Perfluoroalkanesulfonamide Organocatalysts for Asymmetric Conjugate Additions of Branched Aldehydes to Vinyl Sulfones

Abstract
:1. Introduction
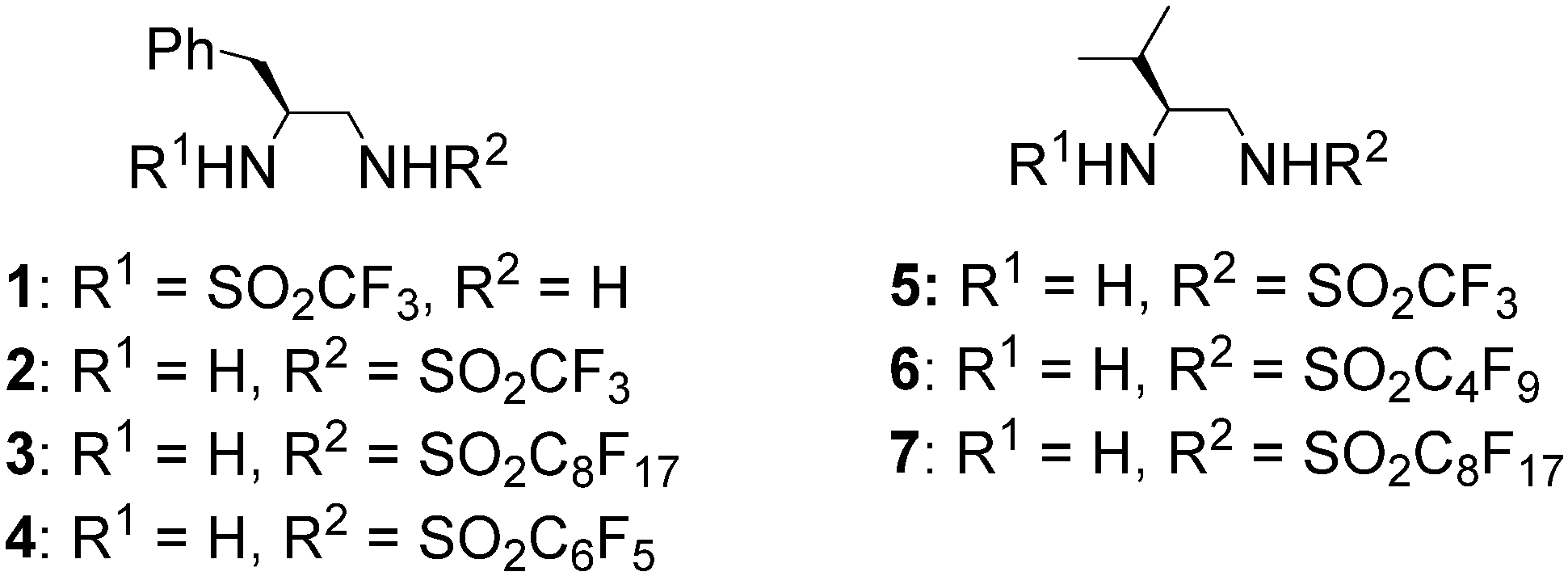
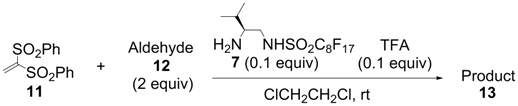
2. Results and Discussion

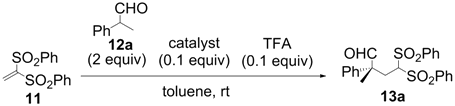
| Entry | Catalyst | Time (h) | Yield a (%) | ee b (%) |
|---|---|---|---|---|
| 1 | 1 | 1 | 95 | −2 |
| 2 | 2 | 1.5 | 99 | 80 |
| 3 | 3 | 2 | 88 | 86 |
| 4 | 4 | 2 | 97 | 79 |
| 5 | 5 | 2 | 95 | 88 |
| 6 | 6 | 2 | 100 | 91 |
| 7 | 7 | 2 | 99 | 89 |
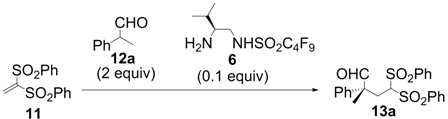
| Entry | Solvent | Temp | Additive (equiv) | Time (h) | Yield a (%) | ee b (%) |
|---|---|---|---|---|---|---|
| 1 | toluene | rt | none | 24 | 75 | 86 |
| 2 | toluene | rt | TFA (0.1) | 2 | 100 | 91 |
| 3 | CH2Cl2 | rt | TFA (0.1) | 2.5 | 97 | 91 |
| 4 | MeOH | rt | TFA (0.1) | 24 | 33 | 32 |
| 5 | Et2O | rt | TFA (0.1) | 5 | 87 | 82 |
| 6 | EtOAc | rt | TFA (0.1) | 2.5 | 98 | 86 |
| 7 | MeCN | rt | TFA (0.1) | 4 | 87 | 77 |
| 8 | CHCl3 | rt | TFA (0.1) | 2 | 99 | 91 |
| 9 | ClCH2CH2Cl | rt | TFA (0.1) | 2 | 98 | 92 |
| 10 | p-xylene | rt | TFA (0.1) | 2 | 94 | 92 |
| 11 | m-xylene | rt | TFA (0.1) | 2 | 95 | 93 |
| 12 | o-xylene | rt | TFA (0.1) | 2 | 97 | 89 |
| 13 | m-xylene | rt | PhCO2H (0.1) | 5.5 | 46 | 80 |
| 14 | m-xylene | rt | 4-NO2C6H4CO2H (0.1) | 24 | 85 | 82 |
| 15 | m-xylene | rt | TfOH (0.1) | 24 | 24 | 83 |
| 16 | m-xylene | rt | TFA (0.2) | 2 | 97 | 91 |
| 17 | m-xylene | rt | TFA (0.05) | 2 | 98 | 91 |
| 18 | m-xylene | 0 °C | TFA (0.1) | 21 | 99 | 95 |
| 19 | m-xylene | −10 °C | TFA (0.1) | 72 | 94 | 95 |
| 20 c | m-xylene | rt | TFA (0.05) | 3 | 99 | 90 |
| 21 d | m-xylene | rt | TFA (0.01) | 20 | 96 | 89 |

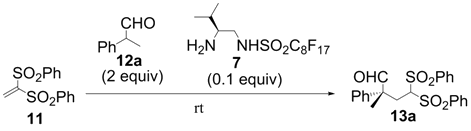
| Entry | Aldehyde | Product | Time (h) | Yield a (%) | ee b (%) |
|---|---|---|---|---|---|
| 1 | 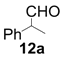 | 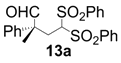 | 2 | 95 | 93 |
| 2 | 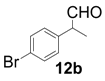 |  | 3 | 97 | 89 |
| 3 | 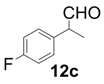 |  | 2 | 95 | 91 |
| 4 | 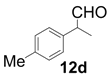 | 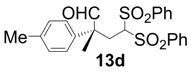 | 2 | 99 | 92 |
| 5 | 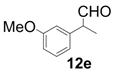 | 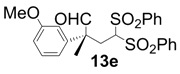 | 4 | 99 | 92 |
| 6 | 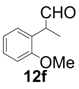 | 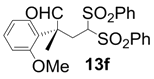 | 10 | 98 | 83 |
| 7 | 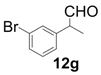 |  | 4 | 99 | 91 |
| 8 | 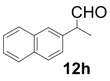 |  | 3 | 99 | 92 |
| 9 | 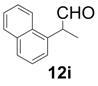 | 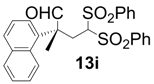 | 4 | 97 | 92 |
| 10 | 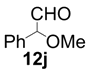 | 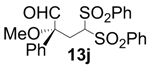 | 77 | 88 | 68 |
| 11 | 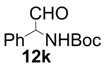 | 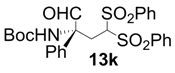 | 6 | 68 | 60 |
| Entry | Solvent | Additive (Equiv) | Time (h) | Yield a (%) | ee b (%) |
|---|---|---|---|---|---|
| 1 | toluene | none | 24 | 85 | 83 |
| 2 | toluene | TFA (0.1) | 2 | 99 | 89 |
| 3 | CH2Cl2 | TFA (0.1) | 2 | 90 | 91 |
| 4 | hexane | TFA (0.1) | 2 | 91 | 86 |
| 5 | Et2O | TFA (0.1) | 2 | 85 | 89 |
| 6 | brine | TFA (0.1) | 24 | 68 | 78 |
| 7 | CHCl3 | TFA (0.1) | 2 | 100 | 91 |
| 8 | ClCH2CH2Cl | TFA (0.1) | 2 | 87 | 92 |
| 9 | m-xylene | TFA (0.1) | 2 | 84 | 91 |
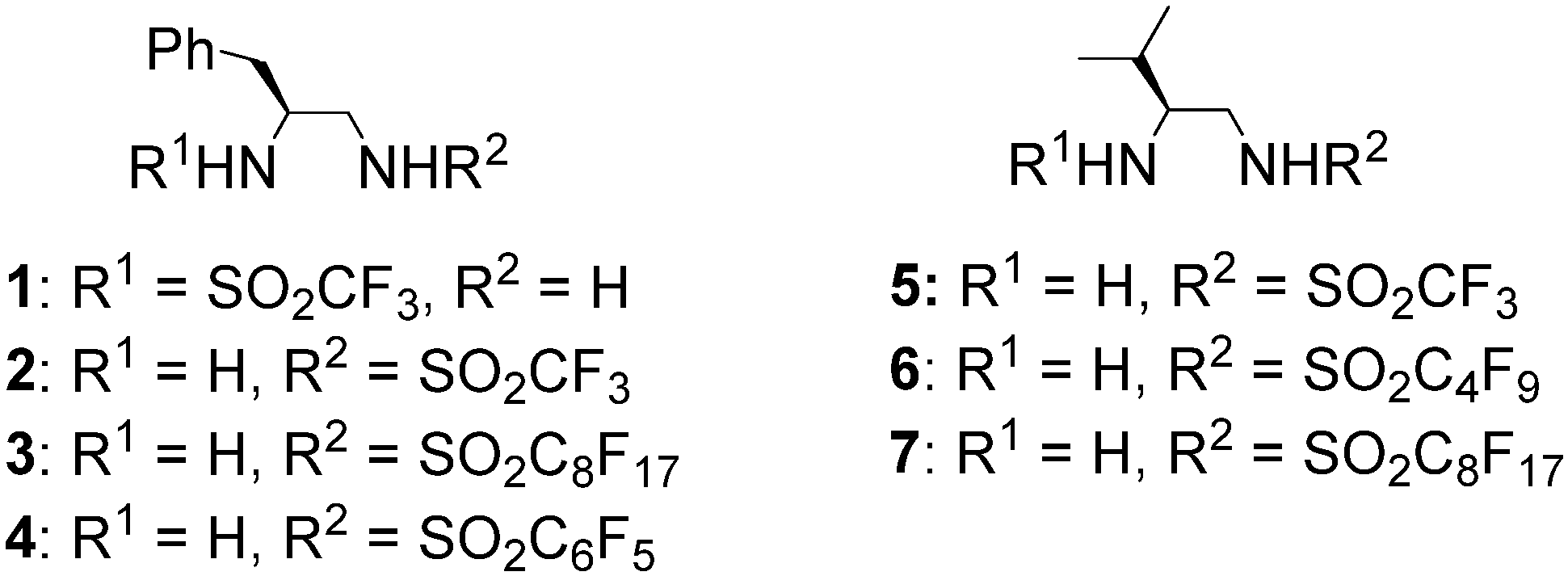{kind=link}
{kind=link}
{kind=link}
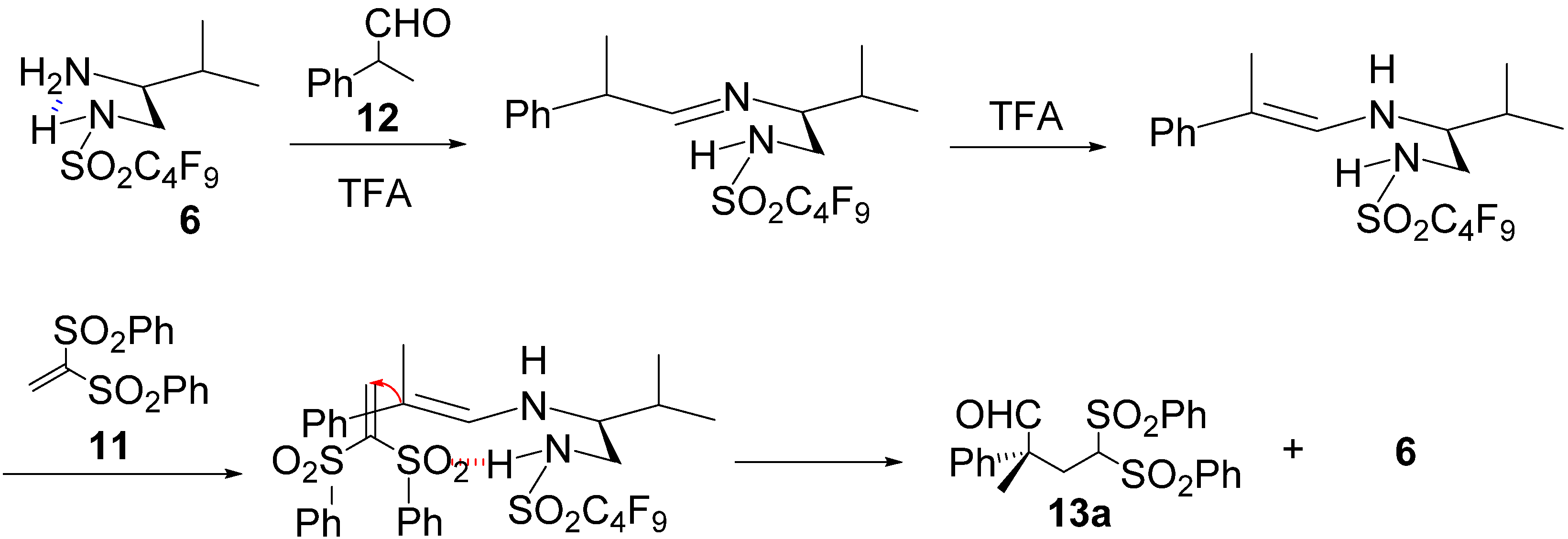{kind=link}
| Entry | Aldehyde | Product | Time (h) | Yield a (%) | ee b (%) |
|---|---|---|---|---|---|
| 1 | 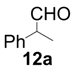 | 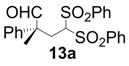 | 2 | 87 | 92 |
| 2 | 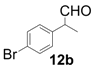 | 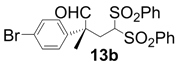 | 4 | 90 | 90 |
| 3 |  | 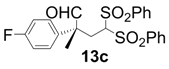 | 6 | 100 | 92 |
| 4 | 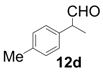 |  | 2 | 92 | 82 |
| 5 | 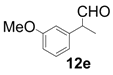 | 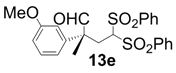 | 3 | 13 | 80 |
| 6 | 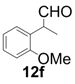 | 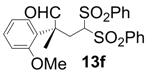 | 5 | 100 | 83 |
| 7 | 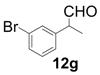 | 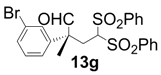 | 4 | 81 | 94 |
| 8 | 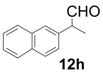 | 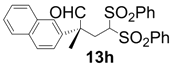 | 3 | 76 | 92 |
| 9 |  | 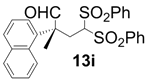 | 24 | 45 | 89 |
| 10 | 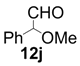 | 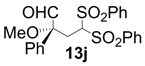 | 24 | 64 | 68 |
| 11 | 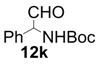 | 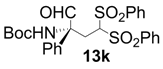 | 6 | 100 | 64 |
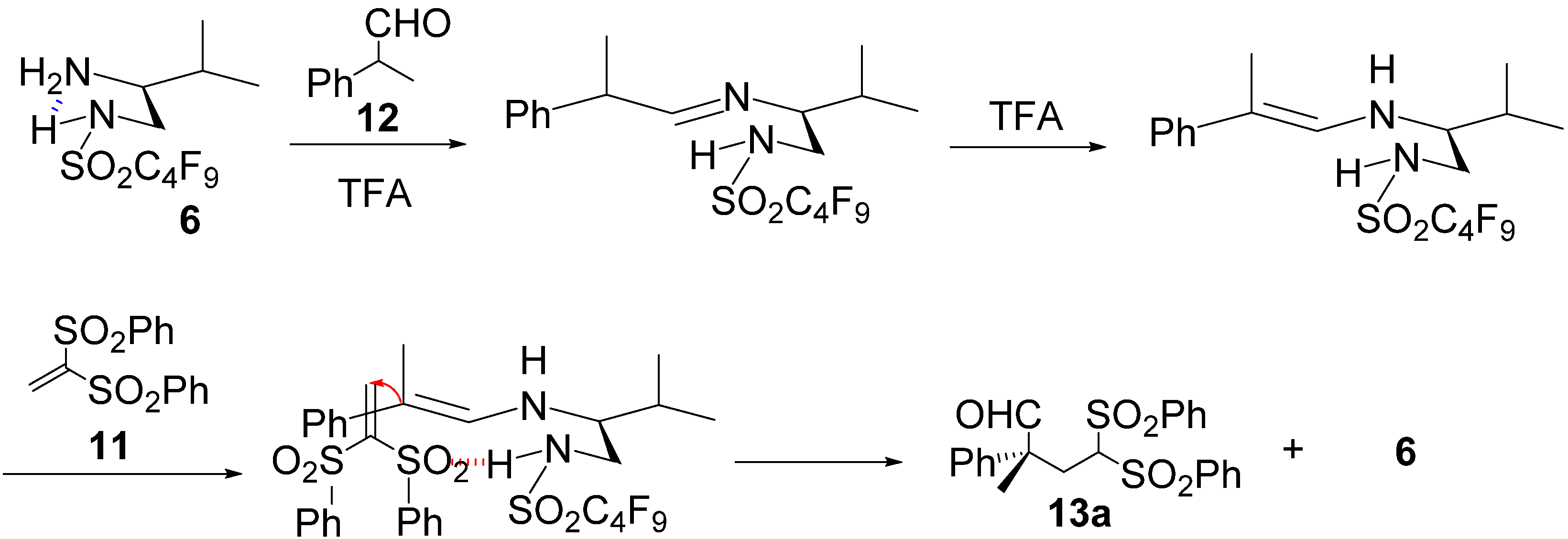
3. Experimental
3.1. General
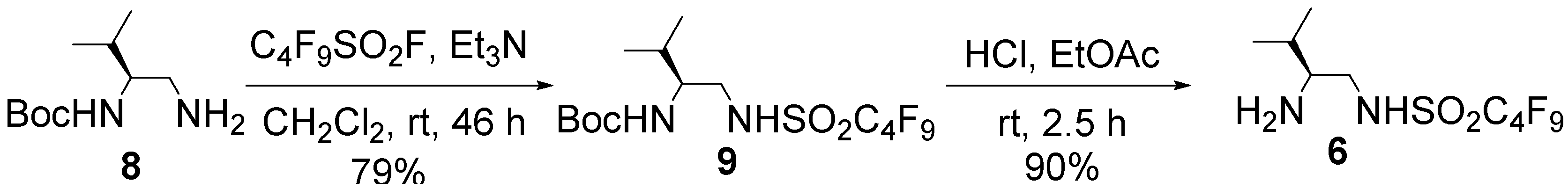
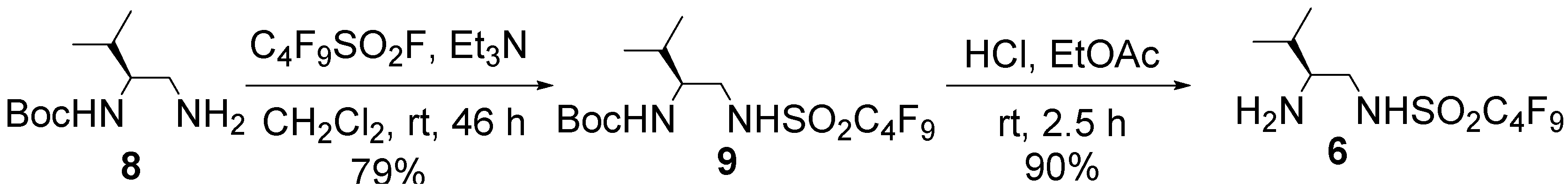
3.2. Preparation of Organocatalyst 6
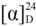 = −5.4° (c = 0.62 in MeOH); 1H-NMR (400 MHz, CD3OD): δ = 0.81 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H), 1.35 (s, 9H), 1.65–1.71 (m, 1H), 3.08 (dd, J = 8.1, 13.5 Hz, 1H), 3.28 (dd, J = 4.5, 13.5 Hz, 1H), 3.31–3.37 (m, 1H); 13C-NMR (125 MHz, CD3OD): δ = 18.2, 19.9, 28.8, 31.0, 47.1, 57.6, 80.2, 110.2–121.0 (complex signals of –CF2 and –CF3), 158.5; HRMS (ESI-TOF): calcd for C14H21F9N2O4SNa (M+Na)+: 507.0976, Found: 507.0991.
= −5.4° (c = 0.62 in MeOH); 1H-NMR (400 MHz, CD3OD): δ = 0.81 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H), 1.35 (s, 9H), 1.65–1.71 (m, 1H), 3.08 (dd, J = 8.1, 13.5 Hz, 1H), 3.28 (dd, J = 4.5, 13.5 Hz, 1H), 3.31–3.37 (m, 1H); 13C-NMR (125 MHz, CD3OD): δ = 18.2, 19.9, 28.8, 31.0, 47.1, 57.6, 80.2, 110.2–121.0 (complex signals of –CF2 and –CF3), 158.5; HRMS (ESI-TOF): calcd for C14H21F9N2O4SNa (M+Na)+: 507.0976, Found: 507.0991.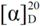 = +7.9° (c = 1.01 in MeOH); 1H-NMR (500 MHz, CD3OD): δ = 1.01 (d, J = 6.9 Hz, 3H), 1.02 (d, J = 6.9 Hz, 3H), 1.90–1.97 (m, 1H), 2.82–2.86 (m, 1H), 3.14 (dd, J = 8.6, 13.1 Hz, 1H), 3.41 (dd, J = 3.5, 13.1 Hz, 1H); 13C-NMR (125 MHz, CD3OD): δ = 18.9, 19.0, 30.1, 47.4, 60.7, 110.2–120.4 (complex signals of –CF2 and –CF3); Anal. Calcd for C9H13F9N2O2S: C, 28.13; H, 3.41; N, 7.29. Found: C, 28.07; H, 3.39; N, 7.26.
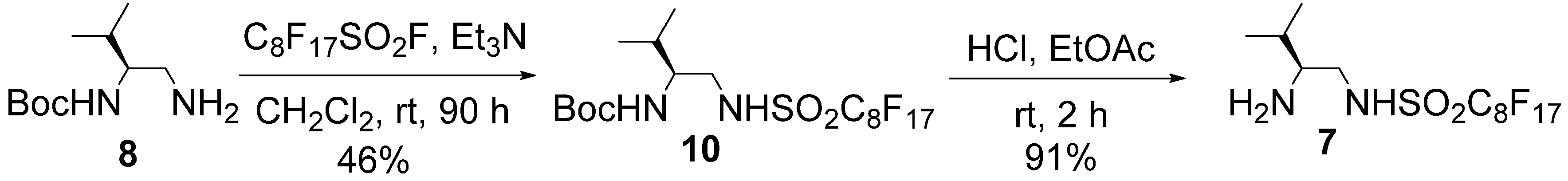
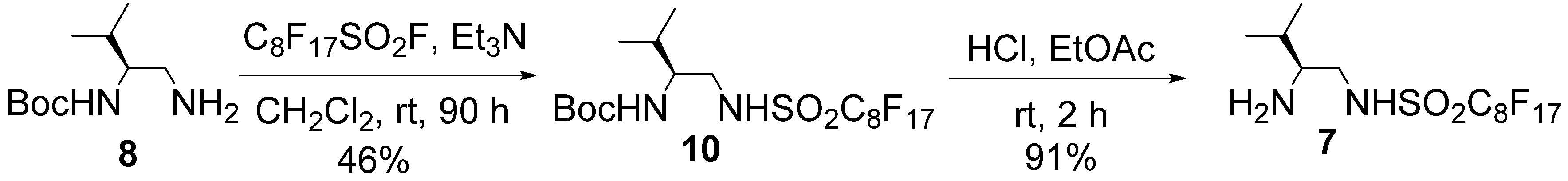
= +7.9° (c = 1.01 in MeOH); 1H-NMR (500 MHz, CD3OD): δ = 1.01 (d, J = 6.9 Hz, 3H), 1.02 (d, J = 6.9 Hz, 3H), 1.90–1.97 (m, 1H), 2.82–2.86 (m, 1H), 3.14 (dd, J = 8.6, 13.1 Hz, 1H), 3.41 (dd, J = 3.5, 13.1 Hz, 1H); 13C-NMR (125 MHz, CD3OD): δ = 18.9, 19.0, 30.1, 47.4, 60.7, 110.2–120.4 (complex signals of –CF2 and –CF3); Anal. Calcd for C9H13F9N2O2S: C, 28.13; H, 3.41; N, 7.29. Found: C, 28.07; H, 3.39; N, 7.26.3.3. Preparation of Organocatalyst 7
= −4.2° (c = 1.28 in MeOH); 1H-NMR (500 MHz, CDCl3): δ = 0.95 (d, J = 7.4 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H), 1.44 (s, 9H), 1.80–1.85 (m, 1H), 3.25 (m, 1H), 3.46 (brd, J = 12.6 Hz, 1H), 3.55 (m, 1H), 4.67 (brd, J = 8.0 Hz, 1H), 7.13 (brs, 1H); 13C-NMR (125 MHz, CDCl3): δ = 18.0, 19.2, 28.2, 30.1, 48.4, 55.7, 80.8, 108.0–113.0 (complex signals of –CF2 and –CF3), 157.6; HRMS (ESI-TOF): calcd for C18H21F17N2O4SNa (M+Na)+: 707.0848, Found: 707.0873. = + 6.9° (c = 1.01 in MeOH); 1H-NMR (500 MHz, CD3OD): δ = 1.01 (d, J = 6.8 Hz, 3H), 1.03 (d, J = 6.8 Hz, 3H), 1.90–1.97 (m, 1H), 2.82–2.86 (m, 1H), 3.15 (dd, J = 8.5, 13.1 Hz, 1H), 3.41 (dd, J = 4.0, 13.1 Hz, 1H); 13C-NMR (125 MHz, CD3OD): δ = 18.9, 19.0, 30.1, 47.5, 60.7, 109.7–121.5 (complex signals of –CF2 and –CF3); Anal. Calcd for C13H13F17N2O2S: C, 26.72; H, 2.24; N, 4.79. Found: C, 26.75; H, 2.41; N, 4.86.3.4. Typical Procedure for Michael Addition (Table 3)
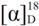 = −25.6° (c = 1.00, CHCl3); 95% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 21.7 min, tminor = 25.9 min.
= −25.6° (c = 1.00, CHCl3); 95% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 21.7 min, tminor = 25.9 min. = −15.2° (c = 1.00, CHCl3); 89% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 27.1 min, tminor = 38.5 min. = +23.5° (c = 1.00, CHCl3); 91% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 25.5 min, tminor = 32.1 min. = +25.4° (c = 1.00, CHCl3); 92% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 21.3 min, tminor = 29.7 min.
= −15.2° (c = 1.00, CHCl3); 89% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 27.1 min, tminor = 38.5 min. = +23.5° (c = 1.00, CHCl3); 91% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 25.5 min, tminor = 32.1 min. = +25.4° (c = 1.00, CHCl3); 92% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 21.3 min, tminor = 29.7 min.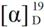 = +10.6° (c = 1.00, CHCl3); 92% ee; enantiomeric excess was determined by HPLC with Chiralcel AD-H column (hexane/2-propanol = 80:20), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 27.4 min, tminor = 38.6 min. = −70.8° (c = 1.00, CHCl3); 83% ee; enantiomeric excess was determined by HPLC with Chiralcel AD-H column (hexane/2-propanol = 80:20), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 18.8 min, tminor = 25.9 min. = −71.7° (c = 1.00, CHCl3); 91% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 80:20), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 41.3 min, tminor = 47.1 min. = +13.0° (c = 1.00, CHCl3); 92% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 32.9 min, tminor = 39.1 min. = −31.8° (c = 1.00, CHCl3); 92% ee; enantiomeric excess was determined by HPLC with Chiralcel AD-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 16.7 min, tminor = 22.4 min.
= +10.6° (c = 1.00, CHCl3); 92% ee; enantiomeric excess was determined by HPLC with Chiralcel AD-H column (hexane/2-propanol = 80:20), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 27.4 min, tminor = 38.6 min. = −70.8° (c = 1.00, CHCl3); 83% ee; enantiomeric excess was determined by HPLC with Chiralcel AD-H column (hexane/2-propanol = 80:20), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 18.8 min, tminor = 25.9 min. = −71.7° (c = 1.00, CHCl3); 91% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 80:20), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 41.3 min, tminor = 47.1 min. = +13.0° (c = 1.00, CHCl3); 92% ee; enantiomeric excess was determined by HPLC with Chiralpak AS-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 32.9 min, tminor = 39.1 min. = −31.8° (c = 1.00, CHCl3); 92% ee; enantiomeric excess was determined by HPLC with Chiralcel AD-H column (hexane/2-propanol = 70:30), flow rate = 1.0 mL/min; λ = 220 nm; tmajor = 16.7 min, tminor = 22.4 min.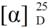 = +10.0° (c = 1.00, CHCl3) 64% ee; enantiomeric excess was determined by HPLC with Chiralcel OD-H column (hexane/2-propanol = 90:10), flow rate = 0.5 mL/min; λ = 220 nm; tmajor = 19.9 min, tminor = 22.2 min.
= +10.0° (c = 1.00, CHCl3) 64% ee; enantiomeric excess was determined by HPLC with Chiralcel OD-H column (hexane/2-propanol = 90:10), flow rate = 0.5 mL/min; λ = 220 nm; tmajor = 19.9 min, tminor = 22.2 min.4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Fuji, K. Asymmetric creation of quaternary carbon centers. Chem. Rev. 1993, 93, 2037–2066. [Google Scholar] [CrossRef]
- Corey, E.J.; Guzman-Perez, A. The catalytic enantioselective construction of molecules with quaternary carbon stereocenters. Angew. Chem. Int. Ed. 1998, 37, 388–401. [Google Scholar] [CrossRef]
- Christoffers, J.; Mann, A. Enantioselective construction of quaternary stereocenters. Angew. Chem. Int. Ed. 2001, 40, 4591–4597. [Google Scholar] [CrossRef]
- Christoffers, J.; Baro, A. Construction of quaternary stereocenters: New perspectives through enantioselective Michael reactions. Angew. Chem. Int. Ed. 2003, 42, 1688–1690. [Google Scholar] [CrossRef]
- Douglas, C.J.; Overman, L.E. Catalytic asymmetric synthesis of all-carbon quaternary stereocenters. Proc. Natl. Acad. Sci. USA 2004, 101, 5363–5367. [Google Scholar] [CrossRef]
- Christoffers, J.; Baro, A. Stereoselective construction of quaternary stereocenters. Adv. Synth. Catal. 2005, 347, 1473–1482. [Google Scholar] [CrossRef]
- Trost, B.M.; Jiang, C. Catalytic enantioselective construction of all-carbon quaternary stereocenters. Synthesis 2006, 2006, 369–396. [Google Scholar] [CrossRef]
- Cozzi, P.G.; Hilgraf, R.; Zimmermann, N. Enantioselective catalytic formation of quaternary stereogenic centers. Eur. J. Org. Chem. 2007, 2007, 5969–5994. [Google Scholar]
- Marek, I.; Sklute, G. Creation of quaternary stereocenters in carbonyl allylation reactions. Chem. Commun. 2007, 2007, 1683–1691. [Google Scholar] [CrossRef]
- Riant, O.; Hannedouche, J. Asymmetric catalysis for the construction of quaternary carbon centres: nucleophilic addition on ketones and ketimines. Org. Biomol. Chem. 2007, 5, 873–888. [Google Scholar] [CrossRef]
- Hawner, C.; Alexakis, A. Metal-catalyzed asymmetric conjugate addition reaction: Formation of quaternary stereocenters. Chem. Commun. (Camb) 2010, 46, 7295–7306. [Google Scholar] [CrossRef]
- Shimizu, M. Construction of asymmetric quaternary carbon centers with high enantioselectivity. Angew. Chem. Int. Ed. 2011, 50, 5998–6000. [Google Scholar] [CrossRef]
- Das, J.P.; Marek, I. Enantioselective synthesis of all-carbon quaternary stereogenic centers in acyclic systems. Chem. Commun. (Camb) 2011, 47, 4593–4623. [Google Scholar] [CrossRef]
- Bella, M.; Gasperi, T. Organocatalytic formation of quaternary stereocenters. Synthesis 2009, 2009, 1583–1614. [Google Scholar] [CrossRef]
- Figueiredo, R.M.; Christmann, M. Organocatalytic synthesis of drugs and bioactive natural products. Eur. J. Org. Chem. 2007, 2007, 2575–2600. [Google Scholar] [CrossRef]
- Mossé, S.; Alexakis, A. First organocatalyzed asymmetric Michael addition of aldehydes to Vinyl sulfones. Org. Lett. 2005, 7, 4361–4364. [Google Scholar] [CrossRef]
- Liu, T.-Y.; Long, J.; Li, B.-J.; Jiang, L.; Li, R.; Wu, Y.; Ding, L.-S.; Chen, Y.-C. Enantioselective construction of quaternary carbon centre catalysed by bifunctional organocatalyst. Org. Biomol. Chem. 2006, 4, 2097–2099. [Google Scholar] [CrossRef]
- Zhu, Q.; Lu, Y. Organocatalytic michael addition of aldehydes to vinyl sulfones: Enantioselective α-alkylations of aldehydes and their derivatives. Org. Lett. 2008, 10, 4803–4806. [Google Scholar] [CrossRef]
- Zhu, Q.; Cheng, L.; Lu, Y. Asymmetric organocatalytic Michael addition of ketones to vinyl sulfone. Chem. Commun. 2008, 2008, 6315–6317. [Google Scholar]
- Quintard, A.; Bournaud, C.; Alexakis, A. Diversity-oriented synthesis towards conceptually new highly modular aminal–pyrrolidine organocatalysts. Chem. Eur. J. 2008, 14, 7504–7507. [Google Scholar] [CrossRef]
- Zhu, Q.; Lu, Y. Enantioselective conjugate addition of nitroalkanes to vinyl sulfone: An organocatalytic access to chiral amines. Org. Lett. 2009, 11, 1721–1724. [Google Scholar] [CrossRef]
- Landa, A.; Maestro, M.; Masdeu, C.; Puente, A.; Vera, S.; Oiarbide, M.; Palomo, C. Highly enantioselective conjugate additions of aldehydes to vinyl sulfones. Chem. Eur. J. 2009, 15, 1562–1565. [Google Scholar] [CrossRef]
- Quintard, A.; Alexakis, A. Organocatalytic addition on 1,2-bis(sulfone)vinylenes leading to an unprecedented rearrangement. Chem. Eur. J. 2009, 15, 11109–11113. [Google Scholar] [CrossRef]
- Xiao, J.; Liu, Y.-L.; Loh, T.-P. The organocatalytic enantioselective Michael addition of aldehydes to vinyl sulfones in water. Synlett 2010, 2010, 2029–2032. [Google Scholar]
- Quintard, A.; Alexakis, A. Asymmetric addition of α-hetero-disubstituted aldehydes to vinyl sulfones; formation of highly functionalized tetrasubstituted carbon centres. Chem. Commun. (Camb) 2010, 46, 4085–4087. [Google Scholar] [CrossRef]
- Alba, A.-N.R.; Conpanyó, X.; Valero, G.; Moyano, A.; Rios, R. Enantioselective organocatalytic addition of oxazolones to 1,1-bis(phenylsulfonyl)ethylene: A convenient asymmetric synthesis of quaternary α-amino acids. Chem. Eur. J. 2010, 16, 5354–5361. [Google Scholar] [CrossRef]
- Chua, P.J.; Tan, B.; Yang, L.; Zeng, X.; Zhu, D.; Zhong, G. Highly stereoselective synthesis of indanes with four stereogenic centers via sequential Michael reaction and [3+2] cycloaddition. Chem. Commun. 2010, 46, 7611–7613. [Google Scholar] [CrossRef]
- Zhu, Q.; Lu, Y. Stereocontrolled creation of all-carbon quaternary stereocenters by organocatalytic conjugate addition of oxindoles to vinyl sulfone. Angew. Chem. Int. Ed. 2010, 49, 7753–7756. [Google Scholar] [CrossRef]
- Quintard, A.; Alexakis, A. 1,2-Sulfone rearrangement in organocatalytic reactions. Org. Biomol. Chem. 2011, 9, 1407–1418. [Google Scholar] [CrossRef]
- Quintard, A.; Alexakis, A. Highly enantioselective organocascade intermolecular iminium/enamine Michael addition on enals. Chem. Commun. (Camb) 2011, 47, 7212–7214. [Google Scholar] [CrossRef]
- Xiao, J.; Lu, Y.-P.; Liu, Y.-L.; Wong, P.-S.; Loh, T.-P. A New class of structurally rigid tricyclic chiral secondary amine organocatalyst: Highly enantioselective organocatalytic michael addition of aldehydes to vinyl sulfones. Org. Lett. 2011, 13, 876–879. [Google Scholar] [CrossRef]
- Kang, J.Y.; Carter, R.G. Primary Amine, Thiourea-based dual catalysis motif for synthesis of stereogenic, all-carbon quaternary center-containing cycloalkanones. Org. Lett. 2012, 14, 3178–3181. [Google Scholar] [CrossRef]
- Dou, X.; Lu, Y. Enantioselective Conjugate addition of 3-fluorooxindoles to vinyl sulfone: An organocatalytic access to chiral 3-fluoro-3-substituted oxindoles. Org. Biomol. Chem. 2013, 11, 5217–5221. [Google Scholar] [CrossRef]
- Mossé, S.; Alexakis, A.; Mareda, J.; Bollot, G.; Bernardinelli, G.; Filinchuk, Y. Enantioselective organocatalytic conjugate addition of aldehydes to vinyl sulfones and vinyl phosphonates as challenging michael acceptors. Chem. Eur. J. 2009, 15, 3204–3220. [Google Scholar] [CrossRef]
- Quintard, A.; Belot, S.; Marchal, E.; Alexakis, A. Aminal–pyrrolidine organocatalysts—highly efficient and modular catalysts for α-functionalization of carbonyl compounds. Eur. J. Org. Chem. 2010, 2010, 927–936. [Google Scholar] [CrossRef]
- Zhu, Q.; Lu, Y. Chiral primary amine mediated conjugate addition of branched aldehydes to vinyl sulfone: Asymmetric generation of quaternary carbon centers. Chem. Commun. (Camb) 2010, 46, 2235–2237. [Google Scholar] [CrossRef]
- Moteki, S.A.; Xu, S.; Arimitsu, S.; Maruoka, K. Design of structurally rigid trans-diamine-based tf-amide organocatalysts with a dihydroanthracene framework for asymmetric conjugate additions of heterosubstituted aldehydes to vinyl sulfones. J. Am. Chem. Soc. 2010, 132, 17074–17076. [Google Scholar] [CrossRef]
- Kanada, Y.; Yuasa, H.; Nakashima, K.; Murahashi, M.; Tada, N.; Itoh, A.; Koseki, Y.; Miura, T. Asymmetric conjugate addition of aldehydes to vinyl sulfone using a diaminomethylenemalononitrile organocatalyst. Tetrahedron Lett. 2013, 54, 4896–4899. [Google Scholar] [CrossRef]
- The Handbook of Fluorous Chemistry; Gladysz, J.A.; Horváth, I.T.; Curran, D.P. (Eds.) Wiley-VCH: Weinheim, Germany, 2004.
- Zhang, W.; Cai, C. New chemical and biological applications of fluorous technologies. Chem. Commun. 2008, 2008, 5686–5694. [Google Scholar] [CrossRef]
- Miura, T.; Imai, K.; Ina, M.; Tada, N.; Imai, N.; Itoh, A. Direct asymmetric aldol reaction with recyclable fluorous organocatalyst. Org. Lett. 2010, 12, 1620–1623. [Google Scholar] [CrossRef]
- Miura, T.; Imai, K.; Kasuga, H.; Ina, M.; Tada, N.; Imai, N.; Itoh, A. Direct asymmetric aldol reactions in brine with recyclable fluorous β-aminosulfonamide organocatalysts. Tetrahedron 2011, 67, 6340–6346. [Google Scholar] [CrossRef]
- Miura, T.; Imai, K.; Kasuga, H.; Ina, M.; Tada, N.; Imai, N.; Itoh, A. Highly efficient asymmetric aldol reaction in brine using fluorous sulfonamide organocatalyst. Org. Biomol. Chem. 2012, 10, 2209–2213. [Google Scholar] [CrossRef]
- Miura, T.; Nishida, S.; Masuda, A.; Tada, N.; Itoh, A. Asymmetric Michael additions of aldehydes to maleimides using a recyclable fluorous thiourea organocatalyst. Tetrahedron Lett. 2011, 52, 4158–4160. [Google Scholar] [CrossRef]
- Miura, T.; Nakashima, K.; Tada, N.; Itoh, A. An Effective and catalytic oxidation using recyclable fluorous IBX. Chem. Commun. 2011, 47, 1875–1877. [Google Scholar] [CrossRef]
- Miura, T.; Ina, M.; Imai, K.; Nakashima, K.; Masuda, A.; Imai, N.; Tada, N.; Itoh, A. β-Aminosulfonamide-catalyzed direct asymmetric Aldol reaction in brine. Synlett 2011, 2011, 410–414. [Google Scholar]
- Miura, T.; Yasaku, Y.; Koyata, N.; Murakami, Y.; Imai, N. Direct asymmetric aldol reactions in brine using novel sulfonamide catalyst. Tetrahedron Lett. 2009, 50, 2632–2635. [Google Scholar] [CrossRef]
- Miura, T.; Ina, M.; Imai, K.; Nakashima, K.; Yasaku, Y.; Koyata, N.; Murakami, Y.; Imai, N.; Tada, N.; Itoh, A. Direct asymmetric aldol reactions in water with a β-aminosulfonamide organocatalyst. Tetrahedron Asymmetry 2011, 22, 1028–1034. [Google Scholar] [CrossRef]
- Miura, T.; Yuasa, H.; Murahashi, M.; Ina, M.; Nakashima, K.; Tada, N.; Itoh, A. Highly efficient asymmetric conjugate additions of aldehydes with vinyl sulfones using sulfonamide organocatalyst. Synlett 2012, 2012, 2385–2388. [Google Scholar]
- Imai, N.; Nokami, J.; Nomura, T.; Ninomiya, Y.; Shinobe, A.; Matsushiro, S. Convenient preparation of various chiral disulfonamides from α-amino acid and alkylation of cinnamaldehyde in the presence of the chiral disulfonamides. Bull. Okayama Univ. Sci. 2002, 47–52. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakashima, K.; Murahashi, M.; Yuasa, H.; Ina, M.; Tada, N.; Itoh, A.; Hirashima, S.-i.; Koseki, Y.; Miura, T. Perfluoroalkanesulfonamide Organocatalysts for Asymmetric Conjugate Additions of Branched Aldehydes to Vinyl Sulfones. Molecules 2013, 18, 14529-14542. https://doi.org/10.3390/molecules181214529
Nakashima K, Murahashi M, Yuasa H, Ina M, Tada N, Itoh A, Hirashima S-i, Koseki Y, Miura T. Perfluoroalkanesulfonamide Organocatalysts for Asymmetric Conjugate Additions of Branched Aldehydes to Vinyl Sulfones. Molecules. 2013; 18(12):14529-14542. https://doi.org/10.3390/molecules181214529
Chicago/Turabian StyleNakashima, Kosuke, Miho Murahashi, Hiroki Yuasa, Mariko Ina, Norihiro Tada, Akichika Itoh, Shin-ichi Hirashima, Yuji Koseki, and Tsuyoshi Miura. 2013. "Perfluoroalkanesulfonamide Organocatalysts for Asymmetric Conjugate Additions of Branched Aldehydes to Vinyl Sulfones" Molecules 18, no. 12: 14529-14542. https://doi.org/10.3390/molecules181214529
APA StyleNakashima, K., Murahashi, M., Yuasa, H., Ina, M., Tada, N., Itoh, A., Hirashima, S.-i., Koseki, Y., & Miura, T. (2013). Perfluoroalkanesulfonamide Organocatalysts for Asymmetric Conjugate Additions of Branched Aldehydes to Vinyl Sulfones. Molecules, 18(12), 14529-14542. https://doi.org/10.3390/molecules181214529