An Efficient Chemoselective Reduction of Furan Series Unsaturated Dinitriles

Abstract
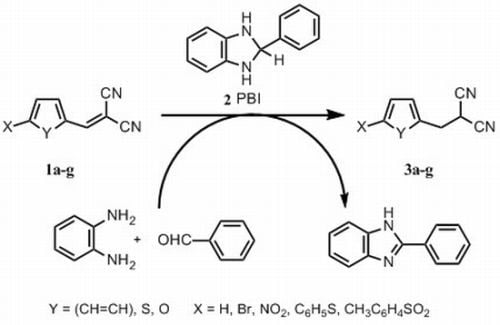
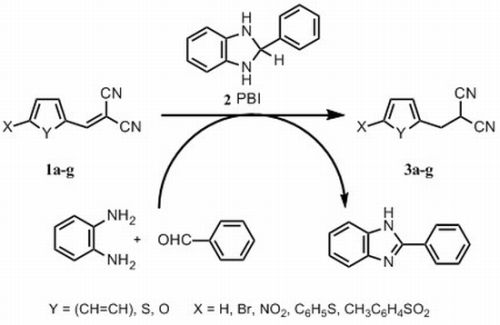
:
1. Introduction
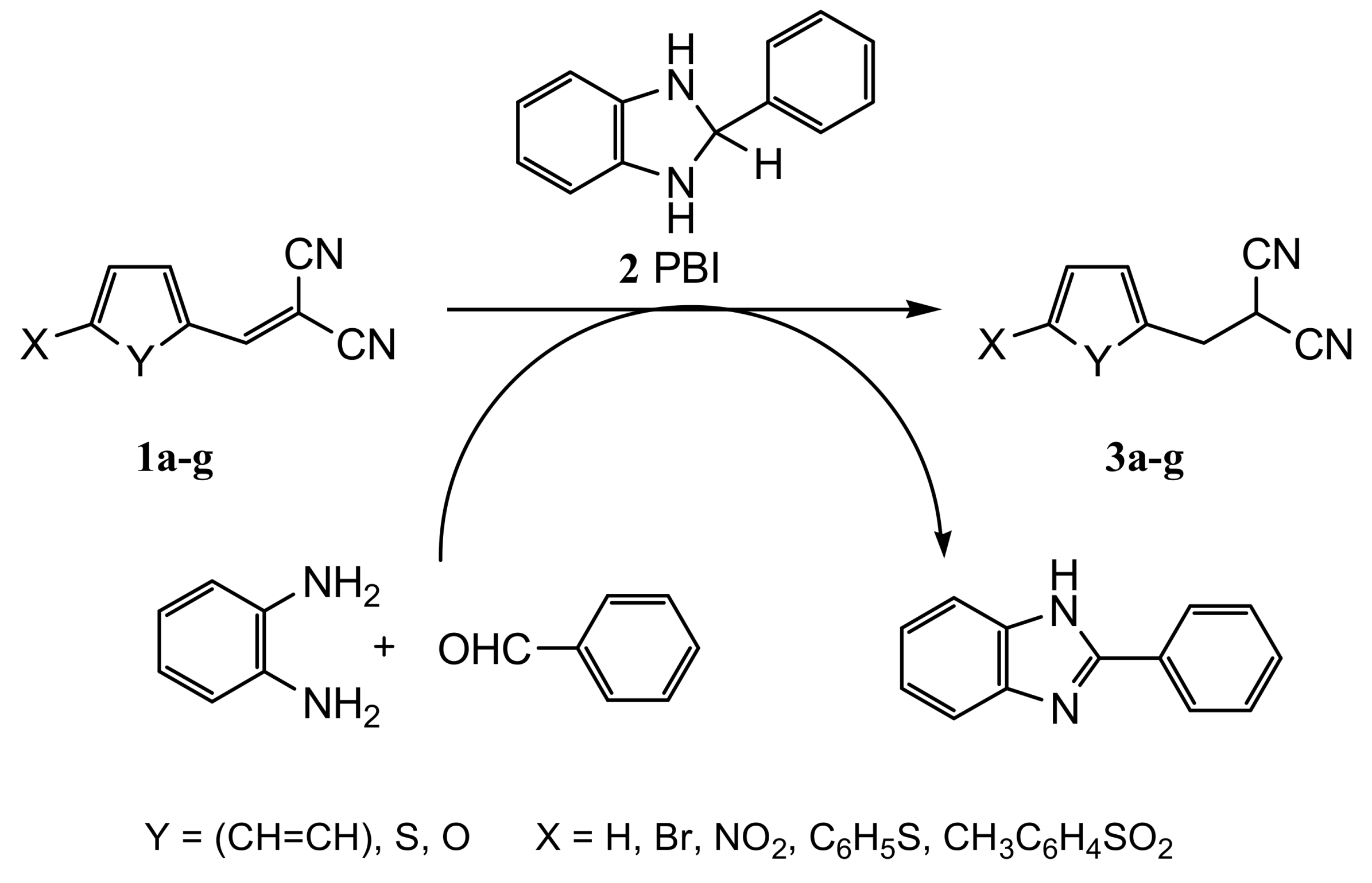
2. Results and Discussion
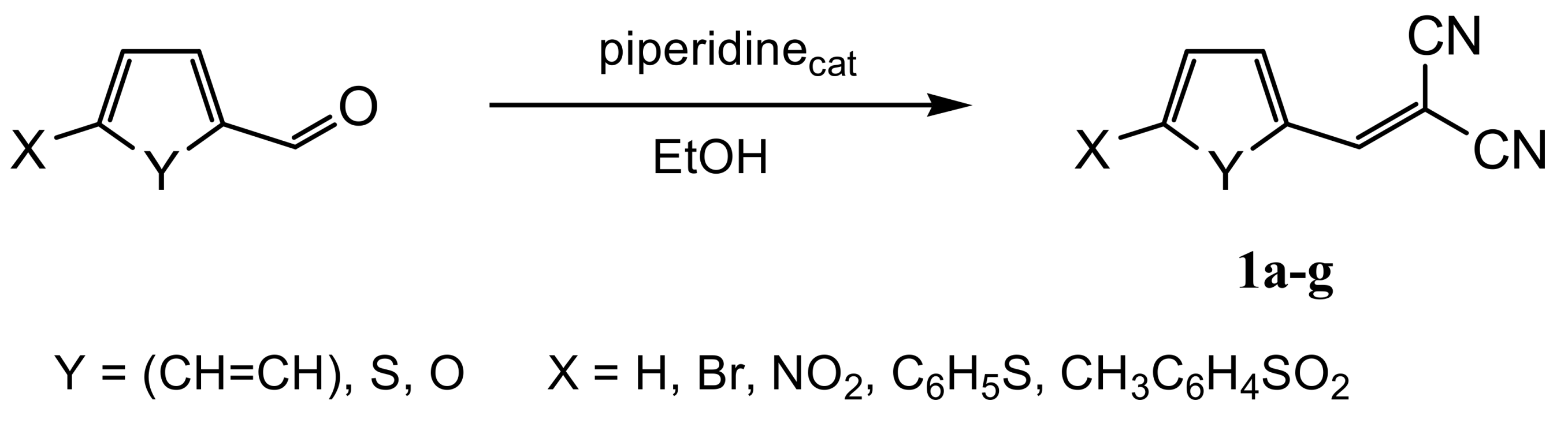
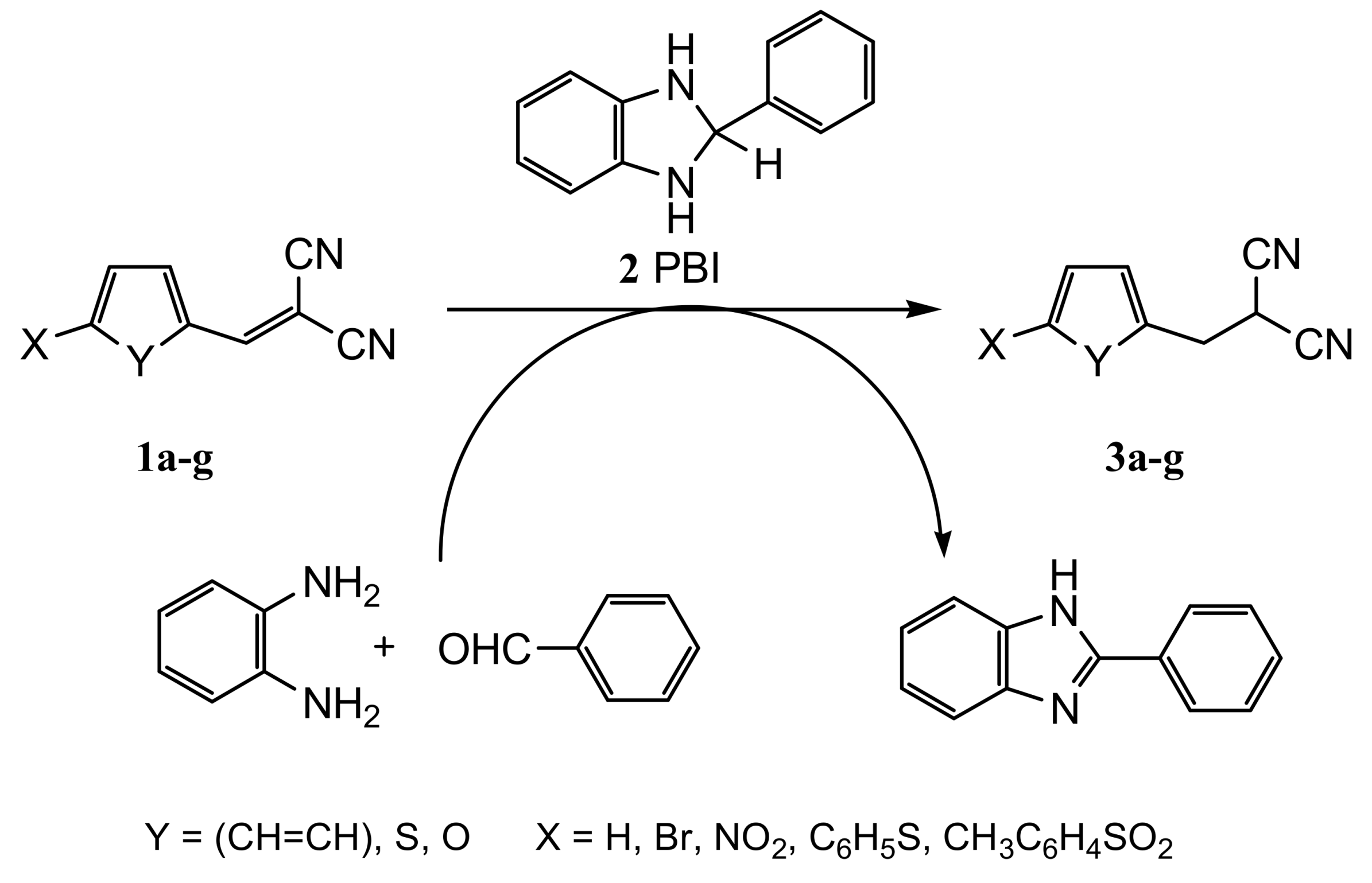
2.1. Chemistry
3. Experimental
3.1. General
3.2. Synthesis
3.2.1. Procedure for the Synthesis of 3-(Phenyl)-2-cyanopropenenitrile (1a) [29]
3.2.2. General Procedure for the Synthesis of 3-(Thiophen-2-yl) and 3-(5-Substituted Furan-2-yl)-2-cyanopropenenitriles 1b–g [28,29]
3.2.3. Procedure for the Synthesis of 2-Benzylpropanedinitrile with NaBH4
3.2.4. General Procedure for the Synthesis of Phenyl-, Thiofen-2-yl and 5-Substituted Furan-2-ylmethylpropanedinitriles 3a–g with 2-Phenylbenzimidazoline (2 PBI)
4. Conclusions
Acknowledgments
References
- Keay, B.A.; Dibble, P.W. Furans and their Benzo Derivatives: Applications. In Comprehensive Heterocyclic Chemistry II; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Elsevier: New York, NY, USA, 1996; Volume 2, pp. 395–436. [Google Scholar]
- Mortensen, D.S.; Rodriguez, A.L.; Carlson, K.E.; Sun, J.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Synthesis and biological evaluation of a novel series of furans: Ligands selective for estrogen receptor α. J. Med. Chem. 2001, 44, 3838–3848. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B.H. Five-membered heteroaromatic rings as intermediates in organic synthesis. Chem. Rev. 1986, 86, 795–819. [Google Scholar] [CrossRef]
- Hou, X.L.; Cheung, H.Y.; Hon, T.Y.; Kwan, P.L.; Lo, T.H.; Tong, S.Y.; Wong, H.N.C. Regioselective syntheses of substituted furans. Tetrahedron 1998, 54, 1955–2020. [Google Scholar] [CrossRef]
- Bosshard, P.; Eugster, C.H. The development of the chemistry of furans, 1952–1963. Adv. Heterocycl. Chem. 1967, 7, 377–490. [Google Scholar]
- Lee, G.H.; Youn, I.K.; Choi, E.B.; Lee, H.K.; Yon, G.H.; Yang, H.C.; Pak, C.S. Magnesium in methanol (Mg/MeOH) in organic syntheses. Curr. Org. Chem. 2004, 8, 1263–1287. [Google Scholar] [CrossRef]
- Dean, F.M. Recent advances in furan chemistry. Part I. Adv. Heterocycl. Chem. 1982, 30, 167–238. [Google Scholar]
- Dean, F.M. Recent advances in furan chemistry. Part II. Adv. Heterocycl. Chem. 1982, 31, 237–344. [Google Scholar]
- Profitt, J.A.; Watt, D.S.; Corey, E.J. A reagent for the α,β reduction of conjugated nitriles. J. Org. Chem. 1975, 40, 127–128. [Google Scholar] [CrossRef]
- Cope, A.C.; Alexander, E.R. A simultaneous condensation-reduction method for the preparation of ethyl monoalkylcyanoacetates. J. Am. Chem. Soc. 1944, 66, 886–888. [Google Scholar]
- Neumann, W.P.; Sommer, R.; Müller, E. Preparation of N-stannylketenimines and ketene ethyl stannyl acetals by 1,4-hydrostannation of conjugated systems. Angew. Chem. Int. Ed. 1966, 514–515. [Google Scholar] [CrossRef]
- Sommer, R.; Müller, E.; Neumann, W.P. Organotin compounds. XXII. Syntheses with N-trialkylstannylketenimines. Liebigs Ann. Chem. 1968, 718, 11–23. [Google Scholar] [CrossRef]
- Nanjo, K.; Suzuki, K.; Sekiya, M. Formic acid reduction. XXVI. α,β-Reduction of conjugated nitriles with formic acid. Chem. Pharm. Bull. 1977, 25, 2396–2400. [Google Scholar] [CrossRef]
- Toda, F.; Kanno, M. Reduction of α,β-unsaturated nitrile to saturated nitrile with sodium borohydride. Bull. Chem. Soc. Jpn. 1976, 49, 2643–2644. [Google Scholar] [CrossRef]
- Fuentes, L.; Lorente, A.; Soto, J.L. Synthesis of heterocyclic compounds. XIII. 2-Amino-3-benzyl-3,5-dicyano-6-methoxy-4-phenyl-3,4-dihydropyridines. J. Heterocycl. Chem. 1979, 16, 273–276. [Google Scholar] [CrossRef]
- Hutchins, R.O.; Rotstein, D.; Natale, N.; Fanelli, J.; Dimmel, D. Selective reduction of α,β-unsaturated esters, nitriles, and nitro compounds with sodium cyanoborohydride. J. Org. Chem. 1976, 41, 3328–3329. [Google Scholar] [CrossRef]
- Deno, N.C.; Peterson, J.; Saines, G.S. The hydride-transfer reaction. Chem. Rev. 1960, 60, 7–14. [Google Scholar] [CrossRef]
- Wallenfels, K.; Ertel, W.; Friedrich, K. Mechanism of hydrogen transfer with pyridine nucleotides. XXXI. Reduction of cyano-activated olefins by direct hydrogen transfer from dihydropyridines. Liebigs Ann. Chem. 1973, 1663–1674. [Google Scholar] [CrossRef]
- Nakamura, K.; Ohno, A.; Oka, S. Reduction by a model of NAD(P)H. 44. Transition metal catalyzed reduction of allylic acetate. Tetrahedron Lett. 1983, 24, 3335–3336. [Google Scholar] [CrossRef]
- Garden, S.J.; Guimaraes, C.R.W.; Correa, M.B.; Fernandes, D.O.C.A.; Pinto, A.D.C.; Bicca, D.A.R. Synthetic and theoretical studies on the reduction of electron withdrawing group conjugated olefins using the hantzsch 1,4-dihydropyridine ester. J. Org. Chem. 2003, 68, 8815–8822. [Google Scholar] [CrossRef] [PubMed]
- Quynh, P.B.N.; Kim, J.N.; Kim, T.H. S-Benzyl isothiouronium chloride as a recoverable organocatalyst for the reduction of conjugated nitroalkenes with Hantzsch ester. Tetrahedron 2012, 68, 6513–6516. [Google Scholar]
- Itoh, K.; Ishida, H.; Chikashita, H. The reactions of benzylidenemalononitriles and β-nitrostyrenes with o-phenylenediamine including the new organic redox reactions between the olefins and 2-phenylbenzimidazolines. Chem. Lett. 1982, 1117–1118. [Google Scholar] [CrossRef]
- Chikashita, H.; Nishida, S.; Miyazaki, M.; Itoh, K. 2-Phenylbenzimidazoline as a reducing agent in the preparation of malononitriles from α,β-unsaturated dinitriles. Synth. Commun. 1983, 13, 1033–1039. [Google Scholar] [CrossRef]
- Chikashita, H.; Nishida, S.; Miyazaki, M.; Morita, Y.; Itoh, K. In situ generation and synthetic application of 2-phenylbenzimidazoline to the selective reduction of carbon-carbon double bonds of electron-deficient olefins. Bull. Chem. Soc. Jpn. 1987, 60, 737–746. [Google Scholar] [CrossRef]
- Feng, Y.S.; Yang, C.Y.; Huang, Q.; Xu, H.J. Study on comparison of reducing ability of three organic hydride compounds. Tetrahedron 2012, 68, 5053–5059. [Google Scholar] [CrossRef]
- Hennawy, I.T. Studies of the Reaction of Triaminophosphines with α,β-unsaturated nitriles. Collect. Czech. Chem. Commun. 1994, 59, 2109–2115. [Google Scholar] [CrossRef]
- Rewcastle, G.W. Pyrimidines and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: New York, NY, USA, 2008; Volume 8, pp. 117–272. [Google Scholar]
- Kada, R.; Ilavsky, D.; Goljer, I.; Gaher, P. 3-(5-X-2-Furyl)-2-cyanoacrylonitriles on reaction with 2-cyanomethylbenzothiazole. Collect. Czech. Chem. Commun. 1991, 56, 418–424. [Google Scholar] [CrossRef]
- Mantri, M.; de Graaf, O.; van Veldhoven, J.; Goeblyoes, A.; von Frijtag Drabbe Kuenzel, J.K.; Mulder-Krieger, T.; Link, R.; de Vries, H.; Beukers, M.W.; Brussee, J.; et al. 2-Amino-6-furan-2-yl-4-substituted nicotinonitriles as A2A adenosine receptor antagonists. J. Med. Chem. 2008, 51, 4449–4455. [Google Scholar] [CrossRef] [PubMed]
- Antonioletti, R.; D’Auria, M.; De, M.A.; Piancatelli, G.; Scettri, A. Photochemical synthesis of 3- and 5-aryl-2-furyl derivatives. J. Chem. Soc. Perkin Trans. 1 1985, 1285–1288. [Google Scholar] [CrossRef]
- Gilman, H.; Wright, G.F. Nitrofurfural and nitrofurylacrylic acid. J. Am. Chem. Soc. 1930, 52, 2550–2554. [Google Scholar] [CrossRef]
- Kada, R.; Kovac, J. Furan derivatives. LXV. Preparation and ultraviolet spectra of 5-arylthio- and 5-heteroarylthio-2-furaldehydes. Chem. Zvesti 1975, 29, 402–407. [Google Scholar]
- Nakamura, S.; Sugimoto, H.; Ohwada, T. Superacid-catalyzed intramolecular cyclization reaction of arylcyanopropionate: Geminal substitution effect on superelectrophilicity. J. Org. Chem. 2008, 73, 4219–4224. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.; Das, S.; Das, P.; Nanda, A.K. Co-immobilized formate anion and palladium on a polymer surface: A novel heterogeneous combination for transfer hydrogenation. Tetrahedron Lett. 2005, 46, 8591–8593. [Google Scholar] [CrossRef]
- Leggans, E.K.; Barker, T.J.; Duncan, K.K.; Boger, D.L. Iron(III)/NaBH4-mediated additions to unactivated alkenes: Synthesis of novel 20'-vinblastine analogues. Org. Lett. 2012, 14, 1428–1431. [Google Scholar] [CrossRef] [PubMed]
- Tayyari, F.; Wood, D.E.; Fanwick, P.E.; Sammelson, R.E. Monosubstituted malononitriles: Efficient one-pot reductive alkylations of malononitrile with aromatic aldehydes. Synthesis 2008, 2, 279–285. [Google Scholar] [CrossRef]
- Shinkai, S.; Kusano, Y.; Ide, T.; Sone, T.; Manabe, O. Coenzyme Models. XII. Kinetics and mechanisms of 1,4-dihydronicotinamide reduction of carbon-carbon double bonds. Influence of added Mg2+ ion and acetic acid. Bull. Chem. Soc. Jpn. 1978, 51, 3544–3548. [Google Scholar] [CrossRef]
- Dunham, J.C.; Richardson, A.D.; Sammelson, R.E. Sodium borohydride as the only reagent for the efficient reductive alkylation of malononitrile with ketones and aldehydes. Synthesis 2006, 4, 680–686. [Google Scholar] [CrossRef]
- Otaka, T.; Ohira, D. Preparation of [(Thienyl, Furyl, or Pyrrolyl)methyl]malononitrile Derivatives as Pesticides. Jpn. Patent JP2004099597A, 2 April 2004. [Google Scholar]
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Product | Yield a (%) | Time (h) |
|---|---|---|---|---|
| 1a | Y = (CH=CH), X = H | 3a | 83 b (85) c | 12 b (2) c |
| 1b | Y = S, X = H | 3b | 78 b (69) c | 8 b (2) c |
| 1c | Y = O, X = H | 3c | 72 b (61) c | 8 b (2) c |
| 1d | Y = O, X = Br | 3d | 80 b (-) c,d | 8 b (1) c |
| 1e | Y = O, X = NO2 | 3e | 62 b (-) c,e | 6 b (1) c |
| 1f | Y = O, X = C6H5S | 3f | 79 b (-) c,e | 12 b (1) c |
| 1g | Y = O, X = CH3C6H4SO2 | 3g | 85 b (-) a,e | 8 b (1) c |
© 2013 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bobal, P.; Bobalova, J. An Efficient Chemoselective Reduction of Furan Series Unsaturated Dinitriles. Molecules 2013, 18, 2212-2221. https://doi.org/10.3390/molecules18022212
Bobal P, Bobalova J. An Efficient Chemoselective Reduction of Furan Series Unsaturated Dinitriles. Molecules. 2013; 18(2):2212-2221. https://doi.org/10.3390/molecules18022212
Chicago/Turabian StyleBobal, Pavel, and Janette Bobalova. 2013. "An Efficient Chemoselective Reduction of Furan Series Unsaturated Dinitriles" Molecules 18, no. 2: 2212-2221. https://doi.org/10.3390/molecules18022212
APA StyleBobal, P., & Bobalova, J. (2013). An Efficient Chemoselective Reduction of Furan Series Unsaturated Dinitriles. Molecules, 18(2), 2212-2221. https://doi.org/10.3390/molecules18022212