3. Experimental
3.1. General
All reagents were purchased from Sigma-Aldrich or Fluka and were used without further purification. Dry solvents were purchased as well from Sigma-Aldrich or Fluka and used as supplied. All the standard phosphoroamidites and ancillary reagents used for oligonucleotide synthesis were purchased from Applied Biosystems or Link Technologies. Flash column chromatography was carried out on silica gel SDS 0.063–0.2 mm/70–230 mesh.
1H- and 13C-NMR spectra were recorded at 25 °C on a Varian Mercury 400 MHz spectrometer and the 31P-NMR spectra were recorded on a Varian Inova 300 MHz spectrometer using CDCl3. Tetramethylsilane (TMS) was used as an internal reference (0 ppm) for 1H spectra and the residual signal of the solvent (77.16 ppm) for 13C and 31P. Chemical shifts are reported in part per million (ppm) in the δ scale, coupling constants in Hz and multiplicity as follows: s (singlet), d (doublet), t (triplet), q (quadruplet), m (multiplet), br (broad signal). HPLC separations were performed using a Waters 2695 Separations Module with a Waters 2998 Photodiode Array Detector. UV analyses and melting curves were performed using a Jasco V-650 instrument equipped with a thermoregulated cell holder. Fluorescent measures were performed using a Jasco FP-6200 spectrofluorometer as well equipped with a thermoregulated cell holder. Electrospray ionization mass spectra (ESI-MS) of compounds 1–3 and 6 were recorded on a Micromass ZQ instrument with single quadrupole detector coupled to a Waters Alliance 2696 HPLC. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra of oligonucleotides were recorded either on a Voyager-DETM RP spectrometer (Applied Biosystems) or in a Perseptive Biosystems Voyager DE-RP instrument (Applied Biosystems) in negative mode by using 2,4,6-trihydroxyacetophenone (THAP)/ammonium citrate (1:1) (THAP-CA as matrix and additive respectively). The mass spectra analysis of oligonucleotide 12 was performed in the negative ion mode on Synapt G1 HDMS travelling wave ion mobility mass spectrometer (Waters-Micromass). The experiment was performed using a 384 well plate and introduced by automated chip-based nanoelectrospray using a Triversa NanoMate (Advion BioSciences).
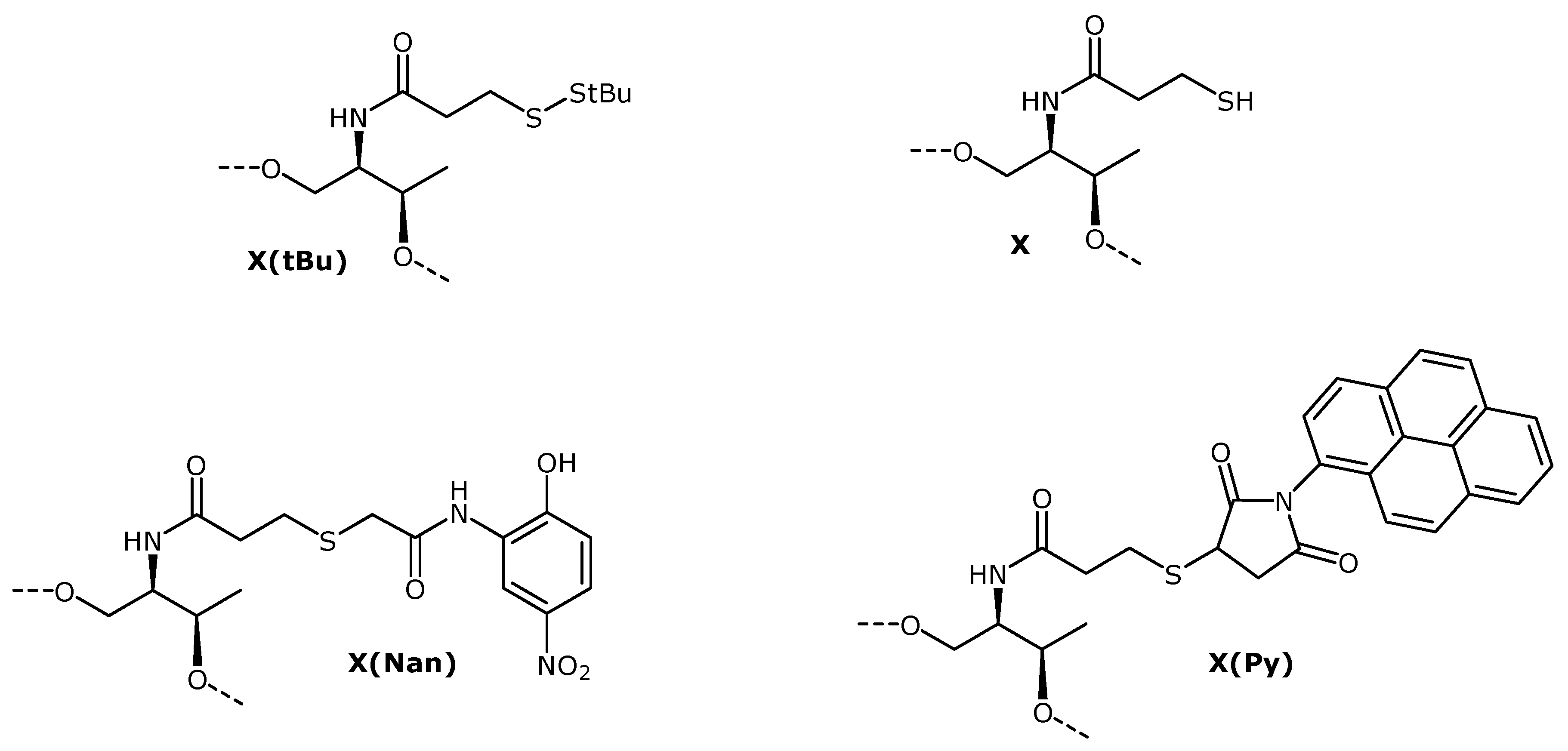
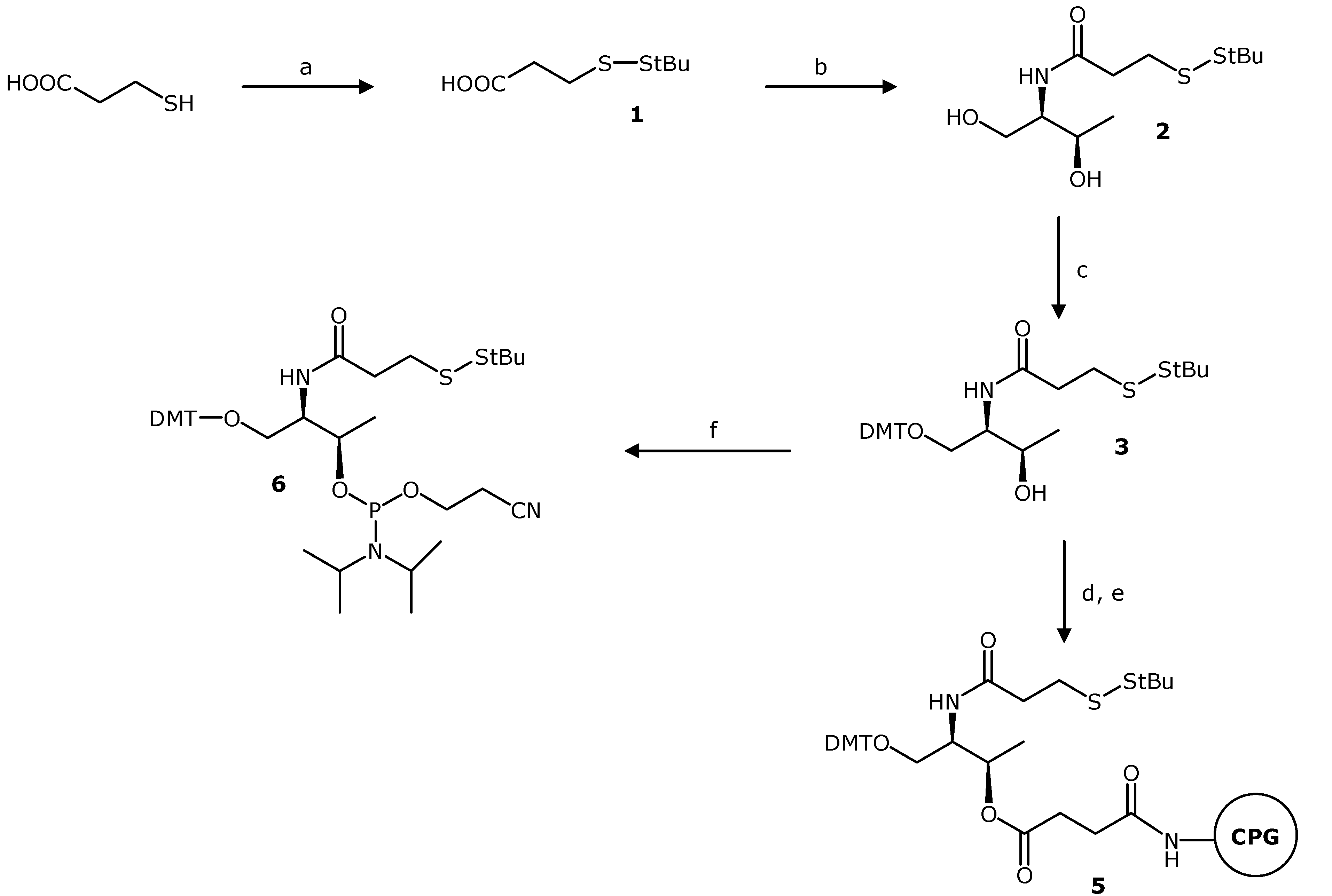
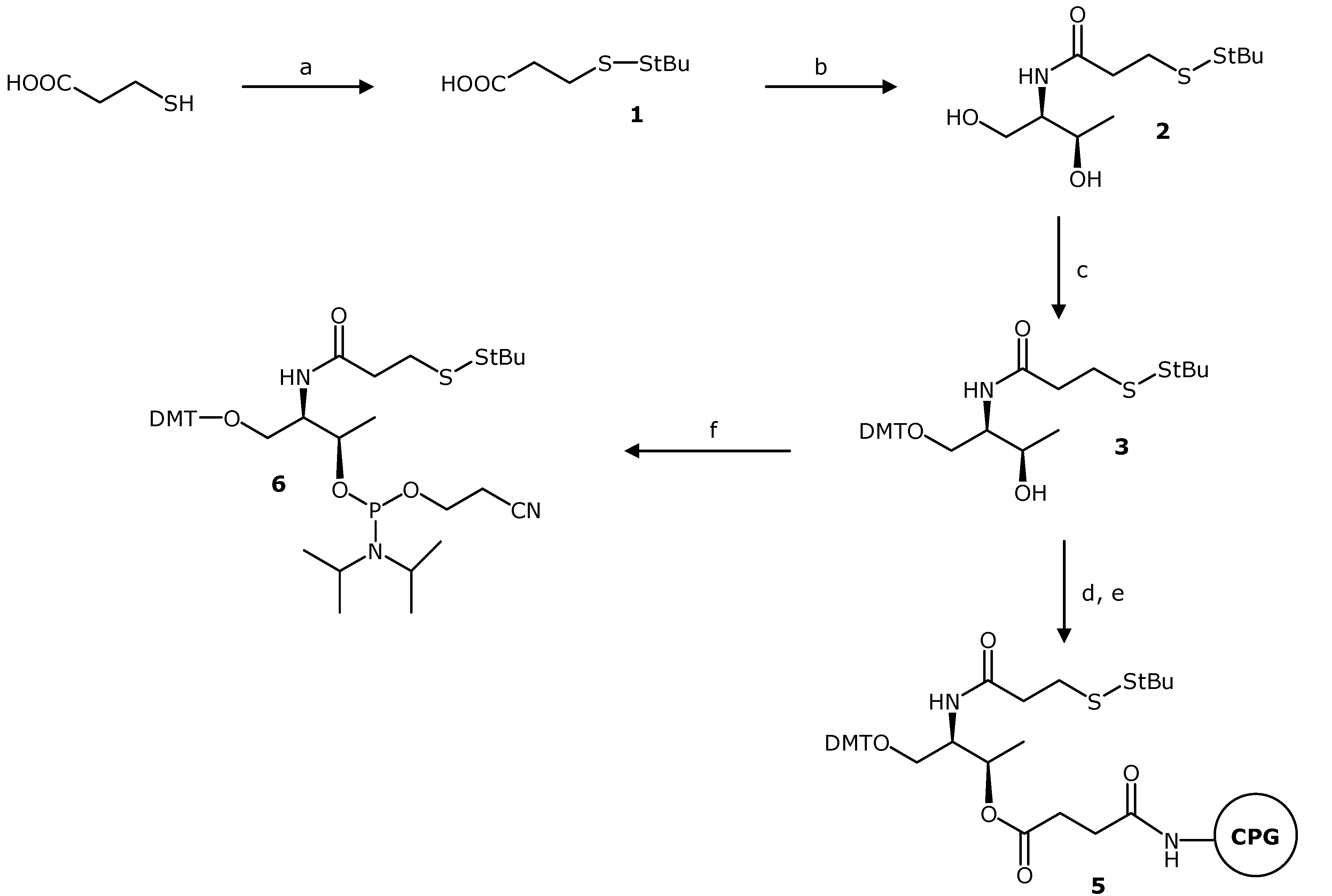
3-(tert-Butyldisulfanyl)propanoic Acid (1). A solution of 3-mercaptopropanoic acid (0.34 mL, 3.9 mmol) and triethylamine (1.1 mL 7.8 mmol) in anhydrous dimethylformamide (DMF) (15 mL) was prepared under argon. Di-tert-butyl-1-(tert-butylthio)-1,2-hydrazinedicarboxylate (2.5 g 7.8 mmol) was dissolved in anhydrous DMF (10 mL) and added dropwise to the stirred solution. The reaction mixture was stirred at room temperature overnight, and the reaction was complete as judged by TLC. The solvent was evaporated under reduced pressure. In order to remove the remaining DMF, the residue was dissolved in toluene and evaporated under reduced pressure. The procedure was repeated three times. We used two different methodologies to purify the carboxylic acid. Method 1: The resulting crude was dissolved with 100 mL 10% aqueous solution of sodium hydroxide and washed three times with 50 mL of hexane. The aquerous solution was acidified with hydrochloric acid fuming 37% until pH = 2–3, and then was extracted with 50 mL of ethyl acetate (×3). The extracts were combined, dried over MgSO4 and concentrated to dryness under reduced pressure. The desired product (pale yellow oil) was obtained with 57% yield (0.36 g). Method 2: The product was purified by chromatography on silica gel. The column was packed with silica gel using a 1% triethylamine solution in ethyl acetate. The byproducts were eluted with a gradient of ethyl acetate to ethyl acetate/methanol 100:10. Then, the product was eluted using a 1% acid acetic solution in ethyl acetate/methanol 100:10. The pure fractions were combined and the solvent was eliminated under reduced pressure. The resulting residue was dissolved with 100 mL of ethyl acetate and the organic phase was washed with 50 mL of water and 50 mL of a saturated NaCl aqueous solution. The organic phase was dried over anhydrous magnesium sulphate and concentrated to dryness under reduced pressure. The desired product (pale yellow oil) was obtained with 34% yield (0.26 g). TLC (ethyl acetate) Rf = 0.35; 1H-NMR, δH (CDCl3): 2.92 (t, J = 7 Hz, 2H), 2.78 (t, J = 7 Hz, 2H), 1.34 (s, 9H); 13C-NMR, δC (CDCl3): 178.20 (CO), 48.27 (C), 34.58 (CH2), 34.26 (CH2), 30.16 (CH3); ESI-MS m/z (negative mode) [M−H]− = 193.04, (M = 194.04 g/mol calculated for C7H14O2S2).
N-[2-(tert-Butyldisulfanyl)ethylcarbonyl]-L-threoninol (2). A solution of compound 1 (300 mg, 1.54 mmol), L-threoninol (162 mg, 1.54 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) (413 mg, 2.31 mmol), hydroxybenzotriazole (HOBt) (313 mg, 2.31 mmol), and N,N-diisopropylethylamine (0.40 mL, 2.31 mmol) in anhydrous DMF (15 mL) was prepared under argon. After stirring the reaction mixture at room temperature overnight, the reaction was complete as judged by TLC and then concentrated to dryness under reduced pressure. In order to remove the remaining DMF, the residue was dissolved in toluene and concentrated to dryness under reduced pressure (×3). The residue was dissolved in 100 mL of dichloromethane (DCM) and the organic phase was washed with 50 mL 10% NaCO3H aqueous solution (×2) and 50 mL of a saturated NaCl aqueous solution. The organic phase was dried over anhydrous magnesium sulphate and concentrated to dryness under reduced pressure. The pure compound was obtained as a white solid (346 mg, 80% yield). TLC (ethyl acetate) Rf = 0.19. 1H-NMR, δH (CDCl3): 6.41 (d, J = 6.8 Hz, 1H), 4.19 (qd, J = 6.4 and 1.6 Hz, 1H), 3.88–3.82 (m, 3H), 2.98 (t, J = 7.0 Hz, 2H), 2.66 (t, J = 7.0 Hz, 2H), 1.34 (s, 9H), 1.23 (d, J = 6.4 Hz, 3H). 13C-NMR, δC (CDCl3): 172.23 (CO), 68.72 (CH), 64.86 (CH2), 55.12 (CH), 48.47 (C), 36.25 (CH2), 35.82 (CH2), 30.18 (CH3), 20.71 (CH3). ESI-MS m/z (positive mode) [M+H]+ = 282.13, (M = 281.11 g/mol calculated for C11H23NO3S2).
O1-(4,4'-Dimethoxytriphenylmethyl)-N-[2-(tert-butyldisulfanyl)ethylcarbonyl]-L-threoninol (3). Compound 2 (242 mg, 0.86 mmol) was dried by evaporation of anhydrous acetonitrile (ACN) under reduced pressure and then dissolved in anhydrous pyridine (10 mL) under argon. 4,4'-Dimethoxytriphenylmethyl chloride (320 mg, 0.94 mmol) was added with stirring and exclusion of moisture, and the reaction was allowed to proceed at room temperature overnight. Then the reaction was quenched with methanol (0.5 mL). The solvent was removed under reduced pressure, and the residue was dissolved in DCM (75 mL). The organic phase was washed with 5% aqueous NaCO3H solution (30 mL) and with saturated NaCl solution (30 mL). The organic phase was dried over anhydrous magnesium sulphate and filtered. After filtration and removal of the solvent under reduced pressure, the product was purified by chromatography on silica gel. The column was packed with silica gel using a 1% triethylamine solution in ethyl acetate/hexane 1:1. The product was eluted with a gradient of hexane/ethyl acetate (1:1) to 100% ethyl acetate. The pure compound was obtained as white foam (309 mg, 62%). TLC (ethyl acetate/hexane 2:1) Rf = 0.35. 1H-NMR, δH (CDCl3): 7.39–6.83 (m, 13H), 6.16 (d, J = 8.8 Hz, 1H), 4.07 (qd, J = 6.4 Hz and 1.6 Hz, 1H), 3.95–3.91 (m, 1H), 3.79 (s, 6H), 3.44 (dd, J = 9.6 Hz and 4.2 Hz, 1H), 3.28 (dd, J = 9.6 Hz and 3.2 Hz, 1H), 2.99–2.95 (m, 2H), 2.64–2.61 (m, 2H), 1.34 (s, 9H), 1.13 (d, J = 6.4 Hz, 3H). 13C-NMR, δC (CDCl3): 171.39 (CO), 158.87 (CH), 158.86 (CH), 144.54 (C), 135.69 (C), 135.50 (C), 130.14 (CH), 128.26 (CH), 128.15 (CH), 127.26 (C), 113.56 (CH), 87.07 (C), 68.91 (CH), 65.52 (CH2), 55.46 (CH3), 53.64 (CH), 48.33 (C), 36.33 (CH2), 35.73 (CH2), 30.20 (CH3), 20.13 (CH3). ESI-MS m/z (negative mode) [M−H]− = 582.24, (M = 583.24 g/mol calculated for C32H41NO5S2).
3.2. Synthesis of CPG Support 5 Functionalized with Compound 3
Compound 3 was incorporated on a long-chain alkylamine-controlled pore glass support (LCAA-CPG) following the standard methodology using the hemisuccinate derivative 4. Compound 3 (50 mg, 0.09 mmol) was dried by evaporation of anhydrous ACN under reduced pressure. Then, compound 3 was dissolved in anhydrous pyridine (5 mL) under argon. Succinic anhydride (21 mg, 0.21 mmol) and 4-dimethylaminopyridine (DMAP) (6 mg, 0.05 mmol) were added to the solution. After four hours of magnetic stirring at room temperature, the reaction was complete as judged by TLC (5% methanol in ethyl acetate, Rf = 0.10). The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane (20 mL). The solution was washed with saturated aqueous sodium chloride (15 mL). After drying the organic phase with magnesium sulphate and filtered, the solvent was evaporated under reduced pressure. The monosuccinate derivative 4, which was used in the next step without further purification, was obtained as a white foam.
Commercial LCAA-CPG (CPG Inc., Lincoln Park , NJ, USA, 300 mg, 73 µmol amino/g) was placed into a polypropylene syringe fitted with a polypropylene disc and washed sequentially with DMF, methanol, THF, DCM, and ACN. Then, a solution of O1-(4,4'-dimethoxytriphenylmethyl)-N-[2-(tert-butyldisulfanyl)ethyl-carbonyl]-O3-(succinyl)-L-threoninol (4) (28 mg 0.045 mmol) and triethylamine (26 µL, 0.36 mmol) in 0.5 mL of anhydrous ACN was prepared. The solution was added to the resin and then a solution of 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) (59 mg 0.36 mmol) in 0.3 mL of anhydrous ACN was incorporated to the mixture and left to react for 30 min. The resin was washed with DMF, methanol, DCM and ACN. The incorporation of compound 4 was determined by DMT quantification. The coupling procedure was repeated once more time and the functionality of the resin was determined by DMT quantification (f = 22 µmol/g). Then, the resin treated with 500 µL of Ac2O/DMF 1:1 to cap free amino groups.
O1-(4,4'-Dimethoxytriphenylmethyl)-N-[2-(tertbutyldisulfanyl)ethyl-carbonyl]-O3-[2-cyanoethyl-N,N-diisopropylaminophosphinyl]-L-threoninol (6). Compound 3 (180 mg 0.31 mmol) was dried by evaporation of anhydrous ACN under reduced pressure. Then, compound 3 and N,N-diisopropylethylamine (215 µL, 1.23 mmol) were dissolved in anhydrous DCM (10 mL) under argon. The solution was cooled on ice and 2-cyanoethoxy-N,N'-diisopropylaminochlorophosphine (112 µL, 0.46 mmol) was added dropwise with a syringe. Afterward, the solution was stirred at room temperature for 1 h and 30 min. After this time the reaction was complete as judged by TLC. Then, 10 mL of DCM were added and the organic layer was washed with 5% NaCO3H aqueous solution (20 mL) and with saturated NaCl aqueous solution (20 mL). The organic phase was dried over magnesium sulphate and filtered, and the solvent was evaporated under reduced pressure. The residue was dissolved in a small amount of ethyl acetate/hexane 1:1 and purified by chromatography on silica gel. The column was packed with silica gel using a 5% triethylamine solution in ethyl acetate/hexane 1:1. The product was eluted with ethyl acetate/hexane 1:1. The pure compound was obtained as pale yellow foam (123 mg 56%). TLC (ethyl acetate/hexane 1:1) Rf = 0.48 and 0.38. 31P-NMR, δP (CDCl3, 81 MHz): 148.13 and 147.93, two isomers. 1H-NMR, δH (CDCl3): 7.42–6.80 (m, 13H), 5.88 and 5.72 (2d, J = 8.8 Hz and 9.2 Hz, respectively, two isomers, 1H), 4.39–4.31 (m, 1H), 4.25–4.11 (m, 1H), 3.79 and 3.78 (2s, two isomers, 6H), 3.57–3.46 (m, 2H), 3.26–3.08 (m, 2H), 3.02–2.90 (m, 2H), 2.60–2.55 (m, 4H), 2.41–2.36 (m, 2H), 1.32 (s, 9H), 1.25–0.99 (m, 15H). 13C-NMR, δC (CDCl3): 170.96 (C), 158.67 (CH), 145.07 and 144.99 (C, two isomers), 136.34 (C), 136.25 and 136.24 (C, two isomers), 130.37 (CH), 130.32 and 130.27 (CH, two isomers), 128.50 and 128.41 (CH, two isomers), 128.00 (CH), 127.00 and 126.96 (C, two isomers), 118.04 (C), 113.33 (CH), 113.30 and 113.28 (CH, two isomers), 86.37 and 86.29 (C, two isomers), 69.73 and 68.96 (CH, 2d, J = 14.4 and 16.4 Hz respectively, two isomers), 63.16 and 62.84 (CH2, two isomers), 58.48 and 58.09 (CH2, 2d, J = 19.5 and 19.5 Hz, respectively, two isomers), 55.44 and 55.41 (CH3, two isomers), 54.46 and 54.16 (CH, 2d, J = 4.9 and 6.3 Hz, respectively, two isomers), 48.25 and 48.21 (C, two isomers), 43.37 and 43.29 (CH, 2d, J = 12.3 and 12.5 Hz, respectively, two isomers), 36.51 and 36.43 (CH2, two isomers), 35.90 and 35.87 (CH2, two isomers), 30.17 (CH3), 24.93 and 24.91 (CH3, 2d, J = 7.7 and 7.8 Hz, respectively, two isomers), 24.76 and 24.57 (CH3, 2d, J = 6.8 and 7.1 Hz, respectively, two isomers), 20.66 and 20.53 (CH3, 2d, J = 6.9 and 6.7 Hz, respectively, two isomers), 19.85–19.79 (CH2, m, two isomers). ESI-MS m/z (positive mode) [M+Na]+ = 806.34, (M = 783.35 g/mol calculated for C41H58N3O6PS2).
3.3. Synthesis of Oligonucleotides
The unmodified oligonucleotides were synthesized on a DNA synthesizer (Applied Biosystems 3400) using 200-nmol scale LV200® polystyrene supports and commercially available chemicals. The benzoyl (Bz) group was used for the protection of the amino group of C and A, and the isobutyryl (iBu) group for the protection of G. The coupling yields were >97%. The last DMT group was removed at the end of the synthesis. Each solid support was treated with aqueous concentrated ammonia at 55 °C for 12 h to cleave the products from the supports and remove the benzoyl and isobutyryl groups. The mixtures were filtered and ammonia solutions were concentrated to dryness. Unmodified oligonucleotides were desalted with Sephadex G-25 (NAP-10 column) G-25 (NAP-10column) and analyzed by HPLC. Column: XBridgeTM OST C18 (4.6 × 50 mm, 2.5 µm). Solvent A: 5% ACN in 100 mM triethylammonium acetate (TEAA) (pH = 7) and solvent B: 70% ACN in 100 mM TEAA (pH = 7). Flow rate: 1 mL/min. Conditions: 10 min of linear gradient from 0 to 30% B. The resulting oligomers were analyzed by mass spectrometry (MALDI-TOF) and UV/Vis spectroscopy (data not shown) and used without further purification. The yields obtained ranged from 72 to 87%.
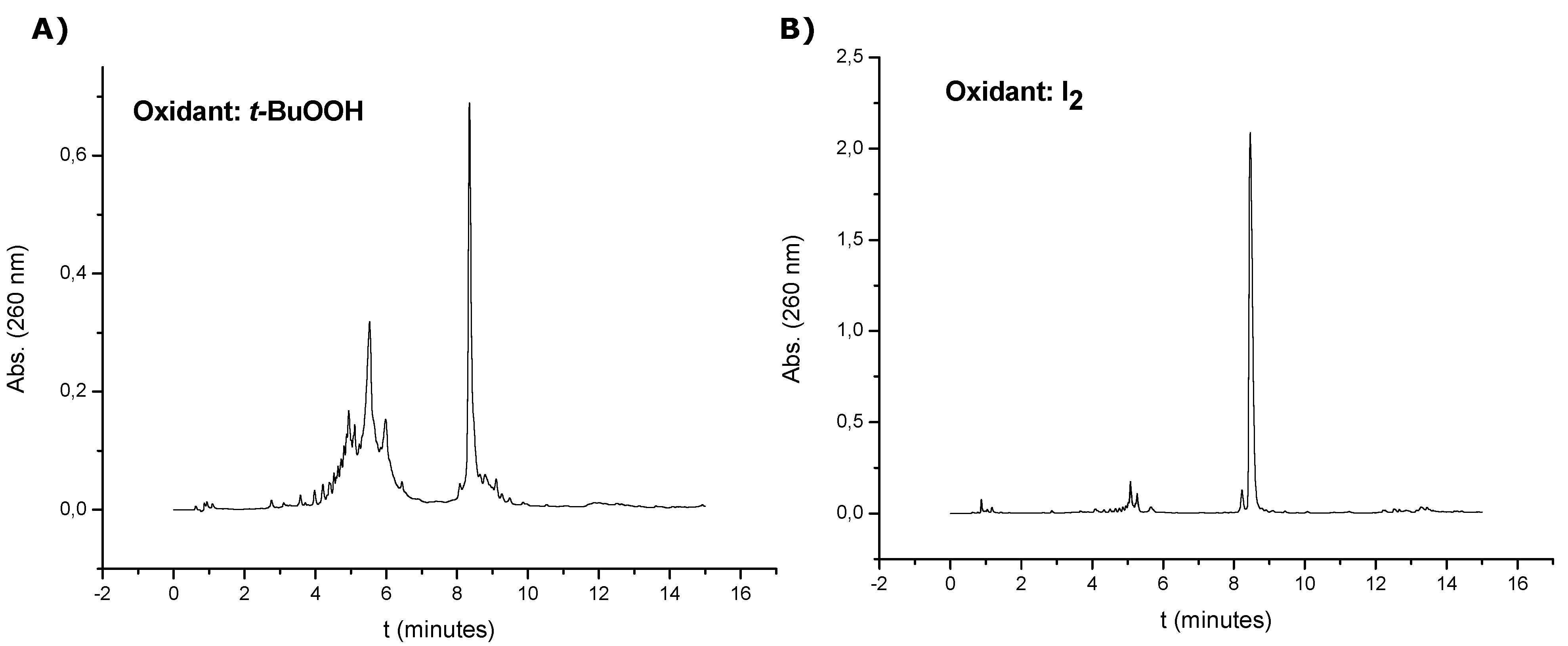
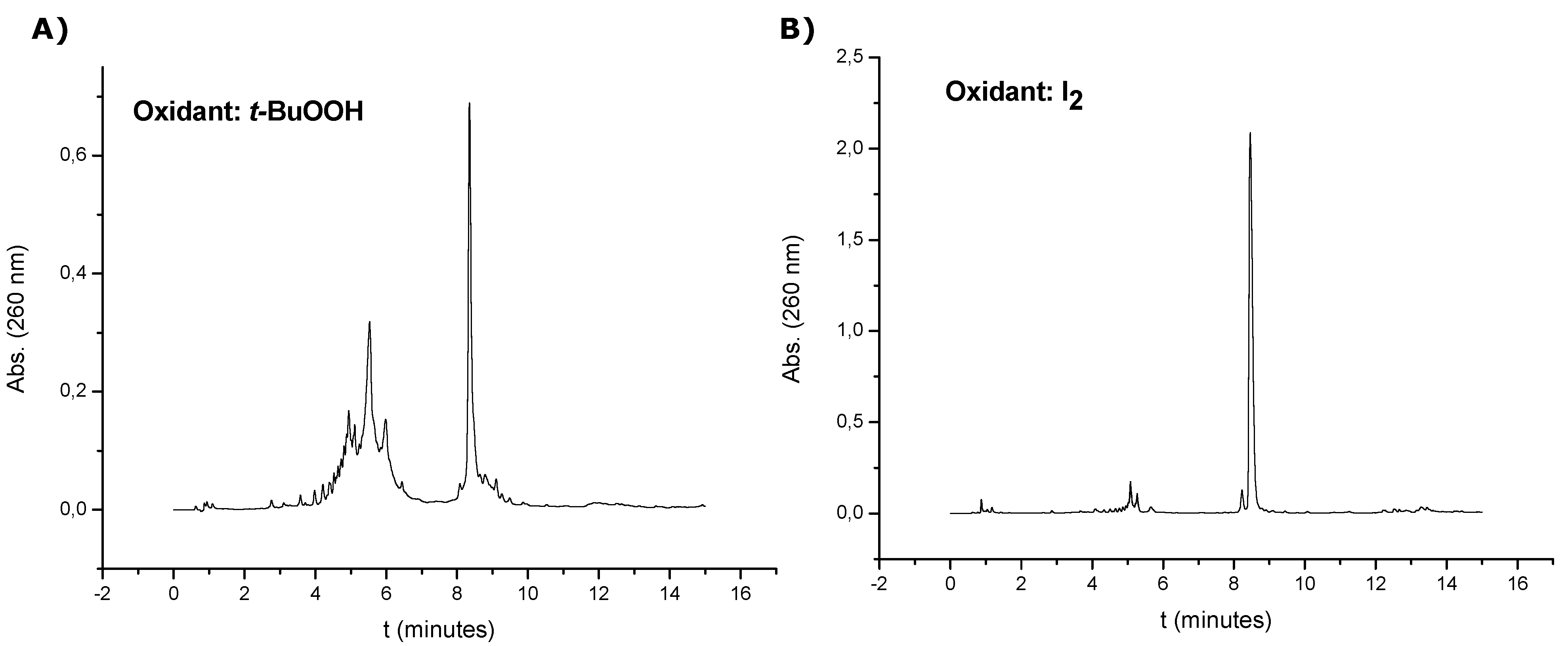
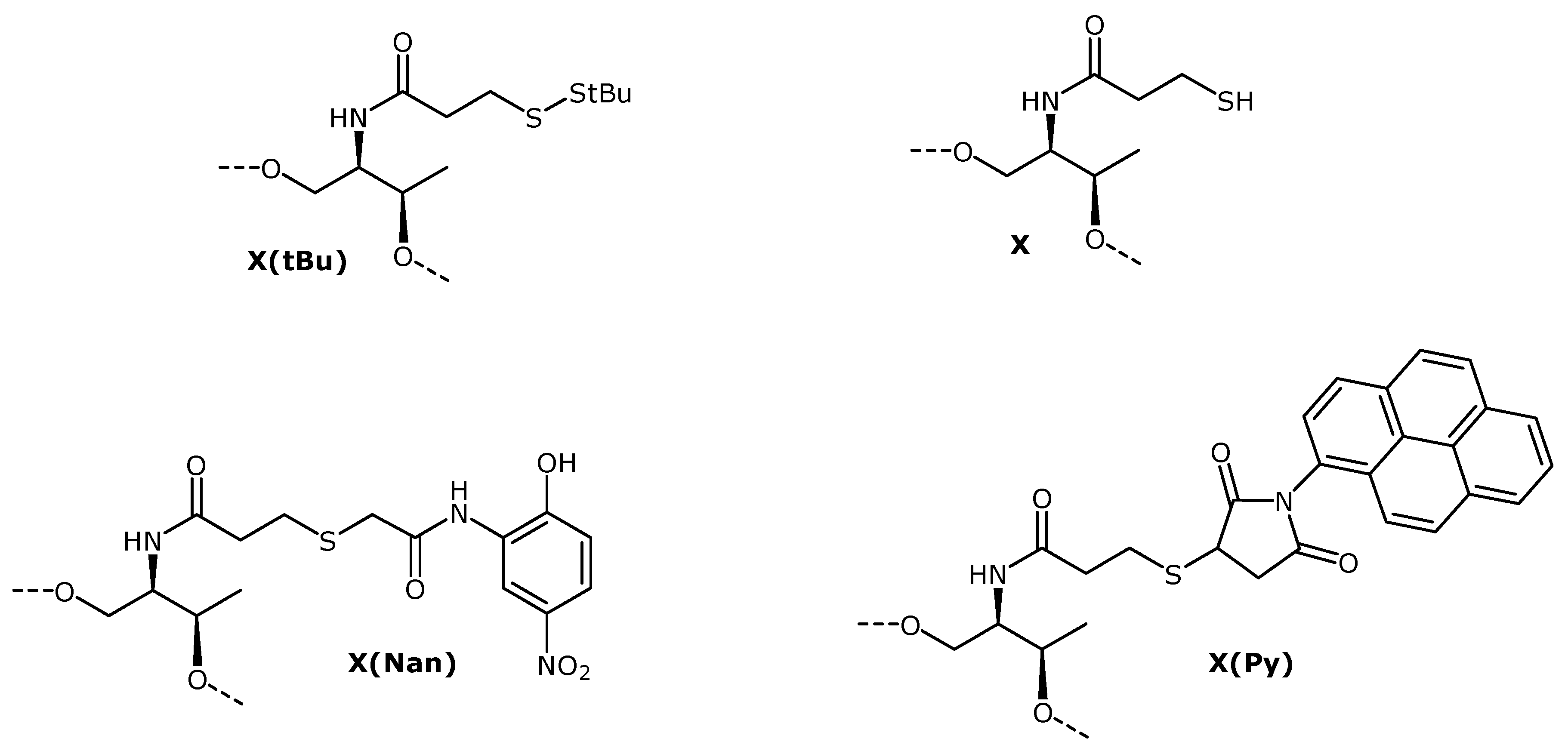
The oligonucleotide sequences containing the threoninol derivative at the 3'-end (7, 8, 13 and 14) were synthesized on a 0.5 µmol scale using the solid support 5. Oligonucleotide sequence 7 was prepared using a tert-butyl hydroperoxide or the standard 0.02 M iodine solution for the oxidation of phosphites. Because better results were obtained using the standard 0.02 M iodine solution, all sequences were prepared in that way. The last DMT group was removed at the end of the synthesis. Each solid support was treated with aqueous concentrated ammonia at 55 °C for 12 h to cleave the products from the supports and remove the benzoyl and isobutyryl groups, except the support coming form the synthesis of oligonucleotide 7 that was treated with aqueous concentrated ammonia at room temperature for 1 h due to the absence of base protecting groups. Work-up was similar as described above. Oligonucleotide 7 (synthesis performed with iodine solution) was used without further purification after it was desalted with Sephadex G-25 (NAP-10 column), (74% yield, 36.2 OD260). Compound 8, 13 and 14 were purified by HPLC on a Nucleosil 120C18 (10 μm, 200 × 10 mm) column. Solvent A: 5% ACN in 100 mM of TEAA (pH = 7) and solvent B: 70% ACN in 100 mM TEAA (pH = 7). Flow rate: 3 mL/min. Conditions: 20 min linear gradient from 0–50% B (oligonucleotides 8 and 13), 20 min linear gradient from 15–38% B (oligonucleotide 14). The resulting products were desalted with Sephadex G-25 (NAP-10 column) and analyzed by HPLC. Column: XBridgeTM OST C18 (4.6 × 50 mm, 2.5 μm). Solvent A: 5% ACN in 100 mM TEAA (pH = 7) and solvent B: 70% ACN in 100 mM TEAA (pH = 7). Flow rate: 1 mL/min. Conditions: 10 min linear gradient from 0–30% B, then 5 min. linear gradient from 30–100% B (oligonucleotides 7, 8 and 13), 4 min linear gradient from 0–12% B, then 1 min linear gradient from 12–40% B, then 5 min linear gradient from 40–45% B, then 5 min linear gradient from 45–100% B (oligonucleotide 14). After HPLC purification the yields obtained ranged from 32 to 48%.
The oligonucleotide sequence 9 (threoninol modification at the 5'-end) and sequences 10–12 (threoninol modification in the middle), were synthesized on a 200 nmol scale employing LV200® polystyrene supports as described above. Phosphoramidite 6 was used to incorporate the threoninol derivative at the 5'-end and in the middle of the corresponding oligonucleotides. The protected phosphoramidite was dissolved in anhydrous DCM instead of ACN to obtain a 0.1 M solution and was allowed to react for 300 s instead of 30 s used for an unmodified phosphoramidite. The DMT determination showed that the efficiency of coupling of the phosphoramidite was >90%. The last DMT group was removed at the end of the synthesis. Each resin was treated with aqueous concentrated ammonia at 55 °C for 12 h to cleave the products from the supports and remove the benzoyl and isobutyryl groups. In the case of oligonucleotide 12 only a small amount of the solid support was treated with ammonia for characterization purposes. Work-up was similar as described above. Oligonucleotide 9 was used without further purification after once desalted with Sephadex G-25 (NAP-10 column), (82% yield, 18.7 OD260). Compound 10–12 were purified by HPLC on a XBridgeTM OST C18 semipreparative column (10 × 50 mm, 2.5 μm). Solvent A: 5% ACN in 100 mM TEAA (pH = 7) and solvent B: 70% ACN in 100 mM TEAA (pH = 7). Flow rate: 3 mL/min. Conditions: 10 min linear gradient from 0–30% B, then 5 min linear gradient 30–100% B, at room temperature for oligonucleotide 10 and at 60 °C for oligonucleotides 11 and 12. The resulting products were desalted with Sephadex G-25 (NAP-10 column) and analyzed by HPLC. Column: XBridgeTM OST C18 (4.6 × 50 mm, 2.5 μm) using the conditions described above except the flow rate that was 1 mL/min. All the purified oligonucleotides were analyzed as well by mass spectrometry. After HPLC purification 12.9 OD260 (50% yield) of compound 10 and 14.5 OD260 (42% yield) of compound 11 were obtained.
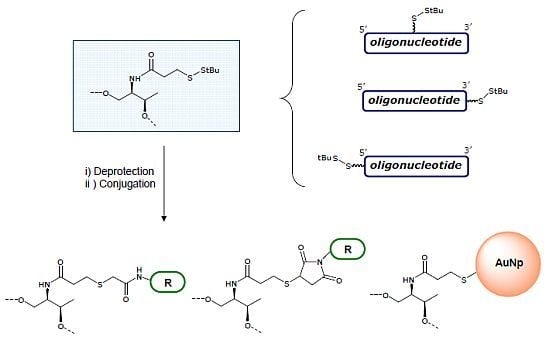
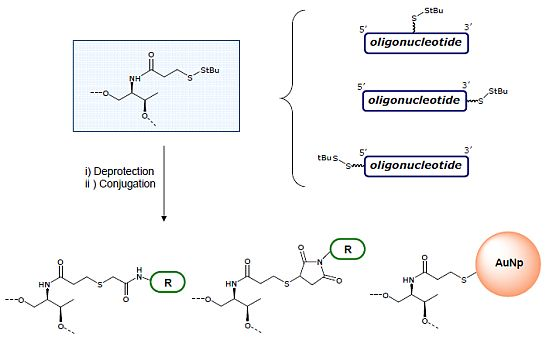
3.4. Removal of StBu Group and Conjugation of the Resulting Thiol Oligonucleotides
Oligonucleotides
7 and
8 (1–3 OD
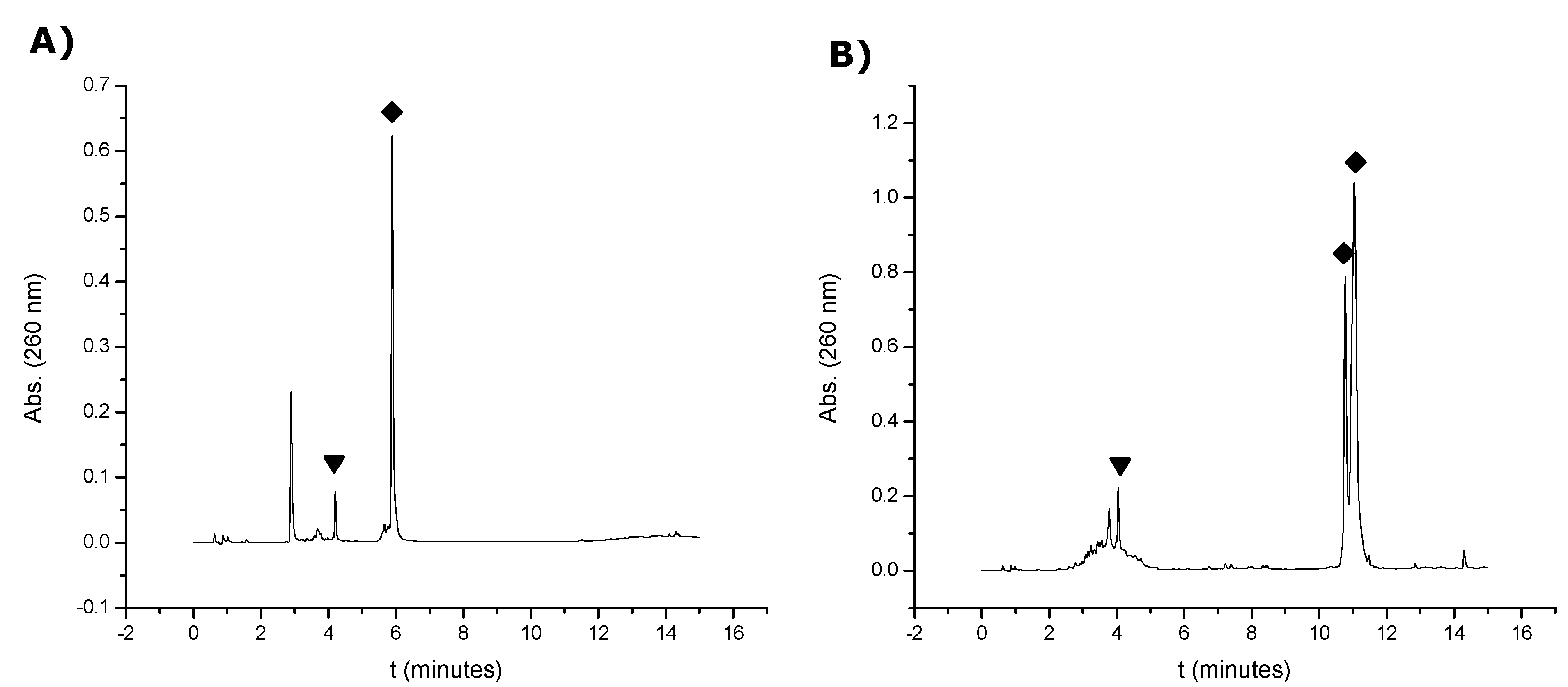
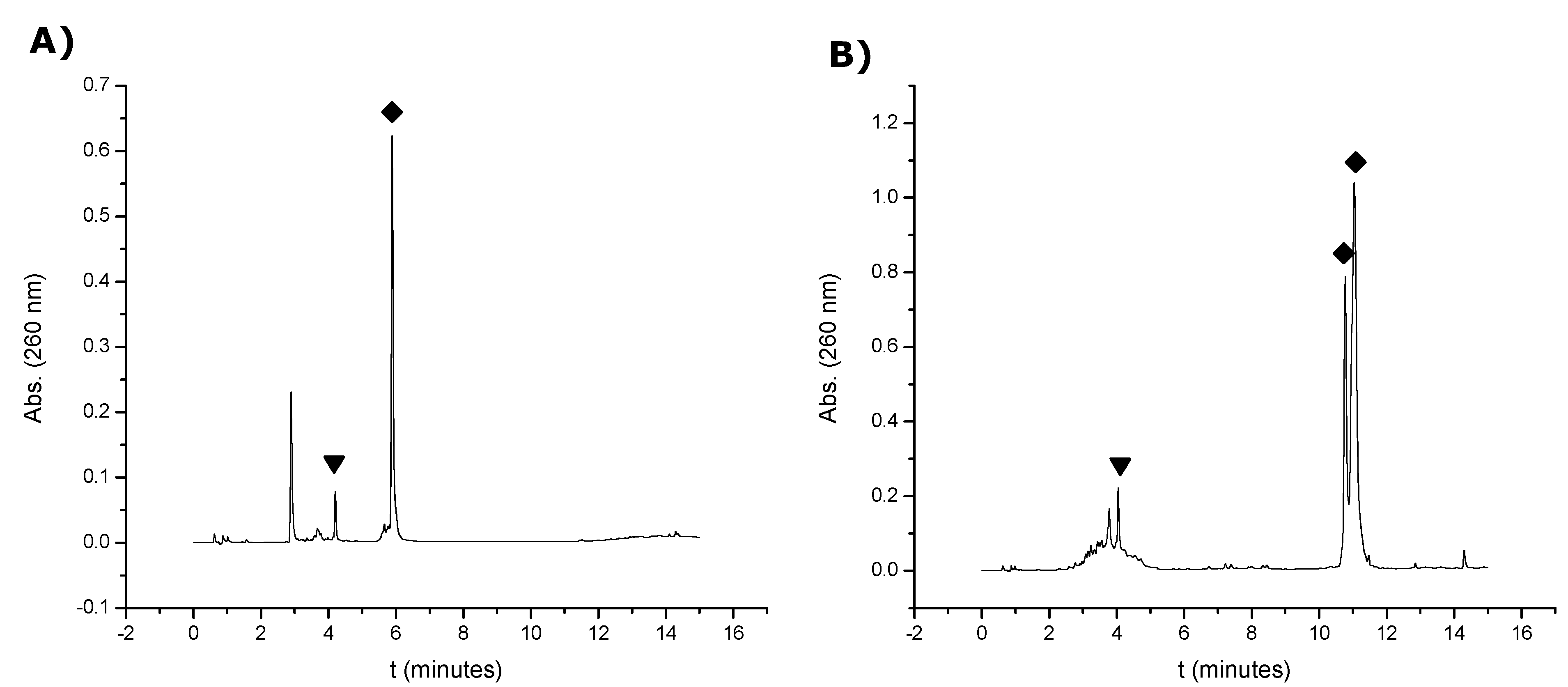
260) were dissolved in 300 μL of an aqueous solution 0.1 M TEAA (pH = 7). Then, 34 μL of an aqueous solution of 0.1 M TCEP were added to the solution and allowed to react at 55 °C. Different samples were analyzed by HPLC at different times of reaction (1 h, 3 h and 4 h) (
Figure S4, Supplementary Information). Column: X-Bridge
TM OST C
18 (2.5 μm 4.6 × 50 mm). Solvent A: 100 mM TEAA (pH = 7) and solvent B: 70% ACN in 100 mM TEAA (pH = 7). Flow rate: 1 mL/min. Conditions: 10 min linear gradient from 0–30% B, then 5 min linear gradient from 30–100% B. The oligonucleotide carrying a free thiol
15 was collected and analyzed by mass spectrometry (MALDI-TOF). The resulting thiol oligonucleotides
15 and
18 were purified with Sephadex G-10 (NAP-5 column). The oligonucleotide was eluted with 1 mL of sterile water.
Conjugation with 2-bromo-2'-hydroxy-5'-nitroacetanilide: The solution from the NAP column was concentrated to 500 μL and 60 μL of an aqueous solution 1 M of hydrogen carbonate (pH = 9) were added. Then, 100 μL of a solution 2.25 mM of 2-bromo-2'-hydroxy-5'-nitroacetanilide in DMF were added and allowed to react at room temperature overnight. The reaction mixtures were desalted on a NAP-5 column. The DNA fractions were analyzed by HPLC. The main compounds were isolated and analyzed by mass spectrometry. Results are summarized in
Table 1.
Conjugation with
N-(1-pyrenyl)maleimide: The solution was concentrated to 500 μL and 60 μL of an aqueous solution 1 M TEAA (pH = 7) were added. Then, 100 μL of a solution 2.25 mM of
N-(1-pyrenyl)maleimide in DMF were added and allowed to react at room temperature overnight. The reaction mixture was desalted on a NAP-5 column. The DNA fractions were purified by HPLC. The main compounds were isolated and analyzed by mass spectrometry and UV/Vis spectroscopy (λ
max = 256 and 342 nm). Results are summarized in
Table 1.
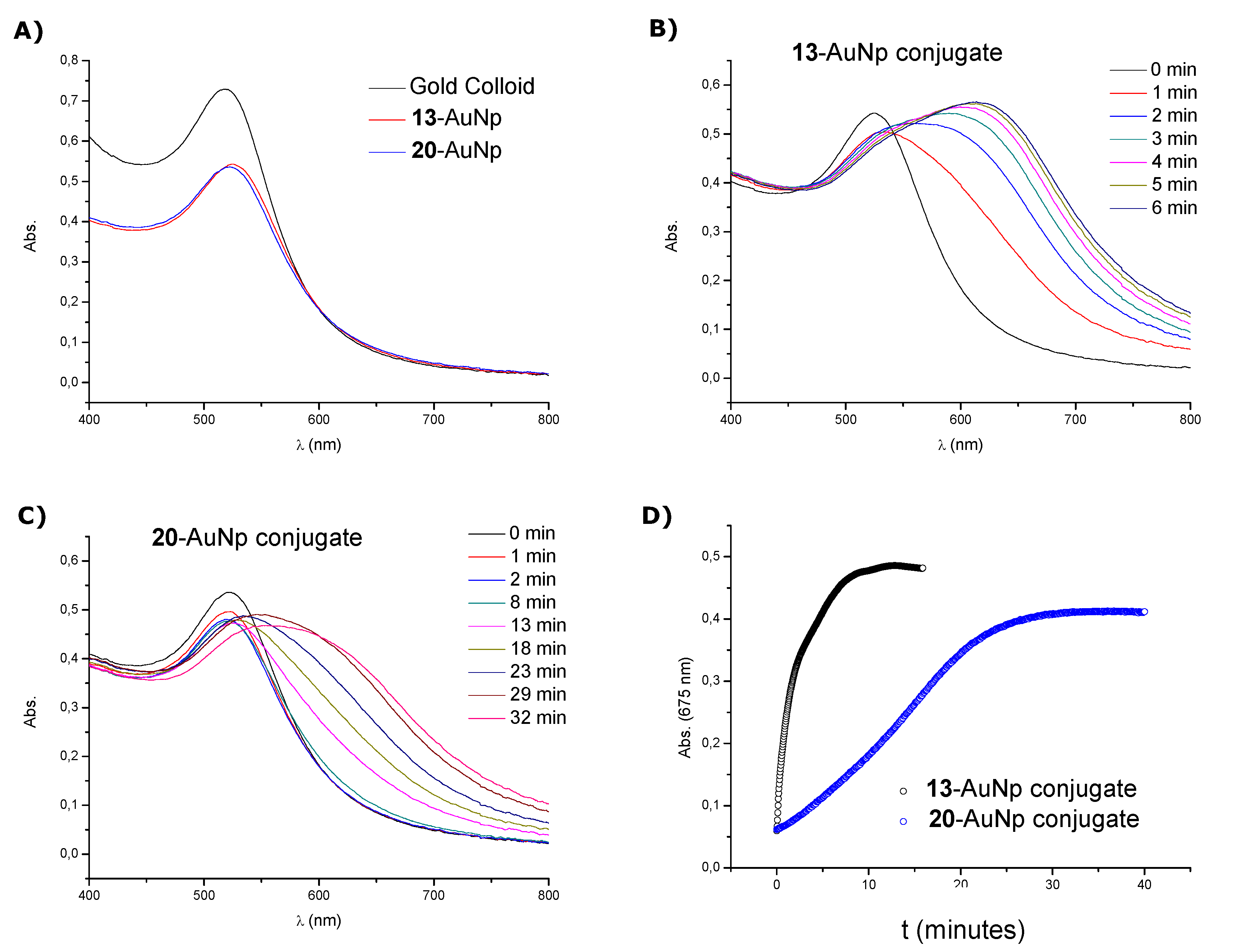
3.5. Preparation of Oligonucleotide-Gold Nanoparticle Conjugates
Citrate stabilized gold nanoparticles (9.7 nm) were purchased from BBI Life Sciences and used as received. To prepare the conjugates, 5 nmol (1 OD260 unit) of disulfide modified-oligonucleotide (13 and 14) were added to 1 mL of the gold nanoparticle solution (9.4 nM). Disulphide oligonucleotides 13 and 14 were converted to their corresponding thiol oligonucleotides (20 and 21 respectively) by reaction with TCEP and as well were used to functionalize gold nanoparticles. Then, 20 nmol (4 OD260) of each oligonucleotide were deprotected following the conditions described above. The resulting thiol oligonucleotides were desalted on a NAP-5 column. Freshly deprotected oligonucleotides (5 nmol, 250 μL) were added to a 1 mL of gold colloid. The mixture was shaken for 16–20 h prior to salt stabilization. Then the solution was brought to a final concentration of 10 mM sodium phosphate (pH = 7.2). The mixture was then allowed to equilibrate for 30 min before bringing the concentration to 0.15 M NaCl over a 7 h 30 min period in a stepwise manner (0.05 M NaCl increments each 2 h 30 min). The solutions were sonicated for 20 s. before each addition to keep the particles dispersed during the salting procedure. The salting process was followed by incubation overnight at room temperature. Finally, to remove all unbound oligonucleotides, the solution was centrifuged at 13,200 rpm (16,100 × g) for 30 min. The supernatant was removed and the reddish solid at the bottom of the centrifuge tube was dispersed in 1 mL of a 0.15 M, 10 mM sodium phosphate buffer (pH = 7.2). This procedure was repeated three times. Approximately 20–30% of the original nanoparticle concentration may be lost during centrifugation and work-up. As we wanted to compare stabilities of conjugates obtained with disulfide and thiol oligonucleotides (13-AuNp and 20-AuNp) disulfide and thiol oligonucleotides we dispersed the reddish residue in 500 μL of 0.15 M NaCl, 10 mM sodium phosphate (pH = 7.2), 0.1% NaN3 solution. Then, each solution was analyzed by UV-visible absorption spectroscopy and the concentration of gold nanoparticle conjugates was adjusted to 7.0–7.2 nM by adding the required volume of buffer. The conjugates obtained with the fluorescent oligonucleotides (14-AunP and 21-AuNp) were directly resuspended in 1 mL of a 0.15 M NaCl, 10 mM sodium phosphate (pH = 7.2), 0.1% NaN3 solution. The extinction coefficient used was the same as that used for unmodified nanoparticles (ε520 = 7.6 × 107 M−1cm−1, data provided by the manufacturer). Each solution was then stored in the fridge (4 °C) prior to use.
3.6. Stability Tests of Oligonucleotide-Gold Nanoparticle Conjugates
The effect of the disulphide or thiol moieties on the stability of oligonucleotide-nanoparticle conjugates was assessed by treating the conjugates with DTT (10 mM final concentration) at 40 °C. UV-vis spectra were recorded for both thiolated conjugates (13 and 20) at 1 min intervals. To give an indication of the durability of the oligonucleotide conjugates, absorbance at 675 nm was monitored against time. Then, the half-life (time to reach half the value for complete aggregation) was calculated for each conjugate.
3.7. Measuring Oligonucleotide Loading on Gold Nanoparticles
To determine the number of oligonucleotides loaded on each particle, the concentration of nanoparticles and the concentration of fluorescent DNA in each sample were measured. The fluorescent oligonucleotides were displaced from the nanoparticle surface using DTT. Then 200 μL of the gold nanoparticle conjugate solution was treated with 200 μL of a 1 M DTT solution in 0.18 M sodium phosphate buffer, (pH = 8) at room temperature for two hours. The sample was centrifuged to remove the sediment from the supernatant and the fluorescence spectra was recorded (conditions: λEx = 495 nm and λEm = 510–550 nm). For each gold nanoparticle conjugate five different aliquots were prepared. Standard curves were prepared with known concentrations of the fluorescein disulfide oligonucleotide 14 using the same buffer, salt and DTT concentrations were prepared. The fluorescents values when correlated with the standard calibration curve gave a concentration for the fluorescent oligonucleotide in solution. The average number of oligonucleotides per particle was obtained by dividing the measured oligonucleotide molar concentration by the original conjugate gold nanoparticle concentration. Normalized surface coverage values were calculated by dividing by the surface area of the sphere which can be easily converted to pmol/cm2.
3.8. Denaturation Studies
Oligonucleotide duplexes (
Table 2, entries 1–6) were dissolved in 0.3 M NaCl, 10 mM sodium phosphate buffer (pH = 7.0). The melting experiments were performed in duplicate at 3.5 μM concentration of oligonucleotide. The samples were heated at 90 °C for 5 min, allowed to cool slowly to room temperature to induce annealing and then kept overnight in a refrigerator at 4 °C. The melting curves were recorded by heating the samples with a temperature controller from 5 to 75 °C at a constant rate of 1 °C/min and monitoring the absorbance at 260 nm. Denaturation curves for hairpin oligonucleotides
11 and the corresponding unmodified hairpin were performed in duplicate at 2.8 μM oligonucleotide concentration in a 50 mM NaCl, 10 mM sodium phosphate buffer (pH = 7.0) (entries 7 and 8). The melting curves were recorded by heating the samples with a temperature controller from 20 to 85 °C at a constant rate of 1 °C/min and monitoring the absorbance at 260 nm. During the experiment, when the temperature was below 25 °C, argon was flushed to prevent water condensation on the cuvettes. The melting experiments were recorded in 1 cm path-length cells. The T
m values were calculated with the
Meltwin 3.2 software [
38].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}