Photo-Induced Cycloaddition Reactions of α-Diketones and Transformations of the Photocycloadducts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
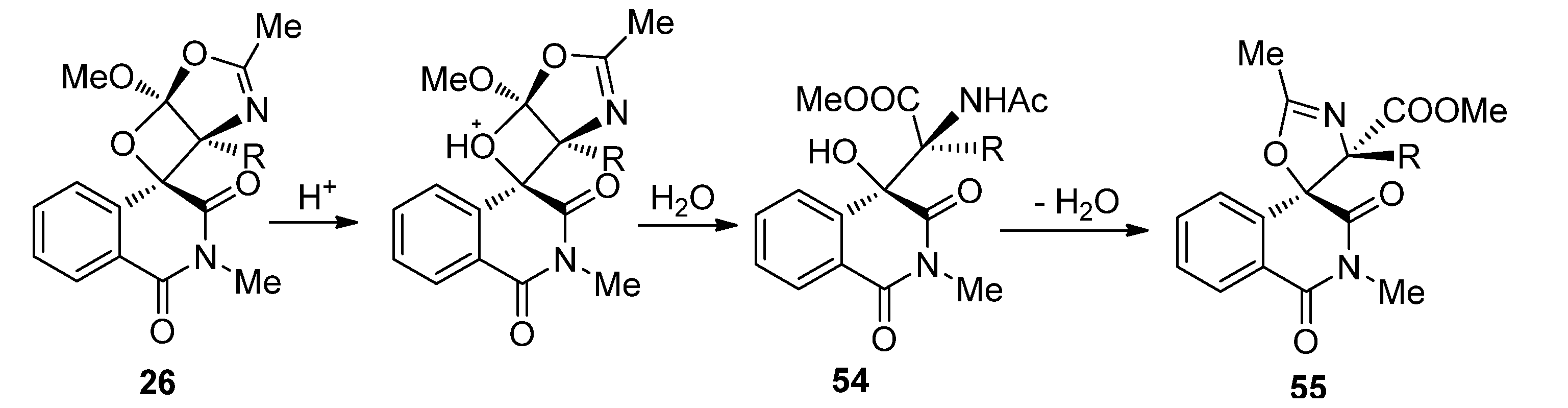{kind=link}
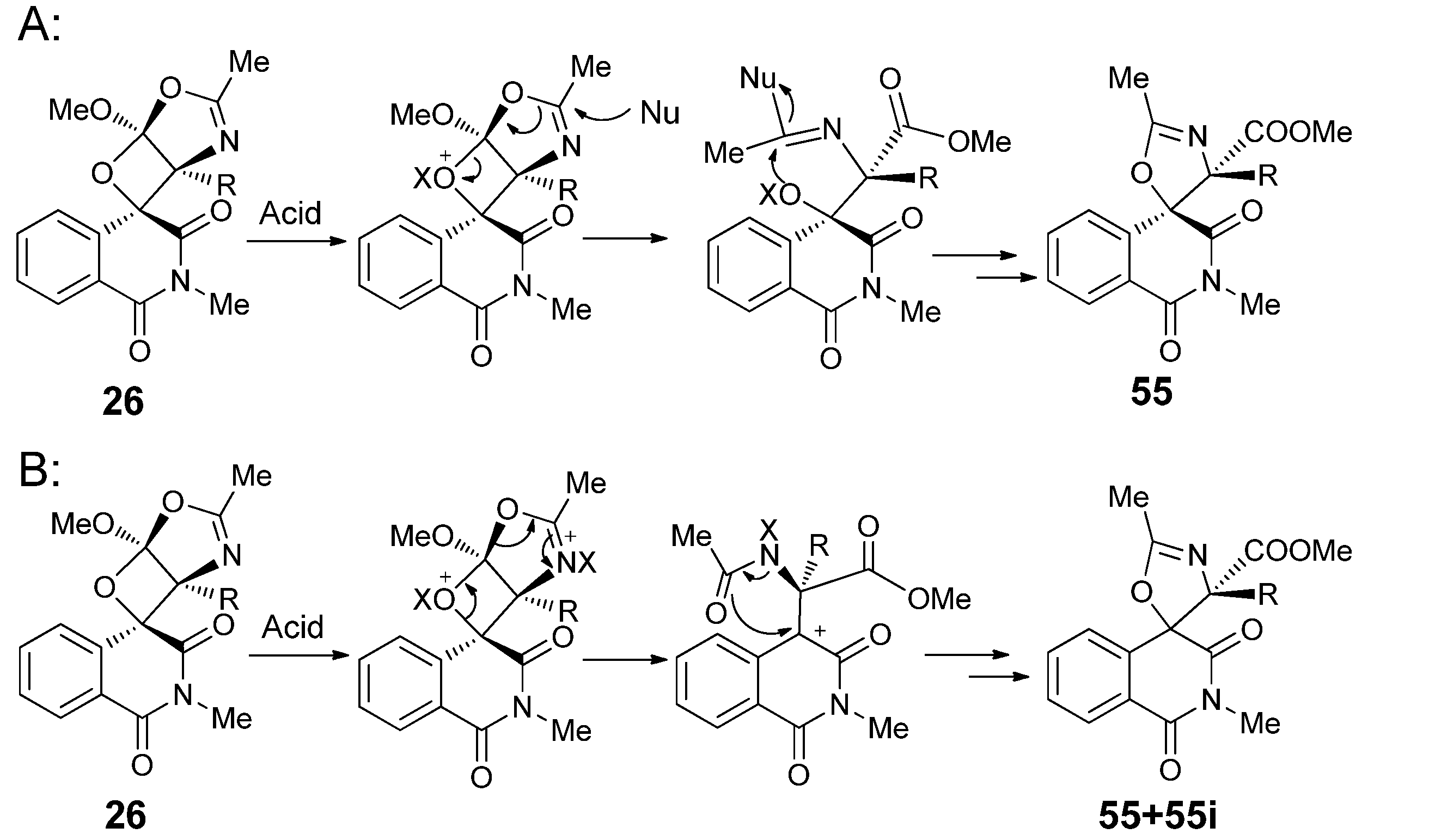{kind=link}
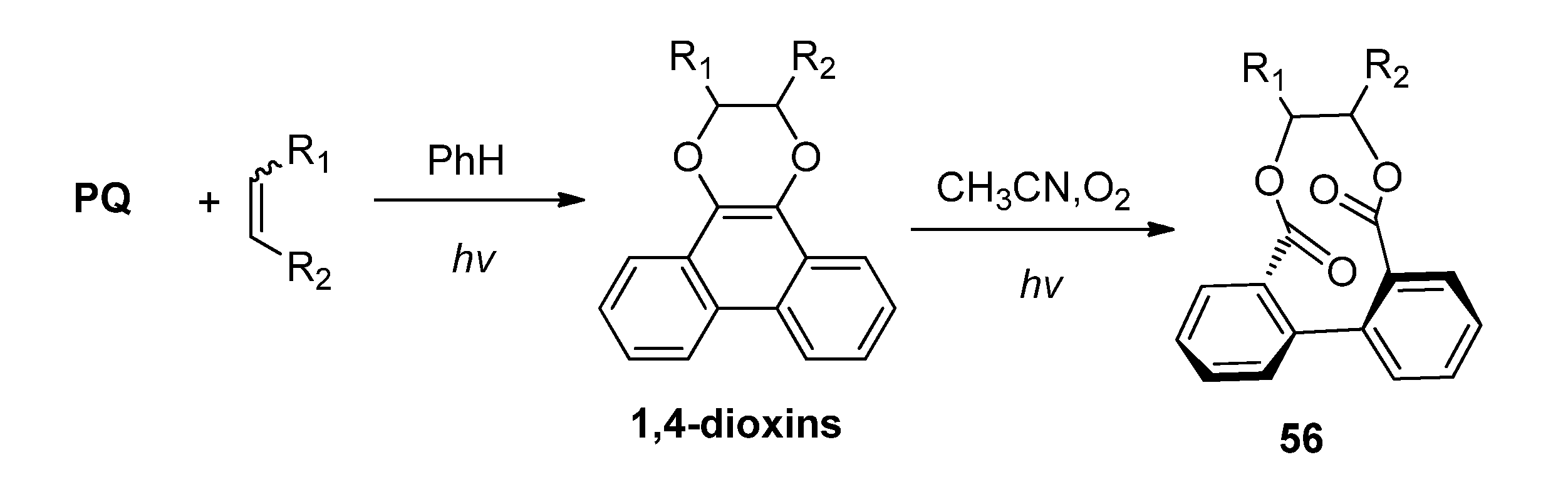{kind=link}
Abstract
:1. Introduction
2. Photo-Induced Cycloaddition Reactions of α-Diketones
2.1. N-Acetylisatin
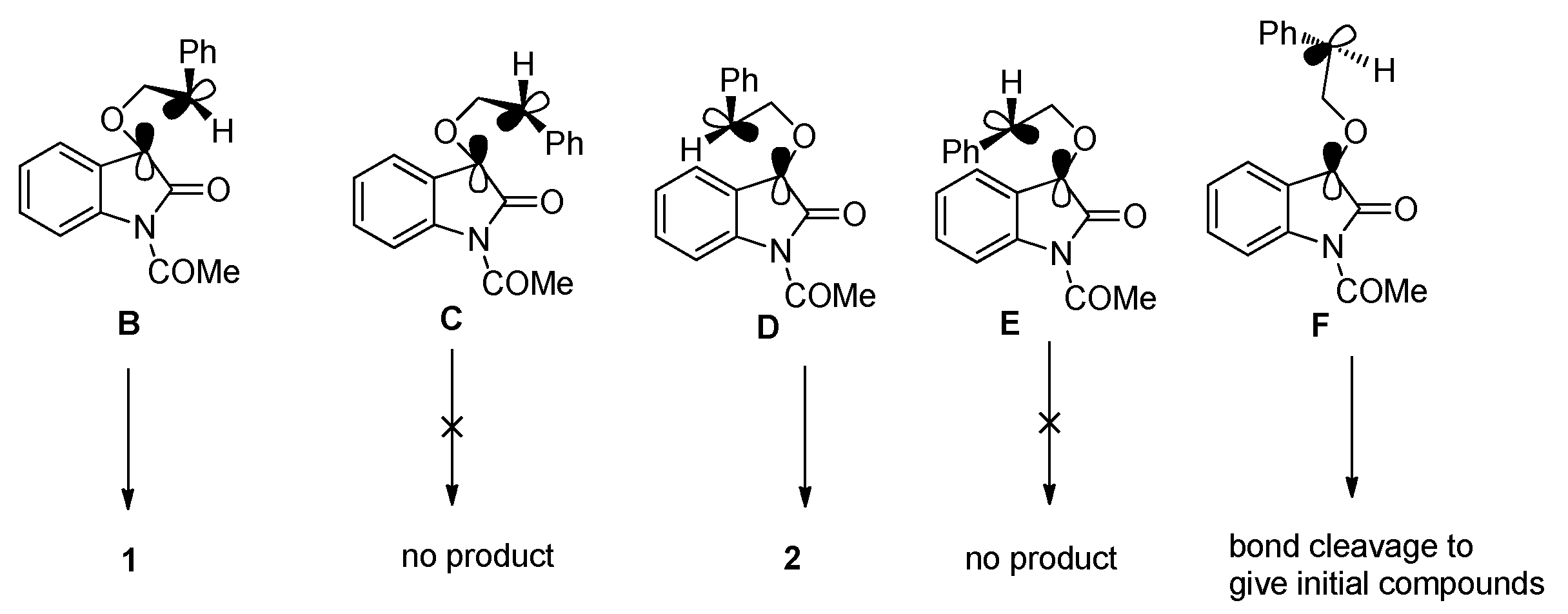
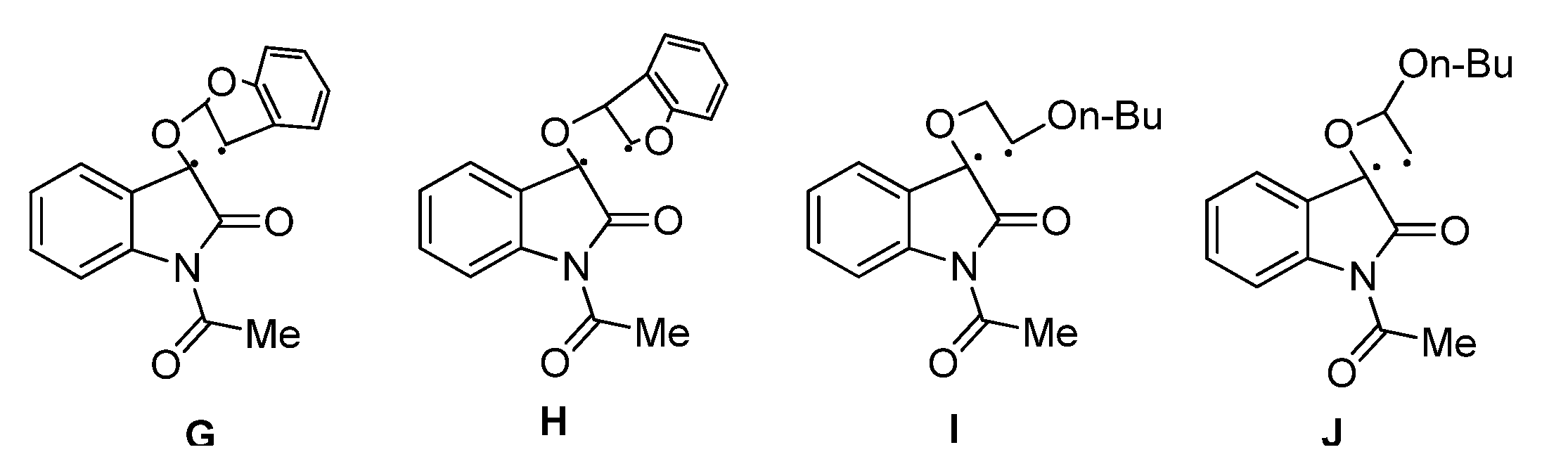
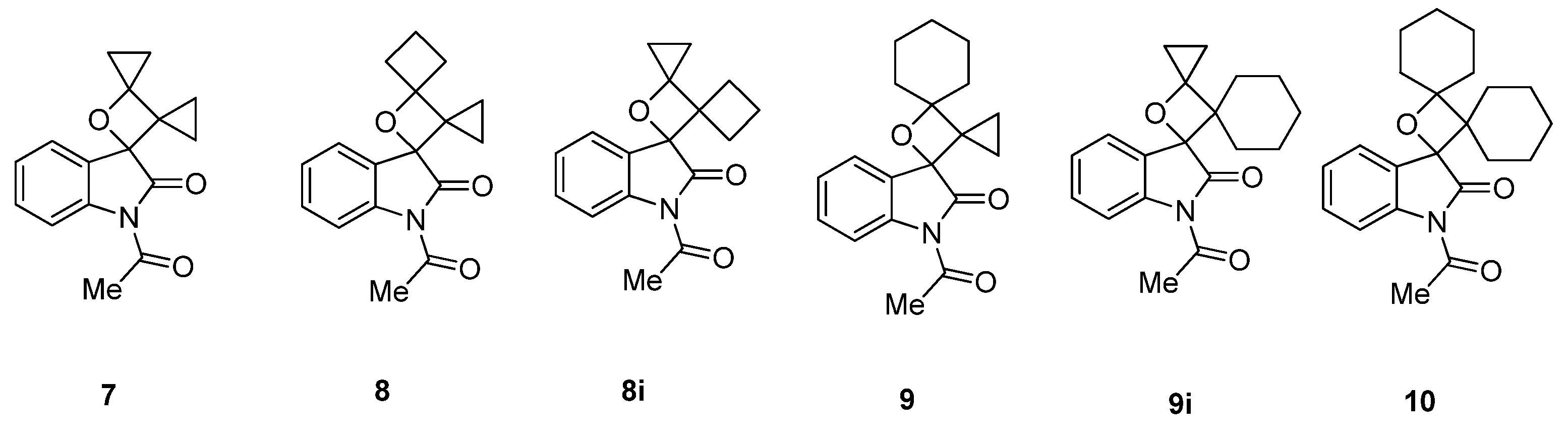
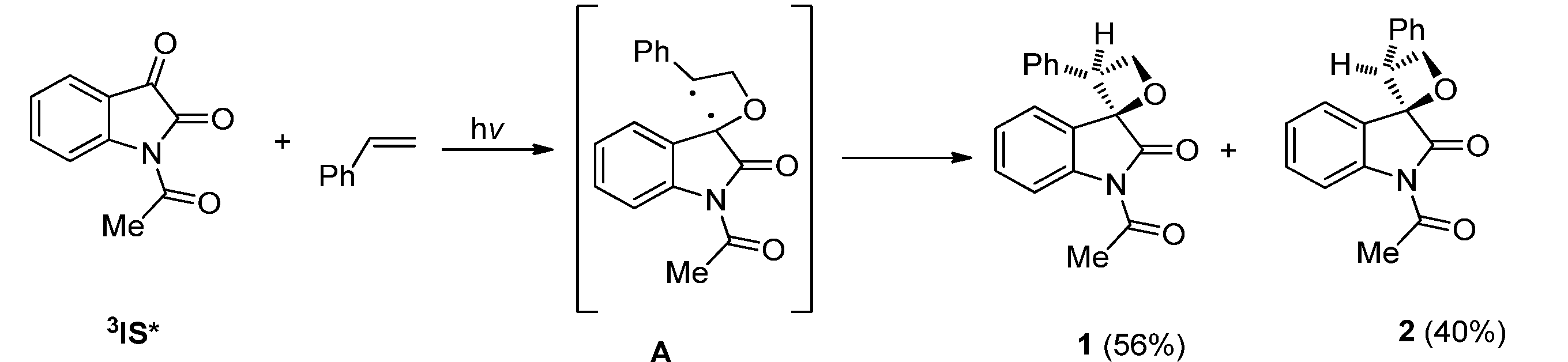
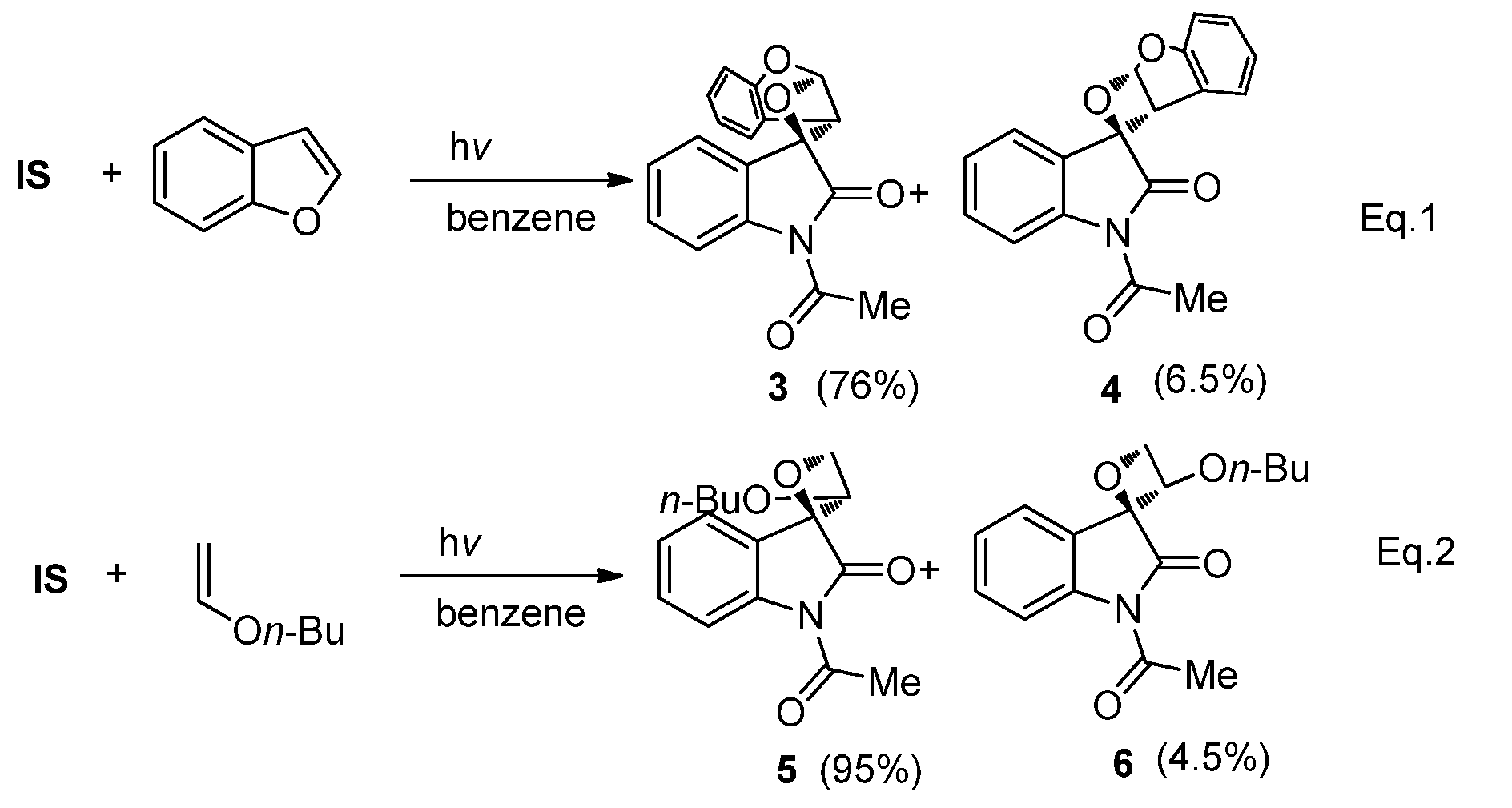
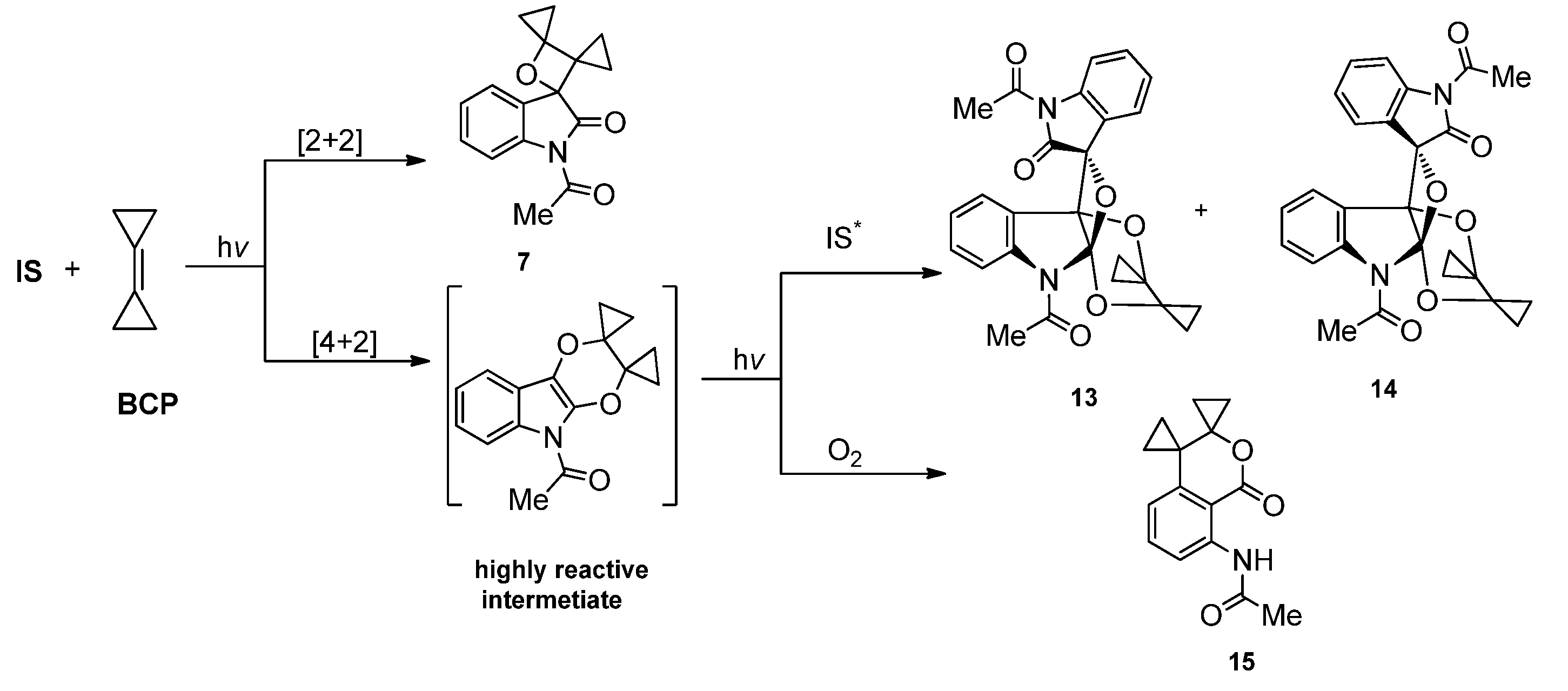
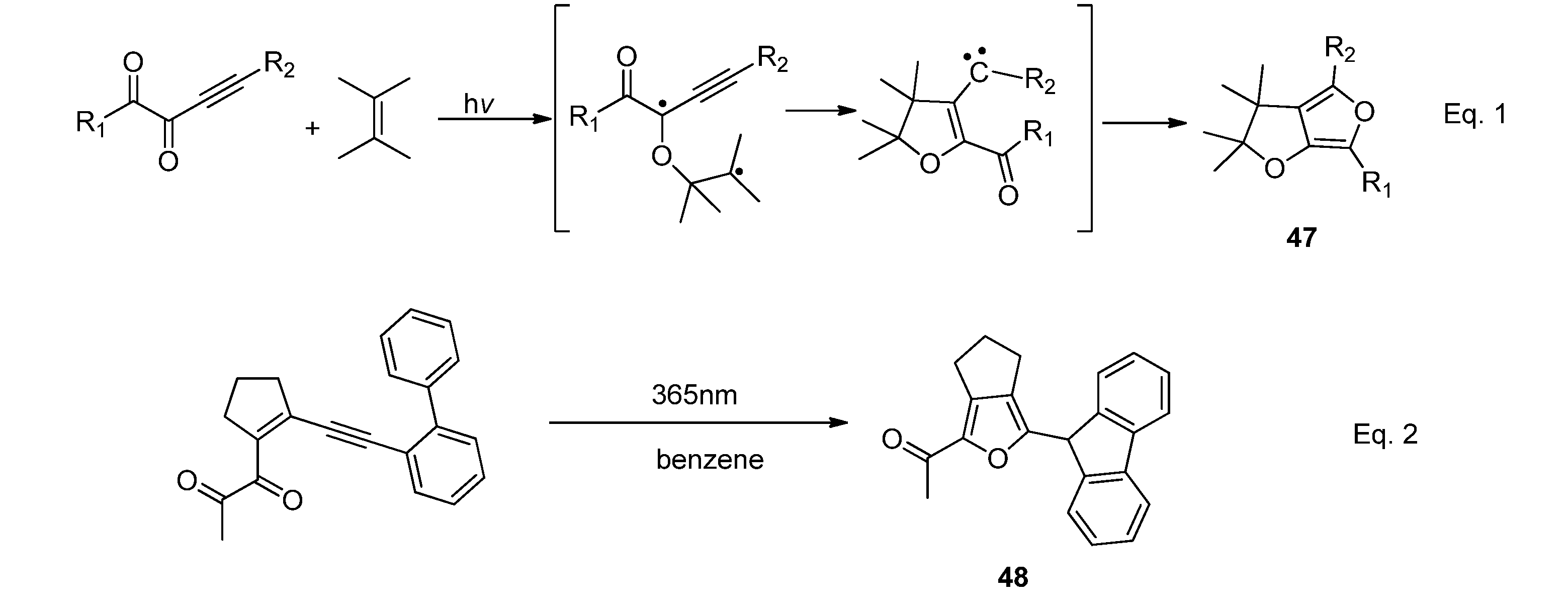
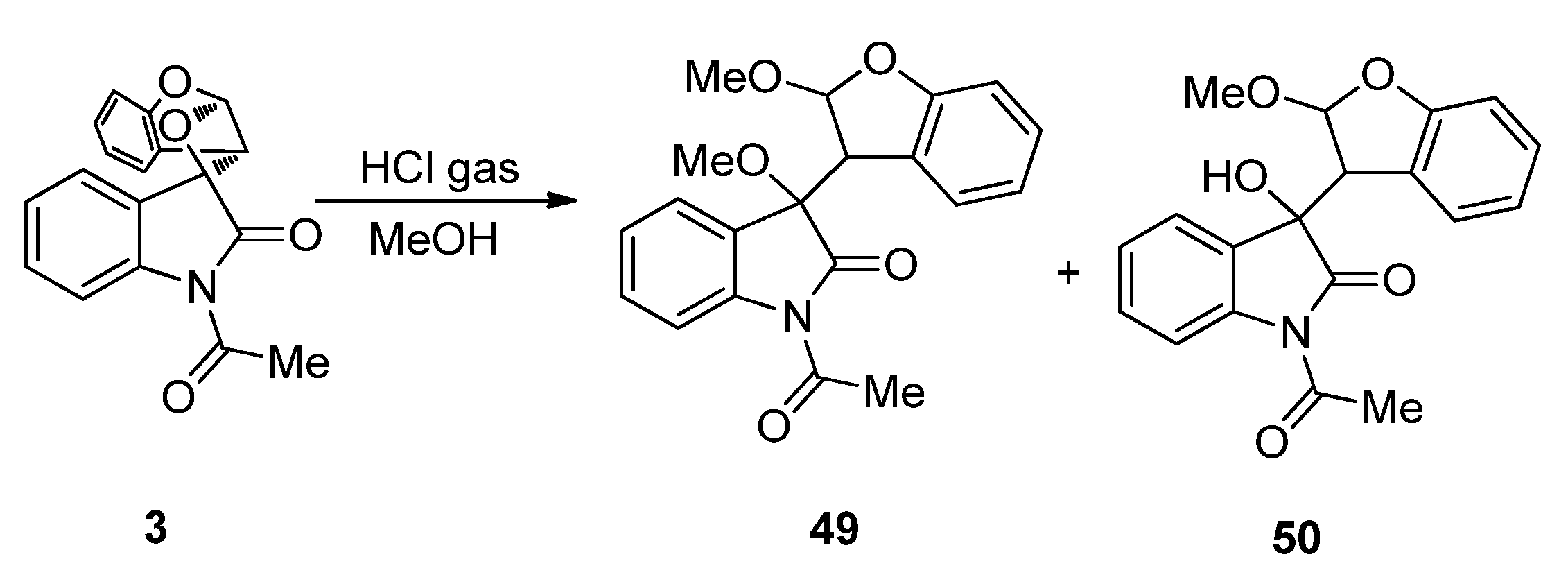
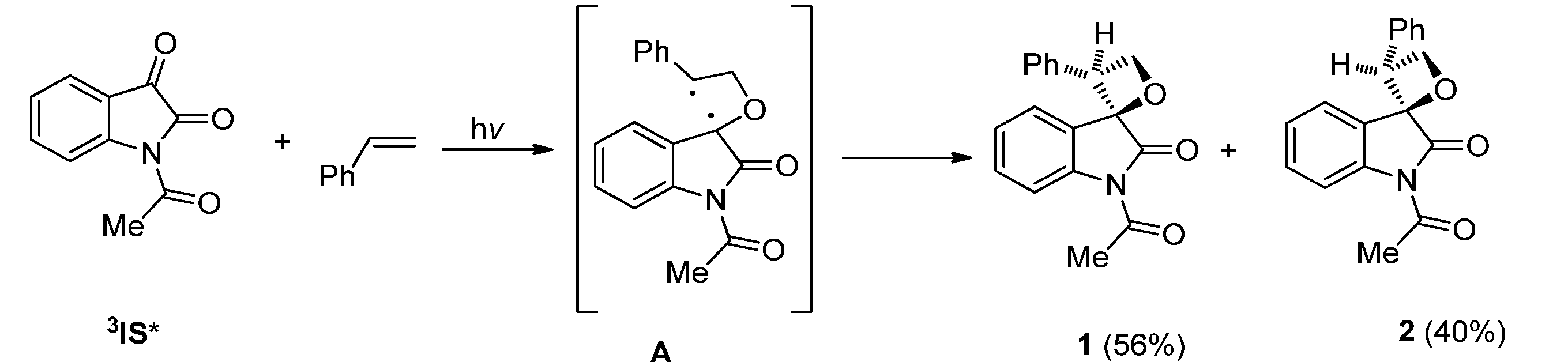
2.1.1. [2 + 2] Photocycloadditions Involving IS
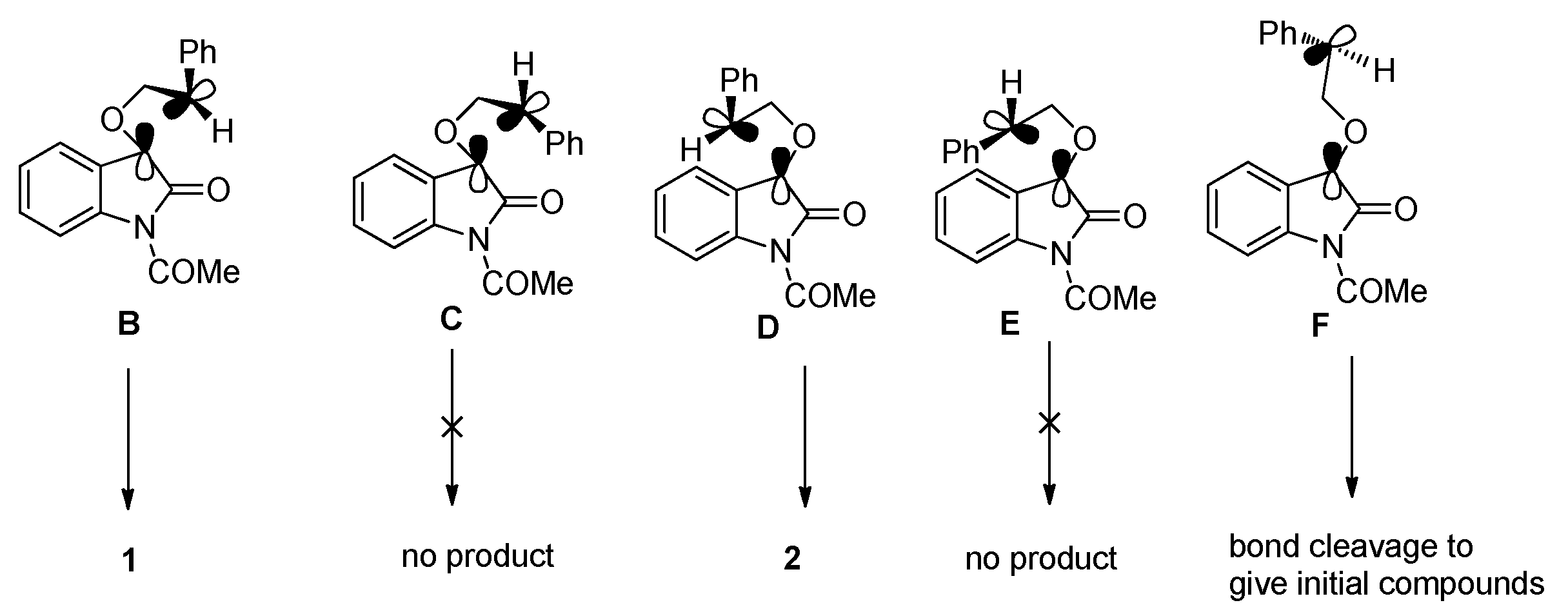
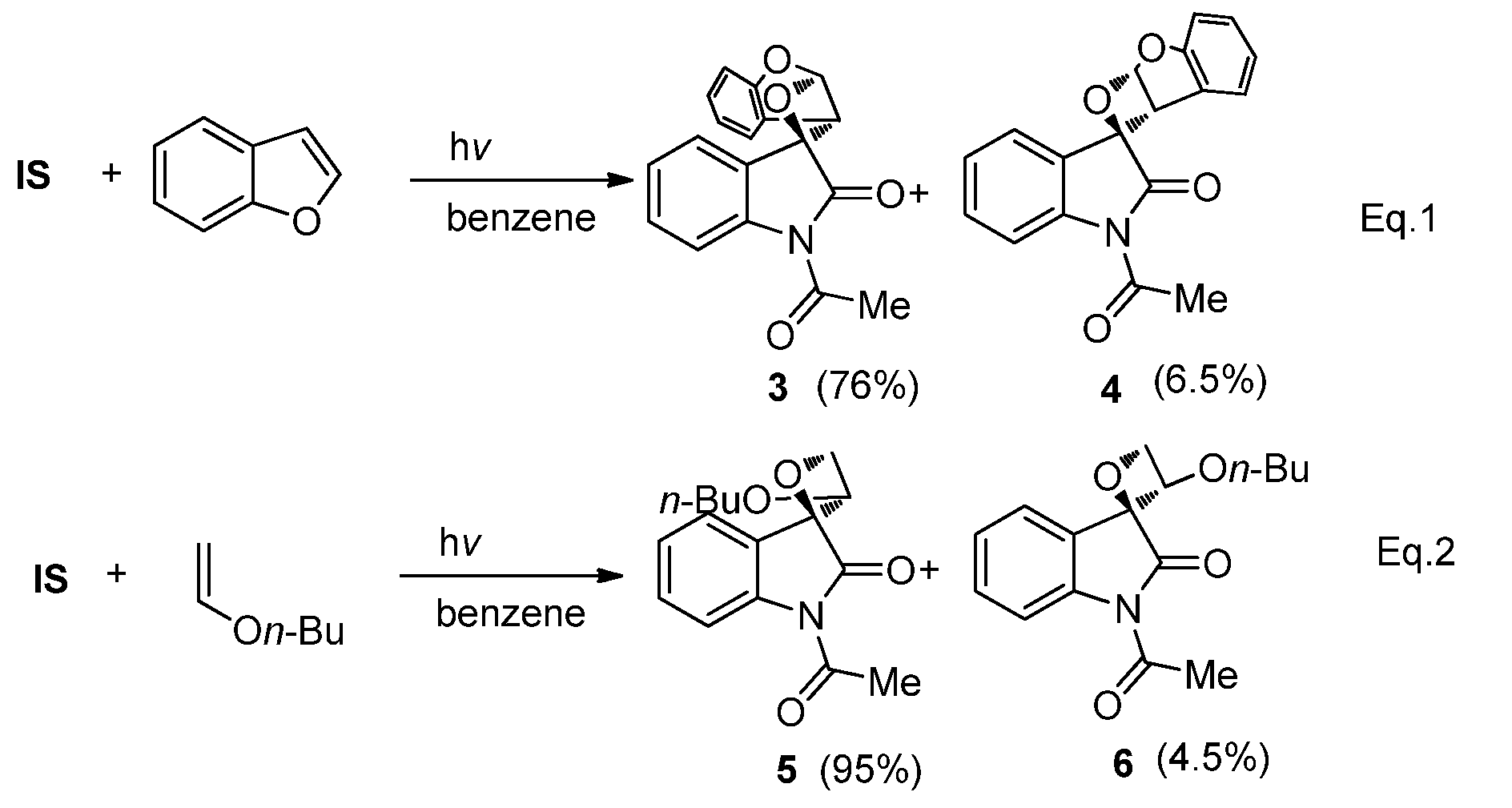
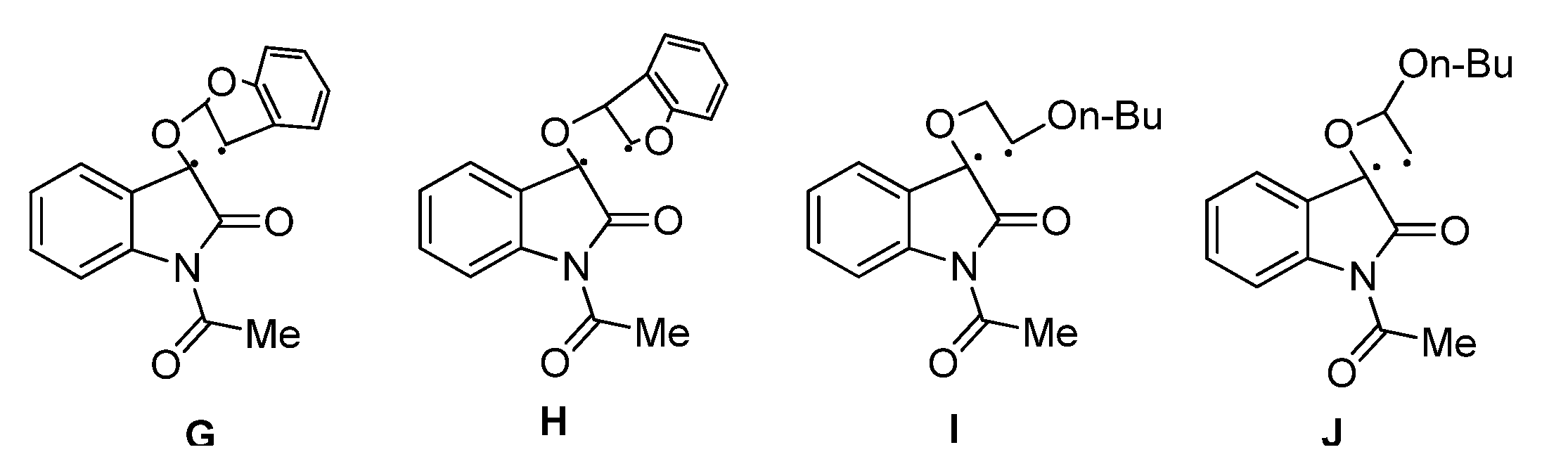
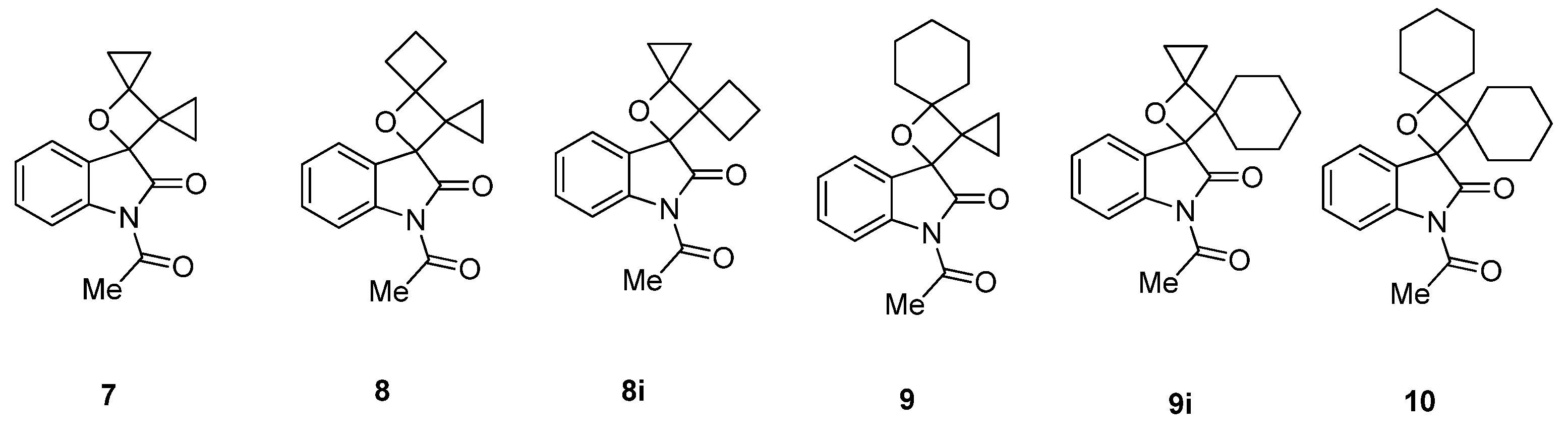
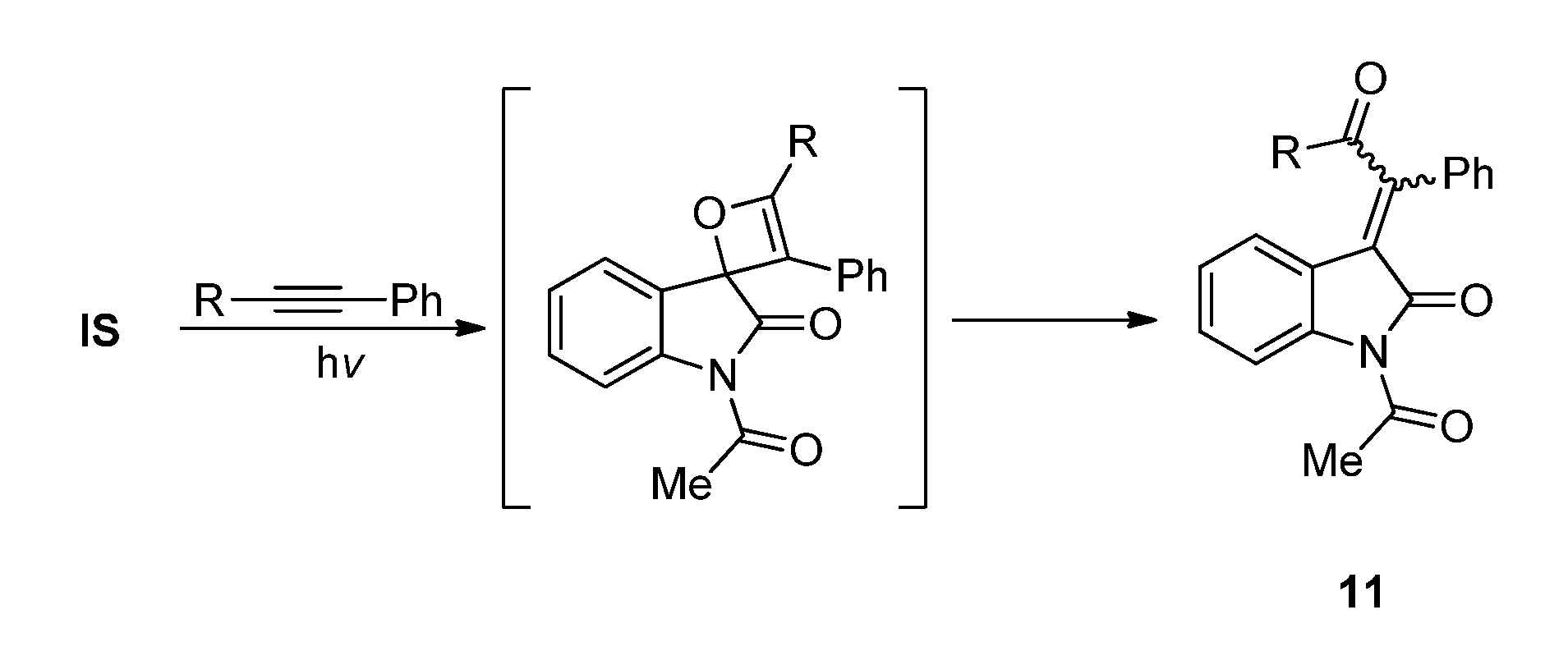
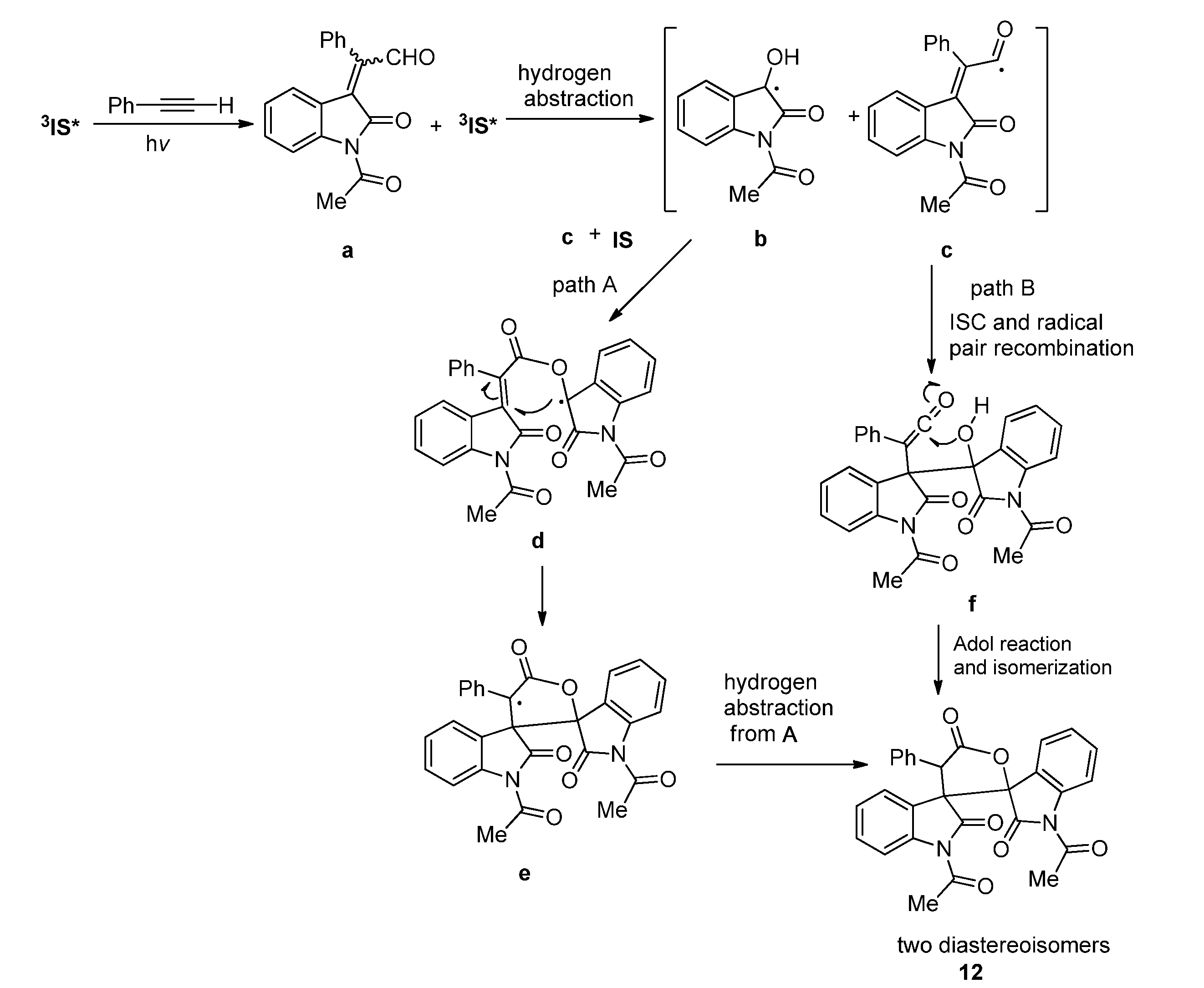
2.1.2. Photoreaction of IS Initiated by [4 + 2] Cycloaddition

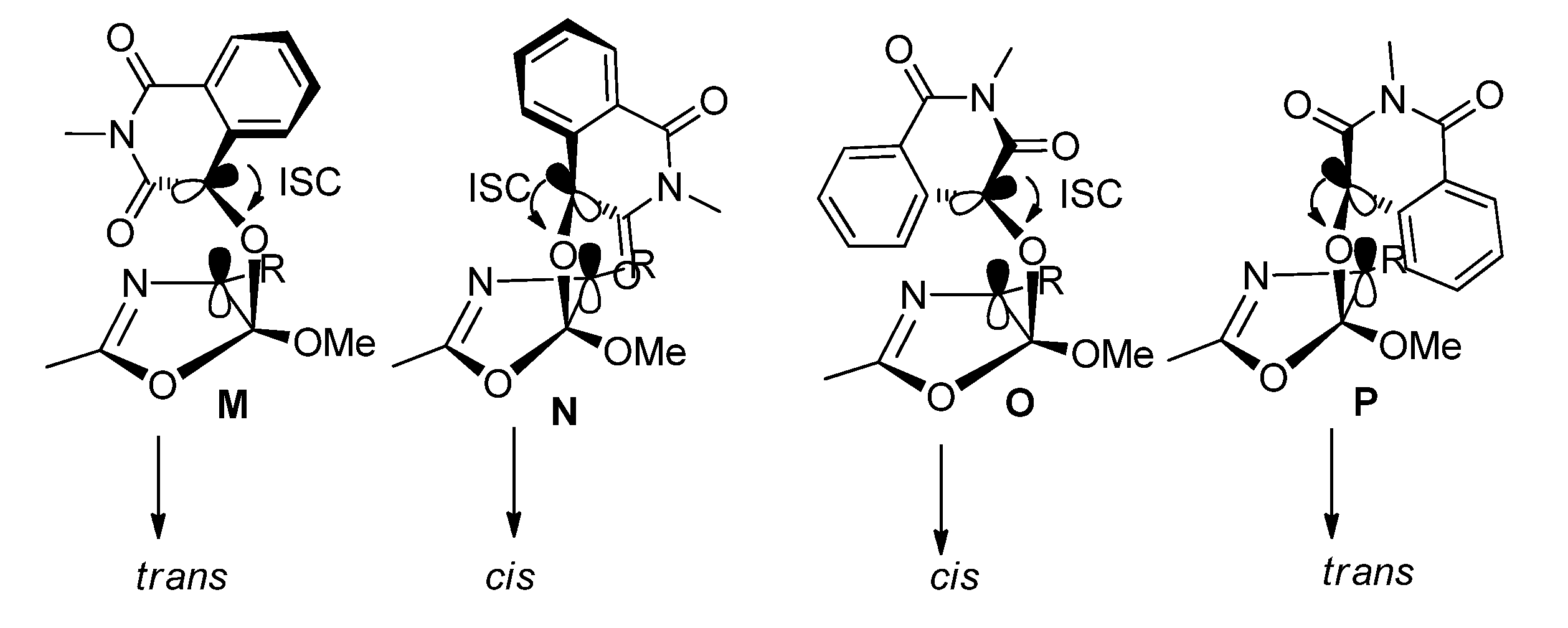
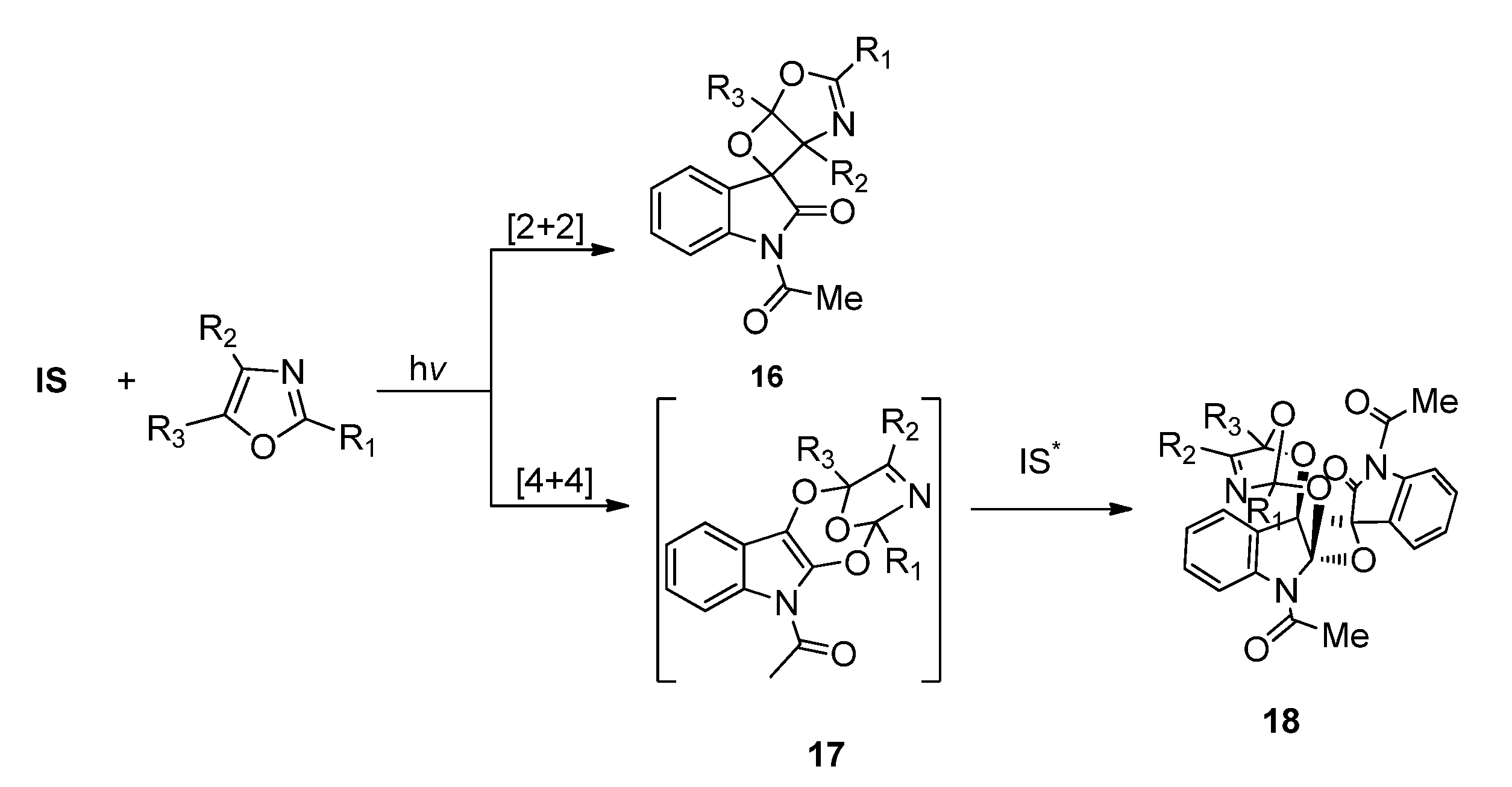
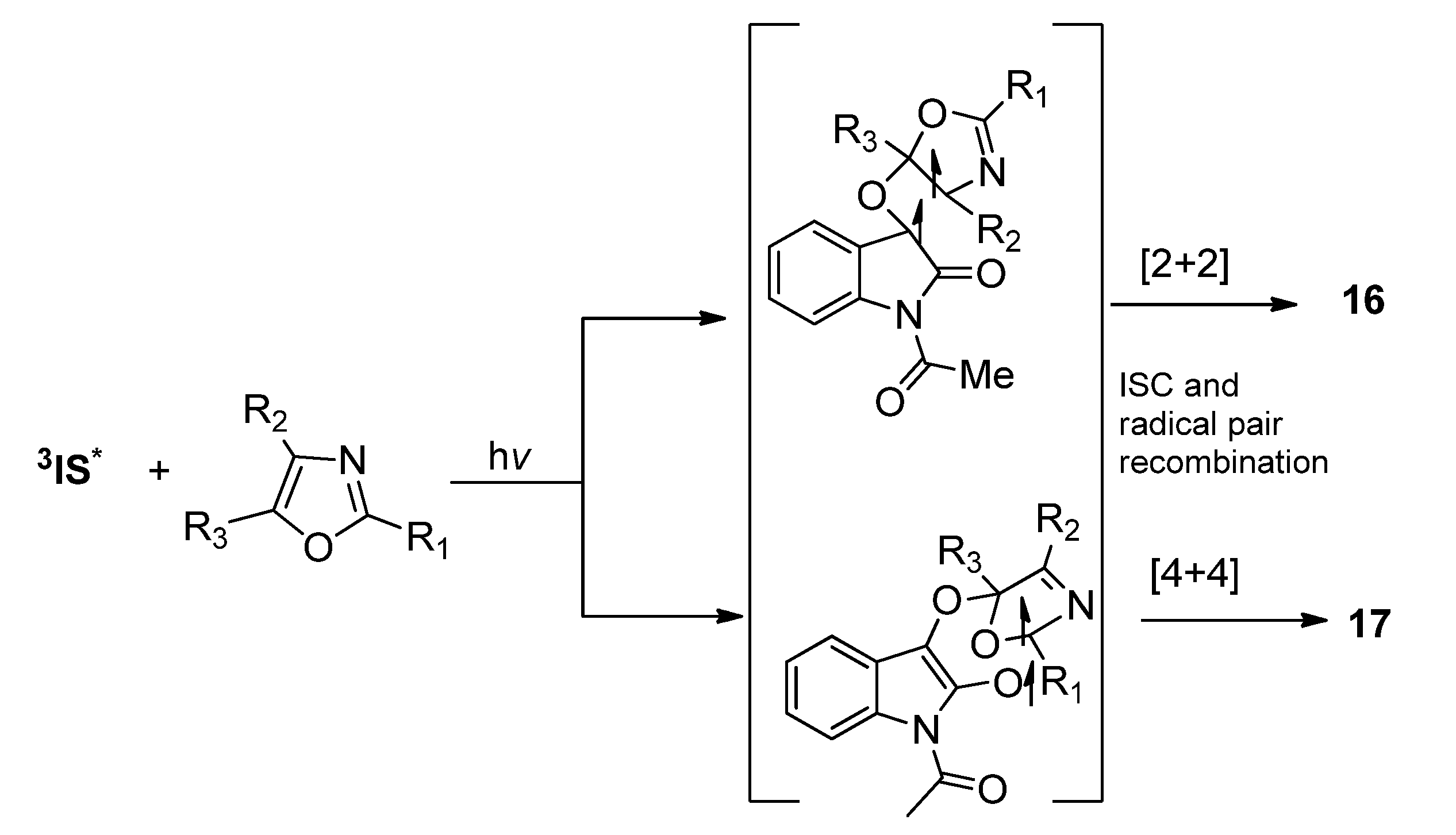
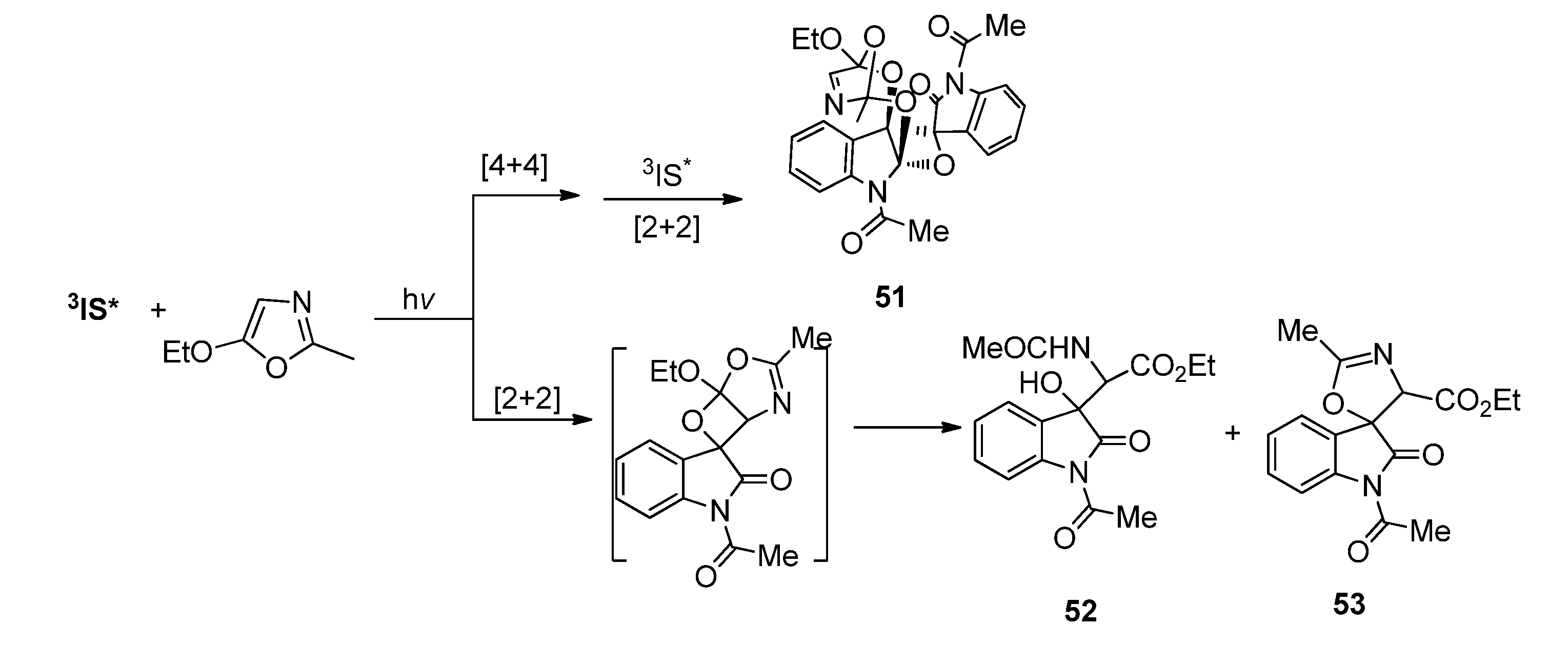
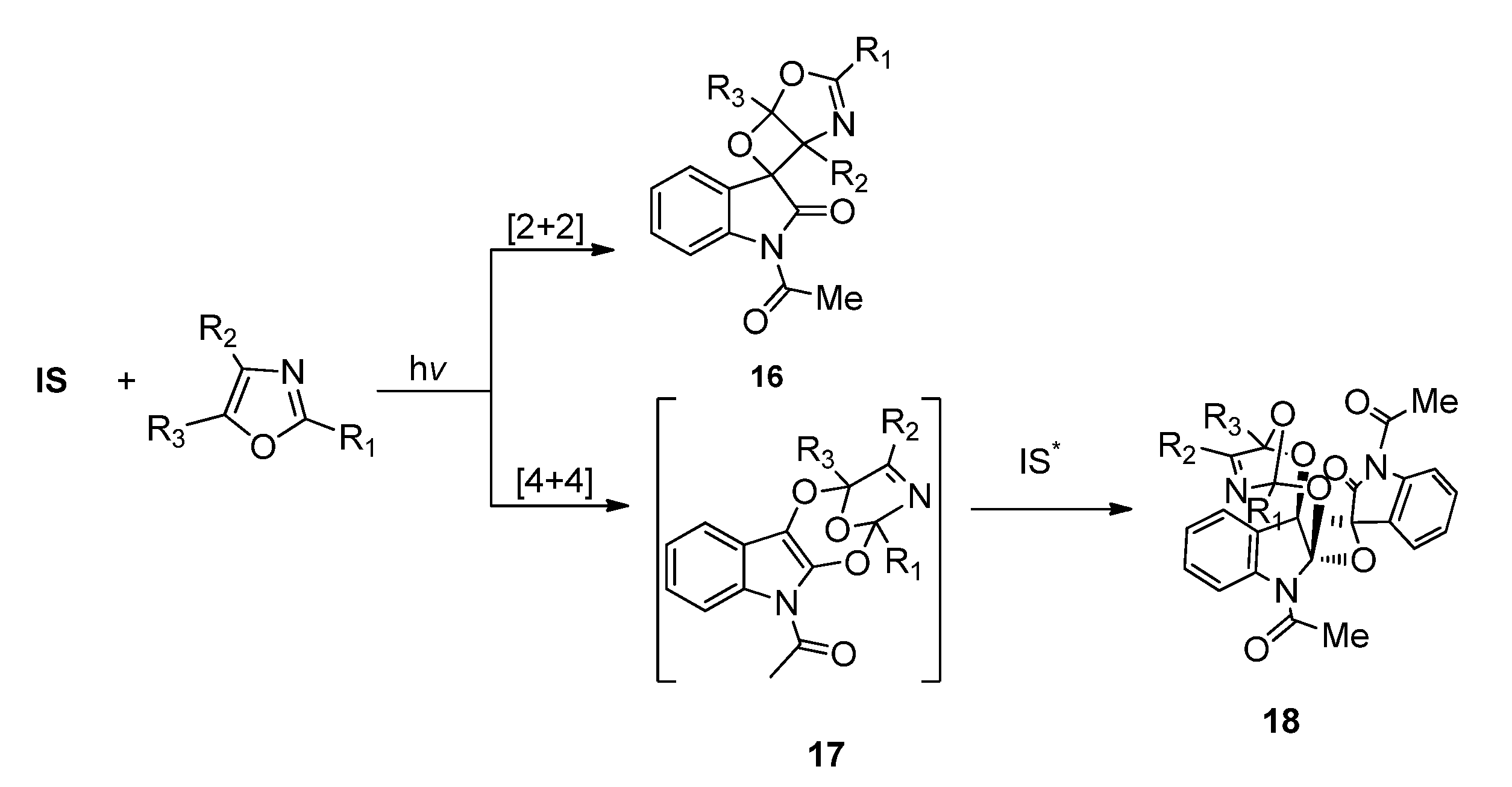
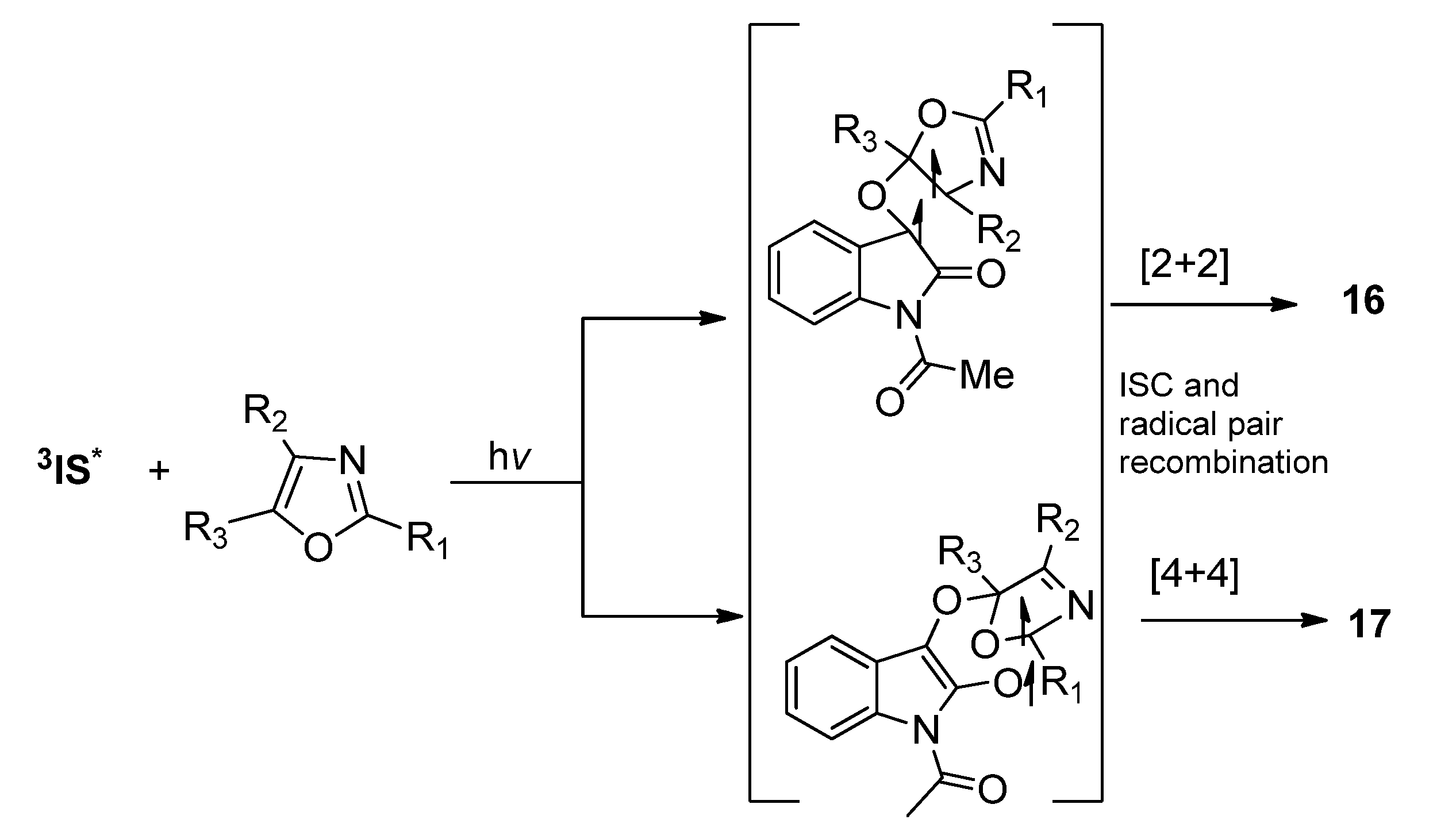
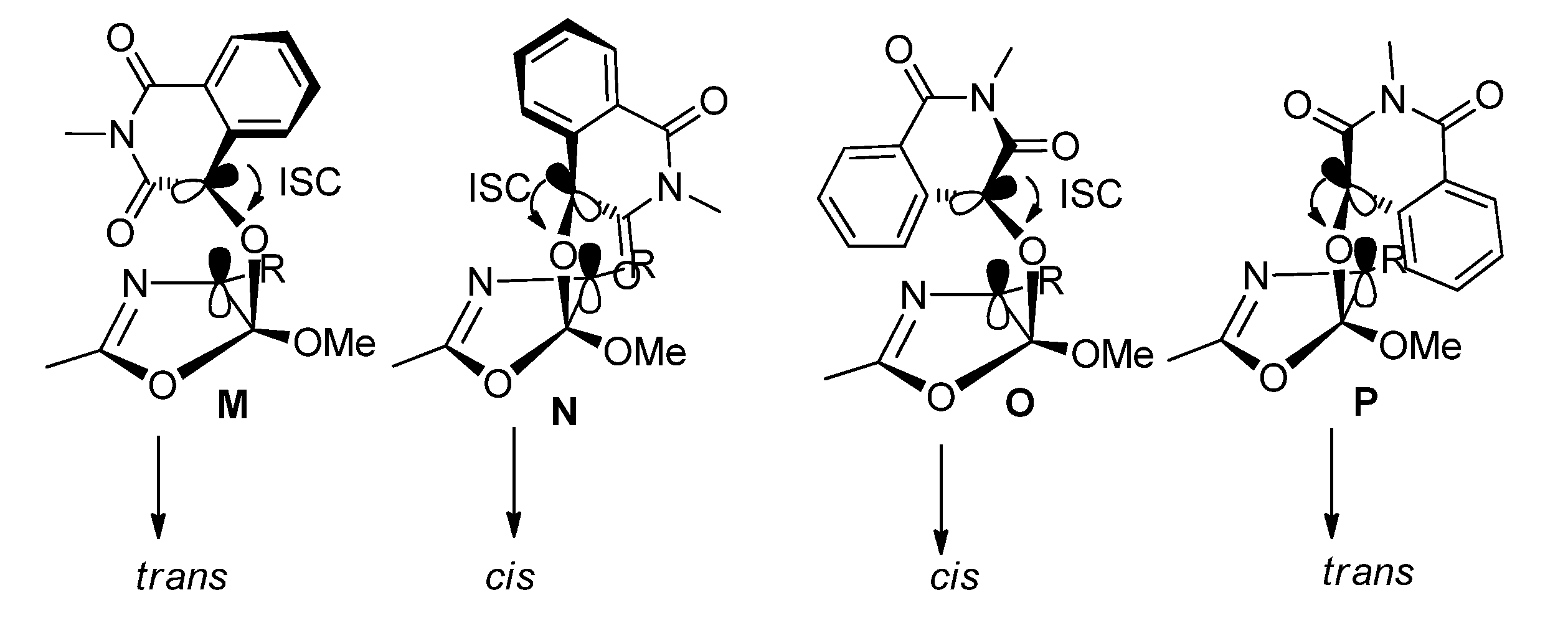
2.1.3. [4 + 4] Photocycloaddition of IS with Oxazoles
2.2. Phenanthrenequinone
2.2.1. [2 + 2] Photocycloaddition of PQ
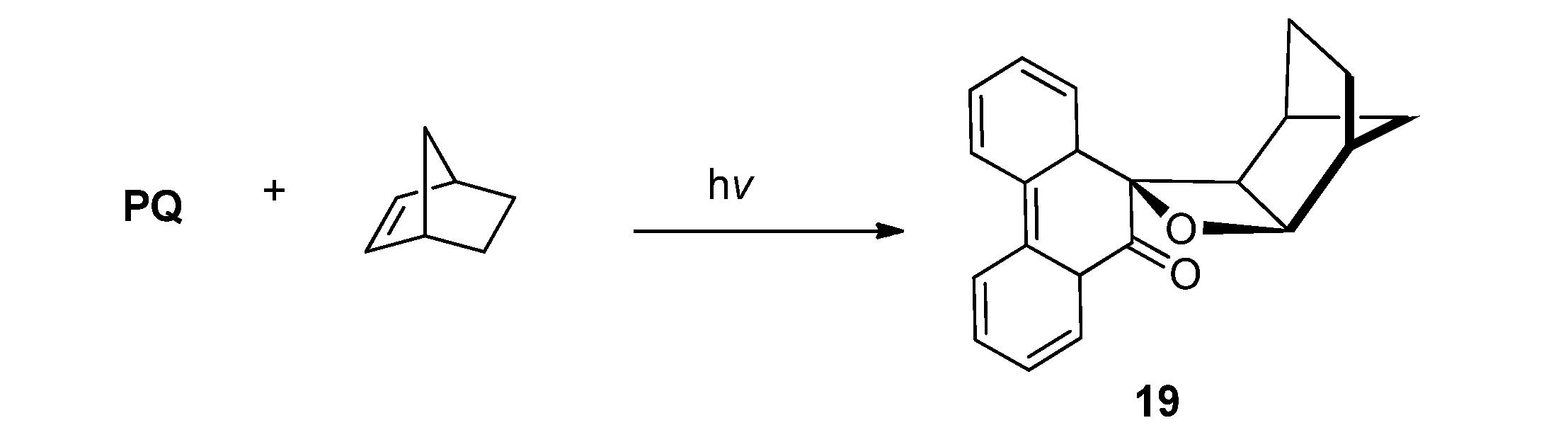
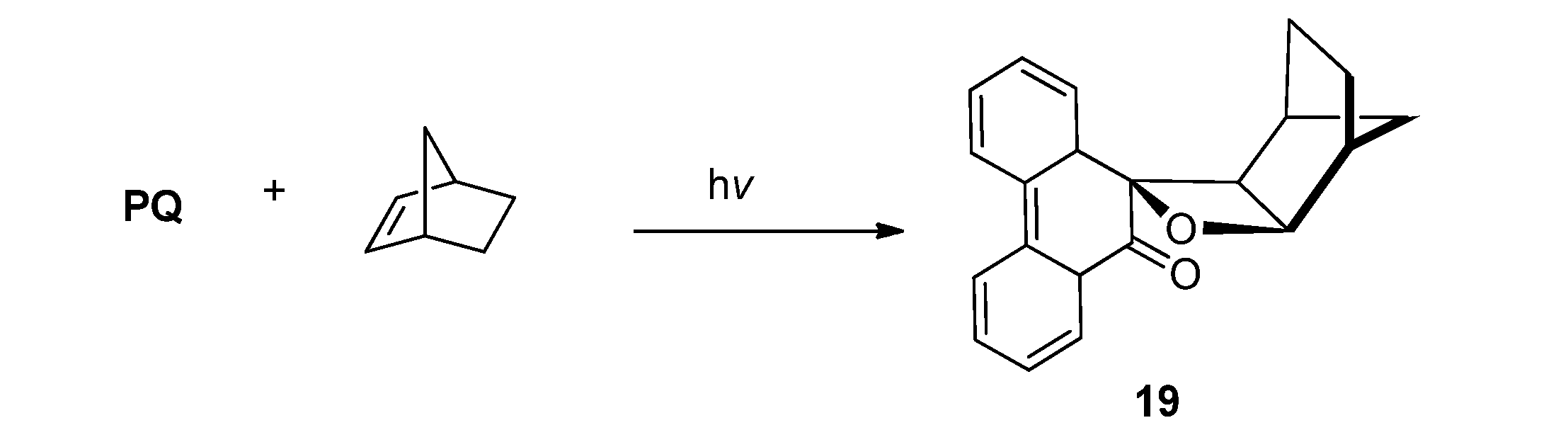
2.2.2. [4 + 2] Photocycloaddition of PQ
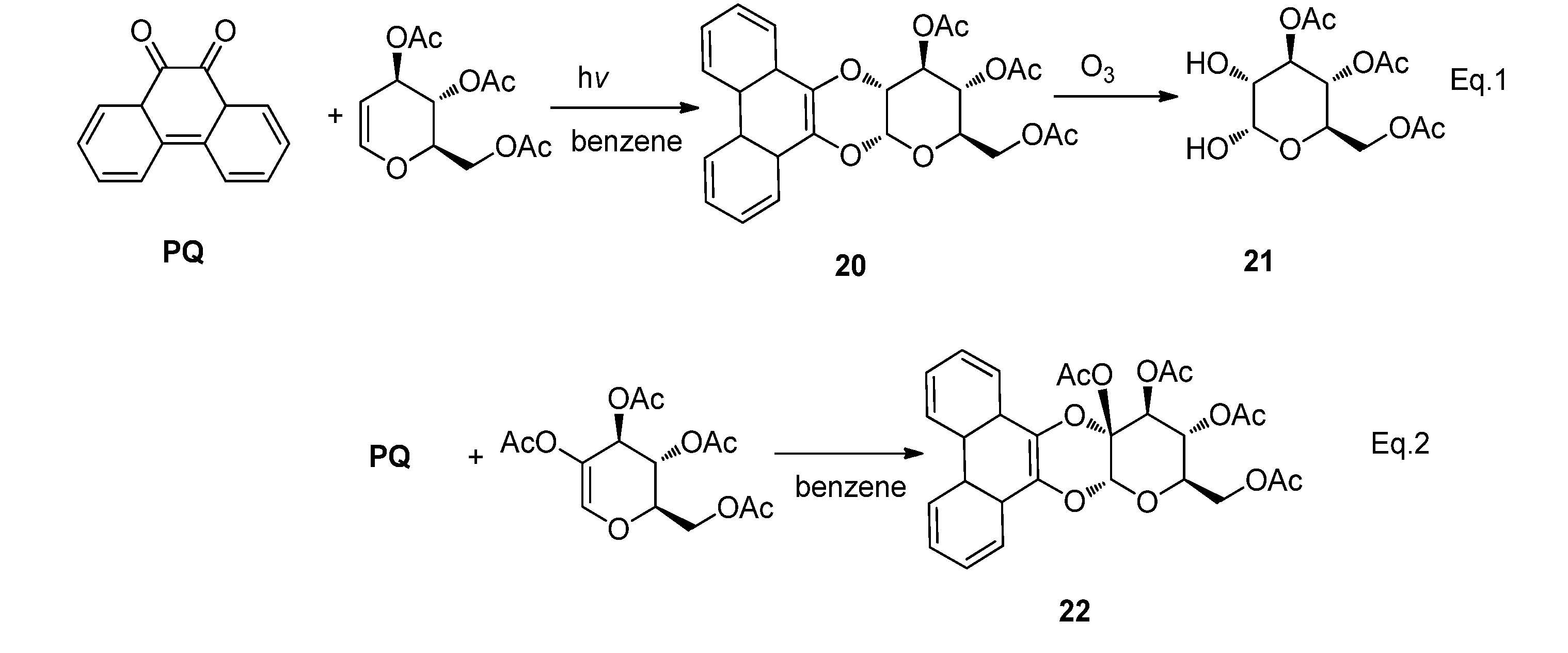
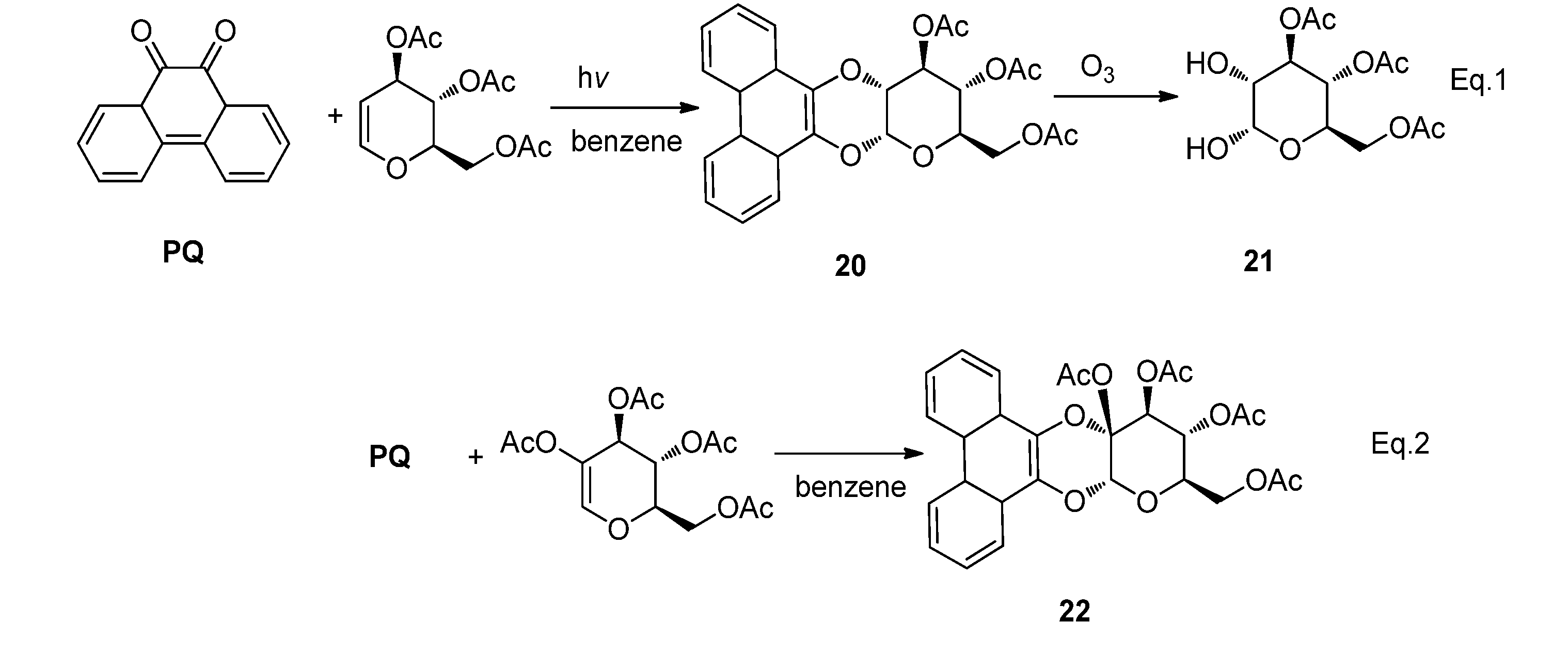
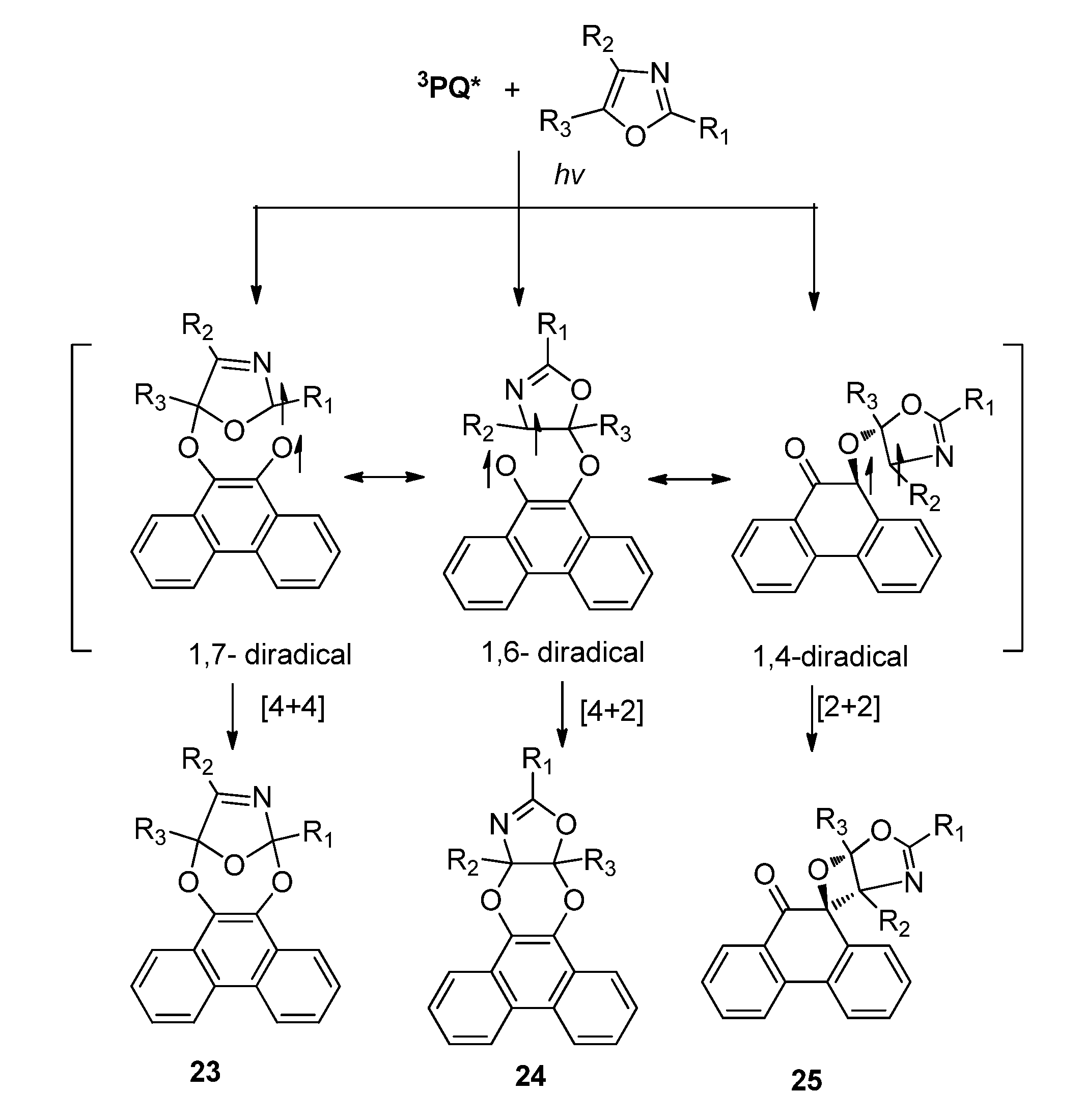
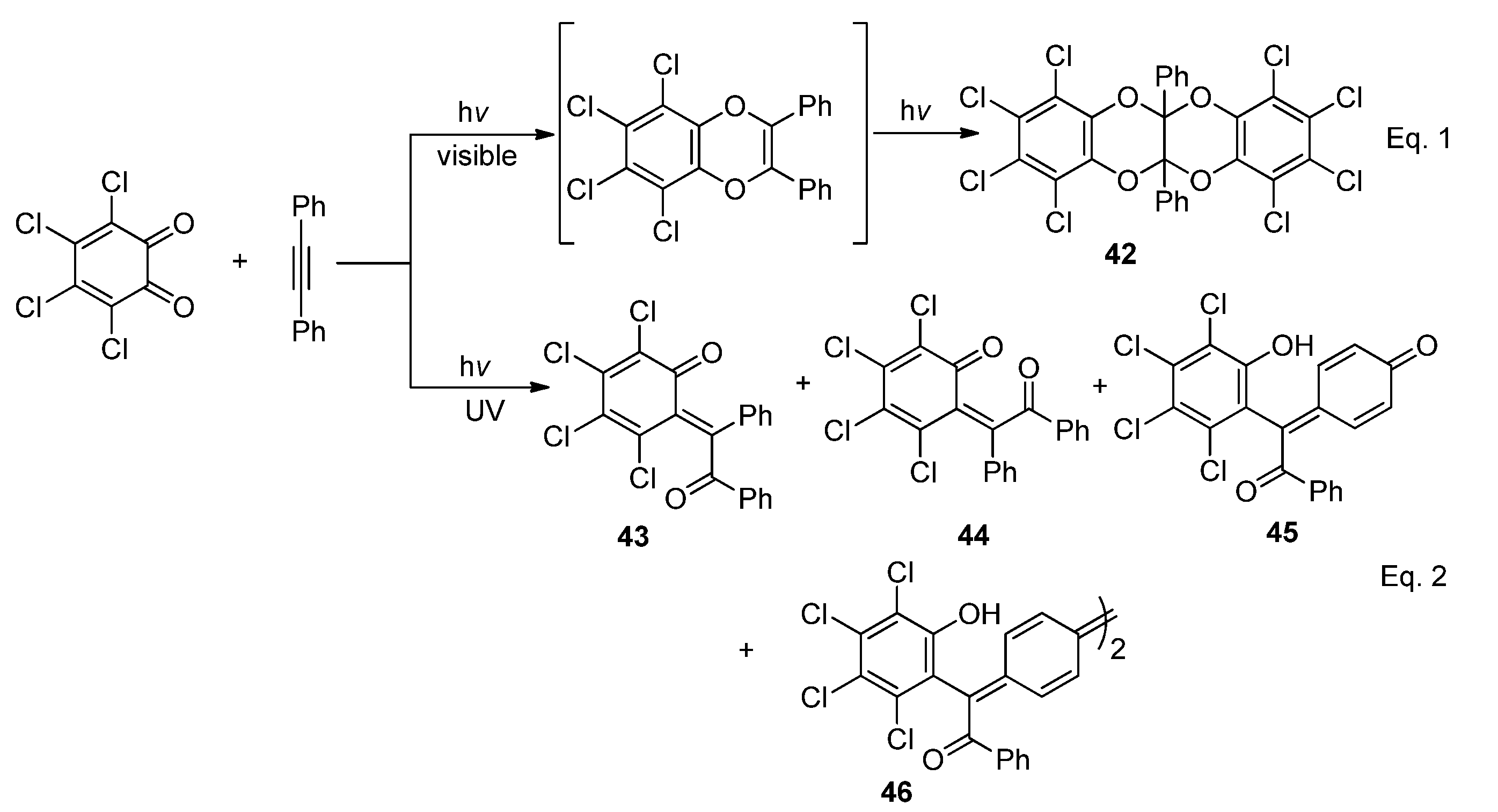
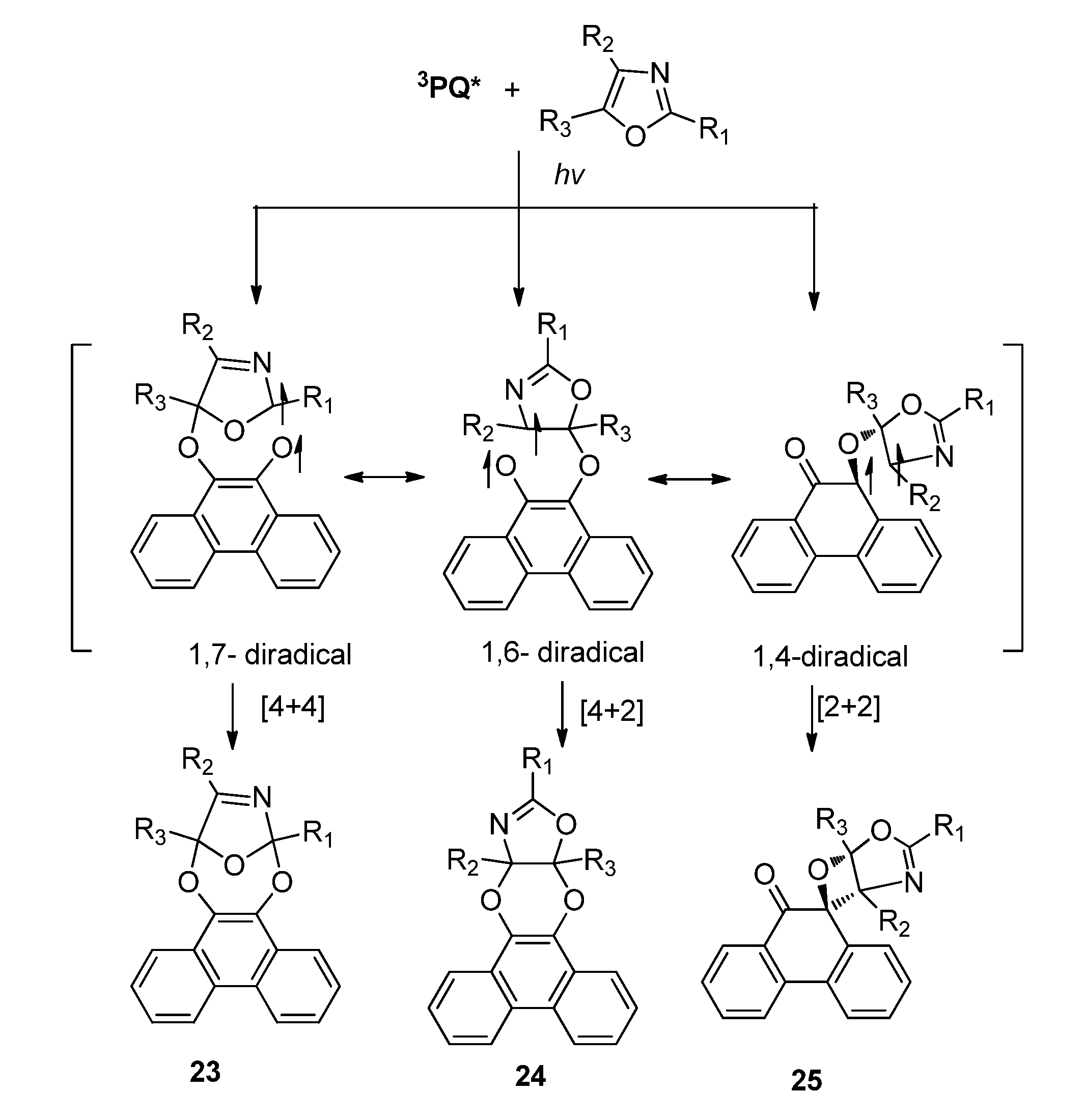
2.2.3. [4 + 4] Photocycloaddition of PQ
2.3. Isoquinoline-1,3,4-trione
2.3.1. Photoreaction with C=C Containing Species
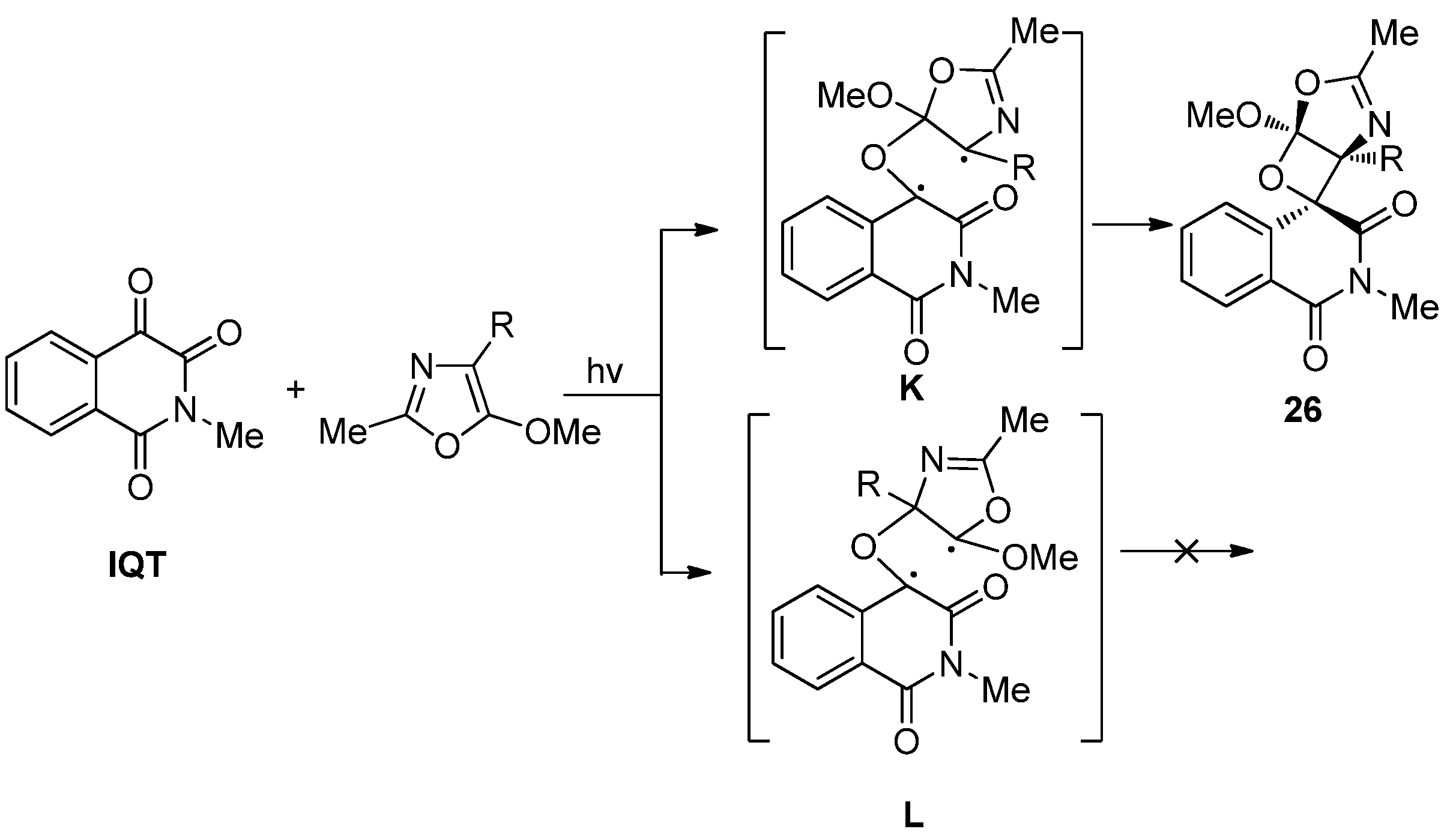
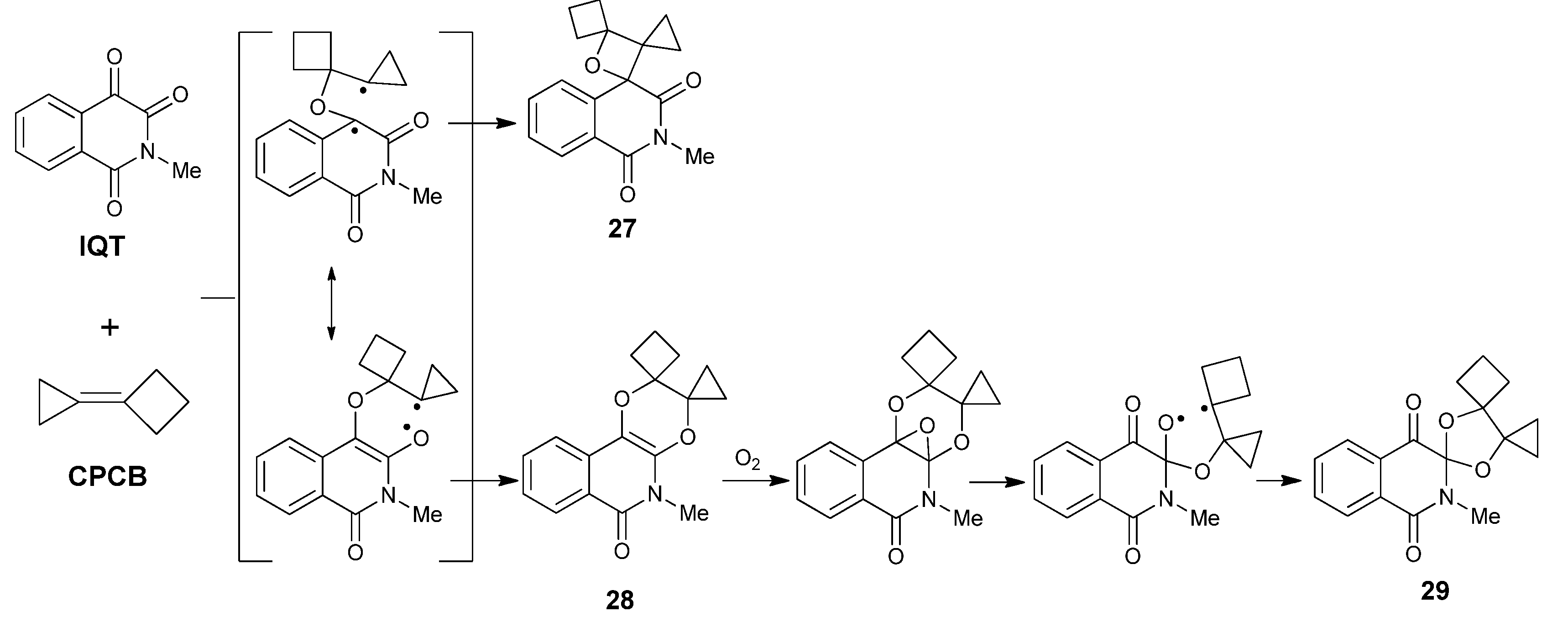
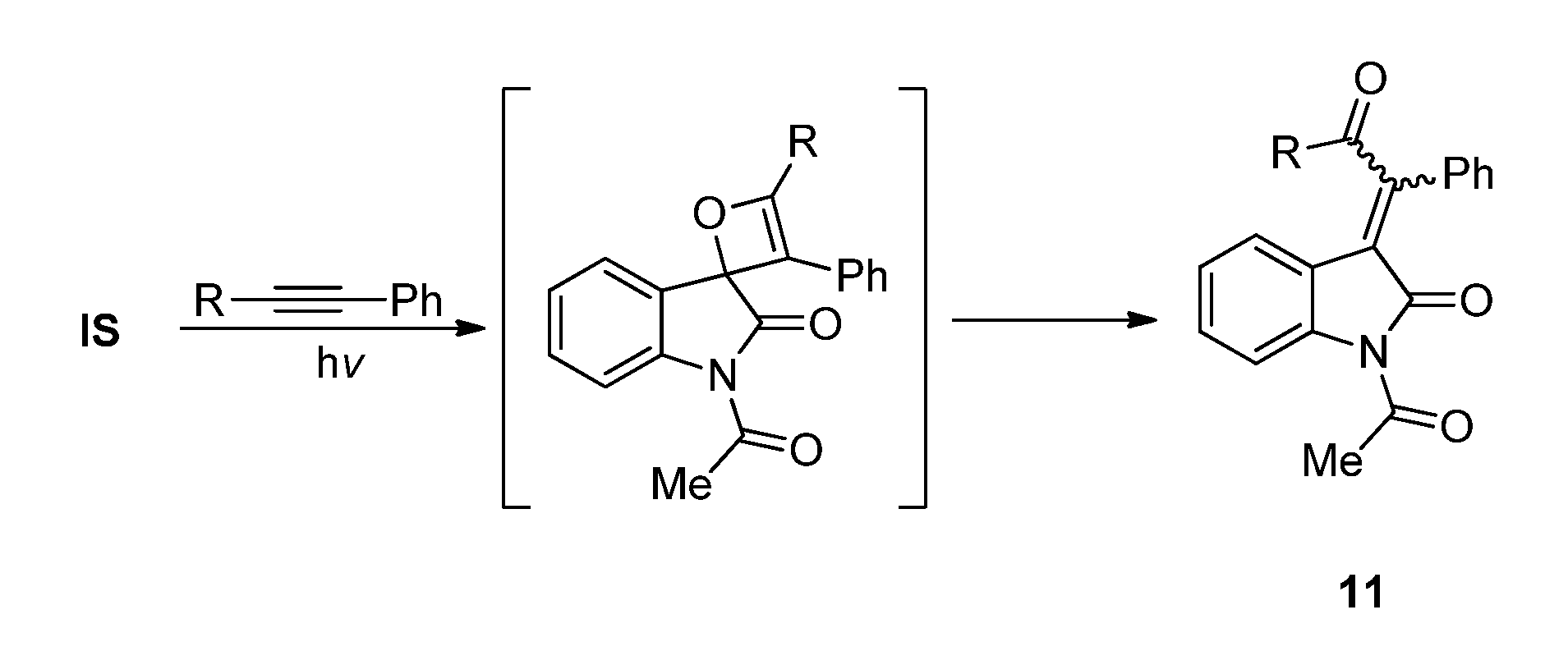
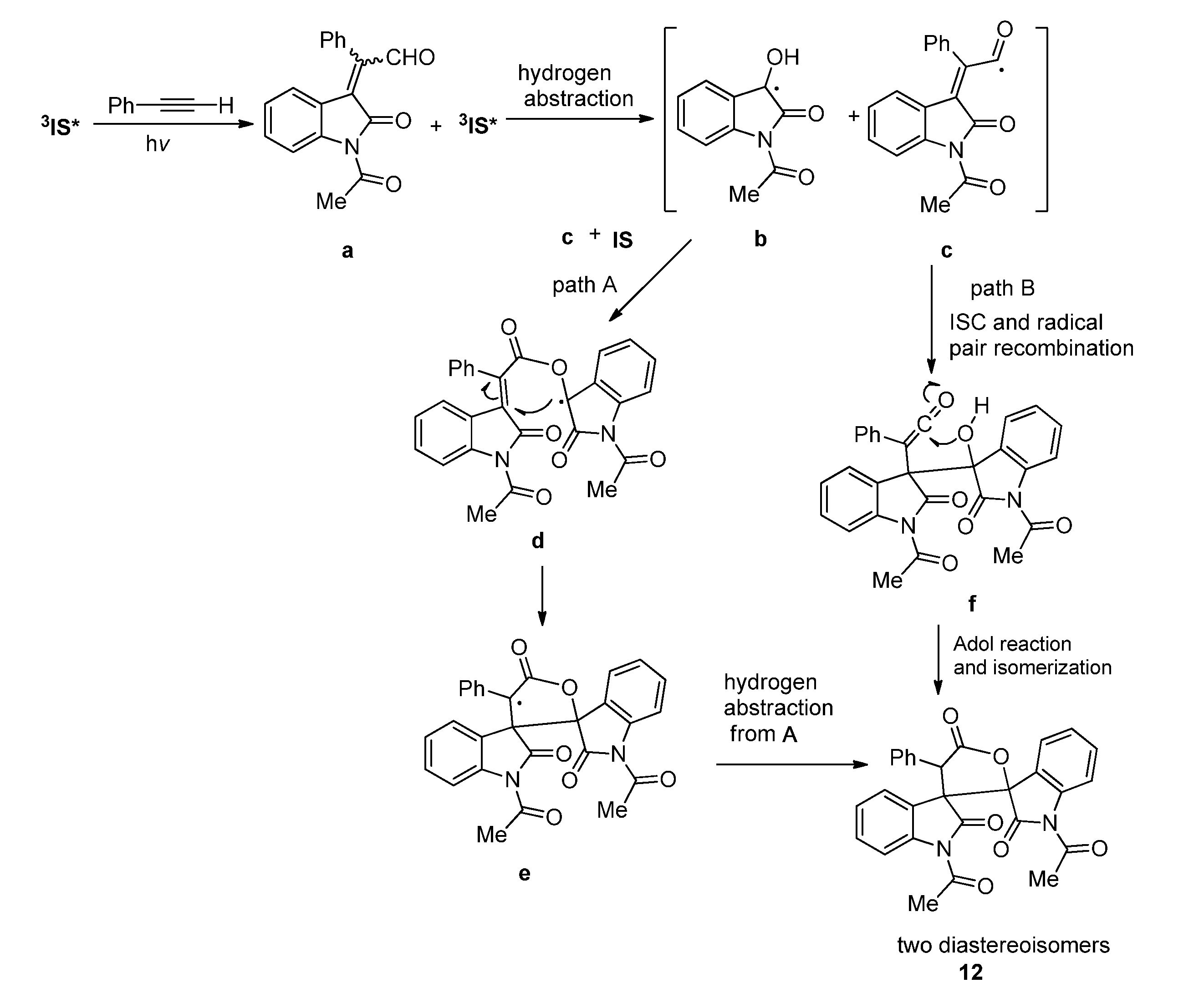
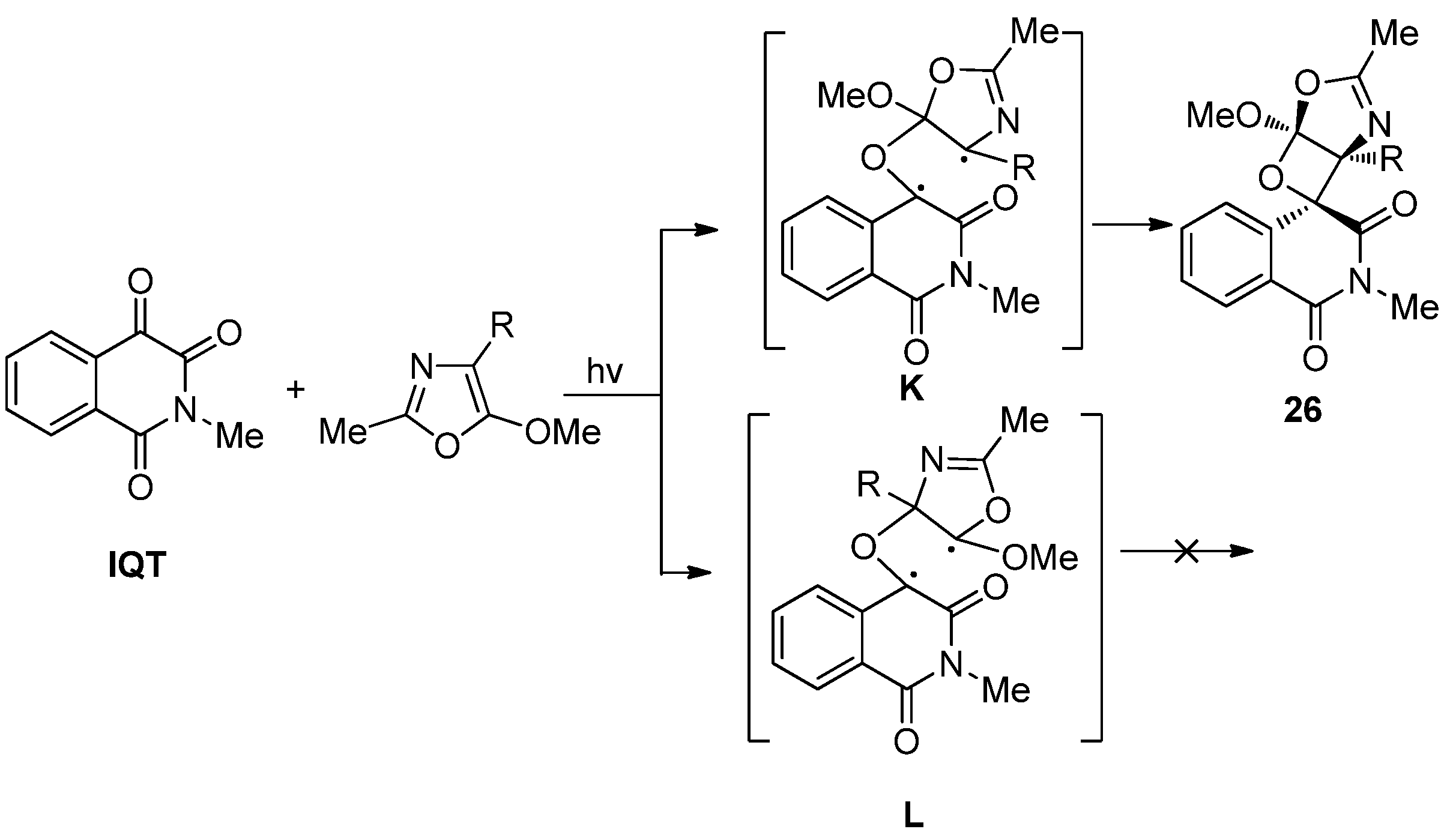
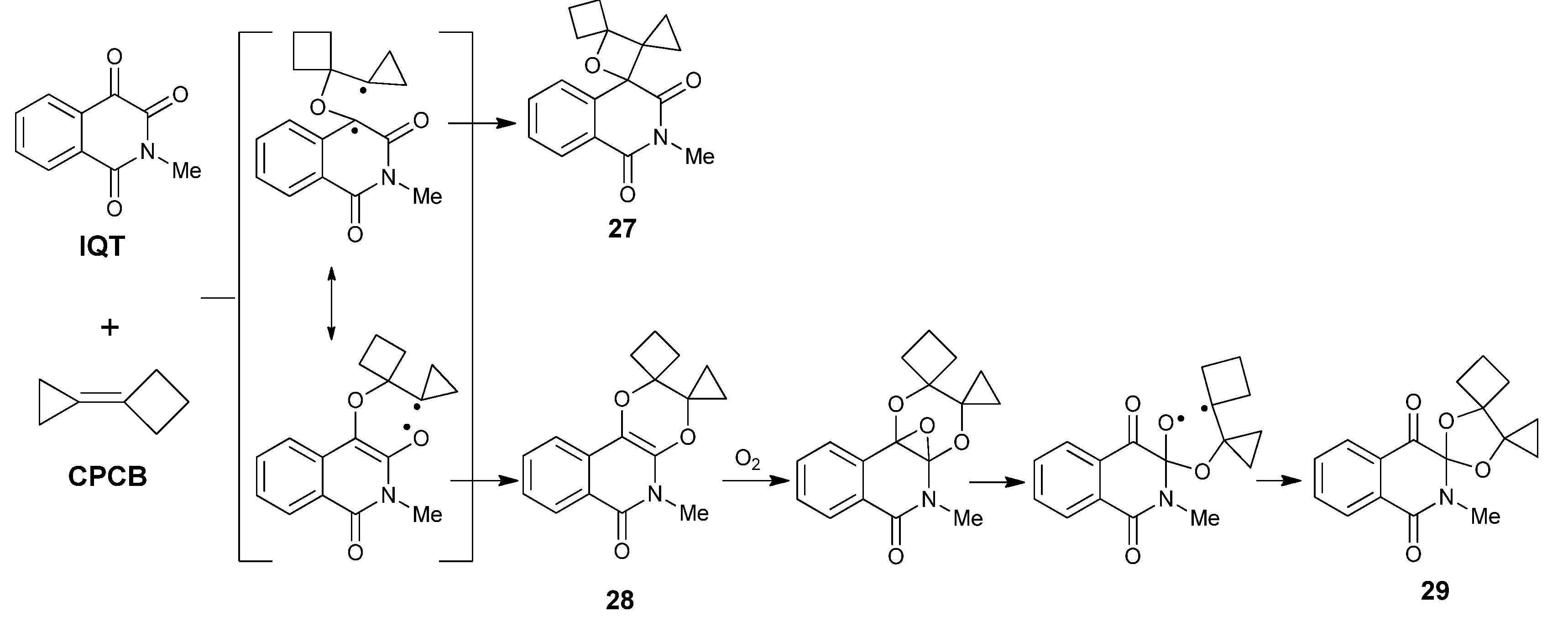
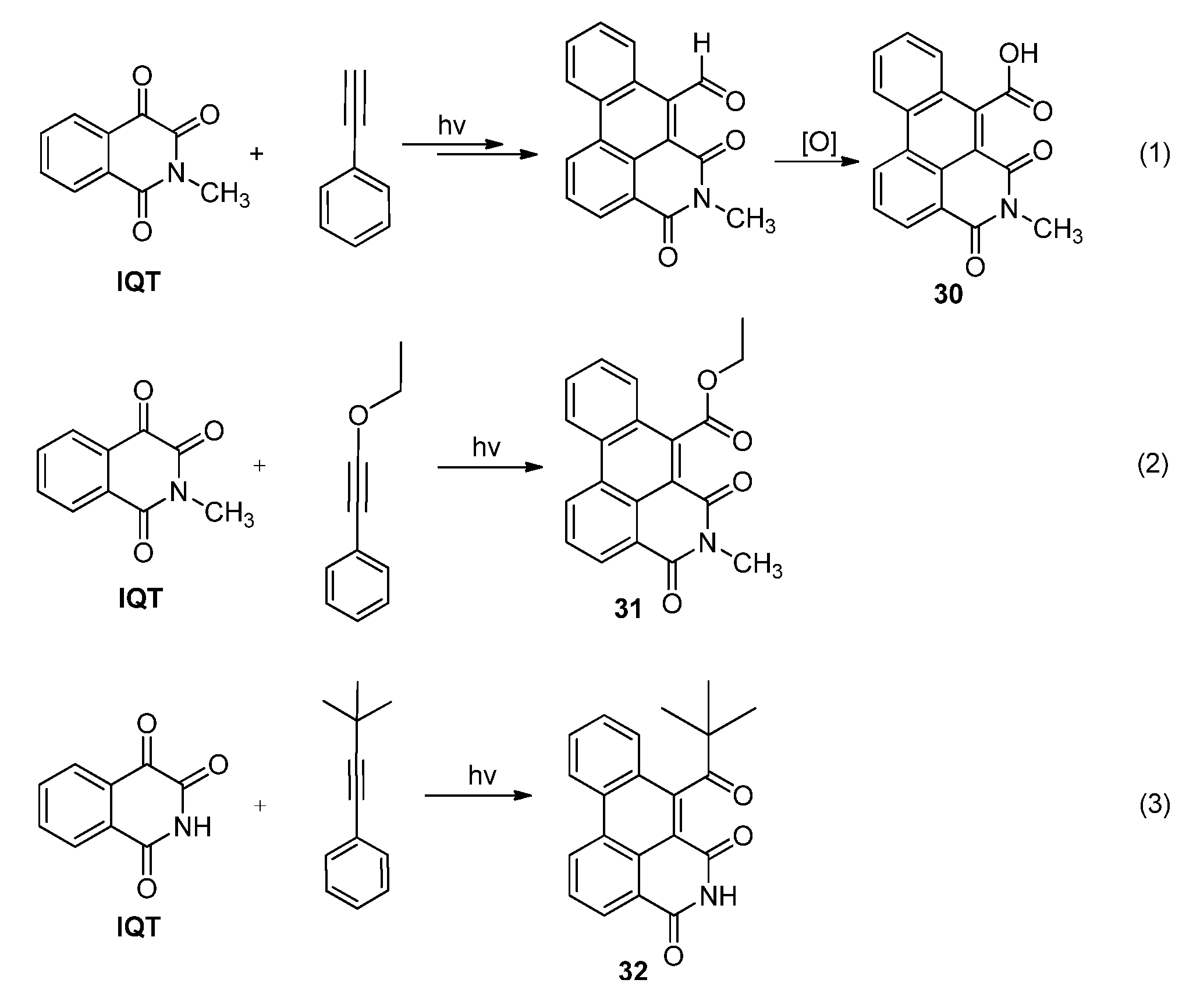
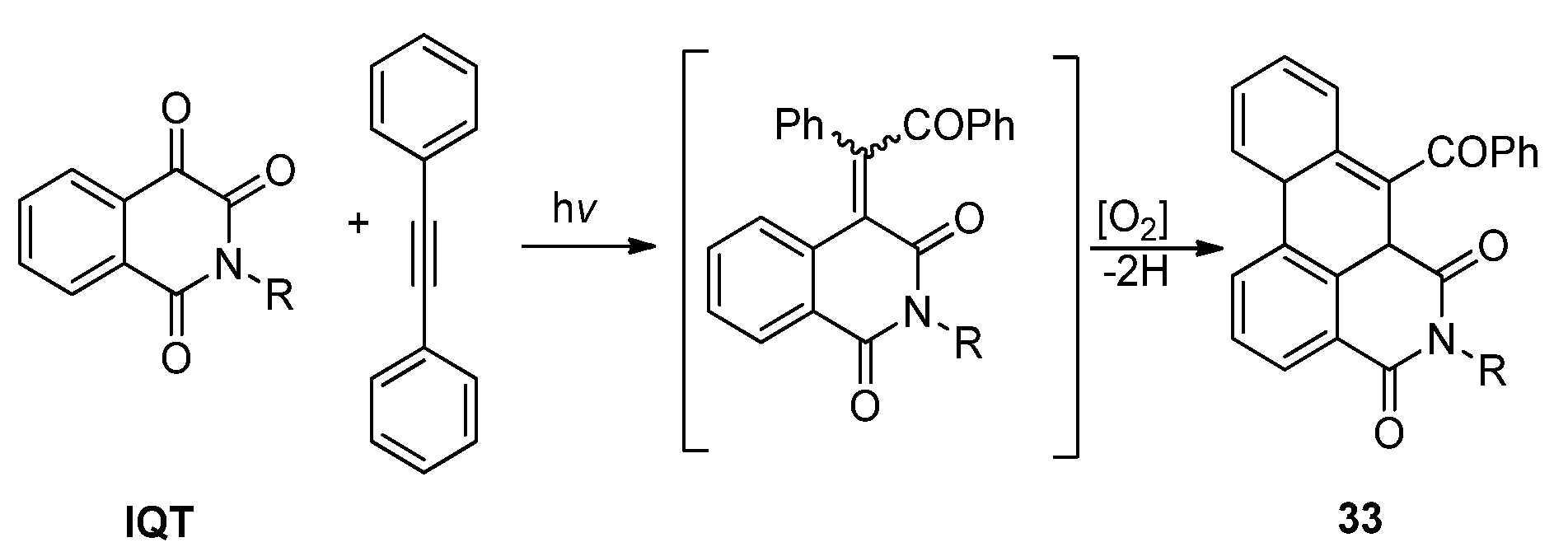
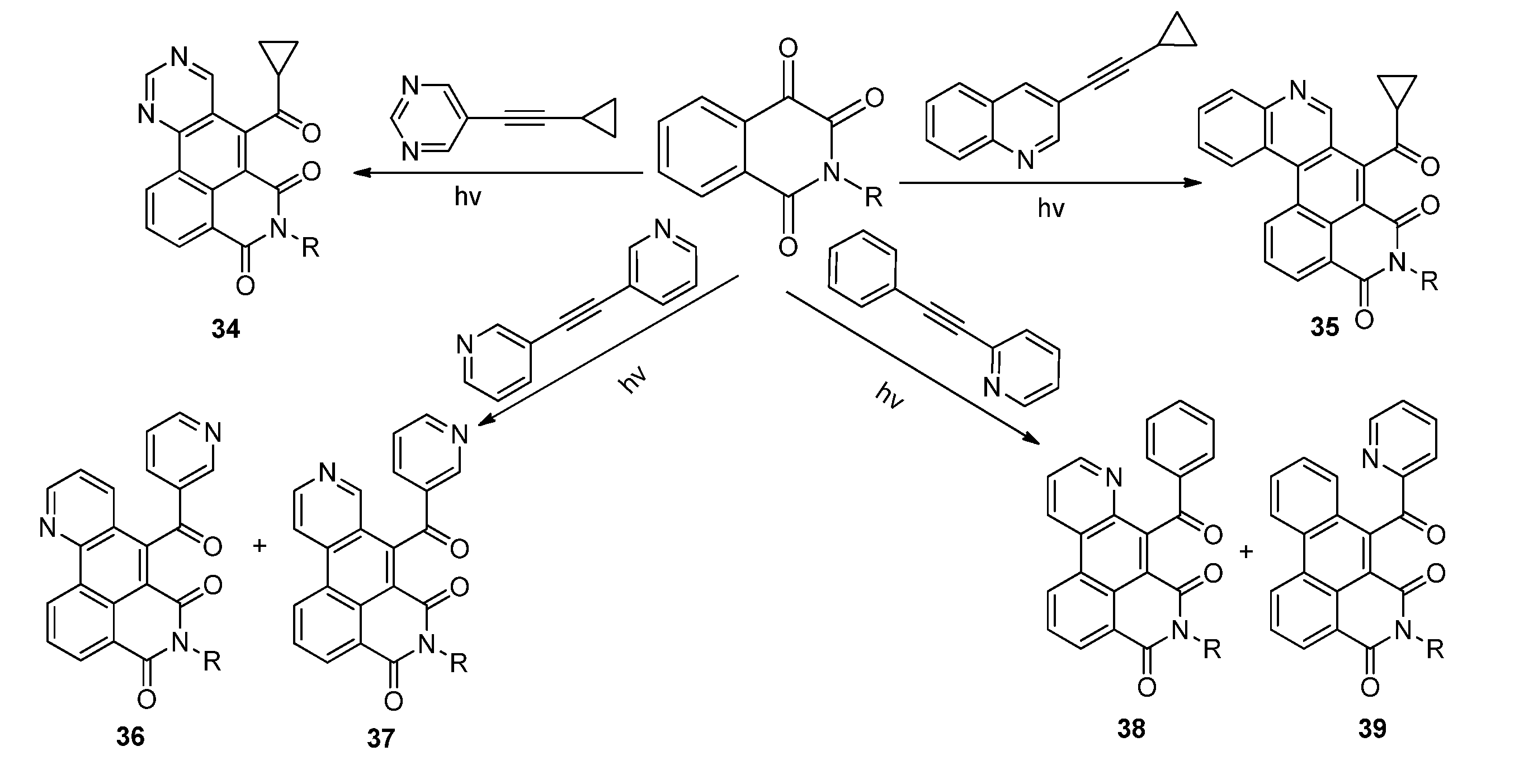
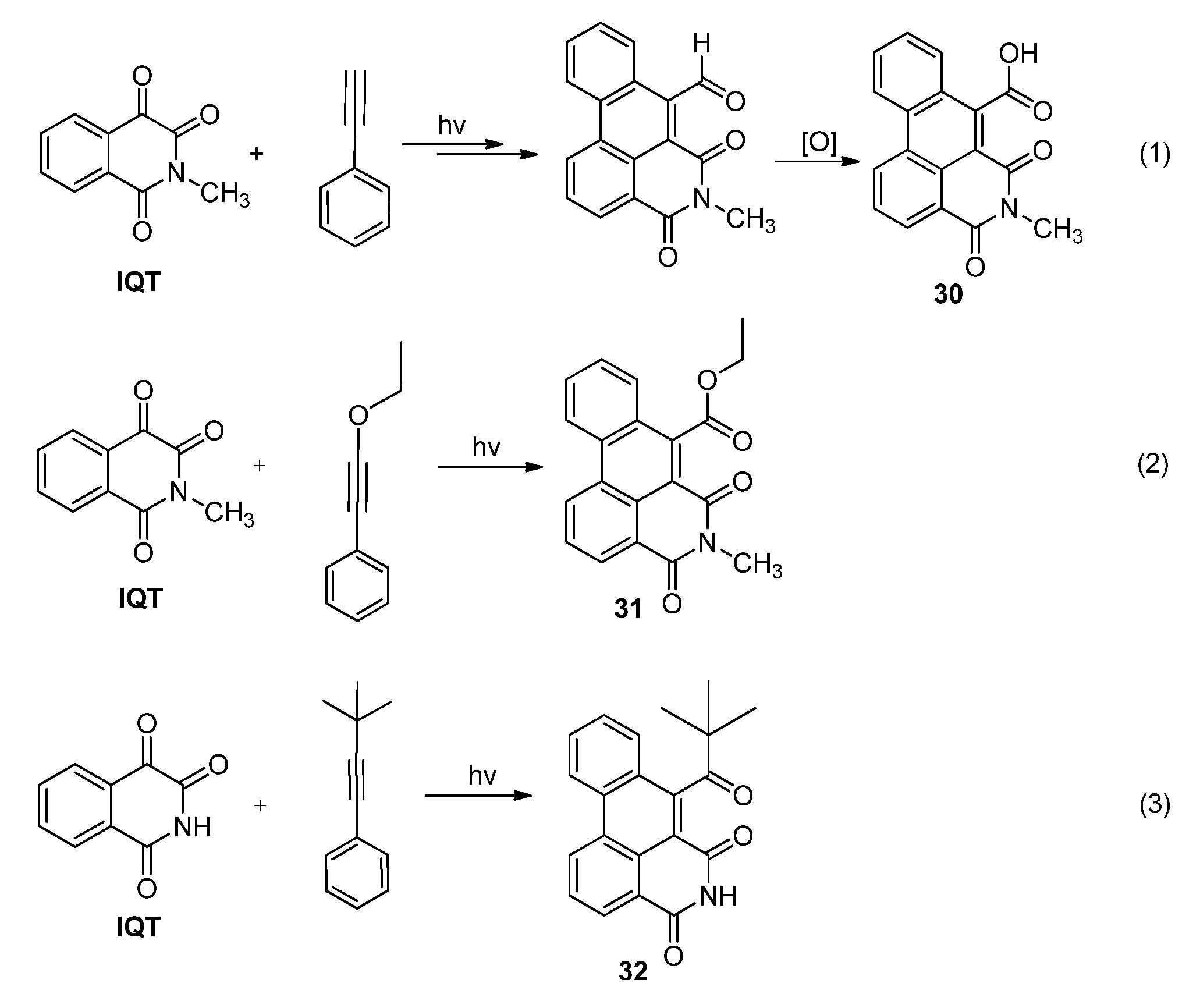
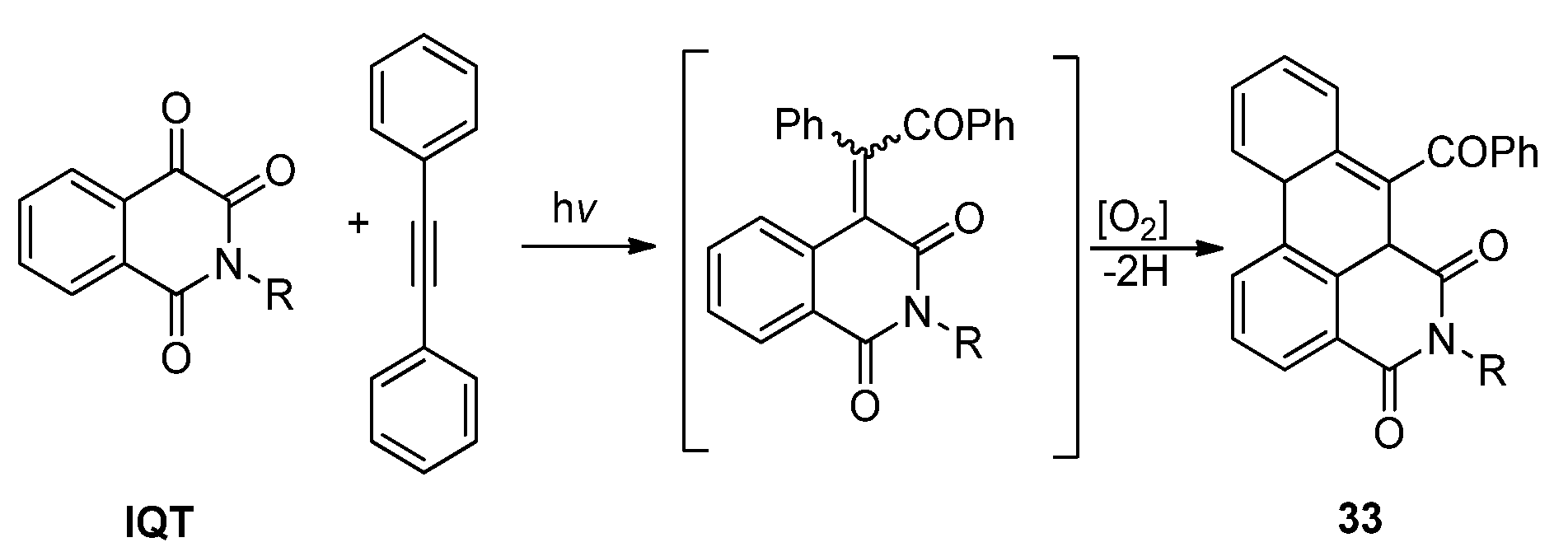
2.3.2. Photoreactions with Acetylenes
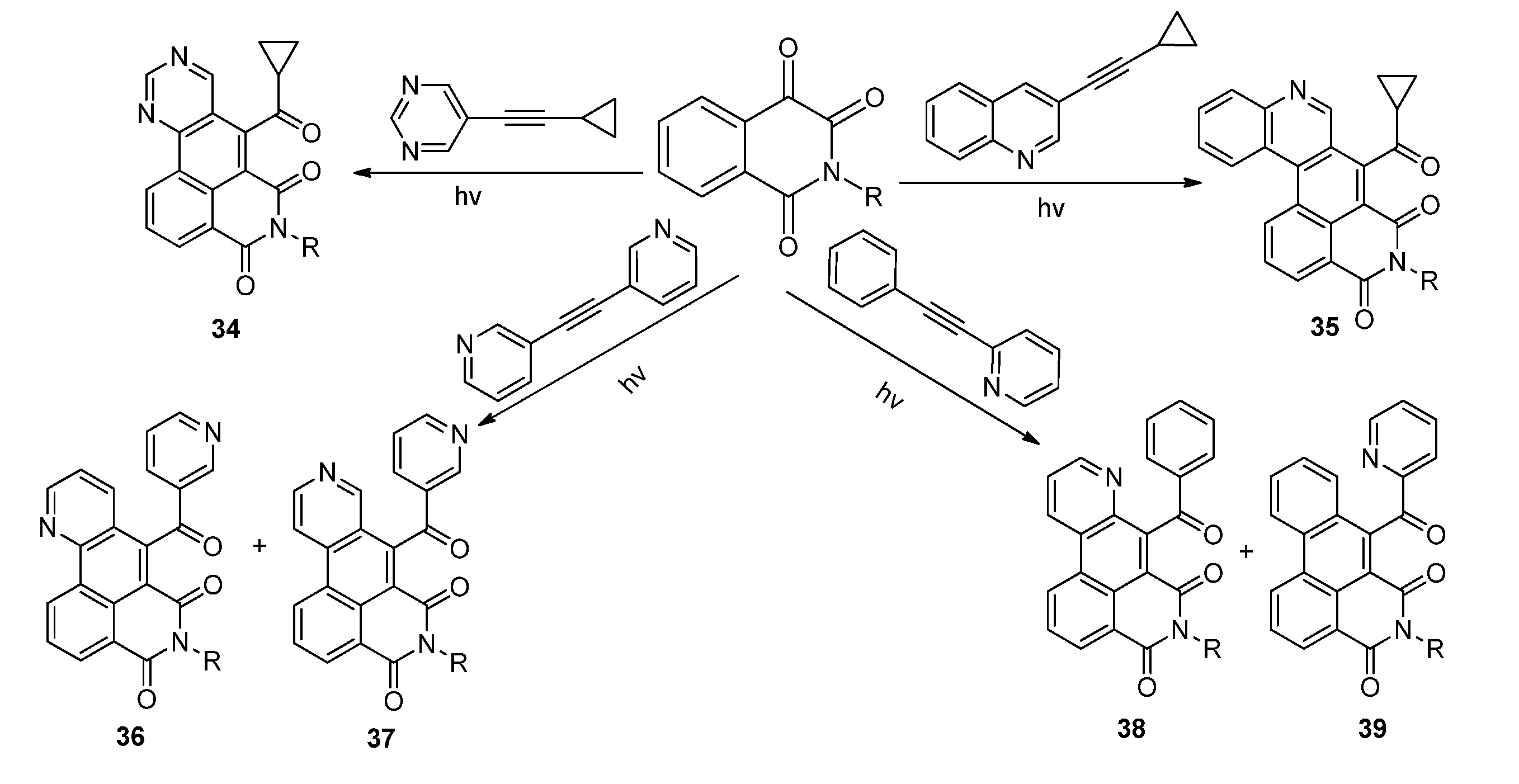
2.4. Photocycloadditions of Other α-Diketones
2.4.1. Cyclic α-Diketones
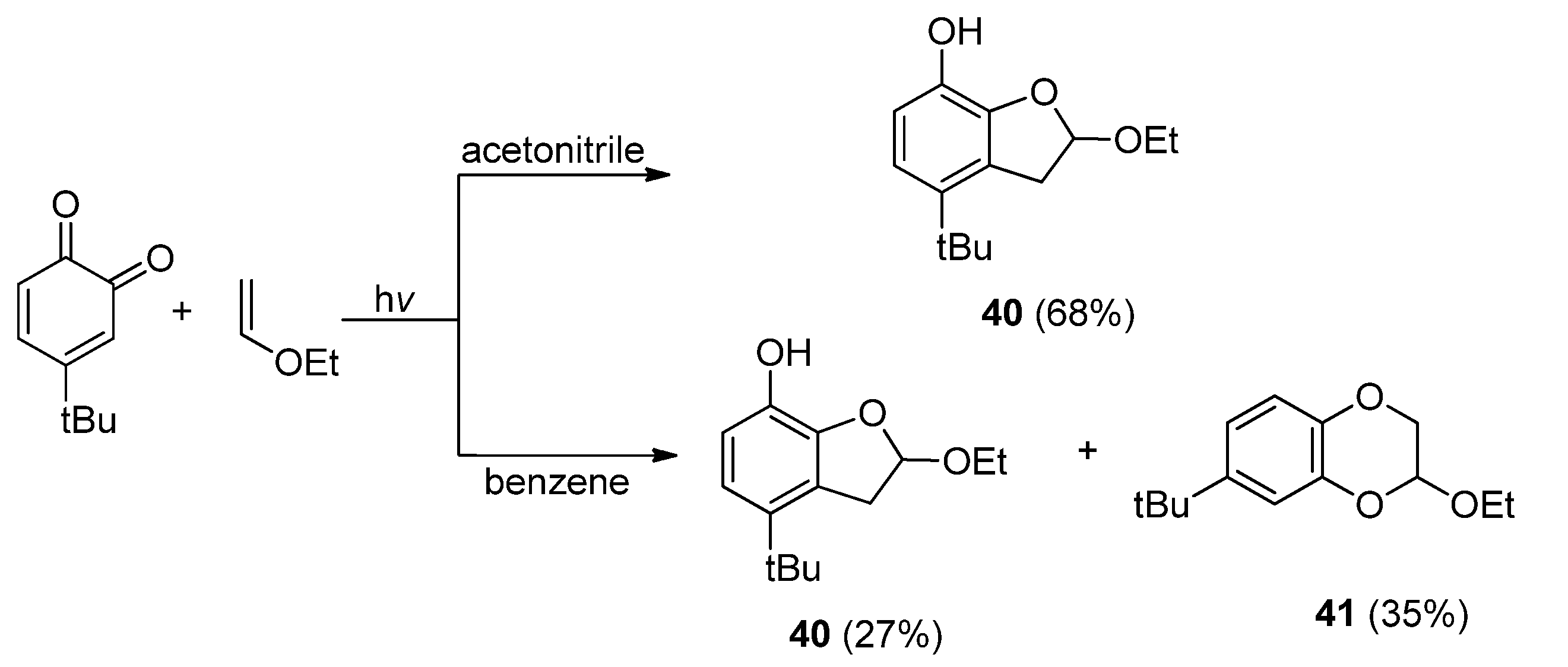
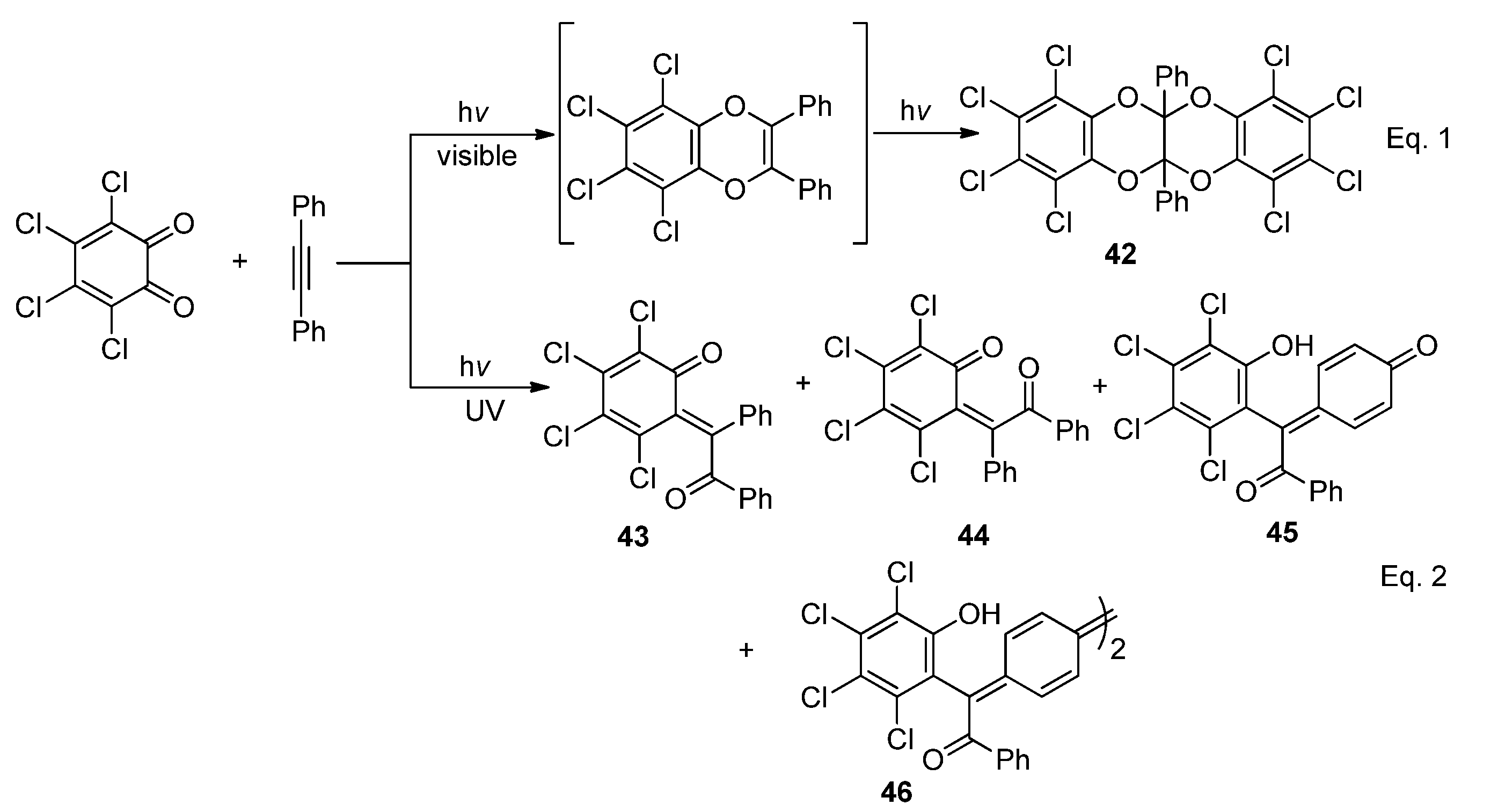
2.4.2. Acyclic α-Diketones
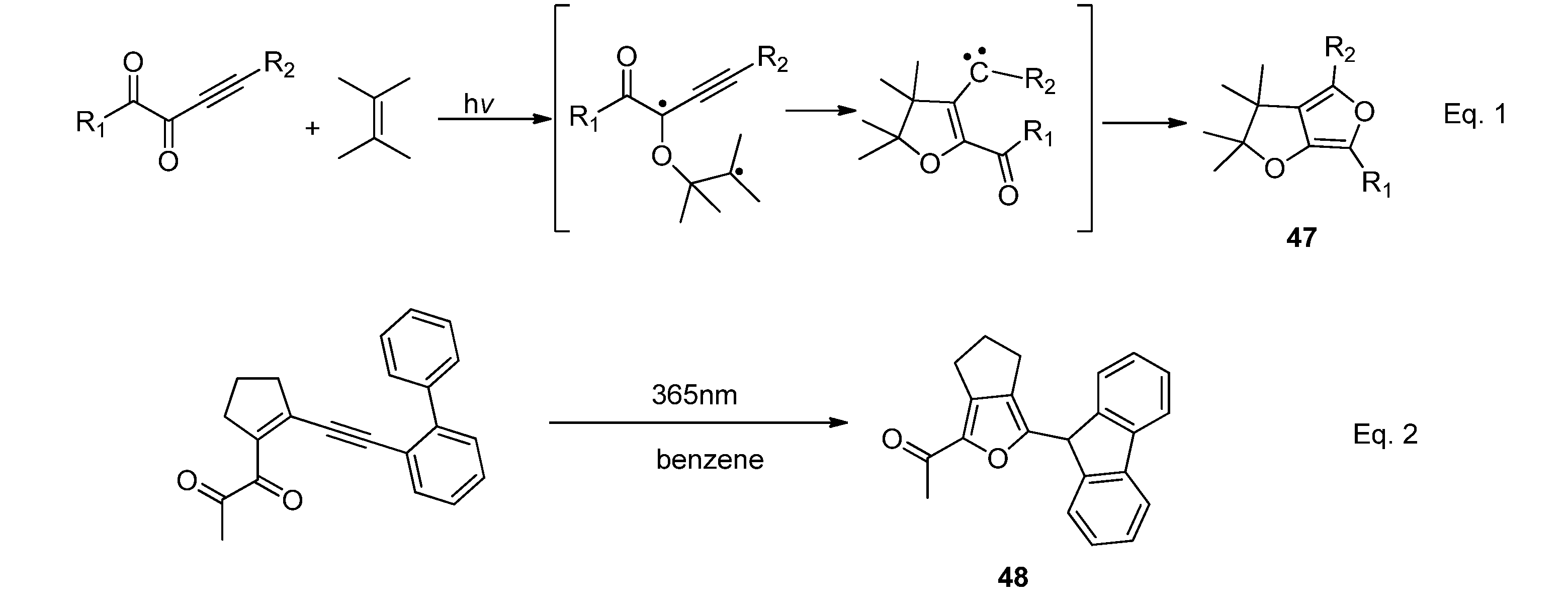
3. Transformations of Photocycloadducts
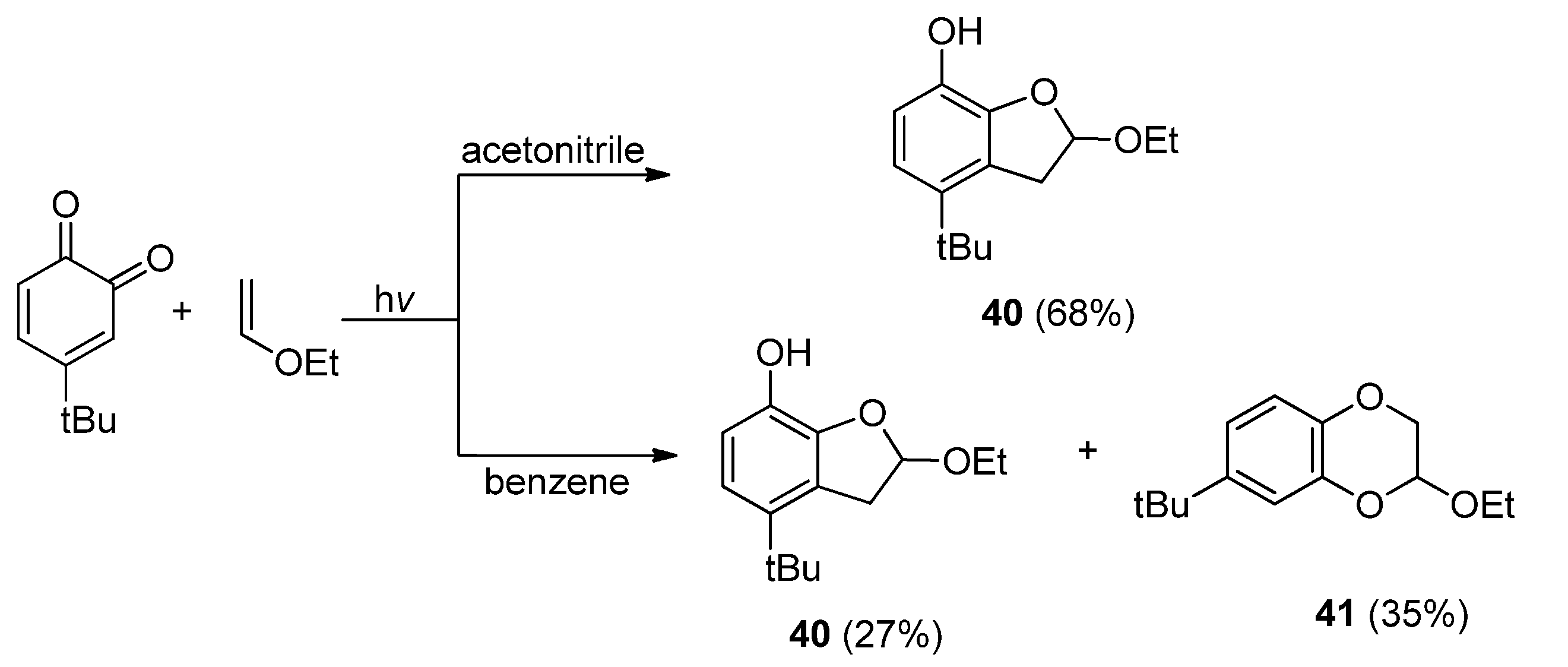
3.1. Transformations of Oxetanes
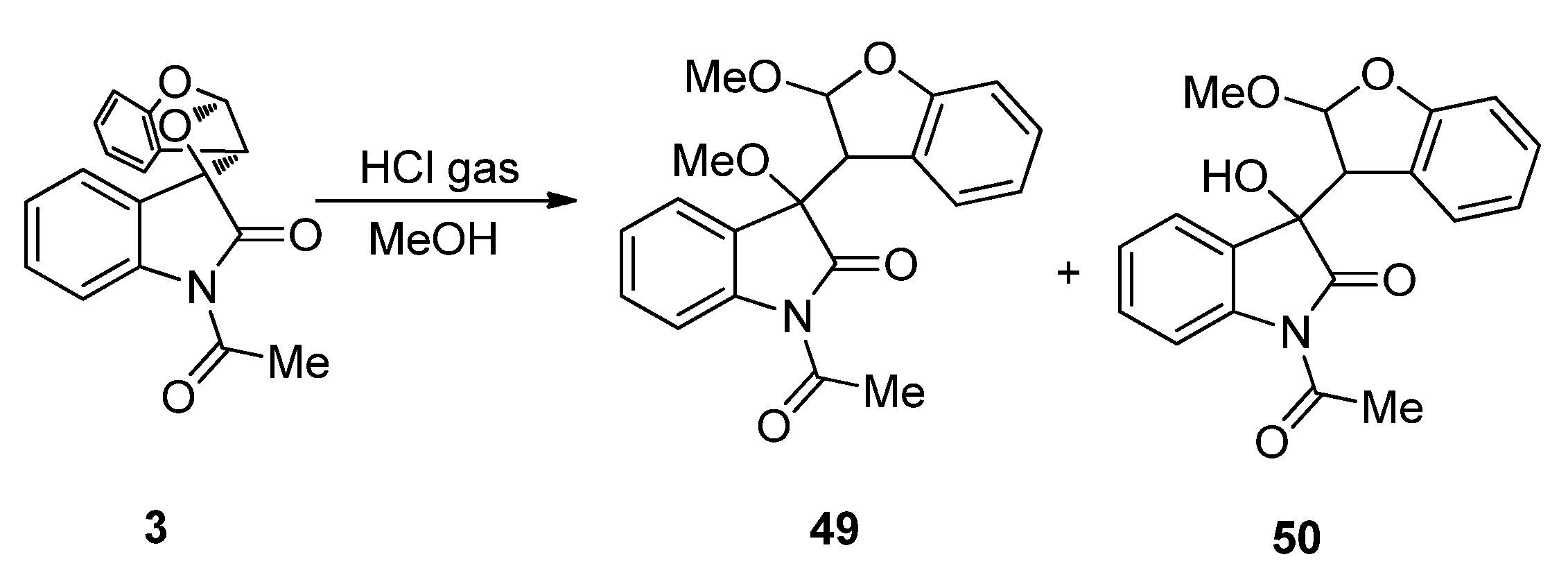
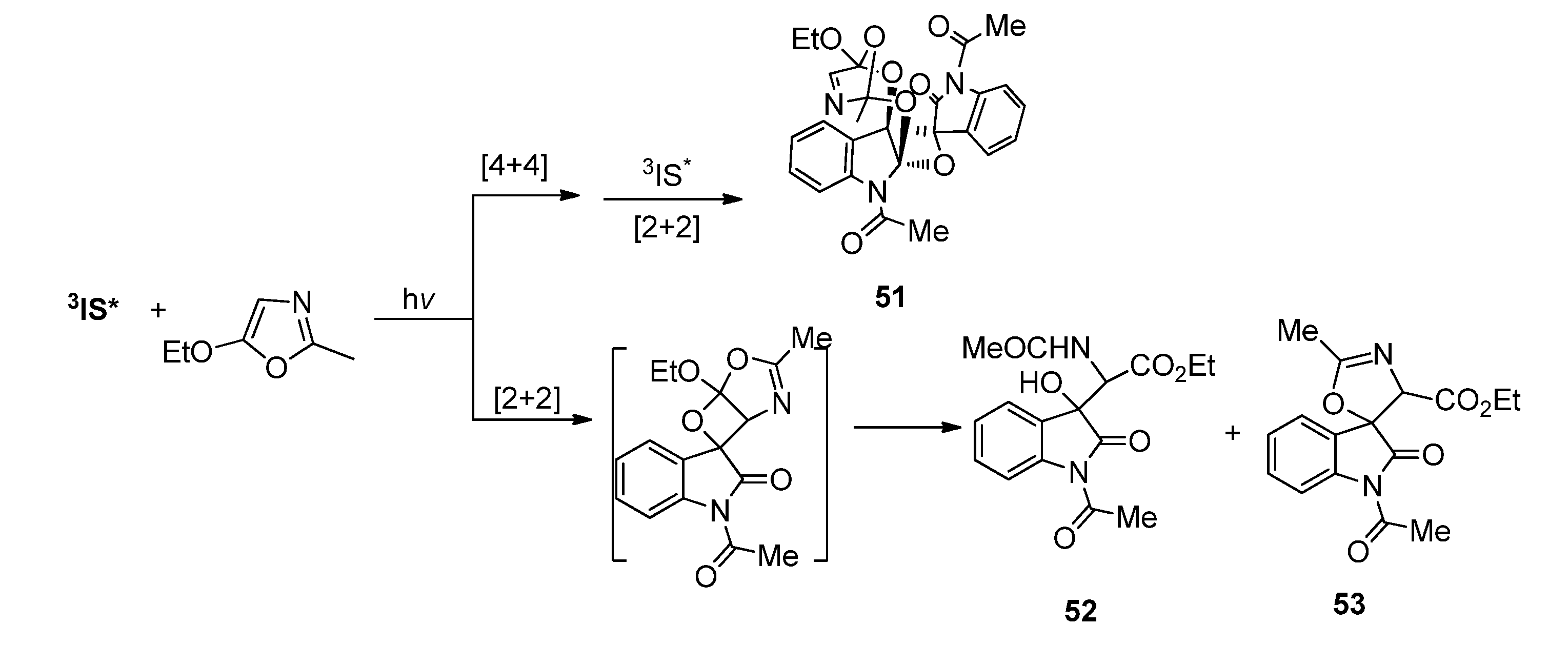
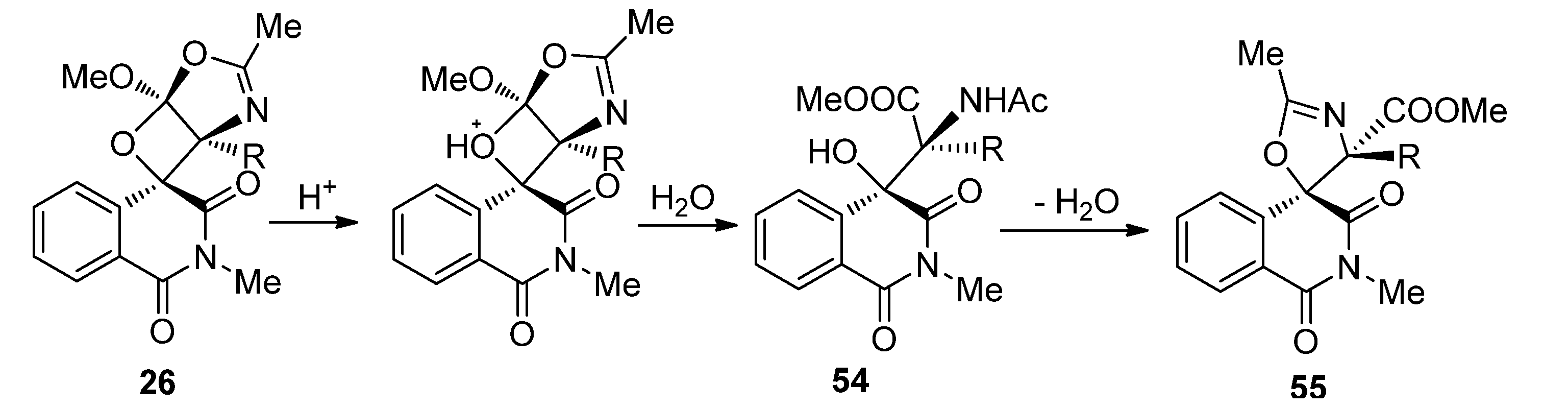
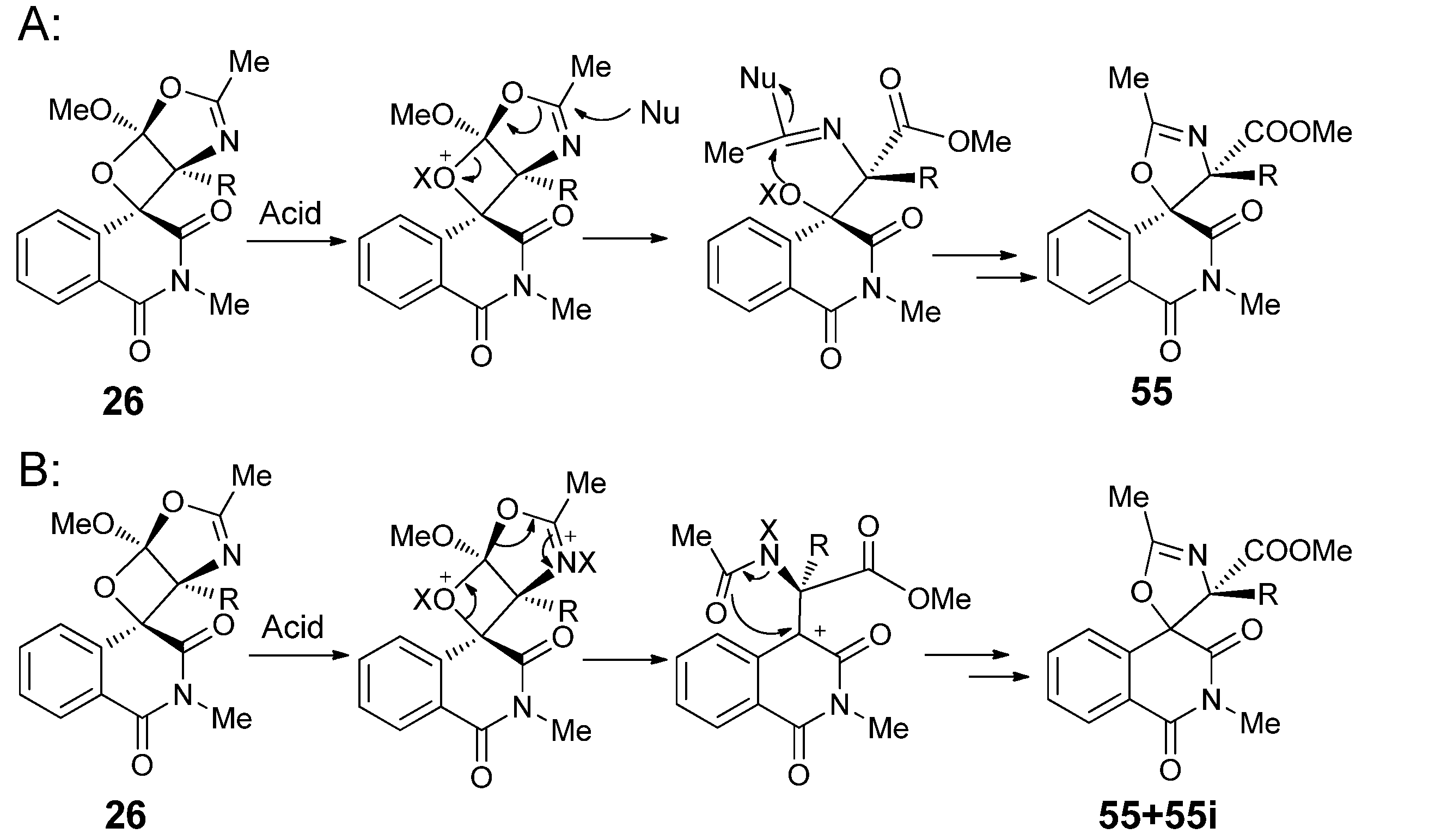
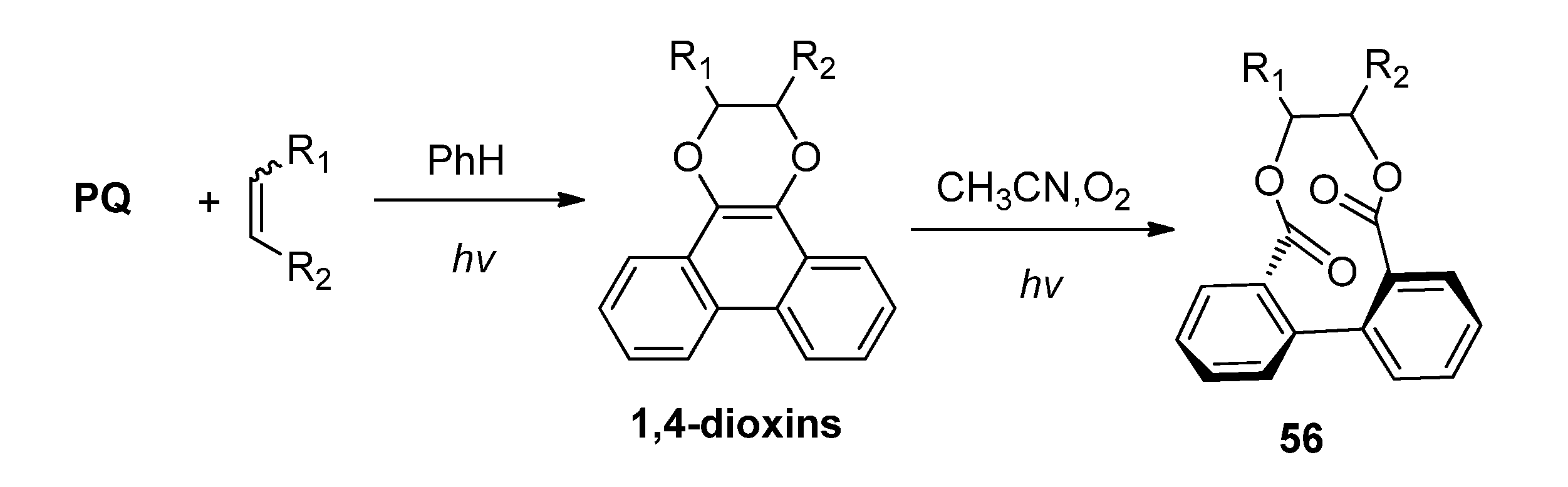
3.2. Transformations of 1,4-Dioxins
4. Conclusions
Acknowledgments
References
- Churruca, F.; Fousteris, M.; Ishikawa, Y.; von Wantoch Rekowski, M.; Hounsou, C.; Surrey, T.; Giannis, A. A Novel Approach to Indoloditerpenes by Nazarov Photocyclization: Synthesis and Biological Investigations of Terpendole E Analogues. Org. Lett. 2010, 12, 2096–2099. [Google Scholar] [CrossRef]
- Fleck, M.; Bach, T. Total Synthesis of Punctaporonin C by a Regio- and Stereoselective [2 + 2]-Photocycloaddition. Chem. Eur. J. 2010, 16, 6015–6032. [Google Scholar]
- Selig, P.; Herdtweck, E.; Bach, T. Total synthesis of meloscine by a [2 + 2]-photocycloaddition/ring-expansion route. Chem. Eur. J. 2009, 15, 3509–3525. [Google Scholar] [CrossRef]
- Morwenna, S.M.P.; David, R.C. Studies toward the photochemical synthesis of functionalized [5]- and [6]carbohelicenes. J. Org. Chem. 2009, 74, 5320–5325. [Google Scholar] [CrossRef]
- Lajkiewicz, N.J.; Roche, S.P.; Gerard, B.; Porco, J.A. Enantioselective photocycloaddition of 3-hydroxyflavones: Total syntheses and absolute configuration assignments of (+)-ponapensin and (+)-elliptifoline. J. Am. Chem. Soc. 2012, 134, 13108–13113. [Google Scholar] [CrossRef]
- Lu, P.; Herdtweck, E.; Bach, T. Intramolecular [2 + 2] photocycloaddition reactions as an entry to the 2-oxatricyclo[4.2.1.04,9]nonan-3-one skeleton of lactiflorin. Chem. Asian J. 2012, 7, 1947–1958. [Google Scholar] [CrossRef]
- Lu, P.; Bach, T. Total Synthesis of (+)-Lactiflorin by an Intramolecular [2 + 2] Photocycloaddition. Angew. Chem. Int. Ed. Engl. 2012, 51, 1261–1264. [Google Scholar] [CrossRef]
- Hehn, J.P.; Herdtweck, E.; Bach, T. A Photocycloaddition/fragmentation approach toward the 3,12-dioxatricyclo[8.2.1.06,13]tridecane skeleton of terpenoid natural products. Org. Lett. 2011, 13, 1892–1895. [Google Scholar] [CrossRef]
- White, J.; Li, Y.; Ihle, D.C. Tandem Intramolecular Photocycloaddition-Retro-Mannich Fragmentation as a Route to Spiro[pyrrolidine-3,3'-oxindoles]. Total synthesis of (±)-coerulescine, (±)-horsfiline, (±)-elacomine, and (±)-6-deoxyelacomine. J. Org. Chem. 2010, 75, 3569–3577. [Google Scholar] [CrossRef]
- Fort, D.A.; Woltering, T.J.; Nettekoven, M.; Knust, H.; Bach, T. Synthesis of Fluorinated Tricyclic Scaffolds by Intramolecular [2 + 2] Photocycloaddition Reactions. Angew. Chem. Int. Ed. Engl. 2012, 51, 10169–10172. [Google Scholar]
- Muller, C.; Bauer, A.; Maturi, M.M.; Cuquerella, M.C.; Miranda, M.A.; Bach, T. Enantioselective intramolecular [2 + 2]-photocycloaddition reactions of 4-substituted quinolones catalyzed by a chiral sensitizer with a hydrogen-bonding Motif. J. Am. Chem. Soc. 2011, 133, 16689–16697. [Google Scholar]
- Pares, S.; de March, P.; Font, J.; Alibes, R.; Figueredo, M. [2 + 2] Photocycloaddition of Symmetrically Disubstituted Alkenes to 2(5H)-Furanones: Diastereoselective Entry to 1,2,3,4-Tetrasubstituted Cyclobutanes. Eur. J. Org. Chem. 2011, 3888–3895. [Google Scholar]
- Mondal, S.; Yadav, R.N.; Ghosh, S. Unprecedented influence of remote substituents on reactivity and stereoselectivity in Cu(I)-catalysed [2 + 2] photocycloaddition. An approach towards the synthesis of tricycloclavulone. Org. Biomol. Chem. 2011, 9, 4903–4913. [Google Scholar] [CrossRef]
- Kulyk, S.; Dougherty, W.G.; Kassel, W.S.; Zdilla, M.J.; Sieburth, S.M. Intramolecular pyridone/enyne photocycloaddition: Partitioning of the [4 + 4] and [2 + 2] pathways. Org. Lett. 2011, 13, 2180–2183. [Google Scholar]
- Yu, W.B.; Han, Y.F.; Lin, Y.J.; Jin, G.X. Construction of tetranuclear macrocycles through C-H activation and structural transformation induced by [2 + 2] photocycloaddition reaction. Chem. Eur. J. 2011, 17, 1863–1871. [Google Scholar] [CrossRef]
- D’Auria, M. CRC Handbook of Organic Photochemistry and Photobiology, 3rd ed.; Griesbeck, A.G., Oelgemoller, M., Ghetti, F., Eds.; CRC Press: Boca Raton, FL, USA, 2012; >Chapter 1; pp. 653–681. [Google Scholar]
- Wu, D.D.; He, M.T.; Liu, Q.D.; Wang, W.; Zhou, J.; Wang, L.; Fun, H.K.; Xu, J.H.; Zhang, Y. Photoinduced reactions of bicycloalkylidenes with isatin and isoquinolinetrione. Org. Biomol. Chem. 2012, 10, 3626–3635. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Hu, H.Y.; Fun, H.K.; Xu, J.H. Photoreactions of 1-acetylisatin with alkynes: Regioselectivity in oxetene formation and easy access to 3-alkylideneoxindoles and dispiro[oxindole[3,2']furan[3',3'']oxindole]s. J. Org. Chem. 2005, 70, 3850–3858. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Zhu, Y.; Xu, J.-H. Mechanism of photoinduced reactions between 1-acetylisatin and aldehydes. Eur. J. Org. Chem. 2004, 2004, 527–534. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Zhang, M.; Fun, H.-K.; Xu, J.-H. Photoinduced [4 + 4] Cycloadditions of o-Quinones with oxazoles. Org. Lett. 2004, 6, 4893–4895. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, J.; Gao, Y.; Fun, H.-K.; Xu, J.-H. Photoinduced [2 + 2] cycloadditions (the Paterno-Büchi reaction) of 1-acetylisatin with enol ethers - regioselectivity, diastereoselectivity and acid catalyzed transformations of the spirooxetane products. J. Chem. Soc. Perkin Trans. 1 2002, 2002, 345–353. [Google Scholar]
- Xue, J.; Zhang, Y.; Wu, T.; Fun, H.-K.; Xu, J.-H. Photoinduced [2 + 2] cycloadditions (the Paterno-Büchi reaction) of 1H-1-acetylindole-2,3-dione with alkenes. J. Chem. Soc. Perkin Trans. 1 2001, 2001, 183–191. [Google Scholar]
- Xue, J.; Zhang, Y.; Wang, X.-L.; Fun, H.-K.; Xu, J.-H. Photoinduced Reactions of 1-Acetylisatin with Phenylacetylenes. Org. Lett. 2000, 2, 2583–2586. [Google Scholar] [CrossRef]
- Wang, L.; Huang, Y.-Z.; Liu, Y.; Fun, H.-K.; Zhang, Y.; Xu, J.-H. Photoinduced [4 + 4], [4 + 2], and [2 + 2] cycloadditions of o-Quinones with oxazoles: Chemo-, regio-, and diastereoselectivity. J. Org. Chem. 2010, 75, 7757–7768. [Google Scholar] [CrossRef]
- Wu, D.D.; Wang, L.; Xu, K.; Song, J.; Fun, H.K.; Xu, J.-H.; Zhang, Y. A facile and highly atom-economic approach to biaryl-containing medium-ring bislactones. Chem. Commun. 2012, 48, 1168–1170. [Google Scholar]
- Yu, H.-T.; Li, J.-B.; Kou, Z.-F.; Du, X.-W.; Wei, Y.; Fun, H.-K.; Xu, J.-H.; Zhang, Y. Photoinduced tandem reactions of isoquinoline-1,3,4-trione with alkynes to build aza-polycycles. J. Org. Chem. 2010, 75, 2989–3001. [Google Scholar] [CrossRef]
- Huang, C.-M.; Yu, H.-T.; Miao, Z.-R.; Zhou, J.; Wang, S.; Fun, H.-K.; Xu, J.-H.; Zhang, Y. Facile synthesis of spiroisoquinolines based on photocycloaddition of isoquinoline-1,3,4-trione with oxazoles. Org. Biomol. Chem. 2011, 9, 3629–3631. [Google Scholar]
- Zhang, Y.; Qian, S.-P.; Fun, H.-K.; Xu, J.-H. Photoinduced reactions of 1,3,4(2H)-isoquinolinetriones with diphenylacetylenes-an efficient one pot syntheses of dibenz[de,g]-(2H)-isoquinoline-4,6-dione derivatives. Tetrahedron Lett. 2000, 41, 8141–8145. [Google Scholar]
- Bieck, P.R.; Antomin, K.H.; Schulz, R. Monoamine Oxidase; Yasuhara, H., Ed.; VSP: Utrecht, The Netherlands, 1993; pp. 177–196. [Google Scholar]
- Popp, F.D. Chemistry of isatin. Adv. Heterocycl. Chem. 1975, 18, 1–58. [Google Scholar] [CrossRef]
- Greisbeck, A.G.; Stadtmueller, S. Photocycloaddition of benzaldehyde to cyclic olefins: Electronic control of endo stereoselectivity. J. Am. Chem. Soc. 1990, 112, 1281–1283. [Google Scholar] [CrossRef]
- Salem, L.; Rowland, C. The Electronic Properties of Diradicals. Angew. Chem. Int. Ed. Engl. 1972, 11, 92–111. [Google Scholar] [CrossRef]
- Salem, L. Diradicals. Pure Appl. Chem. 1973, 33, 317–328. [Google Scholar] [CrossRef]
- Shaik, S.S.; Epiotis, N.D. Spin inversion in triplet Diels-Alder reactions. J. Am. Chem. Soc. 1980, 102, 122–131. [Google Scholar] [CrossRef]
- Doubleday, C., Jr.; Turro, N.J.; Wang, J.F. Dynamics of flexible triplet biradicals. Acc. Chem. Res. 1989, 22, 199–205. [Google Scholar] [CrossRef]
- Adam, W.; Grabowski, S.; Wilson, R.M. Localized cyclic triplet diradicals. Lifetime determination by trapping with oxygen. Acc. Chem. Res. 1990, 23, 165–172. [Google Scholar] [CrossRef]
- Turro, N.J.; Lee, C.; Schore, N.; Barltrop, J.; Carless, H.A.J. Molecular photochemistry. XLV. Structure-reactivity correlations for the quenching of acetone fluorescence by enol ethers and by conjugated unsaturated nitriles. J. Am. Chem. Soc. 1971, 93, 3079–3080. [Google Scholar] [CrossRef]
- De Meijere, A.; Nüske, H.; Es-Sayed, M.; Labahn, T.; Schroen, M.; Bräse, S. Cyclopropyl building blocks in organic synthesis. Part 54. Nitrogen-based linkers. Part 6. New efficient multicomponent reactions with C-C coupling for combinatorial application in liquid and on solid phase. Angew. Chem. Int. Ed. Engl. 1999, 38, 3669–3672. [Google Scholar] [CrossRef]
- De Meijere, A.; von Seebach, M.; Kozhushkov, S.I.; Boese, R.; Bläser, D.; Cicchi, S.; Dimoulas, T.; Brandi, A. Cyclopropyl building blocks for organic synthesis, 72. Cyclobutylidenecyclopropane: New synthesis and use in 1,3-dipolar cycloadditions—A direct route to spirocyclopropane-annulated azepinone derivatives. Eur. J. Org. Chem. 2001, 3789–3795. [Google Scholar]
- Nakamura, I.; Saito, S.; Yamamoto, Y. Hydrofurylation of Alkylidenecyclopropanes Catalyzed by Palladium. J. Am. Chem. Soc. 2000, 122, 2661–2662. [Google Scholar] [CrossRef]
- Rele, S.; Chattopadhyay, S.; Nayak, S.K. Highly active salted low-valent titanium reagents for various SET induced reactions. Tetrahedron Lett. 2001, 42, 9093–9095. [Google Scholar] [CrossRef]
- Zimmerman, H.E.; Craft, L. Photochemical reaction of benzoquinone with tolan. Tetrahedron Lett. 1964, 1964, 2131–2136. [Google Scholar] [CrossRef]
- Bryce-Smith, D.; Fray, G.I.; Gilbert, A. Photo adduct of p-benzoqninone and diphenylacetylene. Tetrahedron Lett. 1964, 1964, 2137–2139. [Google Scholar] [CrossRef]
- Bryce-Smith, D.; Gilbert, A.; Johnson, M.G. Photoadditions of diphenylacetylene, cycloocta-1,3-diene, and cyclooctene to anthraquinone. Tetrahedron Lett. 1968, 1968, 2863–2866. [Google Scholar] [CrossRef]
- Farid, S.; Kothe, W.; Pfundt, G. Competitive photoadditions of acetylenes to the C:C and C:O bonds of p-quinones. Tetrahedron Lett. 1968, 1968, 4147–4150. [Google Scholar] [CrossRef]
- Barltrop, J.A.; Hesp, B. Organic photochemistry. Part V. The illumination of some quinones in the presence of conjugated dienes and other olefinic systems. J. Chem. Soc. C. 1967, 1967, 1625–1635. [Google Scholar] [CrossRef]
- Pappas, S.P.; Portnoy, N.A. Substituent effects on the photoaddition of diphenylacetylene to 1,4-naphthoquinones. J. Org. Chem. 1968, 33, 2200–2203. [Google Scholar] [CrossRef]
- Bos, H.J.T.; Polman, H.; van Montfort, P.F.E. Photoaddition of phenanthraquinone to alkyl-substituted alkoxyacetylenes. Formation of 1,3-dioxoles. J. Chem. Soc. Chem. Commun. 1973, 1973, 188. [Google Scholar]
- Kim, A.R.; Mah, Y.J.; Shim, S.C.; Kim, S.S. Photochemical formation of isomeric quinone methides from o-quinones and one-way isomerization. J. Photosci. 1997, 4, 49–52. [Google Scholar]
- Bosch, E.; Hubig, S.M.; Kochi, J.K. Paterno-Büchi Coupling of (Diaryl)acetylenes and Quinone via Photoinduced Electron Transfer. J. Am. Chem. Soc. 1998, 120, 386–395. [Google Scholar] [CrossRef]
- Noh, T.; Jeong, Y.; Kim, D. Low-temperature irradiation of 1-substituted naphthalenes. J. Chem. Soc. Perkin Trans. 1 1998, 1998, 2501–2504. [Google Scholar]
- Kohmoto, S.; Kobayashi, T.; Minami, J.; Ying, X.; Yamaguchi, K.; Karatsu, T.; Kishikawa, K.; Yamamoto, M. Trapping of 1,8-Biradical Intermediates by Molecular Oxygen in Photocycloaddition of Naphthyl-N-(naphthylcarbonyl)carboxamides; Formation of Novel 1,8-Epidioxides and Evidence of Stepwise Aromatic Cycloaddition. J.Org. Chem. 2001, 2001, 66–73. [Google Scholar]
- Bouas-Laurent, H.; Castellan, A.; Desvergne, J.-P.; Lapouyade, R. Photodimerization of anthracenes in fluid solution: Structural aspects. Chem. Soc. Rev. 2000, 29, 43–55. [Google Scholar]
- Mak, K.T.; Srinivasachar, K.; Yang, N.C. Syntheses of cycloadducts of benzene and naphthalene. J. Chem. Soc. Chem. Commun. 1979, 1979, 1038–1040. [Google Scholar]
- Albini, A.; Fasani, E.; Giavarini, F. Photochemical reaction between naphthalenecarbonitriles and dienes. J. Org. Chem. 1988, 53, 5601–5607. [Google Scholar] [CrossRef]
- Kimura, M.; Kura, H.; Nukada, K.; Okamoto, H.; Satake, K.; Morosawa, S. Synthesis and photochemistry of 3,6-difluoro-10,11-benzopentacyclo [6,4,0.0,0.0]dodeca-4,10-diene. J. Chem. Soc. Perkin Trans. 1. 1988, 1988, 3307–3310. [Google Scholar]
- McSkimming, G.; Tucker, J.H.R.; Bouas-Laurent, H.; Desvergne, J.-P. An anthracene-based photochromic system that responds to two chemical inputs. Angew. Chem. Int. Ed. Engl. 2000, 39, 2167–2169. [Google Scholar] [CrossRef]
- Takeshita, H.; Mori, A.; Funakura, M.; Mametsuka, H.B. Synthetic photochemistry. VII. The addition reaction of acenaphthenequinone and 1,2-naphthoquinone to cycloheptatriene. Bull. Chem.Soc. Jpn. 1977, 50, 315–316. [Google Scholar] [CrossRef]
- Maruyama, K.; Muraok, M.; Naruta, Y. Keto oxetanes produced from photocycloaddition of o-quinone and their thermolysis. Reaction of 9,10-phenanthrenequinone with internally highly strained cyclic olefins. J. Org. Chem. 1981, 46, 983–989. [Google Scholar] [CrossRef]
- Maruyama, K.; Iwai, T.; Naruta, Y. Products and modes of photocycloaddition of 9,10-phenanthraquinone with cyclic olefins. Chem. Lett. 1975, 1975, 1219–1222. [Google Scholar] [CrossRef]
- Sasaki, T.; Kanematsu, K.; Ando, I.; Yamashita, O. Molecular design by cycloaddition reactions. 30. Photochemical cycloadditions of quadricyclane to aromatic hydrocarbons and o-quinones. First example of photochemical pericyclic [4π + 2σ + 2σ] addition. J. Am. Chem. Soc. 1977, 99, 871–877. [Google Scholar] [CrossRef]
- Chow, Y.-L.; Joseph, T.C.; Quon, H.H.; Tam, J.N.S. Photoaddition of α-diketones to olefins. Stereospecificity of the addition reaction. Can. J. Chem. 1970, 48, 3045–3052. [Google Scholar] [CrossRef]
- Helferich, B.; Mulcahy, E.N.; Ziegler, H. The addition of phenanthrenequinone to D-glucal. II. Chem. Ber. 1954, 87, 233–237. [Google Scholar] [CrossRef]
- Lichtenthaler, F.W.; Weimer, T.; Immel, S. [4 + 2] and [2 + 2] Photocycloadditions of 1,2-diketones to glycal and hydroxyglycal esters. Tetrahedron Asymmetry. 2004, 15, 2703–2709. [Google Scholar] [CrossRef]
- Ho, J.-H.; Ho, T.-I.; Chen, T.-H.; Chow, Y.L. Efficientphotocycloadditionof phenanthroquinones with simple olefins. J. Photochem. Photobiol. A Chem. 2001, 138, 111–122. [Google Scholar] [CrossRef]
- Du, J.Q.; Wu, J.; Zhang, H.J.; Zhang, Y.H.; Qiu, B.Y.; Wu, F.; Chen, Y.H.; Li, J.Y.; Nan, F.J.; Ding, J.P.; et al. Isoquinoline-1,3,4-trione derivatives inactivate caspase-3 by generation of reactive oxygen species. J. Biol. Chem. 2008, 283, 30205–30215. [Google Scholar] [CrossRef]
- Chen, Y.H.; Zhang, Y.H.; Zhang, H.J.; Liu, D.Z.; Gu, M.; Li, J.Y.; Wu, F.; Zhu, X.Z.; Li, J.; Nan, F.J. Design, synthesis, and biological evaluation of isoquinoline-1,3,4-trione derivatives as potent caspase-3 inhibitors. J. Med. Chem. 2006, 49, 1613–1623. [Google Scholar] [CrossRef]
- Pollers-Wieers, C.; Vekemans, J.; Toppet, S.; Hoornaert, G. The use of isoquinolinetriones in the synthesis of benzo[c]phenanthridine alkaloids. Tetrahedron 1981, 37, 4321–4326. [Google Scholar] [CrossRef]
- Malamas, M.S.; Hohman, T.C.; Millen, J. Novel Spirosuccinimide Aldose Reductase Inhibitors Derived from Isoquinoline-1,3-diones: 2-[(4-Bromo-2-fluorophenyl)methyl]-6-fluorospiro[isoquinoline-4(11H),3'-pyrrolidine]-1,2',3,5'(2H)-tetrone and Congeners. 1. J. Med. Chem. 1994, 37, 2043–2058. [Google Scholar] [CrossRef]
- Takuwa, A. Evidence for a remarkable difference in the electrophilicity of two carbonyl groups in photoexcited 1,2-naphthoquinone. Photocycloaddition reaction of 1,2-naphthoquinone with olefins. Chem. Lett. 1989, 1989, 5–8. [Google Scholar] [CrossRef]
- Takuwa, A.; Kai, R.; Kawasaki, K.; Nishigaichi, Y.; Iwamoto, H. New formal [3 + 2] photoaddition of vinyl ethers to o-benzoquinones. Chem. Commun. 1996, 1996, 703–704. [Google Scholar]
- Takuwa, A.; Sumikawa, M. Solvent effect on the product distributions in the photocycloaddition of electron-rich olefins to 1,2-naphthoquinone. Chem. Lett. 1989, 1989, 9–12. [Google Scholar] [CrossRef]
- Suginome, H.; Sakurai, H.; Sasaki, A.; Takeuchi, H.; Kobayashi, K. Photoinduced molecular transformation. Part 151. One-pot synthesis of 1H-benz[g]indole-4,5-diones by a regioselective [3 + 2] photoaddition of 4-amino-1,2-naphthoquinones with alkenes. Tetrahedron 1994, 50, 8293–8300. [Google Scholar] [CrossRef]
- Takuwa, A.; Sasaki, T.; Iwamoto, H.; Nishigaichi, Y. Regioselective allylation of 1,2-naphthoquinonesusing photoaddition reaction with allylsilanes: Synthesis of 3-allyl-1,2-naphthoquinones. Synthesis 2001, 2001, 63–68. [Google Scholar] [CrossRef]
- Brace-Smith, D.; Gilbert, A. Photochemical and thermal cycloadditions of cis-stilbene and tolan (diphenylacetylene) to tetrachloro-o-benzoquinone. Photodecarbonylation of an α-diketone. Chem. Comm. 1968, 1968, 1702–1703. [Google Scholar]
- Kim, A.R.; Mah, Y.J.; Kim, S.S. Formation of o-/p-Quinomethanes and p-Quinodimethanes from the photoaddition of diphenylacetylene to o-quinones. Bull. Korean Chem. Soc. 1998, 19, 1295–1297. [Google Scholar]
- Verma, S.; Srivastava, B.; Sharma, K.; Joshi, R.; Pardasani, P.; Pardasani, R.T. Photoinduced [4 + 2] cycloaddition reactions of benzo[b]thiophene-2,3-dione with alkenes. Res. Chem. Intermediat. 2012, 38, 723–731. [Google Scholar] [CrossRef]
- Cho, D.W.; Lee, H.Y.; Oh, S.W.; Choi, J.H.; Park, H.J.; Mariano, P.S.; Yoon, U.C. Photoaddition Reactions of 1,2-Diketones with Silyl Ketene Acetals. Formation of β-Hydroxy-γ-ketoesters. J. Org. Chem. 2008, 73, 4539–4547. [Google Scholar] [CrossRef]
- Gersdorf, J.; Mattay, J.; Görner, H. Radical cations. 3. Photoreactions of biacetyl, benzophenone, and benzil with electron-rich alkenes. J. Am. Chem. Soc. 1987, 109, 1203–1209. [Google Scholar] [CrossRef]
- Maruyama, K.; Ono, K.; Osugi, J. Photochemical reaction of α-diketones. Bull. Chem. Soc. Jpn. 1972, 45, 847–851. [Google Scholar] [CrossRef]
- Nakatani, K.; Adachi, K.; Tanabe, K.; Saito, I. Tandem cyclizations involving carbene as an intermediate: Photochemical reactions of substituted 1,2-diketones conjugated with Ene-Yne. J. Am. Chem. Soc. 1999, 121, 8221–8228. [Google Scholar] [CrossRef]
- Schreiber, S.L.; Porco, J.A., Jr. The Paterno-Büchi reaction. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Paquette, L.A., Eds.; Pergamon Press: New York, NY, USA, 1991; volume 5, p. 168. [Google Scholar]
- Ninomiya, I.; Naito, T. Photochemical Synthesis; Academic Press: London, UK, 1989; Chapter 7; p. 135. [Google Scholar]
- Schreiber, S.L.; Satake, K. Application of the furan carbonyl photocycloaddition reaction to the synthesis of the bis(tetrahydrofuran) moiety of asteltoxin. J. Am. Chem. Soc. 1983, 105, 6723–6724. [Google Scholar] [CrossRef]
- Schreiber, S.L.; Satake, K. Total synthesis of (±)-asteltoxin. J. Am. Chem. Soc. 1984, 106, 4186–4188. [Google Scholar] [CrossRef]
- Schreiber, S.L. [2 + 2] Photocycloadditions in the synthesis of chiral molecules. Science 1985, 227, 857–863. [Google Scholar]
- Griesbeck, A.G.; Bondock, S. Photocycloaddition of 5-methoxyoxazoles to aldehydes and α-keto esters: A comprehensive view on stereoselectivity, triplet biradical conformations, and synthetic applications of Paterno-Büchi adducts. Aust. J. Chem. 2008, 61, 573–580. [Google Scholar] [CrossRef]
- Bondock, S.; Griesbeck, A.G. Diastereoselective photochemical synthesis of α-amino-β-hydroxy ketones by photocycloaddition of carbonyl compounds to 2,5-dimethyl-4-isobutyloxazole. Monatshefte für Chemie 2006, 137, 765–777. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Bondock, S.; Lex, J. Synthesis of erythro-α-Amino β-Hydroxy Carboxylic Acid Esters by Diastereoselective Photocycloaddition of 5-Methoxyoxazoles with Aldehydes. J. Org. Chem. 2003, 68, 9899–9906. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Fiege, M.; Lex, J. Oxazole-Carbonyl photocycloadditions: Selectivity pattern and synthetic route to erythro α-amino, β-hydroxy ketones. Chem. Commun. 2000, 2000, 589–590. [Google Scholar] [CrossRef]
- Schreiber, S.L.; Hoveyda, A.H.; Wu, H.-J. A photochemical route to the formation of threo aldols. J. Am. Chem. Soc. 1983, 105, 660–661. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, C.; Zheng, M.; Xu, J.; Zhang, Y. Photo-Induced Cycloaddition Reactions of α-Diketones and Transformations of the Photocycloadducts. Molecules 2013, 18, 2942-2966. https://doi.org/10.3390/molecules18032942
Huang C, Zheng M, Xu J, Zhang Y. Photo-Induced Cycloaddition Reactions of α-Diketones and Transformations of the Photocycloadducts. Molecules. 2013; 18(3):2942-2966. https://doi.org/10.3390/molecules18032942
Chicago/Turabian StyleHuang, Chengmei, Mengmeng Zheng, Jianhua Xu, and Yan Zhang. 2013. "Photo-Induced Cycloaddition Reactions of α-Diketones and Transformations of the Photocycloadducts" Molecules 18, no. 3: 2942-2966. https://doi.org/10.3390/molecules18032942
APA StyleHuang, C., Zheng, M., Xu, J., & Zhang, Y. (2013). Photo-Induced Cycloaddition Reactions of α-Diketones and Transformations of the Photocycloadducts. Molecules, 18(3), 2942-2966. https://doi.org/10.3390/molecules18032942