Synthesis and Biological Evaluation of Unsymmetrical Curcumin Analogues as Tyrosinase Inhibitors

Abstract
:1. Introduction
2. Results and Discussion
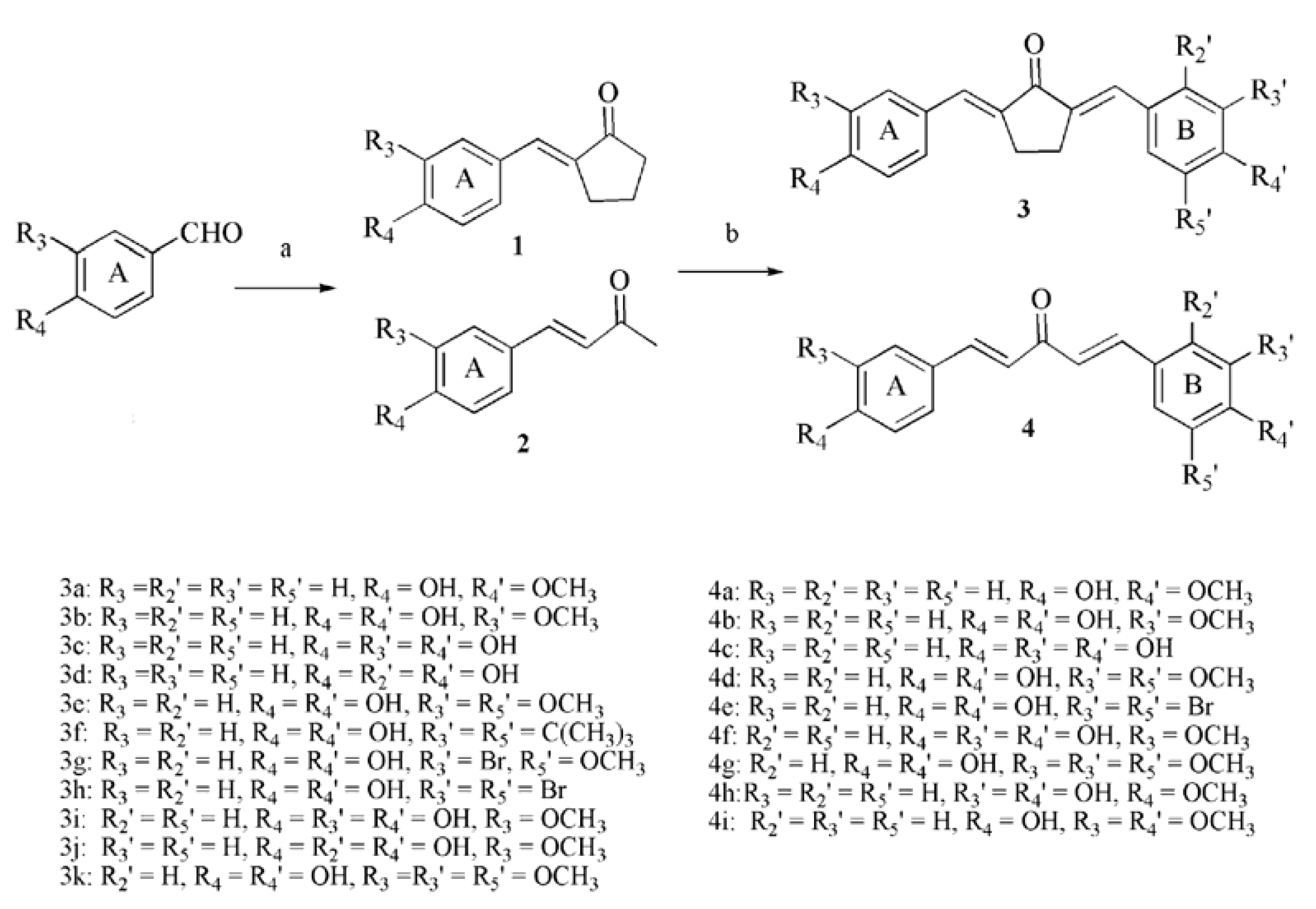
2.1. Chemistry
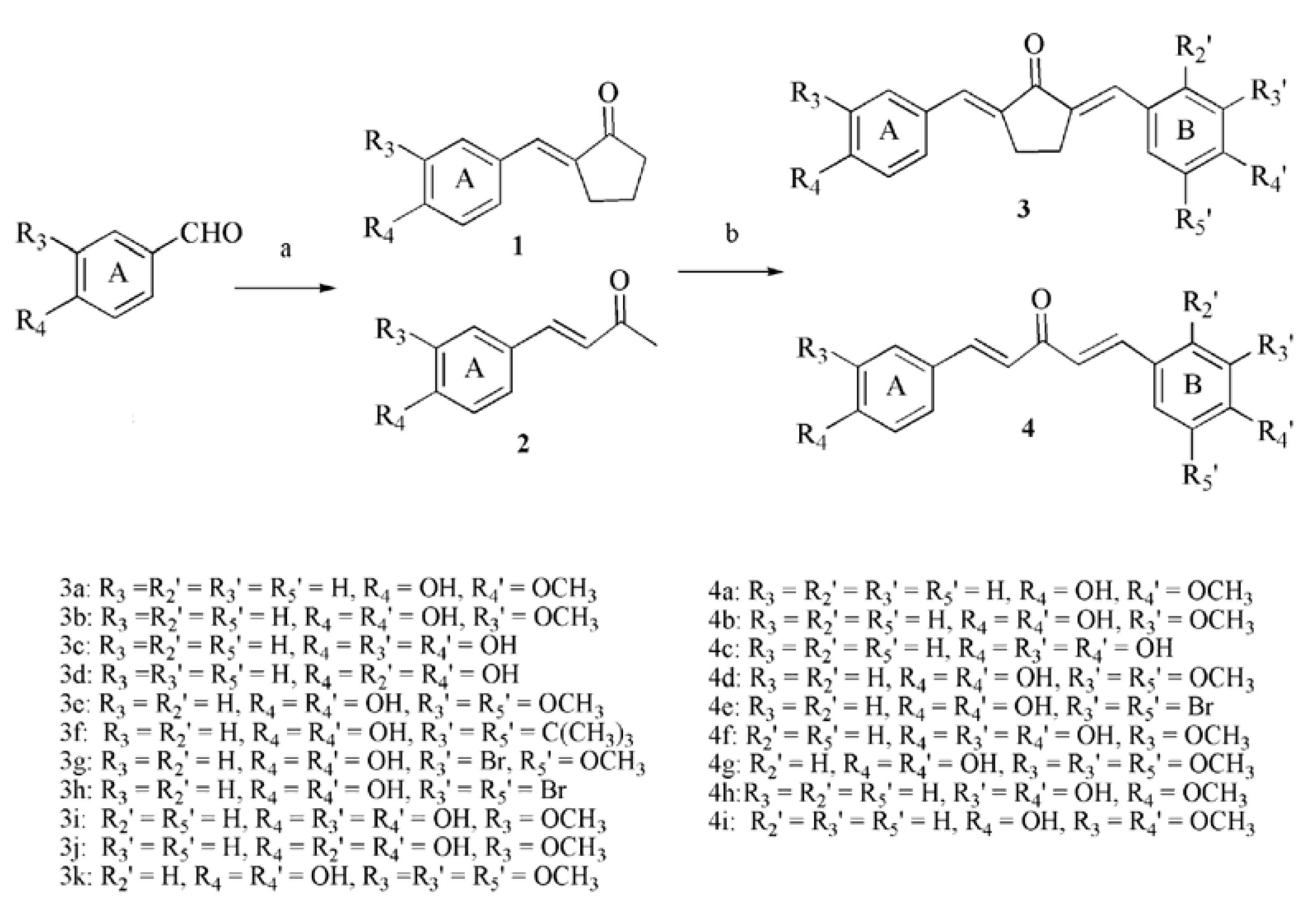
2.2. Inhibitory Effects on the Diphenolase Activity of Mushroom Tyrosinase
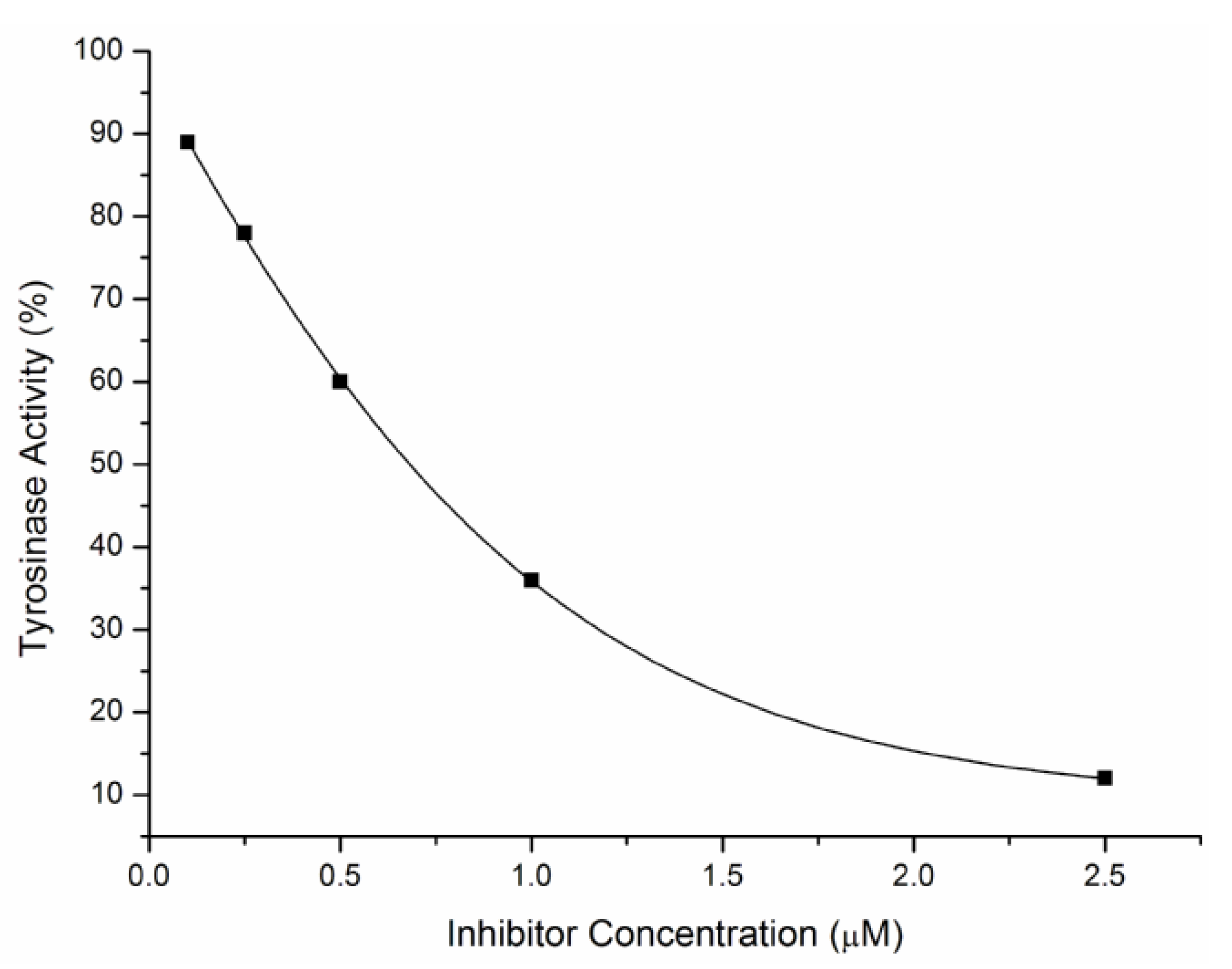

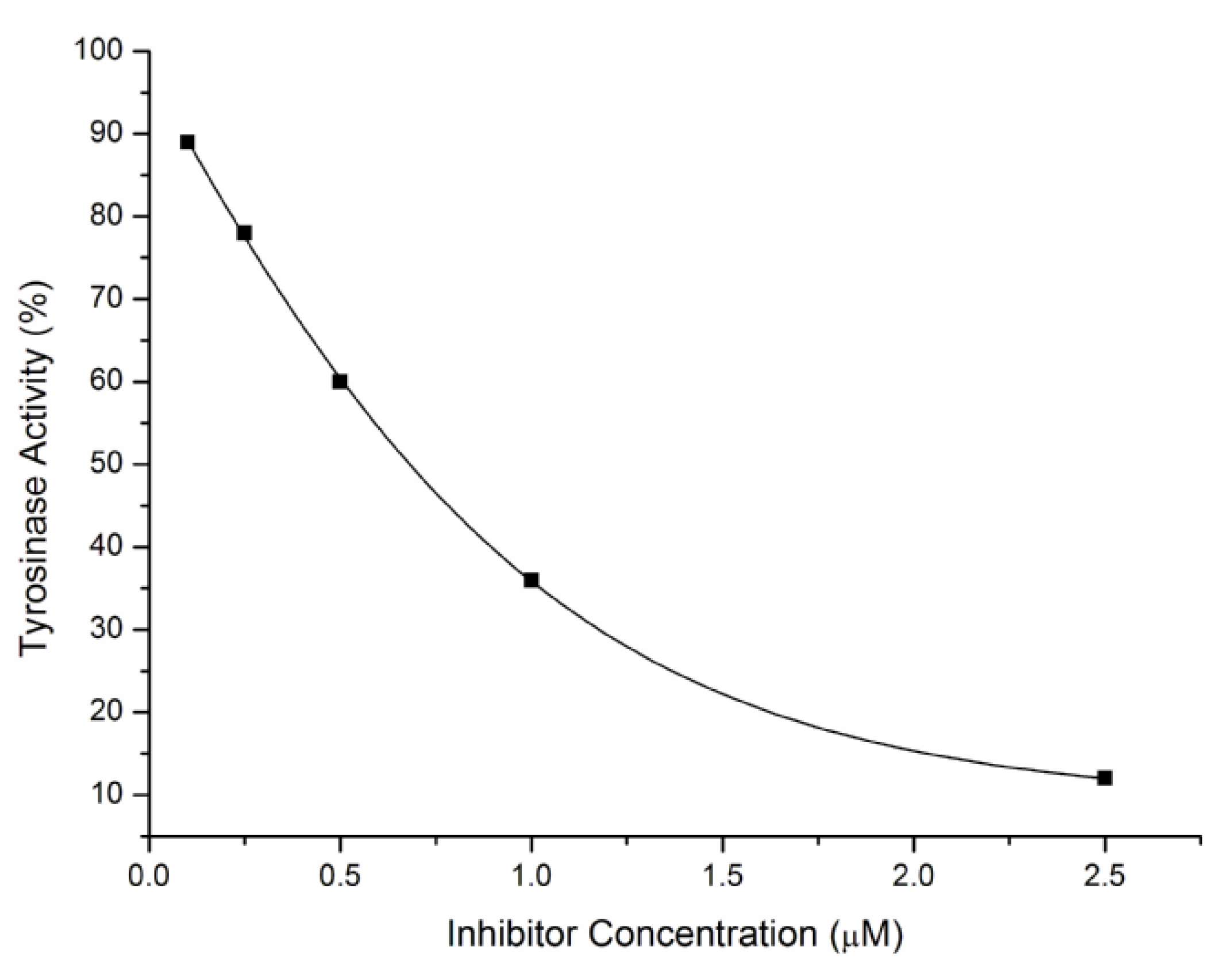{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | Compound | IC50 (μM) |
|---|---|---|---|
| 3a | 189.42 | 4a | 182.86 |
| 3b | 56.64 | 4b | 46.24 |
| 3c | 7.78 | 4c | 4.64 |
| 3d | 1.74 | 4d | 7.20 |
| 3e | 9.66 | 4e | >200 |
| 3f | 168.36 | 4f | 16.34 |
| 3g | 173.48 | 4g | >200 |
| 3h | >200 | 4h | 86.92 |
| 3i | 16.74 | 4i | >200 |
| 3j | 2.78 | 4-Butylresorcinol | 11.27 |
| 3k | >200 | Kojic acid | 28.59 |
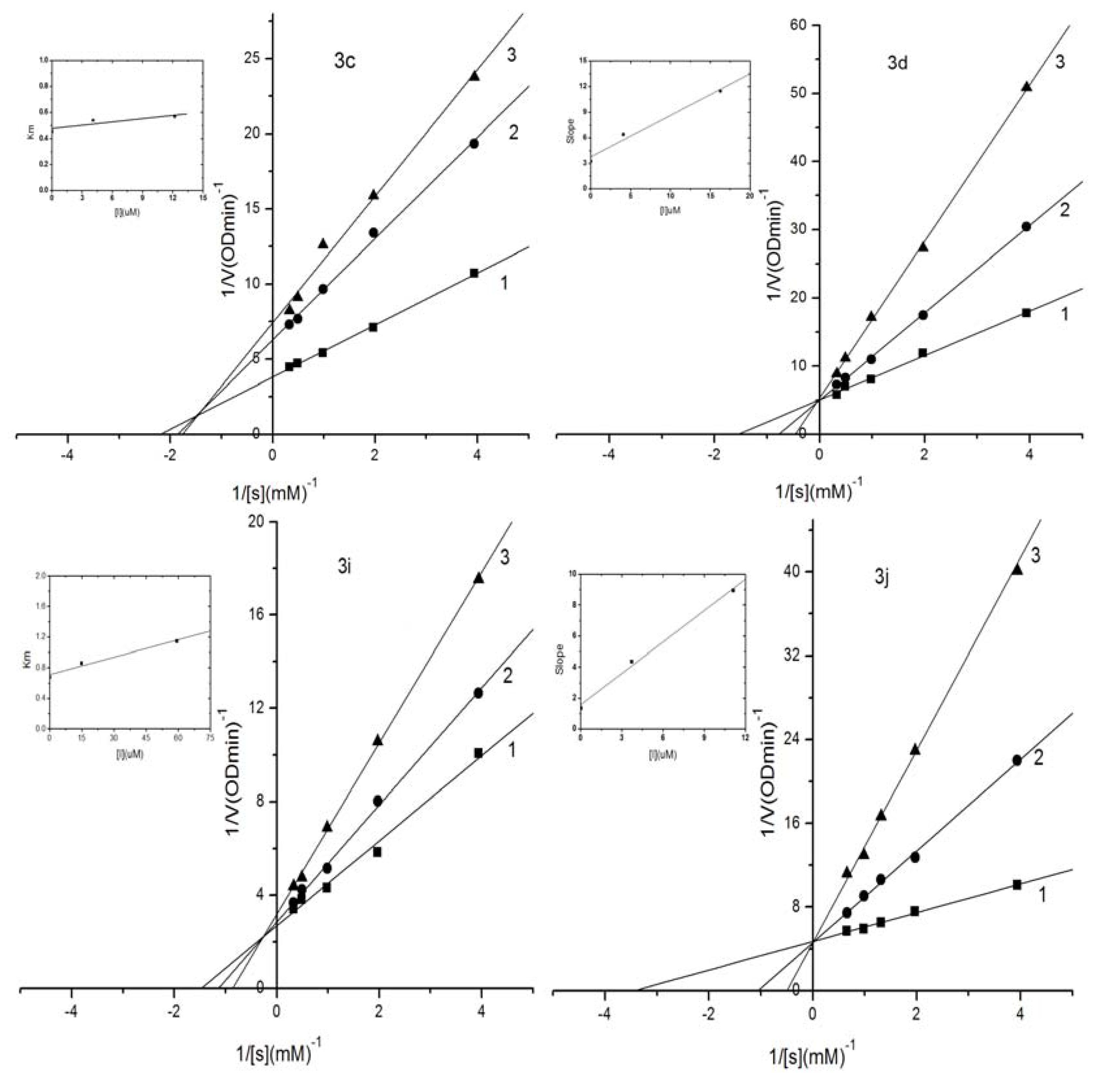
2.3. Kinetic Analysis of Selected Compounds on Mushroom Tyrosinase
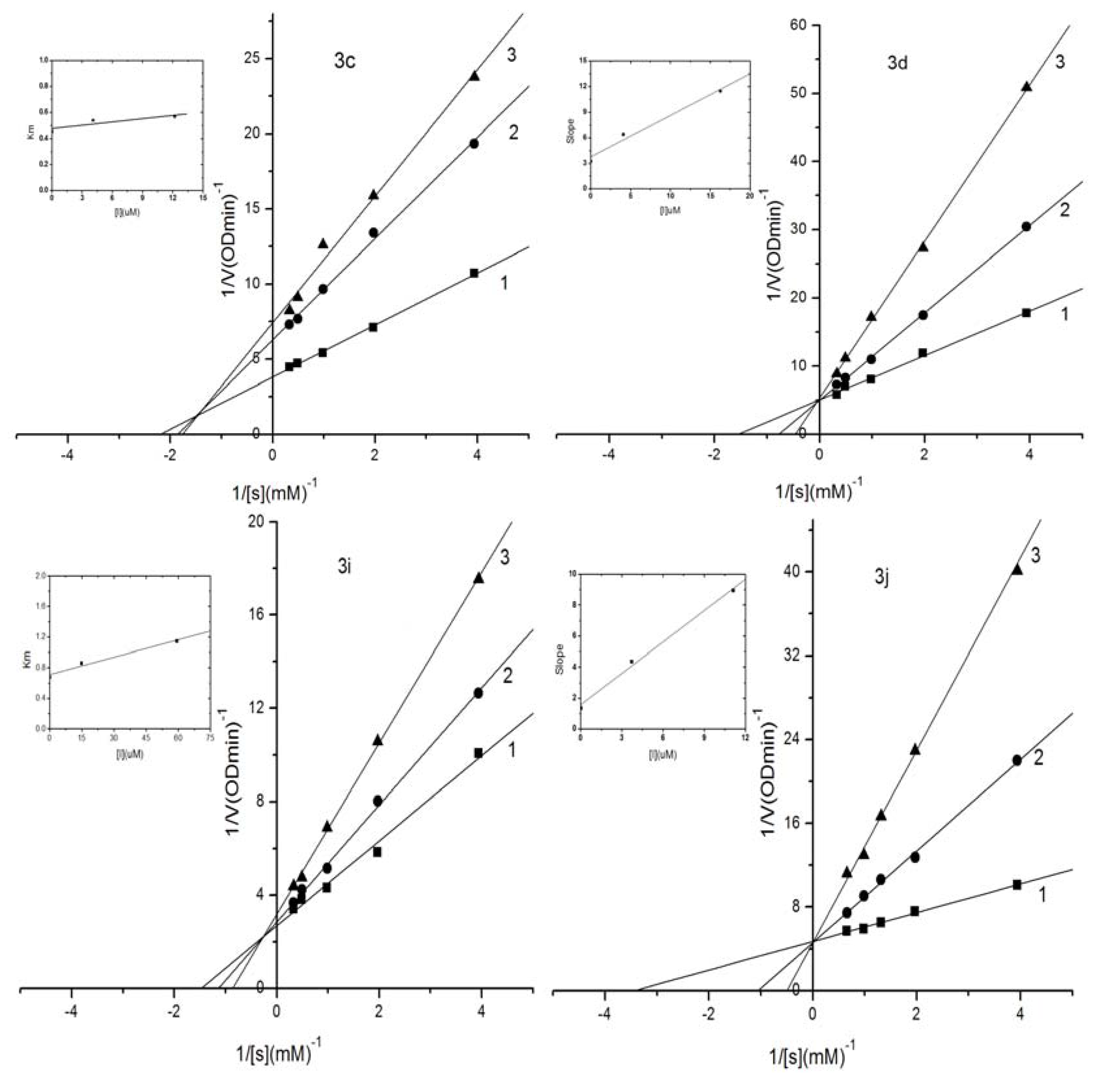
2.4. Evaluation of Acute Toxicity in Mice
| Item | Control | Treated by 3d | Treated by 3j | |||
|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Male | Female | |
| Food Intake (g/day) | 5.1 ± 0.3 | 3.9 ± 0.4 | 5.1 ± 0.5 | 4.0 ± 0.4 | 5.2 ± 0.4 | 3.9 ± 0.5 |
| Drinking Water (mL/day) | 5.5 ± 0.7 | 4.2 ± 0.4 | 5.6 ± 0.6 | 4.2 ± 0.5 | 5.4 ± 0.5 | 4.1 ± 0.4 |
| Body Weight (g) | 31.3 ± 2.5 | 27.1 ± 2.3 | 30.9 ± 2.6 | 27.3 ± 2.9 | 30.1 ± 2.8 | 26.9 ± 2.1 |
| General Appearance | √ | √ | √ | √ | √ | √ |
| Skin and Fur | √ | √ | √ | √ | √ | √ |
| Eyes, Nose | √ | √ | √ | √ | √ | √ |
| Respiration | √ | √ | √ | √ | √ | √ |
| Urine | ∆ | ∆ | ∆ | ∆ | ∆ | ∆ |
| Feces | ∆ | ∆ | ∆ | ∆ | ∆ | ∆ |
| Locomotor | √ | √ | √ | √ | √ | √ |
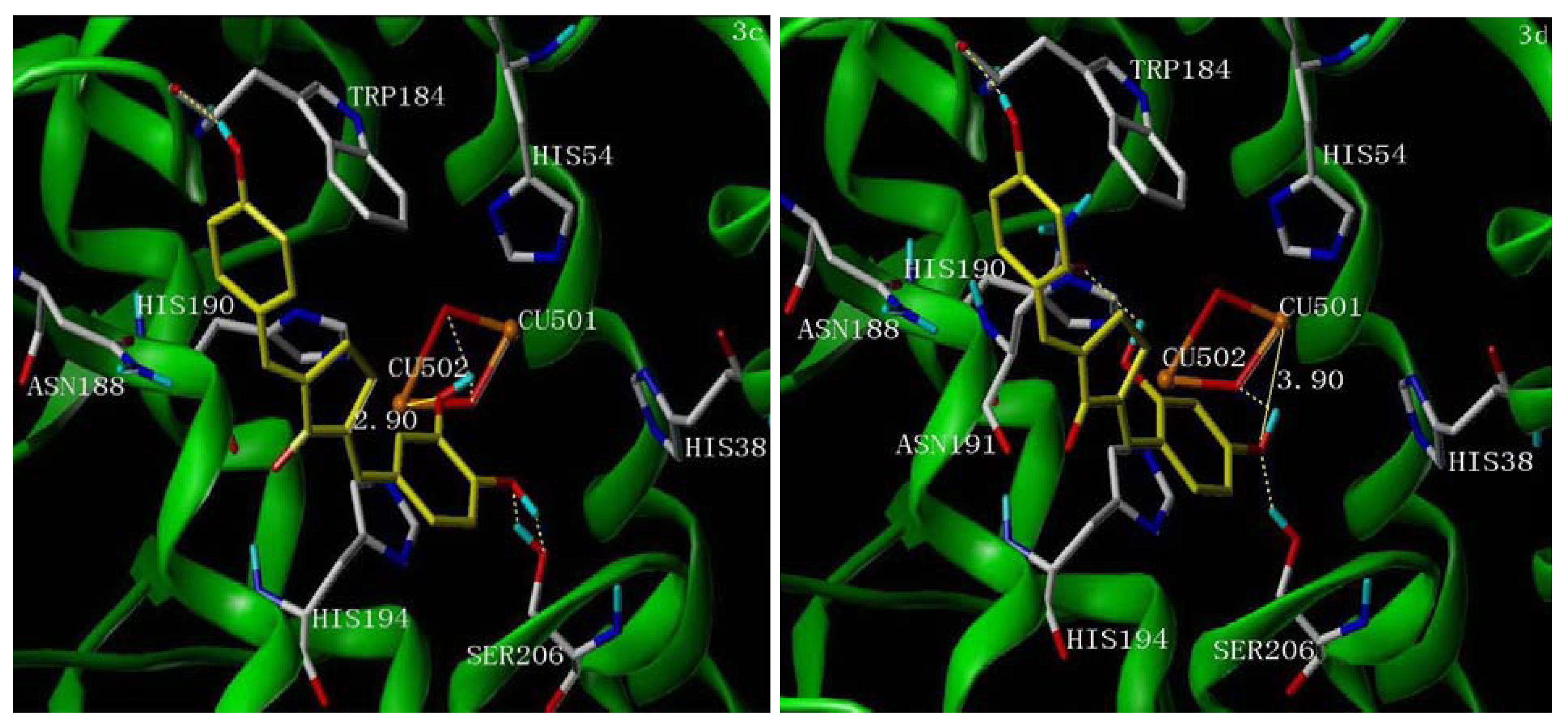
2.5. Molecular Docking Study
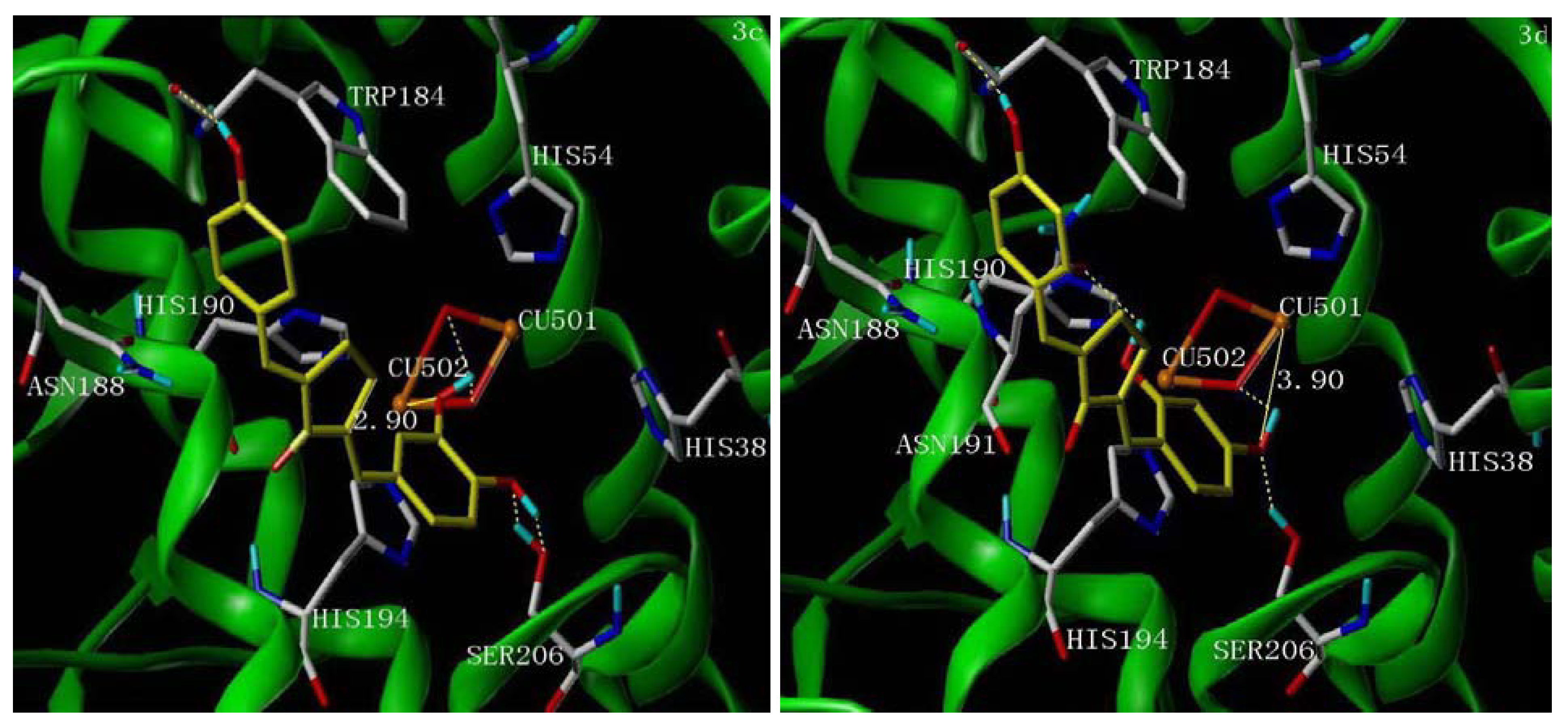
3. Experimental
3.1. Reagents and General Procedures
3.2. Synthesis
3.3. Tyrosinase Assay
3.4. Inhibition Kinetics
3.5. Acute Toxicity Assays in Mice
3.6. Molecular Modeling
4. Conclusions
Acknowledgments
References
- Fistarol, S.K.; Itin, P.H. Disorders of pigmentation. J. Dtsch. Dermatol. Ges. 2010, 8, 187–201. [Google Scholar] [CrossRef]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef]
- Khan, M.T. Novel tyrosinase inhibitors from natural resources—their computational studies. Curr. Med. Chem. 2012, 19, 2262–2672. [Google Scholar]
- Ye, Y.; Chou, G.X.; Mu, D.D.; Wang, H.; Chu, J.H.; Leung, A.K.M.; Fong, W.F.; Yu, Z.L. Screening of Chinese herbal medicines for antityrosinase activity in a cell free system and B16 cells. J. Ethnopharmacol. 2010, 129, 387–390. [Google Scholar] [CrossRef]
- Davis, E.C.; Callender, V.D. Postinflammatory hyperpigmentation: A review of the epidemiology, clinical features, and treatment options in skin of color. J. Clin. Aesthet. Dermatol. 2010, 3, 20–31. [Google Scholar]
- Gillbro, J.M.; Olsson, M.J. The melanogenesis and mechanisms of skin-lightening agents—existing and new approaches. Int. J. Cosmet. Sci. 2011, 33, 210–221. [Google Scholar] [CrossRef]
- Smit, N.; Vicanova, J.; Pavel, S. The hunt for natural skin whitening agents. Int. J. Mol. Sci. 2009, 10, 5326–5349. [Google Scholar] [CrossRef]
- Parvez, S.; Kang, M.; Chung, H.S.; Bae, H. Naturally occurring tyrosinase inhibitors: Mechanism and applications in skin health, cosmetics and agriculture industries. Phytother. Res. 2007, 21, 805–816. [Google Scholar] [CrossRef]
- Kim, Y.J.; Uyama, H. Tyrosinase inhibitors from natural and synthetic sources: Structure, inhibition mechanism and perspective for the future. Cell Mol. Life Sci. 2005, 62, 1707–1723. [Google Scholar] [CrossRef]
- Wang, H.-M.; Chen, C.-Y.; Chen, C.-Y.; Ho, M.-L.; Chou, Y.-T.; Chang, H.-C.; Lee, C.-H.; Wang, C.-Z.; Chu, I.-M. (−)-N-Formylanonaine from Michelia alba as a human tyrosinase inhibitor and antioxidant. Bioorg. Med. Chem. 2010, 18, 5241–5247. [Google Scholar] [CrossRef]
- Wang, H.-M.; Chen, C.-Y.; Wen, Z.-H. Identifying melanogenesis inhibitors from Cinnamomum subavenium with in vitro and in vivo screening systems by targeting the human tyrosinase. Exp. Dermatol. 2011, 20, 242–248. [Google Scholar] [CrossRef]
- Huang, H.-C.; Chang, T.-Y.; Chang, L.-Z; Wang, H.-F.; Yih, K.-H.; Hsieh, W.-Y.; Chang, T.-M. Inhibition of melanogenesis versus antioxidant properties of essential oil extracted from leaves of Vitex negundo Linn and chemical composition analysis by GC-MS. Molecules 2012, 17, 3902–3916. [Google Scholar]
- Burnett, C.L.; Bergfeld, W.F.; Belsito, D.V.; Hill, R.A.; Klaassen, C.D.; Liebler, D.C.; Marks, J.G.; Shank, R.C.; Slaga, T.J.; Snyder, P.W.; et al. Final report of the safety assessment of kojic acid as used in cosmetics. Int. J. Toxicol. 2010, 29, 244S–273S. [Google Scholar] [CrossRef]
- Draelos, Z.D. Skin lightening preparations and the hydroquinone controversy. Dermatol. Ther. 2007, 20, 308–313. [Google Scholar] [CrossRef]
- Lopresti, A.L.; Hood, S.D.; Drummond, P.D. Multiple antidepressant potential modes of action of curcumin: A review of its anti-inflammatory, monoaminergic, antioxidant, immune-modulating and neuroprotective effects. J. Psychopharmacol. 2012, 26, 1512–1524. [Google Scholar] [CrossRef]
- Goel, A.; Aggarwal, B.B. Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr. Cancer 2010, 62, 919–930. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Targeting inflammation-induced obesity and metabolic diseases by curcumin and other nutraceuticals. Annu. Rev. Nutr. 2010, 30, 173–199. [Google Scholar] [CrossRef]
- Zhou, H.; Beevers, C.S.; Huang, S. The targets of curcumin. Curr. Drug Targets 2011, 12, 332–347. [Google Scholar] [CrossRef]
- Marathe, S.A.; Datey, A.A.; Chakravortty, D. Herbal cocktail as anti-infective: Promising therapeutic for the treatment of viral diseases. Recent Pat. Antiinfect. Drug Discov. 2012, 7, 123–132. [Google Scholar] [CrossRef]
- Chainani-Wu, N. Safety and anti-inflammatory activity of curcumin: A component of tumeric (Curcuma longa). J. Altern. Complement. Med. 2003, 9, 161–168. [Google Scholar] [CrossRef]
- Suhaimi, H.; Ahmad, F.B.; Friberg, S.E. Curcumin in a model skin lotion formulation. J. Pharm. Sci. 1995, 84, 376–380. [Google Scholar] [CrossRef]
- Thangapazham, R.L.; Sharad, S.; Maheshwari, R.K. Skin regenerative potentials of curcumin. Biofactors 2013, 39, 141–149. [Google Scholar] [CrossRef]
- Lima, C.F.; Pereira-Wilson, C.; Rattan, S.I. Curcumin induces heme oxygenase-1 in normal human skin fibroblasts through redox signaling: Relevance for anti-aging intervention. Mol. Nutr. Food Res. 2010, 55, 430–442. [Google Scholar]
- Asawanonda, P.; Klahan, S.O. Tetrahydrocurcuminoid cream plus targeted narrowband UVB phototherapy for vitiligo: A preliminary randomized controlled study. Photomed. Laser Surg. 2010, 28, 679–684. [Google Scholar] [CrossRef]
- Lee, K.H.; Ab Aziz, F.H.; Syahida, A.; Abas, F.; Shaari, K.; Israf, D.A.; Lajis, N.H. Synthesis and biological evaluation of curcumin-like diarylpentanoid analogues for anti-inflammatory, antioxidant and anti-tyrosinase activities. Eur. J. Med. Chem. 2009, 44, 3195–3200. [Google Scholar] [CrossRef]
- Bao, K.; Dai, Y.; Zhu, Z.B.; Tu, F.J.; Zhang, W.G.; Yao, X.S. Design and synthesis of biphenyl derivatives as mushroom tyrosinase inhibitors. Bioorg. Med. Chem. 2010, 18, 6708–6714. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kawano, Y. Inhibitory effect of hydroxyindoles and their analogues on human melanoma tyrosinase. Z. Naturforsch. C 2010, 65, 49–54. [Google Scholar]
- Yi, W.; Cao, R.; Peng, W.; Wen, H.; Yan, Q.; Zhou, B.; Song, H. Synthesis and biological evaluation of novel 4-hydroxybenzaldehyde derivatives as tyrosinase inhibitors. Eur. J. Med. Chem. 2010, 45, 639–646. [Google Scholar] [CrossRef]
- OECD. Testing of Chemicals Acute Oral Toxicity–Acute Toxic Class Method. In OECD Guidelines for the Testing of Chemicals; (E-book); OECD Publishing: Paris, France, 2001; pp. 1–14. [Google Scholar]
- Matoba, Y.; Kumagai, T.; Yamamoto, A.; Yoshitsu, H.; Sugiyama, M. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 2006, 281, 8981–8990. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3a–k and 4a–i are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, Y.; Du, Z.; Xue, G.; Chen, Q.; Lu, Y.; Zheng, X.; Conney, A.H.; Zhang, K. Synthesis and Biological Evaluation of Unsymmetrical Curcumin Analogues as Tyrosinase Inhibitors. Molecules 2013, 18, 3948-3961. https://doi.org/10.3390/molecules18043948
Jiang Y, Du Z, Xue G, Chen Q, Lu Y, Zheng X, Conney AH, Zhang K. Synthesis and Biological Evaluation of Unsymmetrical Curcumin Analogues as Tyrosinase Inhibitors. Molecules. 2013; 18(4):3948-3961. https://doi.org/10.3390/molecules18043948
Chicago/Turabian StyleJiang, Yongfu, Zhiyun Du, Guihua Xue, Qian Chen, Yujing Lu, Xi Zheng, Allan H. Conney, and Kun Zhang. 2013. "Synthesis and Biological Evaluation of Unsymmetrical Curcumin Analogues as Tyrosinase Inhibitors" Molecules 18, no. 4: 3948-3961. https://doi.org/10.3390/molecules18043948
APA StyleJiang, Y., Du, Z., Xue, G., Chen, Q., Lu, Y., Zheng, X., Conney, A. H., & Zhang, K. (2013). Synthesis and Biological Evaluation of Unsymmetrical Curcumin Analogues as Tyrosinase Inhibitors. Molecules, 18(4), 3948-3961. https://doi.org/10.3390/molecules18043948