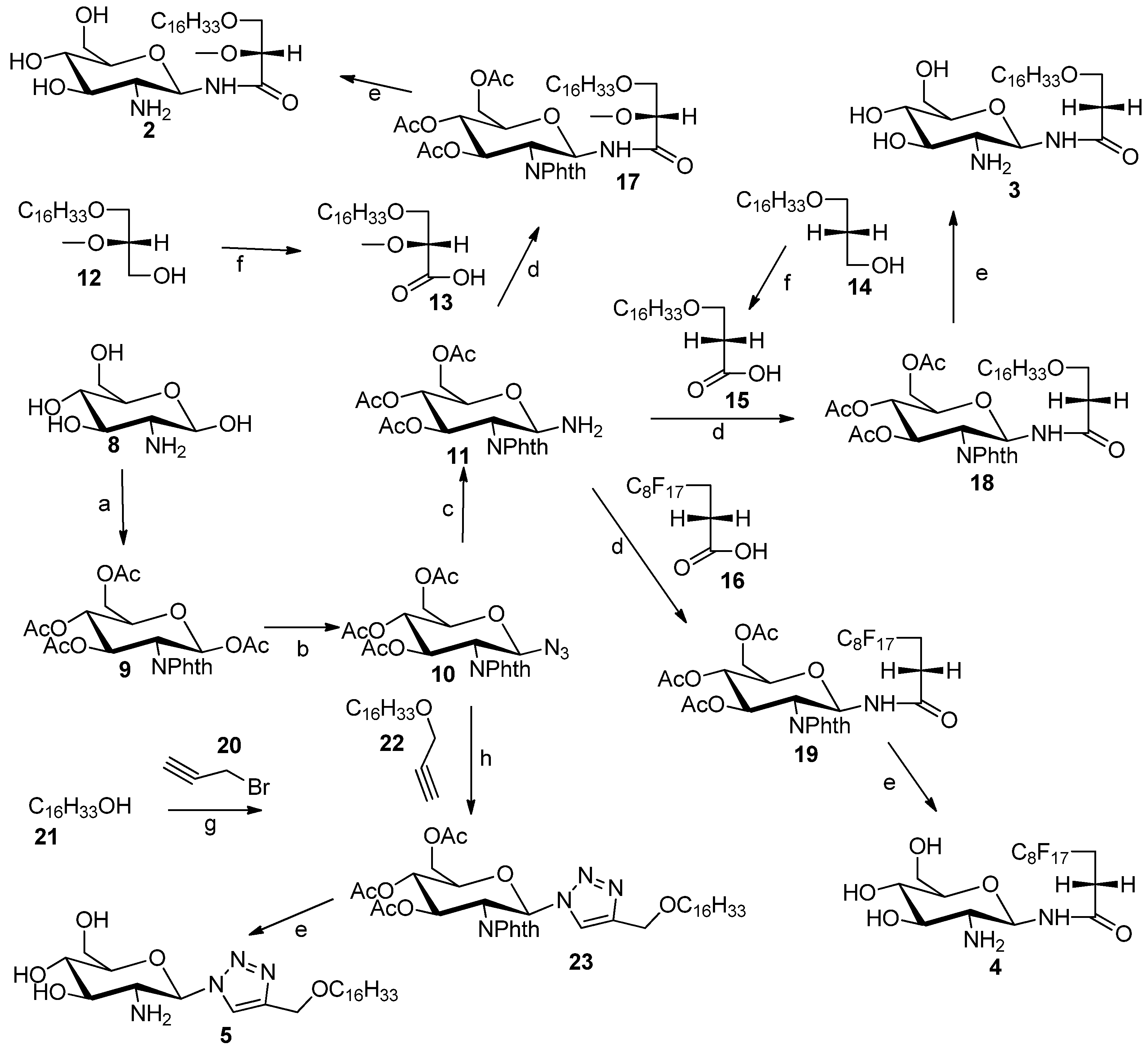
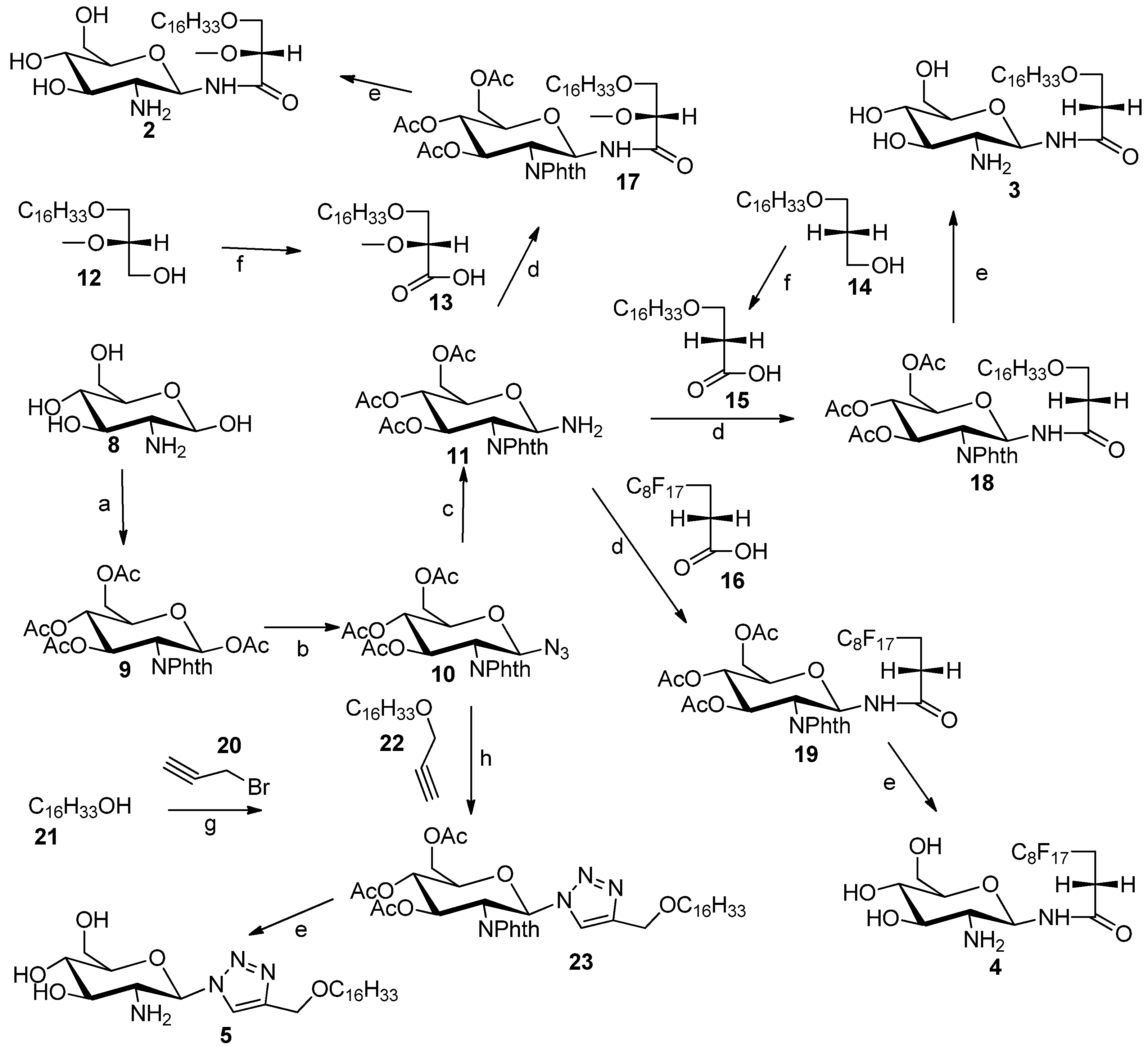
3.2. General Procedure for the Synthesis of N-Linked Compounds 2–5
1,3,4,6-Tetra-O-acetyl-2-deoxy-2-N-phthalimido-β-d-glucopyranoside (
9). Glucosamine hydro-chloride
8 (3.016 g, 14 mmol) and NaOH (28 mmol) were dissolved in water (50 mL). The resulting mixture was stirred at room temperature for 30 min. Phthalic anhydride (2.34 g, 0.0157 mol) was added to the solution. The mixture was stirred vigorously at room temperature for 16 h. The mixture was concentrated and dried using rotary evaporator. The residue was dissolved in pyridine (30 mL), and then Ac
2O (19.8 mL) was added to the solution. The resulting solution was allowed to stir vigorously overnight. The reaction was checked by the TLC. Methanol (6 mL) was used to quench the excess of Ac
2O, and then excess pyridine was removed under high vacuum. The remaining solid was dissolved in CH
2Cl
2 (40 mL), and then the solution was washed with 10% HCl (40 mL×1), Saturated NaHCO
3 solution (40 mL×3), H
2O (40 mL×1) and brine (40 mL×1) and dried over anhydrous MgSO
4. The final solution was concentrated under reduced pressure, and the obtained product
8 (3.3g, 49.4%) was dried overnight. NMR data were consistent with data in the literature [
23].
3,4,6-tri-O-Acetyl-2-deoxy-2-N-phthalimido-β-d-glucopyranosyl azide (
10). Compound
9 (1.8 g, 8 mmol) and trimethylsilyl azide (1.7 g, 148 mmol) were both dissolved in CH
2Cl
2 (20 mL) in a 100 mL-round bottom flask with vigorous stirring. Then FeCl
3 (1.77 g) was added to the reacting mixture. The reaction was allowed to stir for 24 h and then progress was checked by TLC. The dispersion solution was made by mixing hexane and ethyl acetate in 1:1 ratio. The solution was concentrated under reduced pressure by rotary evaporator. The product
10 was isolated and purified by column chromatography (1:2 ethyl acetate/hexane). The obtained product
10 (1.3 g, 76.53%) was a light yellow solid. NMR data were consistent with previously published data [
16].
3,4,6-tri-O-Acetyl-1-amino-2-deoxy-2-N-phthalimido-β-d-glucopyranose (11). Compound 10 (0.11 g, 0.24 mmol) was dissolved in methanol (2 mL) in a 25 mL-round bottom flask with vigorous stirring, and then Pd/C (0.29 g) was added. After that, round bottom flask was connected to a hydrogen balloon. The reaction was allowed to take place for half hour, and checked by TLC. The reaction was stopped when all starting material has disappeared, and the solution was concentrated to provide compound 11 (100 mg, 96%). This compound was not characterised by 1H-NMR, because it is chemically unstable. Therefore it was directly used for the next step, i.e., coupling of carboxylic acids using TBTU as coupling reagent.
3-(Hexadecyloxy)-2-methoxypropanoic acid (13). A 2.7 M solution of Jones reagent (0.5 mL) was added dropwise into a stirred solution of commercially available 3-O-hexadecyloxy-2-O-methyl-sn-glycerol 12 (23 mg) in acetone (5 mL) at 0 °C. The reaction was monitored by TLC and was completed after 1 h. Isopropyl alcohol was added dropwise until a green color remained, to remove excess of CrO3 totally. The organic solvent was removed under reduced pressure. The solid was dissolved in water, and then 3-(hexadecyloxy)-2-methoxypropanoic acid was extracted using ethyl acetate. The combined organic layers were washed with brine, dried using MgSO4 and concentrated under vacuum. The 3-(hexadecyloxy)-2-methoxypropanoic acid (13) was purified by silica gel column flash chromatography (4:1 hexane/ethyl acetate) to yield white solid (16 mg, 70%). 1H-NMR (CDCl3): δ = 4.00–3.75 (m, 3H), 3.55 (s, 3H, –OCH3), 2.66 (t, J = 6.4 Hz, 2H, –OCH2–CH2–(CH2)13), 1.59 (m, 2H, –CH2–(CH2)13), 1.23–1.34 (brs, 26H, –(CH2)13), 0.90 (t, J = 6.9 Hz, 3H, –CH3). ESMS: calcd for C20H40O4Na+m/z 367.3; found: m/z [M+Na]+ 367.3.
3-(Hexadecyloxy)-propanoic acid (15). A 2.7 M solution of Jones reagent (2.0 mL) was added dropwise into a stirred solution of compound 14 (0.124 g, 0.4 mmol) in acetone (20 mL) at 0 °C. The reaction was monitored by TLC and was completed after 1 h. Isopropyl alcohol was added dropwise until a stable green color appeared, to remove the excess of CrO3. The organic solvent was removed under reduced pressure. The solid was dissolved in water, and then compound 15 was extracted using ethyl acetate. The combined organic layers were washed by brine, dried using MgSO4 and concentrated under reduced pressure. The compound 15 was purified by silica gel column flash chromatography (4:1 hexane/ethyl acetate) to yield white solid (63 mg, 50.1%). 1H-NMR (CDCl3): δ = 3.72 (t, J = 6.3 Hz, 2H, –CH2–COOH), 3.48 (t, J = 6.7 Hz, 2H, –OCH2–CH2–COOH), 2.65 (t, J = 6.3 Hz, 2H, –OCH2–CH2–(CH2)13), 1.59 (m, 2H, –CH2–(CH2)13), 1.23–1.34 (brs, 26H, –(CH2)13 0.90 (t, J = 6.9 Hz, 3H, CH3). ESMS: calcd for C19H38O3Na+m/z 337.3; found: m/z [M+Na]+ 337.4
3-Hexadecyloxy-2-methoxyl-1-N-(3,4,6-tri-O-acetyl-2-deoxy-2-N-phthalimido-β-d-glucopyranosyl)-propanamide (17). Glucosylamine 11 (110 mg, 0.25 mmol), carboxylic acid 13 (110 mg, 0.32 mmol), DIPEA (0.40 mL) and TBTU (0.245 g) were dissolved in DMF (10 mL) in a round bottom flask with vigorous stirring overnight and was monitored by TLC. At the completion of the reaction DMF was removed under reduced pressure to obtain a solid residue. The solid residue was dissolved in water, and the product was extracted by ethyl acetate. The combined organic layers were concentrated under reduced pressure to yield a light yellow compound 17 (130 mg, 68%). 1H-NMR (CDCl3): δ = 8.84 (s, br. 1H), 7.93–7.67 (m, 4H, phthalimido), 7.11(d, J = 9.0 Hz, 1H, H–1), 6.14–5.96 (m, 2H, H–2, H–3), 5.19 (dd, J1 = J2 = 12 Hz, 1H, H–4), 4.44–3.54 (m, 8H), 3.26 (s, 3H, –OCH3), 2.12(s, 3H, acetate–CH3), 2.06 (s, 3H, acetate–CH3), 1.89 (s, 3H, acetate–CH3), 1.53 (m, 2H–CH2–(CH2)13), 1.39–1.15 (brs, 26H, (CH2)13), 0.90 (t, J = 6.0 Hz, 3H, CH3). ES-MS: calcd for C40H60N2O12Na+m/z 783.4: found: m/z [M+Na]+ 783.6.
3-Hexadecyloxy-1-N-(3,4,6-tri-O-acetyl-2-deoxy-2-N-phthalimido-β-d-glucopyranosyl)-propanamide (18). Glucosylamine 11 (100 mg, 0.23 mmol) and carboxylic acid 15 (43.7 mg, 0.15 mmol) and DIPEA (0.04 g) and TBTU (0.04 g) and DMF (4 mL) were added into a 25-mL round bottom flask with vigorous stirring. The reaction allowed to stir for overnight and was monitored by TLC. DMF was removed under reduced pressure to obtain solid. The solid was dissolved in water, and the product was extracted by ethyl acetate. The combined organic layers were concentrated by reduced pressure rotary evaporator to yield a light yellow compound 18 (50 mg, 45.5%). 1H-NMR (CDCl3): δ = 8.84 (s, br. 1H), 7.89–7.76 (m, 4H, phthalimido), 7.03 (d, J = 9.0 Hz, 1H, H–1), 6.08 (dd, J1 = J2 = 9.8 Hz, 1H, H-3), 6.02 (dd, J1 = J2 = 9.6 Hz, 1H, H–2), 5.18 (dd, J1 = J2 = 9.8 Hz, 1H, H–4), 4.36 (m, 1H, H–5), 4.28 (dd, J1 = J2 = 10.2 Hz, 2H, ), 4.09 (dd, J1 = J2 = 10.0 Hz, 2H, H–6), 3.51 (m, 1H), 3.34 (m, 2H), 2.26 (m, 2H,), 2.12 (s, 3H, acetate–CH3), 2.05 (s, 3H, acetate–CH3), 1.88 (s, 3H, acetate–CH3), 1.53 (m, 2H, CH2–(CH2)13), 1.31–1.28 (brs, 26H, –(CH2)13), 0.90 (t, J = 6.9 Hz, 3H, –CH3). ES-MS: calcd for C39H58N2O11Na+m/z 753.4: found: m/z [M+Na]+ 753.8.
Heptadecylfluoro-1-N-(3,4,6-tri-O-acetyl-2-deoxy-2-N-phthalimido-β-d-glucopyranosyl)-undecanamide (19). Glucosylamine 11 (110 mg, 0.25 mmol), fluorinated carboxylic acid (250 mg, 0.51 mmol), DIPEA (0.04 g), TBTU (0.245 g) and DMF (10 mL) were added into a 25-mL round bottom flask with vigorous stirring. The reaction allowed to stir for overnight and was monitored by TLC. DMF was removed under reduced pressure to obtain solid. The solid was dissolved in water, and the product was extracted by ethyl acetate. The combined organic layers were concentrated by reduced pressure rotary evaporator to yield a light yellow compound 19 (105 mg, 45%). 1H-NMR (CDCl3): δ = 8.93 (s, br. 1H), 7.93–7.69 (m, 4H, phthalimido), 7.05 (d, J = 9.0 Hz, 1H, H–1), 6.15–5.99 (m, 2H, H–2, H–3), 5.17 (dd, J1 = J2 = 9.0 Hz, 1H, H–4), 4.45–3.98 (m, 5H), 2.38 (m, 2H), 2.13 (s, 3H, acetate–CH3), 2.06 (s, 3H, acetate–CH3), 1.89 (s, 3H, acetate–CH3). ES-MS: calcd for C31H25F17N2O10Na+m/z 931.1: found: m/z [M+Na]+ 931.5.
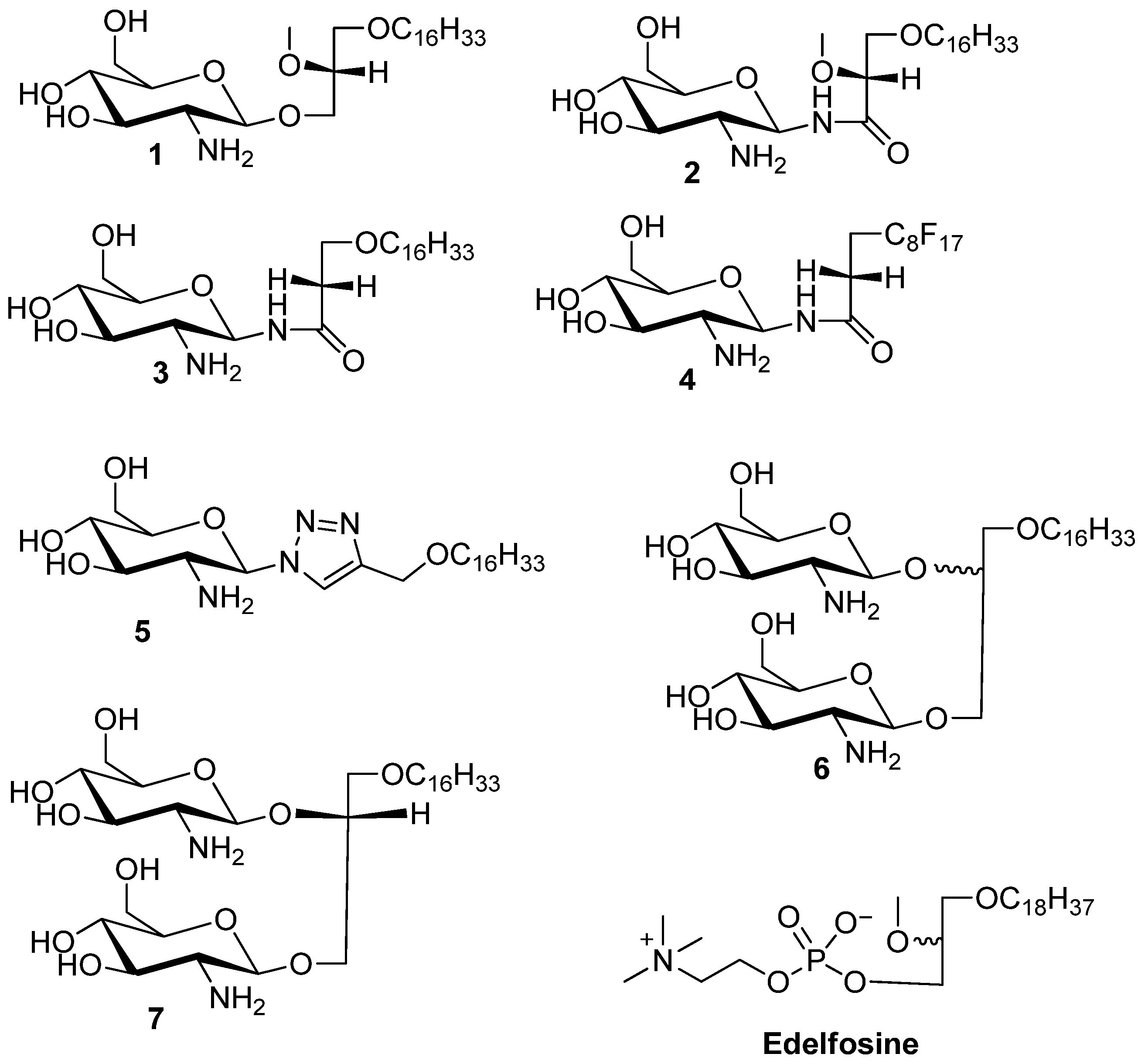
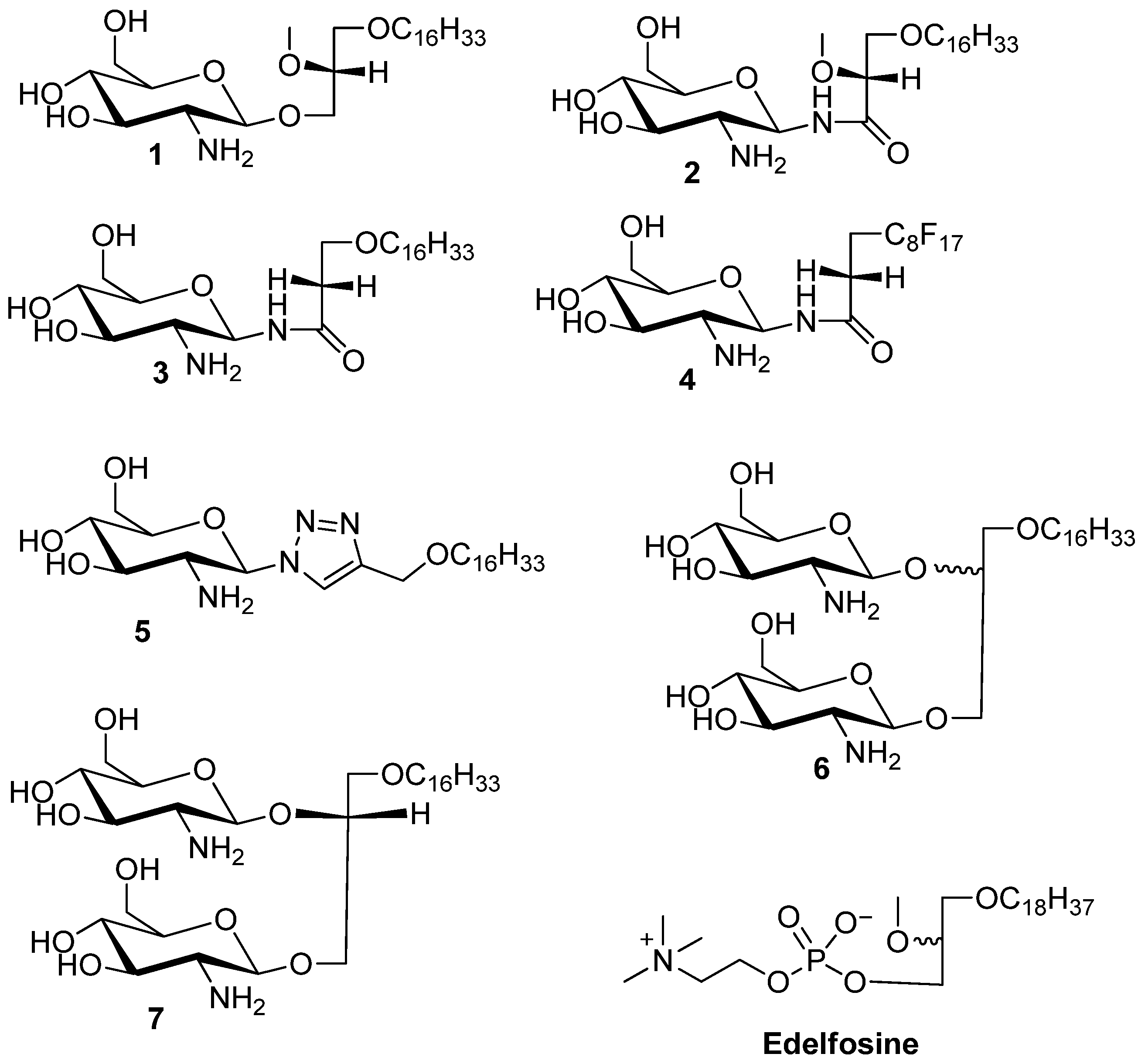
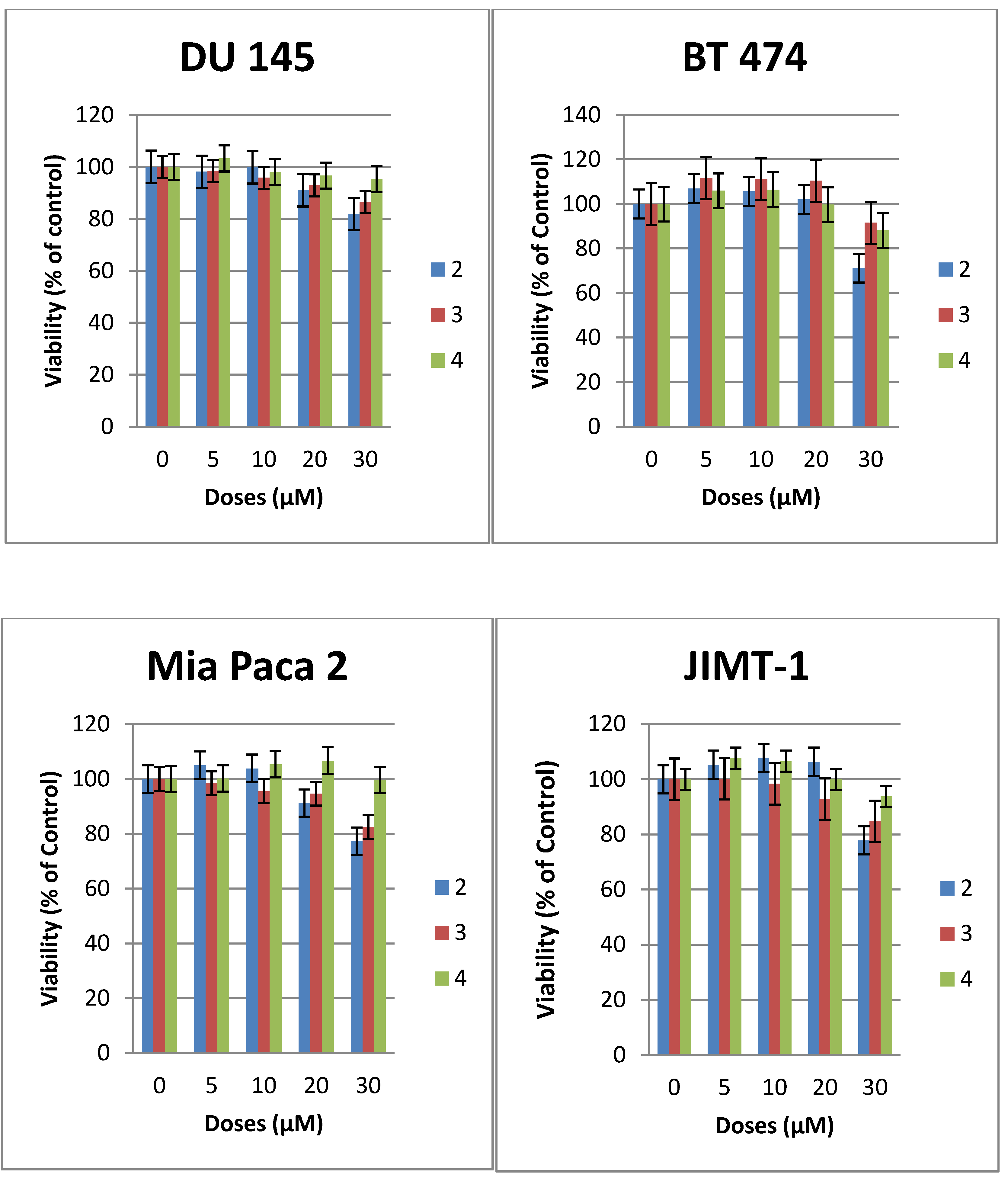
3-Hexadecyloxy-2-methoxy-1-N-(-2-deoxy-2-amino-β-d-glucopyranosyl)-propanamide (2). Compound 17 (130 mg, 0.17 mmol) was dissolved in mixture of butanol (2 mL) and ethylenediamine (2 mL). The resulting mixture was heated to 90 °C and stirred for 3 h. The reaction was monitored by TLC plate. The solution was concentrated to dryness. The resulting reside was purified by flash chromatography (reverse-phase C18 silica gel). The collected product was 20 mg (yield 23%). The compound was acidified with TFA to convert it into the TFA salt. 1H-NMR (CD3OD): δ = 5.14 (d, J = 9.6 Hz, 1H, H–1), 3.87–3.52 (m, 6H), 3.50–3.24 (m, 7H, H–3), 3.08 (dd, J = 9.6 Hz, 10 Hz, 1H, H–2), 1.56–1.36 (m, 2H, CH2–(CH2)13), 1.25–1.13 (brs, 26H, –(CH2)13), 0.80 (t, J = 6.9 Hz, 3H, –CH3). 13C-NMR (75 MHz, CD3OD) characteristic data: 174.03 (C–1), 82.50, 80.41, 78.00, 74.87, 72.88, 71.49, 71.25, 62.11, 58.94 56.46, + multiple methylene carbons, 14.40 (CH3). ES-HRMS: calcd for C26H52N2O7Na+m/z 527.3667, found: m/z [M+Na]+ 527.3677.
3-Hexadecyloxy-1-N-(-2-deoxy-2-amino-β-d-glucopyranosyl)-propanamide (3). Compound 8 (50 mg, 0.07 mmol) was dissolved in mixture of butanol (2 mL) and ethylenediamine (2 mL). The resulting mixture was heated to 90 °C and stirred for 3 h. The reaction was monitored by TLC. The solution was concentrated to dryness. The resulting reside was purified by flash chromatography (reverse-phase C18 silica gel). The collected product was 18 mg (yield 54%). The compound 3 was acidified with TFA to enhance solubility in methanol. 1H-NMR (CD3OD): δ = 5.14 (d, J = 10 Hz, 1H, H–1), 3.74–3.56 (m, 4H), 3.48–3.42 (m, 1H), 3.40–3.32 (m, 2H), 3.30–3.25 (m, 2H), 2.89 (dd, J1 = J2 = 10.0 Hz, 1H, H–2), 2.44 (t, J = 6 Hz, 2H,), 1.51–1.42 (m, 2H, –CH2–(CH2)13), 1.25–1.18 (brs, 26 H, –(CH2)13), 0.81 (t, J = 7 Hz, 3H, –CH3). 13C-NMR (CD3OD) characteristic data: 174.50 (amide), 80.30 77.83, 74.87, 72.43, 71.43, 67.11, 62.18, 56.90, 37.54, (multiple methylene carbons), 14.43 (CH3). ES-HRMS: calcd for C25H50N2O6Na+m/z 497.3567: found: m/z [M+Na]+ 497.3554.
Heptadecylfluoro-1-N-(-2-deoxy-2-amino-β-d-glucopyranosyl)-dodecanamide (4). Compound 19 (105 mg, 0.11 mmol) was dissolved in mixture of butanol (2 mL) and ethylenediamine (2 mL). The resulting mixture was heated to 90 °C and stirred for 3 h. The reaction was monitored by TLC plate. The solution was concentrated to dryness. The resulting residue was purified by flash chromatography (reverse-phase C18 silica gel) to yield product 4 (50 mg, 64% yield). The compound 3 was acidified with TFA to enhance solubility in methanol. 1H-NMR (CD3OD): δ = 5.16 (d, J = 9.9 Hz, 1H, H–1), 3.80–3.73 (m, 1H, H–6a), 3.65–3.55 (m, 1H, H–6b), 3.52–3.43 (m, 1H, H–5), 3.34–3.17 (m, 4H), 2.91 (dd, J1 = 9.9 Hz, J2 = 9.9 Hz, 1H, H–2), 2.59–2.33 (m, 2H). 13C-NMR (CD3OD) for sugar carbons 173.32 (amide), 80.33, 77.96, 74.88, 71.45, 62.21, 56.87. ES-HRMS: calcd for C17H17F17N2O5Na+m/z 675.0758: found: m/z [M+Na]+ 675.0786.
3-Hexadecyloxyl-propy-1-ne (
22). Compound
20 (8.25 mmol, 2.0 g) and
21 (9.9 mmol, 0.884 mL) were dissolved in dry DMF (20 mL) under nitrogen atmosphere, then NaH (9.9 mmol, 237 mg) was added and the reaction mixture was heated to 90 °C overnight. The reaction was stopped by addition of water (8 mL). The mixture was concentrated under high vacuum and the residue was purified by flash chromatography using hexane/ethyl acetate mixture (9.5:0.5) to give compound
22 as a white sticky solid yield 60%. The NMR data correspond to the previously reported data [
24].
4-(Hexadecyloxy)methyl)-1-(3,4,6-tri-O-acetyl-2'-deoxy-2'-N-phthalimido-β-d-glucopyranosyl)-1,2,3-triazole (23). Compound 10 (0.09 mmol, 40 mg) and compound 22 (0.1 mmol, 29 mg) were dissolved in DCM (5 mL), copper-I-iodide (0.009 mmol, 1.65 mg), acetic acid (0.17 mmol, 12.88 mg), and diisopropylethylamine (0.17 mmol, 22.48 mg) were added at the same time and the reaction mixture were left stirring for 6 h. At the end of the reaction (TLC monitoring), the reaction mixture was concentrated under vacuum and purified by flash chromatography using a hexane/ethyl acetate mixture (6:4) to give compound 23 as a white solid. Yield 58 mg (90%). 1H-NMR (CDCl3): δ = 7.81–7.71 (m, 5H, phthalimido and triazole), 6.79 (d, J = 9.9 Hz, 1H, H–1), 6.03 (dd, J1 = 10.2 Hz, J2= 9.6 Hz, 1H, H–3), 5.34 (dd, J1 = J2= 9.6Hz, 1H, H–4), 4.85 (dd, J1 = 9.9 Hz, J2 = 10.2 Hz, 1H, H–2), 4.55 (s, 2H, –CH2–O–CH2), 4.37 (m, 1H, H–5), 4.26–4.05 (m, 2H, H–6), 3.42 (t, J = 6.7 Hz, 2H), 2.14 (s, 3H, acetate–CH3), 2.05 (s, 3H, acetate–CH3), 2.01 (s, 3H, acetate–CH3), 1.45–1.55 (m, 2H, CH2–(CH2)13), 1.25 (brs, 26 H, –(CH2)13), 0.88 (t, J = 6.6 Hz, 3H, –(CH2)13–CH3). 13C-NMR (CDCl3) δ 171.18, 170.59, 169.91, 169.43, 146.16, 134.56, 123.90, 120.75, 83.03, 77.50, 77.28, 77.08, 76.65, 75.05, 73.58, 70.96, 70.47, 68.24, 64.05, 61.69, 60.40, 54.03, 31.92, 29.69, 29.65, 29.59, 29.49, 29.35, 26.07, 22.68, 21.03, 20.71, 20.66, 20.58, 20.34, 14.19, 14.11. ESMS: calcd for C39H56N4O10Na +m/z: 763.4, found m/z [M+Na]+ 763.7.
4-(Hexadecyloxy)methyl)-1-(2'-deoxy-2'-amino-β-d-glucopyranosyl)-1,2,3-triazole (5). Compound 23 (58 mg) was dissolved in mixture of butanol (2 mL) and ethylenediamine (2 mL). The resulting mixture was heated to 90 °C and stirred for 3 h. The reaction was monitored by TLC. The solution was concentrated to dryness. The resulting residue was purified by flash gradient chromatography (reverse-phase C18 silica gel, 100 water to 100 methanol) to give compound 5 as a white solid in a yield 22 mg (60%). 1H-NMR (CD3OD) δ = 8.21 (s, 1H, triazole H), 5.60 (d, J = 9.2 Hz, 1H, H–1), 3.91 (dd, J = 12.2, 12.1 Hz, 1H H–3), 3.74 (dd, J = 12.2, 5.0 Hz, 1H, H–4), 3.67–3.25 (m, 8H), 1.69–1.53 (m, 2H, –CH2–(CH2)13), 1.32 (brs, 26H, –(CH2)13), 0.93 (t, J = 6.9 Hz, 3H, –CH3). 13C-NMR (CD3OD) δ = 124.58 (triazole–CH), 81.27 (–C1), 71.85, 64.63, 62.43, 49.89, 49.60, 49.32, 49.04, 48.75, 48.47, 48.19, 33.09, 30.79, 30.75, 30.62, 30.48, 27.24, 23.75, 14.47. ES-HRMS: calcd for C25H48N4O5Na+m/z 507.3517: found: m/z [M+Na]+ 507.3532.
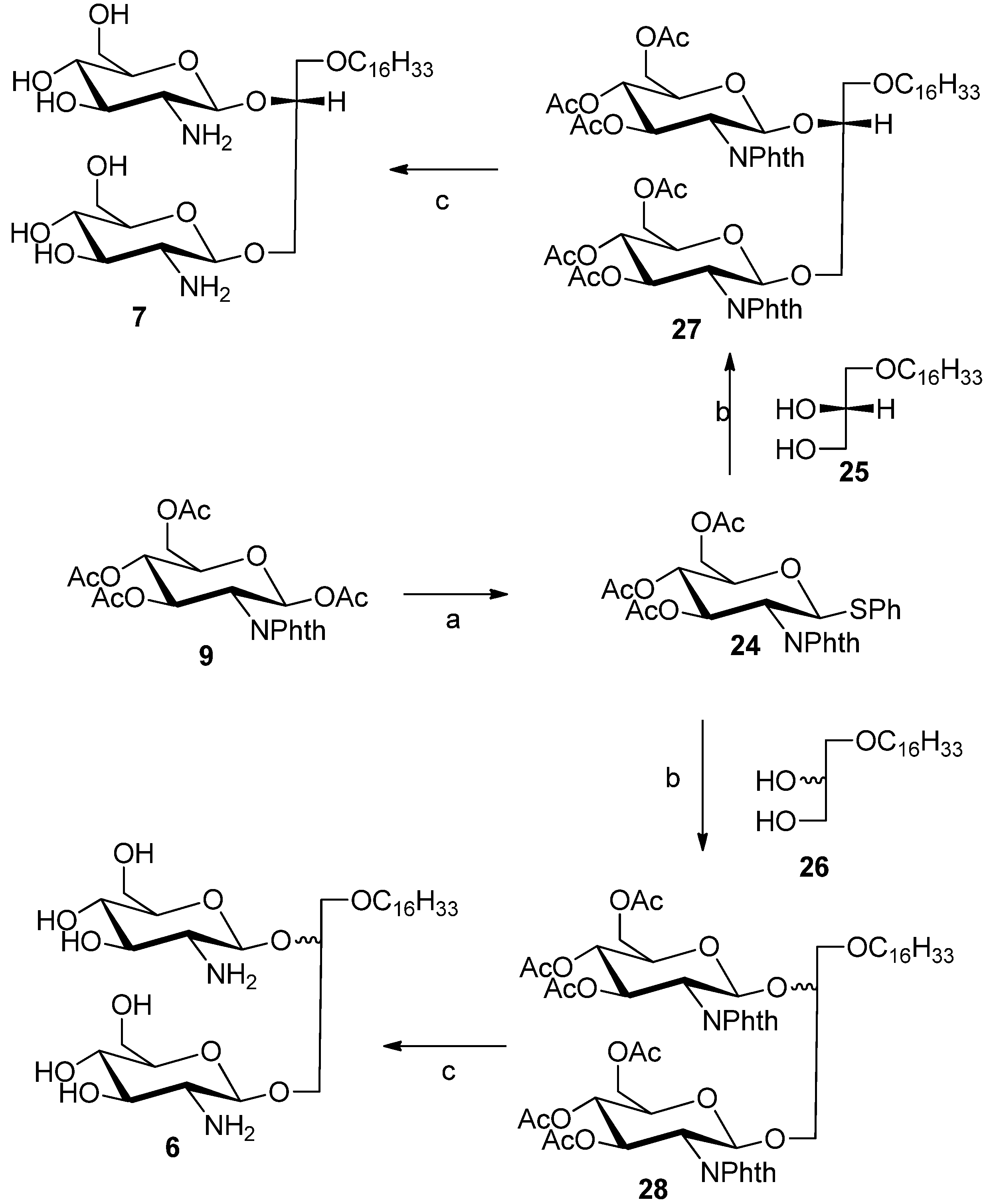
3.3. General Procedure for the Synthesis of Diglycosylated Compounds 6 and 7
Compounds
6 and
7 were synthesized by diglycosylation of commercially available lipids
25 and racemic alcohol
26, respectively. The previously reported thioglycoside donor
24 [
19] was synthesized from compound
9 and thiophenol using BF
3.Et
2O as promoter in DCM at 60 °C for 16 h. The glycosylation reaction was carried out using silver triflate and
N-Iodosuccinimide in anhydrous DCM under argon atmosphere for 5 h. The reaction was stopped by addition of saturated sodium thiosulphate solution followed by washing with saturated NaHCO
3 solution (×3). The organic layer was concentrated under vacuum to give a brownish residue which was purified with flash chromatography using hexane/ethyl acetate mixture (6:4) to give protected diglycosylated glycolipids
27 and
28 as a white foam (yield 45%). Compounds
27 and
28 were subsequently deprotected using a 1:1 mixture of ethylenediamine and butanol for 4 h, followed by removal of solvent under vacuum and purified with Ethyl acetate/methanol mixture (7:3) to give compounds
6 and
7, respectively (yield 70%).
3-Hexadecyloxy-1,2R-di(3,4,6-tri-O-acetyl-2'-deoxy-2'-N-phthalimido-β-d-glucopyranosyl)-glycerol (27). 1H-NMR (CDCl3) δ = 7.93–7.71 (m, 8H, phthalimido), 5.75–5.62 (m, 2H), 5.53 (d, J = 8.5 Hz, 1H), 5.17 (d, J = 8.7 Hz, 1H), 5.14–5.00 (m, 2H), 4.41–4.38 (m, 2H), 4.26–4.09 (m, 3H), 3.91–3.83 (m, 1H), 3.82–3.66 (m, 4H), 3.50–3.33 (m, 2H), 3.09–3.15 (m, 3H), 2.17 (s, 3H), 2.12 (s, 3H), 1.95 (d, J = 2.8 Hz, 6H, acetate–CH3), 1.91 (d, J = 2.8 Hz, 6H, acetate–CH3), 1.64 (m, 2H), 1.43–1.05 (brs, 26H, –(CH3)13), 0.88 (t, J = 6.6 Hz, 3H). 13C-NMR (CDCl3) δ = 171.22, 170.27, 169.74, 168.66, 165.86, 134.16, 123.51, 123.49, 98.76, 96.97, 77.56, 76.71, 71.64, 71.61, 70.66, 70.47, 69.89, 69.16, 68.91, 62.18, 54.61, 54.40, 37.02, 31.94, 29.72, 29.51, 29.38, 25.98, 22.70, 22.70, 20.80, 14.13. ESMS: Calcd for C59H78N2O21Na+m/z 1173.5, found m/z [M+Na]+ 1173.6.
3-Hexadecyloxy-1,2S/R-di(3,4,6-triacetyl-2'-deoxy-2'-N-phthalimido-β-d-glucopyranosyl)-glycerol (28). The NMR data of compound 28 shown here are for the 2R and 2S 1:1 diastereomeric mixture and the four anomeric protons were identified and labeled accordingly. 1H-NMR (CDCl3) characteristic data: δ = 7.90–7.68 (m, 16H aromatic protons), 5.76–5.63 (m, 2H, H-3 (sugar 1), H-3 (sugar 2), 5.53 (d, J = 8.5 Hz, 1H, H–1 (sugar 1-2R-isomer), 5.39 (d, J = 8.5 Hz, 1H, H–1 (sugar 1 2S isomer), 5.33 (d, J = 8.3 Hz, 1H, H–1 (sugar 2-2S isomer), 5.17 (d, J = 8.7 Hz, 1H, H–1 (sugar 2-2R-isomer), 1.43–1.05 (brs, 52H, –(CH3)13), 0.85 (t, J = 6.6 Hz, 6H). 13C-NMR (CDCl3) characteristic data: δ = 170.72, 170.05, 169.55, 167.80, 134.25, 131.47, 123.70, 123.41, 98.77, 98.36, 78.65, 71.76, 71.64, 71.37, 70.76, 70.71, 69.90, 69.17, 69.00, 68.91, 62.19, 62.02, 54.63, 54.42, 31.95, 29.63, 29.38, 28.94, 22.71, 20.80, 14.13. ESMS: Calcd for C59H78N2O21Na+m/z 1173.5, found m/z [M+Na]+ 1173.5.
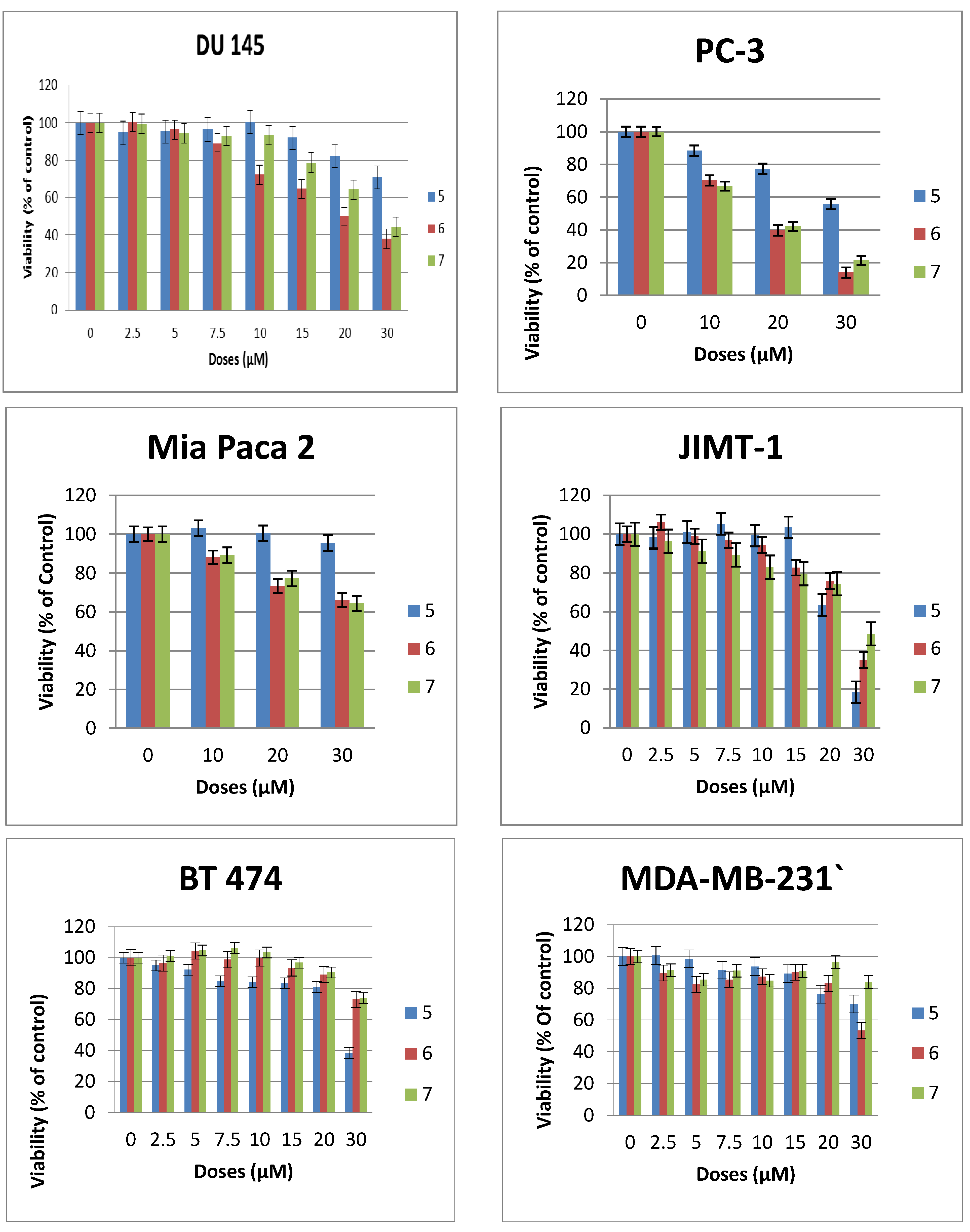
3-Hexadecyloxy-1,2S/R-di(-2'-deoxy-2'-amino-β-d-glucopyranosyl)-glycerol (6). 1H-NMR (CD3OD) characteristic data for the diastereomeric mixture: δ 4.83–4.73 (m, 2H, H–1 (sugar 1, 2R/2S), 4.71–4.57 (m, 2H, H–1 (sugar 2, 2R/2S), 1.59 (m, 4H, –CH2–(CH)13), 1.32 (brs, 52H, (CH2)13), 0.93 (2xt, 6H, −CH3). 13C-NMR (MeOD) δ 102.75, 102.01, 101.47, 100.89 (anomeric carbons) 78.82, 78.75, 78.69, 78.53, 78.12, 75.45, 75.16, 74.95, 73.18, 72.18, 72.14, 71.85, 69.97, 62.80, 62.71, 62.12, 61.93, 58.33, 58.16, 50.24, 49.67, 34.98, 34.86, 33.45, 31.16, 31.14, 31.06, 31.02, 30.84, 27.57, 24.65, 24.11, 19.13, 14.82. ES-HRMS: calcd for C31H62N2O11Na+m/z 661.4246,found: m/z [M+Na]+ 661.4241.
3-Hexadecyloxy-1,2R-di(-2'-deoxy-2'-amino-β-d-glucopyranosyl)-glycerol (7). 1H-NMR (methanol-d4) δ = 4.78 (dd, J = 8.3, 7.9 Hz, 1H, H–1a), 4.68 (dd, J = 8.3 Hz, 7.8 Hz, 1H, H–1b) 4.27–4.19 (m, 1H), 4.09–3.90 (m, 4H), 3.85–3.62 (m, 4H), 3.62–3.45 (m, 4H), 3.45–3.25 (m, 4H), 2.98–2.79 (m, 2H, H–2a, H–2b), 1.66–1.53 (m, 2H, –CH2–CH2–(CH2)13), 1.32 (brs, 26H, –(CH2)13–), 0.93 (t, J = 6.8 Hz, 3H, –CH3). 13C-NMR (CD3OD) δ = 101.92, 100.38, 78.17, 77.74, 74.40, 71.83, 71.69, 71.35, 71.24, 69.52, 67.17, 62.37, 57.95, 57.71, 35.99, 33.08, 30.80, 27.19, 23.74, 14.44. ES-HRMS: calcd for C31H62N2O11Na+m/z 661.4246, found: m/z [M+Na]+ 661.4222.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}