Towards Recyclable NAD(P)H Regeneration Catalysts



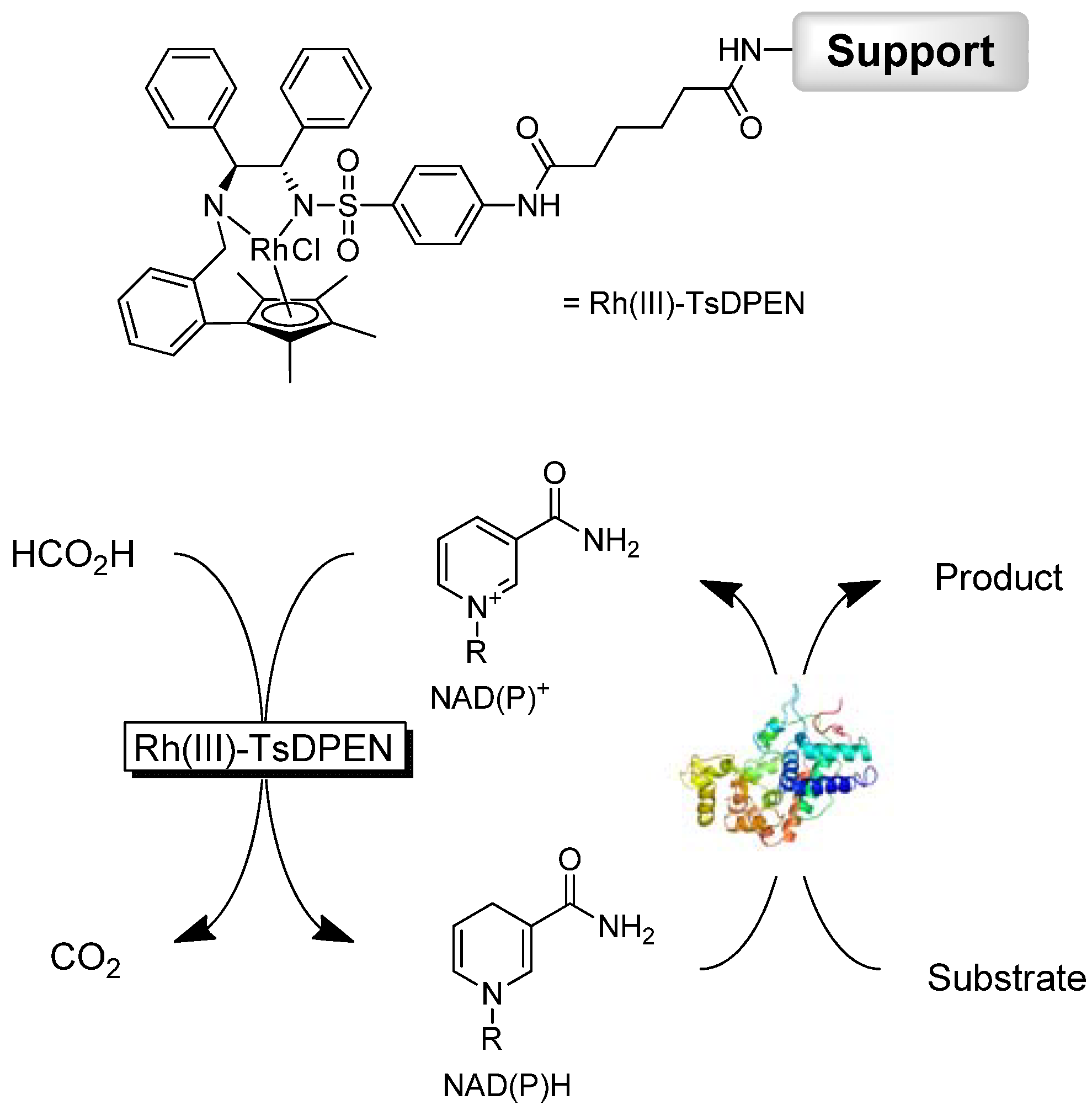
{kind=link}
{kind=link}
{kind=link}
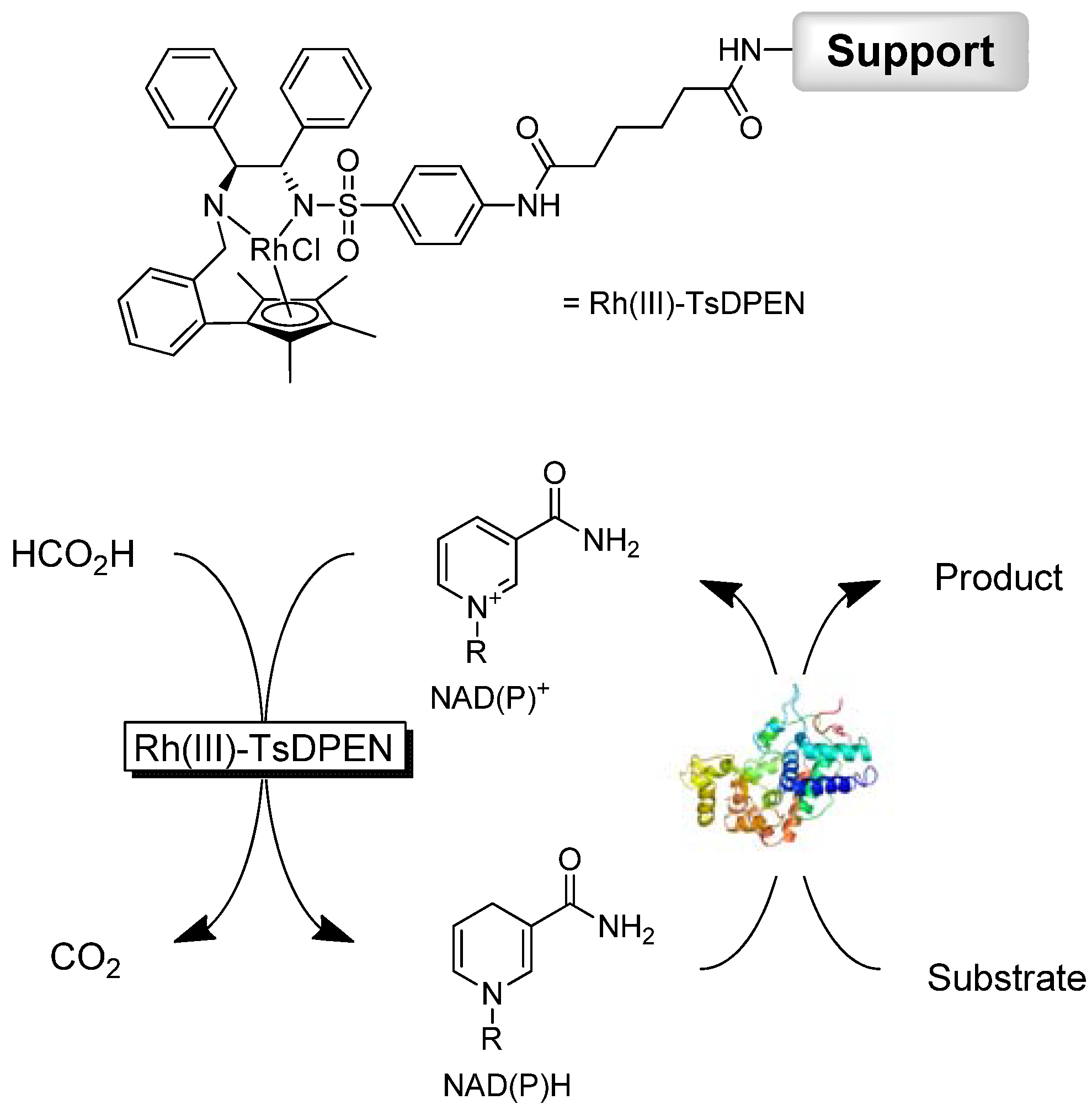{kind=link}
Abstract
:1. Introduction
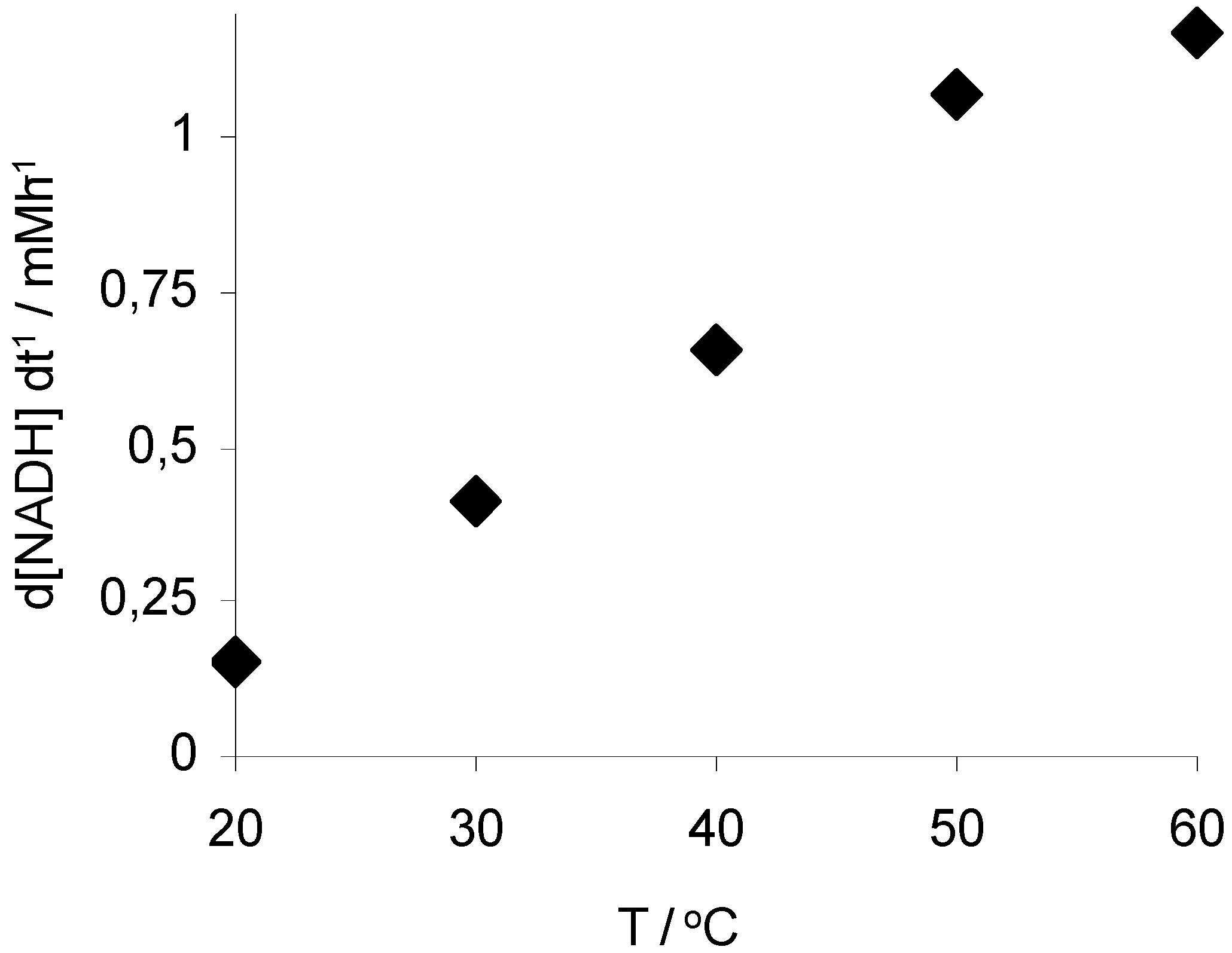
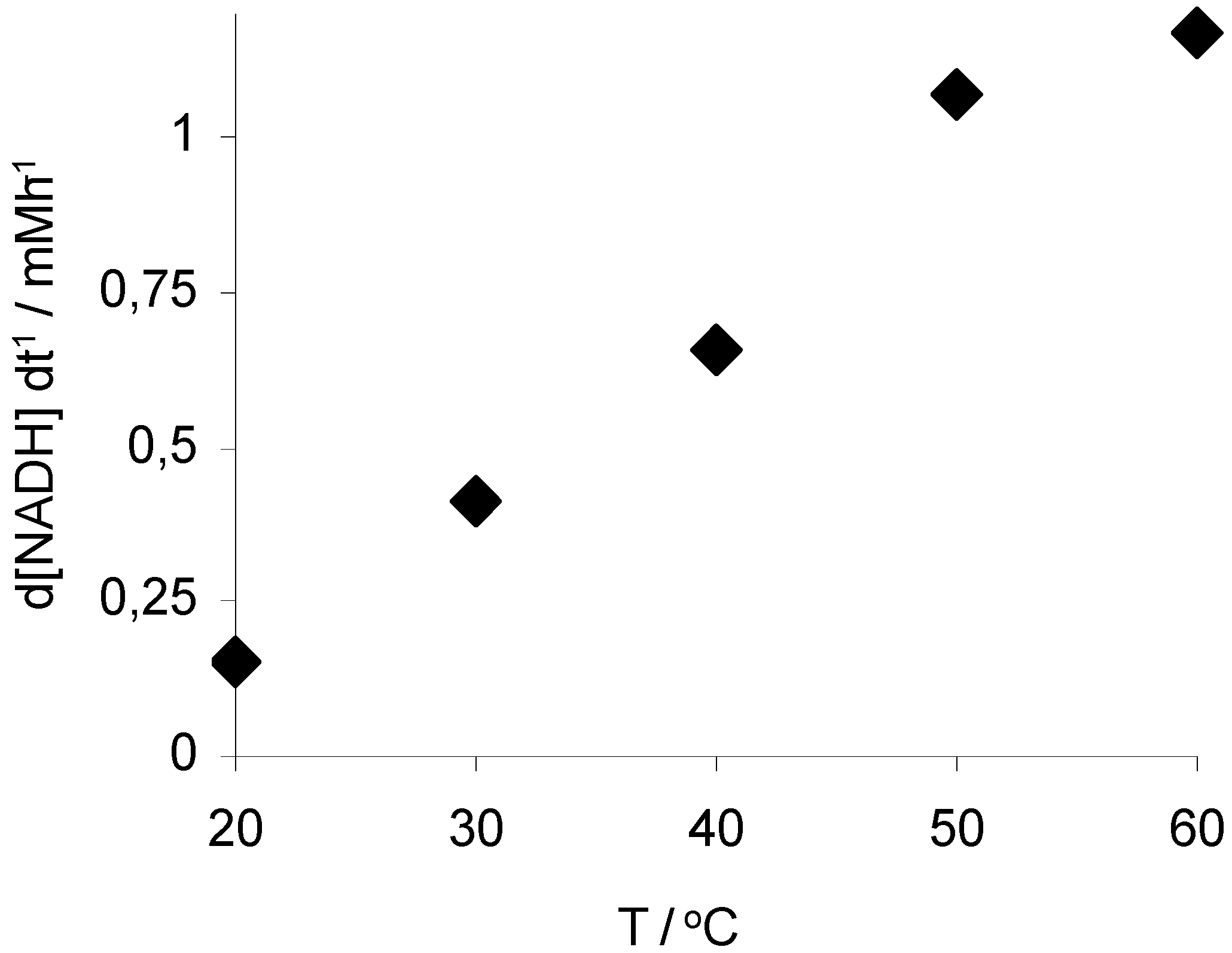
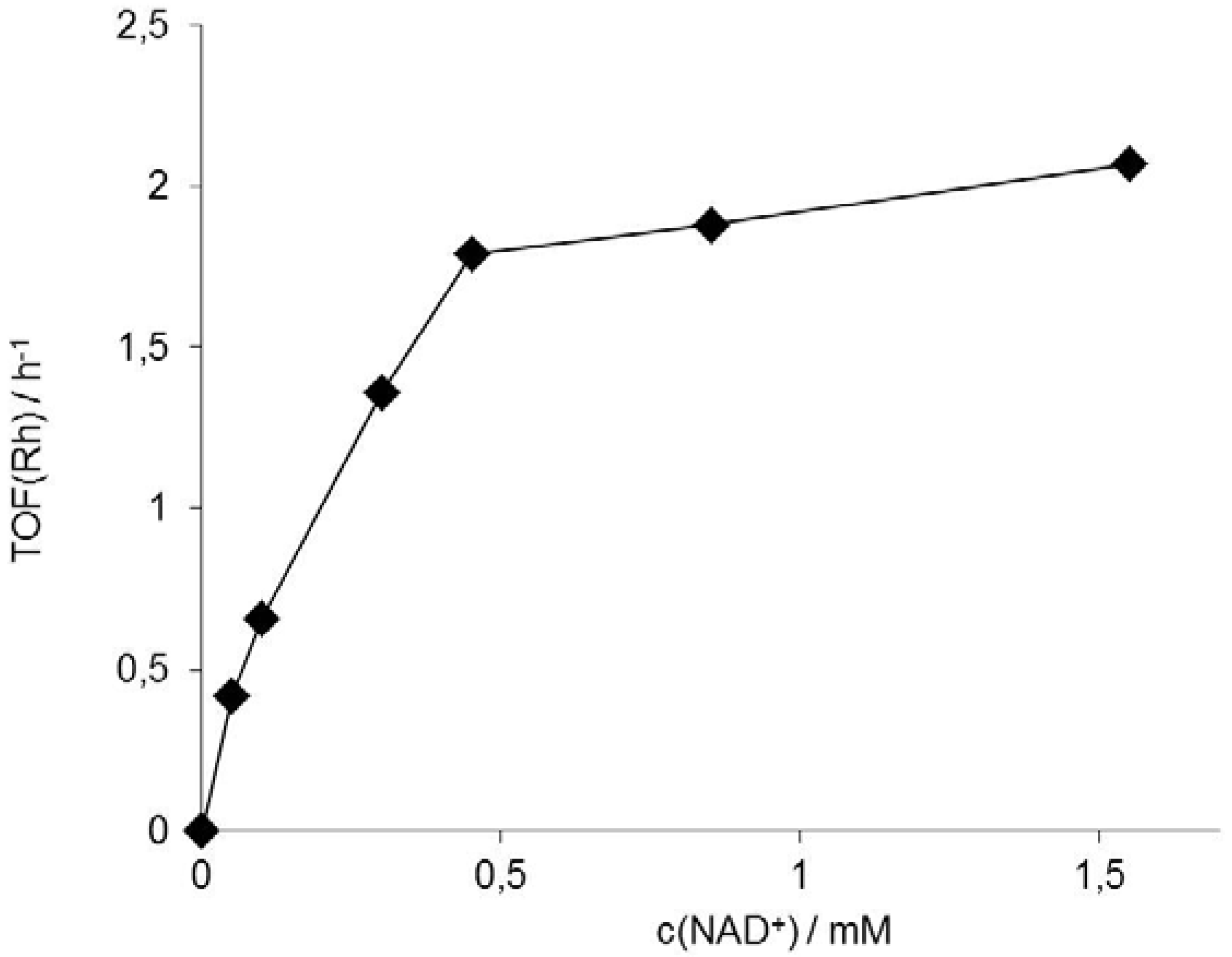
2. Results and Discussion
 ; 2:
; 2:  ; 3:
; 3:  ; 4:
; 4:  ; 5:
; 5:  ; 6:
; 6:  ; 7:
; 7:  ; 8:
; 8:  ; 9:
; 9:  ; 10:
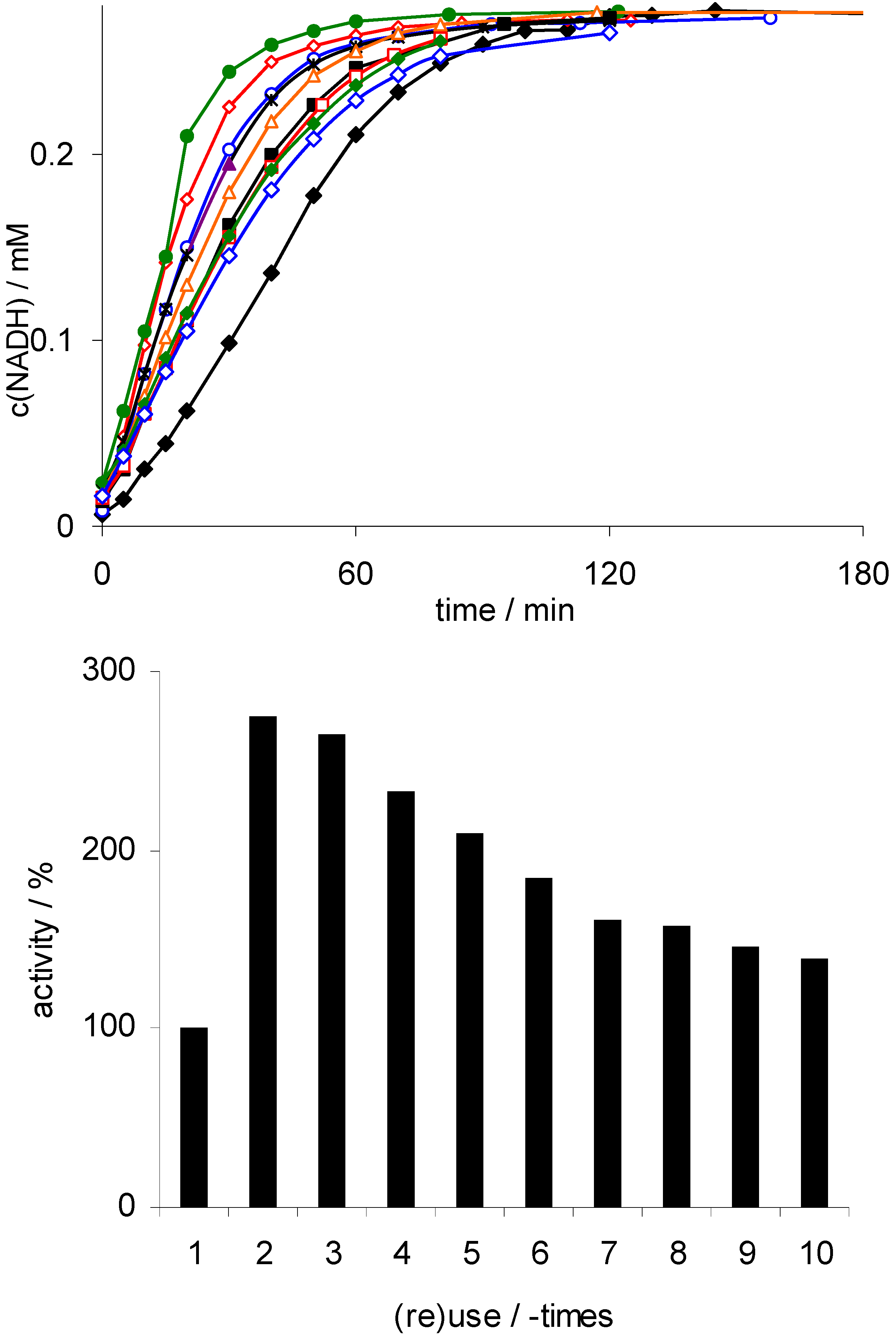
; 10:  ); lower: initial rates. Conditions: 50 mg Rh(III)-TsDPEN (0.35 μmol), c(NAD+)0 = 0.25 mM, 50 mM phosphate buffer pH 7, T = 30 °C, c(NaHCO2) = 150 mM, 1,000 rpm; 100% corresponds to an NADH-generation rate of 0.21 mM·h−1.
; 2: ; 3: ; 4: ; 5: ; 6: ; 7: ; 8: ; 9: ; 10: ); lower: initial rates. Conditions: 50 mg Rh(III)-TsDPEN (0.35 μmol), c(NAD+)0 = 0.25 mM, 50 mM phosphate buffer pH 7, T = 30 °C, c(NaHCO2) = 150 mM, 1,000 rpm; 100% corresponds to an NADH-generation rate of 0.21 mM·h−1.
); lower: initial rates. Conditions: 50 mg Rh(III)-TsDPEN (0.35 μmol), c(NAD+)0 = 0.25 mM, 50 mM phosphate buffer pH 7, T = 30 °C, c(NaHCO2) = 150 mM, 1,000 rpm; 100% corresponds to an NADH-generation rate of 0.21 mM·h−1.
; 2: ; 3: ; 4: ; 5: ; 6: ; 7: ; 8: ; 9: ; 10: ); lower: initial rates. Conditions: 50 mg Rh(III)-TsDPEN (0.35 μmol), c(NAD+)0 = 0.25 mM, 50 mM phosphate buffer pH 7, T = 30 °C, c(NaHCO2) = 150 mM, 1,000 rpm; 100% corresponds to an NADH-generation rate of 0.21 mM·h−1.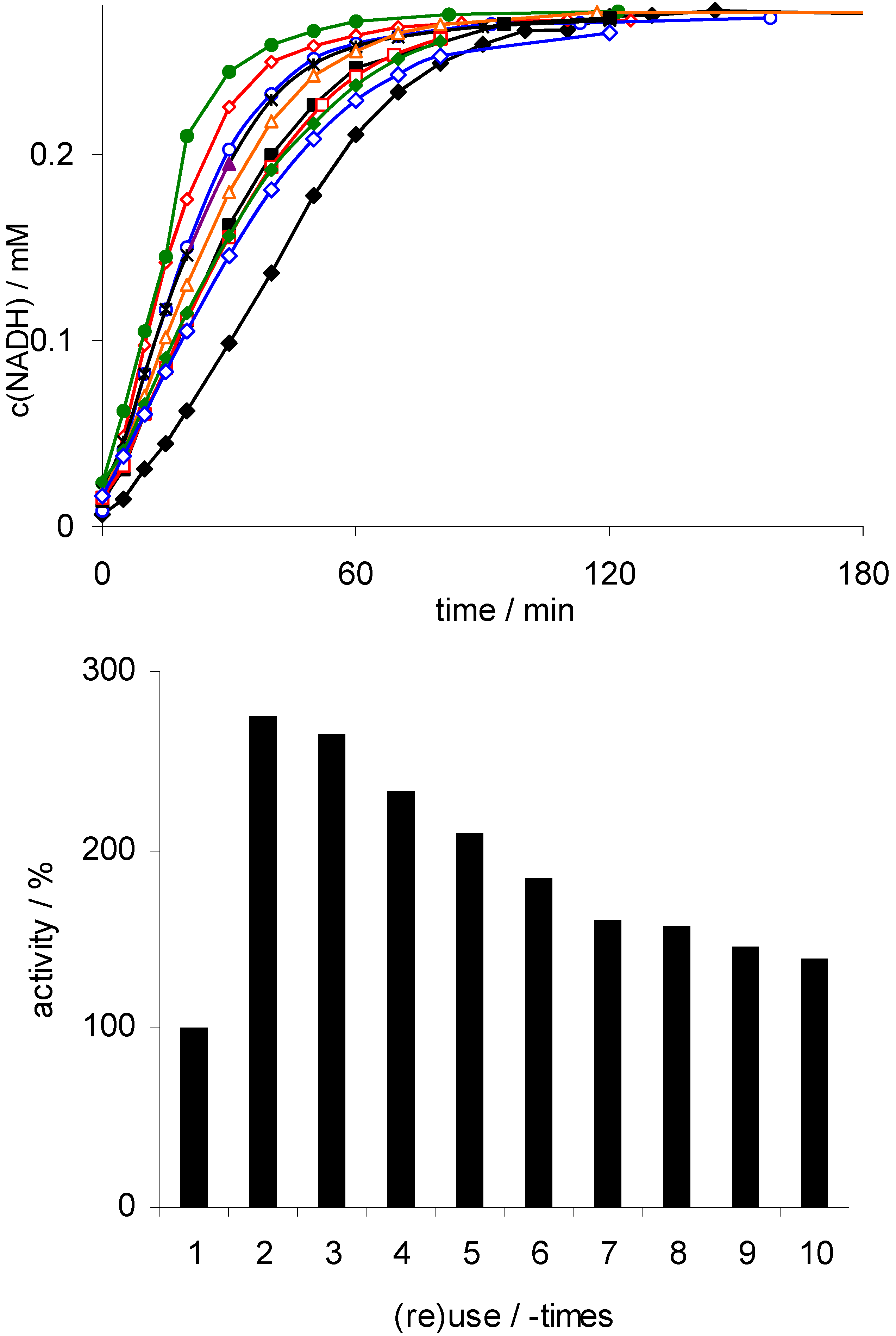
3. Experimental
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Hollmann, F.; Arends, I.W.C.E.; Buehler, K. Biocatalytic Redox Reactions for Organic Synthesis: Nonconventional Regeneration Methods. ChemCatChem 2010, 2, 762–782. [Google Scholar] [CrossRef]
- Lee, S.H.; Nam, D.H.; Kim, J.H.; Baeg, J.-O.; Park, C.B. Eosin Y-Sensitized Artificial Photosynthesis by Highly Efficient Visible-Light-Driven Regeneration of Nicotinamide Cofactor. ChemBioChem 2009, 10, 1621–1624. [Google Scholar] [CrossRef]
- Song, H.K.; Lee, S.H.; Won, K.; Park, J.H.; Kim, J.K.; Lee, H.; Moon, S.J.; Kim, D.K.; Park, C.B. Electrochemical regeneration of NADH enhanced by platinum nanoparticles. Angew. Chem. Int. Ed. 2008, 47, 1749–1752. [Google Scholar]
- Kim, J.H.; Lee, S.H.; Lee, J.S.; Lee, M.; Park, C.B. Zn-containing porphyrin as a biomimetic light-harvesting molecule for biocatalyzed artificial photosynthesis. Chem. Commun. 2011, 47, 10227–10229. [Google Scholar]
- Kim, J.H.; Lee, M.; Lee, J.S.; Park, C.B. Self-Assembled Light-Harvesting Peptide Nanotubes for Mimicking Natural Photosynthesis. Angew. Chem. Int. Ed. 2012, 51, 517–520. [Google Scholar]
- Kohlmann, C.; Greiner, L.; Leitner, W.; Wandrey, C.; Lütz, S. Ionic Liquids as Performance Additives for Electroenzymatic Syntheses. Chem. Eur. J. 2009, 15, 11692–11700. [Google Scholar]
- Hildebrand, F.; Lütz, S. Electroenzymatic synthesis of chiral alcohols in an aqueous-organic two-phase system. Tetrahedron: Asymmetry 2007, 18, 1187–1193. [Google Scholar] [CrossRef]
- de Gonzalo, G.; Ottolina, G.; Carrea, G.; Fraaije, M.W. [Cp*Rh(bpy)(H2O)]2+ as a coenzyme substitute in enzymatic oxidations catalyzed by Baeyer-Villiger monooxygenases. Chem. Commun. 2005, 3724–3726. [Google Scholar]
- Bernard, J.; van Heerden, E.; Arends, I.W.C.E.; Opperman, D.J.; Hollmann, F. Chemoenzymatic reduction of conjugated C=C-bonds. ChemCatChem 2012, 4, 196–199. [Google Scholar] [CrossRef]
- Mifsud Grau, M.; Poizat, M.; Arends, I.W.C.E.; Hollmann, F. Phosphite-driven regeneration of reduced enzyme cofactors. Appl. Organometal. Chem. 2010, 24, 380–385. [Google Scholar]
- Hollmann, F.; Schmid, A. Towards [Cp*Rh(bpy)(H2O)]2+-promoted P450 catalysis: Direct regeneration of CytC. J. Inorg. Biochem. 2009, 103, 313–315. [Google Scholar] [CrossRef]
- Hollmann, F.; Witholt, B.; Schmid, A. [Cp*Rh(bpy)(H2O)]2+: A versatile tool for efficient and non-enzymatic regeneration of nicotinamide and flavin coenzymes. J. Mol. Catal. B Enzym. 2002, 19-20, 167–176. [Google Scholar] [CrossRef]
- Poizat, M.; Arends, I.W.C.E.; Hollmann, F. On the nature of mutual interaction between [Cp*Rh(bpy)(H2O)]2+ and enzymes—Analysis and potential remedies. J. Mol. Catal. B Enzym. 2010, 63, 149–156. [Google Scholar] [CrossRef]
- Hildebrand, F.; Lütz, S. Stable Electroenzymatic Processes by Catalyst Separation. Chem. Eur. J. 2009, 15, 4998–5001. [Google Scholar] [CrossRef]
- Haquette, P.; Talbi, B.; Barilleau, L.; Madern, N.; Fosse, C.; Salmain, M. Chemically engineered papain as artificial formate dehydrogenase for NAD(P)H regeneration. Org. Biomol. Chem. 2011, 9, 5720–5727. [Google Scholar]
- Dimroth, J.; Schedler, U.; Keilitz, J.; Haag, R.; Schomäcker, R. New Polymer-Supported Catalysts for the Asymmetric Transfer Hydrogenation of Acetophenone in Water—Kinetic and Mechanistic Investigations. Adv. Synth. Catal. 2011, 353, 1335–1344. [Google Scholar] [CrossRef]
- Dimroth, J.; Keilitz, J.; Schedler, U.; Schomäcker, R.; Haag, R. Immobilization of a Modified Tethered Rhodium(III)-p-Toluenesulfonyl-1,2-diphenylethylenediamine Catalyst on Soluble and Solid Polymeric Supports and Successful Application to Asymmetric Transfer Hydrogenation of Ketones. Adv. Synth. Catal. 2010, 352, 2497–2506. [Google Scholar]
- Köhler, V.; Wilson, Y.M.; Dürrenberger, M.; Ghislieri, D.; Churakova, E.; Quinto, T.; Knörr, L.; Häussinger, D.; Hollmann, F.; Turner, N.J.; et al. New Synthetic Cascades by Combining Biocatalysts with Artificial Metalloenzymes. Nat. Chem. 2012. submitted. [Google Scholar]
- Einstein, A. The motion of elements suspended in static liquids as claimed in the molecular kinetic theory of heat. Ann. Phys.-Berlin 1905, 17, 549–560. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compound Rh(III)-TsDPEN are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Torres, M.; Dimroth, J.; Arends, I.W.C.E.; Keilitz, J.; Hollmann, F. Towards Recyclable NAD(P)H Regeneration Catalysts. Molecules 2012, 17, 9835-9841. https://doi.org/10.3390/molecules17089835
De Torres M, Dimroth J, Arends IWCE, Keilitz J, Hollmann F. Towards Recyclable NAD(P)H Regeneration Catalysts. Molecules. 2012; 17(8):9835-9841. https://doi.org/10.3390/molecules17089835
Chicago/Turabian StyleDe Torres, Miriam, Jonas Dimroth, Isabel W. C. E. Arends, Juliane Keilitz, and Frank Hollmann. 2012. "Towards Recyclable NAD(P)H Regeneration Catalysts" Molecules 17, no. 8: 9835-9841. https://doi.org/10.3390/molecules17089835
APA StyleDe Torres, M., Dimroth, J., Arends, I. W. C. E., Keilitz, J., & Hollmann, F. (2012). Towards Recyclable NAD(P)H Regeneration Catalysts. Molecules, 17(8), 9835-9841. https://doi.org/10.3390/molecules17089835