G-Quadruplexes as Potential Therapeutic Targets for Embryonal Tumors

 ,
,
Abstract
:
1. Introduction
2. DNA as a Target for Anticancer Therapy

{kind=link}
{kind=link}
| Drug | Mode of Binding | |||
|---|---|---|---|---|
| Covalent | Non covalent | |||
| Alkylating agents (irreversible and leads to complete inhibition of DNA processes and subsequent cell death) | Groove binders | Intercalators | ||
| Minor groove binders | Major groove binders | |||
| Cisplatin (DNA crosslinker) | √ | |||
| Doxorubicin (Stabilizes topoisomerase-II–DNA cleavable) | √ | |||
| Etoposide (Topoisomerase inhibitor) | √ | |||
| Methotrexate (Antimetabolite, a folic acid antagonist) | √ | |||
| (TFOs) Triplex-forming oligonucleotides (oligomers that bind in the major groove and form hydrogen bond with bases of the purine strand) | √ | |||
| (PNAs) peptide nucleic acids (with peptide-like backbone that invade the helix to form a triplex which results in the displacement of noncomplementary oligopyrimidine DNA strand) | √ | |||
| (Daunomycin) combilexins | √ | |||
| Quinacrine | √ | |||
| Ethidium bromide | √ | |||
| Netropsin | √ | |||
| Distamycin | √ | |||
| DAPI | √ | |||
3. G-Quadruplexes
4. G-Quadruplexes as a Potential Cancer Therapeutic Targets
4.1. Telomere Structure and Function
4.2. Biological Significance of Telomeres and Telomerase during Development
4.3. Telomeres and Telomerase Activity during Tumor Development
4.4. Significance of Telomere Biology in Embryonal Tumors of the Nervous System
4.4.1. Neuroblastoma
4.4.2. Medulloblastoma
4.5. Significance of the myc Oncogene Family in Embryonal Tumors of the Nervous System
4.5.1. MYC in neuroblastoma
4.5.2. MYC in medulloblastoma
5. Targeting G-Quadruplex as a Novel Anticancer Strategy
5.1. Targeting Telomere Maintenance
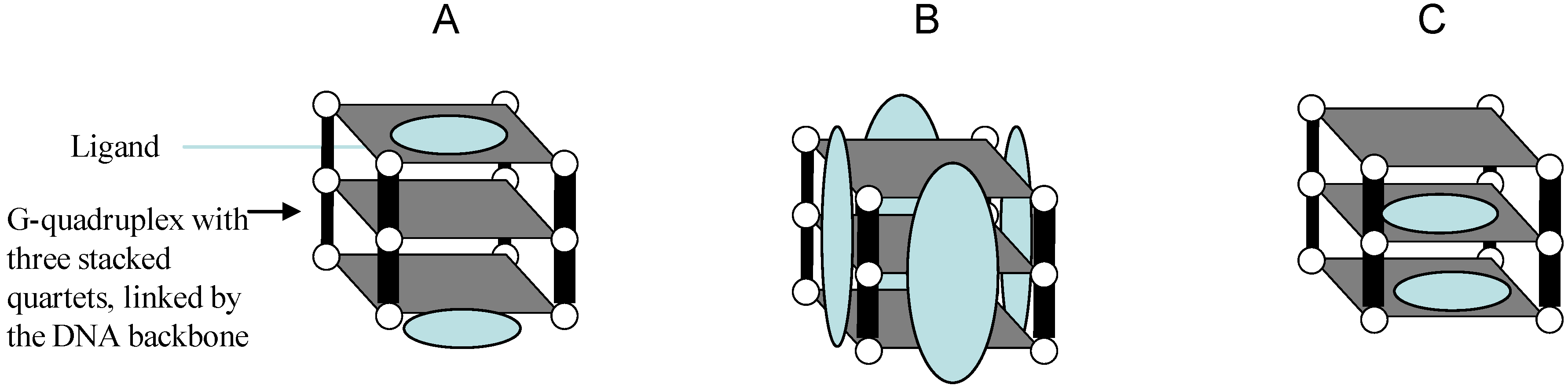
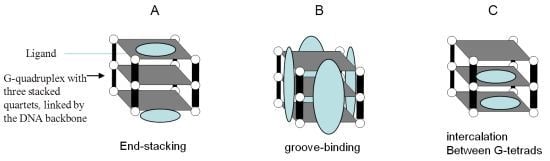
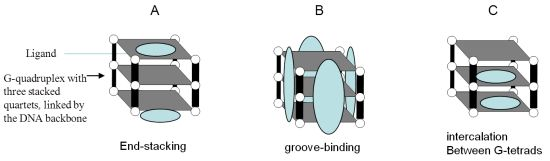
5.2. Targeting G-quadruplexes in Oncogene Promoters
6. G-Quadruplex-Interactive Small-Molecules
| Ligand | Tumor model tested | Antitumor activity | Reference |
|---|---|---|---|
| Telomestatin | Neuroblastoma, myeloma, acute leukemia and glioma stem cells |
| [231,232,233,234,235,236,237,238,239,240] |
| S2T1-6OTD (telomestatin synthetic Derivative) | Paediatric brain cancer (Medulloblastoma and atypical teratoid/rhabdoid) |
| [182] |
| HXDV (telomestatin syntheticDerivative) | A panel of normal/cancer telomerase- and ALT-positive cell lines |
| [241] |
| TMPyP4 (Cationic porphyrin) | Myeloma, cervical, pancreatic, breast, colon, prostate cancer and osteosarcoma, neuroblastoma and retinoblastoma |
| [242,243,244,245,246] |
| SYUIQ-5 and other quindoline derivatives | Leukemia, Burkitt’s lymphoma, human epithelial carcinoma, nasopharyngeal carcinoma |
| [2,41,58,226,247,248] |
| Tetrasubstituted napthalene diimides ligands | Brest, prostate cancer , and lung adenocarcinoma |
| [249] |
| Triazine derivatives | Melanoma, mouth, lung, colon cancer as well as, lung adenocarcinoma |
| [250,251,252,253,254] |
| Trisubstituted acridine (AS1410) | Breast and lung cancer |
| [255] |
| BRACO-19 3,6,9-trisubstituted acridine | Breast and prostate cancer, uterus and vulval carcinoma |
| [252,253,256,257,258,259,260] |
| Pentacyclic acridines (RHPS4) | Melanoma, breast and vulval cancer |
| [253,259,261,262,263,264,265,266] |
| 4,5-di-substituted acridone | Breast and lung cancer |
| [267] |
| Anthracene derivatives | Melanoma, colon cancer and osteogenic sarcoma |
| [268] |
| Amidoanthraquinone derivatives | 60 different human cancer cell lines |
| [269] |
| Perylene derivatives | Melanoma, colon and breast carcinomas and osteosarcoma and colorectal carcinoma cell |
| [270,271] |
| Macrocyclic pyridyl polyoxazoles | Oral carcinoma and breast cancer |
| [272,273] |
| Triethylene tetramine (TETA) | Brest cancer and human epithelial carcinoma |
| [274,275] |
| Bisquinolinium pyridine dicarboxamide compound (360A) | Cervical cancer and colorectal carcinoma |
| [276,277] |
| 307A 2,6-pyridin-dicarboxamide derivative | Glioma and osteosarcoma |
| [278] |
| Bisantrene derivatives (An1,5) | Melanoma and osteogenic sarcoma |
| [268] |
G-Quadruplex-Targeting Drugs in Clinical Trials
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Legler, J.M.; Ries, L.A.; Smith, M.A.; Warren, J.L.; Heineman, E.F.; Kaplan, R.S.; Linet, M.S. Cancer surveillance series [corrected]: Brain and other central nervous system cancers: Recent trends in incidence and mortality. J. Natl. Cancer Inst. 1999, 91, 1382–1390. [Google Scholar] [CrossRef]
- Scotting, P.J.; Walker, D.A.; Perilongo, G. Childhood solid tumours: A developmental disorder. Nat. Rev. Cancer 2005, 5, 481–488. [Google Scholar] [CrossRef]
- Grimmer, M.R.; Weiss, W.A. Childhood tumors of the nervous system as disorders of normal development. Curr. Opin. Pediatr. 2006, 18, 634–638. [Google Scholar] [CrossRef]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Gilbertson, R.J. Medulloblastoma: Signalling a change in treatment. Lancet Oncol. 2004, 5, 209–218. [Google Scholar] [CrossRef]
- Macy, M.E.; Sawczyn, K.K.; Garrington, T.P.; Graham, D.K.; Gore, L. Pediatric developmental therapies: Interesting new drugs now in early-stage clinical trials. Curr. Oncol. Rep. 2008, 10, 477–490. [Google Scholar] [CrossRef]
- Vassal, G. Has chemotherapy reached its limits in pediatric cancers? Eur. J. Cancer 2005, 41, 564–575. [Google Scholar] [CrossRef]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Lane, A.N.; Chaires, J.B.; Gray, R.D.; Trent, J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008, 36, 5482–5515. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef]
- Oganesian, L.; Bryan, T.M. Physiological relevance of telomeric G-quadruplex formation: A potential drug target. Bioessays 2007, 29, 155–165. [Google Scholar] [CrossRef]
- Qin, Y.; Hurley, L.H. Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie 2008, 90, 1149–1171. [Google Scholar] [CrossRef]
- Shukla, A.K.; Roy, K.B. Rec A-independent homologous recombination induced by a putative fold-back tetraplex DNA. Biol. Chem. 2006, 387, 251–256. [Google Scholar]
- van Dieijen-Visser, M.P.; Go, P.M.; Brombacher, P.J. The value of laboratory tests in patients suspected of acute appendicitis. Eur. J. Clin. Chem. Clin. Biochem. 1991, 29, 749–752. [Google Scholar]
- Gutierrez, M.L.; Crooke, S.T. Pediatric cancer chemotherapy: An updated review. I. Cis-Diammine--dichloroplatinum II (cisplatin), VM-26 (teniposide), VP-16 (etoposide), mitomycin C. Cancer Treat. Rev. 1979, 6, 153–164. [Google Scholar] [CrossRef]
- Gurova, K. New hopes from old drugs: Revisiting DNA-binding small molecules as anticancer agents. Future Oncol. 2009, 5, 1685–1704. [Google Scholar] [CrossRef]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–84. [Google Scholar] [CrossRef]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef]
- Gilbert, D.E.; Feigon, J. Multistranded DNA structures. Curr. Opin. Struct. Biol. 1999, 9, 305–314. [Google Scholar] [CrossRef]
- Jing, N.; Li, Y.; Xiong, W.; Sha, W.; Jing, L.; Tweardy, D.J. G-quartet oligonucleotides: a new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 2004, 64, 6603–6609. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Lee, M.P.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef]
- Phan, A.T.; Modi, Y.S.; Patel, D.J. Two-repeat Tetrahymena telomeric d(TGGGGTTGGGGT) Sequence interconverts between asymmetric dimeric G-quadruplexes in solution. J. Mol. Biol. 2004, 338, 93–102. [Google Scholar] [CrossRef]
- Kerwin, S.M. G-Quadruplex DNA as a target for drug design. Curr. Pharm. Des. 2000, 6, 441–478. [Google Scholar] [CrossRef]
- Phan, A.T.; Modi, Y.S.; Patel, D.J. Propeller-type parallel-stranded G-quadruplexes in the human c-myc promoter. J. Am. Chem. Soc. 2004, 126, 8710–8716. [Google Scholar] [CrossRef]
- Wang, Y.; Patel, D.J. Guanine residues in d(T2AG3) and d(T2G4) form parallel-stranded potassium cation stabilized G-quadruplexes with anti glycosidic torsion angles in solution. Biochemistry 1992, 31, 8112–8119. [Google Scholar] [CrossRef]
- Risitano, A.; Fox, K.R. Inosine substitutions demonstrate that intramolecular DNA quadruplexes adopt different conformations in the presence of sodium and potassium. Bioorg. Med. Chem. Lett. 2005, 15, 2047–2050. [Google Scholar] [CrossRef]
- Fry, M. Tetraplex DNA and its interacting proteins. Front. Biosci. 2007, 12, 4336–4351. [Google Scholar] [CrossRef]
- Huang, W.; Smaldino, P.J.; Zhang, Q.; Miller, L.D.; Cao, P.; Stadelman, K.; Wan, M.; Giri, B.; Lei, M.; Nagamine, Y.; et al. Yin Yang 1 contains G-quadruplex structures in its promoter and 5′-UTR and its expression is modulated by G4 resolvase 1. Nucleic Acids Res. 2012, 40, 1033–1049. [Google Scholar] [CrossRef]
- Maizels, N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat. Struct. Mol. Biol. 2006, 13, 1055–1059. [Google Scholar] [CrossRef]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human telomere, oncogenic promoter and 5′-UTR G-quadruplexes: Diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef]
- Schaffitzel, C.; Berger, I.; Postberg, J.; Hanes, J.; Lipps, H.J.; Pluckthun, A. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc Natl Acad Sci USA 2001, 98, 8572–8577. [Google Scholar]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Grand, C.L.; Han, H.; Munoz, R.M.; Weitman, S.; Von Hoff, D.D.; Hurley, L.H.; Bearss, D.J. The cationic porphyrin TMPyP4 down-regulates c-MYC and human telomerase reverse transcriptase expression and inhibits tumor growth in vivo. Mol. Cancer Ther. 2002, 1, 565–573. [Google Scholar]
- Seenisamy, J.; Bashyam, S.; Gokhale, V.; Vankayalapati, H.; Sun, D.; Siddiqui-Jain, A.; Streiner, N.; Shin-Ya, K.; White, E.; Wilson, W.D.; et al. Design and synthesis of an expanded porphyrin that has selectivity for the c-MYC G-quadruplex structure. J. Am. Chem. Soc. 2005, 127, 2944–2959. [Google Scholar] [CrossRef]
- Kostadinov, R.; Malhotra, N.; Viotti, M.; Shine, R.; D'Antonio, L.; Bagga, P. GRSDB: A database of quadruplex forming G-rich sequences in alternatively processed mammalian pre-mRNA sequences. Nucleic Acids Res. 2006, 34, D119–D124. [Google Scholar] [CrossRef]
- Zarudnaya, M.I.; Kolomiets, I.M.; Potyahaylo, A.L.; Hovorun, D.M. Downstream elements of mammalian pre-mRNA polyadenylation signals: Primary, secondary and higher-order structures. Nucleic Acids Res. 2003, 31, 1375–1386. [Google Scholar] [CrossRef]
- Davis, T.L.; Firulli, A.B.; Kinniburgh, A.J. Ribonucleoprotein and protein factors bind to an H-DNA-forming c-myc DNA element: Possible regulators of the c-myc gene. Proc. Natl. Acad. Sci. USA 1989, 86, 9682–9686. [Google Scholar] [CrossRef]
- Ou, T.M.; Lin, J.; Lu, Y.J.; Hou, J.Q.; Tan, J.H.; Chen, S.H.; Li, Z.; Li, Y.P.; Li, D.; Gu, L.Q.; et al. Inhibition of cell proliferation by quindoline derivative (SYUIQ-05) through its preferential interaction with c-myc promoter G-quadruplex. J. Med. Chem. 2011, 54, 5671–5679. [Google Scholar] [CrossRef]
- Murat, P.; Singh, Y.; Defrancq, E. Methods for investigating G-quadruplex DNA/ligand interactions. Chem. Soc. Rev. 2011, 40, 5293–5307. [Google Scholar] [CrossRef]
- Yuan, L.; Tian, T.; Chen, Y.; Yan, S.; Xing, X.; Zhang, Z.; Zhai, Q.; Xu, L.; Wang, S.; Weng, X.; et al. Existence of G-quadruplex structures in promoter region of oncogenes confirmed by G-quadruplex DNA cross-linking strategy. Sci. Rep. 2013, 3, 1811. [Google Scholar]
- Blackburn, E.H. Telomere states and cell fates. Nature 2000, 408, 53–56. [Google Scholar] [CrossRef]
- Hackett, J.A.; Feldser, D.M.; Greider, C.W. Telomere dysfunction increases mutation rate and genomic instability. Cell 2001, 106, 275–286. [Google Scholar] [CrossRef]
- Van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef]
- Chai, W.; Du, Q.; Shay, J.W.; Wright, W.E. Human telomeres have different overhang sizes at leading versus lagging strands. Mol. Cell 2006, 21, 427–435. [Google Scholar] [CrossRef]
- Zhao, Y.; Hoshiyama, H.; Shay, J.W.; Wright, W.E. Quantitative telomeric overhang determination using a double-strand specific nuclease. Nucleic Acids Res. 2008, 36, e14. [Google Scholar] [CrossRef]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–22. [Google Scholar] [CrossRef]
- Shin, J.S.; Hong, A.; Solomon, M.J.; Lee, C.S. The role of telomeres and telomerase in the pathology of human cancer and aging. Pathology 2006, 38, 103–113. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–10. [Google Scholar] [CrossRef]
- Wang, F.; Podell, E.R.; Zaug, A.J.; Yang, Y.; Baciu, P.; Cech, T.R.; Lei, M. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 2007, 445, 506–510. [Google Scholar] [CrossRef]
- Morin, G.B. Telomere control of replicative lifespan. Exp. Gerontol. 1997, 32, 375–382. [Google Scholar] [CrossRef]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef]
- Bosoy, D.; Peng, Y.; Mian, I.S.; Lue, N.F. Conserved N-terminal Motifs of Telomerase Reverse Transcriptase Required for Ribonucleoprotein Assembly in Vivo. J. Biol. Chem. 2003, 278, 3882–3890. [Google Scholar] [CrossRef]
- Masutomi, K.; Yu, E.Y.; Khurts, S.; Ben-Porath, I.; Currier, J.L.; Metz, G.B.; Brooks, M.W.; Kaneko, S.; Murakami, S.; de Caprio, J.A.; et al. Telomerase maintains telomere structure in normal human cells. Cell 2003, 114, 241–253. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Greider, C.W.; Szostak, J.W. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006, 12, 1133–1138. [Google Scholar]
- Forsyth, N.R.; Wright, W.E.; Shay, J.W. Telomerase and differentiation in multicellular organisms: Turn it off, turn it on, and turn it off again. Differentiation 2002, 69, 188–197. [Google Scholar] [CrossRef]
- Harrington, L. Does the reservoir for self-renewal stem from the ends? Oncogene 2004, 23, 7283–7289. [Google Scholar] [CrossRef]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef]
- Neumann, A.A.; Reddel, R.R. Telomere maintenance and cancer? Look, no telomerase. Nat. Rev. Cancer 2002, 2, 879–884. [Google Scholar] [CrossRef]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef]
- Schaetzlein, S.; Lucas-Hahn, A.; Lemme, E.; Kues, W.A.; Dorsch, M.; Manns, M.P.; Niemann, H.; Rudolph, K.L. Telomere length is reset during early mammalian embryogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 8034–8038. [Google Scholar] [CrossRef]
- De Lange, T.; Shiue, L.; Myers, R.M.; Cox, D.R.; Naylor, S.L.; Killery, A.M.; Varmus, H.E. Structure and variability of human chromosome ends. Mol. Cell. Biol. 1990, 10, 518–527. [Google Scholar]
- Achi, M.V.; Ravindranath, N.; Dym, M. Telomere length in male germ cells is inversely correlated with telomerase activity. Biol. Reprod. 2000, 63, 591–598. [Google Scholar] [CrossRef]
- Hemann, M.T.; Rudolph, K.L.; Strong, M.A.; DePinho, R.A.; Chin, L.; Greider, C.W. Telomere dysfunction triggers developmentally regulated germ cell apoptosis. Mol. Biol. Cell. 2001, 12, 2023–2030. [Google Scholar] [CrossRef]
- Stindl, R. Is telomere erosion a mechanism of species extinction? J. Exp. Zoolog. B Mol. Dev. Evol. 2004, 302, 111–120. [Google Scholar]
- Allsopp, R.C.; Vaziri, H.; Patterson, C.; Goldstein, S.; Younglai, E.V.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere length predicts replicative capacity of human fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10114–1018. [Google Scholar] [CrossRef]
- Cooke, H.J.; Smith, B.A. Variability at the telomeres of the human X/Y pseudoautosomal region. Cold Spring Harb. Symp. Quant. Biol. 1986, 51 (Pt 1), 213–219. [Google Scholar] [CrossRef]
- Dahse, R.; Fiedler, W.; Ernst, G. Telomeres and telomerase: Biological and clinical importance. Clin. Chem. 1997, 43, 708–714. [Google Scholar]
- Blasco, M.A.; Funk, W.; Villeponteau, B.; Greider, C.W. Functional characterization and developmental regulation of mouse telomerase RNA. Science 1995, 269, 1267–1270. [Google Scholar] [CrossRef]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Yashima, K.; Maitra, A.; Rogers, B.B.; Timmons, C.F.; Rathi, A.; Pinar, H.; Wright, W.E.; Shay, J.W.; Gazdar, A.F. Expression of the RNA component of telomerase during human development and differentiation. Cell. Growth Differ. 1998, 9, 805–813. [Google Scholar]
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 1998, 58, 4168–4172. [Google Scholar]
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024. [Google Scholar] [CrossRef]
- Sugihara, M.; Ohshima, K.; Nakamura, H.; Suzumiya, J.; Nakayama, Y.; Kanda, M.; Haraoka, S.; Kikuchi, M. Decreased expression of telomerase-associated RNAs in the proliferation of stem cells in comparison with continuous expression in malignant tumors. Int. J. Oncol. 1999, 15, 1075–1080. [Google Scholar]
- Haas, O.A.; Seyger, M. Hypothesis: meiotic origin of trisomic neoplasms. Cancer Genet. Cytogenet. 1993, 70, 112–116. [Google Scholar] [CrossRef]
- Lampert, F. On the “keimversprengungs” origin of embryonic tumors. Cancer Genet. Cytogenet. 1994, 78, 242. [Google Scholar] [CrossRef]
- Trosko, J.E. Review paper: Cancer stem cells and cancer nonstem cells: From adult stem cells or from reprogramming of differentiated somatic cells. Vet. Pathol. 2009, 46, 176–193. [Google Scholar]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar]
- Counter, C.M.; Avilion, A.A.; LeFeuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar]
- Hiyama, E.; Hiyama, K. Clinical utility of telomerase in cancer. Oncogene 2002, 21, 643–649. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Hiyama, E.; Hiyama, K.; Yokoyama, T.; Matsuura, Y.; Piatyszek, M.A.; Shay, J.W. Correlating telomerase activity levels with human neuroblastoma outcomes. Nat. Med. 1995, 1, 249–255. [Google Scholar] [CrossRef]
- Greaves, M. Is telomerase activity in cancer due to selection of stem cells and differentiation arrest? Trends Genet. 1996, 12, 127–128. [Google Scholar] [CrossRef]
- Armstrong, L.; Saretzki, G.; Peters, H.; Wappler, I.; Evans, J.; Hole, N.; von Zglinicki, T.; Lako, M. Overexpression of telomerase confers growth advantage, stress resistance, and enhanced differentiation of ESCs toward the hematopoietic lineage. Stem Cells 2005, 23, 516–529. [Google Scholar] [CrossRef]
- O'Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Mishra, L.; Banker, T.; Murray, J.; Byers, S.; Thenappan, A.; He, A.R.; Shetty, K.; Johnson, L.; Reddy, E.P. Liver stem cells and hepatocellular carcinoma. Hepatology 2009, 49, 318–329. [Google Scholar] [CrossRef]
- Grichnik, J.M. Melanoma, nevogenesis, and stem cell biology. J. Invest. Dermatol. 2008, 128, 2365–2380. [Google Scholar] [CrossRef]
- Trumpp, A.; Wiestler, O.D. Mechanisms of Disease: Cancer stem cells--targeting the evil twin. Nat. Clin. Pract. Oncol. 2008, 5, 337–347. [Google Scholar]
- Read, T.A.; Fogarty, M.P.; Markant, S.L.; McLendon, R.E.; Wei, Z.; Ellison, D.W.; Febbo, P.G.; Wechsler-Reya, R.J. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009, 15, 135–147. [Google Scholar] [CrossRef]
- Cairo, S.; Armengol, C.; de Reynies, A.; Wei, Y.; Thomas, E.; Renard, C.A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008, 14, 471–484. [Google Scholar] [CrossRef]
- Charytonowicz, E.; Cordon-Cardo, C.; Matushansky, I.; Ziman, M. Alveolar rhabdomyosarcoma: Is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009, 279, 126–136. [Google Scholar] [CrossRef]
- Fan, X.; Eberhart, C.G. Medulloblastoma stem cells. J. Clin. Oncol. 2008, 26, 2821–2827. [Google Scholar] [CrossRef]
- Ross, R.A.; Spengler, B.A. Human neuroblastoma stem cells. Semin. Cancer Biol. 2007, 17, 241–247. [Google Scholar] [CrossRef]
- Suva, M.L.; Riggi, N.; Stehle, J.C.; Baumer, K.; Tercier, S.; Joseph, J.M.; Suva, D.; Clement, V.; Provero, P.; Cironi, L.; Osterheld, M.C.; Guillou, L.; Stamenkovic, I. Identification of cancer stem cells in Ewing's sarcoma. Cancer Res. 2009, 69, 1776–1781. [Google Scholar] [CrossRef]
- Allen, N.D.; Baird, D.M. Telomere length maintenance in stem cell populations. Biochim. Biophys. Acta 2009, 1792, 324–328. [Google Scholar]
- Sabatier, L.; Ricoul, M.; Pottier, G.; Murnane, J.P. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol. Cancer Res. 2005, 3, 139–150. [Google Scholar] [CrossRef]
- Brunori, M.; Mathieu, N.; Ricoul, M.; Bauwens, S.; Koering, C.E.; Roborel de Climens, A.; Belleville, A.; Wang, Q.; Puisieux, I.; Decimo, D.; Puisieux, A.; Sabatier, L.; Gilson, E. TRF2 inhibition promotes anchorage-independent growth of telomerase-positive human fibroblasts. Oncogene 2006, 25, 990–997. [Google Scholar] [CrossRef]
- Blackburn, E.H. Structure and function of telomeres. Nature 1991, 350, 569–573. [Google Scholar] [CrossRef]
- Hande, M.P.; Samper, E.; Lansdorp, P.; Blasco, M.A. Telomere length dynamics and chromosomal instability in cells derived from telomerase null mice. J. Cell. Biol. 1999, 144, 589–601. [Google Scholar] [CrossRef]
- Velicescu, M.; Yu, J.; Herbert, B.S.; Shay, J.W.; Granada, E.; Dubeau, L. Aneuploidy and telomere attrition are independent determinants of crisis in SV40-transformed epithelial cells. Cancer Res. 2003, 63, 5813–5820. [Google Scholar]
- Gisselsson, D.; Pettersson, L.; Hoglund, M.; Heidenblad, M.; Gorunova, L.; Wiegant, J.; Mertens, F.; Dal Cin, P.; Mitelman, F.; Mandahl, N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc. Natl. Acad. Sci. USA 2000, 97, 5357–5362. [Google Scholar] [CrossRef]
- Stewenius, Y.; Gorunova, L.; Jonson, T.; Larsson, N.; Hoglund, M.; Mandahl, N.; Mertens, F.; Mitelman, F.; Gisselsson, D. Structural and numerical chromosome changes in colon cancer develop through telomere-mediated anaphase bridges, not through mitotic multipolarity. Proc. Natl. Acad. Sci. USA 2005, 102, 5541–5546. [Google Scholar] [CrossRef]
- Fett-Conte, A.C.; Liedtke Junior, H.; Chaves, H.; Thome, J.A.; Tajara, E.H. Telomeric fusions in a Wilms' tumor. Cancer Genet. Cytogenet. 1993, 69, 141–145. [Google Scholar] [CrossRef]
- Stewenius, Y.; Jin, Y.; Ora, I.; de Kraker, J.; Bras, J.; Frigyesi, A.; Alumets, J.; Sandstedt, B.; Meeker, A.K.; Gisselsson, D. Defective chromosome segregation and telomere dysfunction in aggressive Wilms' tumors. Clin. Cancer Res. 2007, 13, 6593–6602. [Google Scholar] [CrossRef]
- Frappart, P.O.; Lee, Y.; Russell, H.R.; Chalhoub, N.; Wang, Y.D.; Orii, K.E.; Zhao, J.; Kondo, N.; Baker, S.J.; McKinnon, P.J. Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc. Natl. Acad. Sci. USA 2009, 106, 1880–1885. [Google Scholar] [CrossRef]
- Shakhova, O.; Leung, C.; van Montfort, E.; Berns, A.; Marino, S. Lack of Rb and p53 delays cerebellar development and predisposes to large cell anaplastic medulloblastoma through amplification of N-Myc and Ptch2. Cancer Res. 2006, 66, 5190–5200. [Google Scholar]
- Schulte, J.H.; Kuhfittig-Kulle, S.; Klein-Hitpass, L.; Schramm, A.; Biard, D.S.; Pfeiffer, P.; Eggert, A. Expression of the TrkA or TrkB receptor tyrosine kinase alters the double-strand break (DSB) repair capacity of SY5Y neuroblastoma cells. DNA Repair 2008, 7, 1757–1764. [Google Scholar] [CrossRef]
- Sugihara, E.; Kanai, M.; Matsui, A.; Onodera, M.; Schwab, M.; Miwa, M. Enhanced expression of MYCN leads to centrosome hyperamplification after DNA damage in neuroblastoma cells. Oncogene 2004, 23, 1005–1009. [Google Scholar] [CrossRef]
- Goldstein, M.; Meller, I.; Issakov, J.; Orr-Urtreger, A. Novel genes implicated in embryonal, alveolar, and pleomorphic rhabdomyosarcoma: A cytogenetic and molecular analysis of primary tumors. Neoplasia. 2006, 8, 332–343. [Google Scholar] [CrossRef]
- Wang, Z.; Velagaleti, G.V.; Eltorky, M.A.; Tang, W.W.; Hawkins, H.K.; Jones, E.A.; Northup, J.; Panova, N.; Qiu, S. Cytogenetic and molecular studies of an unusual case of multiple primary alveolar rhabdomyosarcomas: Low-level chromosomal instability and reciprocal translocation t(6;11). Exp. Mol. Pathol. 2007, 82, 58–62. [Google Scholar] [CrossRef]
- Rahman, R.; Heath, R.; Grundy, R. Cellular immortality in brain tumours: An integration of the cancer stem cell paradigm. Biochim. Biophys. Acta. 2009, 1792, 280–288. [Google Scholar]
- Polychronopoulou, S.; Koutroumba, P. Telomere length and telomerase activity: Variations with advancing age and potential role in childhood malignancies. J. Pediatr. Hematol. Oncol. 2004, 26, 342–350. [Google Scholar] [CrossRef]
- Bianchi, A.; Shore, D. How telomerase reaches its end: Mechanism of telomerase regulation by the telomeric complex. Mol. Cell. 2008, 31, 153–165. [Google Scholar] [CrossRef]
- Guittat, L.; Alberti, P.; Gomez, D.; De Cian, A.; Pennarun, G.; Lemarteleur, T.; Belmokhtar, C.; Paterski, R.; Morjani, H.; Trentesaux, C.; et al. Targeting human telomerase for cancer therapeutics. Cytotechnology. 2004, 45, 75–90. [Google Scholar] [CrossRef]
- Harley, C.B. Telomerase and cancer therapeutics. Nat. Rev. Cancer 2008, 8, 167–79. [Google Scholar] [CrossRef]
- Rankin, A.M.; Faller, D.V.; Spanjaard, R.A. Telomerase inhibitors and 'T-oligo' as cancer therapeutics: Contrasting molecular mechanisms of cytotoxicity. Anticancer Drugs 2008, 19, 329–338. [Google Scholar] [CrossRef]
- Sawyer, J.R.; Goosen, L.S.; Stine, K.C.; Thomas, J.R. Telomere fusion as a mechanism for the progressive loss of the short arm of chromosome 11 in an anaplastic Wilms' tumor. Cancer 1994, 74, 767–773. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Hoffman, A.R.; Otero, J.; Huang, H.Y.; Zhao, Z.; Mazumdar, M.; Gorlick, R.; Meyers, P.; Healey, J.H.; Ladanyi, M. Divergent patterns of telomere maintenance mechanisms among human sarcomas: Sharply contrasting prevalence of the alternative lengthening of telomeres mechanism in Ewing's sarcomas and osteosarcomas. Genes Chromosomes Cancer 2004, 41, 155–162. [Google Scholar] [CrossRef]
- Yan, P.; Benhattar, J.; Coindre, J.M.; Guillou, L. Telomerase activity and hTERT mRNA expression can be heterogeneous and does not correlate with telomere length in soft tissue sarcomas. Int. J. Cancer 2002, 98, 851–856. [Google Scholar] [CrossRef]
- Ohali, A.; Avigad, S.; Cohen, I.J.; Meller, I.; Kollender, Y.; Issakov, J.; Gelernter, I.; Goshen, Y.; Yaniv, I.; Zaizov, R. Association between telomerase activity and outcome in patients with nonmetastatic Ewing family of tumors. J. Clin. Oncol. 2003, 21, 3836–3843. [Google Scholar] [CrossRef]
- Hiyama, E.; Yamaoka, H.; Matsunaga, T.; Hayashi, Y.; Ando, H.; Suita, S.; Horie, H.; Kaneko, M.; Sasaki, F.; Hashizume, K.; Nakagawara, A.; Ohnuma, N.; Yokoyama, T. High expression of telomerase is an independent prognostic indicator of poor outcome in hepatoblastoma. Br. J. Cancer 2004, 91, 972–979. [Google Scholar]
- Yan, C.T.; Kaushal, D.; Murphy, M.; Zhang, Y.; Datta, A.; Chen, C.; Monroe, B.; Mostoslavsky, G.; Coakley, K.; Gao, Y.; et al. RCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7378–7383. [Google Scholar] [CrossRef]
- Zindy, F.; Uziel, T.; Ayrault, O.; Calabrese, C.; Valentine, M.; Rehg, J.E.; Gilbertson, R.J.; Sherr, C.J.; Roussel, M.F. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007, 67, 2676–2684. [Google Scholar] [CrossRef]
- Hiyama, E.; Hiyama, K.; Ohtsu, K.; Yamaoka, H.; Ichikawa, T.; Shay, J.W.; Yokoyama, T. Telomerase activity in neuroblastoma: Is it a prognostic indicator of clinical behaviour? Eur. J. Cancer 1997, 33, 1932–1936. [Google Scholar] [CrossRef]
- Dome, J.S.; Chung, S.; Bergemann, T.; Umbricht, C.B.; Saji, M.; Carey, L.A.; Grundy, P.E.; Perlman, E.J.; Breslow, N.E.; Sukumar, S. High telomerase reverse transcriptase (hTERT) messenger RNA level correlates with tumor recurrence in patients with favorable histology Wilms' tumor. Cancer Res. 1999, 59, 4301–4307. [Google Scholar]
- Montgomery, E.; Argani, P.; Hicks, J.L.; DeMarzo, A.M.; Meeker, A.K. Telomere lengths of translocation-associated and nontranslocation-associated sarcomas differ dramatically. Am. J. Pathol. 2004, 164, 1523–1529. [Google Scholar] [CrossRef]
- Sawada, T.; Kidowaki, T.; Sakamoto, I.; Hashida, T.; Matsumura, T.; Nakagawa, M.; Kusunoki, T. Neuroblastoma. Mass screening for early detection and its prognosis. Cancer 1984, 53, 2731–2735. [Google Scholar] [CrossRef]
- Schwab, M.; Westermann, F.; Hero, B.; Berthold, F. Neuroblastoma: Biology and molecular and chromosomal pathology. Lancet Oncol. 2003, 4, 472–480. [Google Scholar] [CrossRef]
- Ponzielli, R.; Katz, S.; Barsyte-Lovejoy, D.; Penn, L.Z. Cancer therapeutics: Targeting the dark side of Myc. Eur. J. Cancer 2005, 41, 2485–2501. [Google Scholar] [CrossRef]
- Westermann, F.; Schwab, M. Genetic parameters of neuroblastomas. Cancer Lett. 2002, 184, 127–147. [Google Scholar] [CrossRef]
- Riley, R.D.; Heney, D.; Jones, D.R.; Sutton, A.J.; Lambert, P.C.; Abrams, K.R.; Young, B.; Wailoo, A.J.; Burchill, S.A. A systematic review of molecular and biological tumor markers in neuroblastoma. Clin. Cancer Res. 2004, 10, 4–12. [Google Scholar] [CrossRef]
- Hiyama, E.; Hiyama, K.; Yokoyama, T.; Ichikawa, T.; Matsuura, Y. Length of telomeric repeats in neuroblastoma: Correlation with prognosis and other biological characteristics. Jpn. J. Cancer Res. 1992, 83, 159–164. [Google Scholar] [CrossRef]
- Poremba, C.; Willenbring, H.; Hero, B.; Christiansen, H.; Schafer, K.L.; Brinkschmidt, C.; Jurgens, H.; Bocker, W.; Dockhorn-Dworniczak, B. Telomerase activity distinguishes between neuroblastomas with good and poor prognosis. Ann. Oncol. 1999, 10, 715–721. [Google Scholar] [CrossRef]
- Brinkschmidt, C.; Poremba, C.; Christiansen, H.; Simon, R.; Schafer, K.L.; Terpe, H.J.; Lampert, F.; Boecker, W.; Dockhorn-Dworniczak, B. Comparative genomic hybridization and telomerase activity analysis identify two biologically different groups of 4s neuroblastomas. Br. J. Cancer 1998, 77, 2223–2229. [Google Scholar] [CrossRef]
- Reynolds, C.P.; Zuo, J.J.; Kim, N.W.; Wang, H.; Lukens, J.N.; Matthay, K.K.; Seeger, R.C. Telomerase expression in primary neuroblastomas. Eur. J. Cancer 1997, 33, 1929–1931. [Google Scholar]
- Streutker, C.J.; Thorner, P.; Fabricius, N.; Weitzman, S.; Zielenska, M. Telomerase activity as a prognostic factor in neuroblastomas. Pediatr. Dev. Pathol. 2001, 4, 62–67. [Google Scholar] [CrossRef]
- Poremba, C.; Hero, B.; Heine, B.; Scheel, C.; Schaefer, K.L.; Christiansen, H.; Berthold, F.; Kneif, S.; Stein, H.; Juergens, H.; Boecker, W.; Dockhorn-Dworniczak, B. Telomerase is a strong indicator for assessing the proneness to progression in neuroblastomas. Med. Pediatr. Oncol. 2000, 35, 651–655. [Google Scholar] [CrossRef]
- Maitra, A.; Yashima, K.; Rathi, A.; Timmons, C.F.; Rogers, B.B.; Shay, J.W.; Gazdar, A.F. The RNA component of telomerase as a marker of biologic potential and clinical outcome in childhood neuroblastic tumors. Cancer 1999, 85, 741–749. [Google Scholar] [CrossRef]
- Krams, M.; Hero, B.; Berthold, F.; Parwaresch, R.; Harms, D.; Rudolph, P. Full-length telomerase reverse transcriptase messenger RNA is an independent prognostic factor in neuroblastoma. Am. J. Pathol. 2003, 162, 1019–1026. [Google Scholar] [CrossRef]
- Binz, N.; Shalaby, T.; Rivera, P.; Shin-ya, K.; Grotzer, M.A. Telomerase inhibition, telomere shortening, cell growth suppression and induction of apoptosis by telomestatin in childhood neuroblastoma cells. Eur. J. Cancer 2005, 41, 2873–2881. [Google Scholar] [CrossRef]
- Byrd, T.; Grossman, R.G.; Ahmed, N. Medulloblastoma-biology and microenvironment: A review. Pediatr. Hematol. Oncol. 2012, 29, 495–506. [Google Scholar] [CrossRef]
- Eberhart, C.G.; Kratz, J.E.; Schuster, A.; Goldthwaite, P.; Cohen, K.J.; Perlman, E.J.; Burger, P.C. Comparative genomic hybridization detects an increased number of chromosomal alterations in large cell/anaplastic medulloblastomas. Brain Pathol. 2002, 12, 36–44. [Google Scholar]
- Reardon, D.A.; Michalkiewicz, E.; Boyett, J.M.; Sublett, J.E.; Entrekin, R.E.; Ragsdale, S.T.; Valentine, M.B.; Behm, F.G.; Li, H.; Heidemann, R.L.; Kun, L.E.; Shapiro, D.N.; Look, A.T. Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Research 1997, 57, 4042–4047. [Google Scholar]
- Fan, X.; Wang, Y.; Kratz, J.; Brat, D.J.; Robitaille, Y.; Moghrabi, A.; Perlman, E.J.; Dang, C.V.; Burger, P.C.; Eberhart, C.G. hTERT gene amplification and increased mRNA expression in central nervous system embryonal tumors. Am. J. Pathol. 2003, 162, 1763–1769. [Google Scholar] [CrossRef]
- Didiano, D.; Shalaby, T.; Lang, D.; Grotzer, M.A. Telomere maintenance in childhood primitive neuroectodermal brain tumors. J. Neurooncol. 2004, 6, 1–8. [Google Scholar]
- Liu, J.; Guo, L.; Luo, Y.; Li, J.W.; Li, H. All trans-retinoic acid suppresses in vitro growth and down-regulates LIF gene expression as well as telomerase activity of human medulloblastoma cells. Anticancer Res. 2000, 20, 2659–2664. [Google Scholar]
- Hiraga, S.; Ohnishi, T.; Izumoto, S.; Miyahara, E.; Kanemura, Y.; Matsumura, H.; Arita, N. Telomerase activity and alterations in telomere length in human brain tumors. Cancer Research 1998, 58, 2117–2125. [Google Scholar]
- DeMasters, B.K.; Markham, N.; Lillehei, K.O.; Shroyer, K.R. Differential telomerase expression in human primary intracranial tumors. Am. J. Clin. Pathol. 1997, 107, 548–554. [Google Scholar]
- Sugita, Y.; Nakashima, A.; Kato, S.; Sakata, K.; Morimatsu, M.; Shigemori, M. Telomerase activity in gliomas with the use of non-radioisotopic and semi-quantitative procedure for terminal repeat amplification protocol. Oncol Rep. 2000, 7, 1087–1092. [Google Scholar]
- Falchetti, M.L.; Pallini, R.; Larocca, L.M.; Verna, R.; D'Ambrosio, E. Telomerase expression in intracranial tumours: Prognostic potential for malignant gliomas and meningiomas. J. Clin. Pathol. 1999, 52, 234–236. [Google Scholar] [CrossRef]
- Zimmerman, K.; Alt, F.W. Expression and function of myc family genes. Crit. Rev. Oncog. 1990, 2, 75–95. [Google Scholar]
- Caren, H.; Kryh, H.; Nethander, M.; Sjoberg, R.M.; Trager, C.; Nilsson, S.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc. Natl. Acad. Sci. USA 2010, 107, 4323–4328. [Google Scholar] [CrossRef]
- Westermann, F.; Muth, D.; Benner, A.; Bauer, T.; Henrich, K.O.; Oberthuer, A.; Brors, B.; Beissbarth, T.; Vandesompele, J.; Pattyn, F.; Hero, B.; Konig, R.; Fischer, M.; Schwab, M. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008, 9, R150. [Google Scholar] [CrossRef]
- Fredlund, E.; Ringner, M.; Maris, J.M.; Pahlman, S. High Myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc. Natl. Acad. Sci. USA 2008, 105, 14094–14099. [Google Scholar] [CrossRef]
- Westermark, U.K.; Wilhelm, M.; Frenzel, A.; Henriksson, M.A. The MYCN oncogene and differentiation in neuroblastoma. Semin. Cancer Biol. 2011, 21, 256–266. [Google Scholar] [CrossRef]
- Burkhart, C.A.; Cheng, A.J.; Madafiglio, J.; Kavallaris, M.; Mili, M.; Marshall, G.M.; Weiss, W.A.; Khachigian, L.M.; Norris, M.D.; Haber, M. Effects of MYCN antisense oligonucleotide administration on tumorigenesis in a murine model of neuroblastoma. J. Natl. Cancer Inst. 2003, 95, 1394–1403. [Google Scholar] [CrossRef]
- Kang, J.H.; Rychahou, P.G.; Ishola, T.A.; Qiao, J.; Evers, B.M.; Chung, D.H. MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem. Biophys. Res. Commun. 2006, 351, 192–197. [Google Scholar] [CrossRef]
- Nara, K.; Kusafuka, T.; Yoneda, A.; Oue, T.; Sangkhathat, S.; Fukuzawa, M. Silencing of MYCN by RNA interference induces growth inhibition, apoptotic activity and cell differentiation in a neuroblastoma cell line with MYCN amplification. Int. J. Oncol. 2007, 30, 1189–1196. [Google Scholar]
- Negroni, A.; Scarpa, S.; Romeo, A.; Ferrari, S.; Modesti, A.; Raschella, G. Decrease of proliferation rate and induction of differentiation by a MYCN antisense DNA oligomer in a human neuroblastoma cell line. Cell. Growth Differ. 1991, 2, 511–518. [Google Scholar]
- Pession, A.; Tonelli, R.; Fronza, R.; Sciamanna, E.; Corradini, R.; Sforza, S.; Tedeschi, T.; Marchelli, R.; Montanaro, L.; Camerin, C.; Franzoni, M.; Paolucci, G. Targeted inhibition of NMYC by peptide nucleic acid in N-myc amplified human neuroblastoma cells: Cell-cycle inhibition with induction of neuronal cell differentiation and apoptosis. Int. J. Oncol. 2004, 24, 265–272. [Google Scholar]
- Ponthan, F.; Lindskog, M.; Karnehed, N.; Castro, J.; Kogner, P. Evaluation of anti-tumour effects of oral fenretinide (4-HPR) in rats with human neuroblastoma xenografts. Oncol. Rep. 2003, 10, 1587–1592. [Google Scholar]
- Sun, L.; Fuselier, J.A.; Murphy, W.A.; Coy, D.H. Antisense peptide nucleic acids conjugated to somatostatin analogs and targeted at the n-myc oncogene display enhanced cytotoxity to human neuroblastoma IMR32 cells expressing somatostatin receptors. Peptides 2002, 23, 1557–1565. [Google Scholar] [CrossRef]
- Grotzer, M.A.; Hogarty, M.D.; Janss, A.J.; Liu, X.; Zhao, H.; Eggert, A.; Sutton, L.N.; Rorke, L.B.; Brodeur, G.M.; Phillips, P.C. MYC messenger RNA expression predicts survival outcome in childhood primitive neuroectodermal tumor/medulloblastoma. Clin. Cancer Res. 2001, 7, 2425–2433. [Google Scholar]
- Eberhart, C.G.; Kratz, J.; Wang, Y.; Summers, K.; Stearns, D.; Cohen, K.; Dang, C.V.; Burger, P.C. Histopathological and molecular prognostic markers in medulloblastoma: C-myc, N-myc, TrkC, and anaplasia. J. Neuropathol. Exp. Neurol. 2004, 63, 441–449. [Google Scholar]
- Pession, A.; Tonelli, R. The MYCN oncogene as a specific and selective drug target for peripheral and central nervous system tumors. Curr. Cancer Drug Targets 2005, 5, 273–283. [Google Scholar] [CrossRef]
- Brandes, A.A.; Palmisano, V.; Monfardini, S. Medulloblastoma in adults: Clinical characteristics and treatment. Cancer Treat. Rev. 1999, 25, 3–12. [Google Scholar] [CrossRef]
- Frühwald, M.C.; D'Orisio, M.S.; Dai, Z.; Rush, L.J.; Krahe, R.; Smiraglia, D.J.; Pietsch, T.; Elsea, S.H.; Plass, C. Aberrant hypermethylation of the major breakpoint cluster region in 17p11.2 in medulloblastoma but not supratentorial PNETs. Genes Chromosomes Cancer 2001, 30, 38–47. [Google Scholar] [CrossRef]
- Aldosari, N.; Bigner, S.H.; Burger, P.C.; Becker, L.; Kepner, J.L.; Friedman, H.S.; McLendon, R.E. MYCC and MYCN oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the Children's Oncology Group. Arch. Pathol. Lab. Med. 2002, 126, 540–545. [Google Scholar]
- Rutkowski, S.; von Bueren, A.; von Hoff, K.; Hartmann, W.; Shalaby, T.; Deinlein, F.; Warmuth-Metz, M.; Soerensen, N.; Emser, A.; Bode, U.; Mittler, U.; Urban, C.; Benesch, M.; Kortmann, R.D.; Schlegel, P.G.; Kuehl, J.; Pietsch, T.; Grotzer, M. Prognostic Relevance of Clinical and Biological Risk Factors in Childhood Medulloblastoma: Results of Patients Treated in the Prospective Multicenter Trial HIT'91. Clin. Cancer Res. 2007, 13, 2651–2657. [Google Scholar] [CrossRef]
- Brown, H.G.; Kepner, J.L.; Perlman, E.J.; Friedman, H.S.; Strother, D.R.; Duffner, P.K.; Kun, L.E.; Goldthwaite, P.T.; Burger, P.C. "Large cell/anaplastic" medulloblastomas: A Pediatric Oncology Group Study. J. Neuropathol. Exp. Neurol. 2000, 59, 857–865. [Google Scholar]
- Kenney, A.M.; Cole, M.D.; Rowitch, D.H. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003, 130, 15–28. [Google Scholar] [CrossRef]
- Oliver, T.G.; Grasfeder, L.L.; Carroll, A.L.; Kaiser, C.; Gillingham, C.L.; Lin, S.M.; Wickramasinghe, R.; Scott, M.P.; Wechsler-Reya, R.J. Transcriptional profiling of the Sonic hedgehog response: A critical role for N-myc in proliferation of neuronal precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 7331–7336. [Google Scholar] [CrossRef]
- Rao, G.; Pedone, C.A.; Coffin, C.M.; Holland, E.C.; Fults, D.W. c-Myc enhances sonic hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia. 2003, 5, 198–204. [Google Scholar]
- Guessous, F.; Li, Y.; Abounader, R. Signaling pathways in medulloblastoma. J. Cell. Physiol. 2008, 217, 577–583. [Google Scholar] [CrossRef]
- Zhang, P.; Li, H.; Wu, M.L.; Chen, X.Y.; Kong, Q.Y.; Wang, X.W.; Sun, Y.; Wen, S.; Liu, J. c-Myc downregulation: A critical molecular event in resveratrol-induced cell cycle arrest and apoptosis of human medulloblastoma cells. J. Neurooncol. 2006, 80, 123–131. [Google Scholar] [CrossRef]
- von Bueren, A.O.; Shalaby, T.; Rajtarova, J.; Stearns, D.; Eberhart, C.G.; Helson, L.; Arcaro, A.; Grotzer, M.A. Anti-proliferative activity of the quassinoid NBT-272 in childhood medulloblastoma cells. BMC Cancer 2007, 7, 19. [Google Scholar] [CrossRef]
- Castelletti, D.; Fiaschetti, G.; di Dato, V.; Ziegler, U.; Kumps, C.; de Preter, K.; Zollo, M.; Speleman, F.; Shalaby, T.; de Martino, D.; et al. The quassinoid derivative NBT-272 targets both the AKT and ERK signaling pathways in embryonal tumors. Mol. Cancer Ther. 2010, 9, 3145–3157. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Shalaby, T.; von Bueren, A.O.; Hurlimann, M.L.; Fiaschetti, G.; Castelletti, D.; Masayuki, T.; Nagasawa, K.; Arcaro, A.; Jelesarov, I.; Shin-ya, K.; Grotzer, M. Disabling c-Myc in childhood medulloblastoma and atypical teratoid/rhabdoid tumor cells by the potent G-quadruplex interactive agent S2T1–6OTD. Mol. Cancer Ther. 2010, 9, 167–179. [Google Scholar]
- Mu, J.; Wei, L.X. Telomere and telomerase in oncology. Cell. Res. 2002, 12, 1–7. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; Hanahan, D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell. Biology 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- De Cian, A.; Lacroix, L.; Douarre, C.; Temime-Smaali, N.; Trentesaux, C.; Riou, J.F.; Mergny, J.L. Targeting telomeres and telomerase. Biochimie 2008, 90, 131–155. [Google Scholar] [CrossRef]
- Neidle, S.; Parkinson, G. Telomere maintenance as a target for anticancer drug discovery. Nat. Rev. Drug Discov. 2002, 1, 383–393. [Google Scholar] [CrossRef]
- Saretzki, G. Telomerase inhibition as cancer therapy. Cancer Lett. 2003, 194, 209–219. [Google Scholar] [CrossRef]
- Rossi, A.; Russo, G.; Puca, A.; La Montagna, R.; Caputo, M.; Mattioli, E.; Lopez, M.; Giordano, A.; Pentimalli, F. The antiretroviral nucleoside analogue Abacavir reduces cell growth and promotes differentiation of human medulloblastoma cells. Int. J. Cancer 2009, 125, 235–243. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, J.Q.; Chen, Z.; Zheng, K.W.; Chen, C.Y.; Hao, Y.H.; Tan, Z. G-quadruplex formation at the 3' end of telomere DNA inhibits its extension by telomerase, polymerase and unwinding by helicase. Nucleic Acids Res. 2011, 39, 6229–6237. [Google Scholar] [CrossRef]
- Kramer, R.; Cohen, D. Functional genomics to new drug targets. Nat. Rev. Drug Discov. 2004, 3, 965–972. [Google Scholar] [CrossRef]
- Tan, D.S. Diversity-oriented synthesis: Exploring the intersections between chemistry and biology. Nat. Chem. Biol. 2005, 1, 74–84. [Google Scholar] [CrossRef]
- Neidle, S. Human telomeric G-quadruplex: The current status of telomeric G-quadruplexes as therapeutic targets in human cancer. Febs. J. 2010, 277, 1118–1125. [Google Scholar] [CrossRef]
- Phan, A.T. Human telomeric G-quadruplex: Structures of DNA and RNA sequences. FEBS J. 2010, 277, 1107–1117. [Google Scholar] [CrossRef]
- Neidle, S. The structures of quadruplex nucleic acids and their drug complexes. Curr. Opin. Struct. Biol. 2009, 19, 239–250. [Google Scholar] [CrossRef]
- Bejugam, M.; Sewitz, S.; Shirude, P.S.; Rodriguez, R.; Shahid, R.; Balasubramanian, S. Trisubstituted isoalloxazines as a new class of G-quadruplex binding ligands: Small molecule regulation of c-kit oncogene expression. J. Am. Chem. Soc. 2007, 129, 12926–12927. [Google Scholar]
- Gunaratnam, M.; Greciano, O.; Martins, C.; Reszka, A.P.; Schultes, C.M.; Morjani, H.; Riou, J.F.; Neidle, S. Mechanism of acridine-based telomerase inhibition and telomere shortening. Biochem. Pharmacol. 2007, 74, 679–689. [Google Scholar] [CrossRef]
- Tahara, H.; Shin-Ya, K.; Seimiya, H.; Yamada, H.; Tsuruo, T.; Ide, T. G-Quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3′ telomeric overhang in cancer cells. Oncogene 2006, 25, 1955–1966. [Google Scholar] [CrossRef]
- Yang, D.; Okamoto, K. Structural insights into G-quadruplexes: Towards new anticancer drugs. Future Med. Chem. 2010, 2, 619–646. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef]
- Fernando, H.; Reszka, A.P.; Huppert, J.; Ladame, S.; Rankin, S.; Venkitaraman, A.R.; Neidle, S.; Balasubramanian, S. A conserved quadruplex motif located in a transcription activation site of the human c-kit oncogene. Biochemistry 2006, 45, 7854–7860. [Google Scholar] [CrossRef]
- Rankin, S.; Reszka, A.P.; Huppert, J.; Zloh, M.; Parkinson, G.N.; Todd, A.K.; Ladame, S.; Balasubramanian, S.; Neidle, S. Putative DNA quadruplex formation within the human c-kit oncogene. J. Am. Chem. Soc. 2005, 127, 10584–10589. [Google Scholar] [CrossRef]
- Cogoi, S.; Xodo, L.E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef]
- Lim, K.W.; Alberti, P.; Guedin, A.; Lacroix, L.; Riou, J.F.; Royle, N.J.; Mergny, J.L.; Phan, A.T. Sequence variant (CTAGGG)n in the human telomere favors a G-quadruplex structure containing a G.C.G.C tetrad. Nucleic Acids Res. 2009, 37, 6239–6248. [Google Scholar] [CrossRef]
- Palumbo, S.L.; Ebbinghaus, S.W.; Hurley, L.H. Formation of a unique end-to-end stacked pair of G-quadruplexes in the hTERT core promoter with implications for inhibition of telomerase by G-quadruplex-interactive ligands. J. Am. Chem. Soc. 2009, 131, 10878–10891. [Google Scholar] [CrossRef]
- Dai, J.; Chen, D.; Jones, R.A.; Hurley, L.H.; Yang, D. NMR solution structure of the major G-quadruplex structure formed in the human BCL2 promoter region. Nucleic Acids Res. 2006, 34, 5133–5144. [Google Scholar] [CrossRef]
- Sun, D.; Guo, K.; Rusche, J.J.; Hurley, L.H. Facilitation of a structural transition in the polypurine/polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-quadruplex-interactive agents. Nucleic Acids Res. 2005, 33, 6070–6080. [Google Scholar] [CrossRef]
- De Armond, R.; Wood, S.; Sun, D.; Hurley, L.H.; Ebbinghaus, S.W. Evidence for the presence of a guanine quadruplex forming region within a polypurine tract of the hypoxia inducible factor 1alpha promoter. Biochemistry 2005, 44, 16341–16350. [Google Scholar] [CrossRef]
- Palumbo, S.L.; Memmott, R.M.; Uribe, D.J.; Krotova-Khan, Y.; Hurley, L.H.; Ebbinghaus, S.W. A novel G-quadruplex-forming GGA repeat region in the c-myb promoter is a critical regulator of promoter activity. Nucleic Acids Res. 2008, 36, 1755–1769. [Google Scholar] [CrossRef]
- Mathad, R.I.; Hatzakis, E.; Dai, J.; Yang, D. c-MYC promoter G-quadruplex formed at the 5′-end of NHE III1 element: Insights into biological relevance and parallel-stranded G-quadruplex stability. Nucleic Acids Res. 2011, 39, 9023–9033. [Google Scholar] [CrossRef]
- Yang, D.; Hurley, L.H. Structure of the biologically relevant G-quadruplex in the c-MYC promoter. Nucleosides Nucleotides Nucleic Acids 2006, 25, 951–968. [Google Scholar] [CrossRef]
- Yang, H.; Ma, V.P.; Chan, D.S.; He, H.Z.; Leung, C.H.; Ma, D.L. A cyclometallated iridium(III) complex as a c-myc G-quadruplex stabilizer and down-regulator of c-myc oncogene expression. Curr. Med. Chem. 2013, 20, 576–582. [Google Scholar]
- Qin, Y.; Rezler, E.M.; Gokhale, V.; Sun, D.; Hurley, L.H. Characterization of the G-quadruplexes in the duplex nuclease hypersensitive element of the PDGF-A promoter and modulation of PDGF-A promoter activity by TMPyP4. Nucleic Acids Res. 2007, 35, 7698–7713. [Google Scholar] [CrossRef]
- Xu, Y.; Sugiyama, H. Formation of the G-quadruplex and i-motif structures in retinoblastoma susceptibility genes (Rb). Nucleic Acids Res. 2006, 34, 949–954. [Google Scholar] [CrossRef]
- Collie, G.W.; Parkinson, G.N. The application of DNA and RNA G-quadruplexes to therapeutic medicines. Chem. Soc. Rev. 2011, 40, 5867–5892. [Google Scholar] [CrossRef]
- Eddy, J.; Maizels, N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006, 34, 3887–3896. [Google Scholar] [CrossRef]
- Shirude, P.S.; Ying, L.; Balasubramanian, S. Single molecule conformational analysis of the biologically relevant DNA G-quadruplex in the promoter of the proto-oncogene c-MYC. Chem. Commun. 2008, 2007–2009. [Google Scholar]
- Simonsson, T.; Pecinka, P.; Kubista, M. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998, 26, 1167–1172. [Google Scholar] [CrossRef]
- Grotzer, M.A.; Castelletti, D.; Fiaschetti, G.; Shalaby, T.; Arcaro, A. Targeting Myc in pediatric malignancies of the central and peripheral nervous system. Curr. Cancer Drug Targets 2009, 9, 176–188. [Google Scholar] [CrossRef]
- Fekete, A.; Kenesi, E.; Hunyadi-Gulyas, E.; Durgo, H.; Berko, B.; Dunai, Z.A.; Bauer, P.I. The guanine-quadruplex structure in the human c-myc gene's promoter is converted into B-DNA form by the human poly (ADP-ribose) polymerase-1. PLoS One 2012, 7, e42690. [Google Scholar]
- Gonzalez, V.; Hurley, L.H. The c-MYC NHE III(1): Function and regulation. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 111–129. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef]
- Brooks, T.A.; Hurley, L.H. The role of supercoiling in transcriptional control of MYC and its importance in molecular therapeutics. Nat. Rev. Cancer 2009, 9, 849–861. [Google Scholar]
- Liu, J.N.; Deng, R.; Guo, J.F.; Zhou, J.M.; Feng, G.K.; Huang, Z.S.; Gu, L.Q.; Zeng, Y.X.; Zhu, X.F. Inhibition of myc promoter and telomerase activity and induction of delayed apoptosis by SYUIQ-5, a novel G-quadruplex interactive agent in leukemia cells. Leukemia 2007, 21, 1300–1302. [Google Scholar] [CrossRef]
- Ou, T.M.; Lu, Y.J.; Zhang, C.; Huang, Z.S.; Wang, X.D.; Tan, J.H.; Chen, Y.; Ma, D.L.; Wong, K.Y.; Tang, J.C.; Chan, A.S.; Gu, L.Q. Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J. Med. Chem. 2007, 50, 1465–1474. [Google Scholar] [CrossRef]
- Nicoludis, J.M.; Miller, S.T.; Jeffrey, P.D.; Barrett, S.P.; Rablen, P.R.; Lawton, T.J.; Yatsunyk, L.A. Optimized end-stacking provides specificity of N-methyl mesoporphyrin IX for human telomeric G-quadruplex DNA. J. Am. Chem. Soc. 2012, 134, 20446–20456. [Google Scholar] [CrossRef]
- Folini, M.; Venturini, L.; Cimino-Reale, G.; Zaffaroni, N. Telomeres as targets for anticancer therapies. Expert Opin. Ther. Targets 2011, 15, 579–593. [Google Scholar]
- Duchler, M. G-quadruplexes: Targets and tools in anticancer drug design. J. Drug Target. 2012, 20, 389–400. [Google Scholar] [CrossRef]
- De Cian, A.; Cristofari, G.; Reichenbach, P.; De Lemos, E.; Monchaud, D.; Teulade-Fichou, M.P.; Shin-Ya, K.; Lacroix, L.; Lingner, J.; Mergny, J.L. Reevaluation of telomerase inhibition by quadruplex ligands and their mechanisms of action. Proc. Natl. Acad. Sci. USA 2007, 104, 17347–17352. [Google Scholar] [CrossRef]
- Shin-ya, K.; Wierzba, K.; Matsuo, K.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef]
- Moorhouse, A.D.; Haider, S.; Gunaratnam, M.; Munnur, D.; Neidle, S.; Moses, J.E. Targeting telomerase and telomeres: A click chemistry approach towards highly selective G-quadruplex ligands. Mol. Biosyst. 2008, 4, 629–642. [Google Scholar] [CrossRef]
- Sumi, M.; Tauchi, T.; Sashida, G.; Nakajima, A.; Gotoh, A.; Shin-Ya, K.; Ohyashiki, J.H.; Ohyashiki, K. A G-quadruplex-interactive agent, telomestatin (SOT-095), induces telomere shortening with apoptosis and enhances chemosensitivity in acute myeloid leukemia. Int. J. Oncol. 2004, 24, 1481–1487. [Google Scholar]
- Nakajima, A.; Tauchi, T.; Sashida, G.; Sumi, M.; Abe, K.; Yamamoto, K.; Ohyashiki, J.H.; Ohyashiki, K. Telomerase inhibition enhances apoptosis in human acute leukemia cells: Possibility of antitelomerase therapy. Leukemia 2003, 17, 560–567. [Google Scholar] [CrossRef]
- Shammas, M.A.; Shmookler Reis, R.J.; Li, C.; Koley, H.; Hurley, L.H.; Anderson, K.C.; Munshi, N.C. Telomerase inhibition and cell growth arrest after telomestatin treatment in multiple myeloma. Clin. Cancer Res. 2004, 10, 770–776. [Google Scholar] [CrossRef]
- Miyazaki, T.; Pan, Y.; Joshi, K.; Purohit, D.; Hu, B.; Demir, H.; Mazumder, S.; Okabe, S.; Yamori, T.; Viapiano, M.; Shin-ya, K.; Seimiya, H.; Nakano, I. Telomestatin impairs glioma stem cell survival and growth through the disruption of telomeric G-quadruplex and inhibition of the proto-oncogene, c-Myb. Clin. Cancer Res. 2012, 18, 1268–1280. [Google Scholar] [CrossRef]
- Tauchi, T.; Shin-Ya, K.; Sashida, G.; Sumi, M.; Nakajima, A.; Shimamoto, T.; Ohyashiki, J.H.; Ohyashiki, K. Activity of a novel G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), against human leukemia cells: Involvement of ATM-dependent DNA damage response pathways. Oncogene 2003, 22, 5338–5347. [Google Scholar] [CrossRef]
- Kim, M.Y.; Vankayalapati, H.; Shin-Ya, K.; Wierzba, K.; Hurley, L.H. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular g-quadruplex. J. Am. Chem. Soc. 2002, 124, 2098–2099. [Google Scholar] [CrossRef]
- Tauchi, T.; Shin-ya, K.; Sashida, G.; Sumi, M.; Okabe, S.; Ohyashiki, J.H.; Ohyashiki, K. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: In vitro and in vivo studies in acute leukemia. Oncogene 2006, 25, 5719–5725. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Qi, H.; Lin, C.P.; Lin, R.K.; Kerrigan, J.E.; Rzuczek, S.G.; LaVoie, E.J.; Rice, J.E.; Pilch, D.S.; Lyu, Y.L.; Liu, L.F. A G-quadruplex stabilizer induces M-phase cell cycle arrest. J. Biol. Chem. 2009, 284, 22535–22543. [Google Scholar] [CrossRef]
- Fujimori, J.; Matsuo, T.; Shimose, S.; Kubo, T.; Ishikawa, M.; Yasunaga, Y.; Ochi, M. Antitumor effects of telomerase inhibitor TMPyP4 in osteosarcoma cell lines. J. Orthop. Res. 2011, 29, 1707–1711. [Google Scholar] [CrossRef]
- Kimura, T.; Kawai, K.; Fujitsuka, M.; Majima, T. Detection of the G-quadruplex-TMPyP4 complex by 2-aminopurine modified human telomeric DNA. Chem. Commun. 2006, 2006, 401–402. [Google Scholar]
- Mikami-Terao, Y.; Akiyama, M.; Yuza, Y.; Yanagisawa, T.; Yamada, O.; Kawano, T.; Agawa, M.; Ida, H.; Yamada, H. Antitumor activity of TMPyP4 interacting G-quadruplex in retinoblastoma cell lines. Exp. Eye Res. 2009, 89, 200–208. [Google Scholar] [CrossRef]
- Rha, S.Y.; Izbicka, E.; Lawrence, R.; Davidson, K.; Sun, D.; Moyer, M.P.; Roodman, G.D.; Hurley, L.; Von Hoff, D. Effect of telomere and telomerase interactive agents on human tumor and normal cell lines. Clin. Cancer Res. 2000, 6, 987–993. [Google Scholar]
- Shammas, M.A.; Shmookler Reis, R.J.; Akiyama, M.; Koley, H.; Chauhan, D.; Hideshima, T.; Goyal, R.K.; Hurley, L.H.; Anderson, K.C.; Munshi, N.C. Telomerase inhibition and cell growth arrest by G-quadruplex interactive agent in multiple myeloma. Mol. Cancer Ther. 2003, 2, 825–833. [Google Scholar]
- Zhou, J.M.; Zhu, X.F.; Lu, Y.J.; Deng, R.; Huang, Z.S.; Mei, Y.P.; Wang, Y.; Huang, W.L.; Liu, Z.C.; Gu, L.Q.; Zeng, Y.X. Senescence and telomere shortening induced by novel potent G-quadruplex interactive agents, quindoline derivatives, in human cancer cell lines. Oncogene 2006, 25, 503–511. [Google Scholar]
- Zhou, W.J.; Deng, R.; Zhang, X.Y.; Feng, G.K.; Gu, L.Q.; Zhu, X.F. G-quadruplex ligand SYUIQ-5 induces autophagy by telomere damage and TRF2 delocalization in cancer cells. Mol. Cancer Ther. 2009, 8, 3203–3213. [Google Scholar] [CrossRef]
- Hampel, S.M.; Sidibe, A.; Gunaratnam, M.; Riou, J.F.; Neidle, S. Tetrasubstituted naphthalene diimide ligands with selectivity for telomeric G-quadruplexes and cancer cells. Bioorg. Med. Chem. Lett. 2010, 20, 6459–6463. [Google Scholar] [CrossRef]
- Gomez, D.; Aouali, N.; Renaud, A.; Douarre, C.; Shin-Ya, K.; Tazi, J.; Martinez, S.; Trentesaux, C.; Morjani, H.; Riou, J.F. Resistance to senescence induction and telomere shortening by a G-quadruplex ligand inhibitor of telomerase. Cancer Res. 2003, 63, 6149–6153. [Google Scholar]
- Gomez, D.; Lemarteleur, T.; Lacroix, L.; Mailliet, P.; Mergny, J.L.; Riou, J.F. Telomerase downregulation induced by the G-quadruplex ligand 12459 in A549 cells is mediated by hTERT RNA alternative splicing. Nucleic Acids Res. 2004, 32, 371–379. [Google Scholar] [CrossRef]
- Kelland, L. Targeting the limitless replicative potential of cancer: The telomerase/telomere pathway. Clin. Cancer Res. 2007, 13, 4960–4963. [Google Scholar] [CrossRef]
- Kelland, L.R. Overcoming the immortality of tumour cells by telomere and telomerase based cancer therapeutics--current status and future prospects. Eur. J. Cancer 2005, 41, 971–979. [Google Scholar] [CrossRef]
- Riou, J.F.; Guittat, L.; Mailliet, P.; Laoui, A.; Renou, E.; Petitgenet, O.; Megnin-Chanet, F.; Helene, C.; Mergny, J.L. Cell senescence and telomere shortening induced by a new series of specific G-quadruplex DNA ligands. Proc. Natl. Acad. Sci. USA 2002, 99, 2672–2677. [Google Scholar] [CrossRef]
- Gunaratnam, M.; Green, C.; Moreira, J.B.; Moorhouse, A.D.; Kelland, L.R.; Moses, J.E.; Neidle, S. G-quadruplex compounds and cis-platin act synergistically to inhibit cancer cell growth in vitro and in vivo. Biochem. Pharmacol. 2009, 78, 115–122. [Google Scholar] [CrossRef]
- Collie, G.; Reszka, A.P.; Haider, S.M.; Gabelica, V.; Parkinson, G.N.; Neidle, S. Selectivity in small molecule binding to human telomeric RNA and DNA quadruplexes. Chem. Commun. 2009, 2009, 7482–7484. [Google Scholar]
- Gowan, S.M.; Harrison, J.R.; Patterson, L.; Valenti, M.; Read, M.A.; Neidle, S.; Kelland, L.R. A G-quadruplex-interactive potent small-molecule inhibitor of telomerase exhibiting in vitro and in vivo antitumor activity. Mol. Pharmacol. 2002, 61, 1154–1162. [Google Scholar] [CrossRef]
- Incles, C.M.; Schultes, C.M.; Kempski, H.; Koehler, H.; Kelland, L.R.; Neidle, S. A G-quadruplex telomere targeting agent produces p16-associated senescence and chromosomal fusions in human prostate cancer cells. Mol. Cancer Ther. 2004, 3, 1201–1206. [Google Scholar]
- Phatak, P.; Cookson, J.C.; Dai, F.; Smith, V.; Gartenhaus, R.B.; Stevens, M.F.; Burger, A.M. Telomere uncapping by the G-quadruplex ligand RHPS4 inhibits clonogenic tumour cell growth in vitro and in vivo consistent with a cancer stem cell targeting mechanism. Br. J. Cancer 2007, 96, 1223–1233. [Google Scholar] [CrossRef]
- Read, M.; Harrison, R.J.; Romagnoli, B.; Tanious, F.A.; Gowan, S.H.; Reszka, A.P.; Wilson, W.D.; Kelland, L.R.; Neidle, S. Structure-based design of selective and potent G quadruplex-mediated telomerase inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 4844–4849. [Google Scholar] [CrossRef]
- Cookson, J.C.; Dai, F.; Smith, V.; Heald, R.A.; Laughton, C.A.; Stevens, M.F.; Burger, A.M. Pharmacodynamics of the G-quadruplex-stabilizing telomerase inhibitor 3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate (RHPS4) in vitro: Activity in human tumor cells correlates with telomere length and can be enhanced, or antagonized, with cytotoxic agents. Mol. Pharmacol. 2005, 68, 1551–1558. [Google Scholar]
- Gavathiotis, E.; Heald, R.A.; Stevens, M.F.; Searle, M.S. Drug recognition and stabilisation of the parallel-stranded DNA quadruplex d(TTAGGGT)4 containing the human telomeric repeat. J. Mol. Biol. 2003, 334, 25–36. [Google Scholar] [CrossRef]
- Leonetti, C.; Amodei, S.; D'Angelo, C.; Rizzo, A.; Benassi, B.; Antonelli, A.; Elli, R.; Stevens, M.F.; D'Incalci, M.; Zupi, G.; Biroccio, A. Biological activity of the G-quadruplex ligand RHPS4 (3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate) is associated with telomere capping alteration. Mol. Pharmacol. 2004, 66, 1138–1146. [Google Scholar] [CrossRef]
- Leonetti, C.; Scarsella, M.; Riggio, G.; Rizzo, A.; Salvati, E.; D'Incalci, M.; Staszewsky, L.; Frapolli, R.; Stevens, M.F.; Stoppacciaro, A.; Mottolese, M.; Antoniani, B.; Gilson, E.; Zupi, G.; Biroccio, A. G-quadruplex ligand RHPS4 potentiates the antitumor activity of camptothecins in preclinical models of solid tumors. Clin. Cancer Res. 2008, 14, 7284–7291. [Google Scholar] [CrossRef]
- Rizzo, A.; Salvati, E.; Porru, M.; D'Angelo, C.; Stevens, M.F.; D'Incalci, M.; Leonetti, C.; Gilson, E.; Zupi, G.; Biroccio, A. Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res. 2009, 37, 5353–5364. [Google Scholar] [CrossRef]
- Salvati, E.; Scarsella, M.; Porru, M.; Rizzo, A.; Iachettini, S.; Tentori, L.; Graziani, G.; D'Incalci, M.; Stevens, M.F.; Orlandi, A.; Passeri, D.; Gilson, E.; Zupi, G.; Leonetti, C.; Biroccio, A. PARP1 is activated at telomeres upon G4 stabilization: Possible target for telomere-based therapy. Oncogene 2010, 29, 6280–6293. [Google Scholar] [CrossRef]
- Cuenca, F.; Moore, M.J.; Johnson, K.; Guyen, B.; De Cian, A.; Neidle, S. Design, synthesis and evaluation of 4,5-di-substituted acridone ligands with high G-quadruplex affinity and selectivity, together with low toxicity to normal cells. Bioorg. Med. Chem. Lett. 2009, 19, 5109–5113. [Google Scholar] [CrossRef]
- Folini, M.; Pivetta, C.; Zagotto, G.; De Marco, C.; Palumbo, M.; Zaffaroni, N.; Sissi, C. Remarkable interference with telomeric function by a G-quadruplex selective bisantrene regioisomer. Biochem. Pharmacol. 2010, 79, 1781–1790. [Google Scholar] [CrossRef]
- Huang, H.S.; Huang, K.F.; Li, C.L.; Huang, Y.Y.; Chiang, Y.H.; Huang, F.C.; Lin, J.J. Synthesis, human telomerase inhibition and anti-proliferative studies of a series of 2,7-bis-substituted amido-anthraquinone derivatives. Bioorg. Med. Chem. 2008, 16, 6976–6986. [Google Scholar] [CrossRef]
- Franceschin, M.; Rizzo, A.; Casagrande, V.; Salvati, E.; Alvino, A.; Altieri, A.; Ciammaichella, A.; Iachettini, S.; Leonetti, C.; Ortaggi, G.; Porru, M.; Bianco, A.; Biroccio, A. Aromatic core extension in the series of N-cyclic bay-substituted perylene G-quadruplex ligands: Increased telomere damage, antitumor activity, and strong selectivity for neoplastic over healthy cells. ChemMedChem 2012, 7, 2144–2154. [Google Scholar] [CrossRef]
- Micheli, E.; D'Ambrosio, D.; Franceschin, M.; Savino, M. Water soluble cationic perylene derivatives as possible telomerase inhibitors: The search for selective G-quadruplex targeting. Mini Rev. Med. Chem. 2009, 9, 1622–1632. [Google Scholar] [CrossRef]
- Rzuczek, S.G.; Pilch, D.S.; Liu, A.; Liu, L.; LaVoie, E.J.; Rice, J.E. Macrocyclic pyridyl polyoxazoles: Selective RNA and DNA G-quadruplex ligands as antitumor agents. J. Med. Chem. 2010, 53, 3632–3644. [Google Scholar] [CrossRef]
- Satyanarayana, M.; Kim, Y.A.; Rzuczek, S.G.; Pilch, D.S.; Liu, A.A.; Liu, L.F.; Rice, J.E.; LaVoie, E.J. Macrocyclic hexaoxazoles: Influence of aminoalkyl substituents on RNA and DNA G-quadruplex stabilization and cytotoxicity. Bioorg. Med. Chem. Lett. 2010, 20, 3150–154. [Google Scholar] [CrossRef]
- Liu, J.; Guo, L.; Yin, F.; Zheng, X.; Chen, G.; Wang, Y. Characterization and antitumor activity of triethylene tetramine, a novel telomerase inhibitor. Biomed. Pharmacother. 2008, 62, 480–485. [Google Scholar] [CrossRef]
- Lixia, G.; Fei, Y.; Jiajia, J.; Jianhui, L. Triethylene tetramine, a novel ligand of G-quadruplex, induces senescence of MCF-7 cells. Biotechnol. Lett. 2008, 30, 47–53. [Google Scholar]
- De Cian, A.; Mergny, J.L. Quadruplex ligands may act as molecular chaperones for tetramolecular quadruplex formation. Nucleic Acids Res. 2007, 35, 2483–2493. [Google Scholar] [CrossRef]
- Tang, K.T.; Ho, L.T.; Jap, T.S.; Chang, T.C.; Ching, K.N.; Lam, H.C.; Wu, M.S.; Lee, S.P. Comparison of insulin secretion and insulin sensitivity between normal and impaired glucose tolerance subjects with normal fasting plasma glucose. Zhonghua Yi Xue Za Zhi. 1990, 46, 73–78. [Google Scholar]
- Pennarun, G.; Granotier, C.; Gauthier, L.R.; Gomez, D.; Hoffschir, F.; Mandine, E.; Riou, J.F.; Mergny, J.L.; Mailliet, P.; Boussin, F.D. Apoptosis related to telomere instability and cell cycle alterations in human glioma cells treated by new highly selective G-quadruplex ligands. Oncogene 2005, 24, 2917–2928. [Google Scholar] [CrossRef]
- Guliaev, A.B.; Leontis, N.B. Cationic 5,10,15,20-tetrakis(N-methylpyridinium-4-yl)porphyrin fully intercalates at 5′-CG-3′ steps of duplex DNA in solution. Biochemistry 1999, 38, 15425–15437. [Google Scholar] [CrossRef]
- Lee, Y.A.; Kim, J.O.; Cho, T.S.; Song, R.; Kim, S.K. Binding of meso-tetrakis(N-methylpyridium-4-yl)porphyrin to triplex oligonucleotides: Evidence for the porphyrin stacking in the major groove. J. Am. Chem. Soc. 2003, 125, 8106–8107. [Google Scholar] [CrossRef]
- Dixon, I.M.; Lopez, F.; Tejera, A.M.; Esteve, J.P.; Blasco, M.A.; Pratviel, G.; Meunier, B. A G-quadruplex ligand with 10000-fold selectivity over duplex DNA. J. Am. Chem. Soc. 2007, 129, 1502–1503. [Google Scholar]
- Duan, W.; Rangan, A.; Vankayalapati, H.; Kim, M.Y.; Zeng, Q.; Sun, D.; Han, H.; Fedoroff, O.Y.; Nishioka, D.; Rha, S.Y.; Izbicka, E.; Von Hoff, D.D.; Hurley, L.H. Design and synthesis of fluoroquinophenoxazines that interact with human telomeric G-quadruplexes and their biological effects. Mol. Cancer Ther. 2001, 1, 103–120. [Google Scholar]
- Kim, M.Y.; Duan, W.; Gleason-Guzman, M.; Hurley, L.H. Design, synthesis, and biological evaluation of a series of fluoroquinoanthroxazines with contrasting dual mechanisms of action against topoisomerase II and G-quadruplexes. J. Med. Chem. 2003, 46, 571–583. [Google Scholar] [CrossRef]
- Drygin, D.; Siddiqui-Jain, A.; O'Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; Lim, J.K.; Von Hoff, D.; Anderes, K.; Rice, W.G. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef]
- Fernando, H.; Rodriguez, R.; Balasubramanian, S. Selective recognition of a DNA G-quadruplex by an engineered antibody. Biochemistry 2008, 47, 9365–9371. [Google Scholar] [CrossRef]
- Ou, T.M.; Lu, Y.J.; Tan, J.H.; Huang, Z.S.; Wong, K.Y.; Gu, L.Q. G-quadruplexes: Targets in anticancer drug design. Chem.Med.Chem. 2008, 3, 690–713. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shalaby, T.; Fiaschetti, G.; Nagasawa, K.; Shin-ya, K.; Baumgartner, M.; Grotzer, M. G-Quadruplexes as Potential Therapeutic Targets for Embryonal Tumors. Molecules 2013, 18, 12500-12537. https://doi.org/10.3390/molecules181012500
Shalaby T, Fiaschetti G, Nagasawa K, Shin-ya K, Baumgartner M, Grotzer M. G-Quadruplexes as Potential Therapeutic Targets for Embryonal Tumors. Molecules. 2013; 18(10):12500-12537. https://doi.org/10.3390/molecules181012500
Chicago/Turabian StyleShalaby, Tarek, Giulio Fiaschetti, Kazuo Nagasawa, Kazuo Shin-ya, Martin Baumgartner, and Michael Grotzer. 2013. "G-Quadruplexes as Potential Therapeutic Targets for Embryonal Tumors" Molecules 18, no. 10: 12500-12537. https://doi.org/10.3390/molecules181012500
APA StyleShalaby, T., Fiaschetti, G., Nagasawa, K., Shin-ya, K., Baumgartner, M., & Grotzer, M. (2013). G-Quadruplexes as Potential Therapeutic Targets for Embryonal Tumors. Molecules, 18(10), 12500-12537. https://doi.org/10.3390/molecules181012500