4,6,8-Triarylquinoline-3-carbaldehyde Derivatives: Synthesis and Photophysical Properties


Abstract
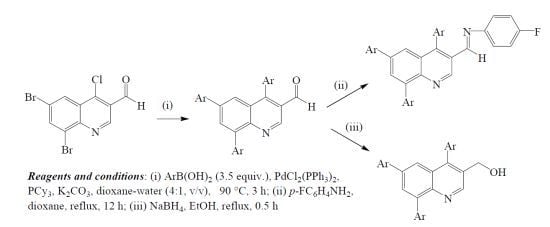
:
1. Introduction
2. Results and Discussion
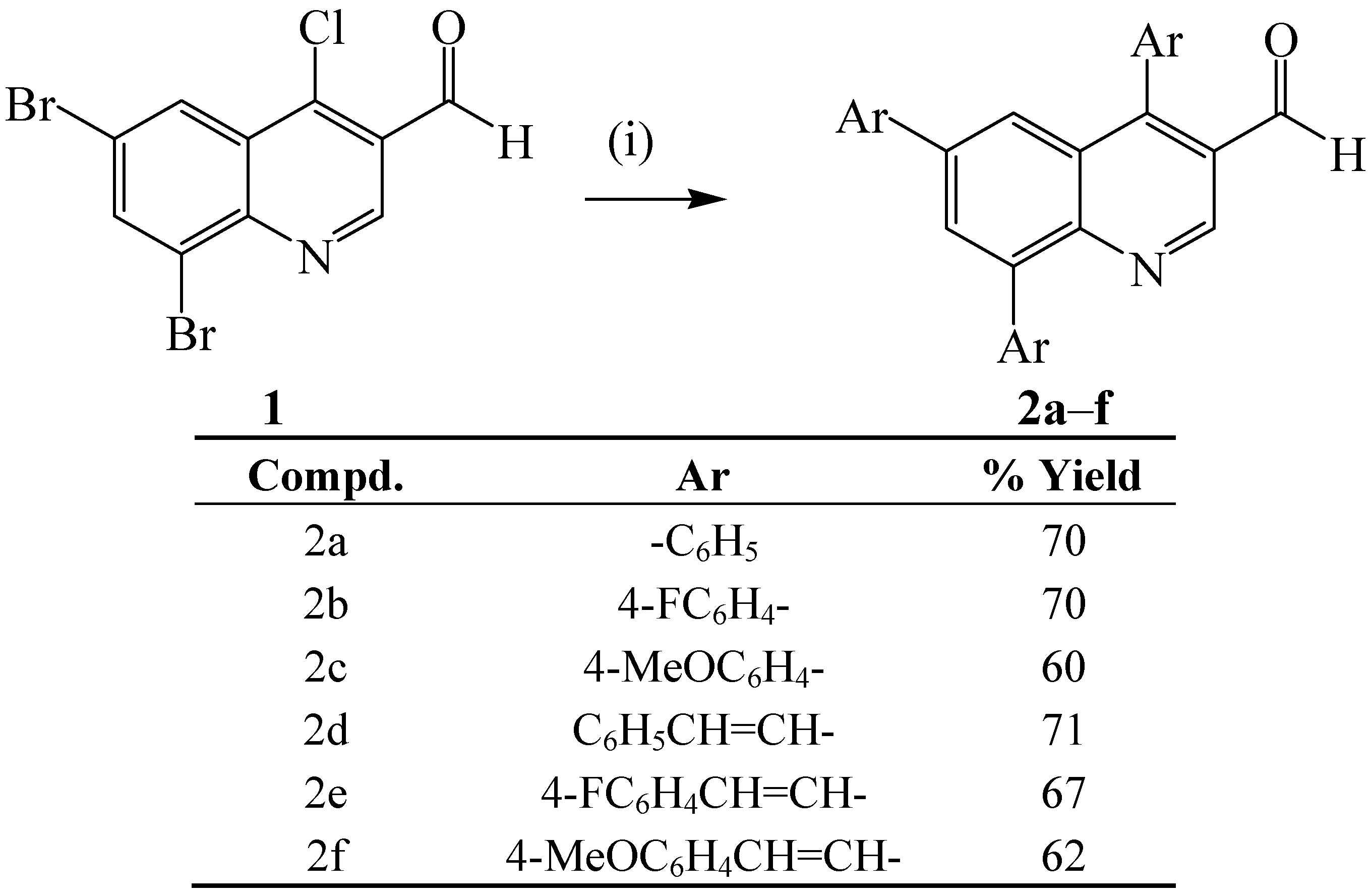
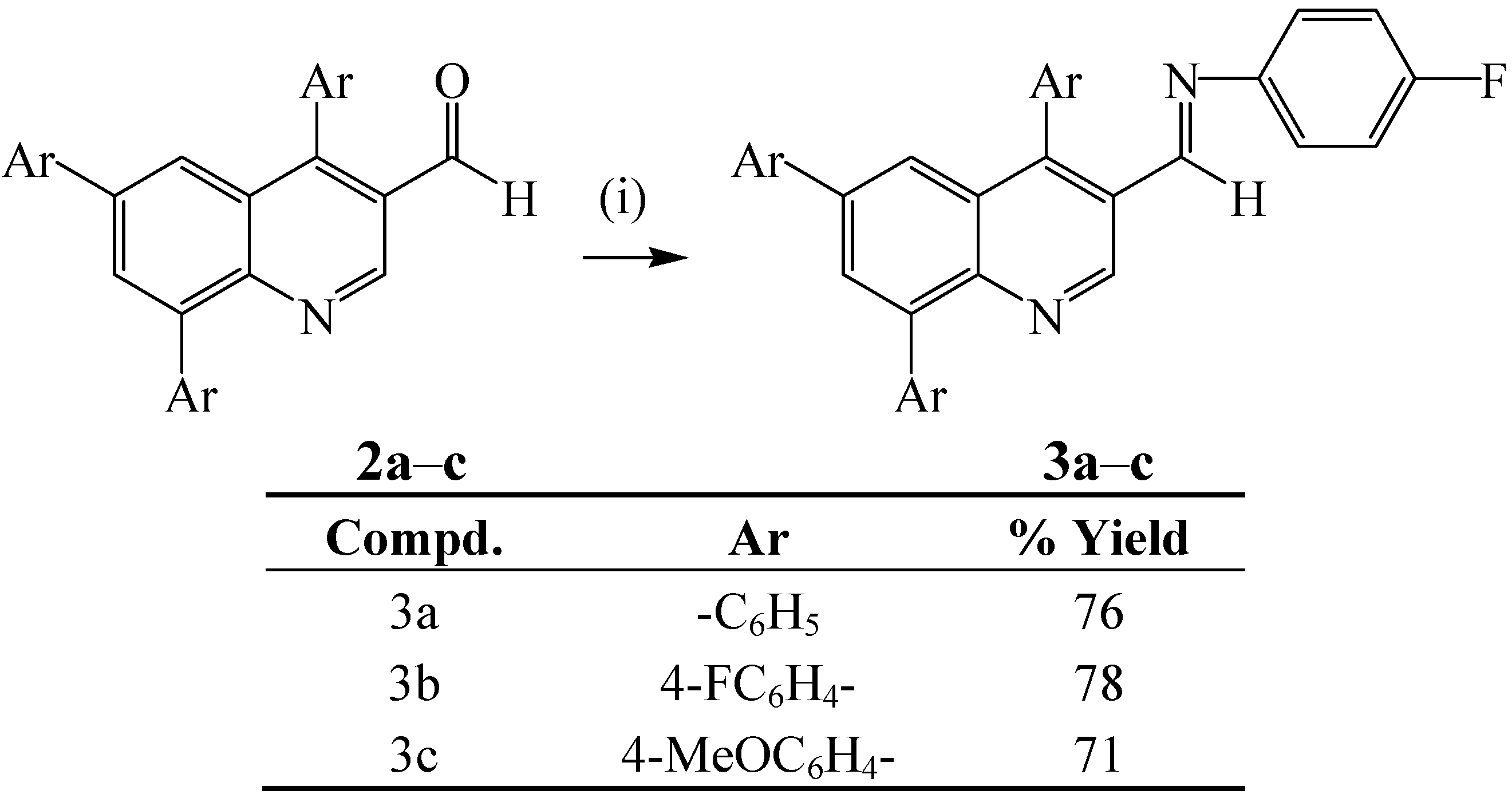

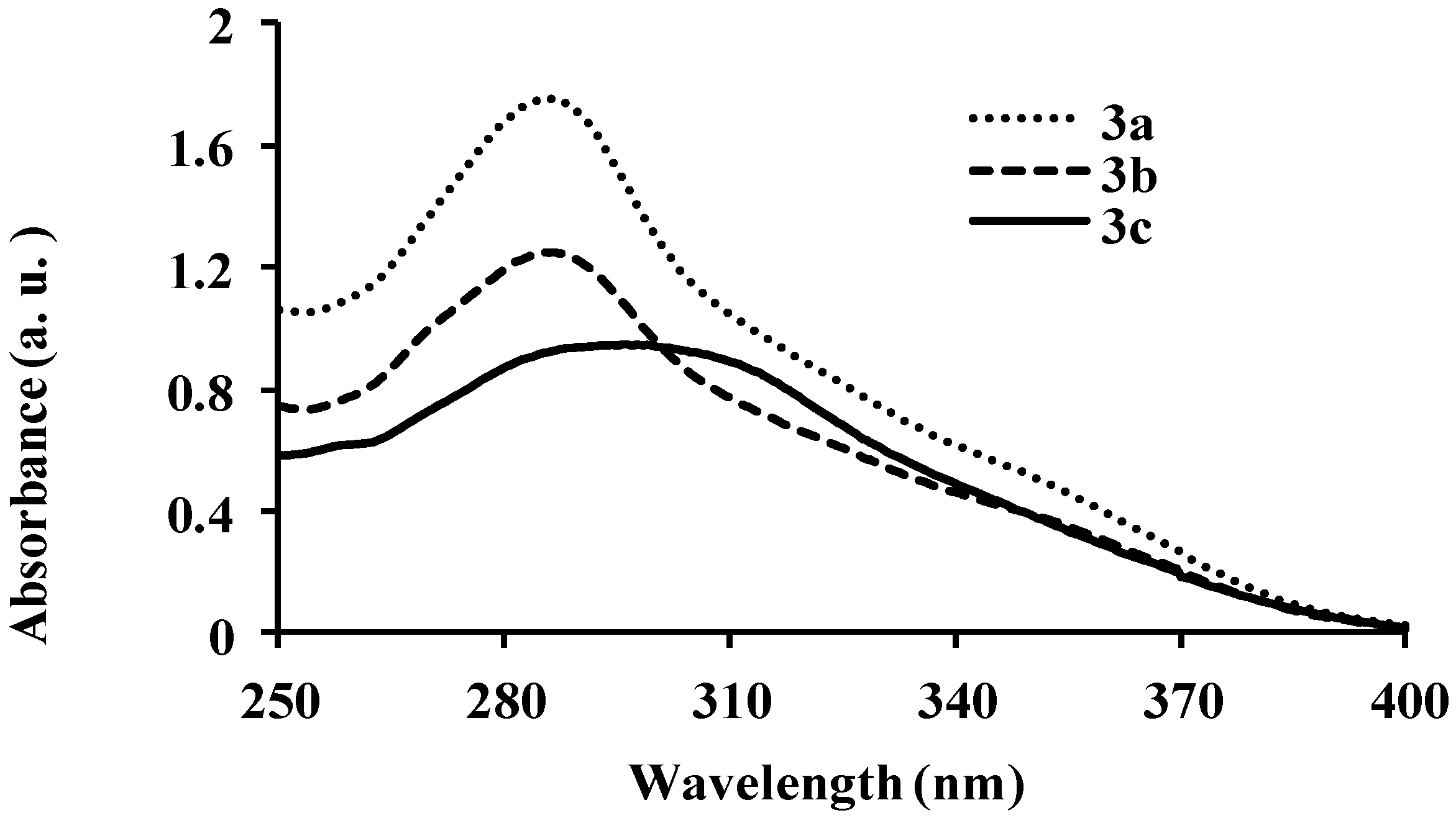
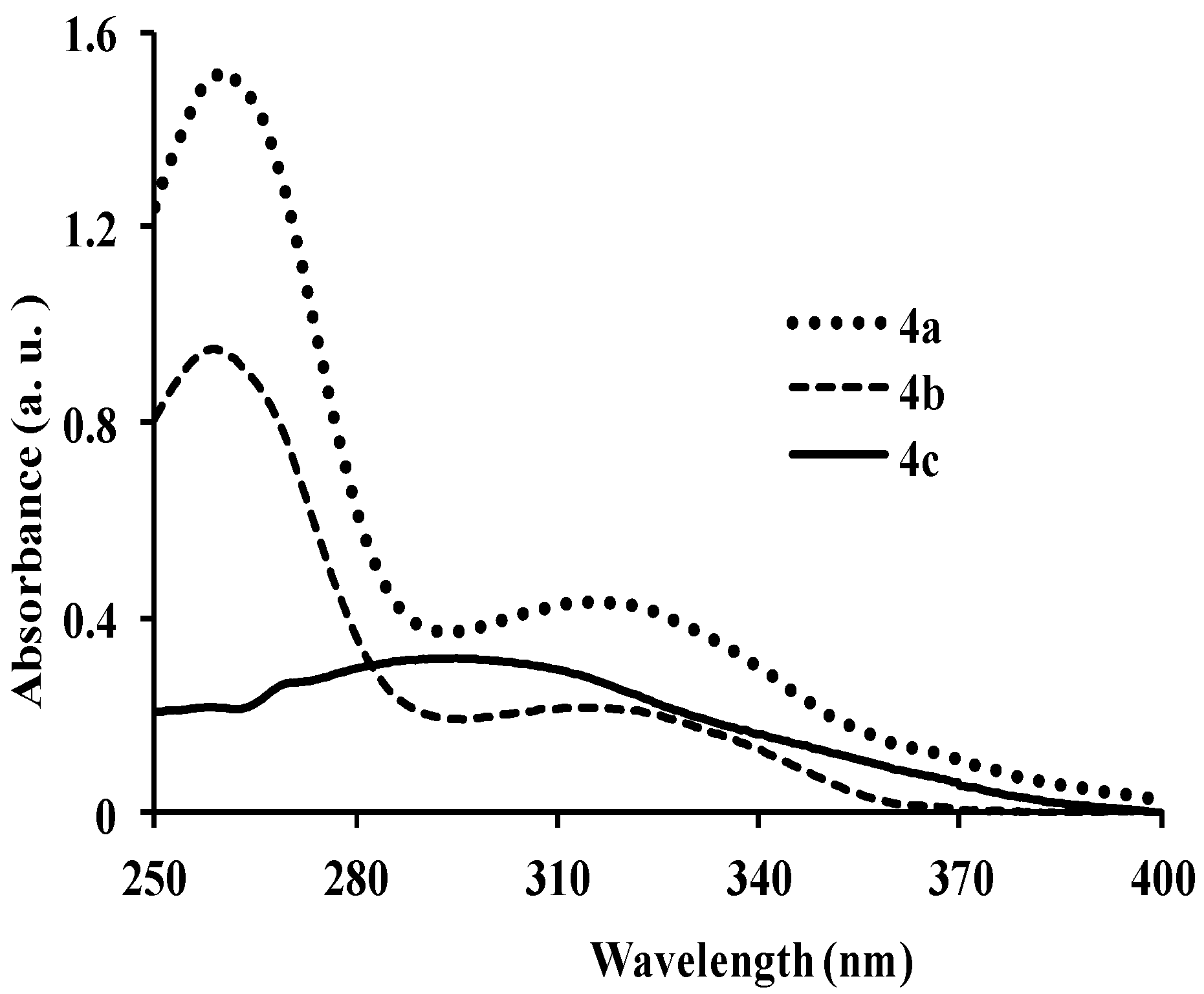

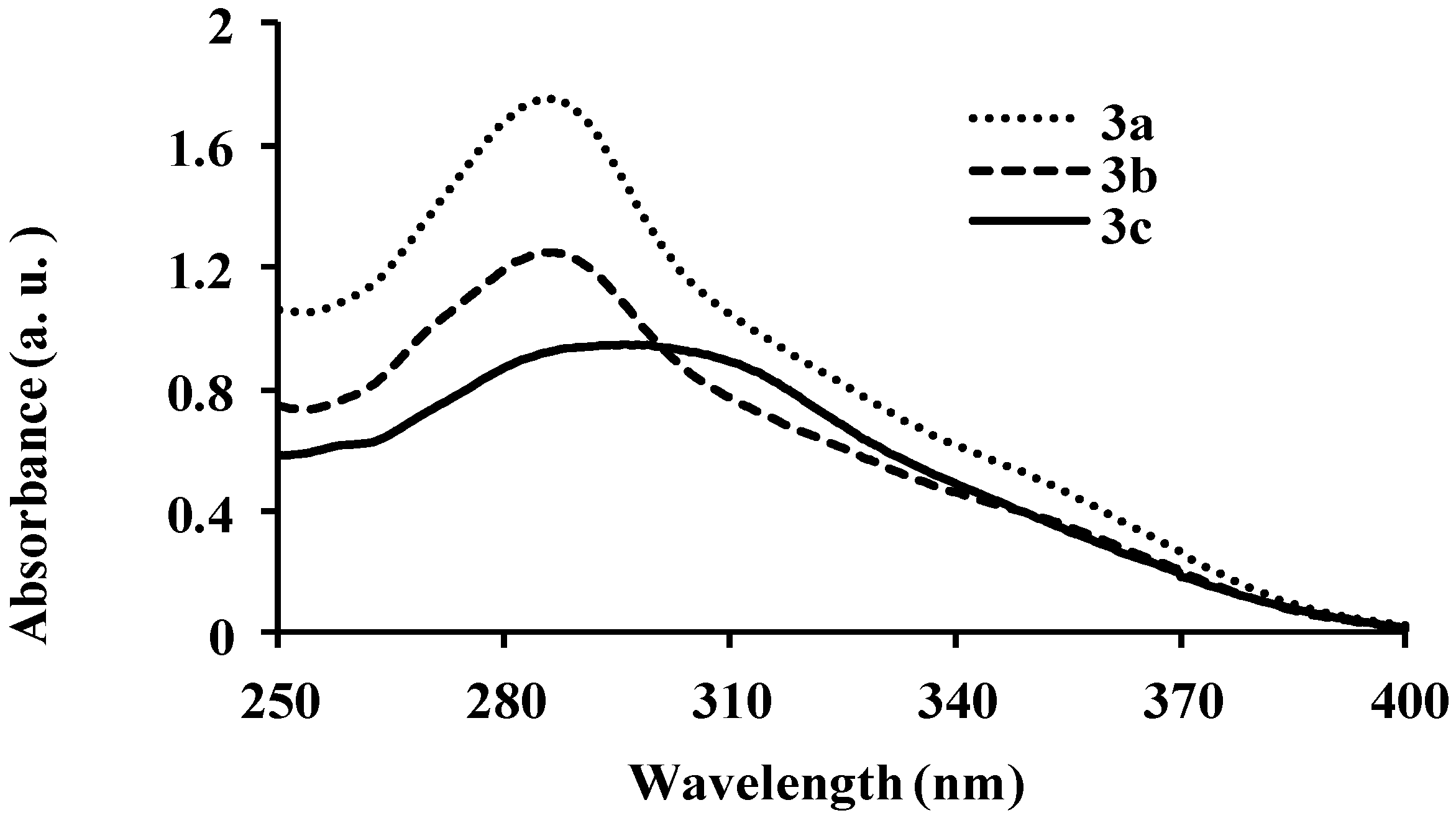
{kind=link}
{kind=link}
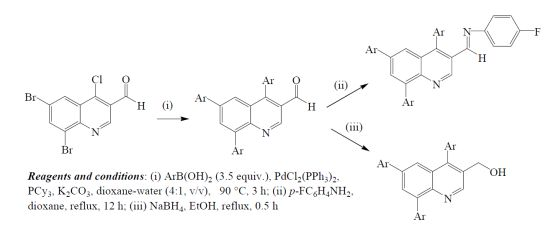
{kind=link}
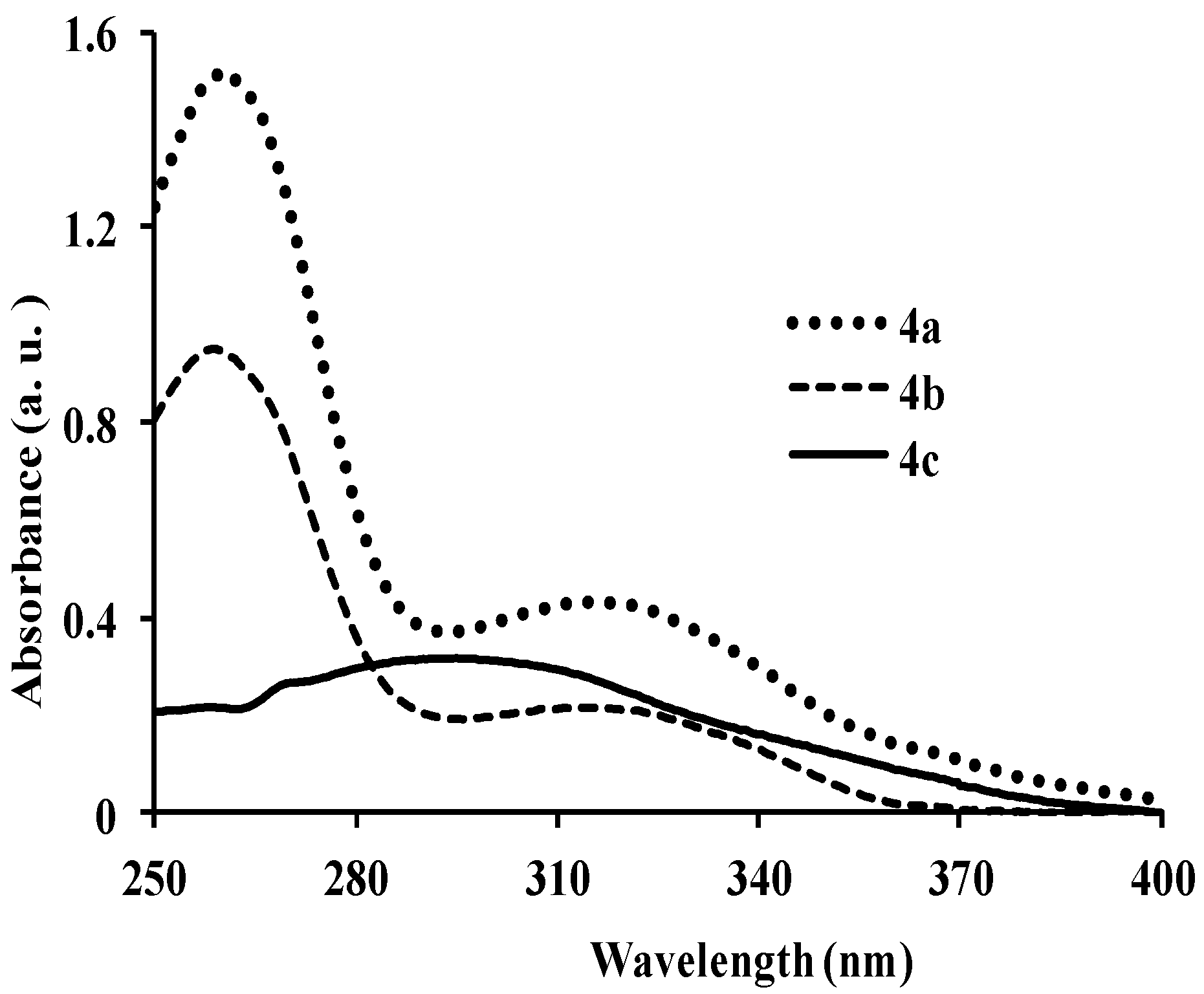{kind=link}
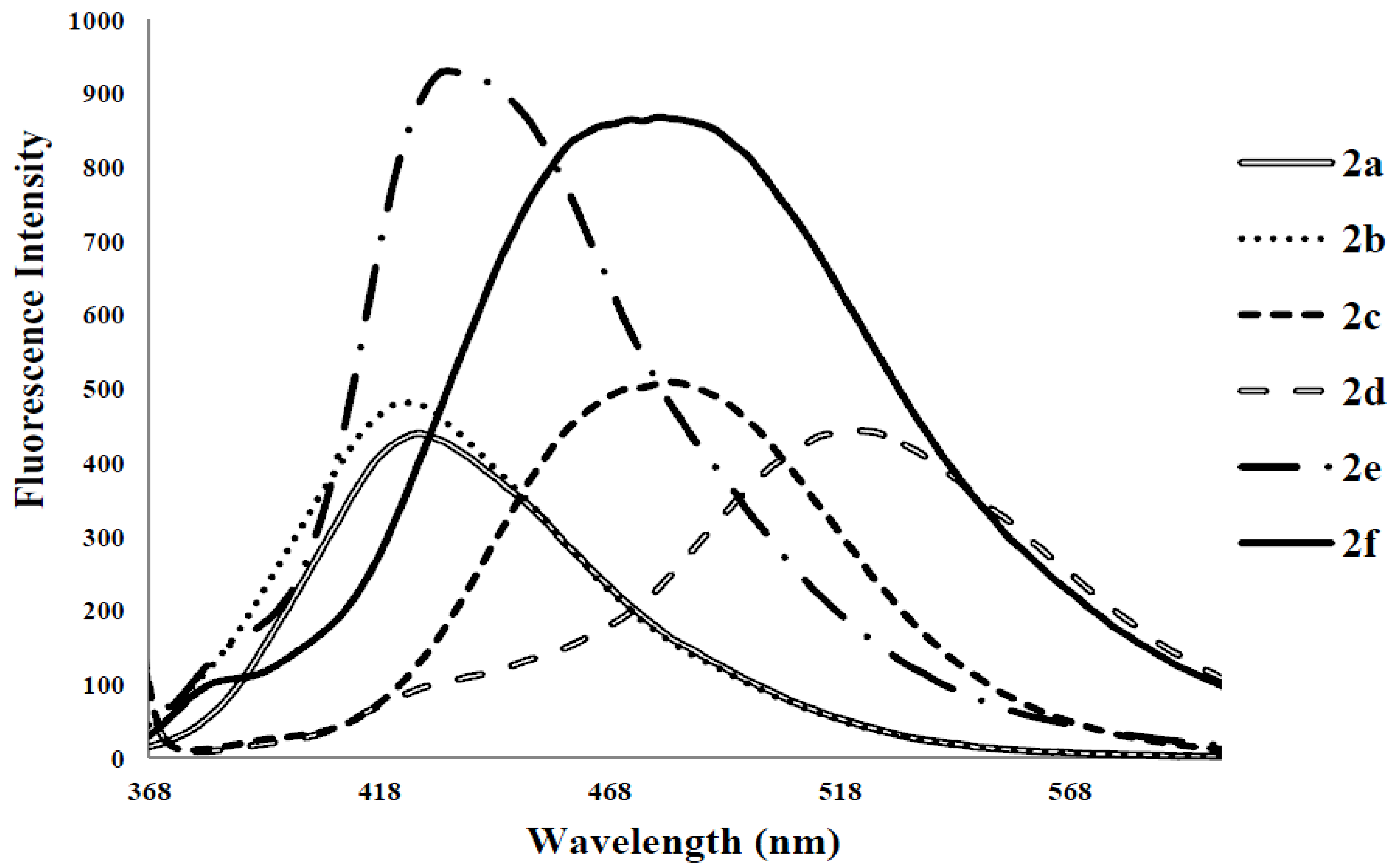{kind=link}
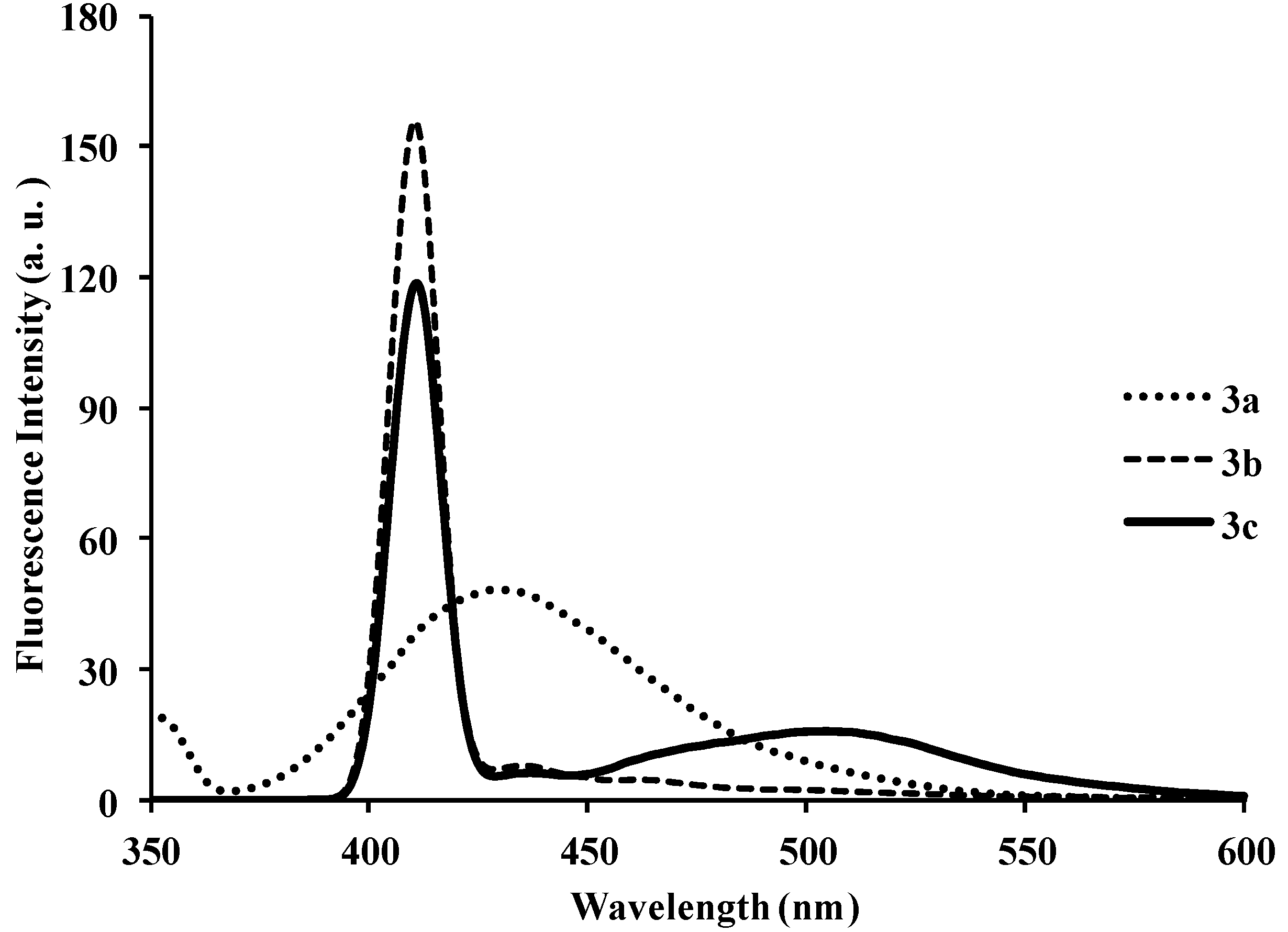{kind=link}
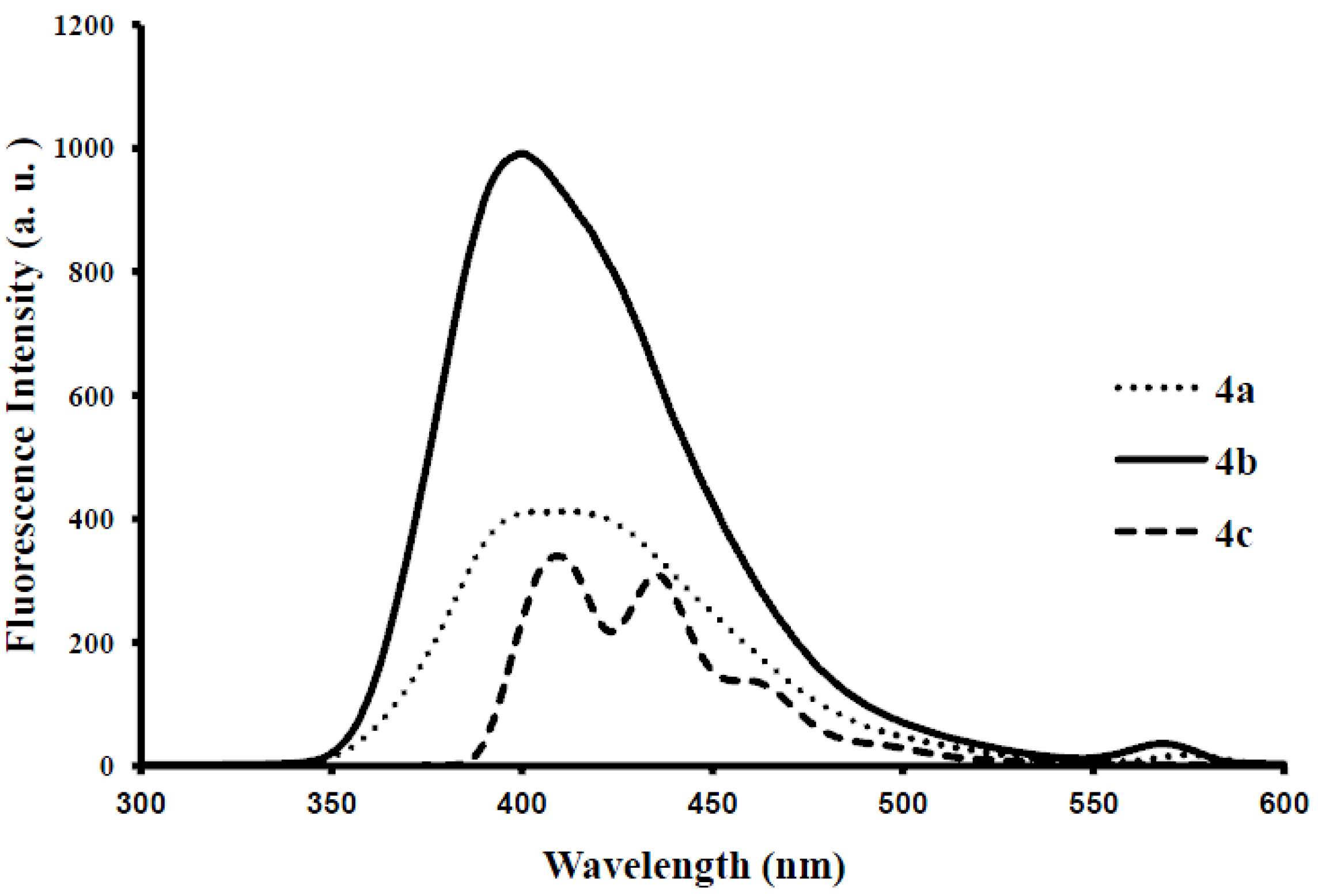{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
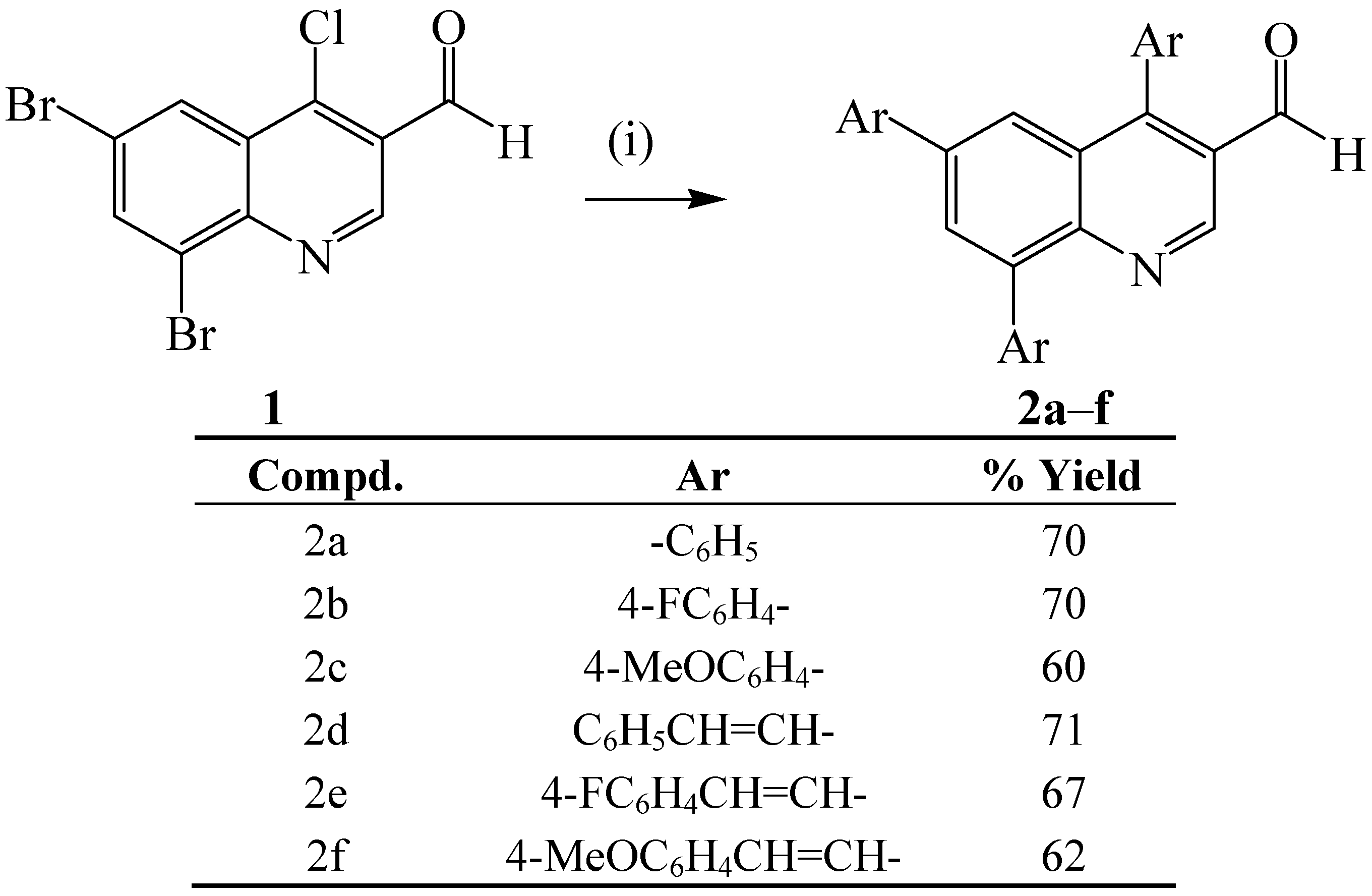{kind=link}
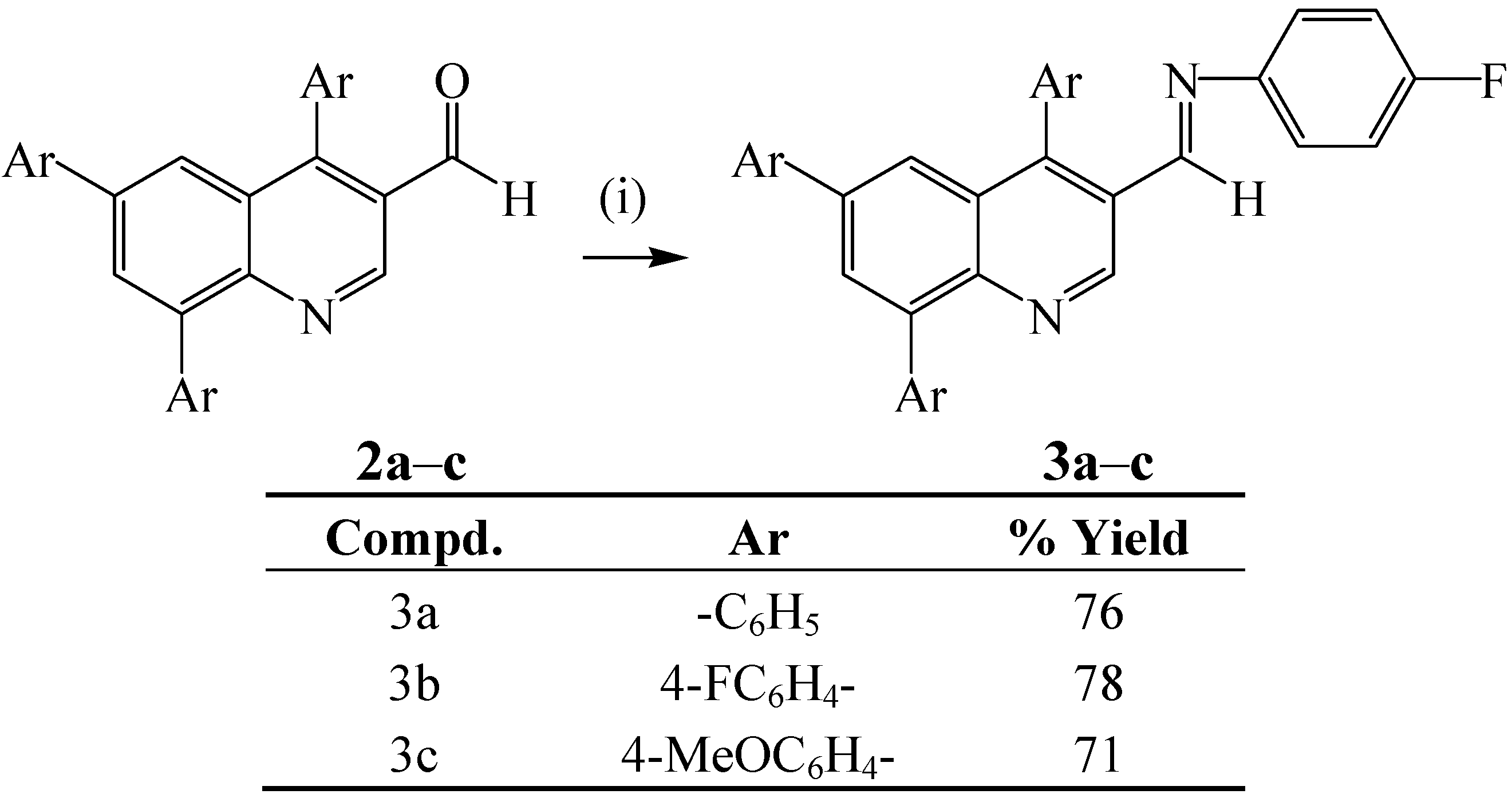{kind=link}
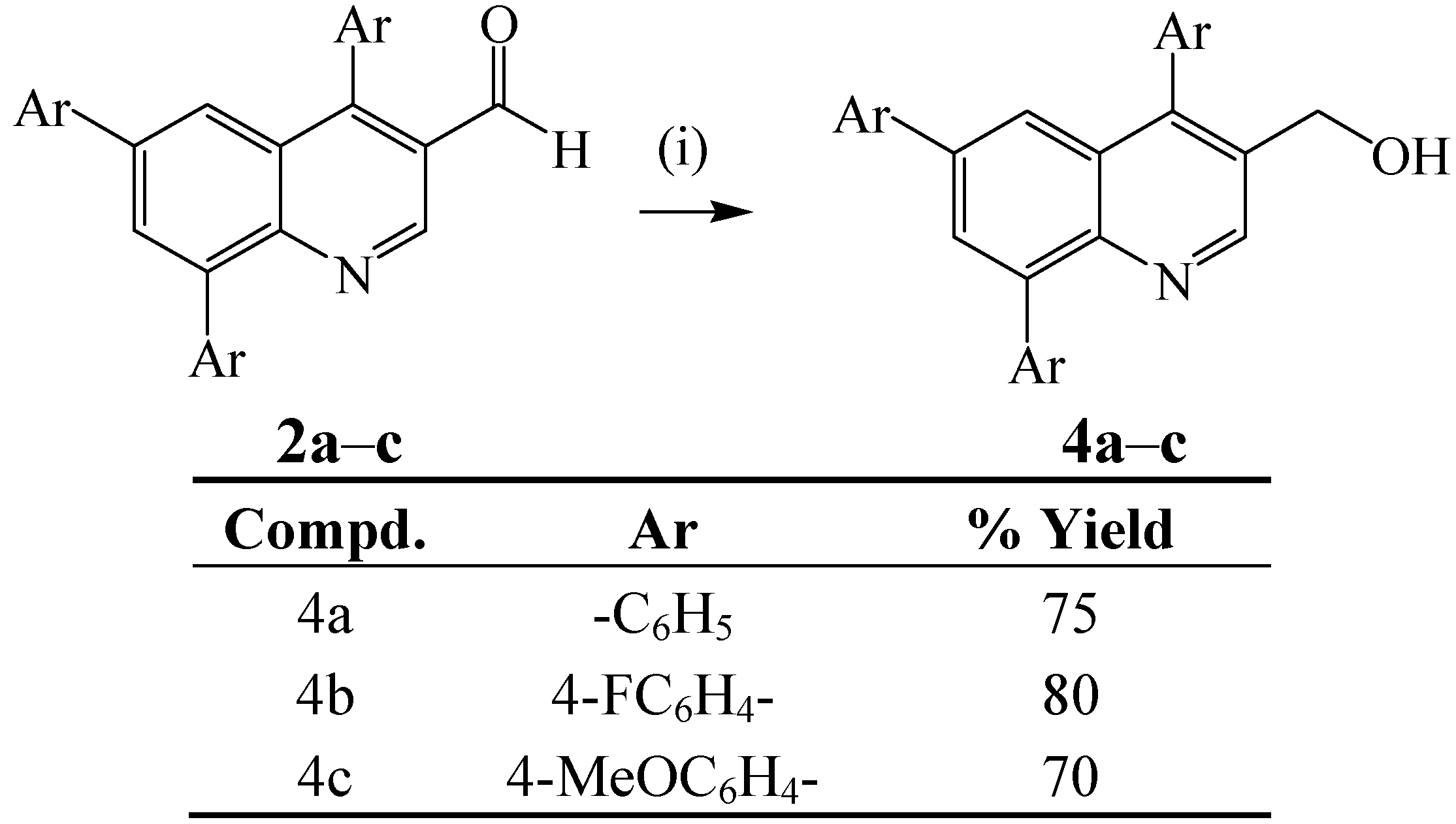{kind=link}
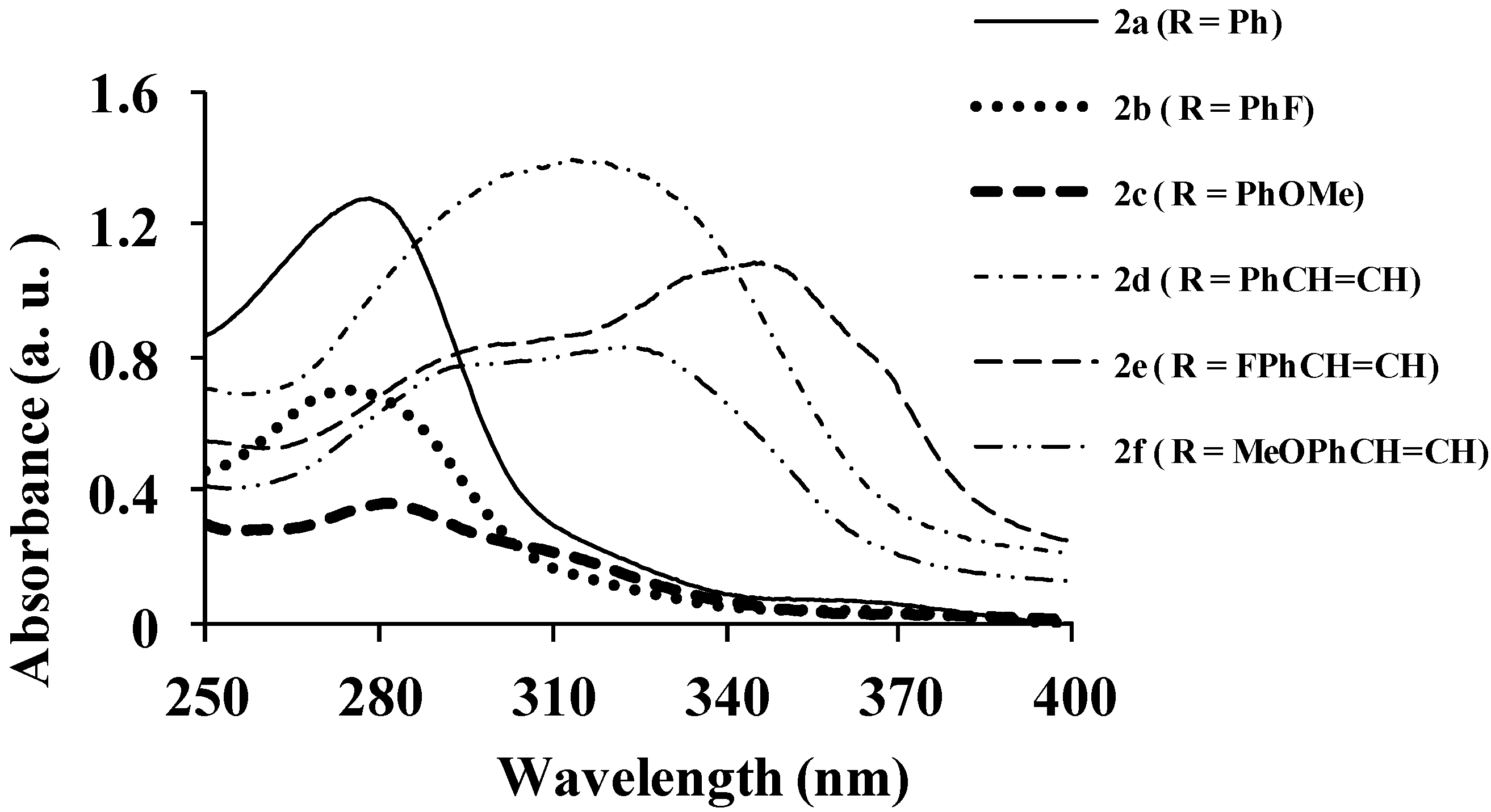
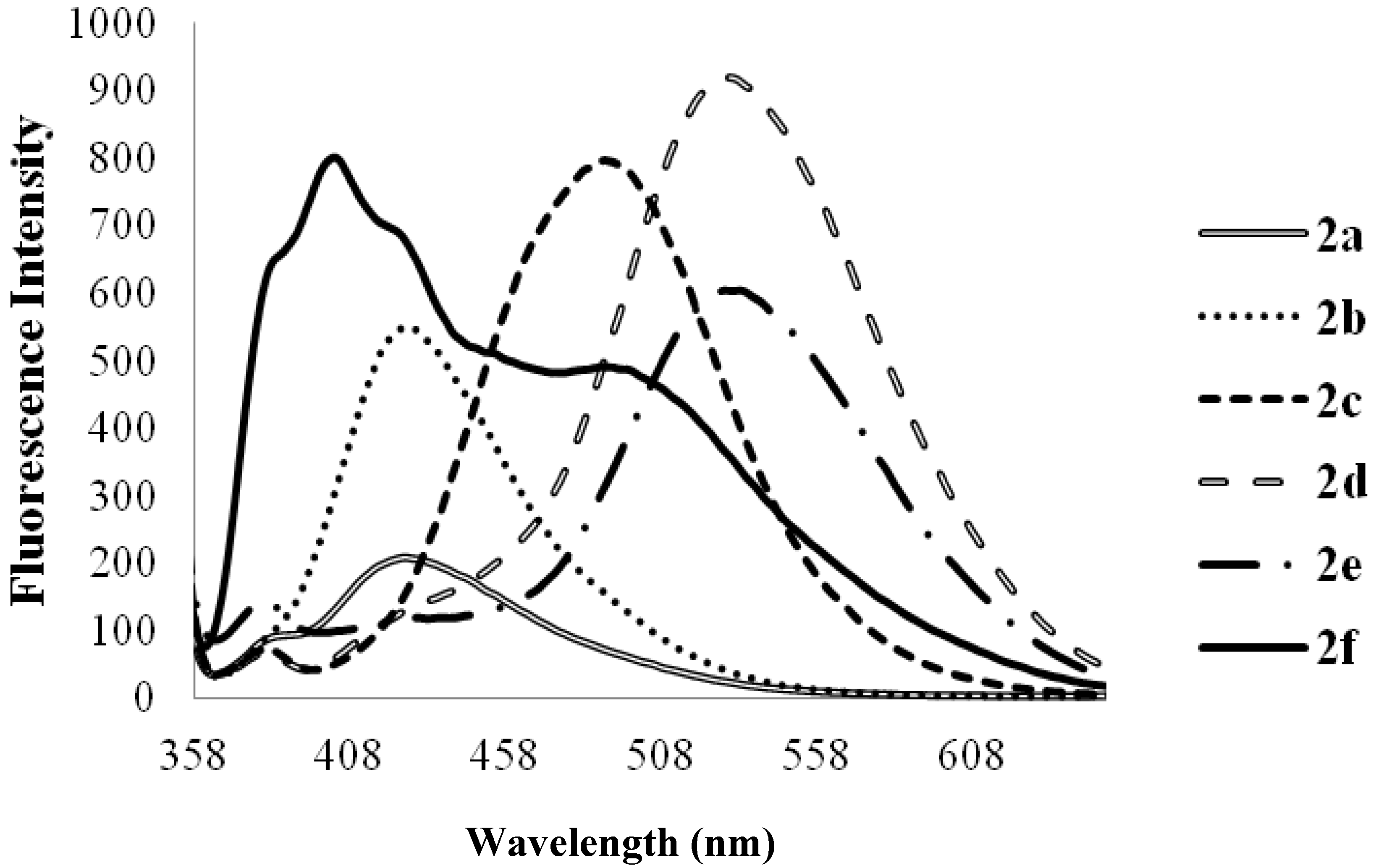
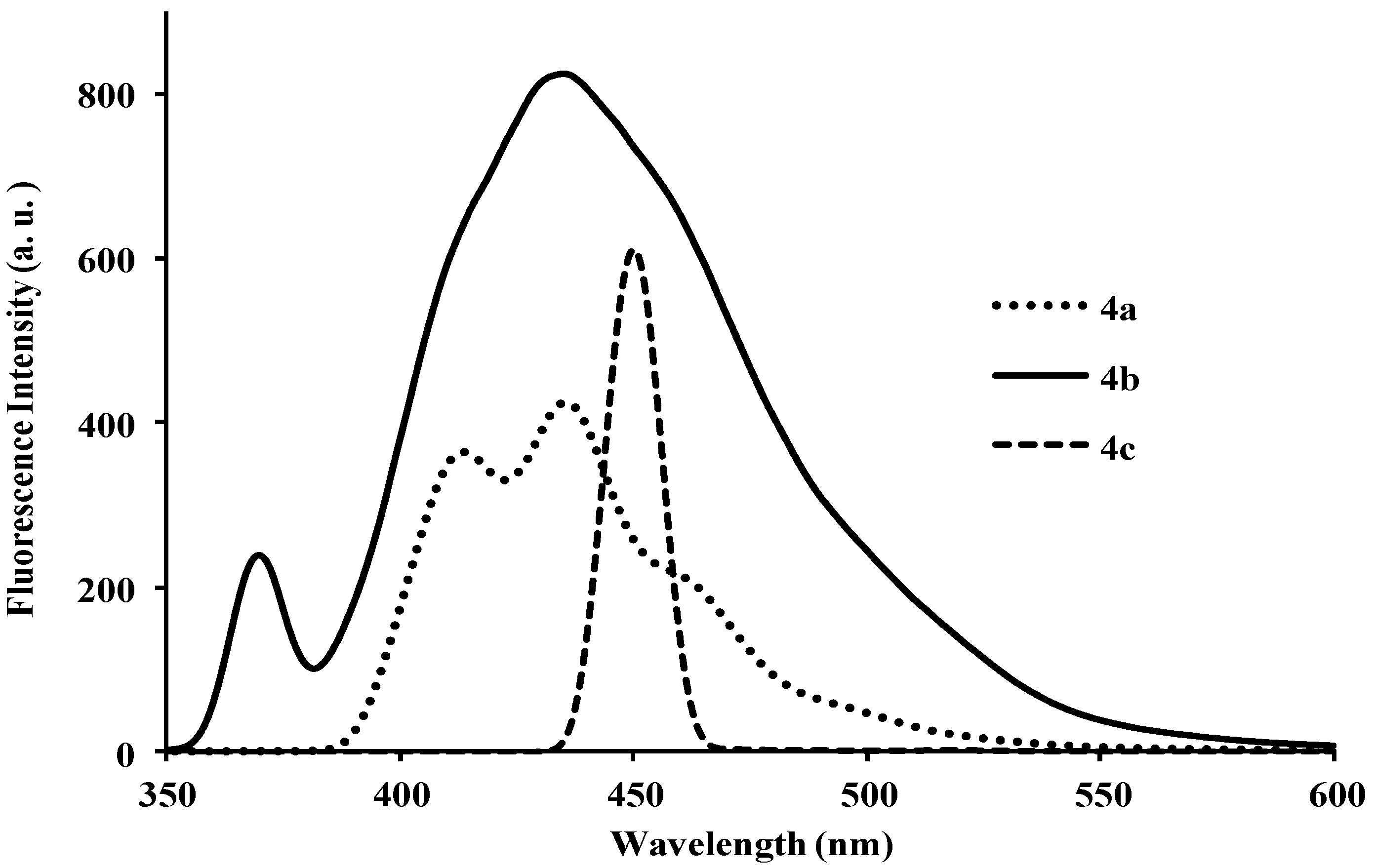
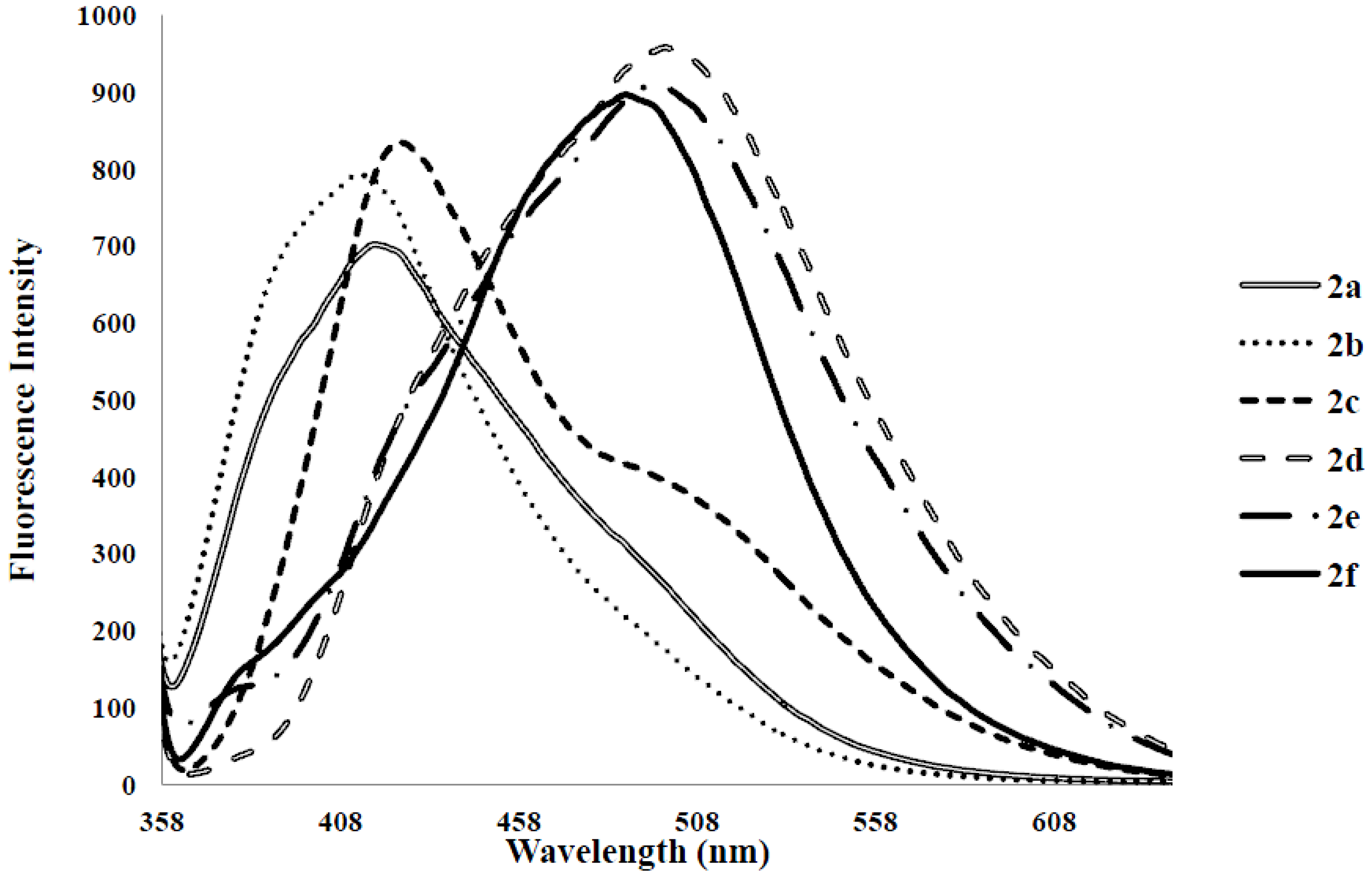
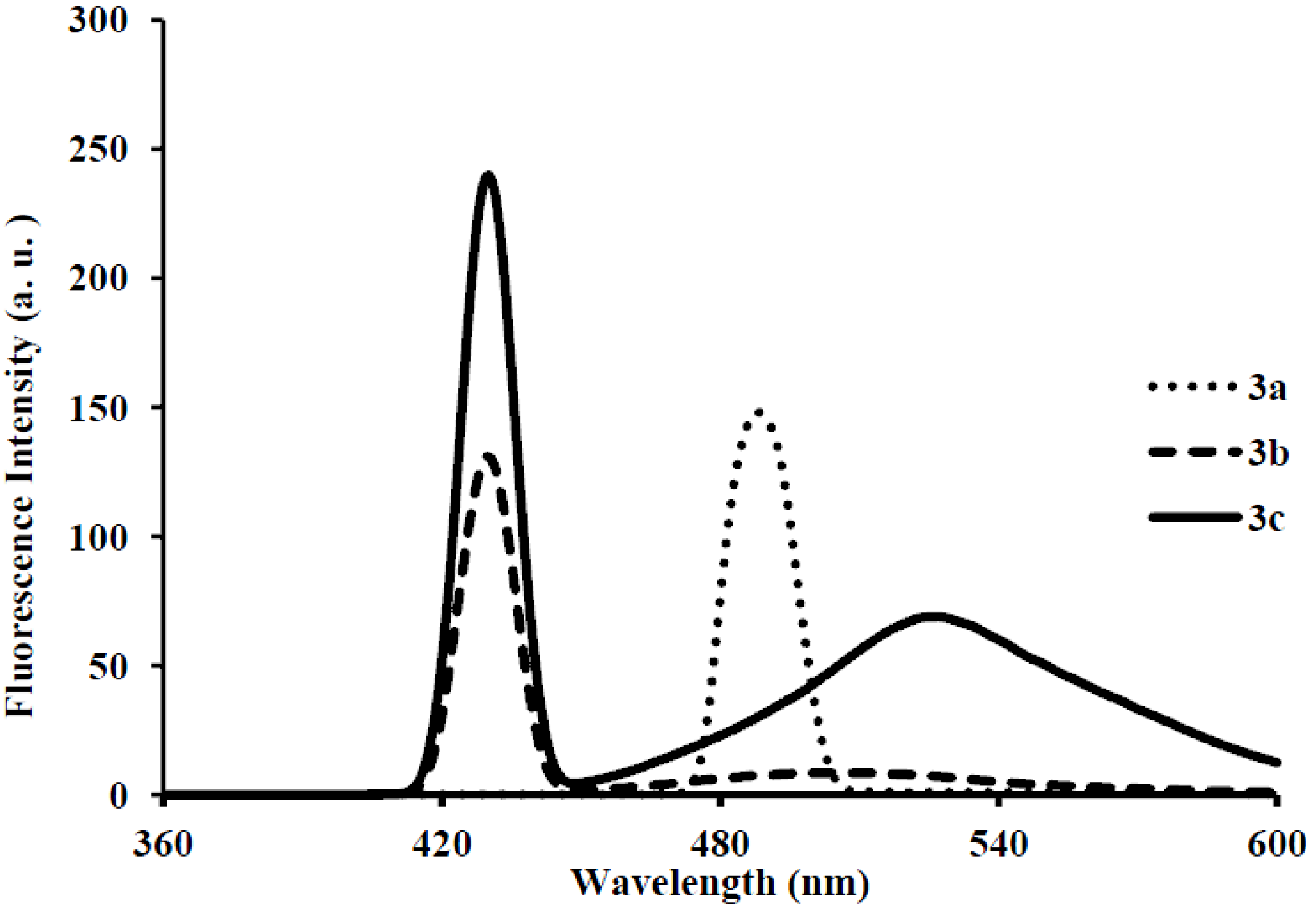
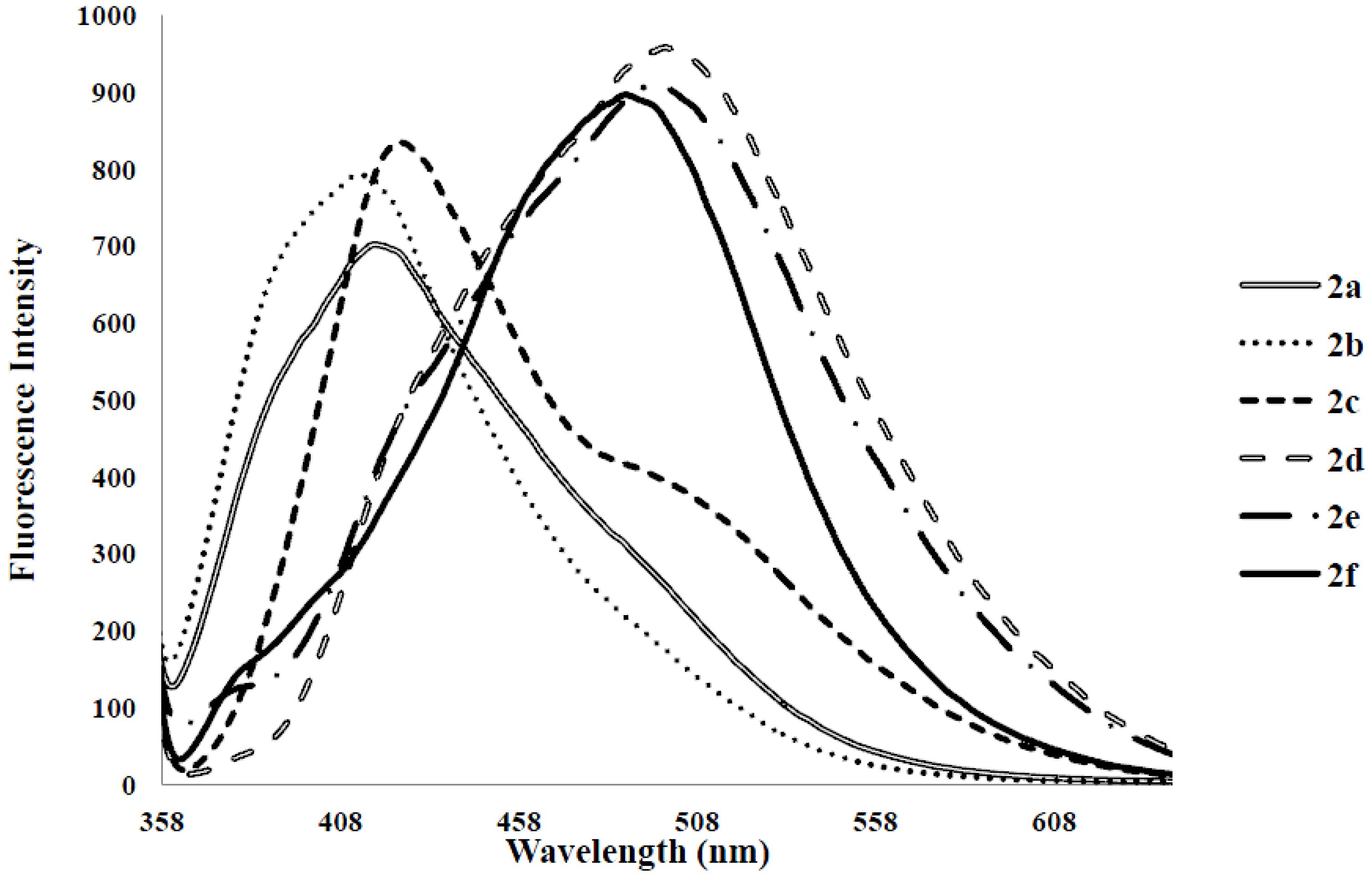
| Compounds | λmax (nm) CHCl3 | (ε) × 104 Mol−1cm−1 | λem (nm) CHCl3 | λem (nm) DMF | λem (nm) CHCl3-CH3OH (1:1) | (a) Quantum Yield (Φ) | Stokes Shift (CHCl3) |
|---|---|---|---|---|---|---|---|
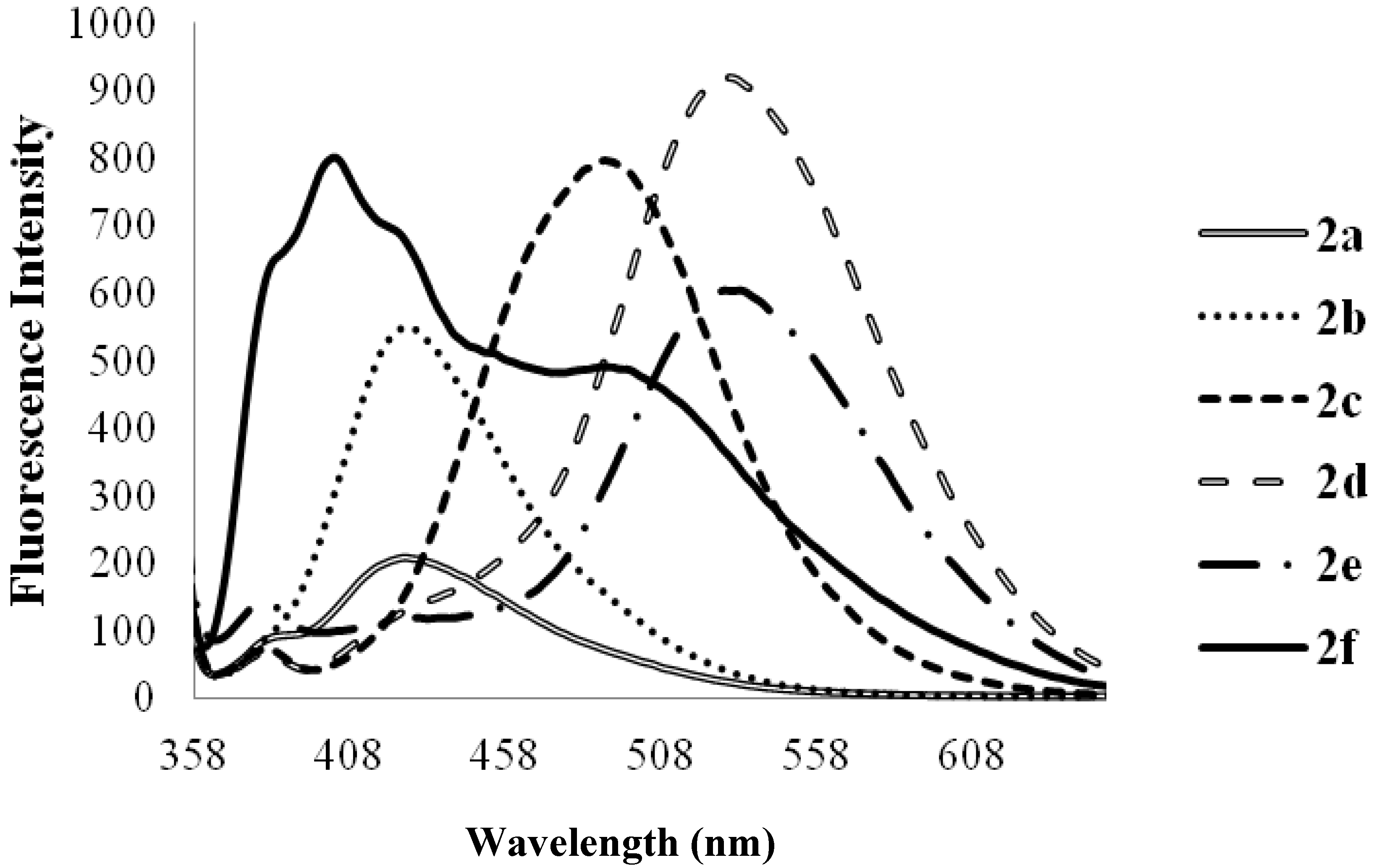
| 2a | 278 | 3.63 | 424 | 425 | 408 | 0.07 | 146 |
| 2b | 275 | 3.06 | 424 | 430 | 406 | 0.14 | 149 |
| 2c | 282 | 1.70 | 474 | 483 | 415 | 0.28 | 192 |
| 2d | 315 | 6.43 | 529 | 543 | 508 | 0.06 | 214 |
| 2e | 347 | 5.99 | 420, 430 | 543 | 504 | 0.17 | 53, 73 |
| 2f | 323 | 4.57 | 370, 470 | 405, 508 | 490 | 0.21 | 47, 147 |
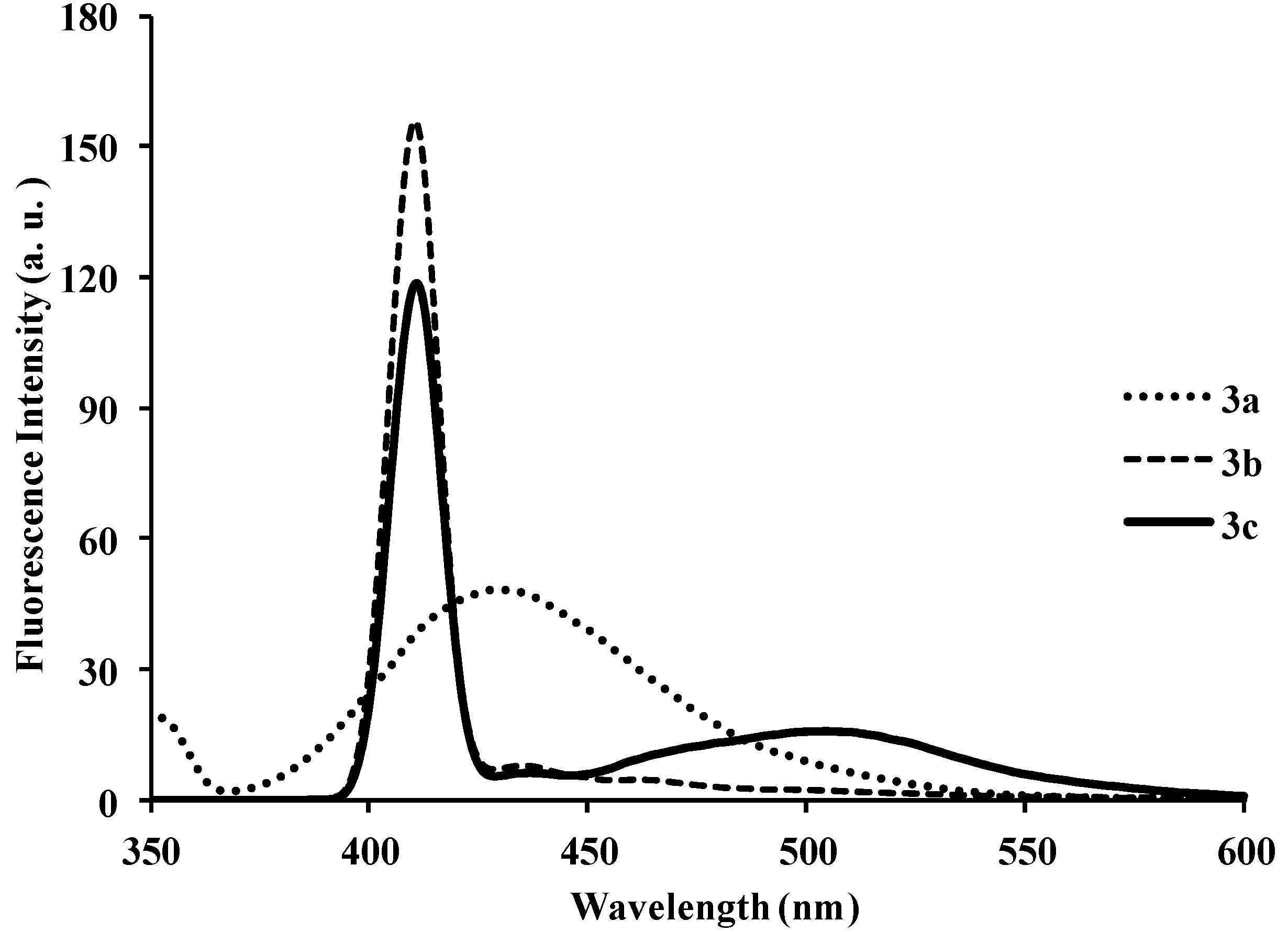
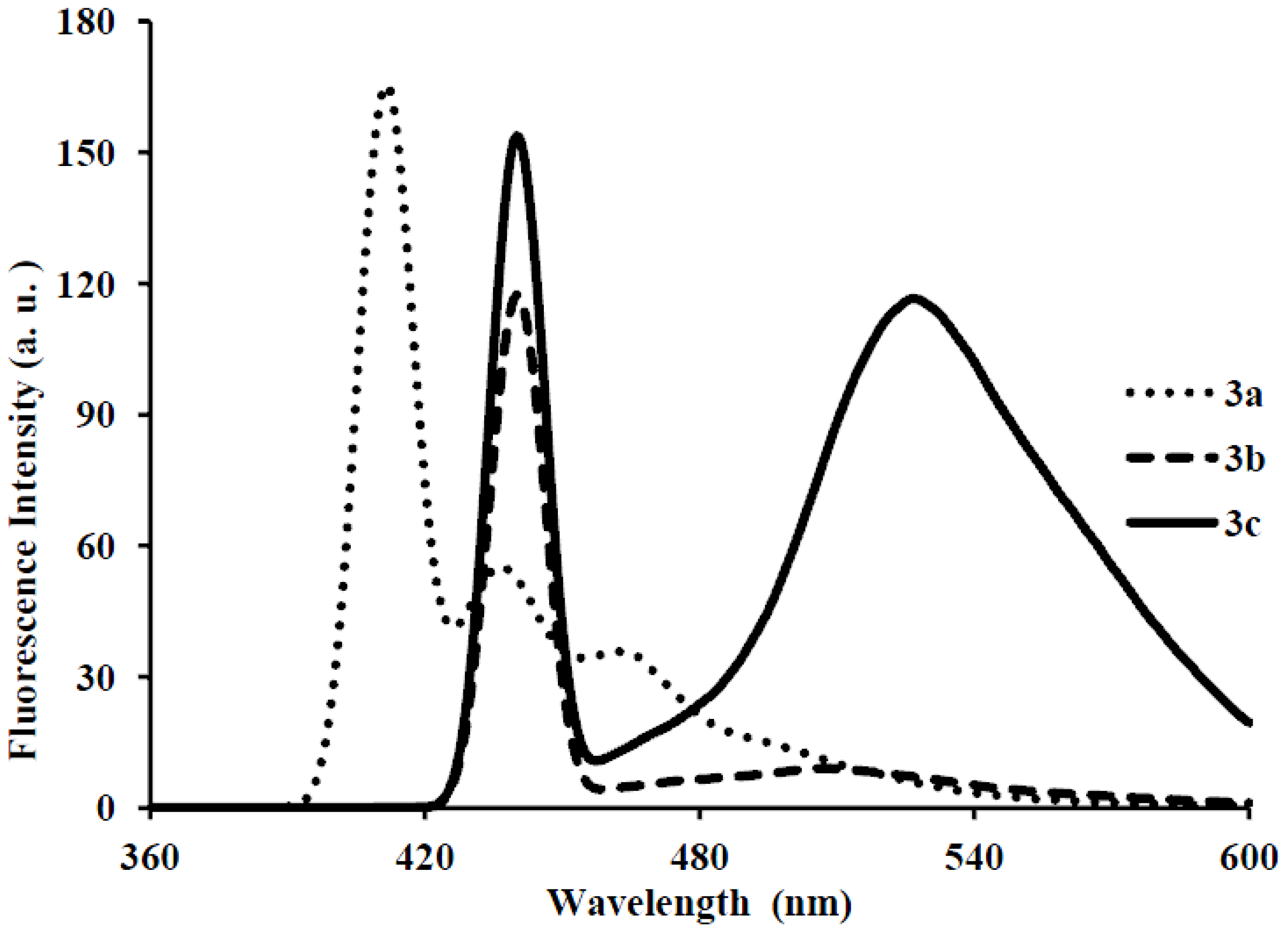
| 3a | 286 | 8.40 | 429 | 411 | 483 | 0.01 | 143 |
| 3b | 286 | 6.64 | 429 | 440 | 430 | 0.03 | 143 |
| 3c | 295 | 5.37 | 410 | 440, 527 | 430, 528 | 0.03 | 115 |
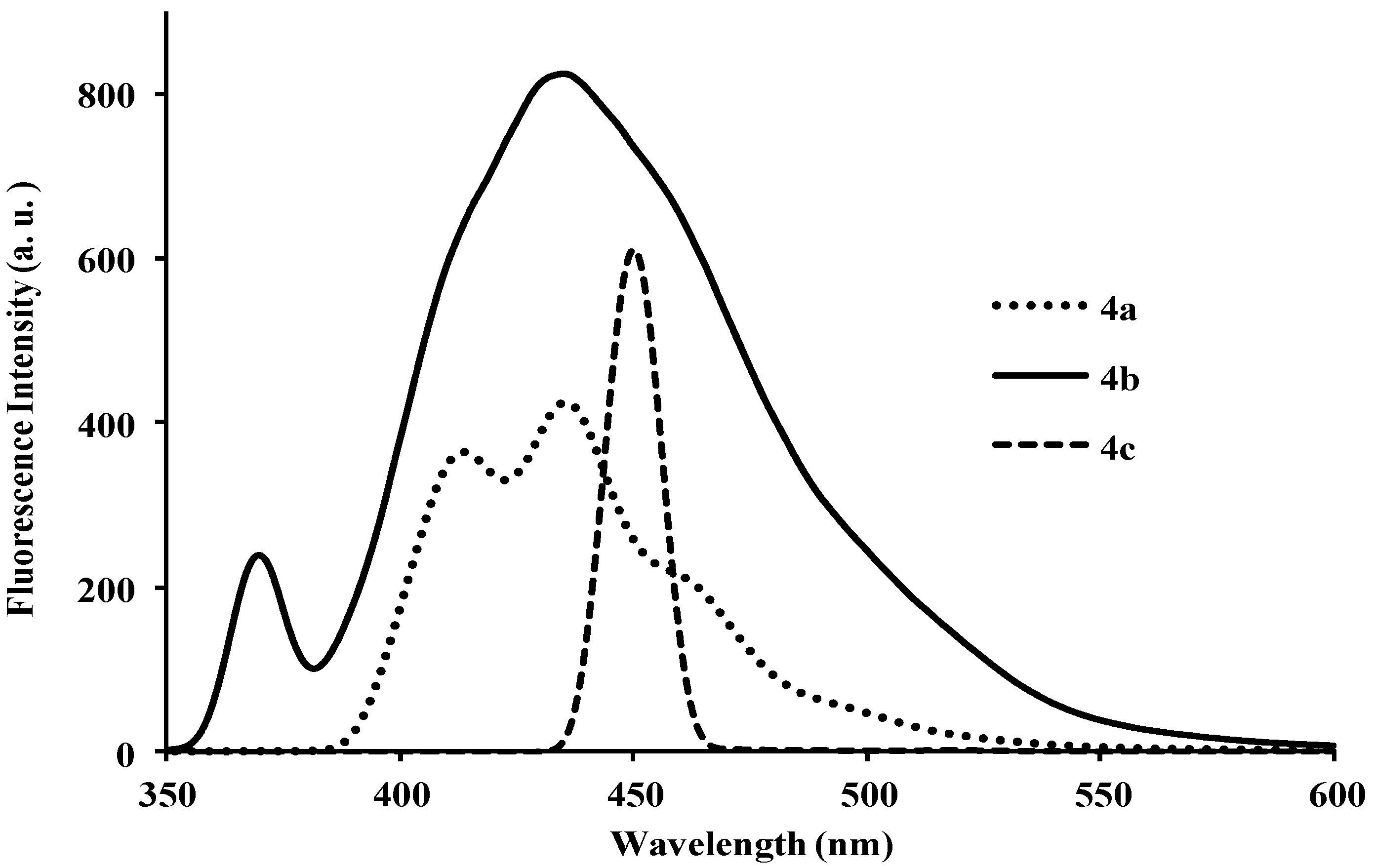
| 4a | 260, 318 | 5.85 | 410 | 464 | 415 | 0.19 | 92, 150 |
| 4b | 260, 320 | 0.92 | 399 | 372, 436 | 400 | 0.21 | 139, 79 |
| 4c | 297 | 1.50 | 461 | 451 | 429 | 0.22 | 164 |
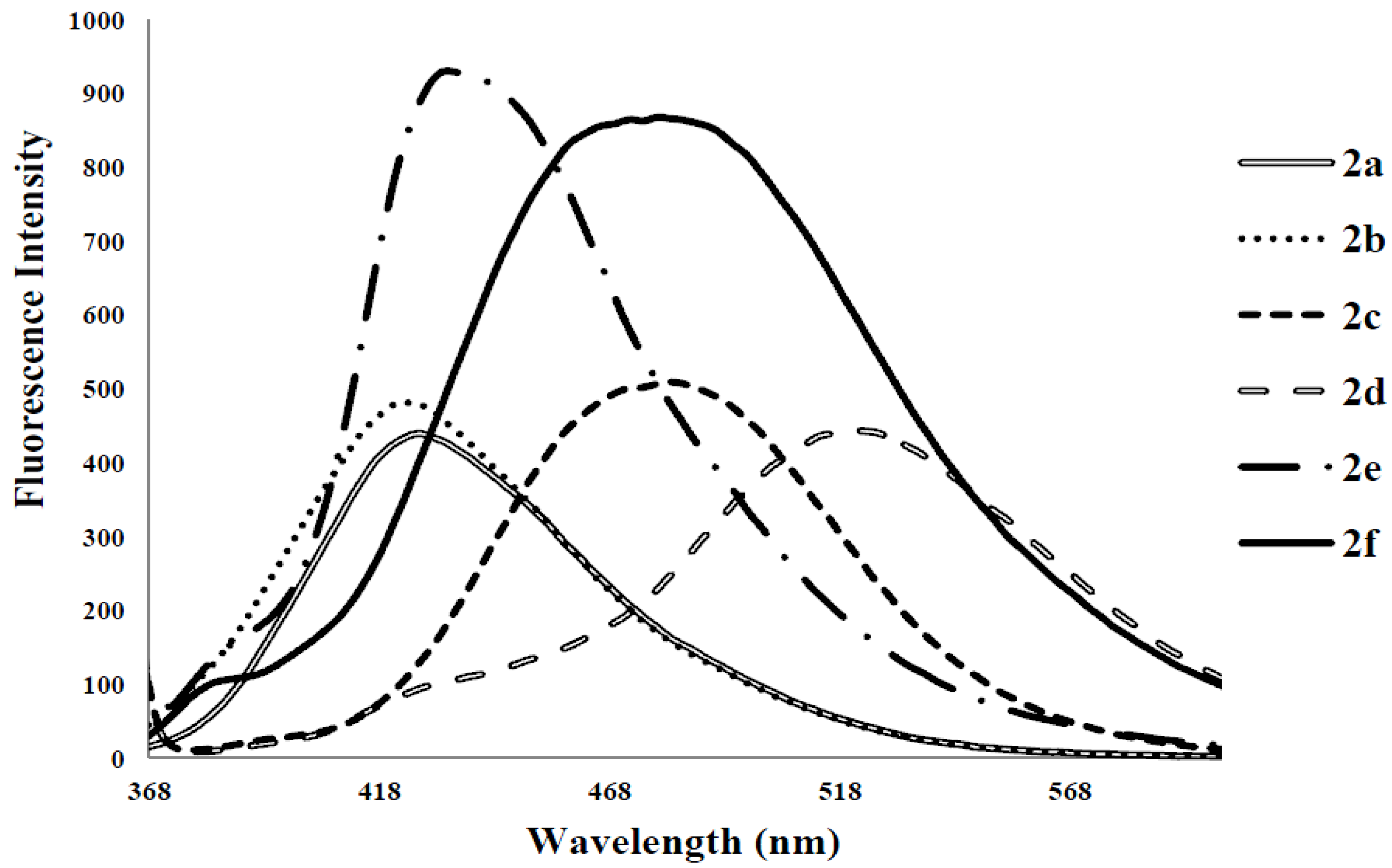



3. Experimental
3.1. General
3.2. Suzuki-Miyaura Cross-Coupling of 1 with Arylboronic Acids. Typical Procedure
3.3. Typical Procedure for the Synthesis of 4,6,8-Triaryl-N-[(quinolin-3-yl)methylene]-3-benzene-amines 3a–c
3.4. Typical Procedure for the Synthesis of 4,6,8-Triarylquinoline-3-methanol 4
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Rotzoll, S.; Willy, B.; Schoenhaber, R.F.; Müller, T.J.J. Regiospecific three-component access to fluorescent 2,4-disubstituted quinolines via one-pot coupling-addition-cyclocondensation-sulfur extrusion sequence. Eur. J. Org. Chem. 2010, 18, 3516–3524. [Google Scholar]
- Seo, J.H.; Kim, Y.K.; Kim, Y.S. New red electrophosphorescent organic light-emitting devices based on Ir(III) complex of 2,3,4-triphenylquinoline. Mol. Cryst. Liq. Cryst. 2008, 491, 194–202. [Google Scholar] [CrossRef]
- Grabowski, Z.R. Electron transfer in flexible molecules and molecular ions. Pure Appl. Chem. 1993, 65, 1751–1756. [Google Scholar] [CrossRef]
- Scherer, T.; van Stokkum, I.H.M.; Brouwer, A.M.; Verhoeven, J.W. Excited-state conformational dynamics of flexibly and semirigidly bridged electron donor-acceptor systems in solution. Influence of temperature and solvent viscosity. J. Phys. Chem. 1994, 98, 10539–10549. [Google Scholar] [CrossRef]
- Lipunova, G.N.; Nosova, E.V.; Trashakhova, T.V.; Charushin, V.N. Azinylarylethenes: Synthesis and photophysical and photochemical properties. Russ. Chem. Rev. 2011, 80, 1115–1133. [Google Scholar] [CrossRef]
- Oevering, H.; Verhoeven, J.W.; Paddon-Row, M.N.; Warman, J.M. Charge-transfer absorption and emission resulting from long-range through-bond interaction; exploring the relation between electronic coupling and electron-transfer in bridged donor-acceptor systems. Tetrahedron 1989, 45, 4751–4766. [Google Scholar] [CrossRef]
- Zamboni, R.; Belley, M.; Champion, E.; Charette, L.; DeHaven, R.; Frenette, R.; Gauthier, J.Y.; Jones, T.R.; Leger, S.; Masson, P.; et al. Development of a novel series of styrylquinoline compounds as high-affinity leukotriene D4 receptor antagonists: Synthetic and structure-activity studies leading to the discovery of (±)-3-[[[3-[2-(7-chloro-2-quinolinyl)-(E)-ethenyl]phenyl][[3-(dimethylamino)-3-oxopropyl]thio]methyl]thio]propionic acid. J. Med. Chem. 1992, 35, 3832–3844. [Google Scholar] [CrossRef]
- Kim, H.M.; Jin, J.H.; Lee, C.J.; Kim, N.; Park, K.H. Synthesis and characterization of nonlinear optical polymers having quinoline-based chromophores. Bull. Korean Chem. Soc. 2002, 23, 964–970. [Google Scholar] [CrossRef]
- Wu, F.-I.; Su, S.-J.; Shu, C.-F.; Luo, L.; Diau, W.-G.; Cheng, C.-H.; Duan, J.-P.; Lee, G.-H. Tuning the emission and morphology of cyclometalated iridium complexes and their applications to organic light-emitting diodes. J. Mater. Chem. 2005, 15, 1035–1042. [Google Scholar] [CrossRef]
- Chen, L.; You, H.; Yang, C.; Zhang, X.; Qin, J.; Ma, D. Tuning the saturated red emission: Synthesis, Electrochemistry and photophysics of 2-arylquinoline based iridium(III) complexes and their application in OLEDs. J. Mater. Chem. 2006, 16, 3332–3339. [Google Scholar] [CrossRef]
- Jenekhe, S.A.; Lu, L.; Alam, M.M. New conjugated polymers with donor-acceptor architectures: Synthesis and photophysics of carbazole-quinoline and phenothiazine-quinoline copolymers and oligomers exhibiting large intramolecular charge transfer. Macromolecules 2001, 34, 7315–7324. [Google Scholar] [CrossRef]
- Kimyonok, A.; Wang, X.-Y.; Weck, M. Electroluminescent poly(quinoline)s and metalloquinolates. Polym. Rev. 2006, 46, 47–77. [Google Scholar]
- Qi, S.; Shi, K.S.; Gao, H.; liu, Q.; Wang, H. Synthesis and fluorescence properties of 5,7-diphenylquinoline and 2,5,7-triphenylquinoline derived from m-terphenylamine. Molecules 2007, 12, 988–996. [Google Scholar] [CrossRef]
- Verhoeven, J.W. Electron transport via saturated hydrocarbon bridges: ‘Exciplex’ emission from flexible, rigid and semiflexible bichromophores. Pure Appl. Chem. 1990, 62, 1585–1596. [Google Scholar] [CrossRef]
- Wang, P.; Wu, S. A study on the spectroscopy and photophysical behavior of chalcone derivatives. J. Photochem. Photobiol. A Chem. 1994, 77, 127–137. [Google Scholar] [CrossRef]
- Raju, B.B.; Varadarajan, T.S. Spectroscopic studies of 7-diethylamino-3-styryl coumarins. J. Photochem. Photobiol. A Chem. 1995, 85, 263–267. [Google Scholar] [CrossRef]
- Yang, Y.; Jia, H.-M.; Liu, B.-L. (E)-5-styryl-1H-indole and (E)-6-styrylquinoline derivatives serve as probes for β-amyloid plaques. Molecules 2012, 17, 4252–4265. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A. Mechanistic and kinetic studies of palladium catalytic systems. J. Organomet. Chem. 1999, 576, 254–278. [Google Scholar] [CrossRef]
- Grushin, V.V.; Alper, H. Transformations of chloroarenes, catalyzed by transition-metal complexes. Chem. Rev. 1994, 94, 1047–1062. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Tsvetkov, A.V.; Latyshev, G.V.; Lukashev, N.V. Successive replacement of halogen atoms in 4,6-dihaloquinolines in cross-coupling reactions with arylboronic acids catalyzed by palladium and nickel complexes. Russ. J. Org. Chem. 2003, 39, 1660–1667. [Google Scholar] [CrossRef]
- Garcia, Y.; Schoenebeck, F.; Legault, C.Y.; Merlic, C.A.; Houk, K.N. Theoretical bond dissociation energies of halo-heterocycles: Trends and relationships to regioselectivity in palladium-catalyzed cross-coupling reactions. J. Am. Chem. Soc. 2009, 131, 6632–6639. [Google Scholar] [CrossRef]
- Piala, A.; Mayi, D.; Handy, S.T. Studies of one-pot double couplings on dibromoquinolines. Tetrahedron 2011, 6, 4147–4154. [Google Scholar] [CrossRef]
- Kappaun, S.; Sovic, T.; Stelzer, F.; Pogantsch, A.; Zojer, E.; Slugovc, C. Molecular fluorescent pH-probes based on 8-hydroxyquinoline. Org. Biomol. Chem. 2006, 4, 1503–1511. [Google Scholar] [CrossRef]
- Khoza, T.A.; Maluleka, M.M.; Mama, N.; Mphahlele, M.J. Synthesis and photophysical properties of the 2-aryl-6,8-bis(arylethenyl)-4-methoxyquinolines. Molecules 2012, 17, 14186–14204. [Google Scholar] [CrossRef]
- Akrawi, O.A.; Mohammed, H.H.; Langer, P. Synthesis and Suzuki-Miyaura reactions of 3,6,8-tribromoquinoline: A structural revision. Synlett 2013, 24, 1121–1124. [Google Scholar] [CrossRef]
- Dahlén, K.; Wallén, E.A.A.; Grotli, M.; Luthman, K. Synthesis of 2,3,6,8-tetrasubstituted chromone scaffolds. J. Org. Chem. 2006, 71, 6863–6871. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, J.; Li, W.; Lee, H.; Lu, B.Z.; Senanayake, C.H. Synthesis of 8-arylquinolines via one-pot Pd-catalyzed borylation of quinoline-8-yl halides and subsequent Suzuki–Miyaura coupling. J. Org. Chem. 2011, 76, 6394–6400. [Google Scholar]
- Nayyar, A.; Malde, A.; Coutinho, E.; Jain, R. Synthesis, Anti-tuberculosis activity, and 3D-QSAR study of ring-substituted-2/4-quinolinecarbaldehyde derivatives. Bioorg. Med. Chem. 2006, 14, 7302–7310. [Google Scholar] [CrossRef]
- Moglie, Y.; Alonso, F.; Vitale, C.; Yusb, M.; Radivoy, G. New active-iron based reducing system for carbonyl compounds and imines. Stereoselective reduction of cyclic ketones. Tetrahedron 2006, 62, 2812–2818. [Google Scholar] [CrossRef]
- Miecznikowski, J.R.; Crabtree, R.H. Transfer hydrogenation reduction of ketones, Aldehydes and imines using chelated iridium(III) N-heterocyclic bis-carbene complexes. Polyhedron 2004, 23, 2857–2872. [Google Scholar] [CrossRef]
- Kazemi, F.; Kiasat, A.R.; Sarvestani, F. Practical reduction of imines by NaBH4/alumina under solvent-free conditions: An efficient route to secondary amine. Chin. Chem. Lett. 2008, 19, 1167–1179. [Google Scholar] [CrossRef]
- Chen, G.S.; Talekar, R.S.; Wong, K.-T.; Chi, L.-C.; Chern, J.-W. Physical properties of 8-substituted 5,7-dichloro-2-styrylquinolines as potential light emitting materials. J. Chin. Chem. Soc. 2007, 54, 1387–1394. [Google Scholar]
- Diaz, A.N. Absorption and emission spectroscopy and photochemistry of 1,10-anthraquinone derivatives: A review. J. Photochem. Photobiol. A Chem. 1990, 53, 141–167. [Google Scholar] [CrossRef]
- Kalinowski, J. Excimers and exciplexes in organic electroluminescence. Mater. Sci.-Pol. 2009, 27, 735–756. [Google Scholar]
- Wang, S.-L.; Ho, T.-I. Substituent effects on intramolecular charge-transfer behavior of styrylhetrocycles. J. Photochem. Photobiol. A Chem. 2000, 135, 119–126. [Google Scholar]
- Cossiello, R.F.; Akcelrud, L.; Atvars, T.D.Z. Solvent and molecular weight effects on fluorescence emission of MEH-PPV. J. Braz. Chem. Soc. 2005, 16, 74–86. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Kwong, R.C.; Thompson, M.E.; Schwartz, B.J. Improving the performance of conjugated polymer-based devices by control of interchain interactions and polymer film morphology. Appl. Phys. Lett. 2000, 76, 2454–2456. [Google Scholar] [CrossRef]
- Tian, B.; Zerbi, G.; Schenk, R.; Mullen, K. Optical spectra and structure of oligomeric models of polyparaphenylenevinylene. J. Chem. Phys. 1991, 95, 3191–3197. [Google Scholar] [CrossRef]
- Sunahara, H.; Urano, Y.; Kojima, H.; Nagano, T. Design and synthesis of a library of BODIPY-based environmental polarity sensors utilizing photoinduced electron-transfer-controlled fluorescence on/off switching. J. Am. Chem. Soc. 2007, 129, 5597–5604. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1, 2a–f, 3a–c and 4a–c are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mphahlele, M.J.; Adeloye, A.O. 4,6,8-Triarylquinoline-3-carbaldehyde Derivatives: Synthesis and Photophysical Properties. Molecules 2013, 18, 15769-15787. https://doi.org/10.3390/molecules181215769
Mphahlele MJ, Adeloye AO. 4,6,8-Triarylquinoline-3-carbaldehyde Derivatives: Synthesis and Photophysical Properties. Molecules. 2013; 18(12):15769-15787. https://doi.org/10.3390/molecules181215769
Chicago/Turabian StyleMphahlele, Malose Jack, and Adewale Olufunsho Adeloye. 2013. "4,6,8-Triarylquinoline-3-carbaldehyde Derivatives: Synthesis and Photophysical Properties" Molecules 18, no. 12: 15769-15787. https://doi.org/10.3390/molecules181215769
APA StyleMphahlele, M. J., & Adeloye, A. O. (2013). 4,6,8-Triarylquinoline-3-carbaldehyde Derivatives: Synthesis and Photophysical Properties. Molecules, 18(12), 15769-15787. https://doi.org/10.3390/molecules181215769