Two New Phragmalin-Type Limonoids from Chukrasia tabularis var. velutina

Abstract
:1. Introduction
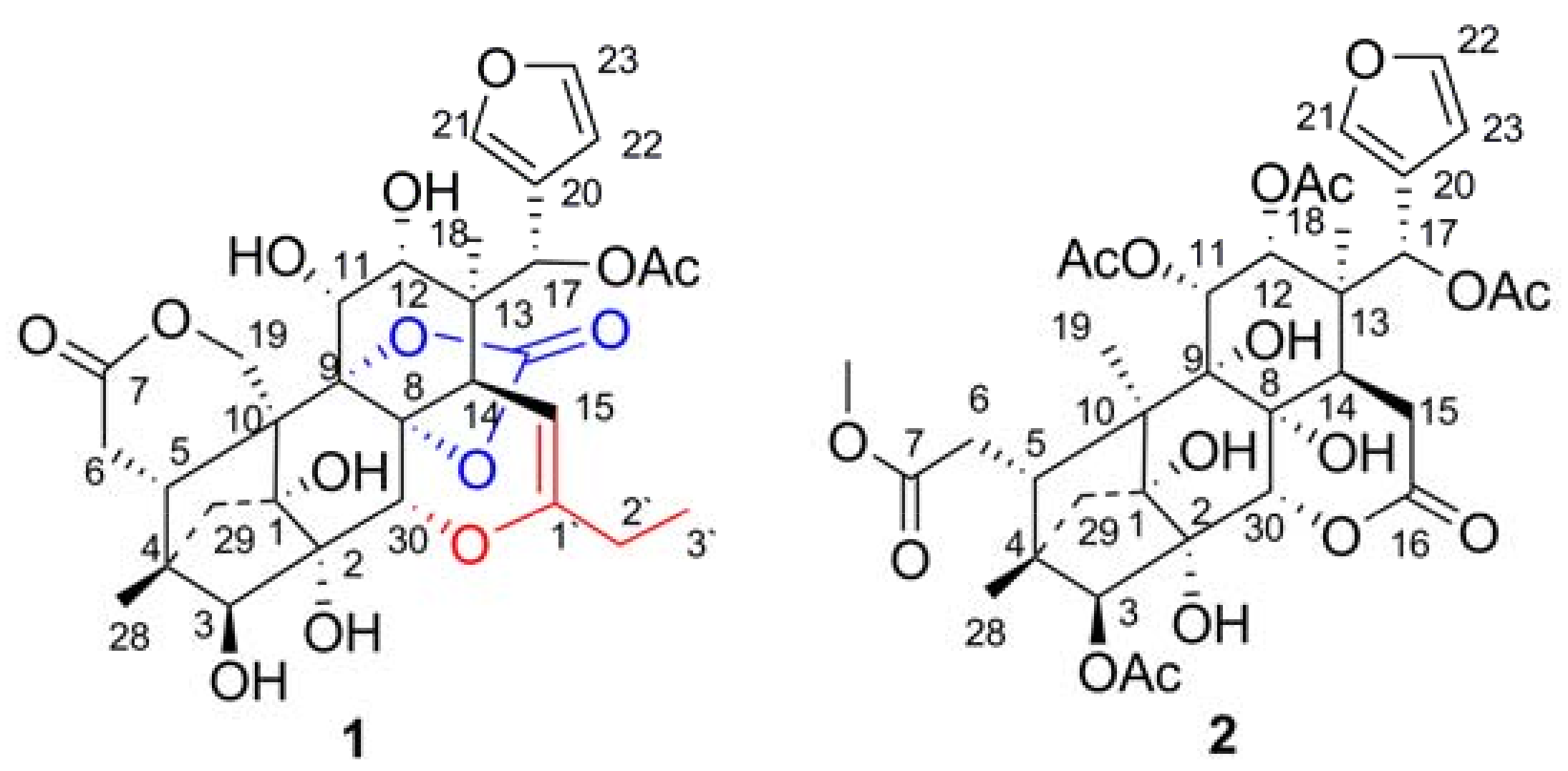
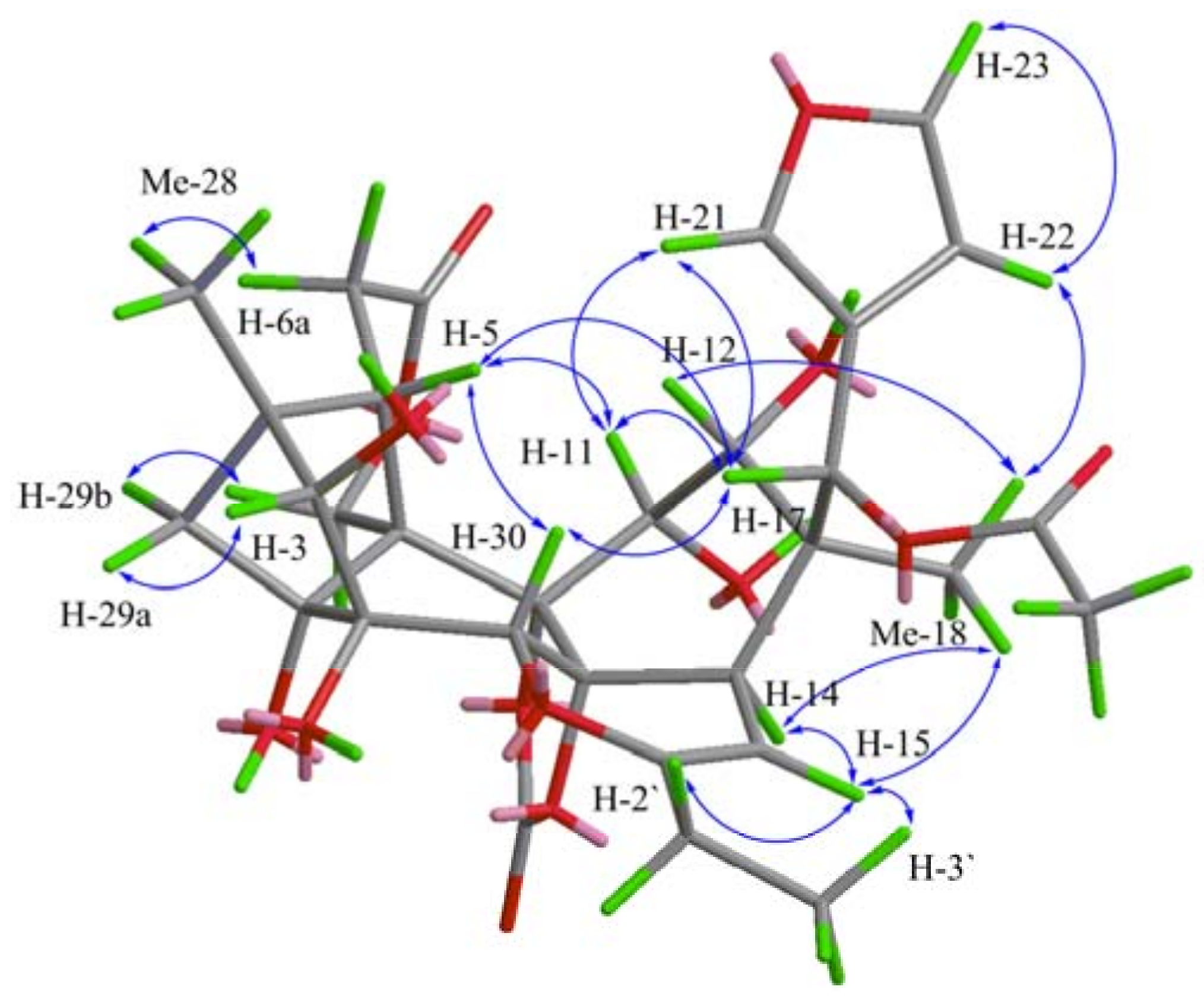
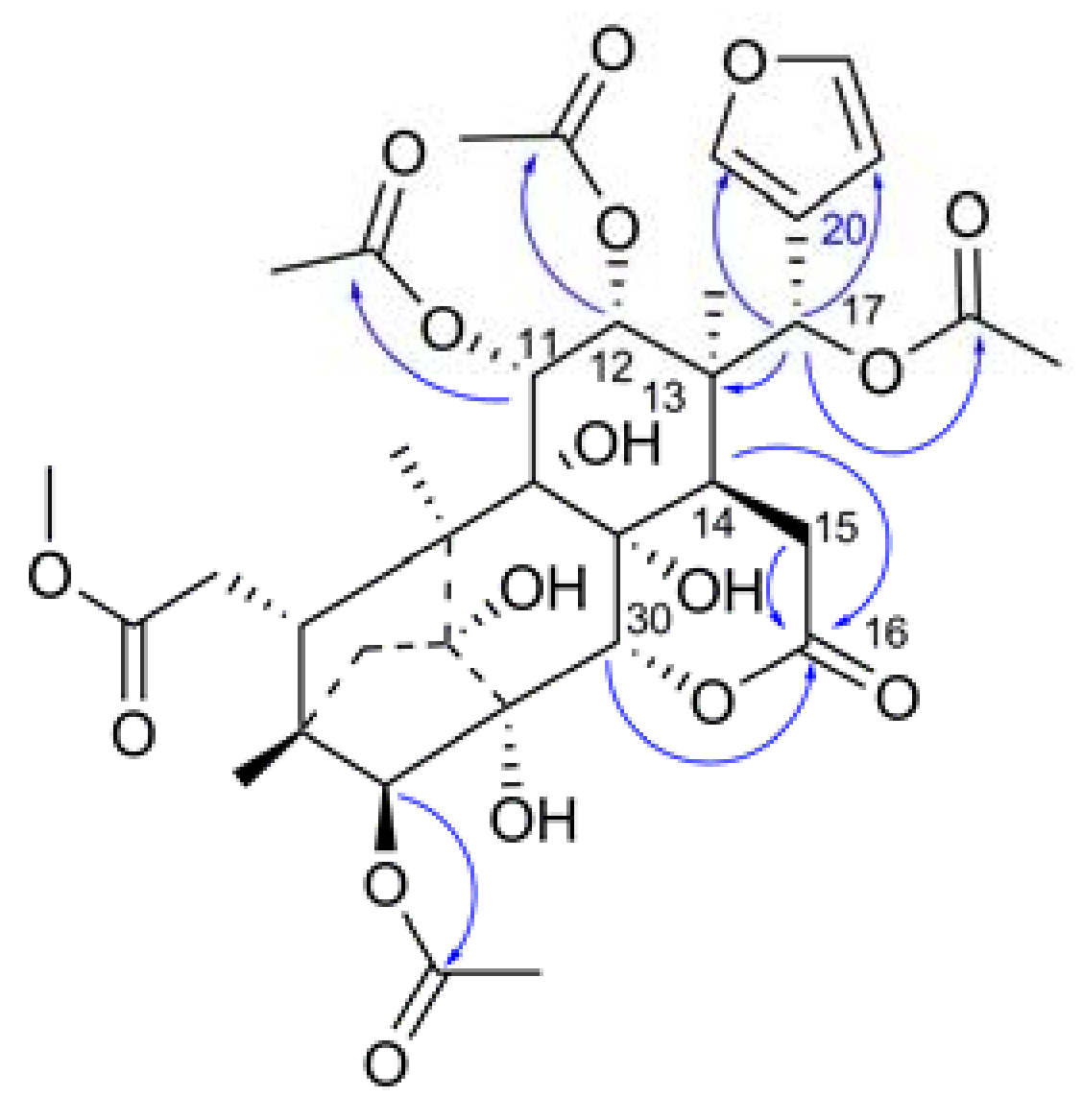
2. Results and Discussion
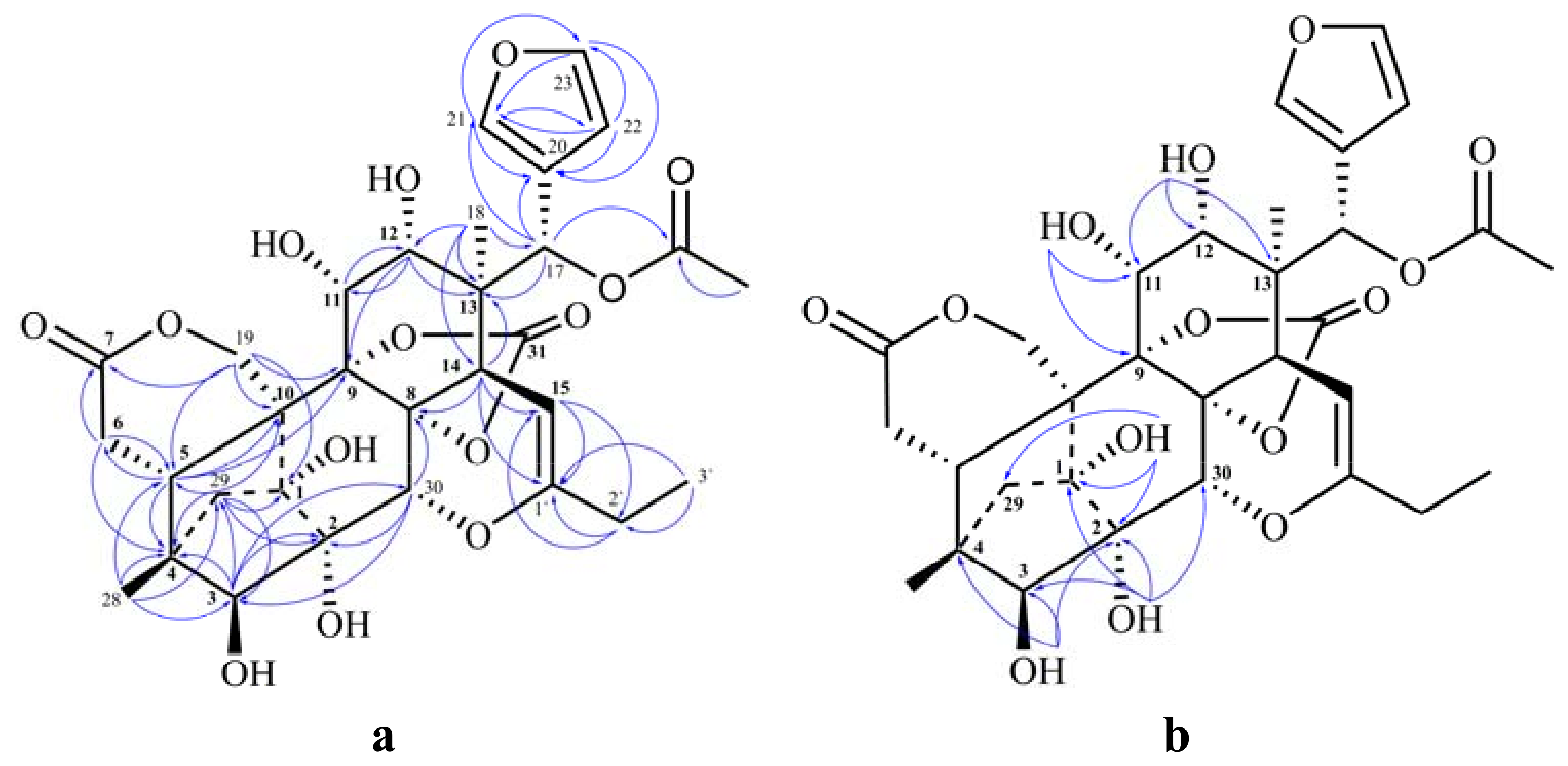

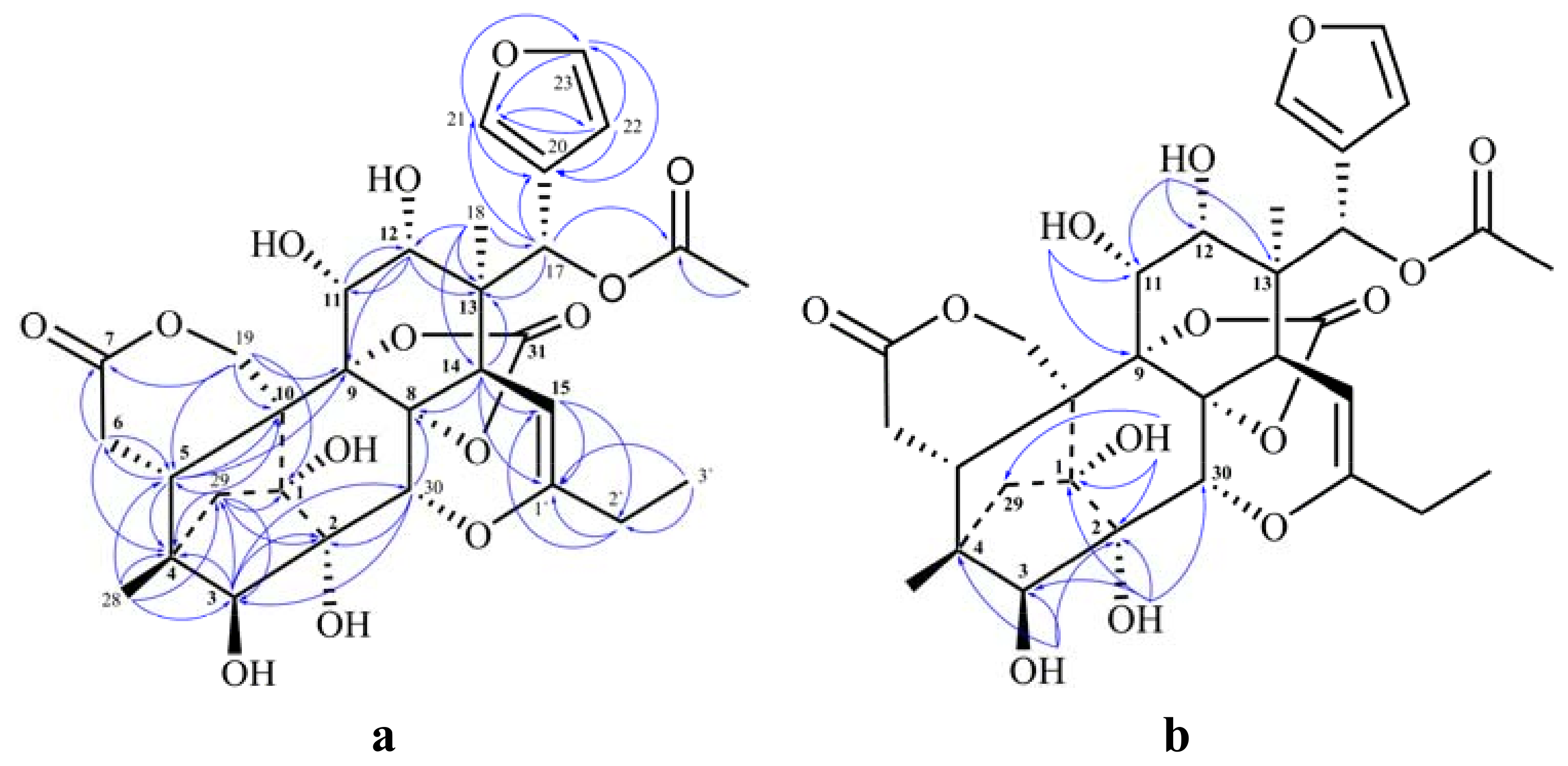
{kind=link}
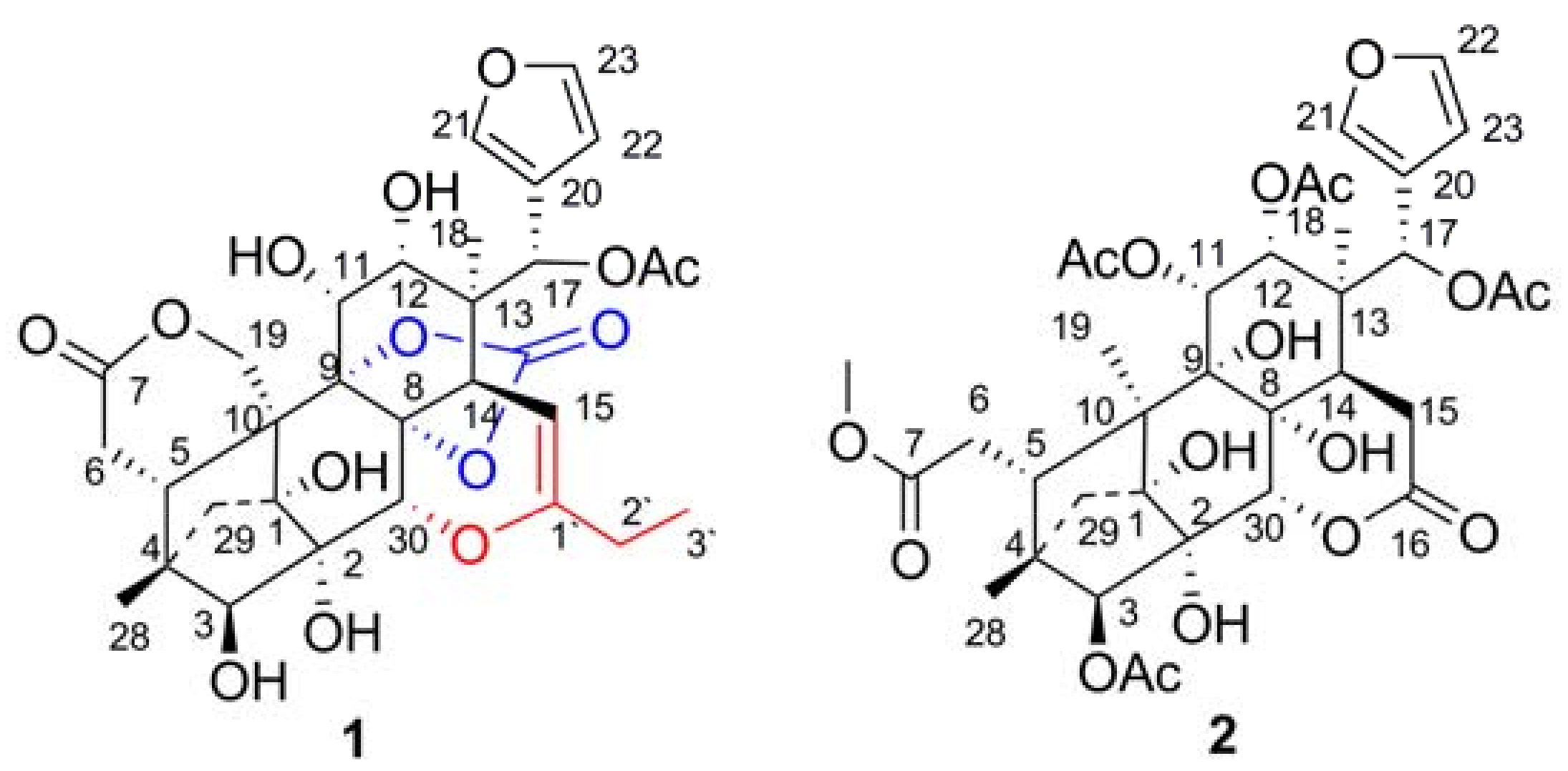
{kind=link}
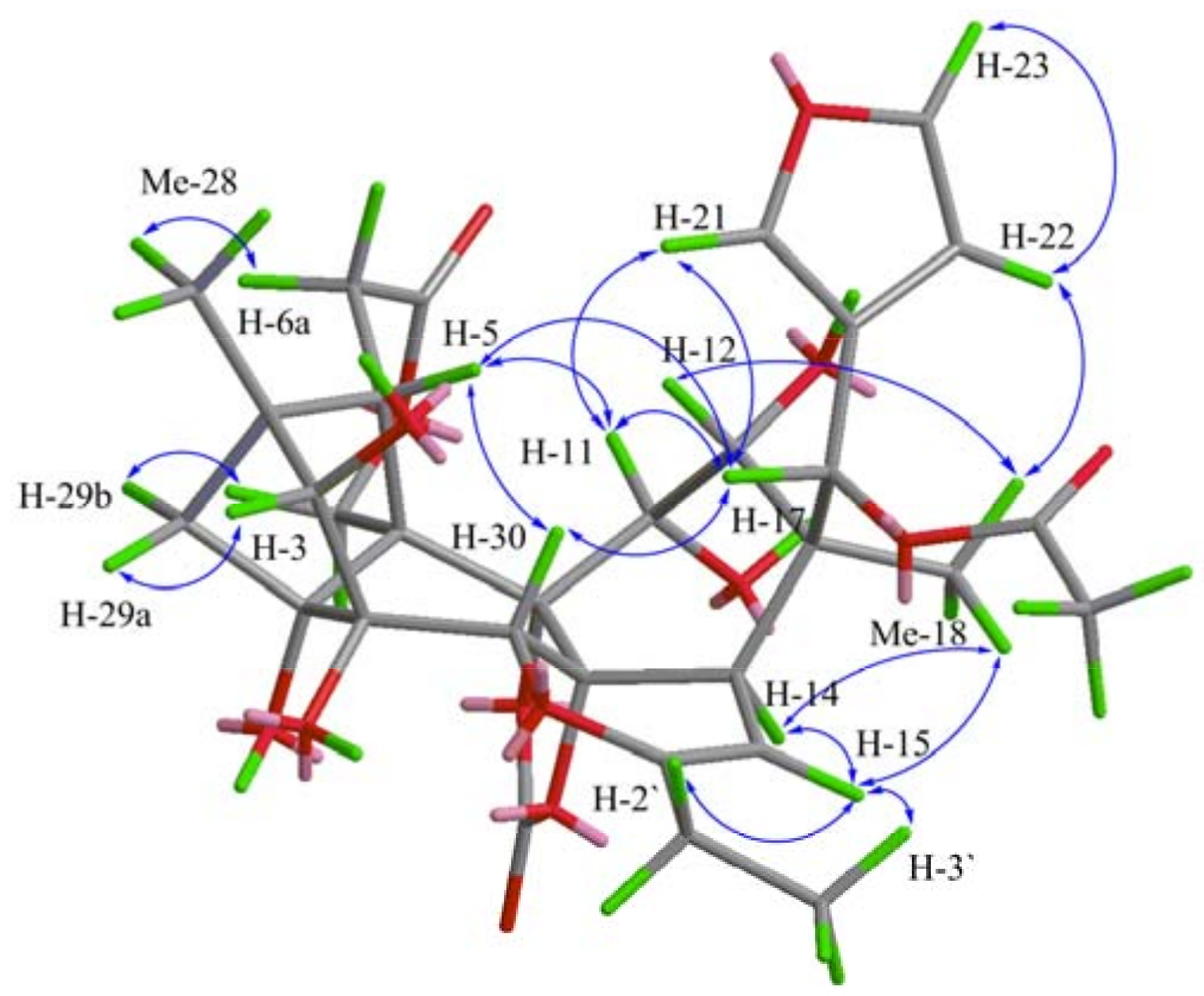{kind=link}
{kind=link}
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δH (multi, J in Hz) | δC | δH (multi, J in Hz) | δC | |
| 1 | 82.3 | 82.9 | ||
| 2 | 74.0 | 74.8 | ||
| 3 | 3.35 (d 5.5) | 85.7 | 4.73 (s) | 86.1 |
| 4 | 43.1 | 43.9 | ||
| 5 | 2.44 (m ) | 36.7 | 2.60 * | 39.2 |
| 6a | 2.38 (dd 16.0, 3.3) | 29.6 | 1.94 * 2.47(dd 18.0, 11.0) | 31.9 |
| 6b | 2.94 (dd 16.0, 6.0) | |||
| 7 | 172.8 | 172.4 | ||
| 8 | 83.2 | 71.7 | ||
| 9 | 86.6 | 76.4 | ||
| 10 | 48.1 | 51.8 | ||
| 11 | 3.88 (dd 3.0, 8.6) | 67.3 | 5.11 (d 3.0) | 71.2 |
| 12 | 3.17 (dd 3.0, 5.0) | 73.4 | 4.96 (d 3.0) | 70.8 |
| 13 | 43.9 | 42.3 | ||
| 14 | 2.52 (d 4.5 ) | 43.4 | 2.60 * | 40.3 |
| 15a | 4.67 (d 4.5) | 92.4 | 2.76 (d 18.5) | 27.8 |
| 15b | 2.88(dd 18.5, 9.0) | |||
| 16 | 168.9 | |||
| 17 | 5.79 (s) | 67.5 | 6.00(s) | 70.2 |
| 18 | 1.29 (s, 3H) | 17.1 | 1.04 (s 3H) | 18.4 |
| 19a | 4.57 (d 12.5) | 67.9 | 1.19 (s 2H) | 15.3 |
| 19b | 5.27(d 12.5) | |||
| 20 | 121.5 | 121.4 | ||
| 21 | 7.59 (s) | 141.1 | 7.75 (s) | 141.7 |
| 22 | 6.42 (s) | 109.9 | 6.57 (t 1.0) | 109.7 |
| 23 | 7.68 (s) | 143.7 | 7.65 (t 1.5) | 143.2 |
| 28 | 0.88 (s 3H) | 14.9 | 0.70 (s 3H) | 14.6 |
| 29a | 1.46 (d 11.0) | 41.4 | 1.51 (d 11.0) | 40.8 |
| 29b | 1.92 (d 11.0) | 1.98(d 11.0) | ||
| 30 | 4.57 (s) | 65.7 | 4.98 (s) | 74.4 |
| 31 | 152.0 | |||
| 1' | 152.8 | |||
| 2' | 2.10 (q 7.5) | 26.0 | ||
| 3' | 1.04 (t, 7.5) | 10.9 | ||
| 7-OMe | 3.62 (s 3H) | 51.4 | ||
| 1-OH | 5.35 (s) | 6.46 (s) | ||
| 2-OH | 4.23 (s) | 5.06 (s) | ||
| 3-OH | 5.92 (d5.5) | |||
| 8-OH | 6.64 (s) | |||
| 9-OH | 4.42 (s) | |||
| 11-OH | 5.67 (d 8.6) | |||
| 12-OH | 5.14 (d 5.0) | |||
| 3-OAc | 170.0 | |||
| 2.22 (s 3H) | 20.4 | |||
| 11-OAc | 170.1 | |||
| 2.03 (s 3H) | 20.5 | |||
| 12-OAc | 168.9 | |||
| 1.91 (s 3H) | 20.4 | |||
| 17-OAc | 167.9 | 168.6 | ||
| 1.91 (s 3H) | 20.7 | 1.97 (s 3H) | 20.8 | |
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
4. Conclusions
Acknowledgments
References
- Editorial Committee of the Administration Bureau of Traditional Chinese Medicine. Chinese Materia Medica (Zhonghua Bencao), 1st ed.; Shanghai Science and Technology Press: Shanghai, China, 1999; Volume 5, p. 31. [Google Scholar]
- Fan, C.Q.; Wang, X.N.; Yin, S.; Zhang, C.R.; Wang, F.D.; Yue, J.M. Tabularisins A–D, phragmalin ortho esters with new skeleton isolated from the seeds of Chukrasia tabularis. Tetrahedron 2007, 63, 6741–6747. [Google Scholar] [CrossRef]
- Zhang, C.R.; Yang, S.P.; Liao, S.G.; Fan, C.Q.; Wu, Y.; Yue, J.M. Chuktabularins A–D, four new limonoids with unprecedented carbon skeletons from the stem bark of Chukrasia tabularis. Org. Lett. 2007, 9, 3383–3386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.R.; Fan, C.Q.; Zhan, L.G.; Yang, S.P.; Wu, Y.; Lu, Y.; Yue, J.M. Chuktabrins A and B, two novel limonoids from the twigs and leaves of Chukrasia tabularis. Org. Lett. 2008, 10, 3183–3186. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, J.S.; Luo, J.G.; Wang, X.B.; Kong, L.Y. Chukvelutins A–C, 16-Norphragmalin Limonoids with Unprecedented Skeletons from Chukrasia tabularis var. velutina. Org. Lett. 2009, 11, 2281–2284. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Zhang, H.; Li, P.; Gao, Z.B.; Yue, J.M. Chukrasones A and B: Potential Kv1.2 Potassium Channel Blockers with New Skeletons from Chukrasia tabularis. Org. Lett. 2012, 14, 4438–4441. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, J.S.; Wang, X.B.; Luo, J.G.; Kong, L.Y. Chuktabularins E–T, 16-Norphragmalin Limonoids from Chukrasia tabularis var. velutina. J. Nat. Prod. 2010, 73, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, Y.; Wang, J.S.; Wang, X.B.; Luo, J.G.; Kong, L.Y. Novel and diverse 16-norphragmalin-type limonoids from Chukrasia tabularis var. velutina. Chem. Pharm. Bull. 2012, 60, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, Y.; Wang, J.S.; Lu, J.; Kong, L.Y. Three new C-15-isobutyryl 16-norphragmalin-type limonoids from Chukrasia tabularis var. velutina. Phytochem. Lett. 2012, 5, 249–252. [Google Scholar] [CrossRef]
- Luo, J.; Wang, J.S.; Wang, X.B.; Huang, X.F.; Luo, J.G.; Kong, L.Y. Chukvelutilides A–F, phragmalin limonoids from the stem barks of Chukrasia tabularis var. velutina. Tetrahedron 2009, 65, 3425–3431. [Google Scholar] [CrossRef]
- Luo, J.; Wang, J.S.; Wang, X.B.; Luo, J.G.; Kong, L.Y. Velutabularins A–J, phragmalin-type limonoids with novel cyclic moiety from Chukrasia tabularis var. velutina. Tetrahedron 2011, 67, 2942–2948. [Google Scholar] [CrossRef]
- Luo, J.; Wang, J.S.; Wang, X.B.; Luo, J.G.; Kong, L.Y. Phragmalin-type limonoid orthoesters from Chukrasia tabularis var. velutina. Chem. Pharm. Bull. 2011, 59, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, Y.; Wang, J.S.; Kong, L.Y. D-Ring opened phragmalin-type limonoids from Chukrasia tabularis var. velutina. Chem. Biodivers. 2011, 8, 2261–2269. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, J.; Wang, J.S.; Kong, L.Y. Two new limonoids from the stem barks of Chukrasia tabularis var. velutina. J. Asian Nat. Prod. Res. 2011, 13, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, J.S.; Cao, W.J.; Kong, L.Y. A new phragmalin-type limonoid from Chukrasia tabularis var. velutina. Chin. J. Nat. Med. 2011, 9, 98–101. [Google Scholar]
- Luo, J.; Wang, J.S.; Wang, L.B.; Kong, L.Y. Two new phragmalin-type limonoid orthesters from Chukrasia tabularis var. velutina. Acta Pharm. Sin. 2011, 46, 183–187. [Google Scholar]
Sample Availability: Samples of the compounds 1 and 2 are available from the authors. |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, Y.; Luo, J.; Li, H.; Kong, L.-Y. Two New Phragmalin-Type Limonoids from Chukrasia tabularis var. velutina. Molecules 2013, 18, 373-380. https://doi.org/10.3390/molecules18010373
Li Y, Luo J, Li H, Kong L-Y. Two New Phragmalin-Type Limonoids from Chukrasia tabularis var. velutina. Molecules. 2013; 18(1):373-380. https://doi.org/10.3390/molecules18010373
Chicago/Turabian StyleLi, Yi, Jun Luo, Hui Li, and Ling-Yi Kong. 2013. "Two New Phragmalin-Type Limonoids from Chukrasia tabularis var. velutina" Molecules 18, no. 1: 373-380. https://doi.org/10.3390/molecules18010373
APA StyleLi, Y., Luo, J., Li, H., & Kong, L.-Y. (2013). Two New Phragmalin-Type Limonoids from Chukrasia tabularis var. velutina. Molecules, 18(1), 373-380. https://doi.org/10.3390/molecules18010373