Synthesis and Dual Histamine H1 and H2 Receptor Antagonist Activity of Cyanoguanidine Derivatives


Abstract
:
1. Introduction
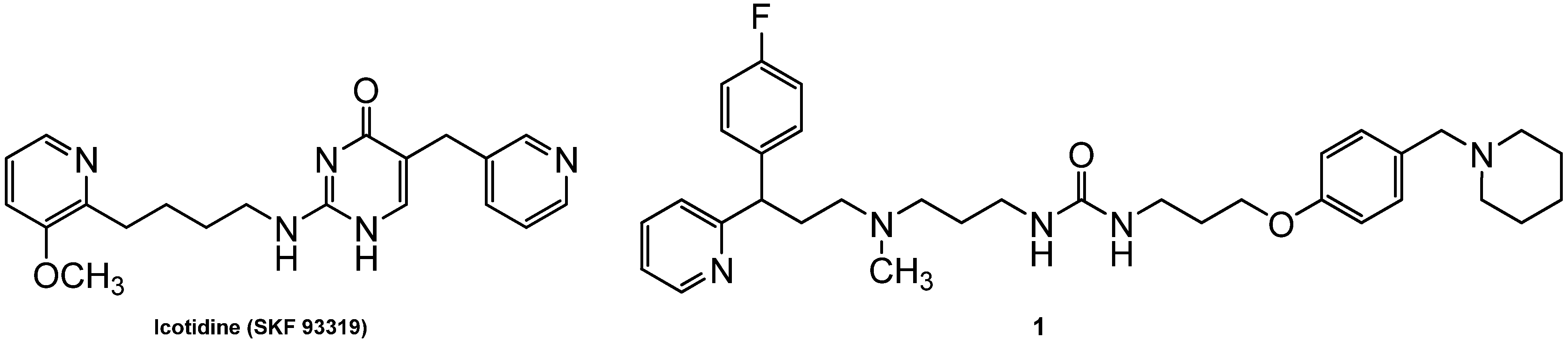
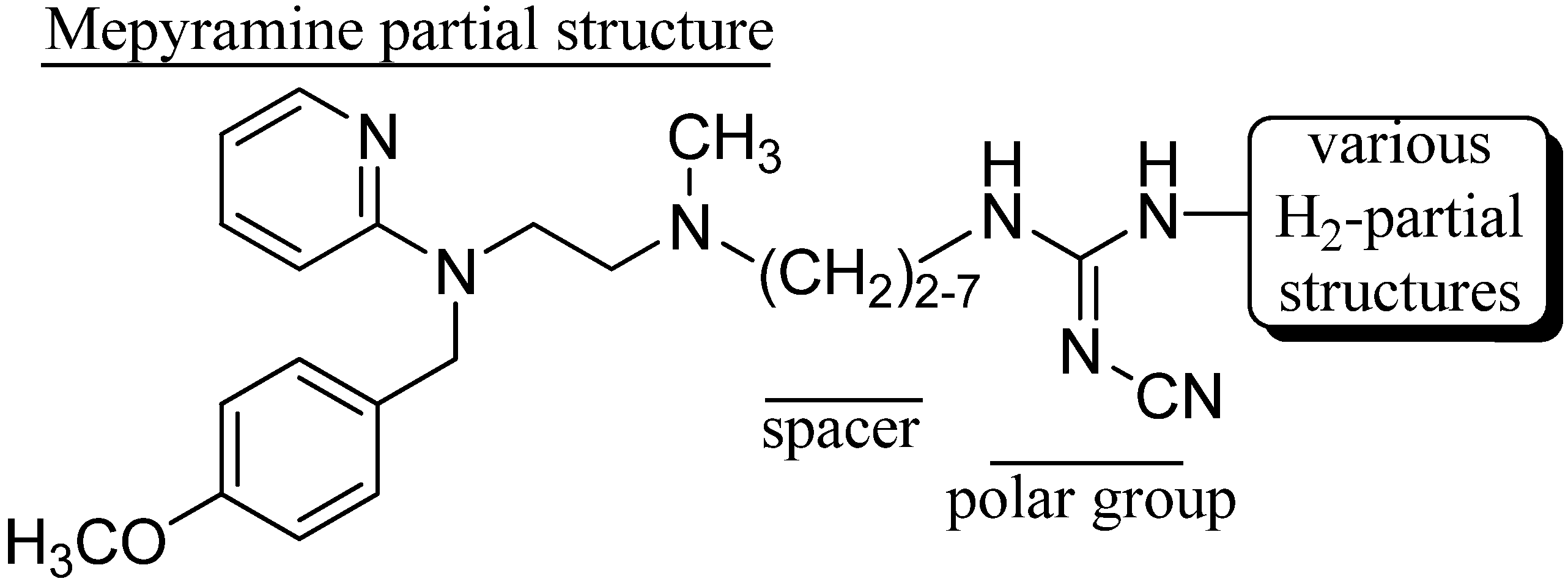
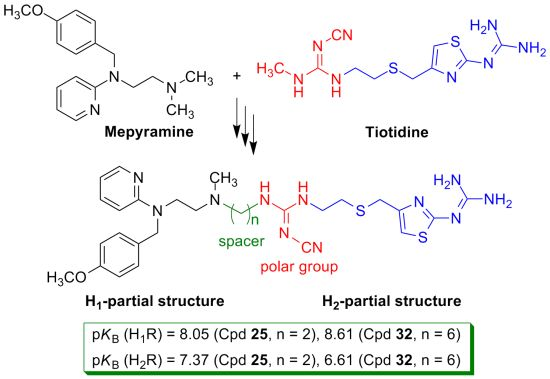
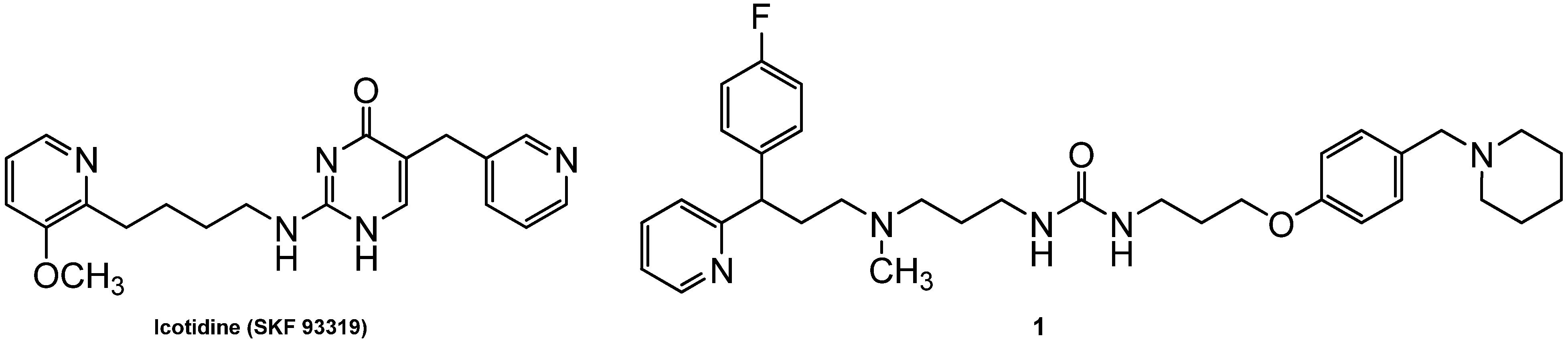
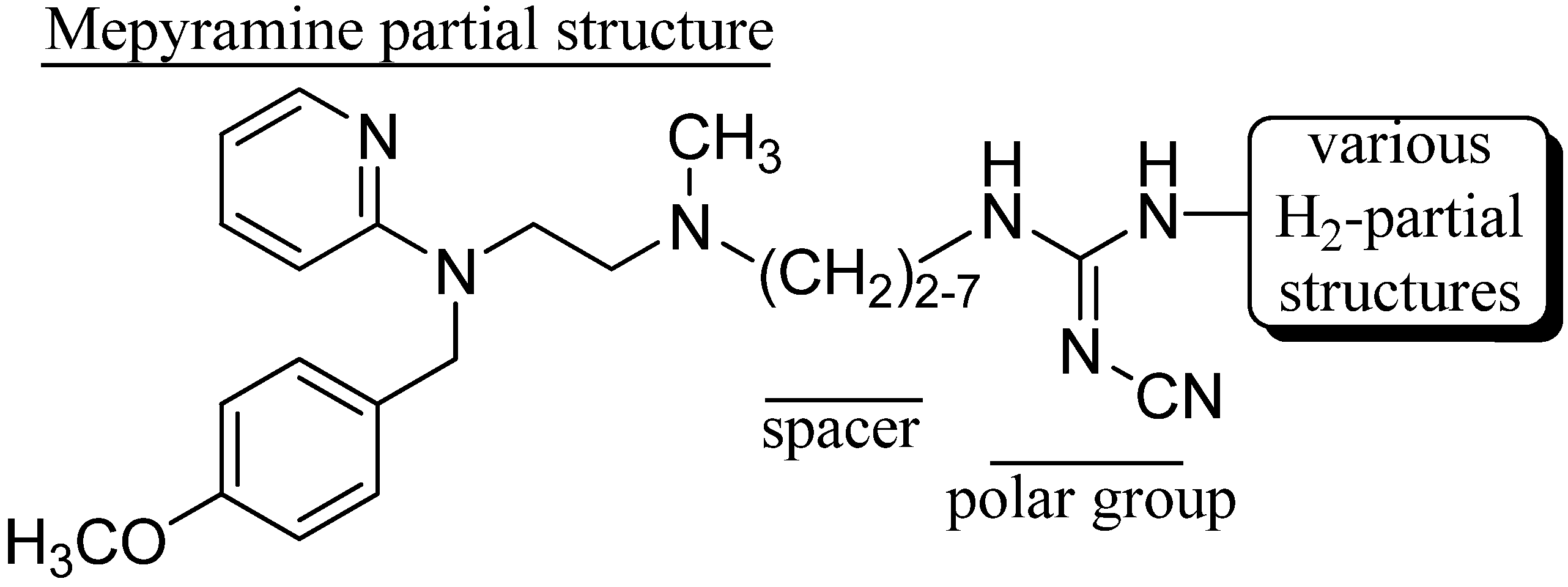
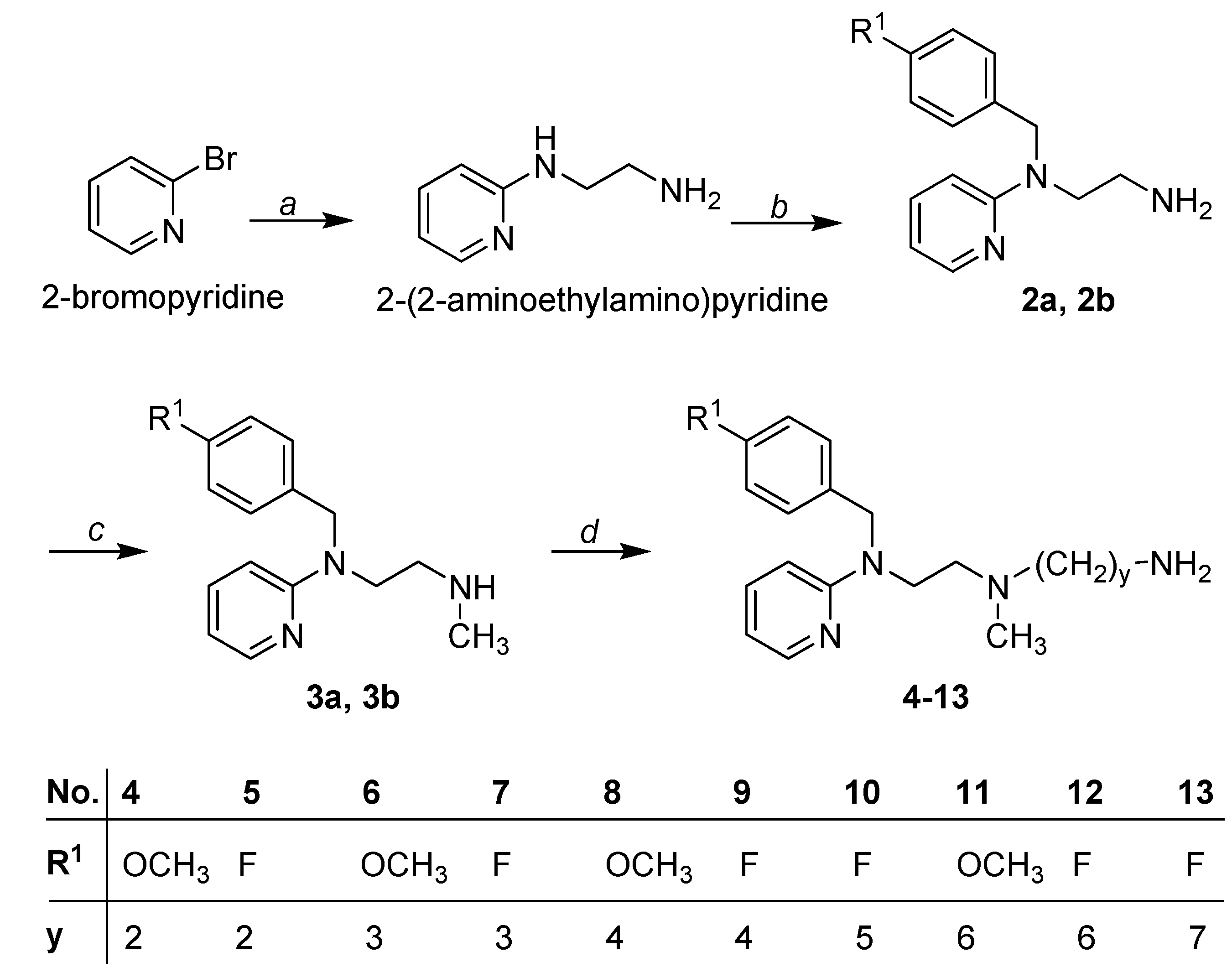
2. Results and Discussion
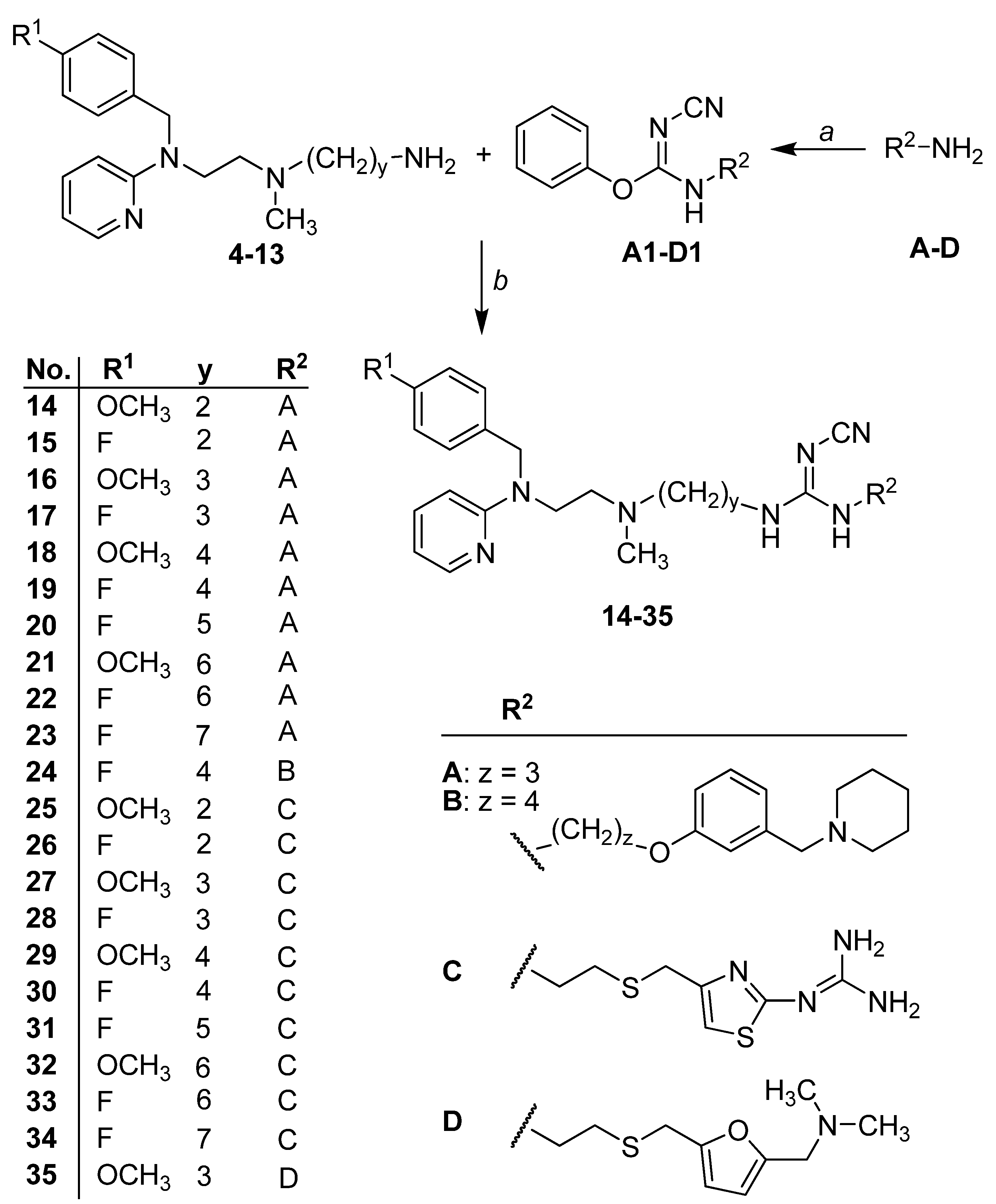
2.1. Chemistry

2.2. Pharmacology
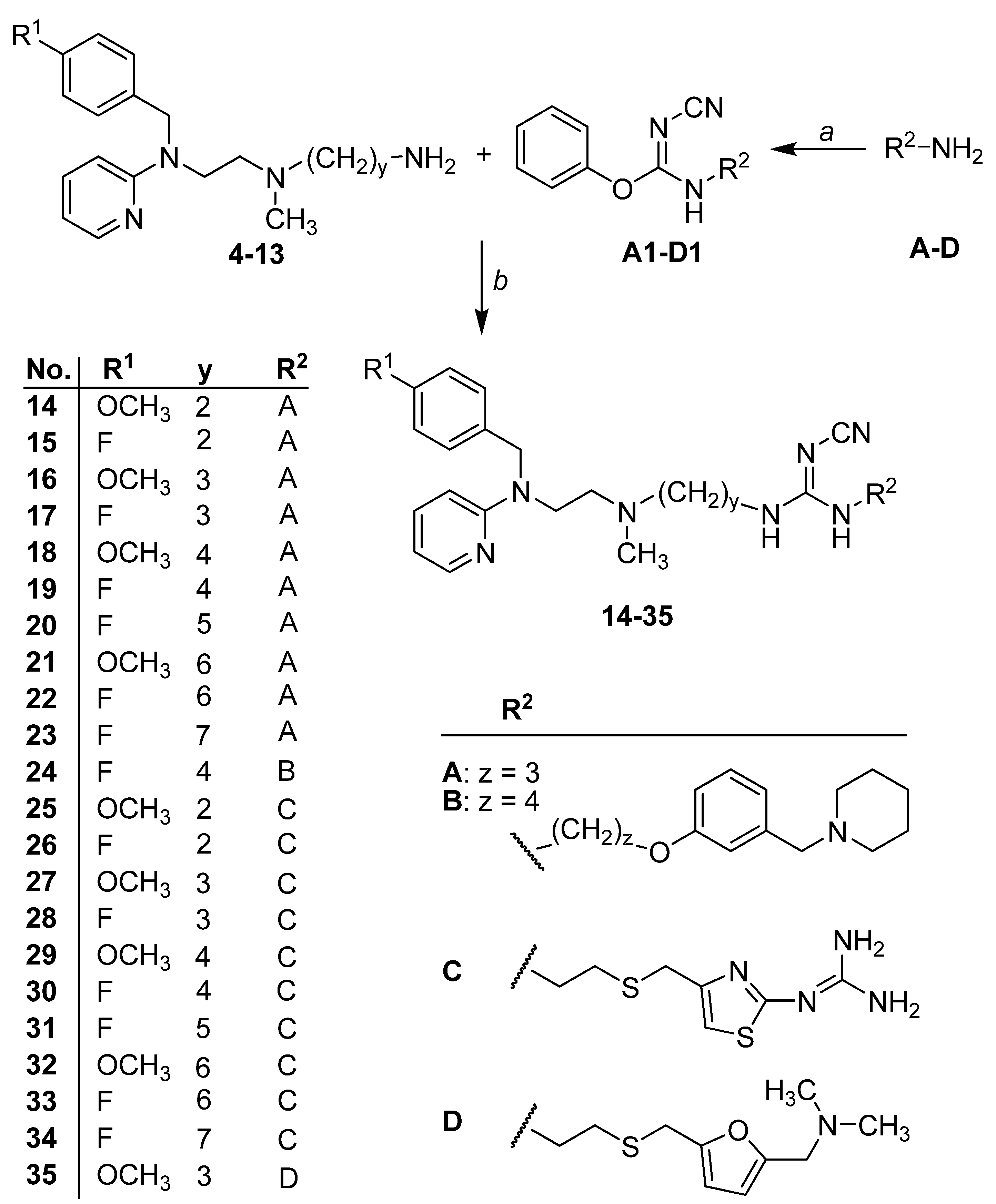

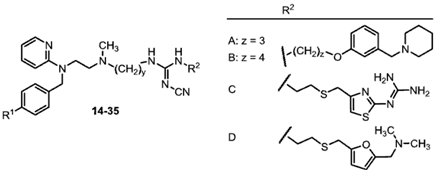
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H1 Receptor Antagonism | H2 Receptor Antagonism | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | y | R2 | p KB a | rel. activity | conc. [M] | p KB a | rel. activity | conc. [M] |
| Mepyramine | - | - | - | 9.07 b | 100 | - | |||
| Cimetidine | - | - | - | - | 6.40 | 100 | |||
| Icotidine c | - | - | - | 7.77 | 5.0 | 7.49 | 1230 | ||
| 1d | 8.21 | 14 | 0.3 × 10−6 | 6.68 | 190 | 10−6–0.3 × 10−5 | |||
| 14 | OCH3 | 2 | A | 6.88 | 1 | 0.3 × 10−6 | 6.63 | 170 | 10−7–0.3 × 10−6 |
| 15 | F | 2 | A | 7.83 | 6 | 10−7 | 5.97 e | 37 | 0.3 × 10−5 |
| 16 | OCH3 | 3 | A | 7.81 | 6 | 10−7–0.3 × 10−6 | 6.37 | 93 | 0.3 × 10−6 |
| 17 | F | 3 | A | 7.77 e | 5 | 10−7–0.3 × 10−6 | 6.0 | 40 | 0.3 × 10−6–10−6 |
| 18 | OCH3 | 4 | A | 7.76 | 5 | 10−8–10−7 | 6.38 | 96 | 0.3 × 10−6 |
| 19 | F | 4 | A | 7.75 | 5 | 10−7–0.3 × 10−7 | 5.76 e | 23 | 10−5–0.3 × 10−5 |
| 20 | F | 5 | A | 7.95 | 7 | 10−7 | 5.67 | 19 | 0.3 × 10−5 |
| 21 | OCH3 | 6 | A | 8.42 | 22 | 10−7–0.3 × 10−7 | 6.43 | 107 | 0.3 × 10−6 |
| 22 | F | 6 | A | 8.06 | 10 | 10−7–0.3 × 10−7 | 6.41 | 102 | 10−6 |
| 23 | F | 7 | A | 7.93 | 7 | 10−7–0.3 × 10−7 | 6.17 | 59 | 10−6–0.3 × 10−5 |
| 24 | F | 4 | B | 7.65 | 4 | 10−7–0.3 × 10−6 | 6.07 | 47 | 10−6 |
| 25 | OCH3 | 2 | C | 8.05 | 10 | 10−7–0.3 × 10−7 | 7.37 | 933 | 10−7 |
| 26 | F | 2 | C | 7.94 | 7 | 10−8–0.3 × 10−7 | 6.62 | 166 | 10−6 |
| 27 | OCH3 | 3 | C | 8.21 | 14 | 10−8–0.3 × 10−7 | 6.52 | 132 | 10−6 |
| 28 | F | 3 | C | 8.62 | 36 | 10−8–0.3 × 10−8 | 6.47 | 118 | 0.3 × 10−6–0.3 × 10−5 |
| 29 | OCH3 | 4 | C | 7.91 | 7 | 0.3 × 10−7 | 7.00 | 398 | 10−7–0.3 × 10−6 |
| 30 | F | 4 | C | 8.32 | 18 | 10−8–0.3 × 10−8 | 6.28 | 76 | 10−6 |
| 31 | F | 5 | C | 8.36 | 20 | 10−8–0.3 × 10−7 | 6.51 | 129 | 10−6–10−5 |
| 32 | OCH3 | 6 | C | 8.61 | 35 | 10−7–0.3 × 10−7 | 6.61 | 162 | 10−7–0.3 × 10−6 |
| 33 | F | 6 | C | 8.44 | 23 | 0.3 × 10−7 | 6.52 | 132 | 10−6 |
| 34 | F | 7 | C | 8.15 | 12 | 10−8 | 7.02 | 417 | 10−6–0.3 × 10−6 |
| 35 | OCH3 | 3 | D | 7.95 | 7 | 10−8–10−7 | 5.22 e | 7 | 10−5 |
3. Experimental
3.1. Chemistry
3.1.1. General Conditions
3.1.2. Synthesis of Primary Amines 2a and 2b
3.1.3. Synthesis of Secondary Amines 3a and 3b
3.1.4. General Procedure for the Synthesis of the Diamines 4–13
3.1.5. General Procedure for the Synthesis of Cyanoguanidines 14–35
3.2. Pharmacology
3.2.1. Histamine H1 Receptor Antagonist Activity on the Isolated Guinea Pig Ileum
3.2.2. Histamine H2 Receptor Antagonist Activity on the Isolated Guinea Pig Right Atrium
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hill, S.J.; Ganellin, C.R.; Timmerman, H.; Schwartz, J.C.; Shankley, N.P.; Young, J.M.; Schunack, W.; Levi, R.; Haas, H.L. International union of pharmacology. XIII. Classification of histamine receptors. Pharmacol. Rev. 1997, 49, 253–278. [Google Scholar]
- Seifert, R.; Strasser, A.; Schneider, E.H.; Neumann, D.; Dove, S.; Buschauer, A. Molecular and cellular analysis of human histamine receptor subtypes. Trends Pharmacol. Sci. 2013, 34, 33–58. [Google Scholar] [CrossRef]
- Strasser, A.; Wittmann, H.-J.; Buschauer, A.; Schneider, E.H.; Seifert, R. Species-dependent activities of GPCR ligands: Lessons from histamine receptor orthologs. Trends Pharmacol. Sci. 2013, 34, 13–32. [Google Scholar] [CrossRef]
- Martinez-Mir, M.I.; Pollard, H.; Moreau, J.; Arrang, J.M.; Ruat, M.; Traiffort, E.; Schwartz, J.C.; Palacios, J.M. Three histamine receptors (H1, H2 and H3) visualized in the brain of human and non-human primates. Brain Res. 1990, 526, 322–327. [Google Scholar] [CrossRef]
- Lim, H.D.; van Rijn, R.M.; Ling, P.; Bakker, R.A.; Thurmond, R.L.; Leurs, R. Evaluation of histamine H1-, H2-, and H3-receptor ligands at the human histamine H4 receptor: identification of 4-methylhistamine as the first potent and selective H4 receptor agonist. J. Pharmacol. Exp. Ther. 2005, 314, 1310–1321. [Google Scholar] [CrossRef]
- Yu, F.; Bonaventure, P.; Thurmond, R.L. The future antihistamines: Histamine H3 and H4 receptor ligands. Adv. Exp. Med. Biol. 2010, 709, 125–40. [Google Scholar]
- Simons, F.E.; Simons, K.J. Histamine and H1-antihistamines: Celebrating a century of progress. J. Allergy Clin. Immunol. 2011, 128, 1139–1150. [Google Scholar] [CrossRef]
- Dajani, E.Z.; Klamut, M.J. Novel therapeutic approaches to gastric and duodenal ulcers: An update. Expert Opin. Investig. Drugs. 2000, 9, 1537–1544. [Google Scholar] [CrossRef]
- Bedarida, G.; Bushell, E.; Blaschke, T.F.; Hoffman, B.B. H1- and H2-histamine receptor-mediated vasodilation varies with aging in humans. Clin. Pharmacol. Ther. 1995, 58, 73–80. [Google Scholar] [CrossRef]
- Doenicke, A.W.; Hoernecke, R.; Celik, I. Premedication with H1 and H2 blocking agents reduces the incidence of postoperative nausea and vomiting. Inflamm. Res. 2004, 53, S154–S158. [Google Scholar]
- Kim, S.H.; Lee, S.H.; Lee, S.M.; Kang, H.R.; Park, H.W.; Kim, S.S.; Cho, S.H.; Min, K.U.; Kim, Y.Y.; Chang, Y.S. Outcomes of premedication for non-ionic radio-contrast media hypersensitivity reactions in Korea. Eur. J. Radiol. 2011, 80, 363–367. [Google Scholar] [CrossRef]
- Zidan, J.; Hussein, O.; Abzah, A.; Tamam, S.; Farraj, Z.; Friedman, E. Oral premedication for the prevention of hypersensitivity reactions to paclitaxel. Med. Oncol. 2008, 25, 274–278. [Google Scholar] [CrossRef]
- Lin, T.K.; Man, M.Q.; Santiago, J.L.; Park, K.; Roelandt, T.; Oda, Y.; Hupe, M.; Crumrine, D.; Lee, H.J.; Gschwandtner, M.; et al. Topical antihistamines display potent anti-inflammatory activity linked in part to enhanced permeability barrier function. J. Invest. Dermatol. 2013, 133, 469–478. [Google Scholar] [CrossRef]
- Navo, M.; Kunthur, A.; Badell, M.L.; Coffer, L.W.; Markman, M.; Brown, J.; Smith, J.A. Evaluation of the incidence of carboplatin hypersensitivity reactions in cancer patients. Gynecol. Oncol. 2006, 103, 608–613. [Google Scholar] [CrossRef]
- Kloover, J.S.; den Bakker, M.A.; Gelderblom, H.; van Meerbeeck, J.P. Fatal outcome of a hypersensitivity reaction to paclitaxel: a critical review of premedication regimens. Brit. J. Cancer 2004, 26, 304–305. [Google Scholar]
- Dietz, W.; Stinner, B.; Lorenz, W.; Dick, W.; Junginger, T.; Menke, H.; Rothmund, M.; Doenicke, A. Histamin H1 + H2-Prophylaxe im perioperativen Zeitraum. Inn. Med. 1990, 17, 129–135. [Google Scholar]
- Wolff, A.A.; Levi, R. Histamine and cardiac arrhythmias. Circ. Res. 1986, 58, 1–16. [Google Scholar] [CrossRef]
- Baller, D.; Huchzermeyer, H. Histamine effects on the heart with special reference to cardiac side effects of H2 receptor antagonists. Klin. Wochenschr. 1989, 67, 743–755. [Google Scholar] [CrossRef]
- Larsen, R. Narkosezwischenfaelle und Narkosekomplikationen, 3rd ed.; Urban and Schwarzenberg: Muenchen, Germany, 1990; pp. 743–754. [Google Scholar]
- Lieberman, P. The use of antihistamines in the prevention and treatment of anaphylaxis and anaphylactoid reactions. J. Allergy Clin. Immunol. 1990, 86, 684–686. [Google Scholar] [CrossRef]
- Blakemore, R.C.; Brown, T.H.; Cooper, D.G.; Durant, G.J.; Ganellin, C.R.; Ife, R.J.; Parsons, M.E.; Rasmussen, A.C.; Sach, G.S. SK&F 93319: A specific antagonist of histamine at H1- and H2-receptors. Br. J. Pharmacol. 1983, 80, 437. [Google Scholar]
- Schulze, F.R.; Alisch, R.A.; Buschauer, A.; Schunack, W. Synthese und kombinierte H1-/H2-antagonistische Aktivitat von Mepyramin-, Pheniramin- und Cyclizin-Derivaten rnit Cyanoguanidin-, Harnstoff- und Nitroethendiamin-Partialstrukturen. Arch. Pharm. (Weinheim) 1994, 327, 455–462. [Google Scholar] [CrossRef]
- Schulze, F.R.; Buschauer, A.; Schunack, W. Combined histamine H /H receptor antagonists: Part I. Pharmacological hybrids with pheniramine- and roxatidine-like substructures. Eur. J. Pharm. Sci. 1998, 6, 177–186. [Google Scholar] [CrossRef]
- Alisch, R.A.; Schulze, F.R.; Buschauer, A.; Schunack, W. Preparation of (Hetero)arylalkylamines as Histamine H1 and -H2 Receptor Antagonists. Eur. Pat. Appl. EP 0526395A1, 1993, pp. 1–36. Chem. Abstr. 1993, 120, 244679. [Google Scholar]
- Elz, S.; Kramer, K.; Pertz, H.H.; Detert, H.; ter Laak, A.M.; Kuehne, R.; Schunack, W. Histaprodifens: synthesis, pharmacological in vitro evaluation, and molecular modeling of a new class of highly active and selective histamine H(1)-receptor agonists. J. Med. Chem. 2000, 43, 1071–1084. [Google Scholar] [CrossRef]
- Durant, G.J.; Emmett, J.C.; Ganellin, C.R.; Miles, P.D.; Parsons, M.E.; Prain, H.D.; White, G.R. Cyanoguanidine-thiourea equivalence in the development of the histamine H2-receptor antagonist, cimetidine. J. Med. Chem. 1977, 20, 901–906. [Google Scholar]
- Young, R.C.; Durant, G.J.; Emmett, J.C.; Ganellin, C.R.; Graham, M.J.; Mitchell, R.C.; Prain, H.D.; Roantree, M.L. Dipole moment in relation to H2 receptor histamine antagonist activity for cimetidine analogues. J. Med. Chem. 1986, 29, 44–49. [Google Scholar] [CrossRef]
- Wolf, C.; Schulze, R.F.R.; Buschauer, A.; Schunack, W. Combined histamine H1/H2 receptor antagonists: part II. Pharmacological hybrids with pheniramine- and tiotidine-like substructures. Eur. J. Pharm. Sci. 1998, 6, 187–196. [Google Scholar] [CrossRef]
- Buschauer, A.; Postius, S.; Szelenyi, I.; Schunack, W. Isohistamine and homologs as components of H2-antagonists. Arzneimittelforschung 1985, 35, 1025–1029. [Google Scholar]
- Horner, J.K.; Skinner, W.A. The synthesis of 5-mercaptotryptamine, the thiol analog of serotonin. Can. J. Chem. 1966, 44, 315–319. [Google Scholar] [CrossRef]
- Pertz, H.H.; Elz, S. In-vitro pharmacology of sarpogrelate and the enantiomers of its major metabolite: 5-HT2A receptor specificity, stereoselectivity and modulation of ritanserin-induced depression of 5-HT contractions in rat tail artery. J. Pharm. Pharmacol. 1995, 47, 310–316. [Google Scholar]
- Kenakin, T.P. Competitive antagonism. In Pharmacolgic Analysis of Drug-receptor Interaction; Kenakin, T.P., Ed.; Raven Press: New York, NY, USA, 1993; pp. 278–322. [Google Scholar]
- Black, J.W.; Duncan, W.; Durant, C.J.; Ganellin, C.R.; Parsons, E.M. Definition and antagonism of histamine H2-receptors. Nature 1972, 236, 385–390. [Google Scholar] [CrossRef]
- Van Rossum, J.M. Cumulative dose-response curves. II. Technique for the making of dose-response curves in isolated organs and the evaluation of drug parameters. Arch. Int. Pharmacodyn. Ther. 1963, 143, 87–94. [Google Scholar]
- Sample Availability: Samples of the compounds 25 and 32 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sadek, B.; Alisch, R.; Buschauer, A.; Elz, S. Synthesis and Dual Histamine H1 and H2 Receptor Antagonist Activity of Cyanoguanidine Derivatives. Molecules 2013, 18, 14186-14202. https://doi.org/10.3390/molecules181114186
Sadek B, Alisch R, Buschauer A, Elz S. Synthesis and Dual Histamine H1 and H2 Receptor Antagonist Activity of Cyanoguanidine Derivatives. Molecules. 2013; 18(11):14186-14202. https://doi.org/10.3390/molecules181114186
Chicago/Turabian StyleSadek, Bassem, Rudi Alisch, Armin Buschauer, and Sigurd Elz. 2013. "Synthesis and Dual Histamine H1 and H2 Receptor Antagonist Activity of Cyanoguanidine Derivatives" Molecules 18, no. 11: 14186-14202. https://doi.org/10.3390/molecules181114186
APA StyleSadek, B., Alisch, R., Buschauer, A., & Elz, S. (2013). Synthesis and Dual Histamine H1 and H2 Receptor Antagonist Activity of Cyanoguanidine Derivatives. Molecules, 18(11), 14186-14202. https://doi.org/10.3390/molecules181114186