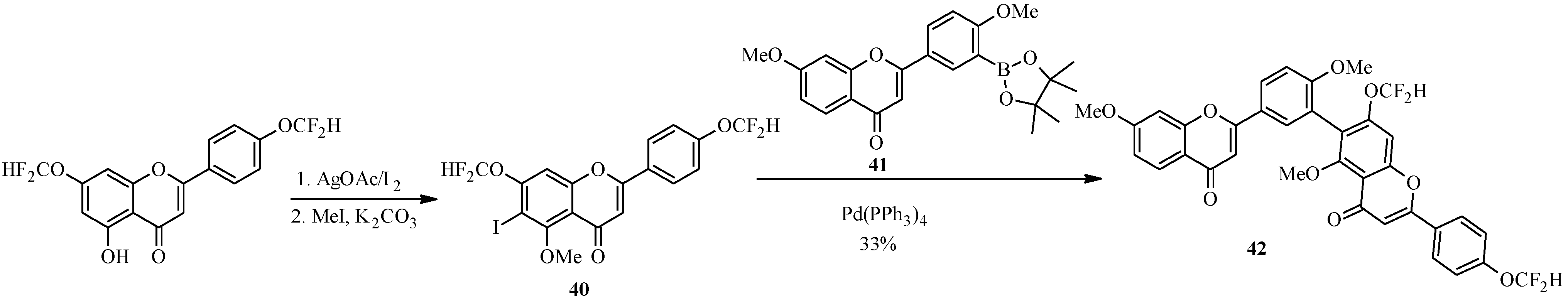
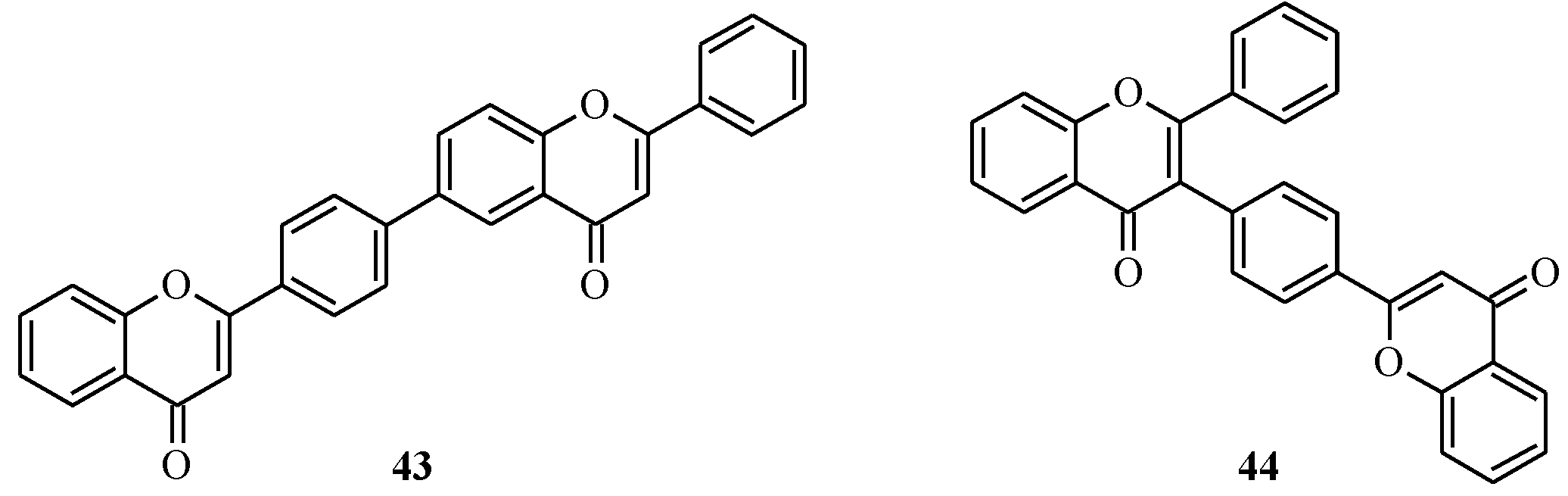
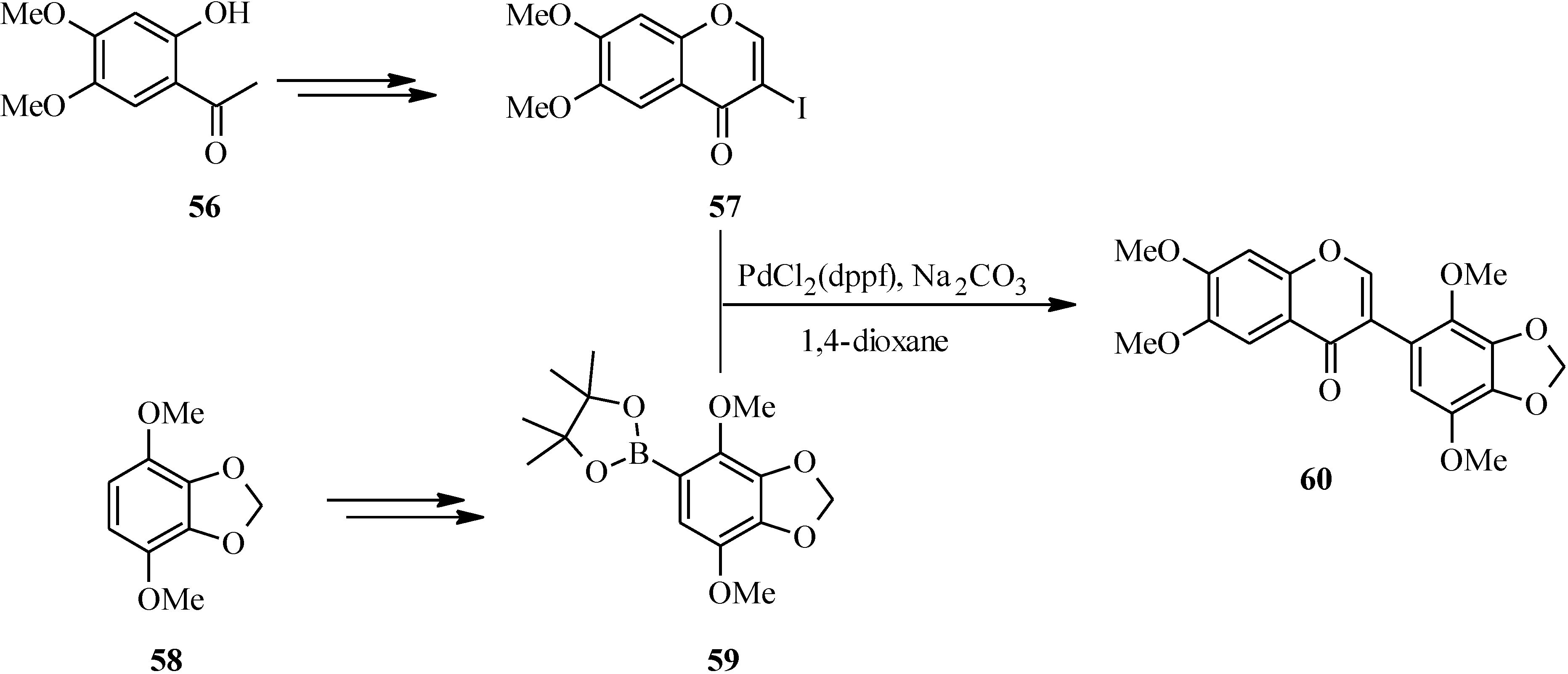
4. Isoflavones
4.1. Isoflavones
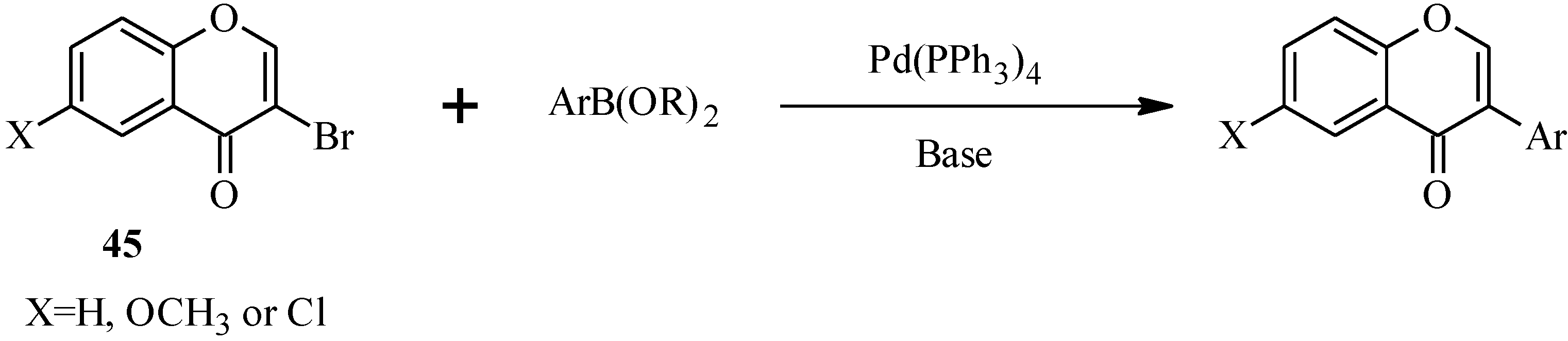
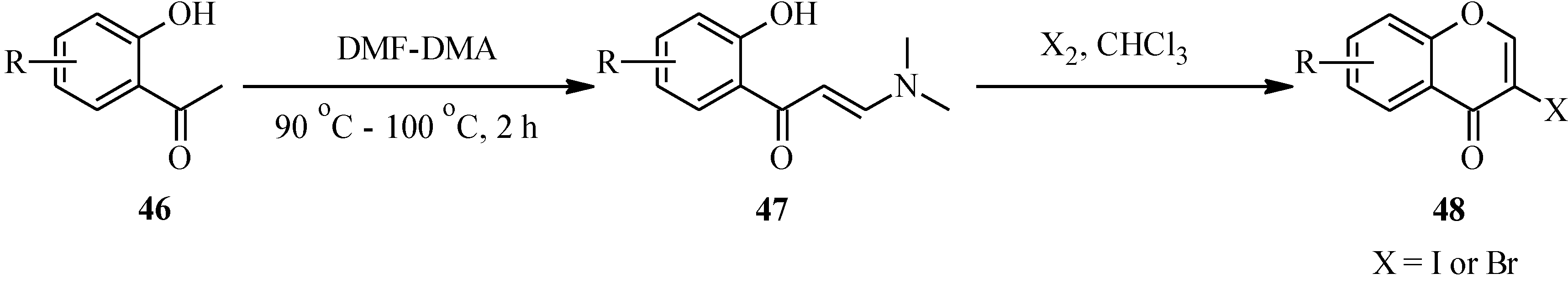
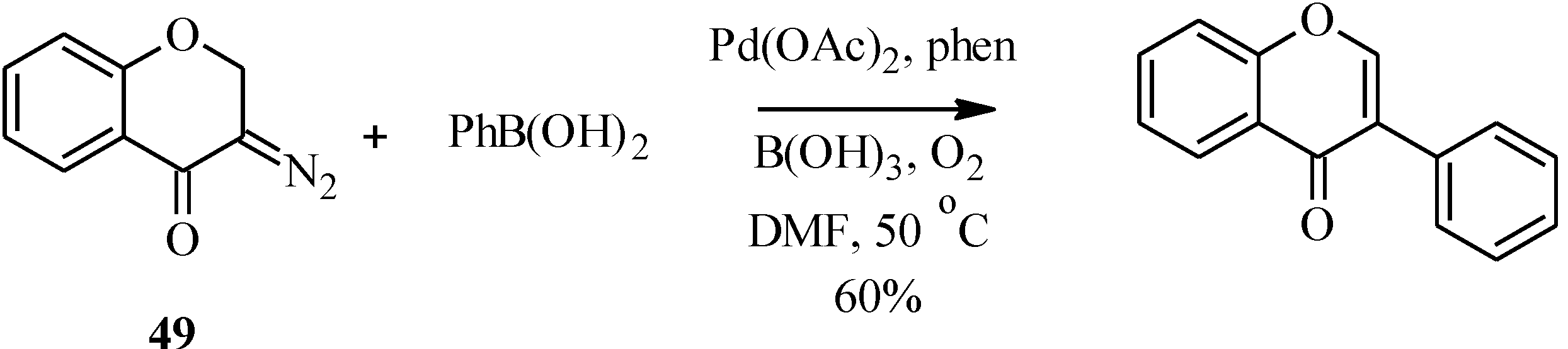
4.2. Preparation of Isoflavone Analogues for Biological Activity Studies
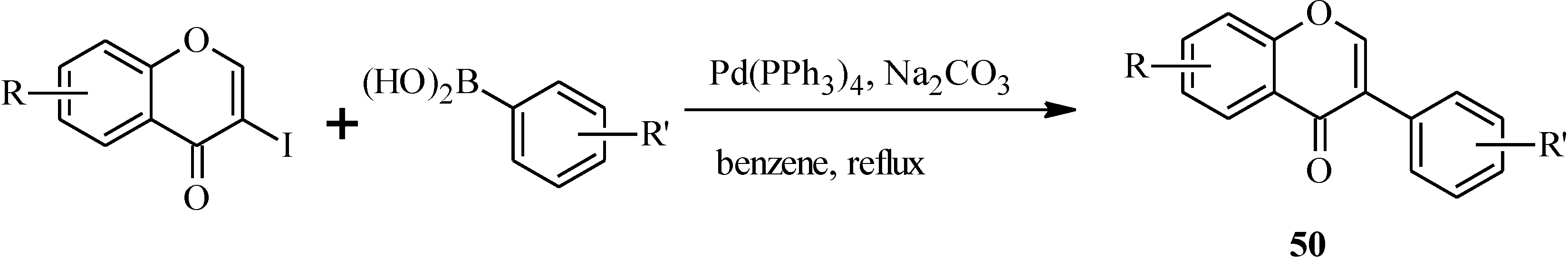
| 50 | R | R' |
|---|---|---|
| a | H | 4-Cl |
| b | H | 4-Br |
| c | H | 3-NO2 |
| d | H | 3-OMe |
| e | H | 4-OMe |
| f | H | 3,4-di-OMe |
| g | 6-F | 4-OMe |
| h | 6-F | 3,4-di-OMe |
| i | 6-F | 3,4-OCH2O |
| j | 7-F | 4-OMe |
| k | 7-F | 3,4-di-OMe |
| l | 8-F | 3,4-di-OMe |
| m | 6,7-di-F | 3,4-di-OMe |
| n | 6,8-di-F | 3,4-di-OMe |
| o | 7,8-di-F | 3,4-di-OMe |
| 62 | R | R' | R'' | IC50 (μM) * |
|---|---|---|---|---|
| a | OH | H | 4-OH | 0.018 |
| b | OH | H | H | 0.040 |
| c | OH | H | 4-OMe | 0.022 |
| d | OH | H | 2-OMe | 0.015 |
| e | OH | H | 3-OMe | 0.006 |
| f | OH | H | 4-Me | 0.035 |
| g | OH | H | 2-Me | 0.020 |
| h | OH | H | 3-Me | 0.050 |
| i | OH | H | 3-OH | 0.017 |
| j | OH | H | 4-F | 0.015 |
| k | OH | H | 4-NMe2 | 0.008 |
| l | OH | H | 4-NHBoc | 0.020 |
| m | OH | H | 4-CF3 | 0.030 |
| n | OH | H | 4-SiMe3 | (35% at 0.050) |
| o | H | OH | H | 10 |
| p | H | OH | 4-OMe | 8 |
| q | H | OH | 2-OMe | 10 |
| r | H | OH | 3-OMe | 10 |
| s | H | OH | 4-Me | 7 |
| t | H | OH | 3-Me | 5 |
| u | H | OH | 4-OH | 15 |
| v | H | OH | 3-OH | (33% at 50) |
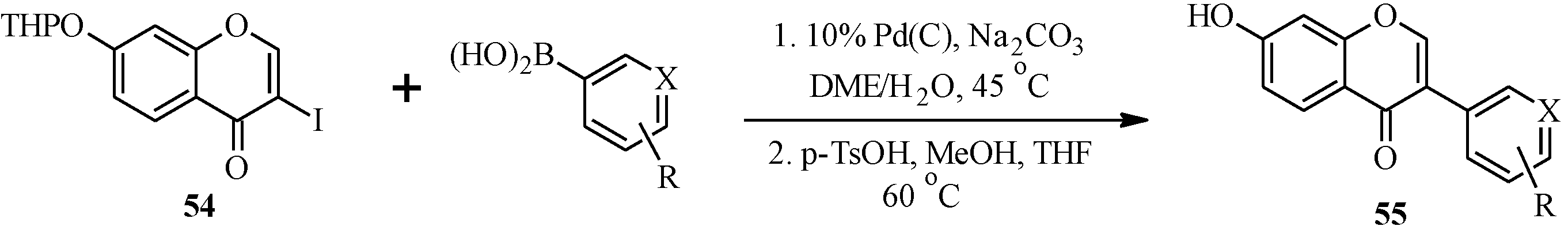
| 55 | X | R | 55 | X | R |
|---|---|---|---|---|---|
| a | C | 3,5-di-OMe | j | C | 2-OMe, 3,5-di-F |
| b | C | 4-OMe | k | C | 3,4,5-tri-F |
| c | C | 3,4-OCH2O- | l | N | 2,4-di-OMe |
| d | C | 4-CF3 | m | C | 4-Cl |
| e | C | 4-F | n | C | 3-F |
| f | C | 3,4-di-OMe | o | C | 2-OMe |
| g | C | 3,4-O(CH2)2-O | p | C | 3-OCF3 |
| h | C | 2,4-di-F | q | C | 3-OBn |
| i | C | 3-OMe | r | C | 3,4,5-tri-OMe |
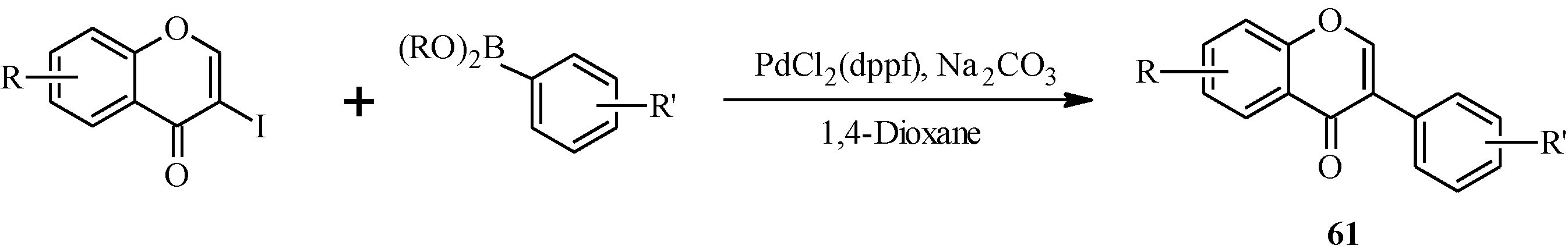
| 61 | R | R' |
|---|---|---|
| a | 6,7-di-OMe | 2',5'-di-OMe, 3',4'-OC(CH3)2O- |
| b | 6,7-di-OMe | 2',3',4',5'-tetra-OMe |
| c | 6,7-di-OMe | 3,4-OCH2O- |
| d | 6-OMe, 7-OTHP | 2',5'-di-OMe, 3',4'-OCH2O- |
| e | 5,6,7-tri-OMe | 2',5'-di-OMe, 3',4'-OCH2O- |
| f | 6-OMe, 7-OH | 2',5'-di-OMe, 3',4'-OCH2O- |
| g | 6-OMe, 7-OBn | 2',5'-di-OMe, 3',4'-OCH2O- |
| h | 6-OMe, 7-O-propargyl | 2',5'-di-OMe, 3',4'-OCH2O- |
| i | 6-OMe, 7-O-allyl | 2',5'-di-OMe, 3',4'-OCH2O- |
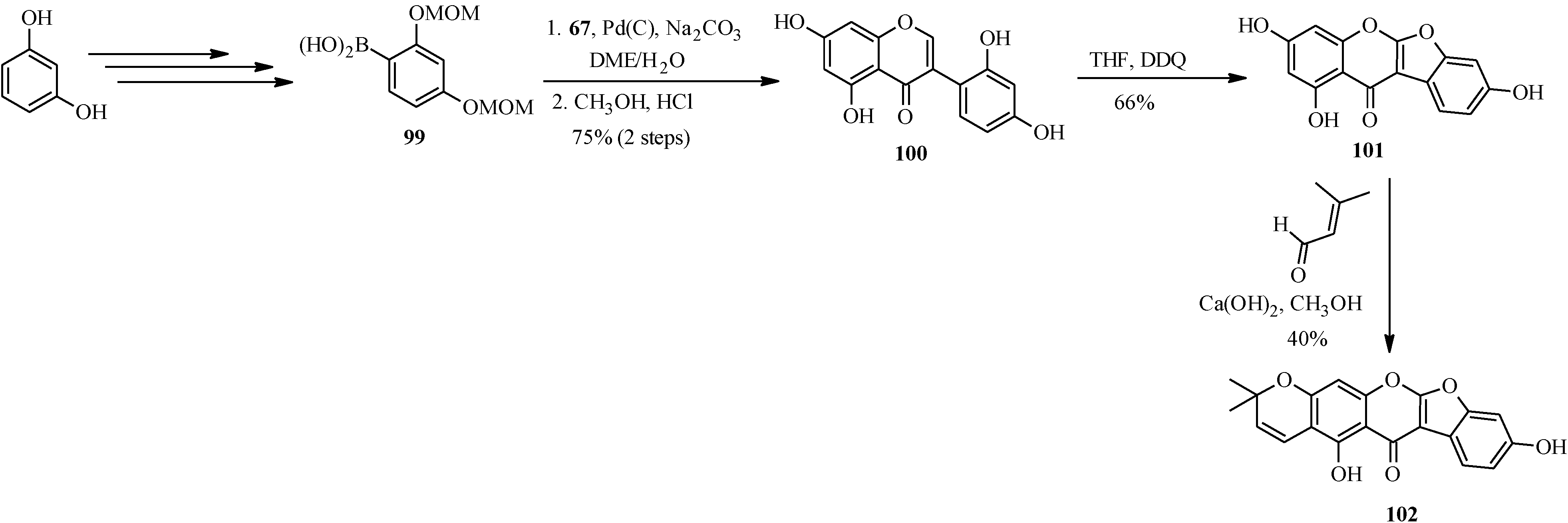
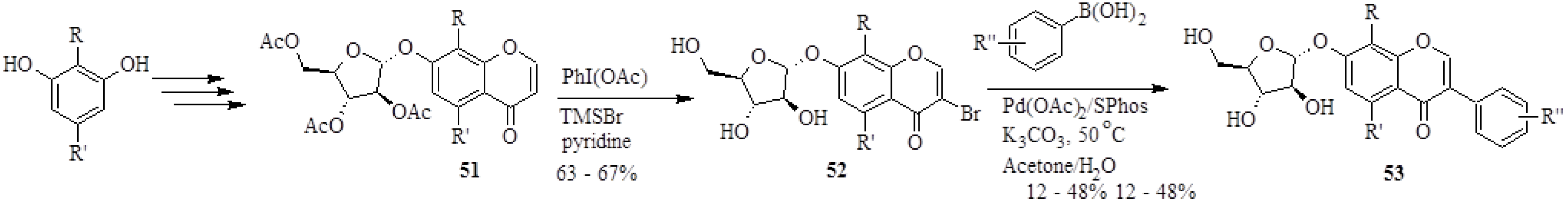
4.3. Synthesis of Soy Isoflavones
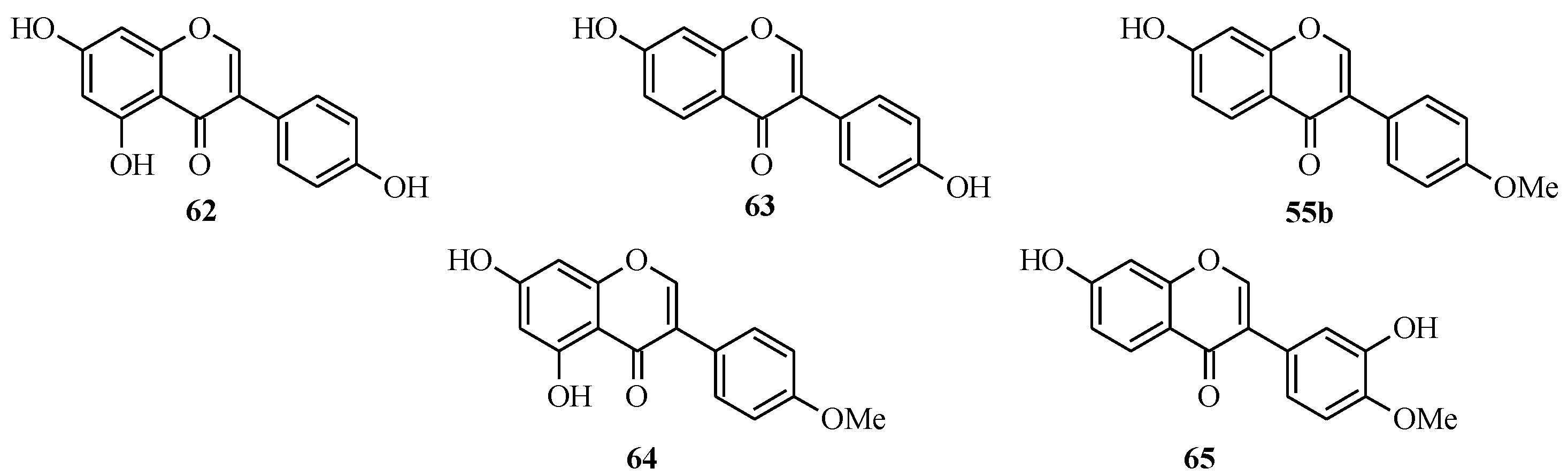
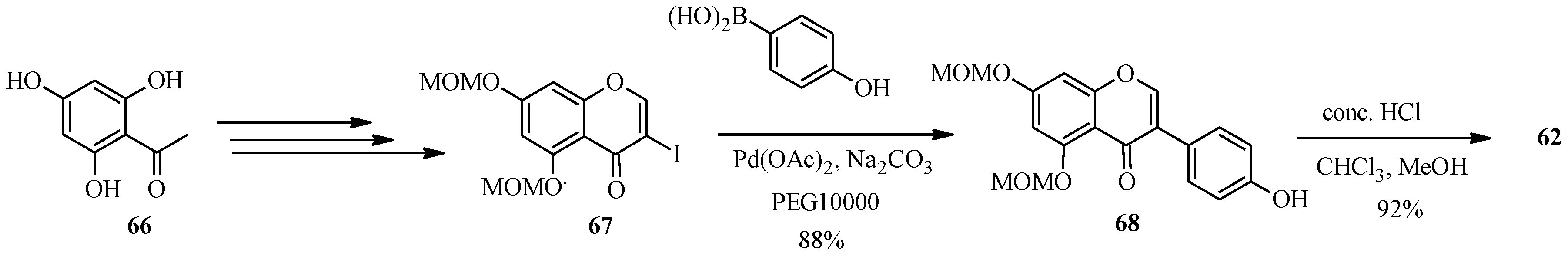
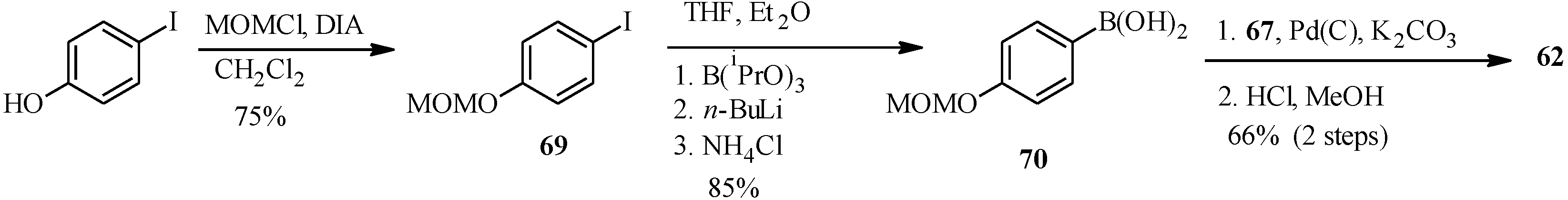
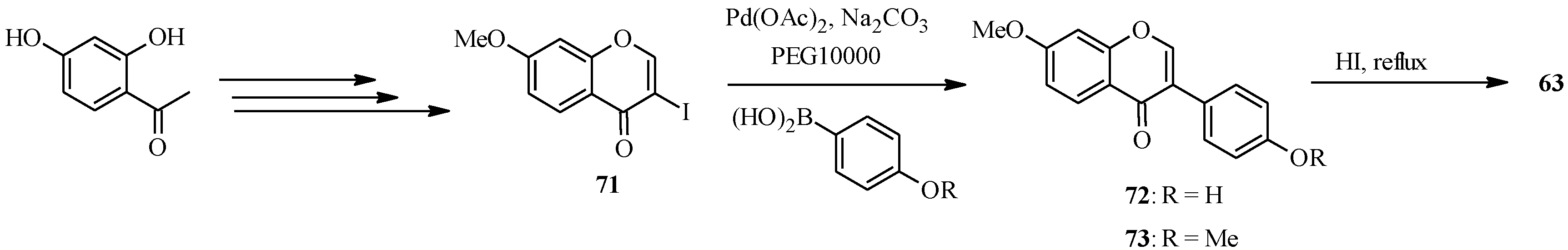
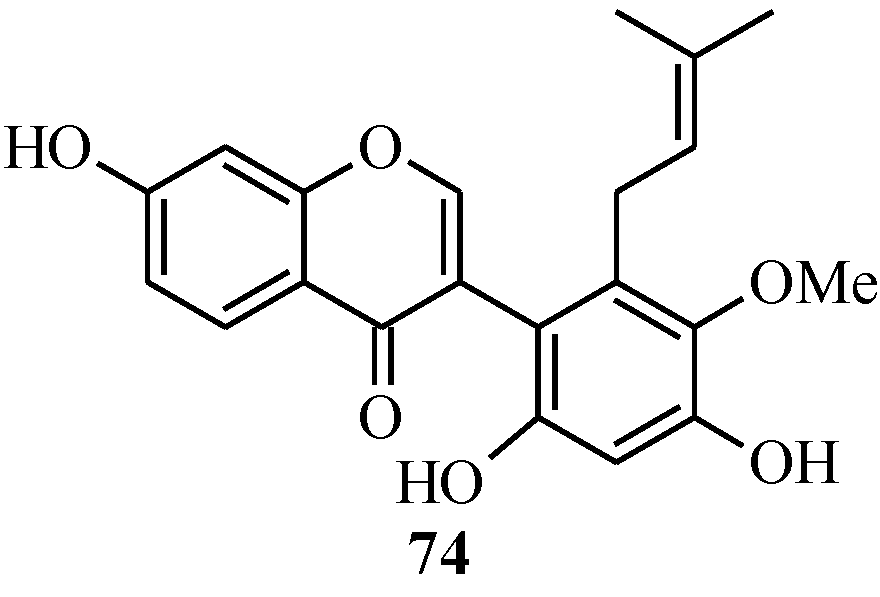
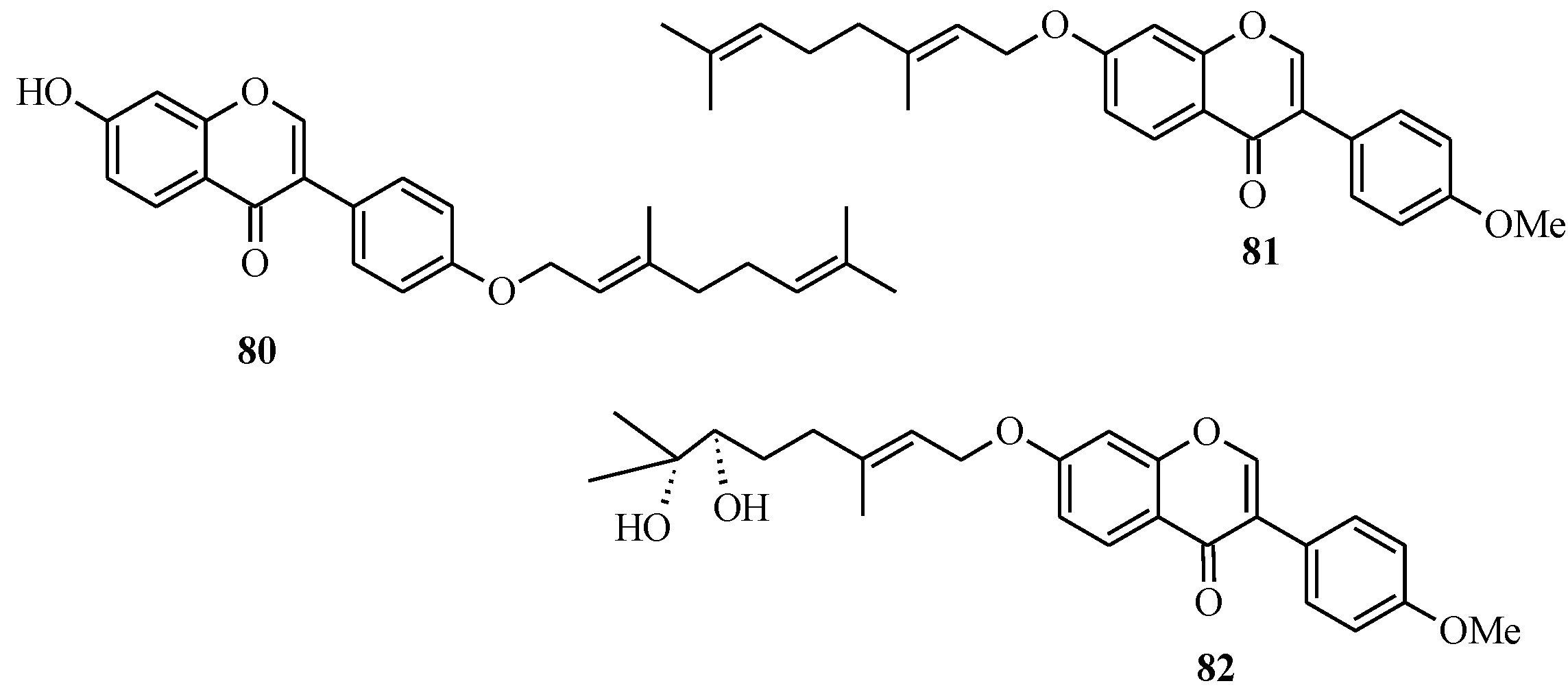
4.4. Synthesis of Prenylated Isoflavonoids
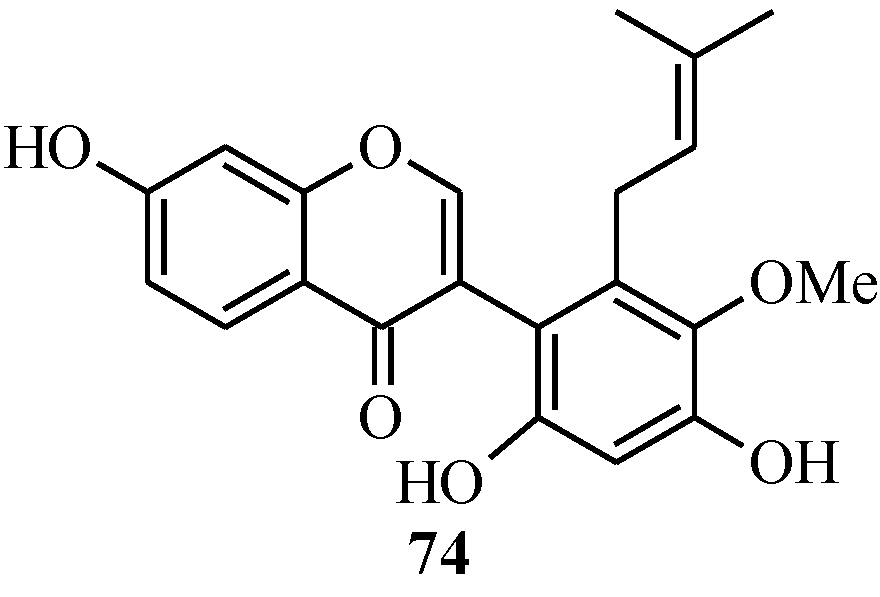
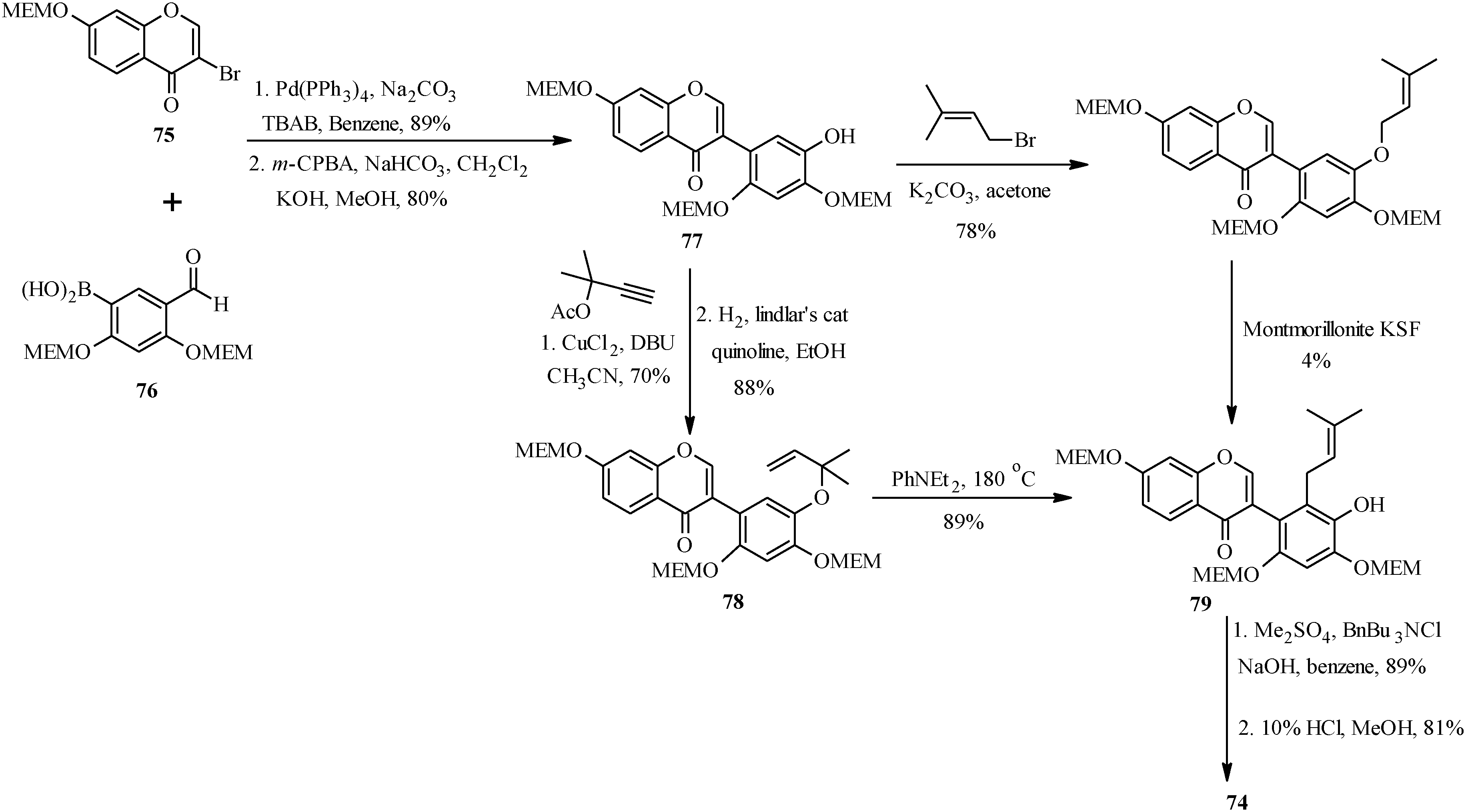
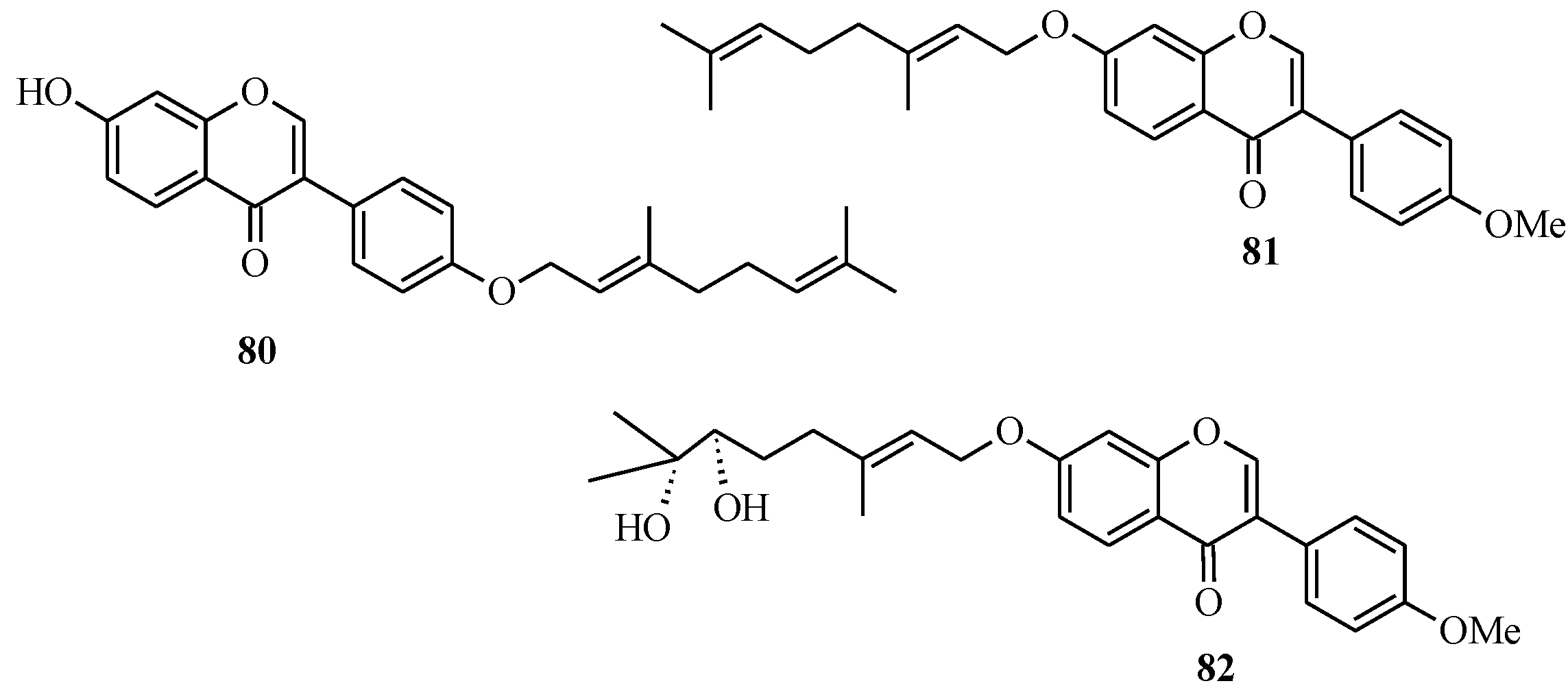
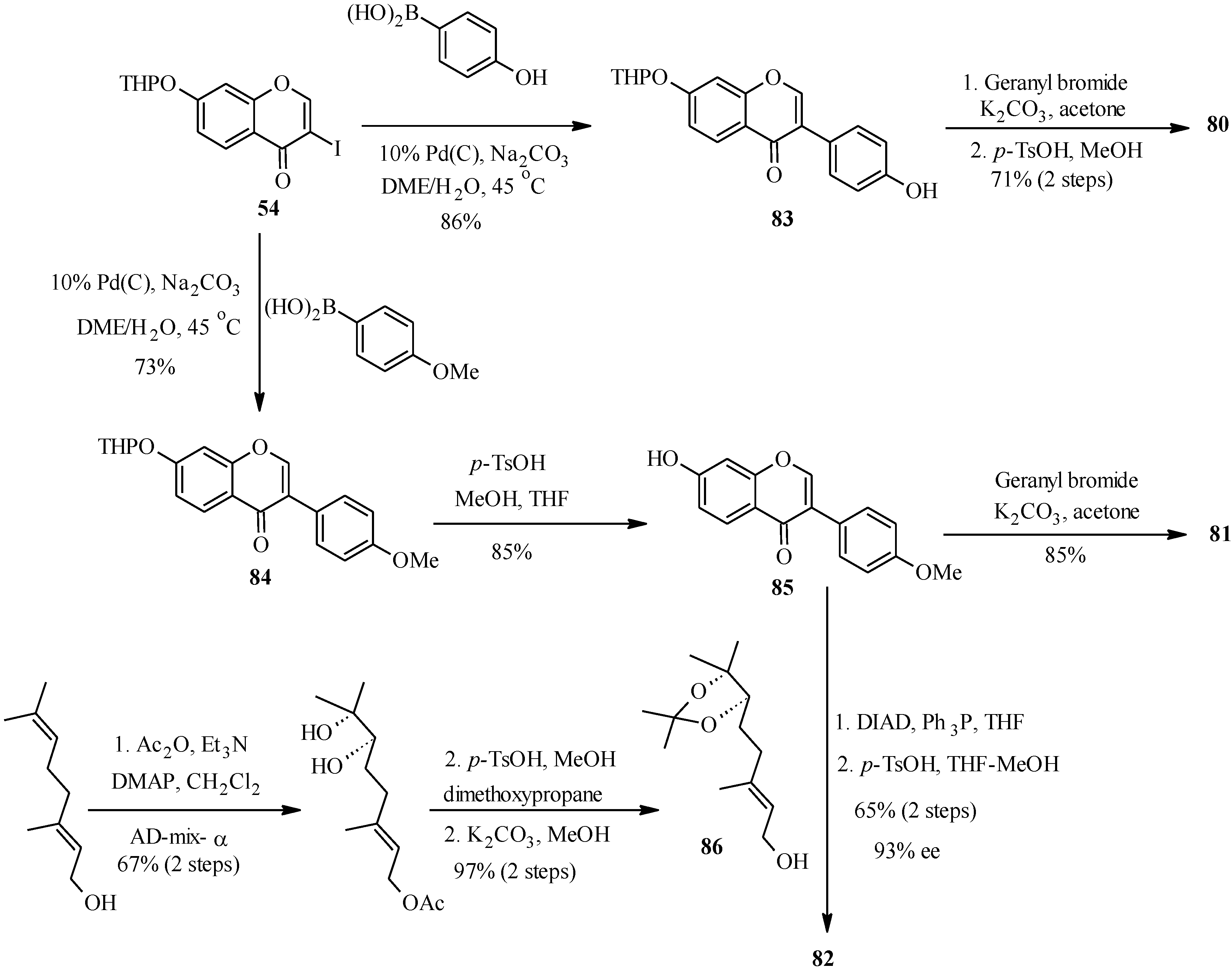
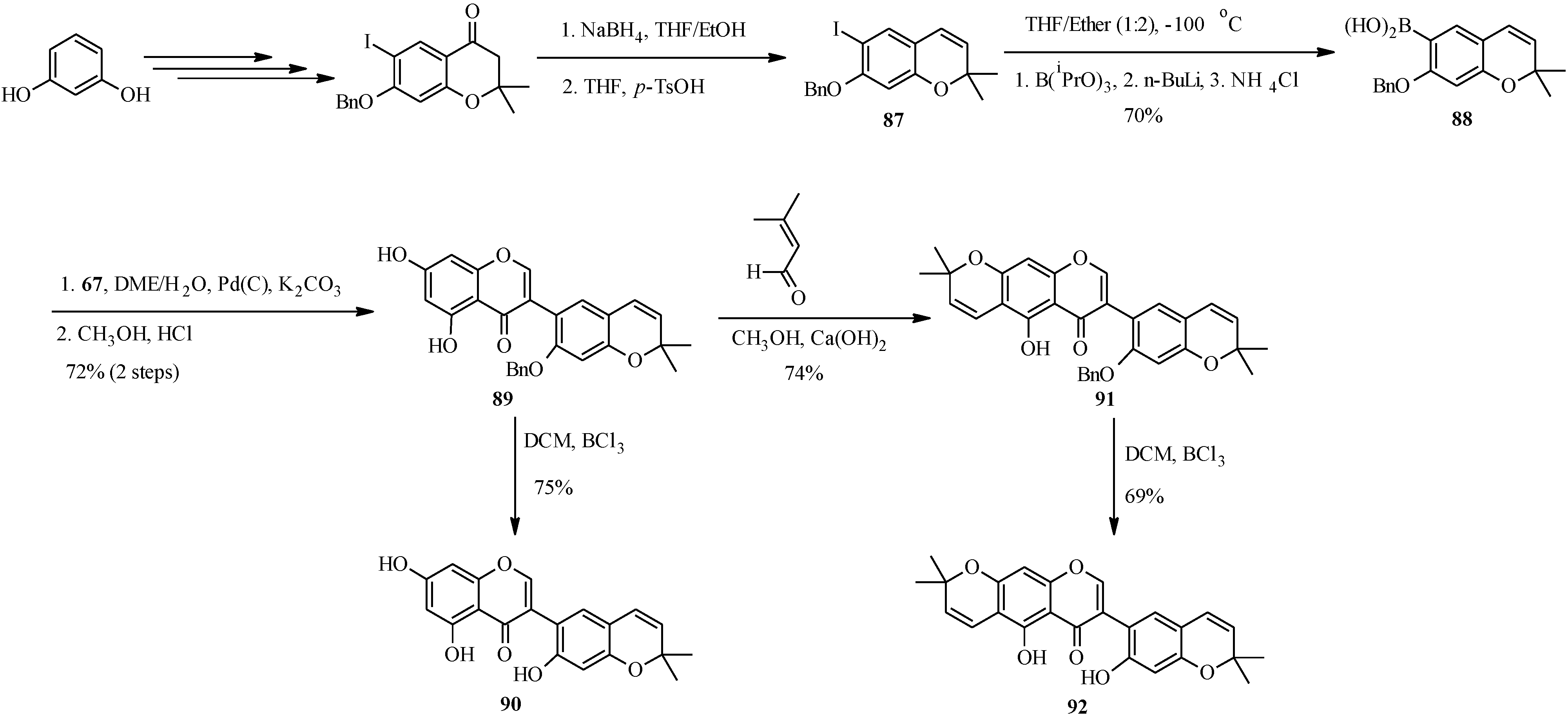
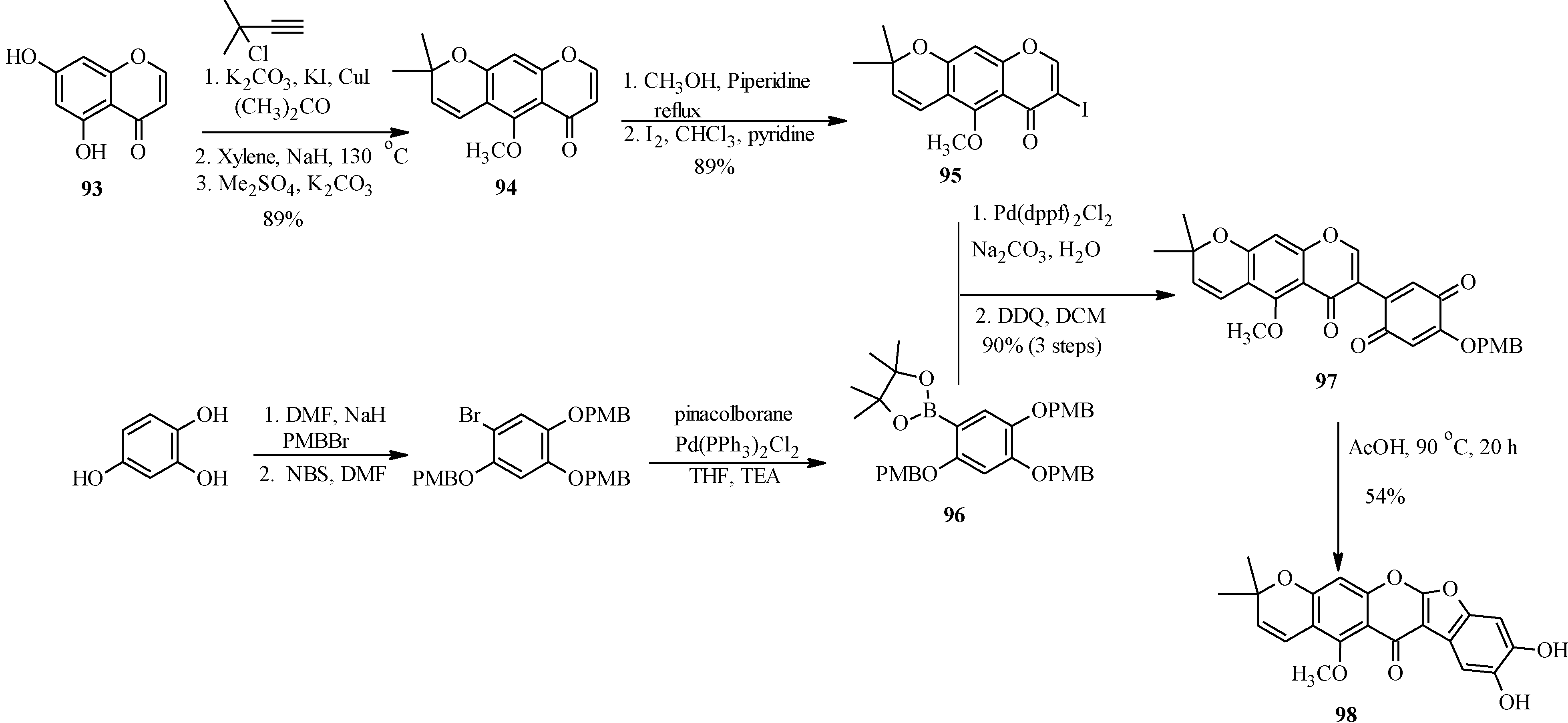
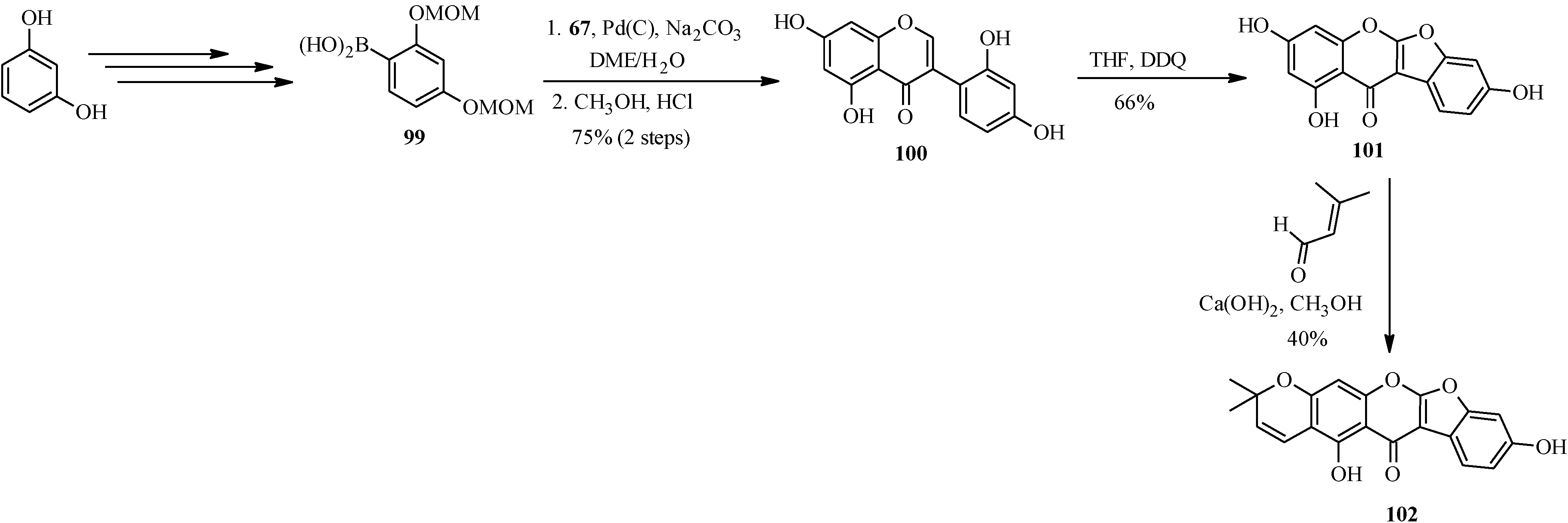
5. Neoflavones
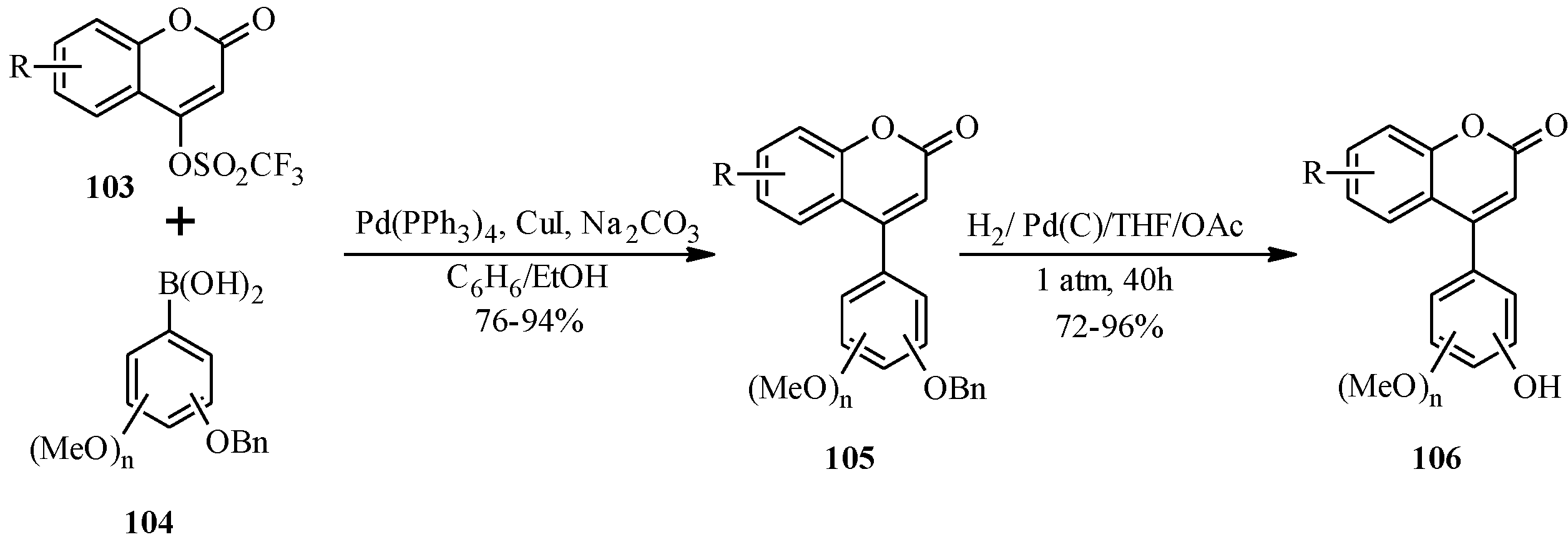
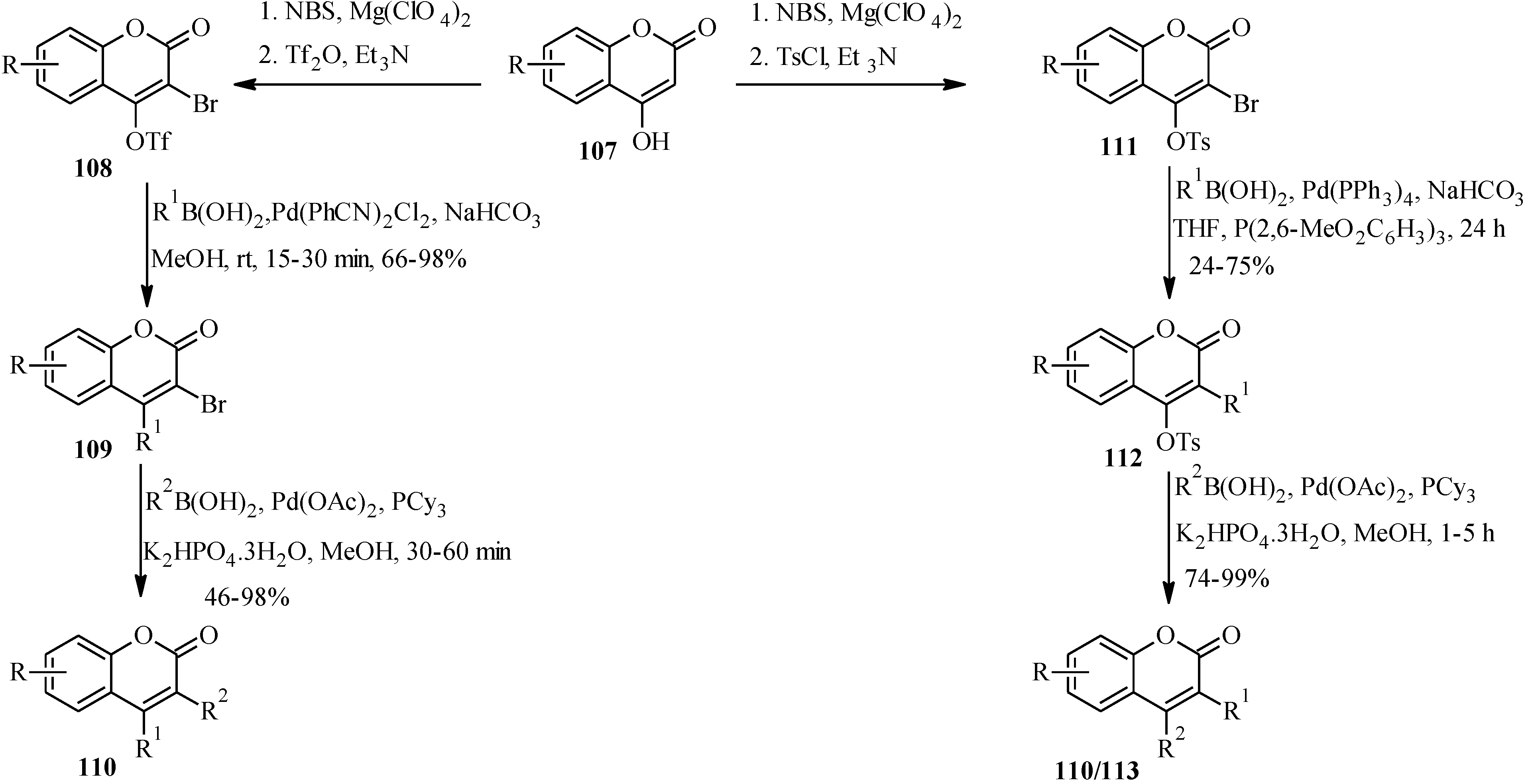
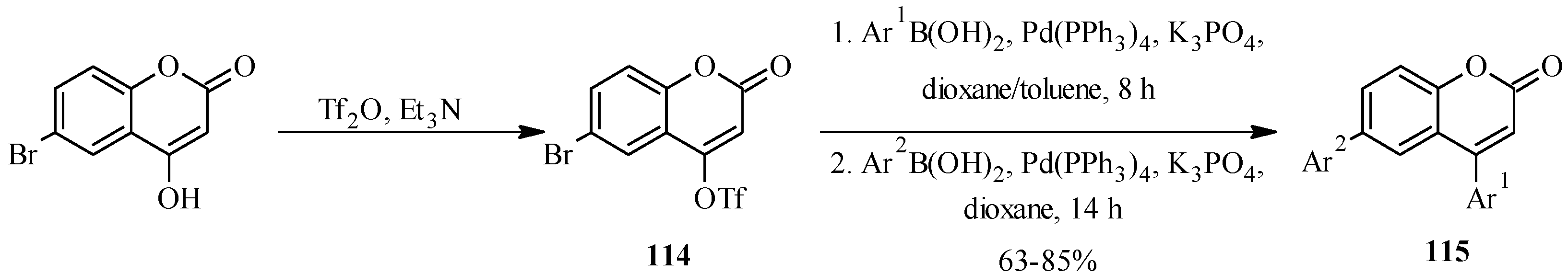
5.1. Synthesis of Neoflavones
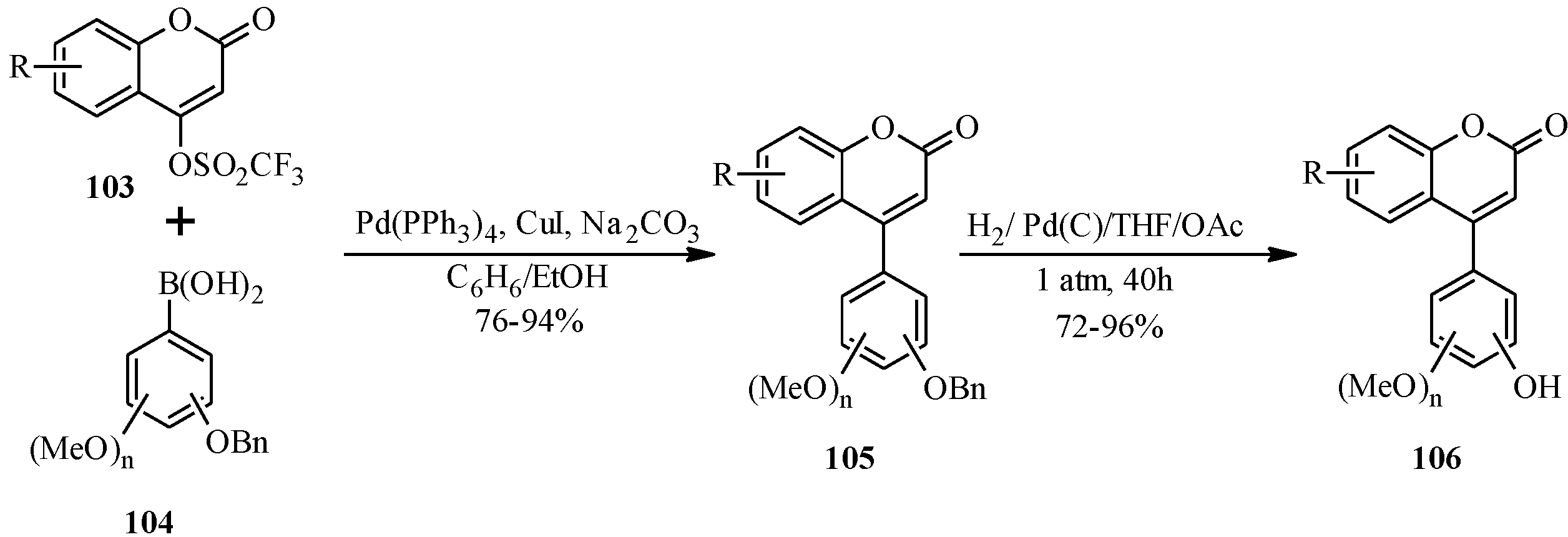
5.2. Synthesis of Arylated Neoflavones
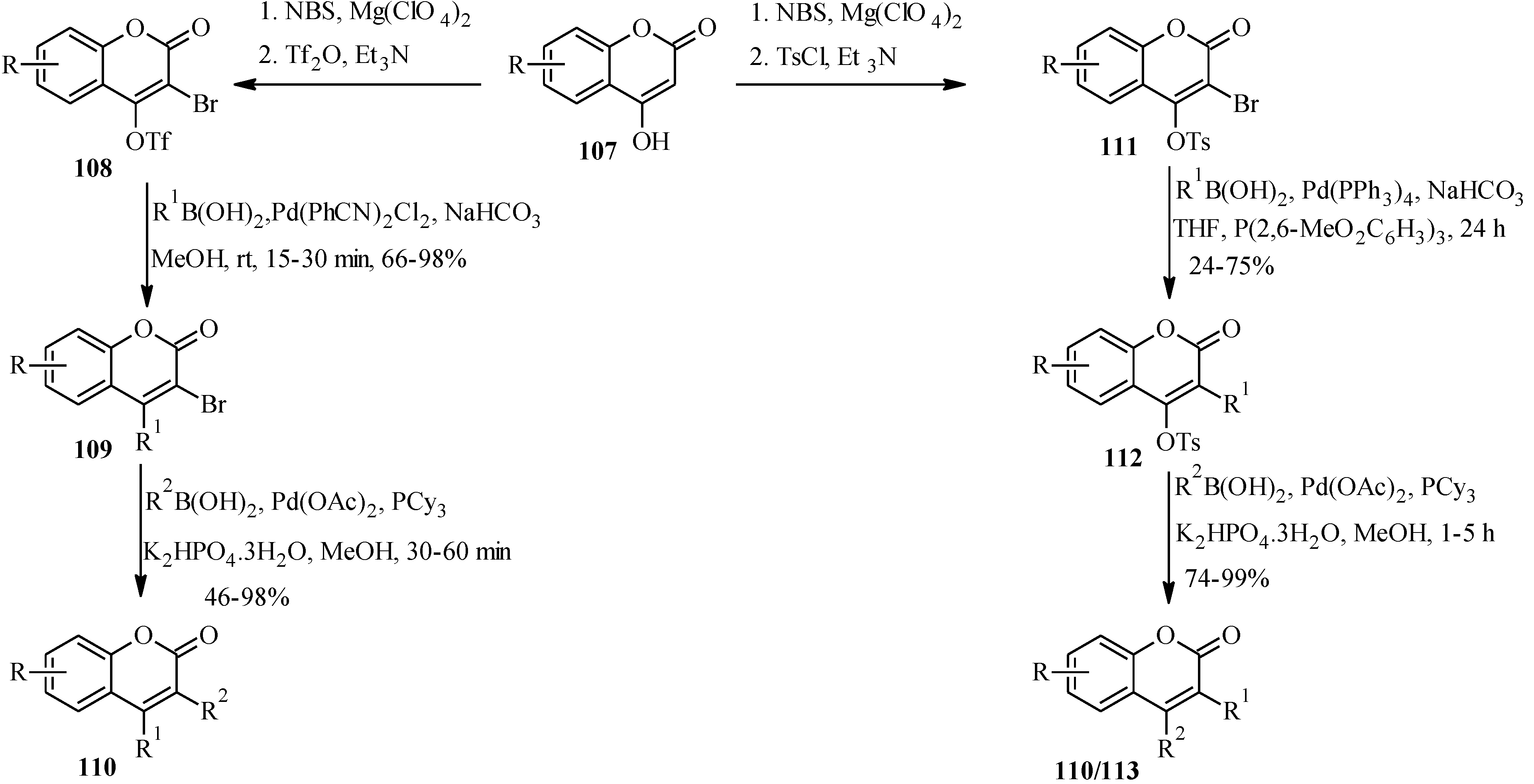
| R1 | R2 | Product |
|---|---|---|
| 4-MeOC6H4 | 4-MeOC6H4 | 110a |
| 4-MeOC6H4 | 4-CF3OC6H4 | 110b |
| 4-MeOC6H4 | 4-MeC6H4 | 110c |
| 4-MeOC6H4 | C6H5 | 110d |
| 4-MeOC6H4 | 4-NO2C6H4 | 110e |
| 4-MeOC6H4 | 4-MeO2CC6H4 | 110f |
| C6H5 | C6H5 | 110g |
| C6H5 | 4-MeOC6H4 | 110h |
| 4-NO2C6H4 | 4-MeOC6H4 | 110i |
| 4-MeO2CC6H4 | 4-MeOC6H4 | 110j |
| 4-MeOC6H4 | 4-MeOC6H4 | 110a |
| 4-MeOC6H4 | 4-CF3OC6H4 | 113a |
| 4-MeOC6H4 | 4-MeC6H4 | 113b |
| 4-MeOC6H4 | C6H5 | 110h |
| 4-MeOC6H4 | 4-MeO2CC4H6 | 110j |
| 4-MeC6H4 | 4-MeC6H4 | 113c |
| 4-MeC6H4 | 4-MeOC6H4 | 110c |
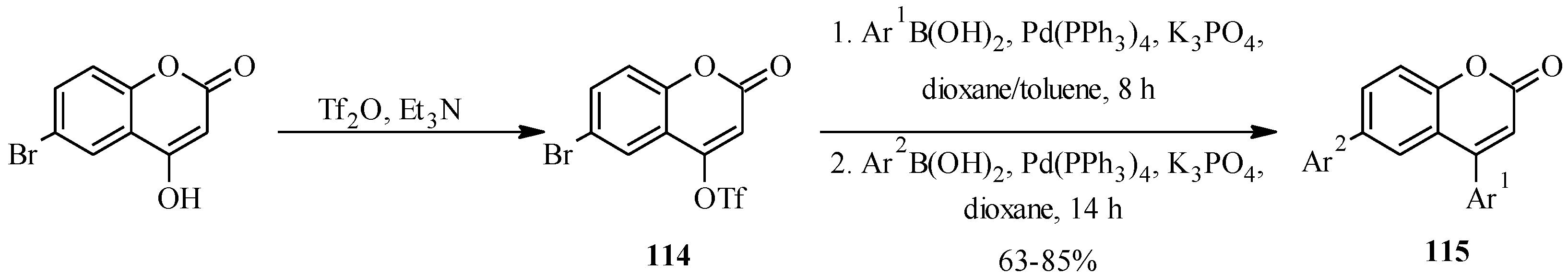
| 115 | Ar1 | Ar2 |
|---|---|---|
| a | 4-MeOC6H4 | 4-ClC6H4 |
| b | 4-MeOC6H4 | 3-ClC6H4 |
| c | 3,5-Me2C6H3 | 4-MeOC6H4 |
| d | 4-ClC6H4 | 4-MeOC6H4 |
| e | 4-ClC6H4 | 3-FC6H4 |
6. Conclusions
Acknowledgments
References
- Ito, F.; Iwasaki, M.; Watanabe, T.; Ishikawa, T.; Higuchi, Y. The first total synthesis of kwakhurin, a characteristic component of a rejuvenating plant, “kwao keur”: toward an efficient synthetic route to phytoestrogenic isoflavones. Org. Biomol. Chem. 2005, 3, 674–681. [Google Scholar] [CrossRef]
- Selepe, M.A.; Drewes, S.E.; van Heerden, F.R. Total synthesis of the pyranoisoflavone kraussianone 1 and related isoflavones. J. Nat. Prod. 2010, 73, 1680–1685. [Google Scholar] [CrossRef]
- Zheng, S.Y.; Shen, Z.W. Total synthesis of hirtellanine A. Tetrahedron Lett. 2010, 51, 2883–2887. [Google Scholar] [CrossRef]
- Wei, G.; Yu, B. Isoflavone glycosides: Synthesis and evaluation as α-glucosidase inhibitors. Eur. J. Org. Chem. 2008, 3156–3163. [Google Scholar]
- Matin, A.; Gavande, N.; Kim, M.S.; Yang, N.X.; Salam, N.K.; Hanrahan, J.R.; Roubin, R.H.; Hibbs, D.E. 7-Hydroxy-benzopyran-4-one derivatives: A novel pharmacophore of peroxisome proliferator-activated receptor α and -γ (PPARα and γ) dual agonists. J. Med. Chem. 2009, 52, 6835–6850. [Google Scholar]
- Mihigo, S.O.; Mammo, W.; Bezabih, M.; Andrae-Marobela, K.; Abegaz, B.M. Total synthesis, antiprotozoal and cytotoxicity activities of rhuschalcone VI and analogs. Bioorg. Med. Chem. 2010, 18, 2464–2473. [Google Scholar] [CrossRef]
- Pierson, J.-T.; Dumetre, A.; Hutter, S.; Delmas, F.; Laget, M.; Finet, J.-P.; Azas, N.; Combes, S. Synthesis and antiprotozoal activity of 4-arylcoumarins. Eur. J. Med. Chem. 2010, 45, 864–869. [Google Scholar] [CrossRef]
- Combes, S.; Barbier, P.; Douillard, S.; McLeer-Florin, A.; Bourgarel-Rey, V.; Pierson, J.-T.; Fedorov, A.Y.; Finet, J.-P.; Boutonnat, J.; Peyrot, V. Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. Part 2. J. Med. Chem. 2011, 54, 3153–3162. [Google Scholar] [CrossRef]
- Ganina, O.G.; Daras, E.; Bourgarel-Rey, V.; Peyrot, V.; Andresyuk, A.N.; Finet, J.-P.; Fedorov, A.Y.; Beletskaya, I.P.; Combes, S. Synthesis and biological evaluation of polymethoxylated 4-heteroarylcoumarins as tubulin assembly inhibitor. Bioorg. Med. Chem. 2008, 16, 8806–8812. [Google Scholar]
- Bailly, C.; Bal, C.; Barbier, P.; Combes, S.; Finet, J.P.; Hildebrand, M.P.; Peyrot, V.; Wattez, N. Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. J. Med. Chem. 2003, 46, 5437–5444. [Google Scholar] [CrossRef]
- Felpin, F.-X. Practical and efficient Suzuki-Miyaura cross-coupling of 2-iodocycloenones with arylboronic acids catalyzed by recyclable Pd(0)/C. J. Org. Chem. 2005, 70, 8575–8578. [Google Scholar] [CrossRef]
- Felpin, F.-X.; Lory, C.; Sow, H.; Acherar, S. Practical and efficient entry to isoflavones by Pd(0)/C-mediated Suzuki-Miyaura reaction. Total synthesis of geranylated isoflavones. Tetrahedron 2007, 63, 3010–3016. [Google Scholar] [CrossRef]
- Claisen, L.; Claparède, A. Condensationen von Ketonen mit Aldehyden. Chem. Ber. 1881, 14, 2460–2468. [Google Scholar] [CrossRef]
- Satyanarayana, M.; Tiwari, P.; Tripathi, B.K.; Srivastava, A.K.; Pratap, R. Synthesis and antihyperglycemic activity of chalcone based aryloxypropanolamines. Bioorg. Med. Chem. 2004, 12, 883–889. [Google Scholar] [CrossRef]
- Daskiewicz, J.B.; Comte, G.; Barron, D.; Di Pietro, A.; Thomasson, F. Organolithium mediated synthesis of prenylchalcones as potential inhibitors of chemoresistance. Tetrahedron Lett. 1999, 40, 7095–7098. [Google Scholar] [CrossRef]
- Wang, Z. Claisen-Schmidt Condensation. Comprehensive Organic Name Reactions and Reagents. Available online: http://dx.doi.org/10.1002/9780470638859.conrr145 (accessed on 15 September 2010).
- Daskiewicz, J.-B.; Barron, D. Substituent effects in the BF3-mediated acylation of phenols with cinnamic acids. Nat. Prod. Res. 2003, 17, 347–350. [Google Scholar] [CrossRef]
- Bianco, A.; Cavarischia, C.; Farina, A.; Guiso, M.; Marra, C. A new synthesis of flavonoids via Heck reaction. Tetrahedron Lett. 2003, 44, 9107–9109. [Google Scholar] [CrossRef]
- Liao, W.-W.; Mueller, T.J.J. Sequential coupling-isomerization-coupling reactions - A novel three-component synthesis of aryl chalcones. Synlett 2006, 3469–3473. [Google Scholar]
- Braun, R.U.; Ansorge, M.; Mueller, T.J.J. Coupling-isomerization synthesis of chalcones. Chem. Eur. J. 2006, 12, 9081–9094. [Google Scholar] [CrossRef]
- Eddarir, S.; Cotelle, N.; Bakkour, Y.; Rolando, C. An efficient synthesis of chalcones based on the Suzuki reaction. Tetrahedron Lett. 2003, 44, 5359–5363. [Google Scholar] [CrossRef]
- Haddach, M.; McCarthy, J.R. A new method for the synthesis of ketones: The palladium-catalyzed cross-coupling of acid chlorides with arylboronic acids. Tetrahedron Lett. 1999, 40, 3109–3112. [Google Scholar] [CrossRef]
- Al-Masum, M.; Ng, E.; Wai, M.C. Palladium-catalyzed direct cross-coupling of potassium styryltrifluoroborates and benzoyl chlorides - a one step method for chalcone synthesis. Tetrahedron Lett. 2011, 52, 1008–1010. [Google Scholar] [CrossRef]
- Xin, B.-W. Synthesis of aryl ketones by cross-coupling reaction of arylboronic acids with carboxylic anhydrides in aqueous phase. Synth. Commun. 2008, 38, 2826–2837. [Google Scholar] [CrossRef]
- Zuo, Y.; Yu, Y.; Wang, S.; Shao, W.; Zhou, B.; Lin, L.; Luo, Z.; Huang, R.; Du, J.; Bu, X. Synthesis and cytotoxicity evaluation of biaryl-based chalcones and their potential in TNF alpha-induced nuclear factor-kappa B activation inhibition. Eur. J. Med. Chem. 2012, 50, 393–404. [Google Scholar] [CrossRef]
- Vieira, L.C.C.; Paixão, M.W.; Corrêa, A.G. Green synthesis of novel chalcone and coumarin derivatives via Suzuki coupling reaction. Tetrahedron Lett. 2012, 53, 2715–2718. [Google Scholar]
- Baker, W. 322. Molecular rearrangement of some o-acyloxyacetophenones and the mechanism of the production of 3-acylchromones. J. Chem. Soc. 1933, 1381–1389. [Google Scholar] [CrossRef]
- Mahal, H.S.; Venkataraman, K. 387. Synthetical experiments in the chromone group. Part XIV. The action of sodamide on 1-acyloxy-2-acetonaphthones. J. Chem. Soc. 1934, 1767–1769. [Google Scholar] [CrossRef]
- Sharma, D.; Kumar, S.; Makrandi, J.K. Solid phase Baker–Venkataraman rearrangement under solvent-free condition using grinding technique. Green Chem. Lett. Rev. 2009, 2, 53–55. [Google Scholar] [CrossRef]
- Li, J.J. Allan-Robinson reaction. In Name Reactions Name Reactions—A Collection of Detailed Reaction Mechanisms; Springer: Berlin/Heidelberg, Germany, 2006; pp. 8–9. [Google Scholar]
- Bennett, M.; Burke, A.J.; O’Sullivan, W.I. Aspects of the Algar-Flynn-Oyamada (AFO) reaction. Tetrahedron 1996, 52, 7163–7178. [Google Scholar] [CrossRef]
- Bose, G.; Mondal, E.; Khan, A.T.; Bordoloi, M.J. An environmentally benign synthesis of aurones and flavones from 2'-acetoxychalcones using n-tetrabutylammonium tribromide. Tetrahedron Lett. 2001, 42, 8907–8909. [Google Scholar] [CrossRef]
- Kraus, G.A.; Gupta, V. Divergent approach to flavones and aurones via dihaloacrylic acids. Unexpected dependence on the halogen atom. Org. Lett. 2010, 12, 5278–5280. [Google Scholar] [CrossRef]
- Dahlen, K.; Wallen, E.A.A.; Grotli, M.; Luthman, K. Synthesis of 2,3,6,8-tetrasubstituted chromone scaffolds. J. Org. Chem. 2006, 71, 6863–6871. [Google Scholar] [CrossRef]
- Arai, M.A.; Sato, M.; Sawada, K.; Hosoya, T.; Ishibashi, M. Efficient synthesis of chromone and flavonoid derivatives with diverse heterocyclic units. Chem.-Asian J. 2008, 3, 2056–2064. [Google Scholar] [CrossRef]
- Larsen, L.; Yoon, D.H.; Weavers, R.T. Synthesis of a range of polyhydroxy 8-aryl flavones. Synth. Commun. 2009, 39, 2935–2948. [Google Scholar] [CrossRef]
- Deodhar, M.; Black, D.S.; Chan, D.S.-H.; Kumar, N. Synthesis of some new biheterocycles by a one-pot Suzuki-Miyaura coupling reaction. Heterocycles 2010, 80, 1267–1274. [Google Scholar] [CrossRef]
- Malik, I.; Hussain, M.; Hung, N.T.; Villinger, A.; Langer, P. Synthesis of 7,8-diarylflavones by site-selective Suzuki-Miyaura reactions. Synlett 2010, 2244–2246. [Google Scholar]
- Che, H.; Lim, H.; Kim, H.P.; Park, H. A chrysin analog exhibited strong inhibitory activities against both PGE(2) and NO production. Eur. J. Med. Chem. 2011, 46, 4657–4660. [Google Scholar] [CrossRef]
- Joshi, V.; Hatim, J.G. Facile synthesis of new substituted 3-aryl/heteroarylflavones by Suzuki-Miyaura coupling of 3-bromoflavone with substituted aryl/heteroarylboronic acids. Indian J. Heterocycl. Chem. 2011, 21, 111–116. [Google Scholar]
- Eleya, N.; Malik, I.; Reimann, S.; Wittler, K.; Hein, M.; Patonay, T.; Villinger, A.; Ludwig, R.; Langer, P. Efficient synthesis of arylated flavones by site-selective Suzuki–Miyaura cross-coupling ceactions of the cis(triflate) of 5,7- and 7,8-dihydroxyflavone. Eur. J. Org. Chem. 2012, 2012, 1639–1652. [Google Scholar]
- Nakazawa, K.; Ito, M. Synthesis of ginkgetin. Tetrahedron Lett. 1962, 317–319. [Google Scholar] [CrossRef]
- Chen, J.J.; Chang, H.W.; Kim, H.P.; Park, H. Synthesis of phospholipase A2 inhibitory biflavonoids. Bioorg. Med. Chem. Lett. 2006, 16, 2373–2375. [Google Scholar] [CrossRef]
- Muller, D.; Fleury, J.P. A new strategy for the synthesis of biflavonoids via arylboronic acids. Tetrahedron Lett. 1991, 32, 2229–2232. [Google Scholar] [CrossRef]
- Zembower, D.E.; Zhang, H.P. Total synthesis of robustaflavone, a potential anti-hepatitis B agent. J. Org. Chem. 1998, 63, 9300–9305. [Google Scholar] [CrossRef]
- Zheng, X.; Meng, W.D.; Qing, F.L. Synthesis of gem-difluoromethylenated biflavonoid via the Suzuki coupling reaction. Tetrahedron Lett. 2004, 45, 8083–8085. [Google Scholar] [CrossRef]
- Ollis, W.D. The Isoflavonoids. In The Chemistry of Flavonoids; Geissman, T.A., Ed.; Pergamon Press Inc.: Oxford, UK, 1962; pp. 353–440. [Google Scholar]
- Baker, W.; Chadderton, J.; Harborne, B.J.; Ollis, D.W. A new synthesis of isoflavones. J. Chem. Soc. 1953, 1852–1854. [Google Scholar]
- Dewick, P.M. Isoflavonoids. In The Flavonoids: Advances in Research since 1986; Harborne, J.B., Ed.; Chapman and Hall: London, UK, 1994; pp. 117–238. [Google Scholar]
- Dixon, R.A.; Ferreira, D. Genistein. Phytochemistry 2002, 60, 205–211. [Google Scholar] [CrossRef]
- Granados-Covarrubias, E.H.; Maldonado, L.A. A Wacker-Cook synthesis of isoflavones: formononetine. Tetrahedron Lett. 2009, 50, 1542–1545. [Google Scholar] [CrossRef]
- Li, Q.-L.; Liu, Q.-L.; Ge, Z.-Y.; Zhu, Y.-M. A novel synthesis of isoflavones via copper(I)-catalyzed intramolecular cyclization reaction. Helv. Chim. Acta 2011, 94, 1304–1309. [Google Scholar] [CrossRef]
- Hoshino, Y.; Miyaura, N.; Suzuki, A. Novel synthesis of isoflavones by the palladium-catalyzed cross-coupling reaction of 3-bromochromones with arylboronic acids or their esters. Bull. Chem. Soc. Jpn. 1988, 61, 3008–3010. [Google Scholar] [CrossRef]
- Dawood, K.M. Microwave-assisted Suzuki-Miyaura and Heck-Mizoroki cross-coupling reactions of aryl chlorides and bromides in water using stable benzothiazole-based palladium(II) precatalysts. Tetrahedron 2007, 63, 9642–9651. [Google Scholar] [CrossRef]
- Eisnor, C.R.; Gossage, R.A.; Yadav, P.N. Oxazoline Chemistry. Part 11: Syntheses of natural and synthetic isoflavones, stilbenes and related species via C-C bond formation promoted by a Pd-oxazoline complex. Tetrahedron 2006, 62, 3395–3401. [Google Scholar] [CrossRef]
- Ding, K.; Wang, S.M. Efficient synthesis of isoflavone analogues via a Suzuki coupling reaction. Tetrahedron Lett. 2005, 46, 3707–3709. [Google Scholar] [CrossRef]
- Biegasiewicz, K.F.; St Denis, J.D.; Carroll, V.M.; Priefer, R. An efficient synthesis of daidzein, dimethyldaidzein, and isoformononetin. Tetrahedron Lett. 2010, 51, 4408–4410. [Google Scholar] [CrossRef]
- Gammill, B.R. A new and efficient synthesis of 3-halogenated 4H-1-benzopyran-4-ones. Synthesis 1979, 901–903. [Google Scholar] [CrossRef]
- Tsoi, Y.-T.; Zhou, Z.; Chan, A.S.C.; Yu, W.-Y. Palladium-catalyzed oxidative cross-coupling reaction of arylboronic acids with diazoesters for stereoselective synthesis of (E)-α,β-diarylacrylates. Org. Lett. 2010, 12, 4506–4509. [Google Scholar] [CrossRef]
- Ikedo, A.; Hayakawa, I.; Usui, T.; Kazami, S.; Osada, H.; Kigoshi, H. Structure-activity relationship study of glaziovianin A against cell cycle progression and spindle formation of HeLa S3 cells. Bioorg. Med. Chem. Lett. 2010, 20, 5402–5404. [Google Scholar] [CrossRef] [Green Version]
- Vasselin, D.A.; Westwell, A.D.; Matthews, C.S.; Bradshaw, T.D.; Stevens, M.F.G. Structural studies on bioactive compounds. 40. Synthesis and biological properties of fluoro-, methoxyl-, and amino-substituted 3-phenyl-4H-1-benzopyran-4-ones and a comparison of their antitumor activities with the activities of related 2-phenylbenzothiazoles. J. Med. Chem. 2006, 49, 3973–3981. [Google Scholar] [CrossRef]
- Hayakawa, I.; Ikedo, A.; Kigoshi, H. Synthesis of glaziovianin A: A potent antitumor isoflavone. Chem. Lett. 2007, 36, 1382–1383. [Google Scholar] [CrossRef]
- Cornwell, T.; Cohick, W.; Raskin, I. Dietary phytoestrogens and health. Phytochemistry 2004, 65, 995–1016. [Google Scholar] [CrossRef]
- St Denis, J.D.; Gordon, J.S.; Carroll, V.M.; Priefer, R. Novel synthesis of the isoflavone genistein. Synthesis 2010, 1590–1592. [Google Scholar]
- Andersen, Ø.M.; Markham, K.R. Flavonoids; Taylor and Francis: New York, NY, USA, 2006. [Google Scholar]
- Rice-Evans, C.A.; Packer, L. Flavonoids in Health and Disease, 2nd ed.; Marcel Dekker: New York, NY, USA, 2003. [Google Scholar]
- Lin, J.-K.; Weng, M.-S. Flavonoids as Nutraceuticals. In The Science of Flavonoids; Grotewold, E., Ed.; Springer Science and Business media: New York, NY, USA, 2006. [Google Scholar]
- Rao, K.S.R.M.; Iyer, C.S.R.; Iyer, P.R. Synthesis of alpinum isoflavone, derrone and related pyranoisoflavones. Tetrahedron 1987, 43, 3015–3019. [Google Scholar] [CrossRef]
- Jain, A.C.; Jain, S.M. Synthesis of derrubone and robustone. Tetrahedron 1972, 28, 5063–5067. [Google Scholar] [CrossRef]
- Hastings, J.M.; Hadden, M.K.; Blagg, B.S.J. Synthesis and evaluation of derrubone and select analogues. J. Org. Chem. 2007, 73, 369–373. [Google Scholar] [CrossRef]
- Botta, B.; Menendez, P.; Zappia, G.; de Lima, R.A.; Torge, R.; Delle Monache, G. Prenylated isoflavonoids: Botanical distribution, Structures, Biological activities and biotechnological studies. An update (1995–2006). Curr. Med. Chem. 2009, 16, 3414–3468. [Google Scholar] [CrossRef]
- Selepe, M.A.; Drewes, S.E.; van Heerden, F.R. Total synthesis of the pyranocoumaronochromone lupinalbin H. Tetrahedron 2011, 67, 8654–8658. [Google Scholar] [CrossRef]
- Veitch, N.C. Isoflavonoids of the Leguminosae. Nat. Prod. Rep. 2007, 24, 417–464. [Google Scholar] [CrossRef]
- Veitch, N.C. Isoflavonoids of the Leguminosae. Nat. Prod. Rep. 2009, 26, 776–802. [Google Scholar] [CrossRef]
- Shou, Q.Y.; Tan, Q.; Shen, Z.W. Hirtellanines A and B, a pair of isomeric isoflavonoid derivatives from Campylotropis hirtella and their immunosuppressive activities. Bioorg. Med. Chem. Lett. 2009, 19, 3389–3391. [Google Scholar] [CrossRef]
- Harfenist, M.; Thom, E. Influence of structure on the rate of thermal rearrangement of aryl propargyl ethers to the chromenes. Gem-dimethyl effect. J. Org. Chem. 1972, 37, 841–848. [Google Scholar] [CrossRef]
- Garazd, M.M.; Garazd, Y.L.; Khilya, V.P. Neoflavones. 2. Methods for synthesizing and modifying 4-arylcoumarins. Chem. Nat. Comp. 2005, 41, 245–271. [Google Scholar] [CrossRef]
- Phapale, V.B.; Cardenas, D.J. Nickel-catalysed Negishi cross-coupling reactions: scope and mechanisms. Chem. Soc. Rev. 2009, 38, 1598–1607. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, T.; Fan, R.; Wu, H. General and efficient route for the synthesis of 3,4-disubstituted coumarins via Pd-catalyzed site-selective cross-coupling reactions. J. Org. Chem. 2007, 72, 7279–7286. [Google Scholar] [CrossRef]
- Akrawi, O.A.; Nagy, G.Z.; Patonay, T.; Villinger, A.; Langer, P. Chemoselective Suzuki-Miyaura reactions of 4-trifluoromethylsulfonyloxy-6-bromocoumarin. Tetrahedron Lett. 2012, 53, 3206–3209. [Google Scholar] [CrossRef]
- Khoobi, M.; Alipour, M.; Zarei, S.; Jafarpour, F.; Shafiee, A. A facile route to flavone and neoflavone backbones via a regioselective palladium catalyzed oxidative Heck reaction. J. Chem. Soc., Chem. Commun. 2012, 48, 2985–2987. [Google Scholar] [CrossRef]
- Li, Y.; Qi, Z.; Wang, H.; Fu, X.; Duan, C. Palladium-catalyzed oxidative Heck coupling reaction for direct synthesis of 4-arylcoumarins using coumarins and arylboronic acids. J. Org. Chem. 2012, 77, 2053–2057. [Google Scholar] [CrossRef]
- Boland, G.M.; Donnelly, D.M.X.; Finet, J.P.; Rea, M.D. Synthesis of neoflavones by Suzuki arylation of 4-substituted coumarins. J. Chem. Soc. Perkin Trans. 1996, 1, 2591–2597. [Google Scholar]
- Donnelly, D.M.X.; Finet, J.P.; Guiry, P.J.; Rea, M.D. Synthesis of C-ring hydroxylated neoflavonoids by ligand coupling reactions. Synth. Commun. 1999, 29, 2719–2730. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Selepe, M.A.; Van Heerden, F.R. Application of the Suzuki-Miyaura Reaction in the Synthesis of Flavonoids. Molecules 2013, 18, 4739-4765. https://doi.org/10.3390/molecules18044739
Selepe MA, Van Heerden FR. Application of the Suzuki-Miyaura Reaction in the Synthesis of Flavonoids. Molecules. 2013; 18(4):4739-4765. https://doi.org/10.3390/molecules18044739
Chicago/Turabian StyleSelepe, Mamoalosi A., and Fanie R. Van Heerden. 2013. "Application of the Suzuki-Miyaura Reaction in the Synthesis of Flavonoids" Molecules 18, no. 4: 4739-4765. https://doi.org/10.3390/molecules18044739
APA StyleSelepe, M. A., & Van Heerden, F. R. (2013). Application of the Suzuki-Miyaura Reaction in the Synthesis of Flavonoids. Molecules, 18(4), 4739-4765. https://doi.org/10.3390/molecules18044739