Synthesis, DNA-Binding and Antiproliferative Properties of Acridine and 5-Methylacridine Derivatives



Abstract
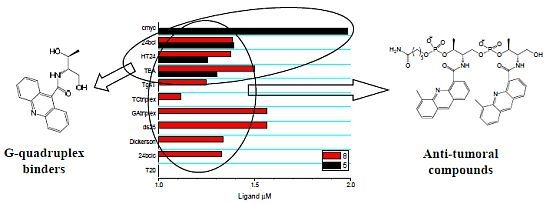
:
1. Introduction
2. Results and Discussion
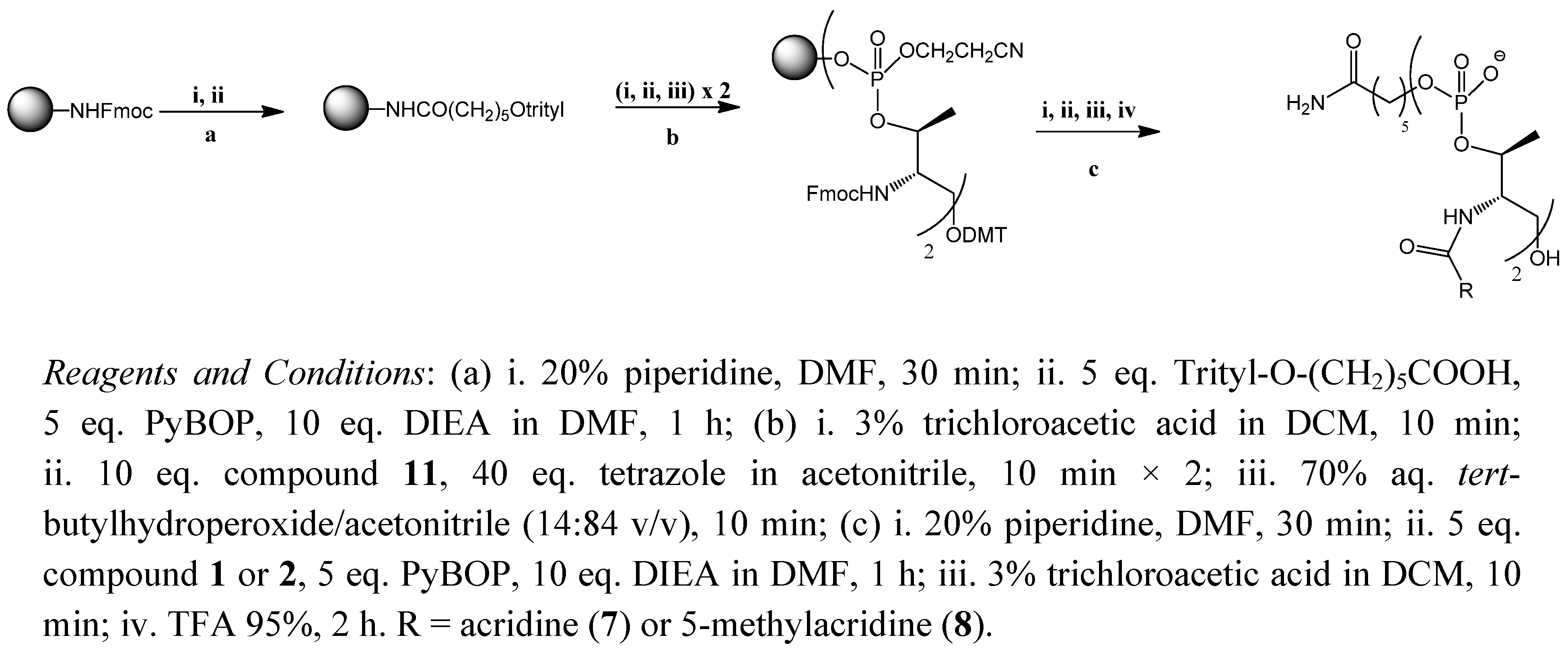
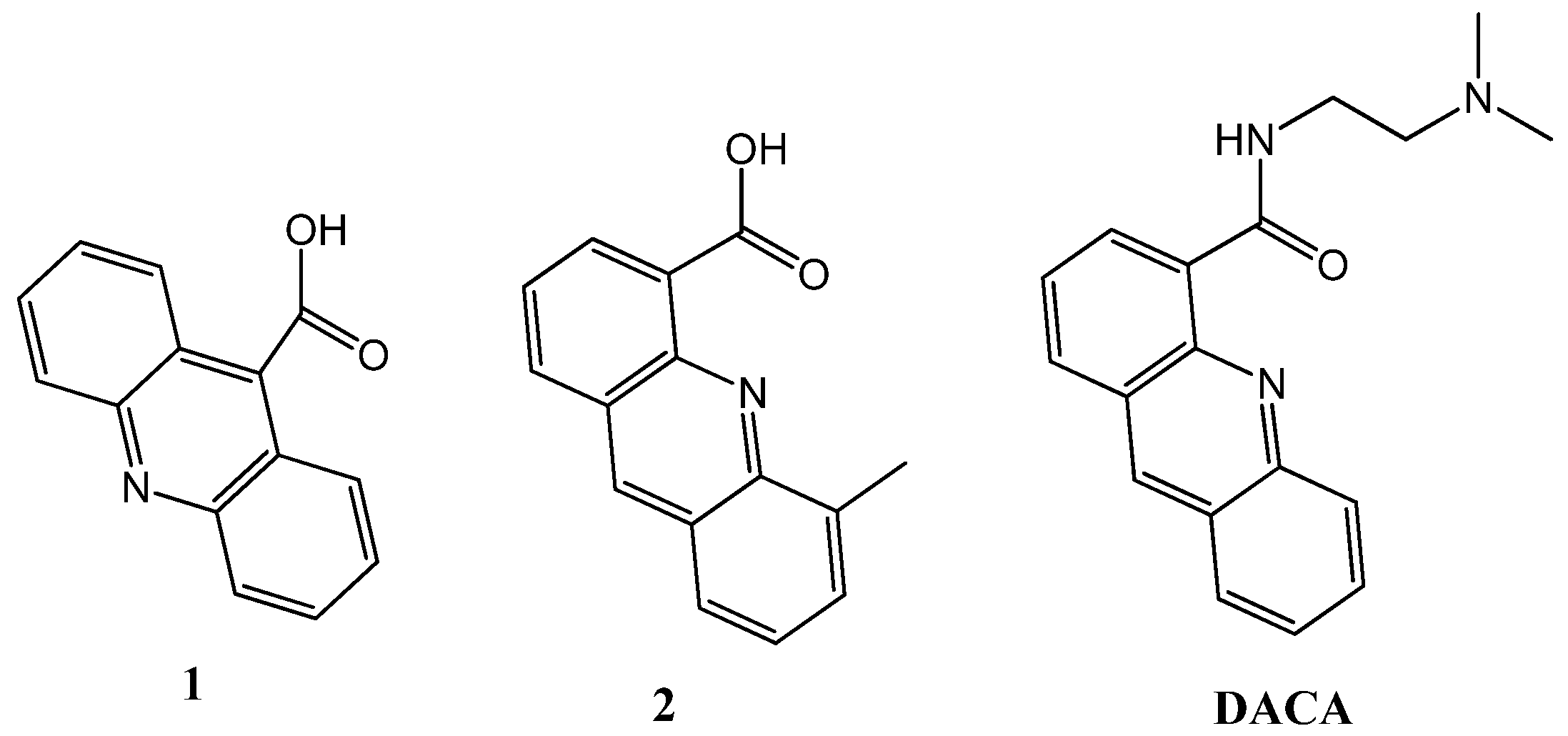
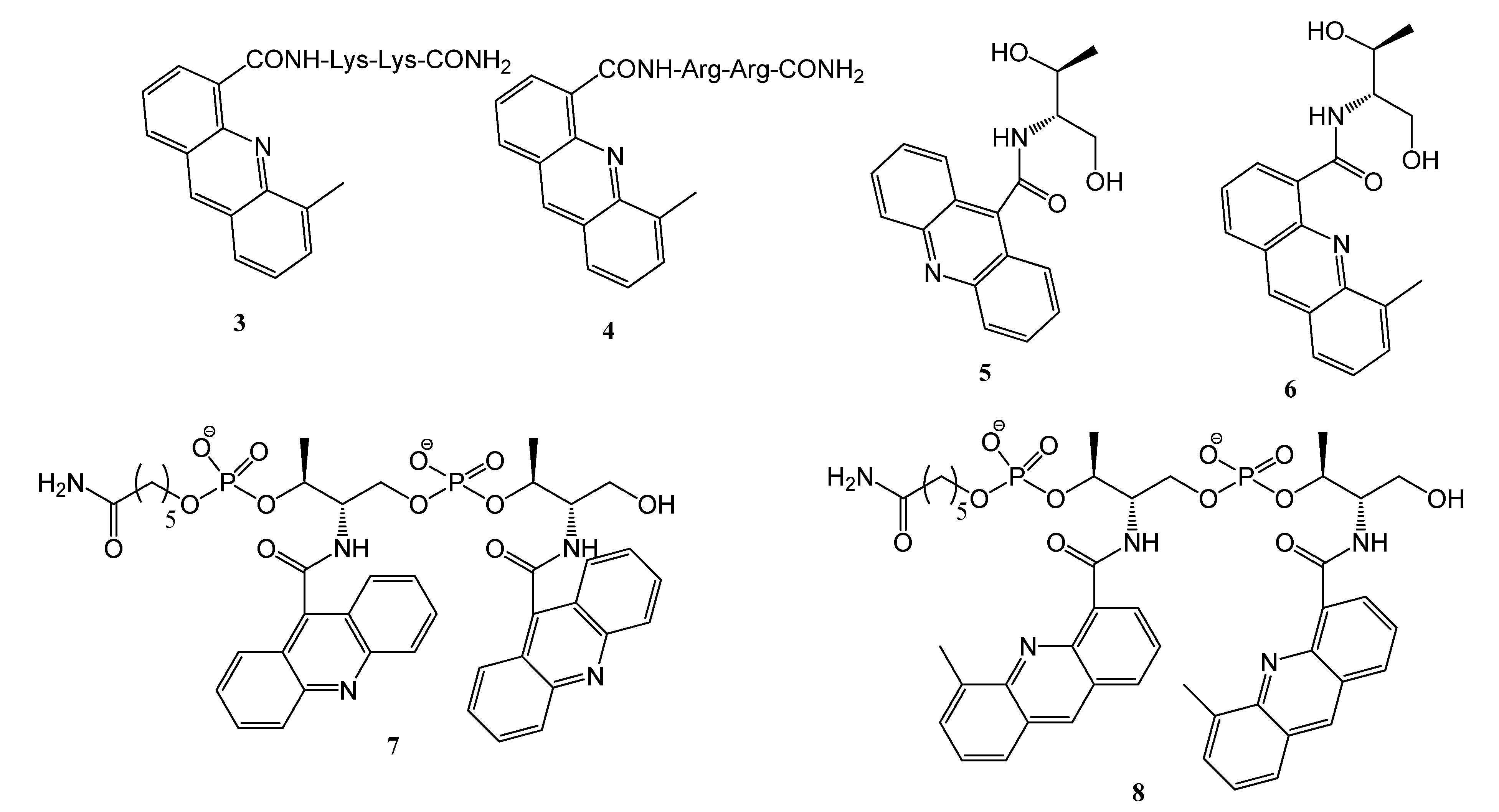
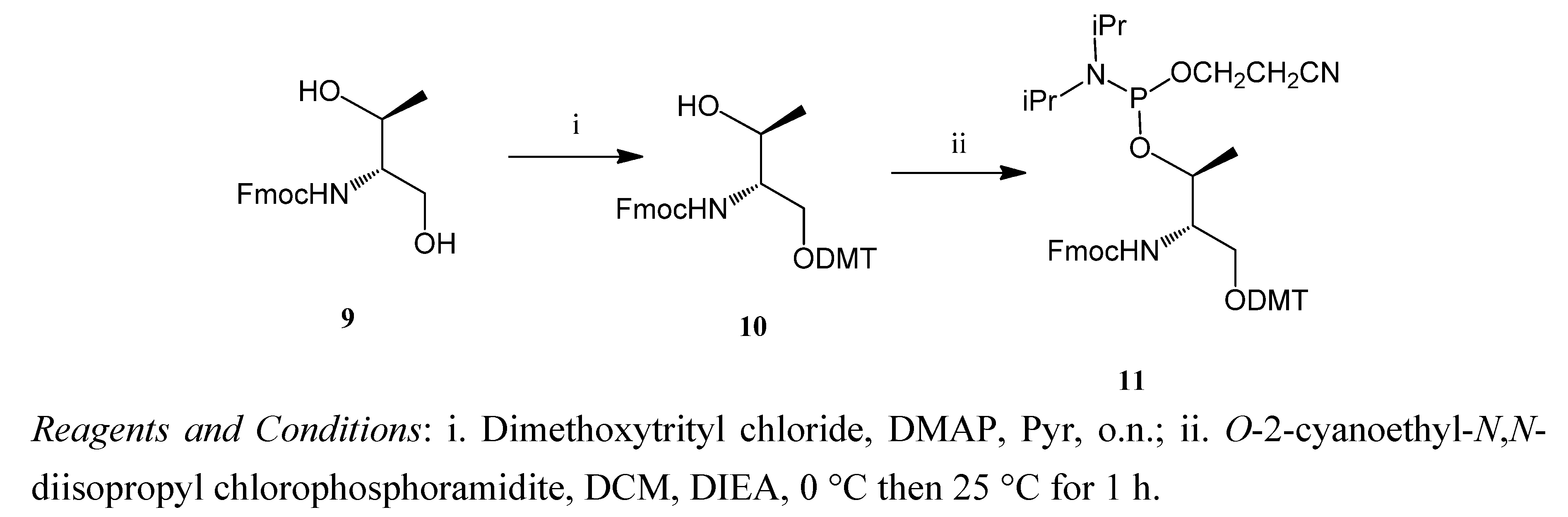
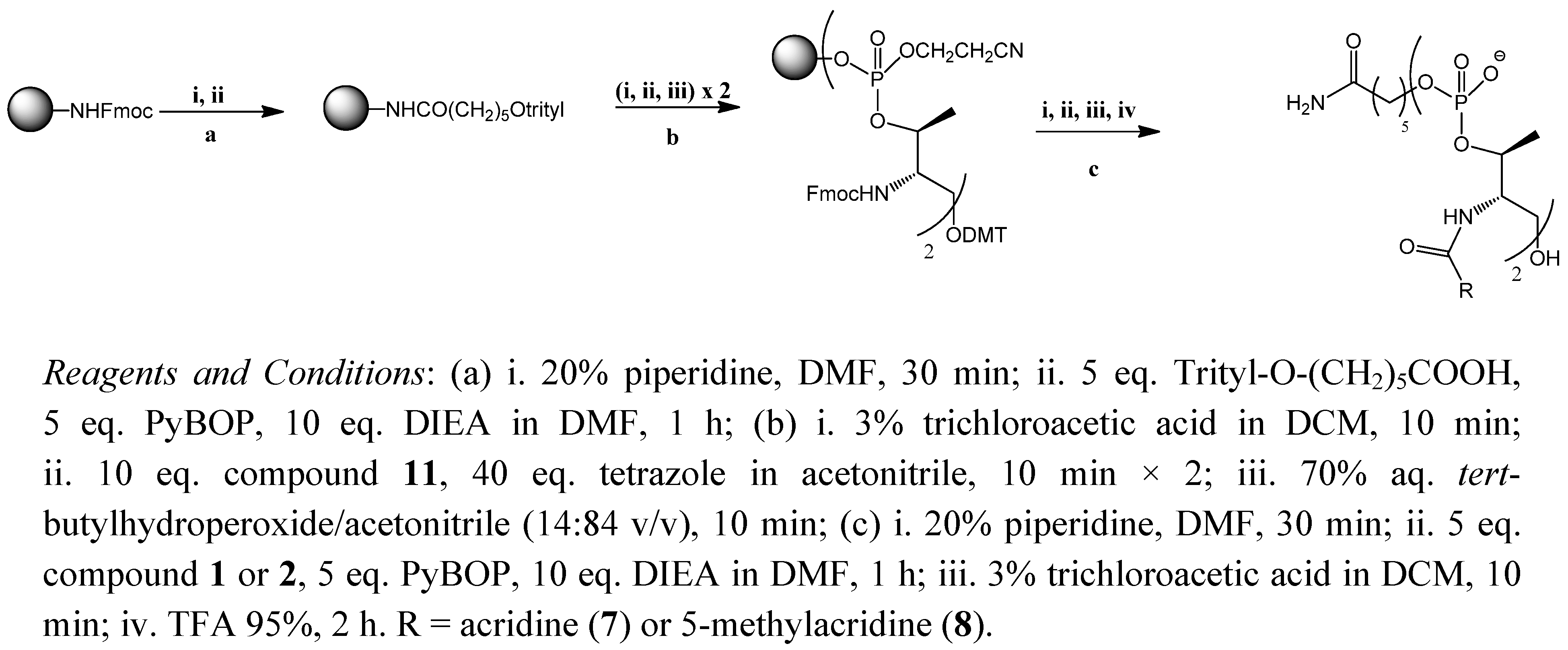
2.1. Synthesis of the Acridine Derivatives
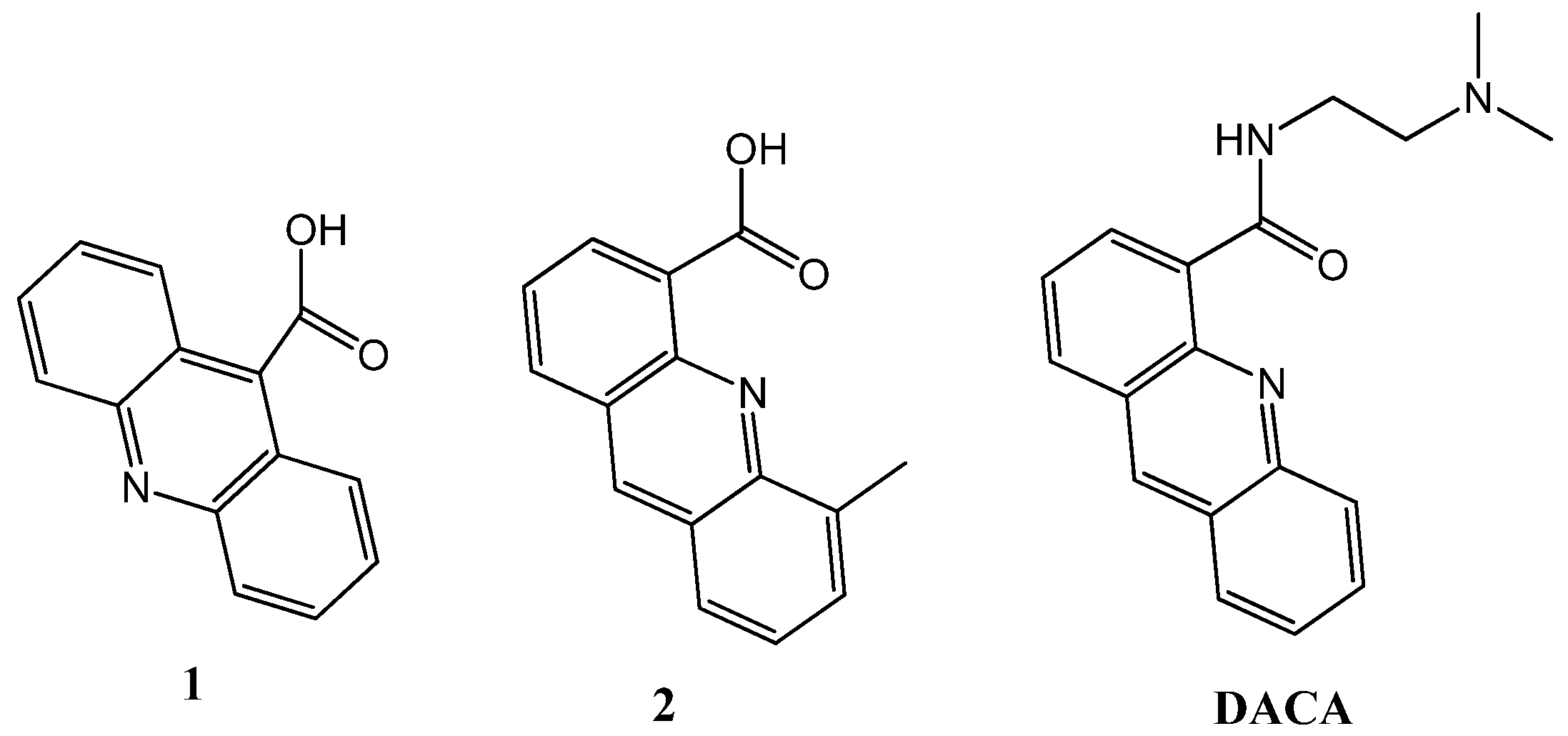
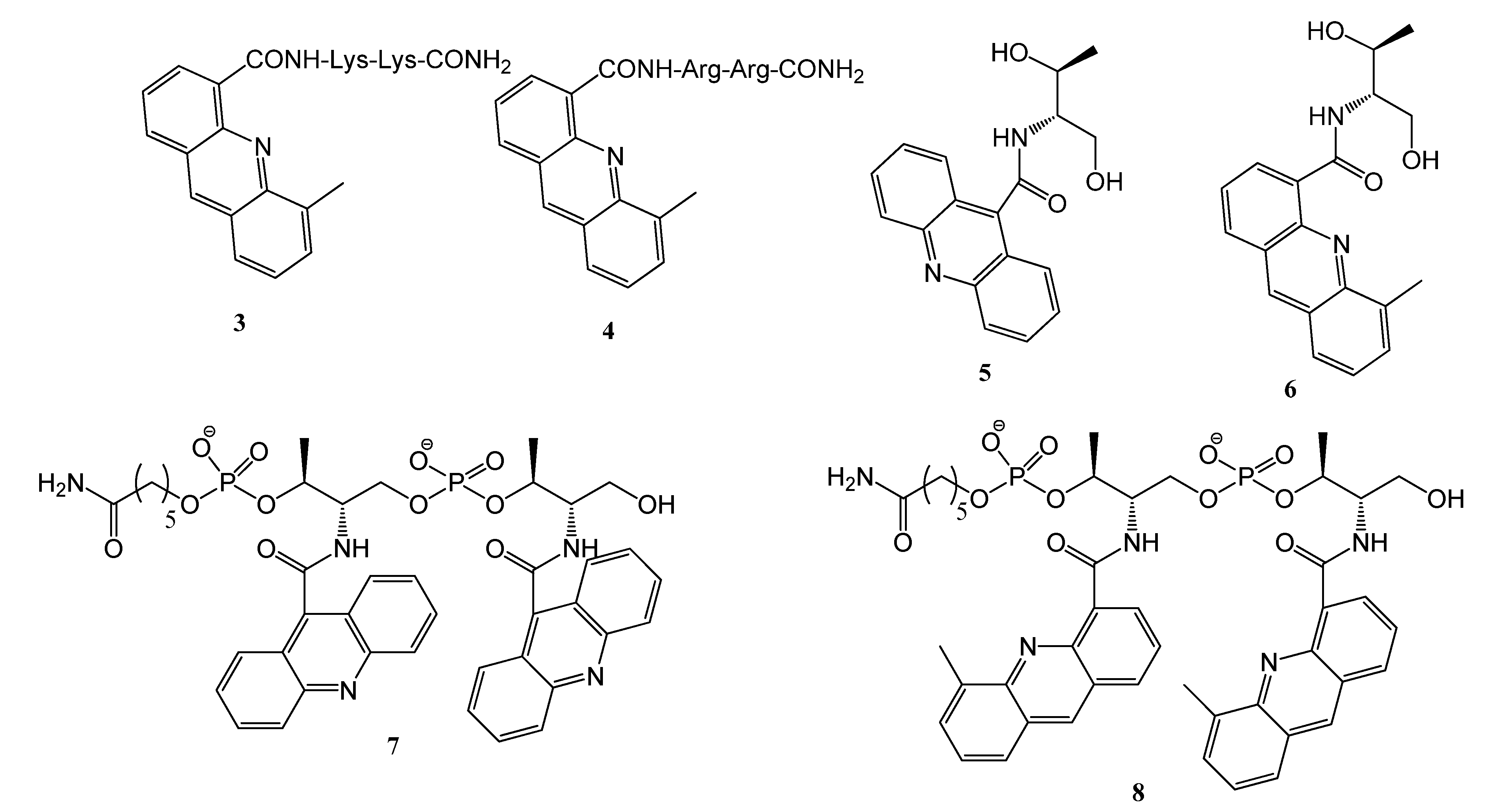
2.2. Cell Viability Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) HBT38 |
|---|---|
| 1 | n.a. |
| 2 | 75 |
| 3 | n.a. |
| 4 | n.a. |
| 5 | n.a. |
| 6 | 60 |
| 7 | n.a. |
| 8 | 25 |
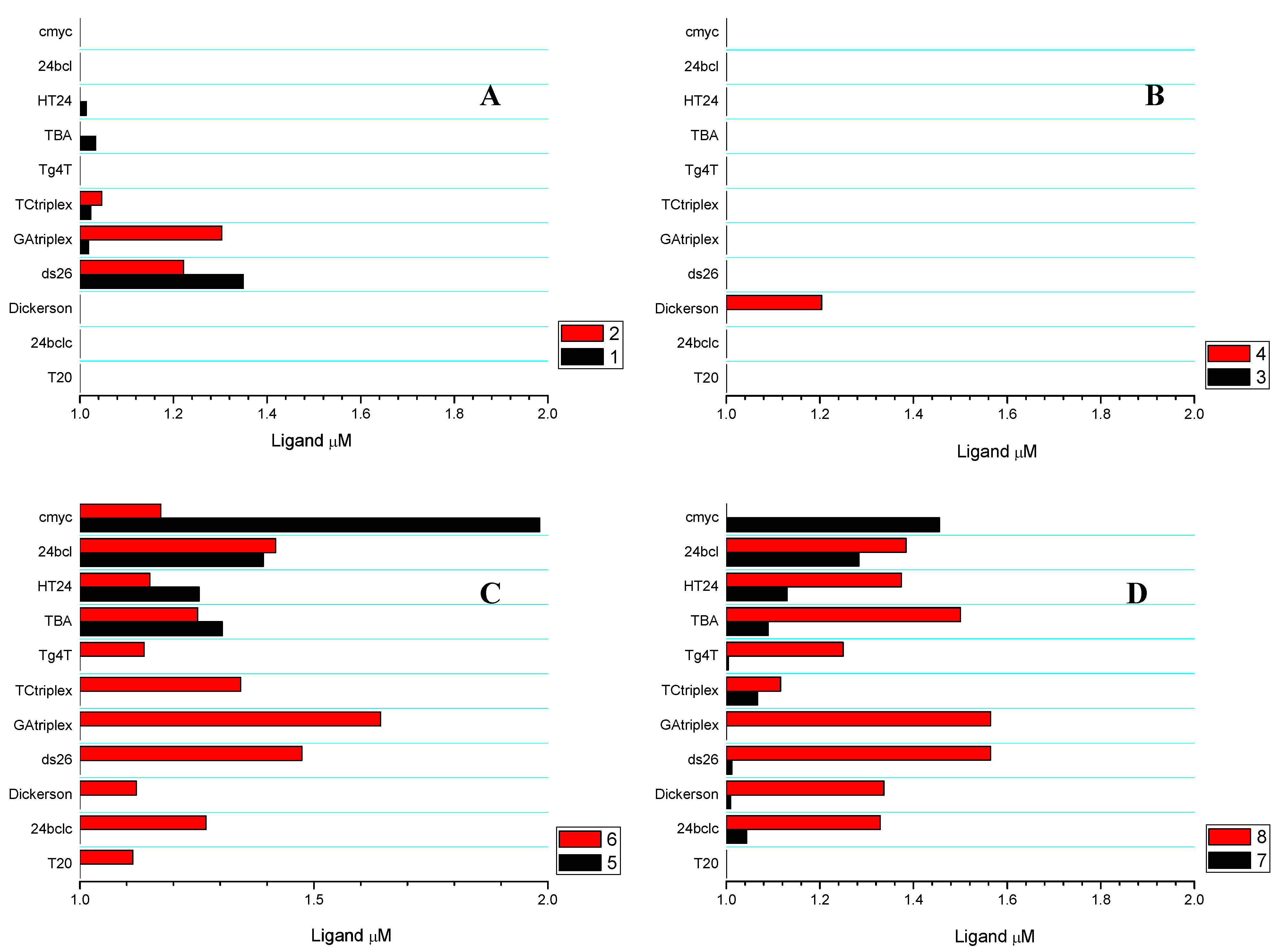
2.3. Competitive Dialysis Studies
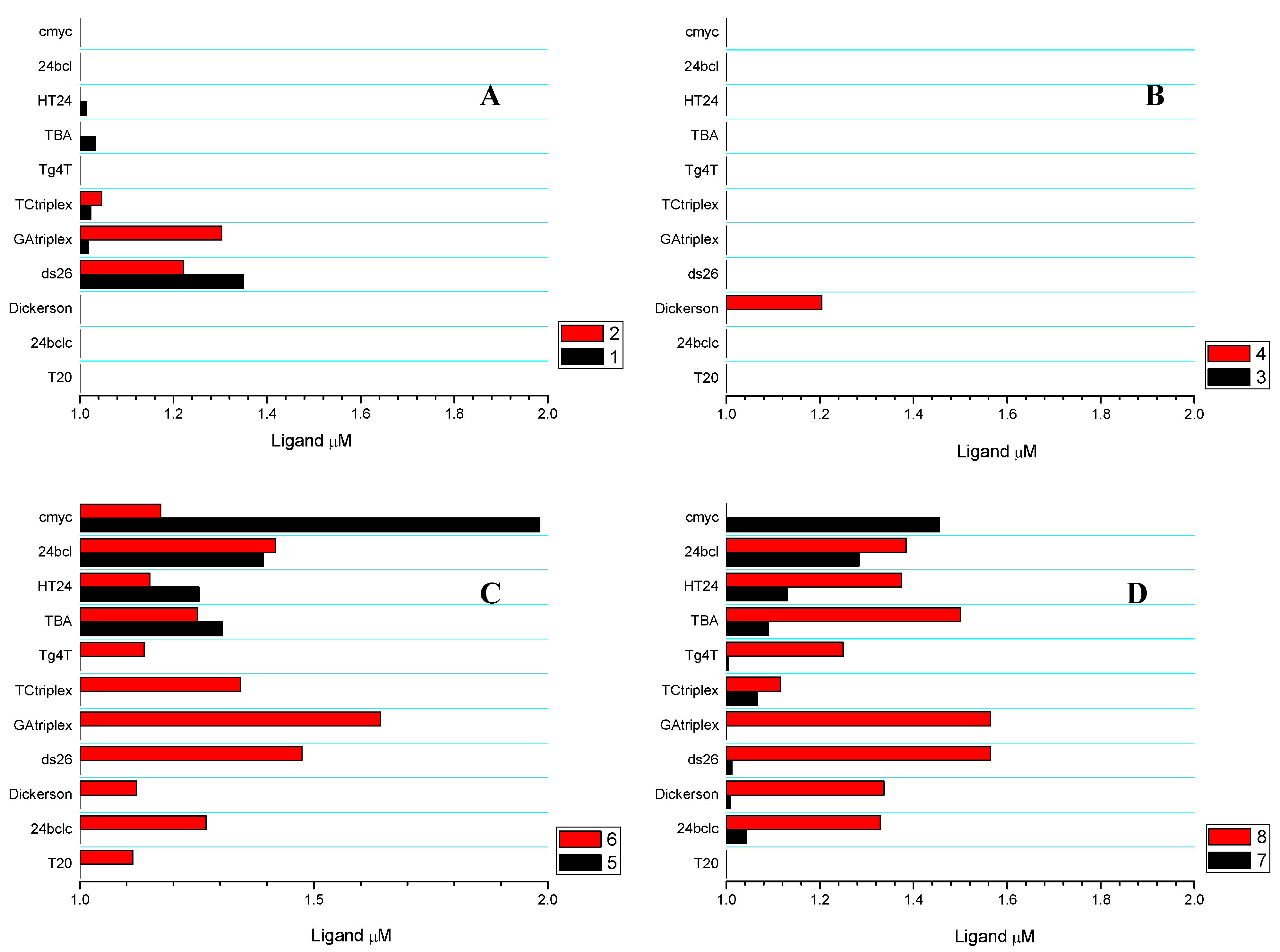
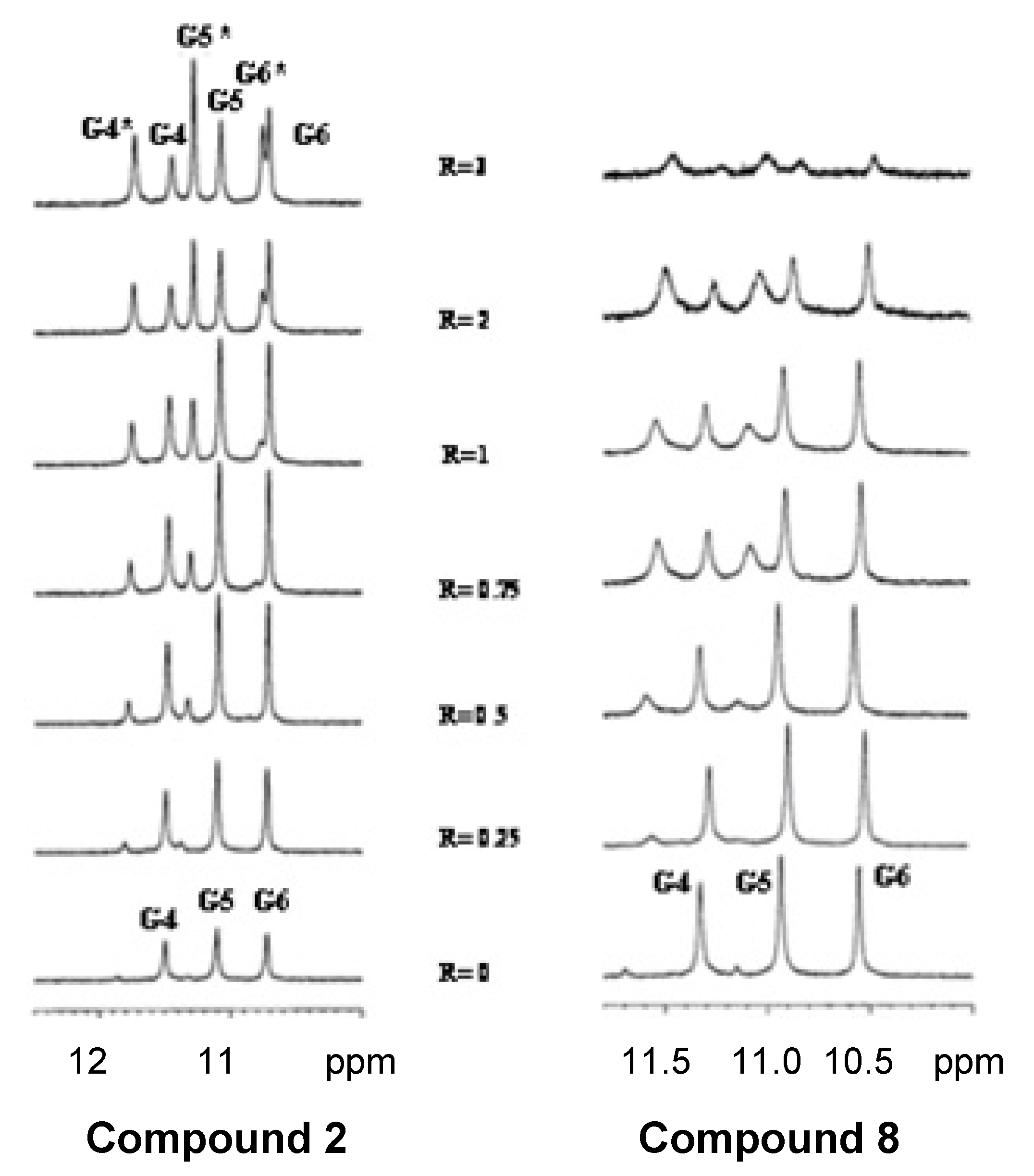
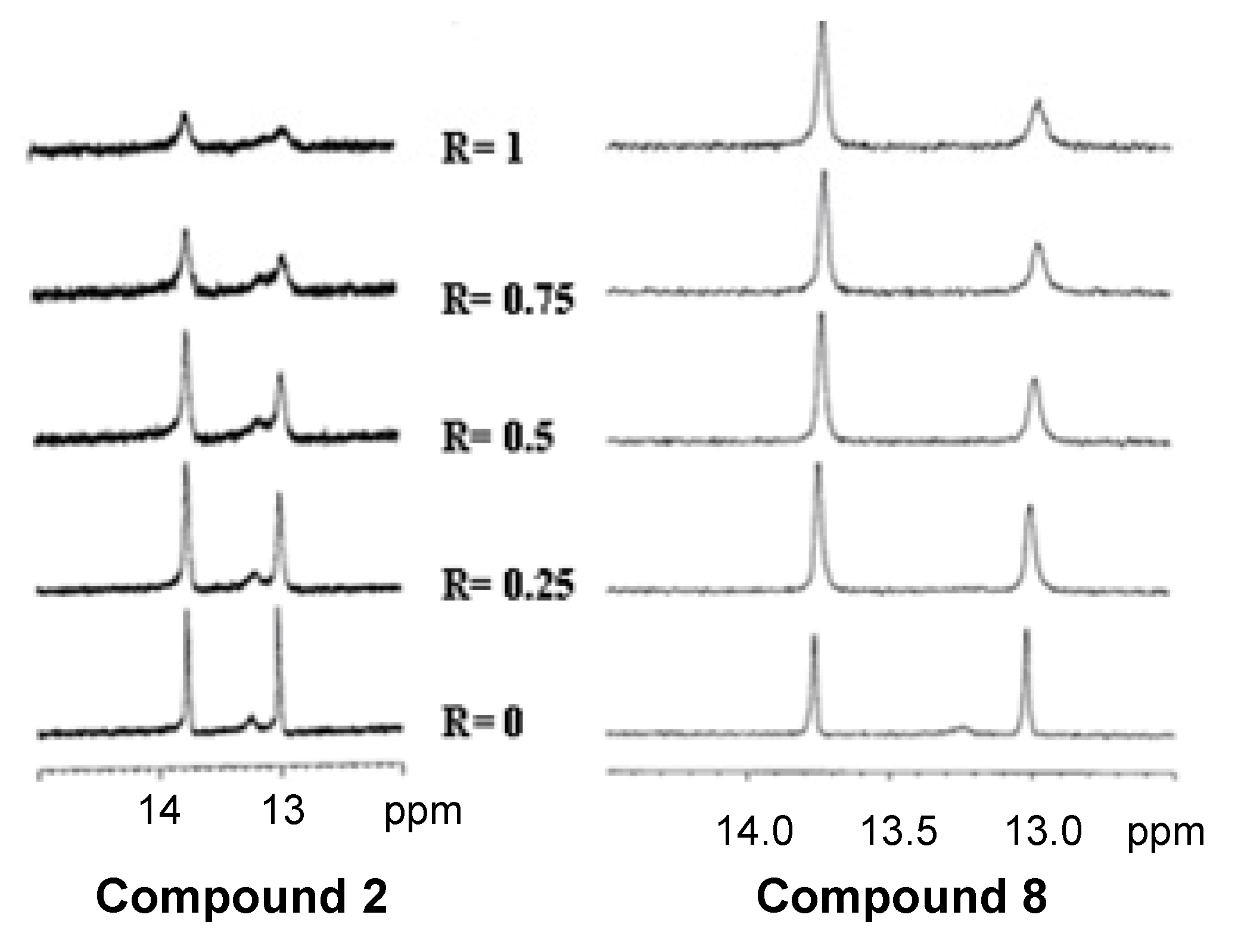
2.4. NMR Spectroscopy
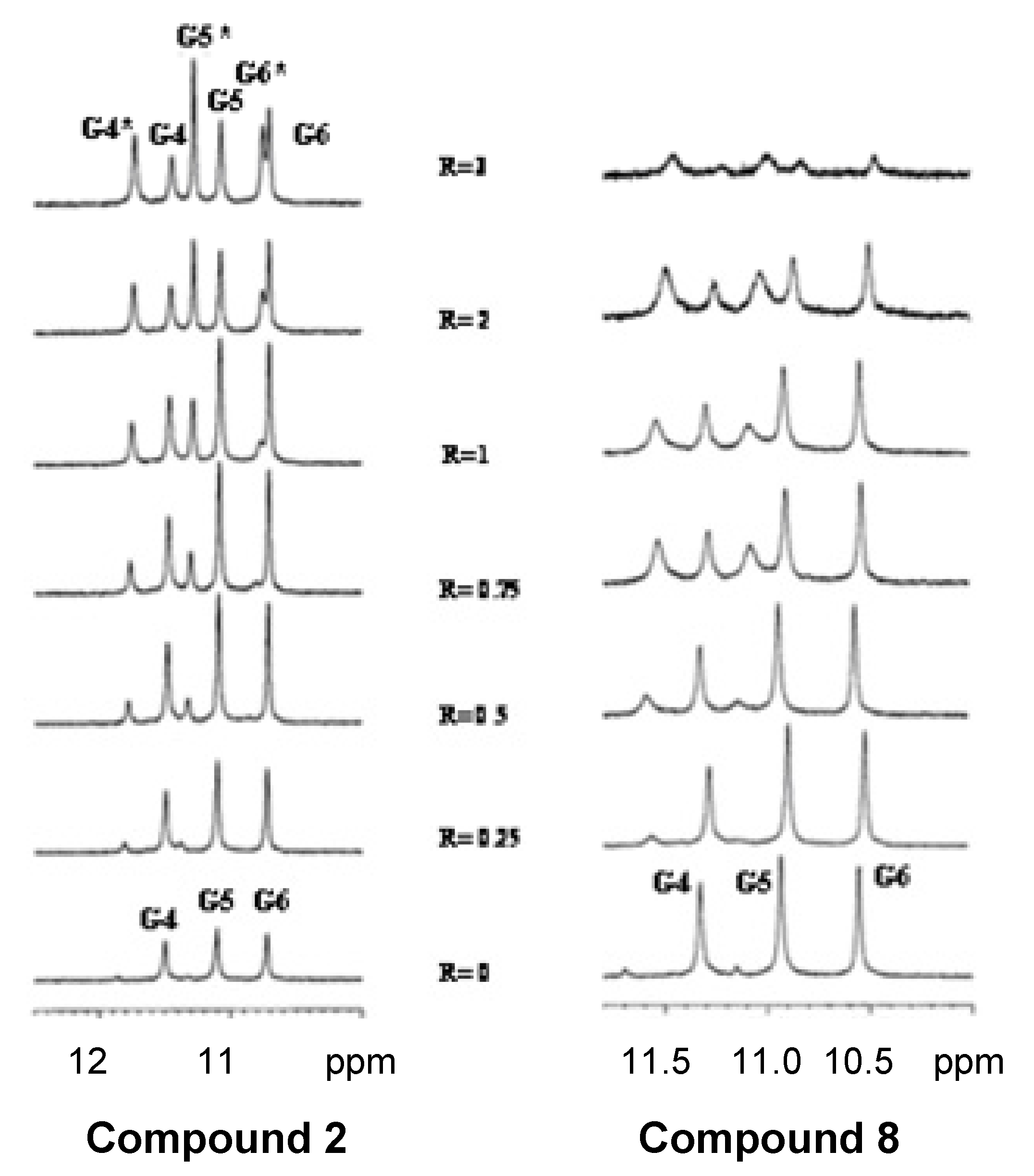
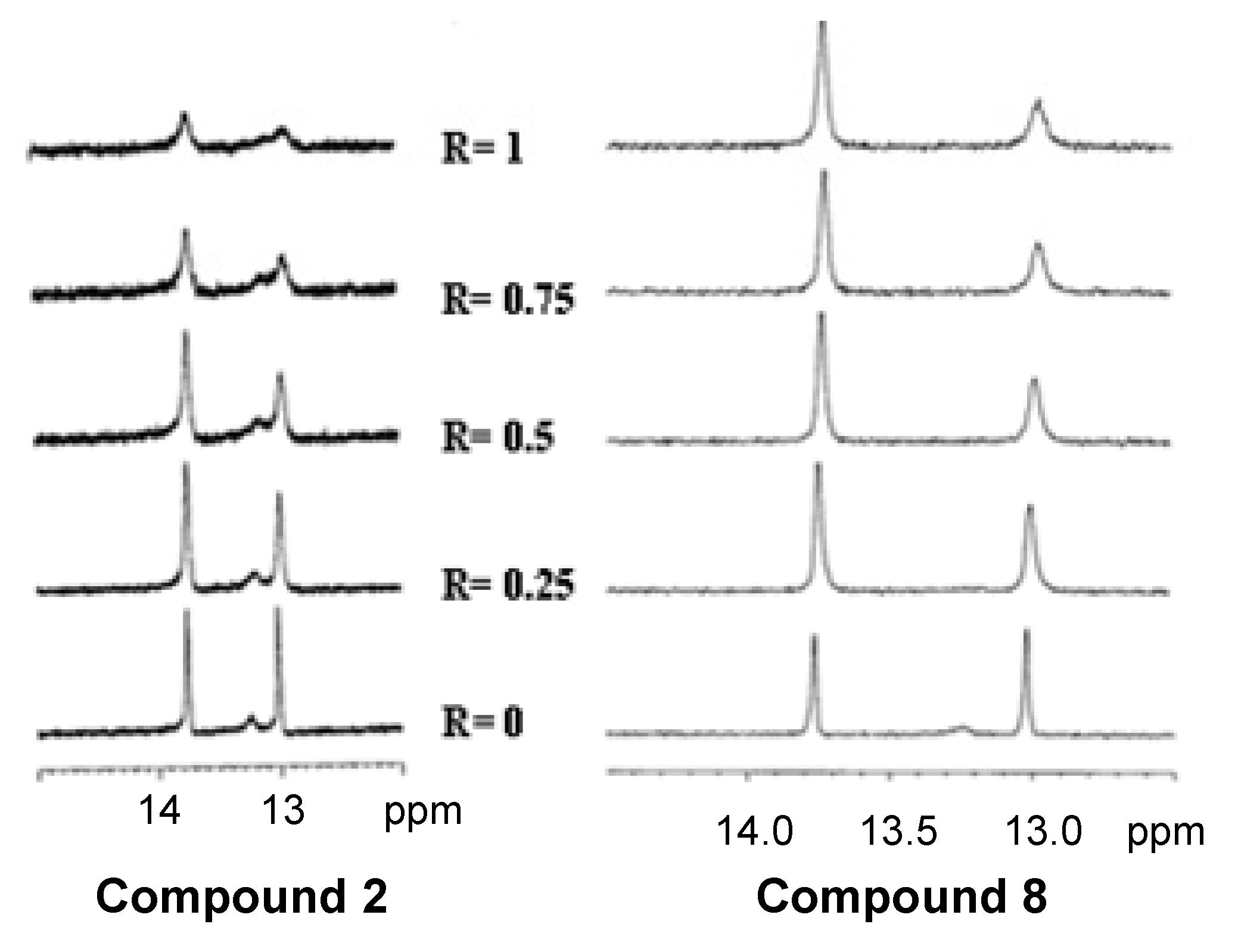
3. Experimental
3.1. Oligonucleotides and General Information
3.2. Solid-Phase Synthesis of Peptide Derivatives 3 and 4
3.3. N-((2S,3S)-1,3-Dihydroxybutan-2-yl)-5-methylacridine-4-carboxamide (6)
3.4. Solid-Phase Synthesis of Acridine Oligomers 7 and 8
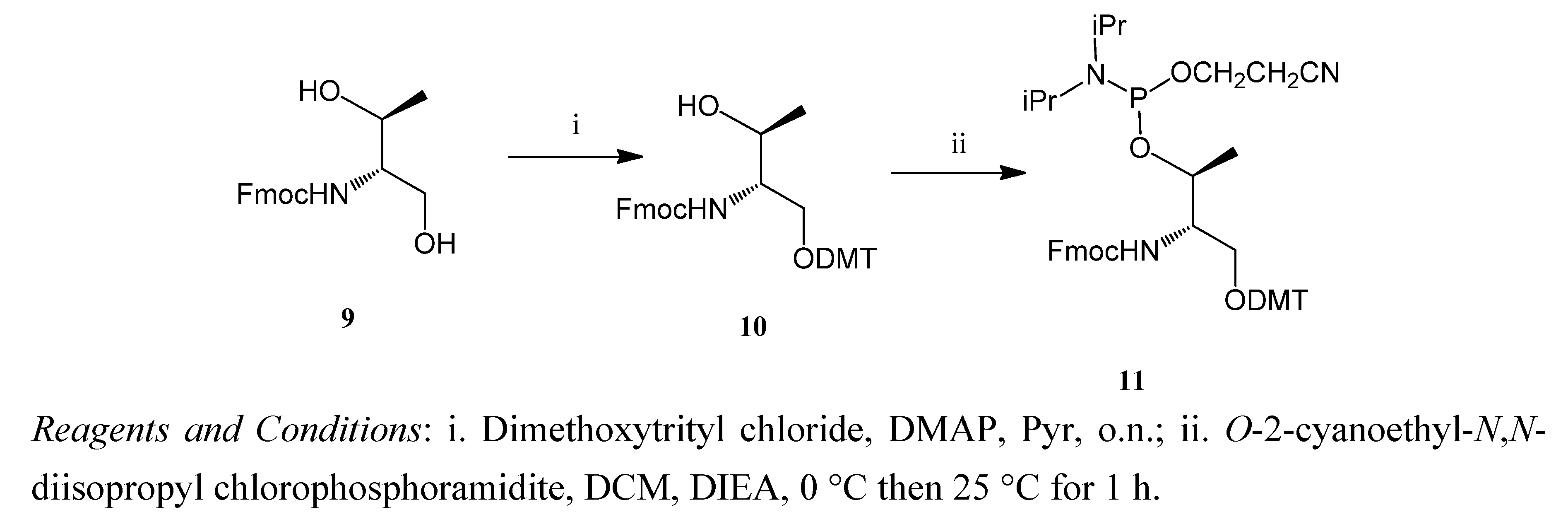
3.5. (9H-Fluoren-9-yl)methyl (2R,3R)-1-(bis(4-methoxyphenyl)(phenyl)methoxy)-3-hydroxybutan-2-ylcarbamate (10)
3.6. (9H-Fluoren-9-yl)methyl (4R,5R)-7-(2-cyanoethoxy)-8-isopropyl-1,1-bis(4-methoxyphenyl)-5,9-dimethyl-1-phenyl-2,6-dioxa-8-aza-7-phosphadecan-4-ylcarbamate (11)
3.7. Competitive Dialysis Assays
3.8. Cell Viability Assays
3.9. NMR Spectroscopy
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds 1, 2, 5 and 6 are available from the authors.
References
- Palchaudhuri, R.; Hergerother, P.J. DNA as a target for anticancer compounds: Methods to determine the mode of binding and the mechanism of action. Curr. Opin. Biotechnol. 2007, 18, 497–503. [Google Scholar] [CrossRef]
- Neto, B.A.; Lapis, A.A. Recent developments in the chemistry of deoxyribonucleic acid (DNA) intercalators: Principles, design, synthesis, applications and trends. Molecules 2009, 14, 1725–1746. [Google Scholar] [CrossRef]
- Neidle, S.; Parkinson, G. Telomere maintenance as a target for anticancer drug discovery. Nat. Rev. Drug Discov. 2002, 1, 383–393. [Google Scholar] [CrossRef]
- Mergny, J.L.; Hélène, C. G-quadruplex DNA: A target for drug design. Nat. Med. 1998, 4, 1366–1367. [Google Scholar]
- Perry, P.J.; Read, M.A.; Davies, R.T.; Gowan, S.M.; Reszka, A.P.; Wood, A.A; Kelland, L.R.; Neidle, S. 2,7-Disubstituted aminofluorenone derivatives as inhibitors of human telomerase. J. Med. Chem. 1999, 42, 2679–2684. [Google Scholar] [CrossRef]
- Harrison, R.J.; Gowan, S.M.; Kelland, L.R.; Neidle, S. Human telomerase inhibition by substituted acridine derivatives. Bioorg. Med. Chem. Lett. 1999, 9, 2463–2468. [Google Scholar]
- Monchaud, D.; Teulade-Fichou, M.P. A hitchhiker’s guide to G-quadruplex ligands. Org. Biomol. Chem. 2008, 6, 627–636. [Google Scholar] [CrossRef]
- Ou, T.M.; Lu, Y.J.; Tan, J.H.; Huang, Z.S.; Wong, K.Y.; Gu, L.Q. G-quadruplexes: Targets in anticancer drug design. ChemMedChem 2008, 3, 690–713. [Google Scholar]
- Campbell, N.H.; Patel, M.; Tofa, A.B.; Ghosh, R.; Parkinson, G.N.; Neidle, S. Selectivity in ligand recognition of G-quadruplex loops. Biochemistry 2009, 48, 1675–1680. [Google Scholar]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol. Ther. 2003, 99, 167–181. [Google Scholar] [CrossRef]
- Corbett, K.D.; Berger, J.M. Structure, molecular mechanism, and evolutionary relatioships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef]
- Topcu, Z. DNA topoisomerases as targets for anticancer drugs. J. Clin. Pharm. Ther. 2001, 26, 405–416. [Google Scholar] [CrossRef]
- Arimondo, P.B.; Hélène, C. Design of new anti-cancer agents based on topoisomerase poisons targeted to specific DNA sequences. Curr. Med. Chem. Anticancer Agents 2001, 1, 219–235. [Google Scholar] [CrossRef]
- Adams, A. Crystal structures of acridine complexed with nucleic acids. Curr. Med. Chem. 2002, 9, 1667–1675. [Google Scholar]
- Arya, D.P.; Willis, B. Reaching into the major groove of B-DNA: Synthesis and nucleic acid binding of a neomycin-Hoechst 33258 conjugate. J. Am. Chem. Soc. 2003, 125, 12398–12399. [Google Scholar] [CrossRef]
- Fechter, E.; Olenyuk, B.; Dervan, P.B. Design of a sequence specific DNA bis-intercalator. Angew. Chem. Int. Ed. 2004, 43, 3591–3594. [Google Scholar] [CrossRef]
- Spicer, J.A.; Gamage, S.A.; Finlay, G.J.; Denny, W.A. Synthesis and evaluation of unsymmetrical bis(arylcarboxamides) designed as topoisomerase-targeted anticancer drugs. Bioorg. Med. Chem. 2002, 10, 19–29. [Google Scholar] [CrossRef]
- Gamage, S.A.; Spiecer, J.A.; Atwell, G.J.; Finlay, G.J.; Baguley, B.C.; Denny, W.A. Structure-activity relationships for substituted bis(acridine-4-carboxamides): A new class of anticancer agents. J. Med. Chem. 1999, 42, 2383–2393. [Google Scholar] [CrossRef]
- Mucsi, I.; Molnár, J.; Tanaka, M.; Santelli-Rouvier, C.; Patelis, A.M.; Galy, J.P.; Barbe, J. Effect of acridine derivatives on the multiplication of herpes simplex virus. Anticancer Res. 1998, 18, 3011–3015. [Google Scholar]
- Wakelin, L.P.; Adams, A.; Denny, W.A. Kinetic studies of the binding of acridine carboxamide topoisomerase poisons to DNA: Implications for mode of binding of ligands with uncharged chromophores. J. Med. Chem. 2002, 45, 894–901. [Google Scholar] [CrossRef]
- Atwell, G.J.; Rewcastle, G.W.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 50. In vivo solid-tumor activity of derivatives of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide. J. Med. Chem. 1987, 30, 664–669. [Google Scholar] [CrossRef]
- Atwell, G.J.; Cain, B.F.; Baguley, B.C.; Finlay, G.J.; Denny, W.A. Potential antitumor agents. 43. Synthesis and biological activity of dibasic 9-aminoacridine-4-carboxamides, a new class of antitumor agents. J. Med. Chem. 1984, 24, 1481–1485. [Google Scholar]
- Aviñó, A.; Navarro, I.; Farrera-Sinfreu, J.; Royo, M.; Aymamí, J.; Delgado, A.; Llebaria, A.; Albericio, F.; Eritja, R. Solid-phase synthesis of oligomers carrying several chromophore units linked by phosphodiester backbones. Bioorg. Med. Chem. Lett. 2008, 18, 2306–2310. [Google Scholar]
- Ferrera-Sinfreu, J.; Aviñó, A.; Navarro, I.; Aymamí, J.; Beteta, N.G.; Varón, S.; Pérez-Tomás, R.; Castillo-Avila, W.; Eritja, R.; Albericio, F.; Royo, M. Design, synthesis and antiproliferative properties of oligomers with chromophore units linked by amide backbones. Bioorg. Med. Chem. Lett. 2008, 18, 2440–2444. [Google Scholar]
- Ferreira, R.; Aviñó, A.; Pérez-Tomás, R.; Gargallo, R.; Eritja, R. Synthesis and G-quadruplex binding properties of defined acridine oligomers. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef]
- Ferreira, R.; Artali, R.; Ferrera-Sinfreu, J.; Albericio, F.; Royo, M.; Eritja, R.; Mazzini, S. Acridine and quindoline oligomers linked through a 4-aminoproline backbone prefer G-quadruplex structures. Biochim. Biophys. Acta 2011, 1810, 769–776. [Google Scholar]
- Asanuma, H.; Shirasuka, K.; Takarada, T.; Kashida, H.; Komiyama, M. DNA-dye conjugates for controllable H* aggregation. J. Am. Chem. Soc. 2003, 125, 2217–2223. [Google Scholar]
- Ocampo, S.; Albericio, F.; Fernández, I.; Vilaseca, M.; Eritja, R. A straightforward synthesis of 5’-peptide oligonucleotide conjugates using Nα-Fmoc-protected amino acids. Org. Lett. 2005, 7, 4349–4352. [Google Scholar] [CrossRef]
- Gros, J.; Rosu, F.; Amrane, S.; De Cian, A.; Gabelica, V.; Lacroix, L.; Mergny, J.L. Guanines are a quartet’s best friends: Impact of base substitutions on the kinetics and stability of tetramolecular quadruplexes. Nucleic Acids Res. 2007, 35, 3064–3075. [Google Scholar] [CrossRef]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef]
- Wang, Y.; Patel, D.J. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure 1993, 1, 263–282. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bears, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-myc transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar]
- Dai, J.; Dexheimer, T.S.; Chen, D.; Carver, M.; Ambrus, A.; Jones, R.A.; Yang, D. An intermolecular G-quadruplex structure with mixed parallel/antiparallel G-strands formed in the BCL-2 promoter region in solution. J. Am. Chem. Soc. 2006, 128, 1096–1098. [Google Scholar]
- Ren, J.; Chaires, J.B. Sequence and structural selectivity of nucleic acid binding ligands. Biochemistry 1999, 38, 16067–16075. [Google Scholar] [CrossRef]
- Ragazzon, P.; Chaires, J.B. Use of competition dialysis in the discovery of G-quadruplex selective ligands. Methods 2007, 43, 313–323. [Google Scholar] [CrossRef]
- De Cian, A.; Guittat, L.; Kaiser, M.; Saccà, B.; Amrane, S.; Bourdoncle, A.; Alberti, P.; Teulade-Fichou, M.P.; Lacroix, L.; Mergny, J.L. Fluorescence-based melting aasyas for studying quadruplex ligands. Methods 2007, 42, 183–195. [Google Scholar] [CrossRef]
- Tran, P.L.; Largy, E.; Hamon, F.; Teulade-Fichou, M.P.; Mergny, J.L. Fluorescence intercalator displacement assay for screening G4 ligands towards a variety of G-quadruplex structures. Biochimie 2011, 93, 1288–1296. [Google Scholar] [CrossRef]
- Guittat, L.; Alberti, P.; Rosu, F.; van Miert, S.; Thetiot, E.; Pieters, L.; Gabelica, V.; De Pauw, E.; Ottaviani, A.; Riou, J.F.; Mergny, J.L. Interactions of cryptolepine and neocryptolepine with unusual DNA structures. Biochimie 2003, 85, 535–547. [Google Scholar] [CrossRef]
- Lu, Y.J.; Ou, T.M.; Tan, J.H.; Hou, J.Q.; Shao, W.Y.; Peng, D.; Sun, N.; Wang, X.D.; Wu, W.B.; Bu, X.Z.; et al. 5-N-Methylated quindoline derivatives as telomeric G-quadruplex stabilizing ligands: Effects of 5-N positive charge on quadruplex binding affinity and cell proliferation. J. Med. Chem. 2008, 51, 6381–6392. [Google Scholar] [CrossRef]
- Spicer, A.; Gamage, S.A.; Atwell, G.J.; Finlay, G.J.; Baguley, B.C.; Denny, W.A. Structure-activity relationships for acridine-substituted analogues of the mixed topoisomerase I/II inhibitor N-[2-(dimethylamino)ethyl]acridine-4-carboxamide. J. Med. Chem. 1997, 40, 1919–1929. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ferreira, R.; Aviñó, A.; Mazzini, S.; Eritja, R. Synthesis, DNA-Binding and Antiproliferative Properties of Acridine and 5-Methylacridine Derivatives. Molecules 2012, 17, 7067-7082. https://doi.org/10.3390/molecules17067067
Ferreira R, Aviñó A, Mazzini S, Eritja R. Synthesis, DNA-Binding and Antiproliferative Properties of Acridine and 5-Methylacridine Derivatives. Molecules. 2012; 17(6):7067-7082. https://doi.org/10.3390/molecules17067067
Chicago/Turabian StyleFerreira, Rubén, Anna Aviñó, Stefania Mazzini, and Ramon Eritja. 2012. "Synthesis, DNA-Binding and Antiproliferative Properties of Acridine and 5-Methylacridine Derivatives" Molecules 17, no. 6: 7067-7082. https://doi.org/10.3390/molecules17067067
APA StyleFerreira, R., Aviñó, A., Mazzini, S., & Eritja, R. (2012). Synthesis, DNA-Binding and Antiproliferative Properties of Acridine and 5-Methylacridine Derivatives. Molecules, 17(6), 7067-7082. https://doi.org/10.3390/molecules17067067