Systematic Study of the Physicochemical Properties of a Homologous Series of Aminobisphosphonates

Abstract
:1. Introduction
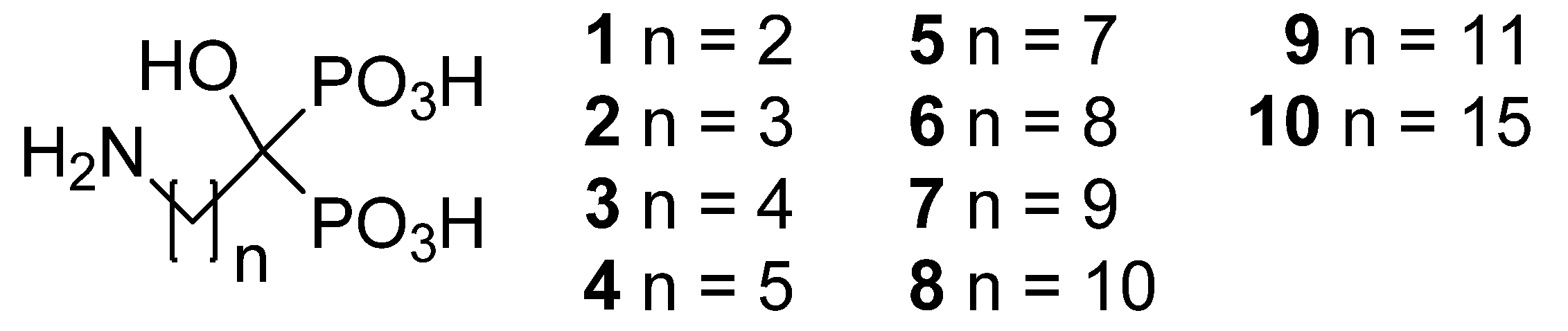
2. Results and Discussion
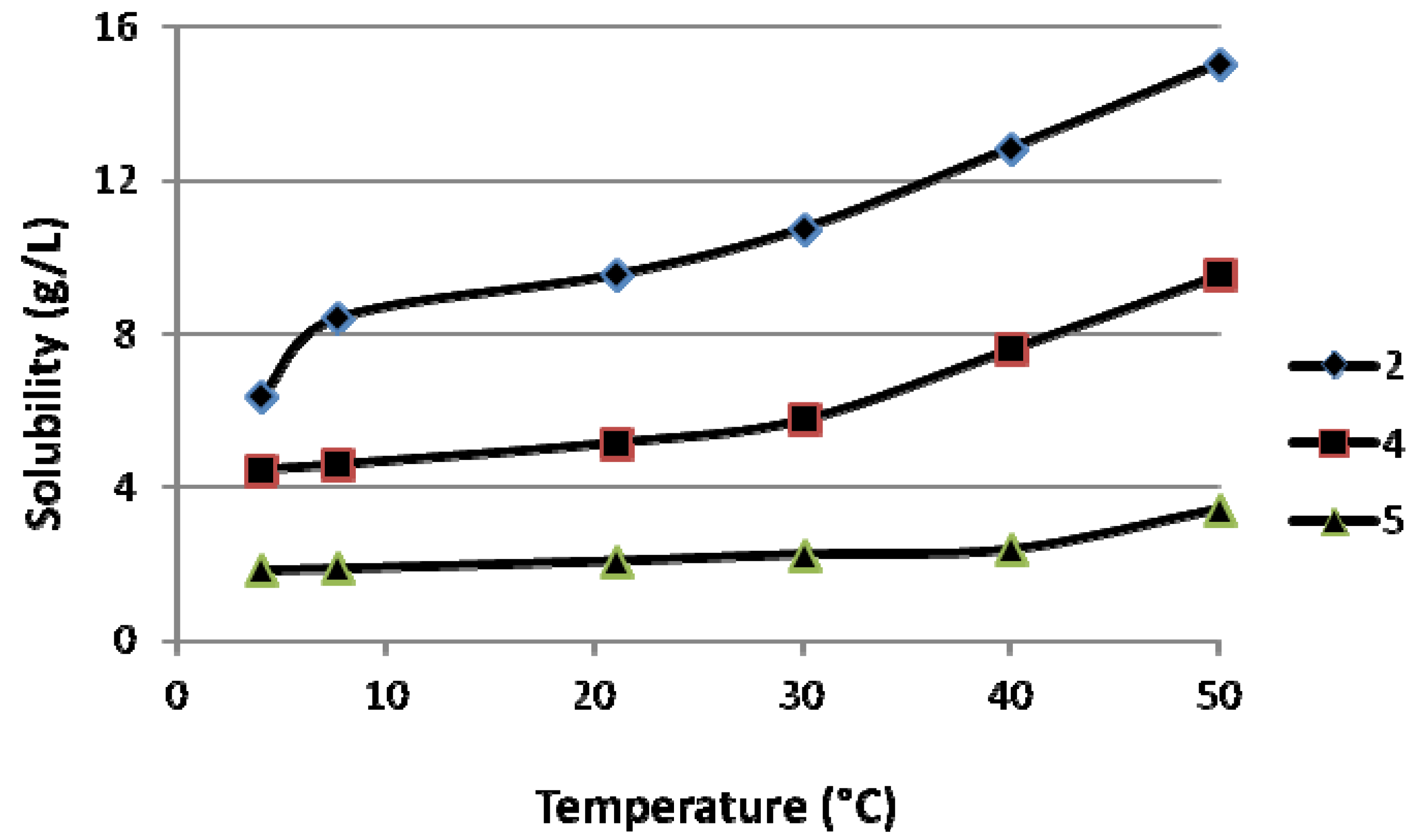
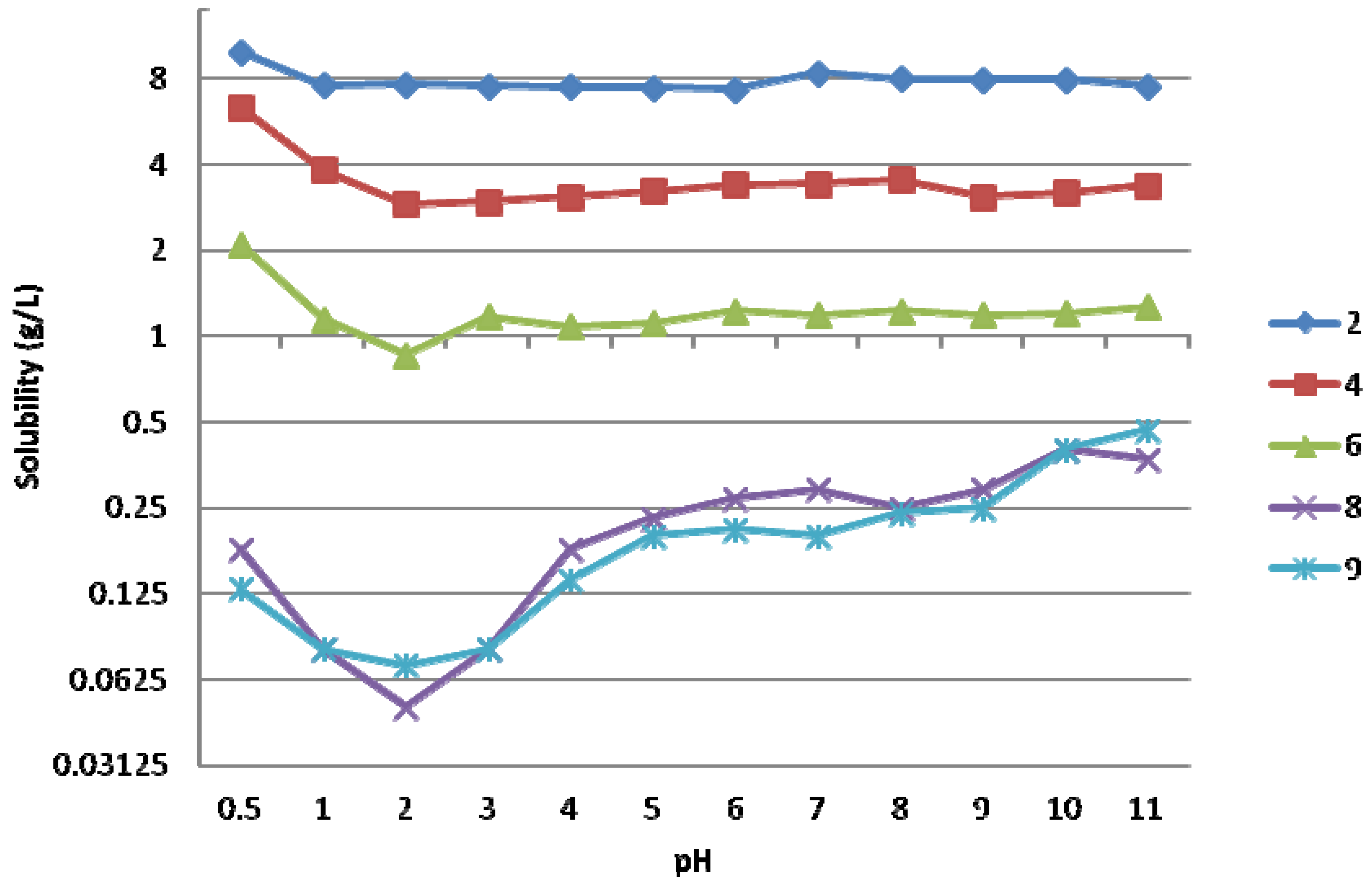
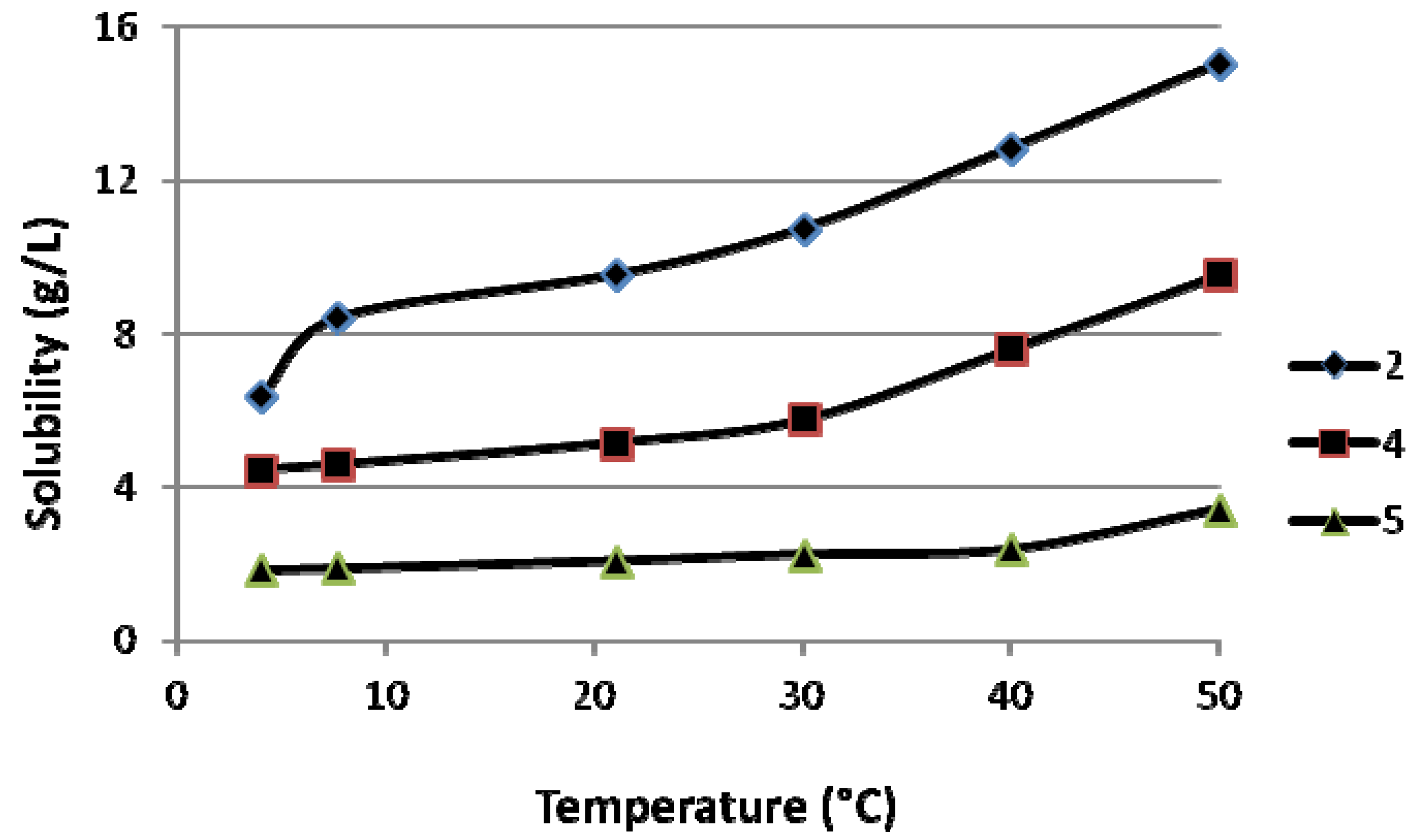
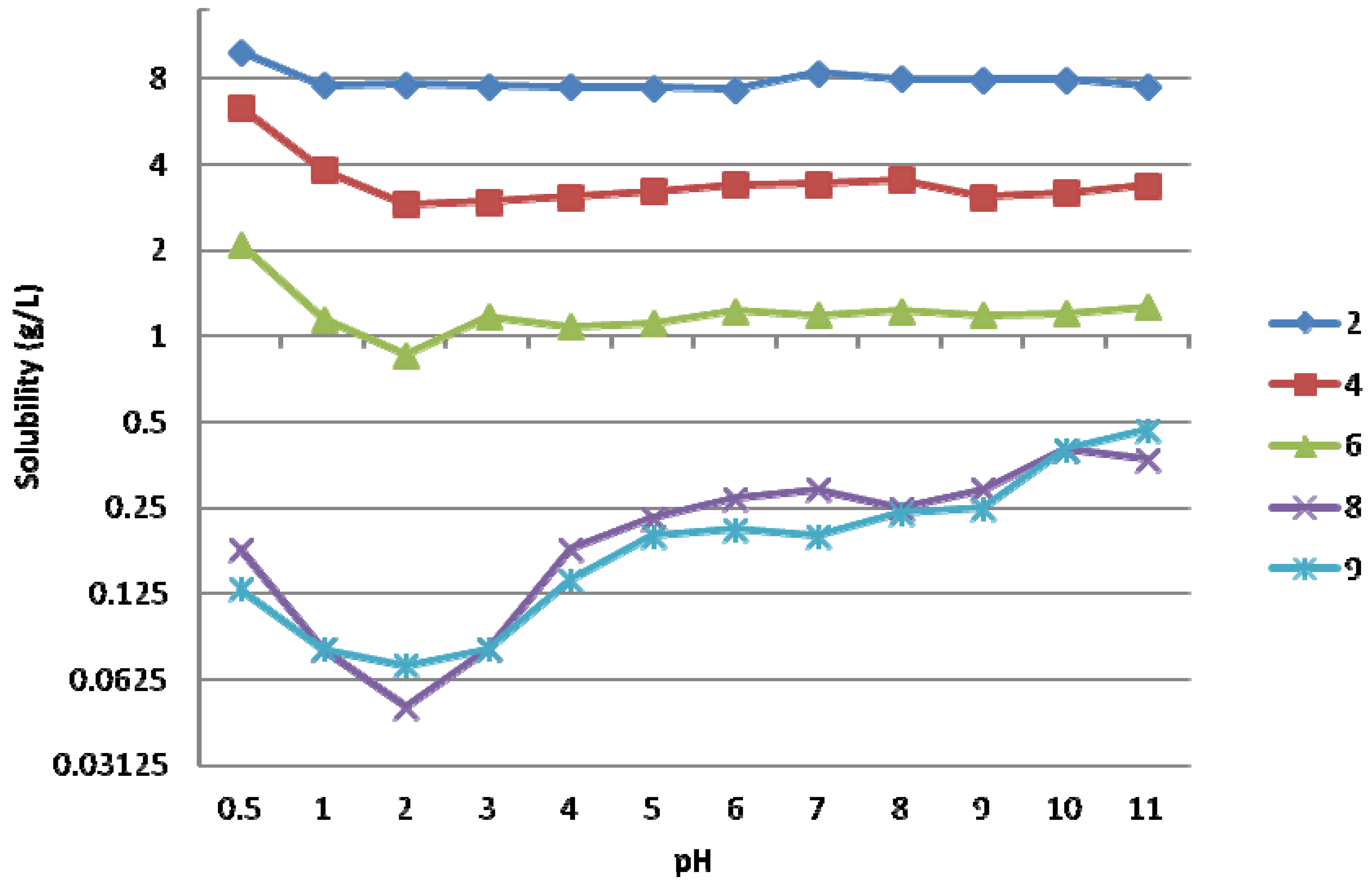
2.1. Solubility of the ABPs

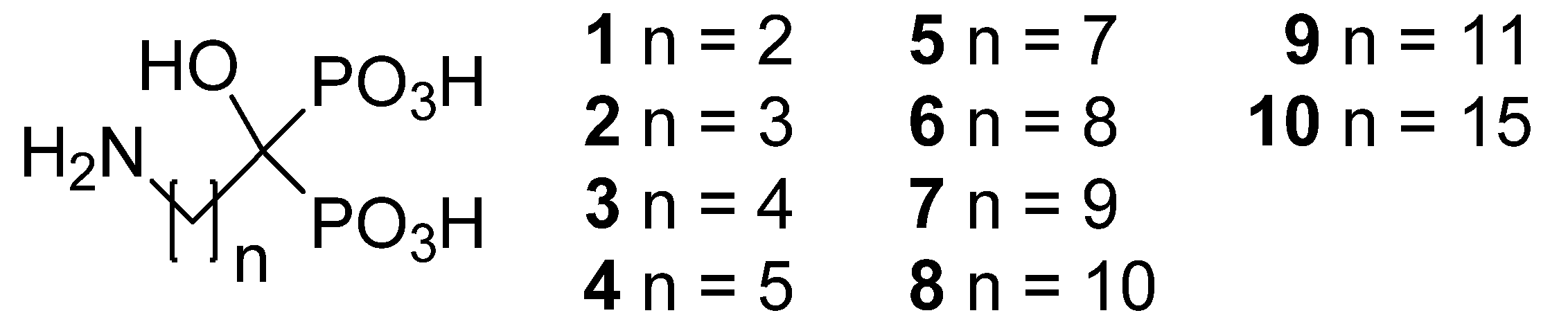{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | n | Formula | MW | pH | Solubility | |
|---|---|---|---|---|---|---|
| (mg P/L) a | (mgL) b | |||||
| 1 | 2 | C3H10NO7P2Na | 257.05 | 4.62 | 2634 | 10928 |
| 2 | 3 | C4H13NO7P2 | 249.10 | 1.94 | 1905 | 7663 |
| 3 | 4 | C5H15NO7P2 | 263.12 | 2.22 | 944 | 4009 |
| 4 | 5 | C6H17NO7P2 | 277.15 | 2.26 | 764 | 3419 |
| 5 | 7 | C8H21NO7P2 | 305.20 | 2.61 | 426 | 2099 |
| 6 | 8 | C9H23NO7P2 · H2O | 337.23 | 2.51 | 400 | 2066 |
| 7 | 9 | C10H25NO7P2 | 333.26 | 3.28 | 40 | 217 |
| 8 | 10 | C11H27NO7P2 · H2O | 365.30 | 4.71 | 10 | 58 |
| 9 | 11 | C12H29NO7P2 | 361.31 | 4.05 | 5 | 30 |
| 10 | 15 | C16H37NO7P2 | 417.42 | 7.79 | 6 | 39 |
2.2. Protonation Constants for Compounds 1–5
| Protonation Reaction | log K | pKa | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| L4− + H+⇋ HL3− | 12.86 a | 12.13 f | 12.05 | 11.94 k | 11.65 | pKa5 |
| HL3− + H+⇋ H2L2− | 10.04 b | 10.69 g | 10.78 | 10.86 l | 10.67 | pKa4 |
| H2L2− + H+⇋ H3L− | 5.90 c | 6.26 h | 6.44 | 6.62 m | 6.75 | pKa3 |
| H3L− + H+⇋ H4L | 1.70 d | 2.12 i | 2.30 | 2.38 n | 2.52 | pKa2 |
| H4L + H+⇋ H5L+ | 1.06 e | 0.60 j | 0.62 | 0.94 | 1.08 | pKa1 |
| Protonation Reaction | compound 1 logK | pKa | ||||||
|---|---|---|---|---|---|---|---|---|
| Ref.13 | Ref.14 | Ref.15 | Ref.4 | Ref.17 | Ref.18 | Ref.18 | ||
| L4− + H+⇋ HL3− | 10.8 | 13.06 | 10.95 | 11.02 | 12.14 | 10.74 | 10.40 | pKa5 |
| HL3– + H+⇋ H2L2− | 9.9 | 10.30 | 9.80 | 9.90 | 10.18 | 9.97 | 9.46 | pKa4 |
| H2L2− + H+⇋ H3L− | 5.83 | 5.85 | 6.01 | 5.86 | 6.04 | 6.01 | 5.39 | pKa3 |
| H3L− + H+⇋ H4L | 2.55 | 1.80 | 2.56 | 2.04 | 1.93 | - | - | pKa2 |
| H4L + H+⇋ H5L+ | - | <1.2 | - | - | 1.24 | - | - | pKa1 |
| Protonation Reaction | Comp. 2 logK | Comp. 4 logK | pKa | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Ref.13 | Ref.14 | Ref.17 | Ref.18 | Ref.18 | Ref.19 | Ref.13 | Ref.4 | ||
| L4− + H+⇋ HL3− | 11.6 | 12.68 | 11.82 | 11.4 | 10.5 | 12.04 | 10.9 | - | pKa5 |
| HL3− + H+⇋ H2L2− | 10.5 | 11.07 | 10.96 | 10.68 | 10.25 | 10.77 | 8.63 | 10.66 | pKa4 |
| H2L2− + H+⇋ H3L− | 8.73 | 6.36 | 6.39 | 6.38 | 5.95 | 6.21 | 5.49 | 6.50 | pKa3 |
| H3L− + H+⇋ H4L | 2.72 | 2.19 | 2.22 | 2.24 | 2.34 | 2.16 | 2.90 | 2.45 | pKa2 |
| H4L + H+⇋ H5L+ | - | <1.2 | 1.33 | - | - | ~1 | - | - | pKa1 |
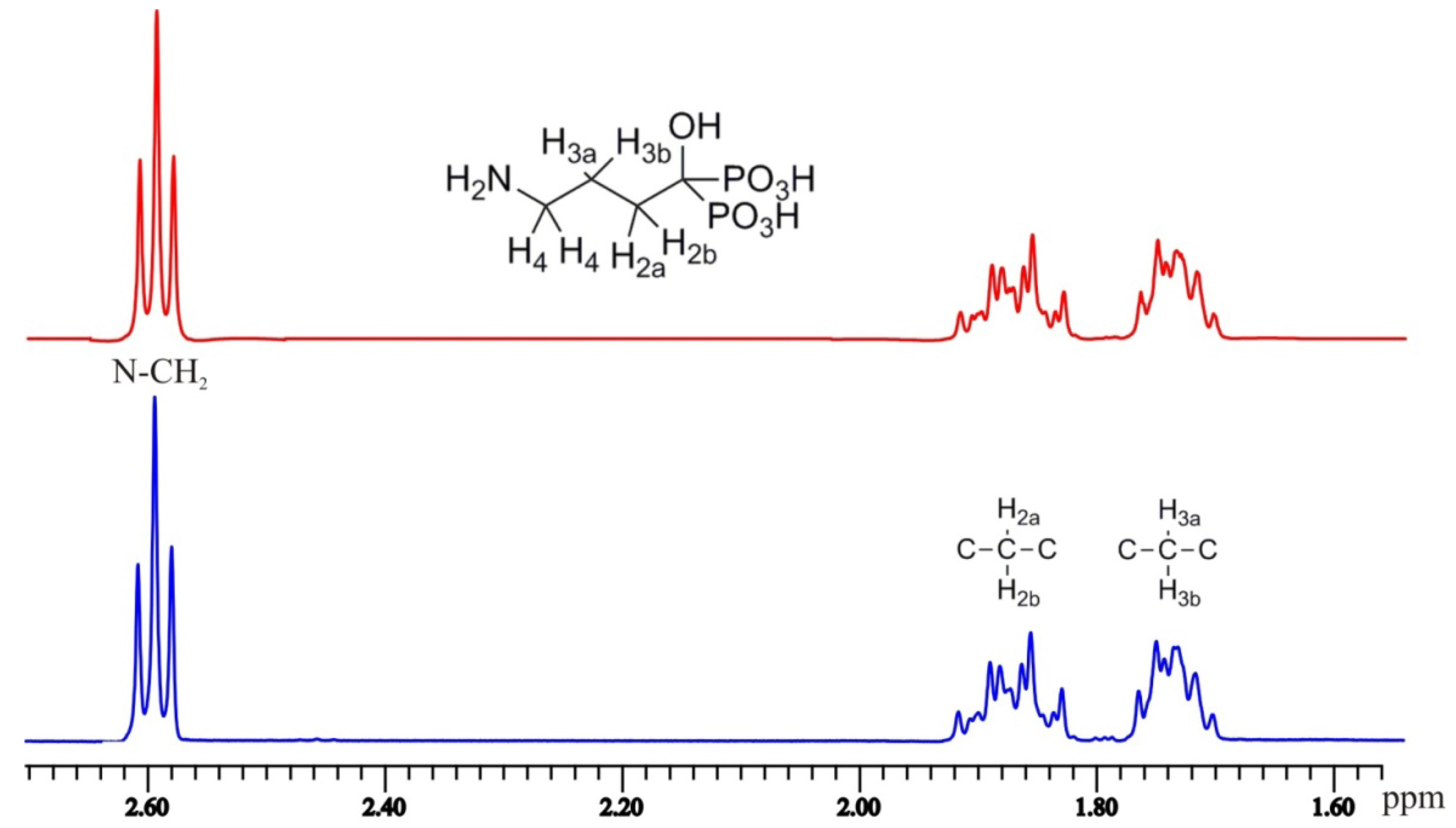
2.3. NMR Spectroscopy
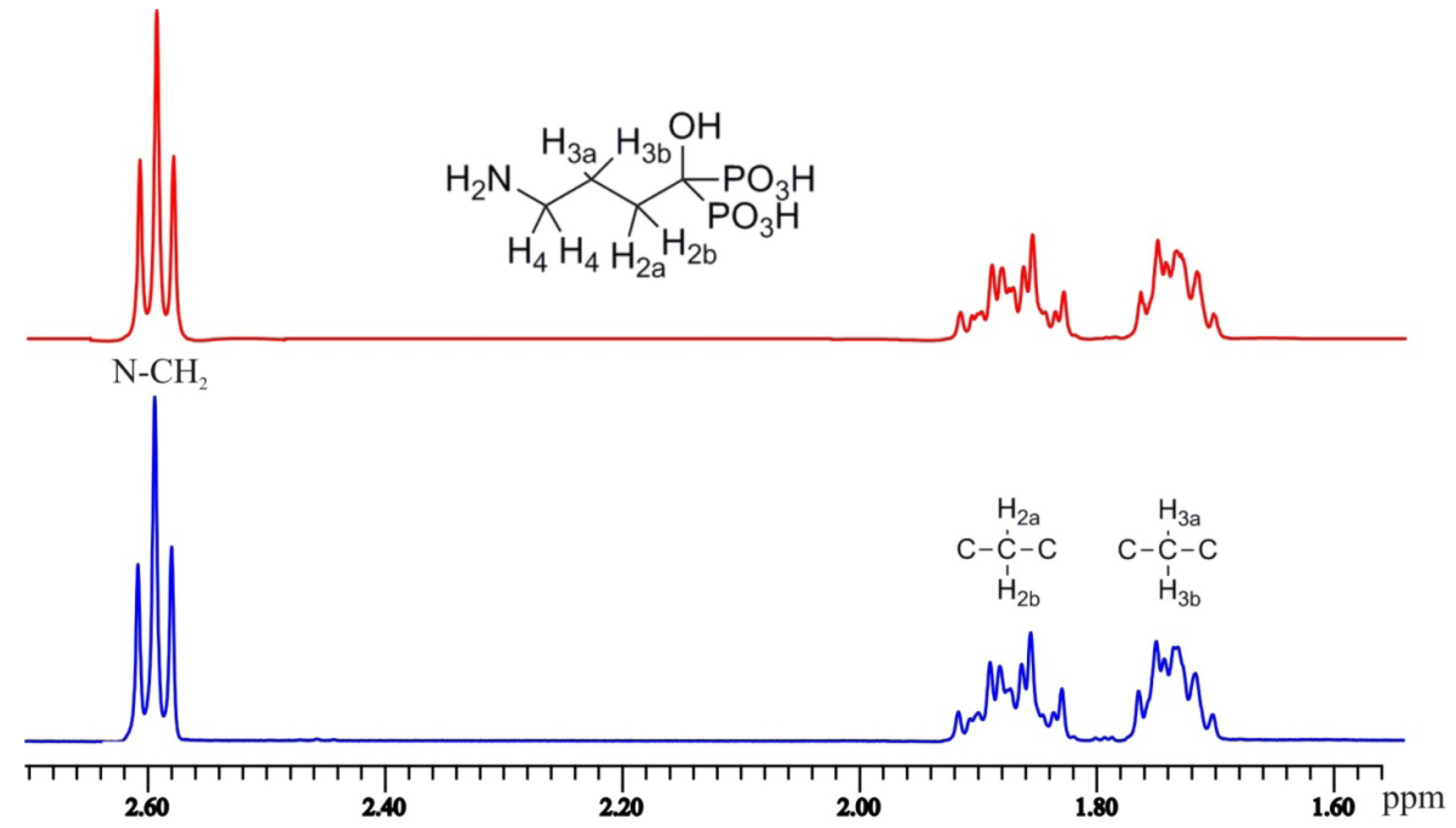
| H-2a | H-2b | H-3a | H-3b | H-4 | H-5 | H-6 | ||
|---|---|---|---|---|---|---|---|---|
| 1 | shift (ppm)3JHH (Hz)3/4JHP (Hz) | 2.04 9.8, 5.8 13.4 | 2.04 10.0, 5.7 13.4 | 2.94 10.0, 5.8 0.9 | 2.94 9.8, 5.7 0.9 | |||
| 2 | shift (ppm)3JHH (Hz)3/4JHP (Hz) | 1.87 13.9, 4.4 13.5 | 1.87 11.7, 4.2 13.5 | 1.73 11.7, 8.8, 4.4 1.0 | 1.73 13.9, 5.4, 4.2 1.0 | 2.59 8.8, 5.4- | ||
| 3 | shift (ppm)3JHH (Hz)3/4JHP (Hz) | 1.88 13.9, 5.6 113.6 | 1.88 11.8, 2.9 13.6 | 1,62 13.9, 5.9, 2.9 1.0 | 1.62 11.8, 9.4, 5.6 1.0 | 1.47 9.4, 6.9, 5.9 - | 2.70 6.9- | |
| 4 | shift (ppm)3JHH (Hz)3/4JHP (Hz) | 1.88 12.4, 4.0 13.3 | 1.88 13.4, 4.5 13.3 | 1.58 13.4, 7.2, 4.0 0.9 | 1.58 12.4, 7.9, 4.5 0.9 | 1.29 7.9, 7.8, 7.2- | 1.47 7.8, 7.1- | 2.61 7.1- |
| 5 | shift (ppm)3JHH (Hz)3/4JHP (Hz) | 1.87 13.0, 4.313.3 | 1.87 13.0, 4.3 13.6 | 1.56 13.0, 6.0, 4.3 1.5 | 1.56 13.0, 7.7, 4.3 1.5 | 2 | 2 | 1.46 7.1, 6.7- |
2.4. Thermal Analysis
| Comp. | MW | Δwt-% (%) | Temp. range b | Tm c | Td d | Ref. melting points | |
|---|---|---|---|---|---|---|---|
| exp. | calc. | ||||||
| 1 | 257.05 | anhydrous | 149 | 152 | - | ||
| 2 | 249.10 | anhydrous | 223 | 224 | 234 (dec.) f | ||
| 3 | 263.12 | anhydrous | 216 | 220 | 212 (dec.) f | ||
| 4 | 277.15 | anhydrous | 205 | 212 | 247 f | ||
| 5 | 305.20 | anhydrous | 193 | 195 | - | ||
| 6 | 337.23 a | 5.00 | 5.34 (1 H2O) | 33–175 (81) | 134 | 191 | - |
| 7 | 333.26 | anhydrous | 181 | 187 | - | ||
| 8 | 365.30 a | 4.43 | 4.93 (1 H2O) | 40–148 (72) | 127 | 202 | - |
| 9 | 361.31 | anhydrous | 181 | 188 | - | ||
| 10 | 417.42 | anhydrous | 175 | 180 | - | ||
3. Experimental
3.1. Materials and Instrumentation
3.2. Synthesis
3.3. Spectrophotometric Analysis
3.4. Determination of Solubility
3.5. Determination of the Protonation Constants
3.6. Elemental Analysis
3.7. Thermal Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Menschutkin, N. Über die Einwirkung des Chloracetyls auf phosphorige Saure. Ann. Chem. Pharmacol. 1865, 133, 317–320. [Google Scholar] [CrossRef]
- Russell, R.G.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar]
- Fleisch, H. Bisphosphonates in Bone Disease: From the Laboratory to the Patient; The Parthenon Publishing Group Inc.: New York, NY, USA, 1995. [Google Scholar]
- Casolaro, M.; Casolaro, I.; Spreafico, A.; Capperucci, C.; Frediani, B.; Marcolongo, R.; Margiotta, N.; Ostuni, R.; Mendichi, R.; Samperi, F.; et al. Novel therapeutic agents for bone resorption. Part 1. Synthesis and protonation thermodynamics of poly(amido-amine)s containing bis-phosphonate residues. Biomacromolecules 2006, 7, 3417–3427. [Google Scholar]
- Hirabayashi, H.; Sawamoto, T.; Fujisaki, J.; Tokunaga, Y.; Kimura, S.; Hata, T. Relationship between physicochemical and osteotropic properties of bisphosphonic derivatives: Rational design for osteotropic drug delivery system (ODDS). Pharm. Res. 2001, 18, 646–651. [Google Scholar] [CrossRef]
- Reddy, G.V.; Jacobs, H.K.; Gopalan, A.S.; Barrans, R.E., Jr.; Dietz, M.L.; Stepinski, D.C.; Herlinger, A.W. Synthesis of symmetrical methylenebis(alkyl hydrogen phosphonates) by selectivecleavage of methylenebis(dialkyl phosphonates) with morpholine. Synt. Commun. 2004, 34, 331–334. [Google Scholar] [CrossRef]
- Chuiko, A.L.; Lozinsky, M.O.; Jasicka-Misiak, I.; Kafarski, P. Herbicidal derivatives of aminomethylenebisphosphonic acid. Part IV. Hydroxyalkylidenebisphosphonates, iminomethylenebisphosphonates and ureidomethylenebisphosphonates J. Plant. Growth Regul. 1999, 18, 171–174. [Google Scholar] [CrossRef]
- Ghosh, S.; Chan, J.M.W.; Lea, C.R.; Meints, G.A.; Lewis, J.C.; Tovian, Z.S.; Flessner, R.M.; Loftus, T.C.; Bruchhaus, I.; Kendrick, H.; et al. Effects of bisphosphonates on the growth of Entamoeba histolytica and Plasmodium species in vitro and in vivo. J. Med. Chem. 2004, 47, 175–187. [Google Scholar] [CrossRef]
- Fu, R.; Hu, S.; Wu, X. Mix-solvothermal syntheses, characterization, and properties of new zinc diphosphonates with zn-o-p chains, layers, and 3d frameworks. Crystal Growth Des. 2007, 7, 1134–1144. [Google Scholar]
- Ylitalo, R. Bisphosphonates and atherosclerosis. Gen. Pharmacol. 2002, 35, 287–296. [Google Scholar]
- Liu, D.; Kramer, S.A.; Huxford-Phillips, R.C.; Wang, S.; Della Rocca, J.; Lin, W. Coercing bisphosphonates to kill cancer cells with nanoscale coordination polymers. Chem. Commun. 2012, 48, 2668–2670. [Google Scholar]
- Rogers, M.J. New insights into the molecular mechanisms of action of bisphosphonates. Curr. Pharm. Des. 2003, 9, 2643–2658. [Google Scholar] [CrossRef]
- Martell, A.E.; Smith, R.M. Critical Stability Constants Database; NIST: Gaithersburg, MD, USA, 2003. [Google Scholar]
- Kubíček, V.; Kotek, J.; Hermann, P.; Lukeš, I. Aminoalkylbis(phosphonates): Their complexation properties in solution and in the solid state. Eur. J. Inorg. Chem. 2007, 333–344. [Google Scholar]
- Zeevaart, J.R.; Jarvis, N.V.; Louw, W.K.A.; Jackson, G.E.; Cukrowski, I.; Mouton, C.J. Metal-ion speciation in blood plasma incorporating the bisphosphonate, 1-hydroxy-4-aminopropilydenediphosphonate (APD), in therapeutic radiopharmaceuticals. J. Inorg. Biochem. 1999, 73, 265–272. [Google Scholar] [CrossRef]
- Yates, A.J.; Rodan, G.A. Alendronate and osteoporosis. DDT 1998, 3, 69–78. [Google Scholar]
- Hägele, G.; Szakács, Z.; Ollig, J.; Hermens, S.; Pfaff, C. NMR-Controlled titrations: Characterizing aminophosphonates and related structures. Heteroatom Chem. 2000, 11, 562–582. [Google Scholar] [CrossRef]
- Boichenko, A.P.; Markov, V.V.; Le Kong, H.; Matveeva, A.G.; Loginova, L.P. Re-evaluated data of dissociation constants of alendronic, pamidronic and olpadronic acids. Cent. Eur. J. Chem. 2009, 7, 8–13. [Google Scholar] [CrossRef]
- Dyba, M.; Jezowska-Bojczuk, M.; Kiss, E.; Kiss, T.; Kozlowski, H.; Leroux, Y.; El Manouni, D. 1-Hydroxyalkane-1,1-diyldiphosphonates as potent chelating agents for metal ions. Potentiometric and spectroscopic studies of copper(II) co-ordination. J. Chem. Soc. Dalton Trans. 1996, 1119–1123. [Google Scholar]
- Kieczykowski, G.; Jobson, R.; Melillo, D.; Reinhold, D.; Grenda, V.; Shinkai, I. Preparation of (4-amino-1-hydroxybutylidene) bisphosphonic acid sodium salt, MK-217 (alendronate sodium). An improved procedure for the preparation of 1-hydroxy-1,l-bisphosphonica acids. J. Org. Chem. 1995, 60, 8310–8312. [Google Scholar] [CrossRef]
- Kivikoski, J.; Garcia-Ruiz, J.M.; Vepsäläinen, J.; Higes, F.; Pohjala, E.; Välisaari, J. Crystal growth and characterization of methylenebisphosphonate partial esters. J. Phys. D Appl. Phys. 1993, 26, B172–B175. [Google Scholar]
- Günther, H. NMR Spectroscopy, 2nd ed; Wiley: Chichester, UK, 1995; p. 207. [Google Scholar]
- Shkol'nikova, L.M.; Sotman, S.S.; Afonin, E.G. Crystal and molecular-structures of 2 complexones of the alkylidenediphosphone family- Monohydrate of dimethylaminomethylidenediphosphonic and alpha-oxy-gamma-aminopropylidendiphosphonic acids. Kristallografiya 1990, 35, 1442–1449. [Google Scholar]
- Ohanessian, J.; Avenel, D.; El Manouni, D.; Benramdane, M. The molecular structure of 4-amino 1-hydroxy butylidene-1 bisphosphonic acid (AHBBPA); an uncommon anhydrous hydroxybisphosphonic acid. Phosphorus Sulfur Silicon Relat. Elem. 1997, 129, 99–110. [Google Scholar] [CrossRef]
- Coiro, V.M.; Lamba, D. Structure of 6-amino-1-hydroxyhexylidenebis(phosphonic acid). Acta Cryst. Sect. C Cryst. Struct. Commun. 1989, 45, 446–448. [Google Scholar] [CrossRef]
- Simmonds, P.G.; Medley, E.E.; Ratcliff, M.A., Jr.; Shulman, G.P. Thermal decomposition of aliphatic monoaminomonocarboxylic acids. Anal. Chem. 1972, 44, 2060–2066. [Google Scholar] [CrossRef]
- Afonin, E.G.; Aleksandrov, G.G. Piperazinium and piperidinium trihydrogen1-hydroxyethane-1,1-diphosphonates. Russ. J. Gen. Chem. 2002, 72, 221–225. [Google Scholar] [CrossRef]
- Widler, L.; Jaeggi, K.A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; et al. Highly potent geminal bisphosphonates. From pamidronate disodium (Aredia) to zoledronic acid (Zometa). J. Med. Chem. 2002, 45, 3721–3788. [Google Scholar]
- Amara, N.; Mashiach, R.; Amar, D.; Krief, P.; Spieser, S.; Bottomley, M.; Aharoni, A.; Meijler, M. Covalent inhibition of bacterial quorum sensing. J. Am. Chem. Soc. 2009, 131, 10610–10619. [Google Scholar]
- 4500-P Phosphorus#1 in Standard Methods for the Examination of Water and Wastewater. American Public Health Association, 1999. Available online: http://www.umass.edu/tei/mwwp/acrobat/sm4500P-E.PDF (accessed on 1 August 2012).
- Sample Availability: Samples of the compounds 1–10 are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alanne, A.-L.; Hyvönen, H.; Lahtinen, M.; Ylisirniö, M.; Turhanen, P.; Kolehmainen, E.; Peräniemi, S.; Vepsäläinen, J. Systematic Study of the Physicochemical Properties of a Homologous Series of Aminobisphosphonates. Molecules 2012, 17, 10928-10945. https://doi.org/10.3390/molecules170910928
Alanne A-L, Hyvönen H, Lahtinen M, Ylisirniö M, Turhanen P, Kolehmainen E, Peräniemi S, Vepsäläinen J. Systematic Study of the Physicochemical Properties of a Homologous Series of Aminobisphosphonates. Molecules. 2012; 17(9):10928-10945. https://doi.org/10.3390/molecules170910928
Chicago/Turabian StyleAlanne, Aino-Liisa, Helena Hyvönen, Manu Lahtinen, Markku Ylisirniö, Petri Turhanen, Erkki Kolehmainen, Sirpa Peräniemi, and Jouko Vepsäläinen. 2012. "Systematic Study of the Physicochemical Properties of a Homologous Series of Aminobisphosphonates" Molecules 17, no. 9: 10928-10945. https://doi.org/10.3390/molecules170910928
APA StyleAlanne, A.-L., Hyvönen, H., Lahtinen, M., Ylisirniö, M., Turhanen, P., Kolehmainen, E., Peräniemi, S., & Vepsäläinen, J. (2012). Systematic Study of the Physicochemical Properties of a Homologous Series of Aminobisphosphonates. Molecules, 17(9), 10928-10945. https://doi.org/10.3390/molecules170910928