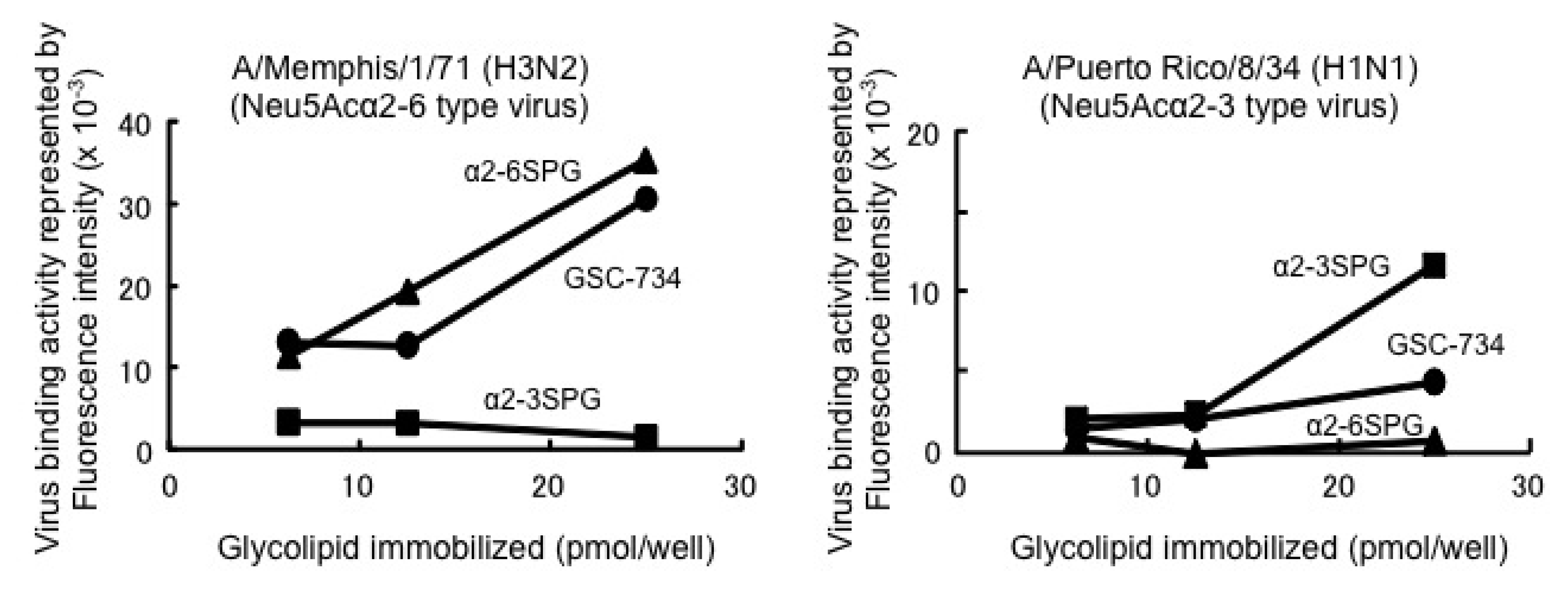
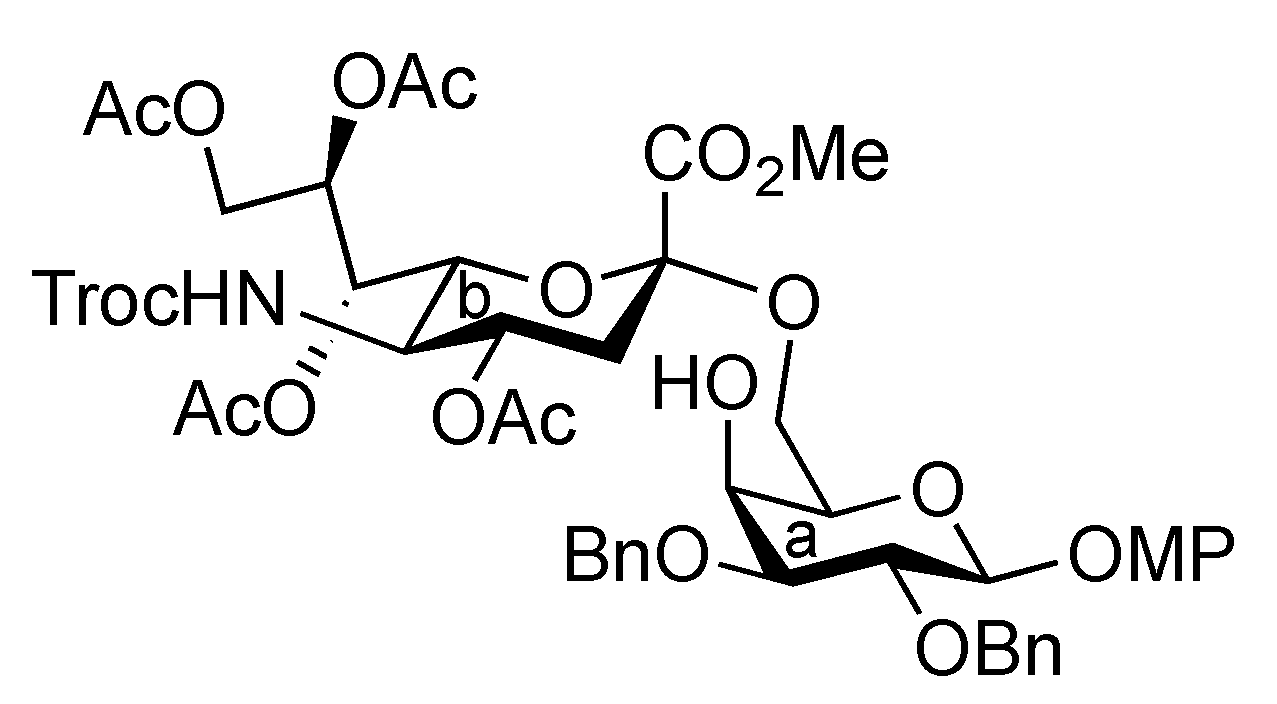
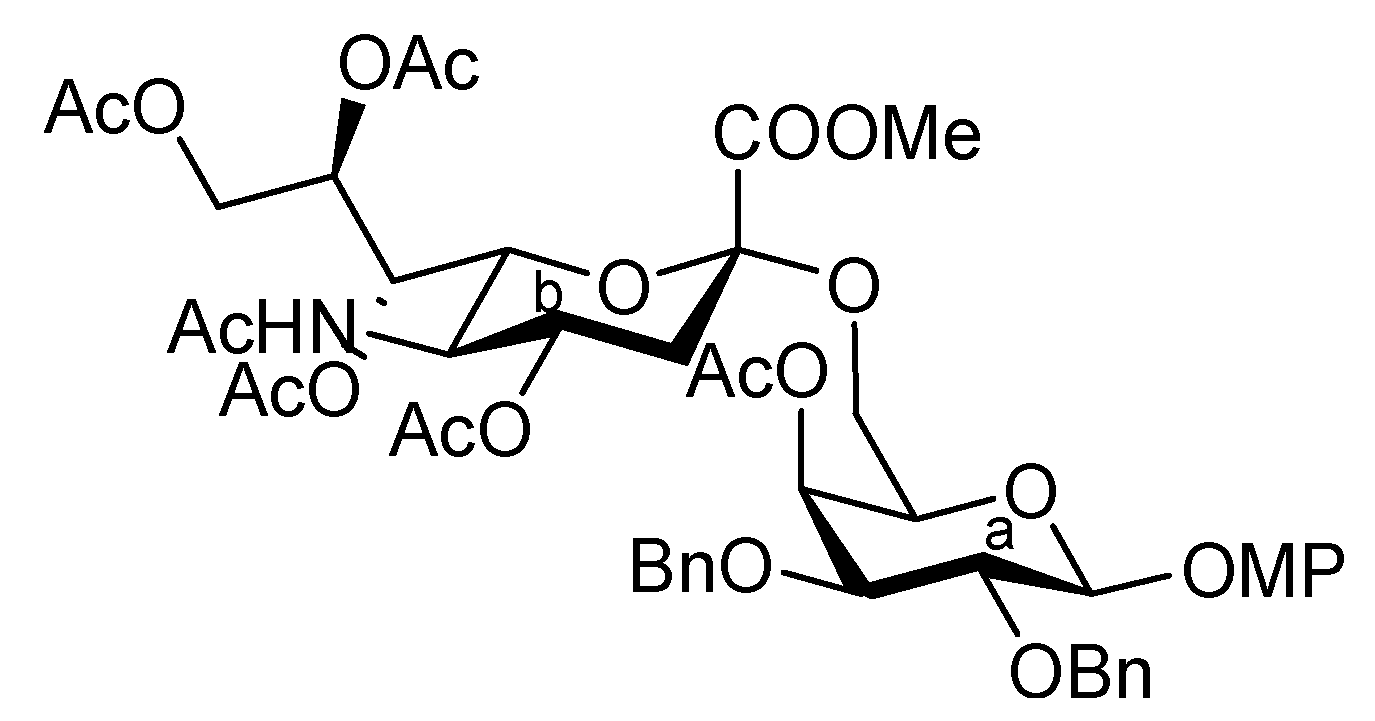
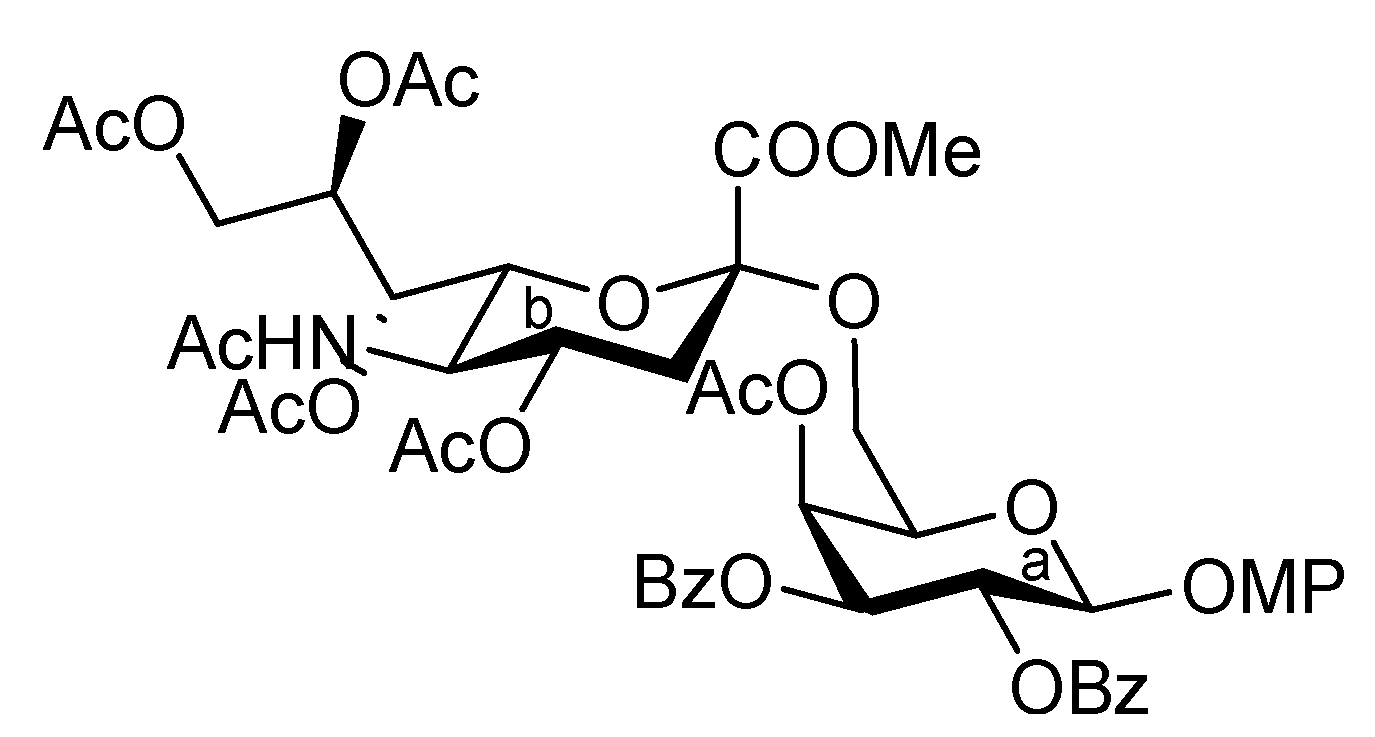
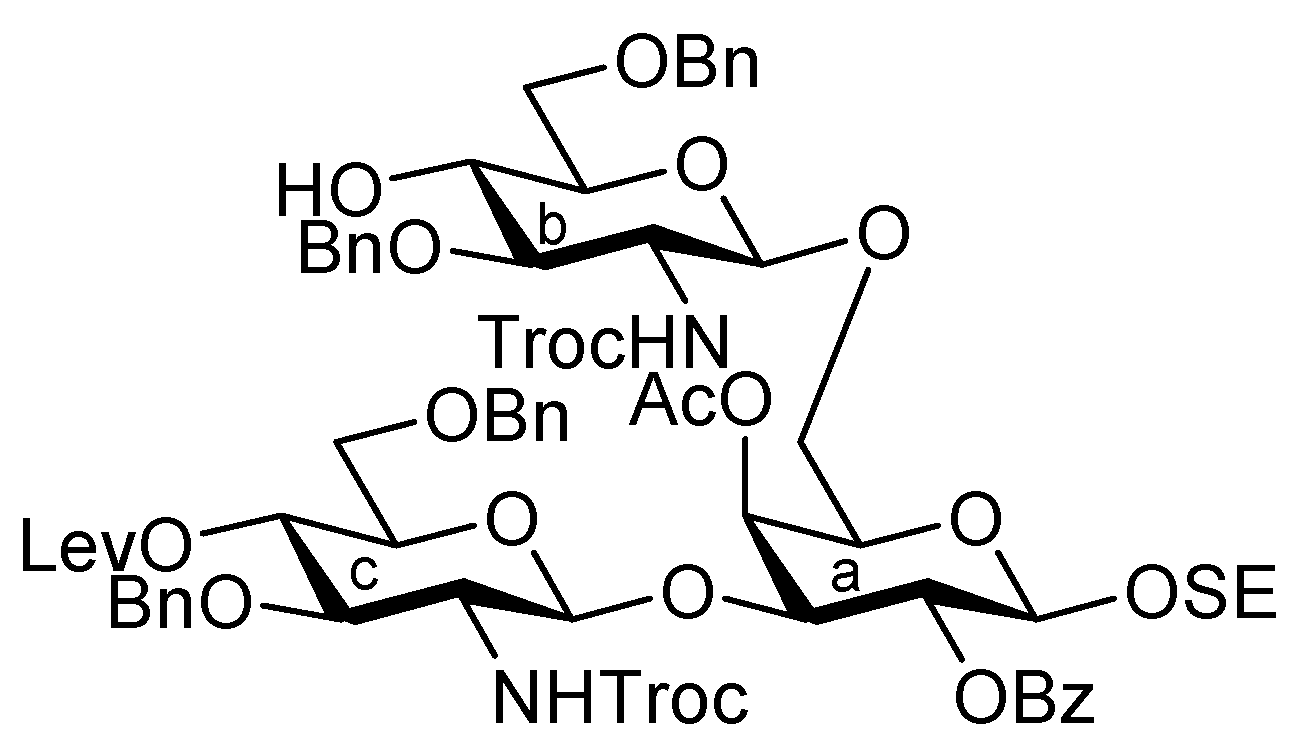
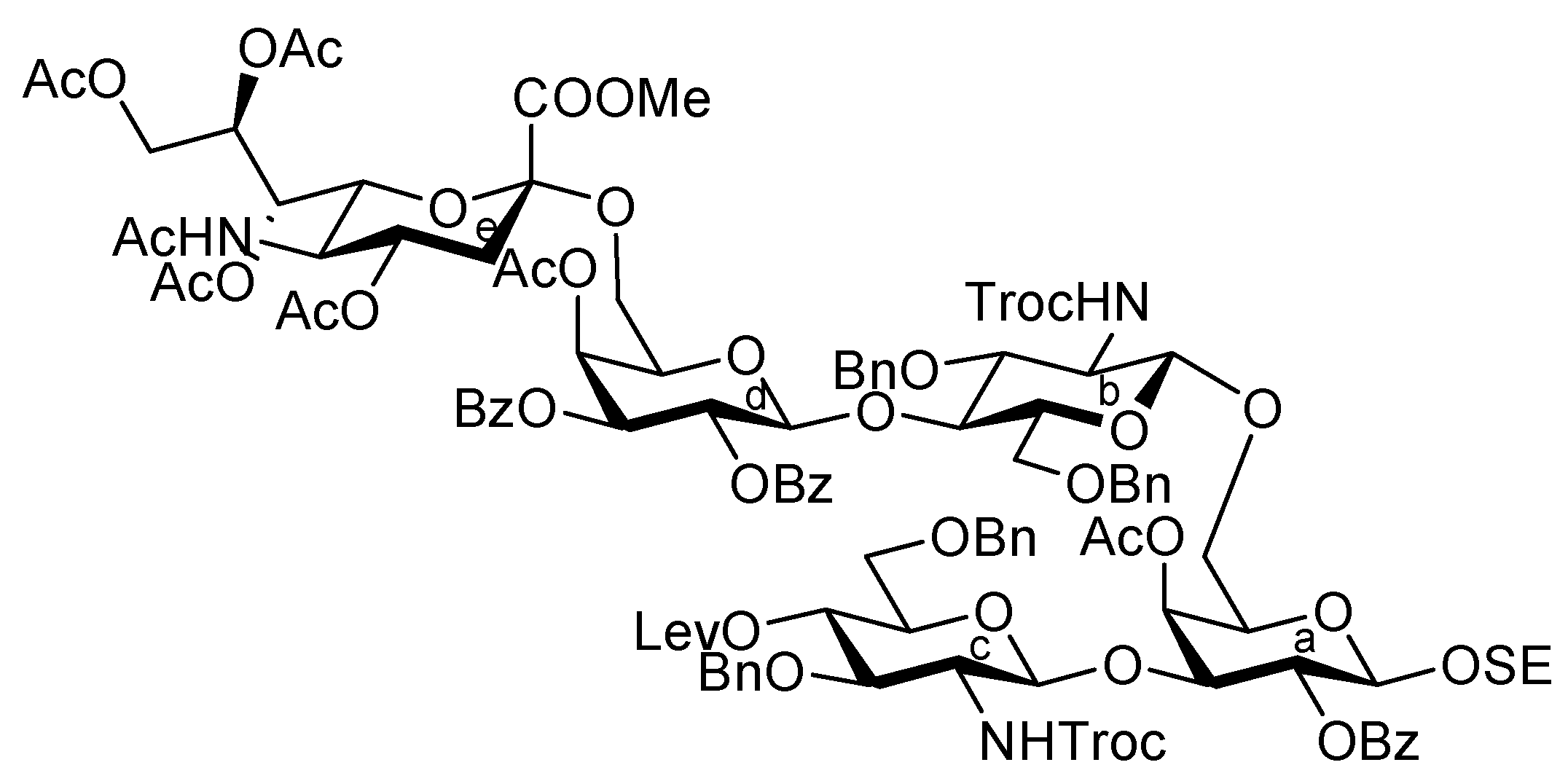
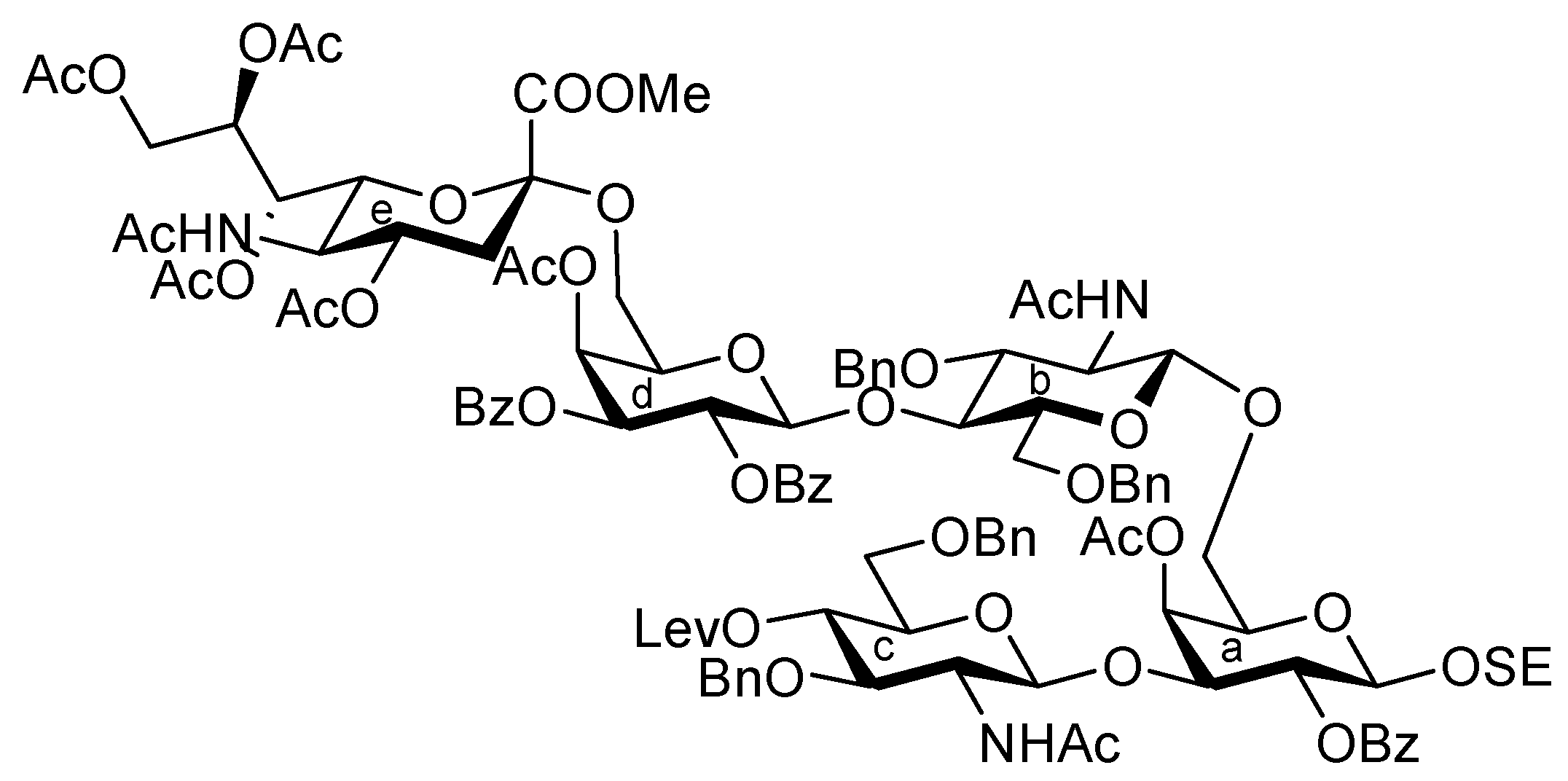
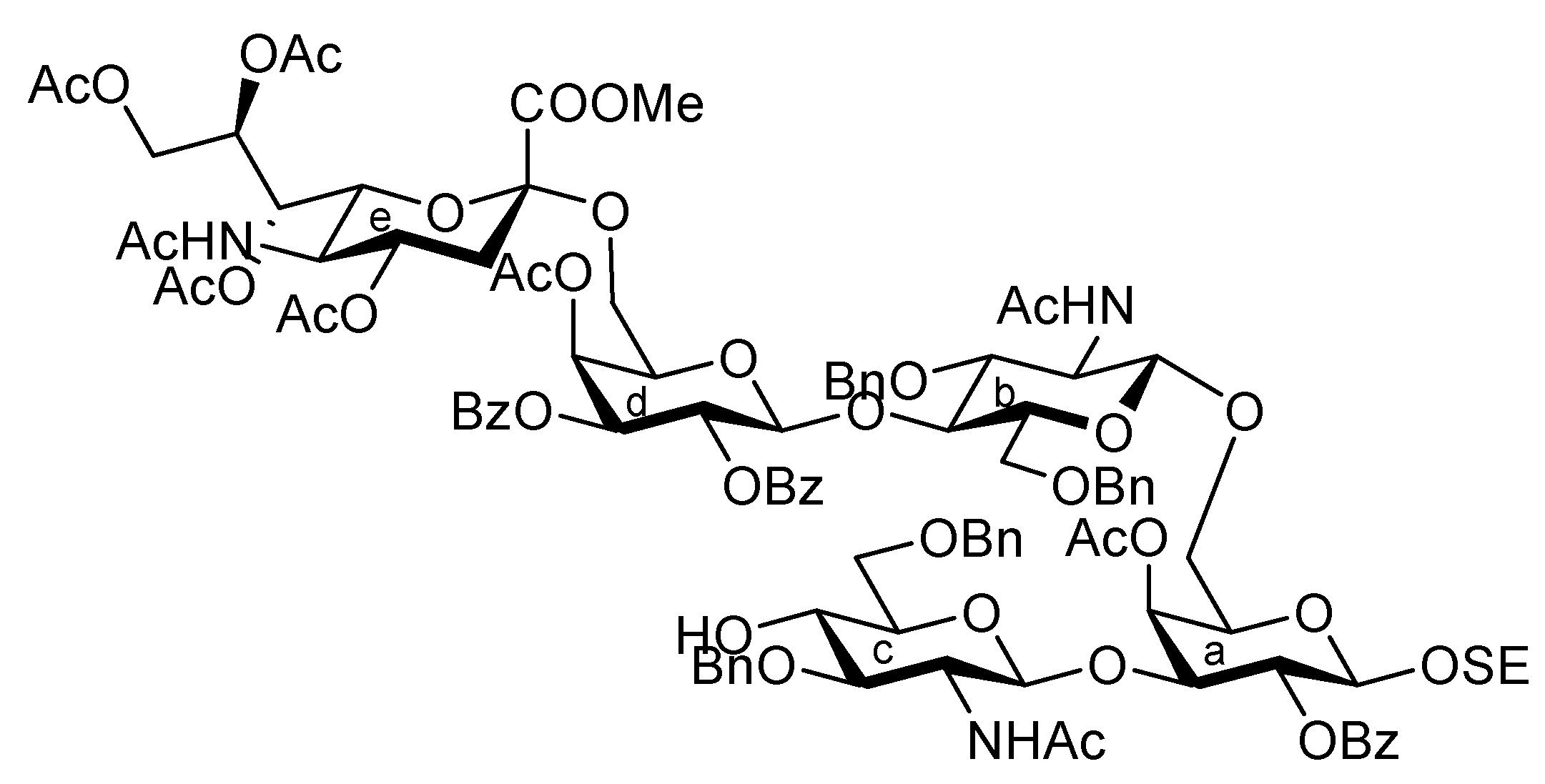
3.1. General Methods for Chemical Synthesis
All reactions were carried out under a positive pressure of argon, unless otherwise noted. All chemicals were purchased from commercial suppliers and used without further purification, unless otherwise noted. Molecular sieves were purchased from Wako Chemicals Inc. (Osaka, Japan) and dried at 300 °C for 2 h in a muffle furnace prior to use. Solvents as reaction media were dried over molecular sieves and used without purification. TLC analysis was performed on Merck TLC (silica gel 60F254 on glass plate, Darmstadt, Germany). Compound detection was either by exposure to UV light (2536 Å) or by soak in a solution of 10% H2SO4 in ethanol followed by heating. Silica gel (80 mesh and 300 mesh) manufactured by Fuji Silysia Co. (Kasugai, Japan) was used for flash column chromatography. Quantity of silica gel was usually estimated as 100 to 150-fold weight of sample to be charged. Solvent systems in chromatography were specified in v/v. Evaporation and concentration were carried out in vacuo. 1H-NMR and 13C-NMR spectra were recorded with JEOL ECA 400/500/600 spectrometers. Chemical shifts in 1H-NMR spectra are expressed in ppm (δ) relative to the signal of Me4Si, adjusted to δ 0.00 ppm. Data are presented as follow: Chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, dd = double of doublet, dt = double of triplet, m = multiplet and/or multiple resonances), integration, coupling constant in Hertz (Hz), position of the corresponding proton. COSY methods were used to confirm the NMR peak assignments. MALDI-TOF mass spectra were run in a Bruker Autoflex (Billerica, MA, USA) and CHCA was used as the matrix. High-resolution mass (ESI-TOF MS) spectra were run in a Bruker micrOTOF. Optical rotations were measured with a ‘Horiba SEPA-300’ high-sensitive polarimeter (Kyoto, Japan).
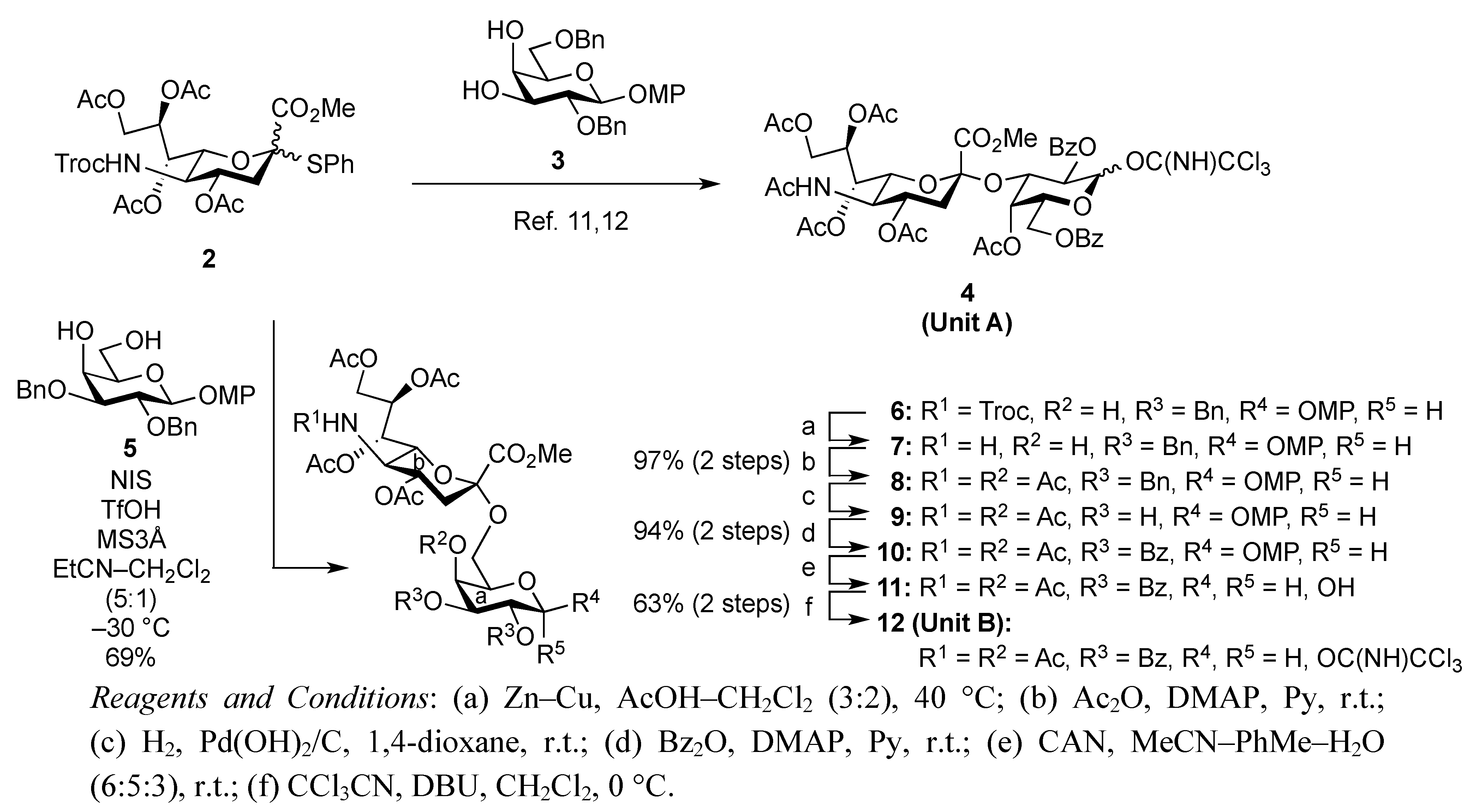
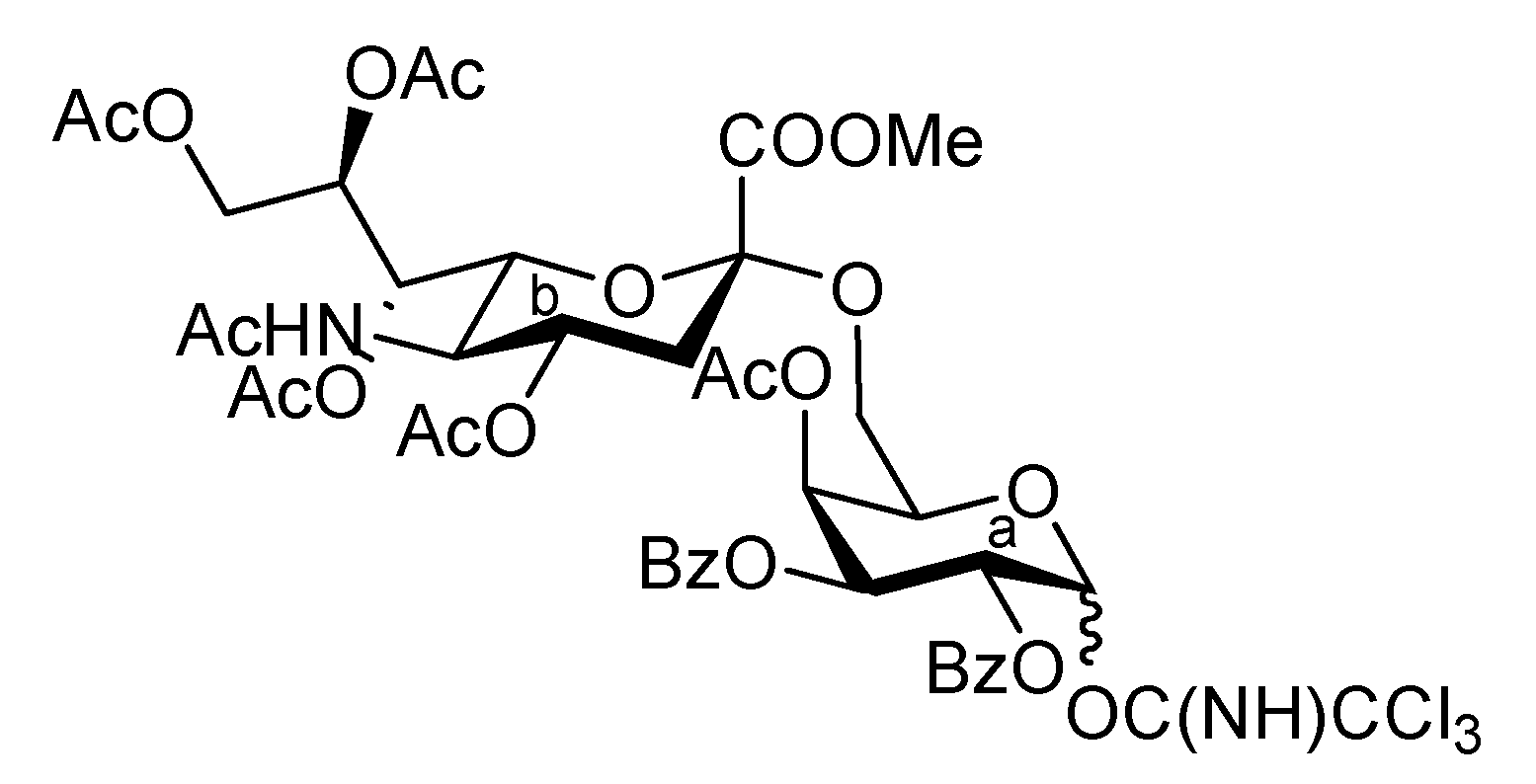
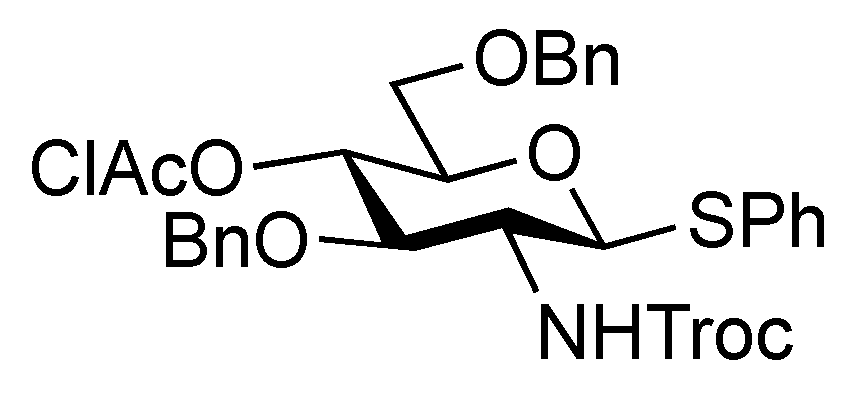
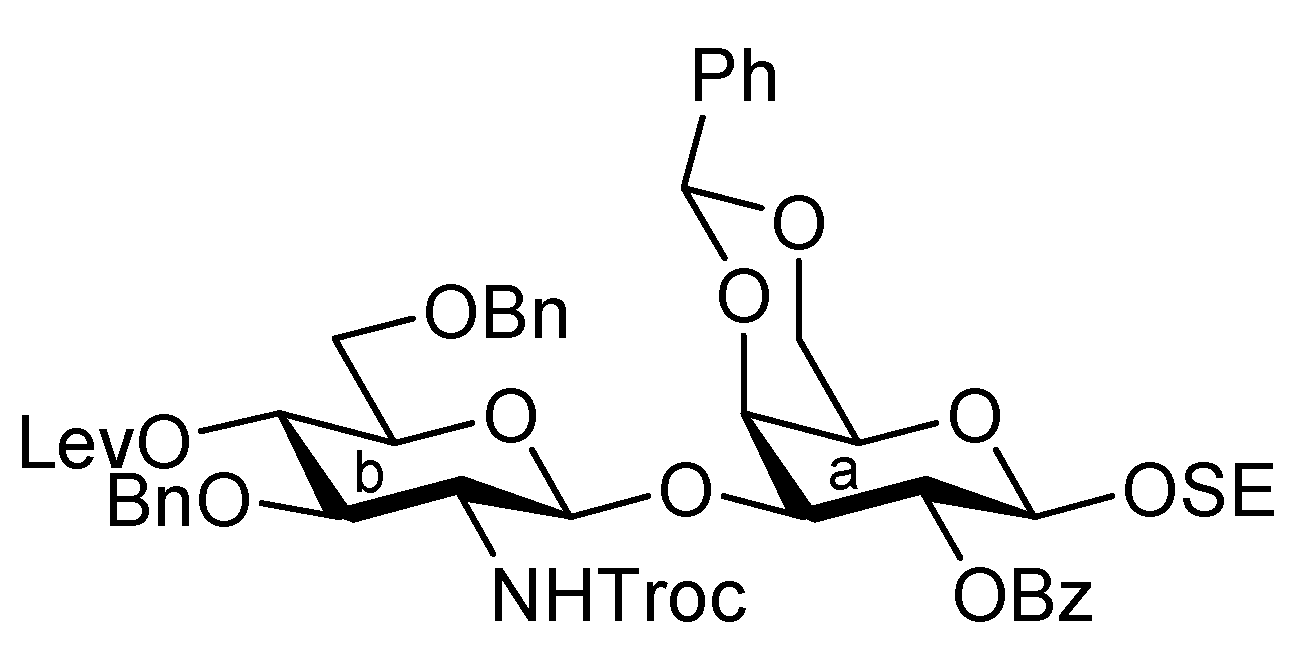
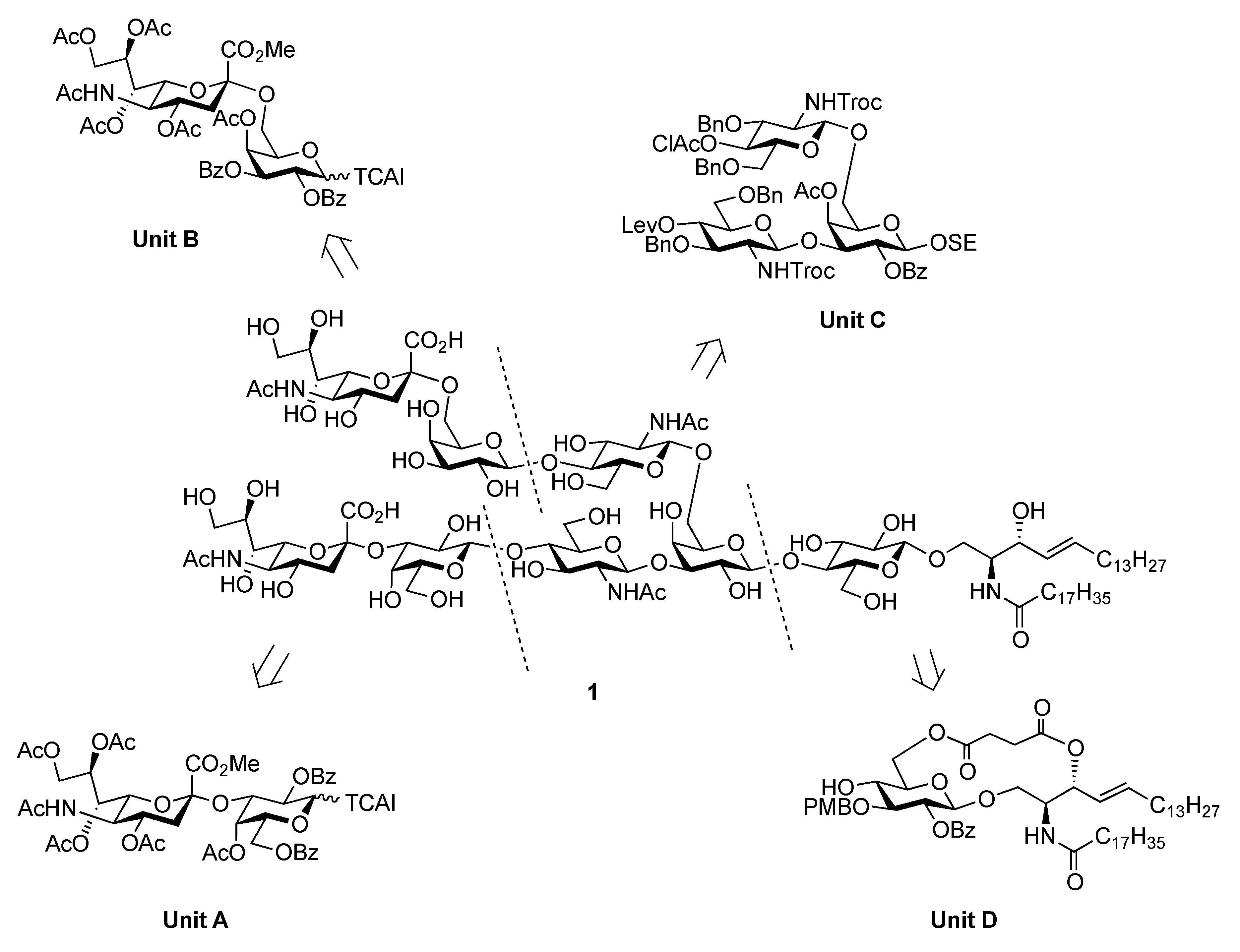
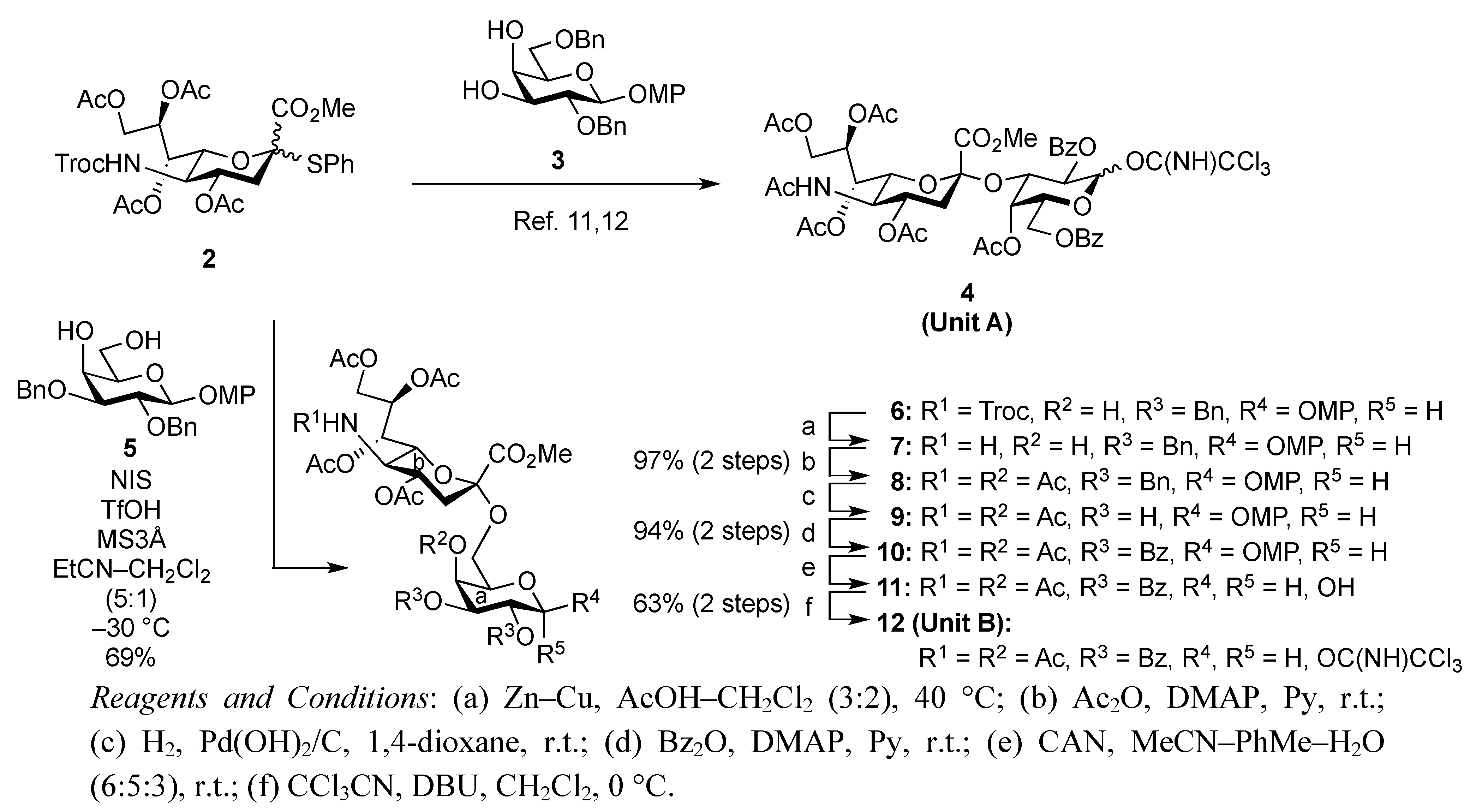
4-Methoxyphenyl (methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-(2,2,2-trichloroethoxycarbamoyl)-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-2,3-di-O-benzyl-β-D-galactopyranoside (6). To a mixture of 2 (691 mg, 0.964 mmol) and 5 (300 mg, 0.643 mmol) in EtCN/CH2Cl2 (5:1, 9.6 mL) was added 3 Å molecular sieves (991 mg) at r.t. After stirring for 1 h and then cooling to −30 °C, NIS (324 mg, 1.44 mmol) and TfOH (12.7 μL, 0.144 mmol) were added to the mixture. After stirring for 45 min at the same temperature as the reaction was monitored by TLC (1:3 EtOAc–toluene, twice development), the reaction was quenched by the addition of triethylamine. The precipitate was filtered through Celite. The filtrate was evaporated to remove EtCN and then diluted with CHCl3, washed with satd aq Na2S2O3 and brine. The organic layer was subsequently dried over Na2SO4, concentrated and the residue was purified by silica gel column chromatography (1:5 EtOAc–toluene) to give 6 (473 mg, 69%) along with its β-isomer (96 mg, 14%). [α]D −13.9° (c 0.4, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.38–6.80 (m, 14 H, Ar), 5.39 (m, 1 H, H-8b), 5.36 (dd, 1 H, J6,7 = 1.7 Hz, H-7b), 5.00 (d, 1 H, Jgem = 10.2 Hz, OCH2), 4.99 (m, 1 H, H-4b), 4.89 (d, 1 H, Jgem = 12.2 Hz, OCH2), 4.86 (d, 1 H, J5,NH = 9.7 Hz, NH), 4.84 (d, 1 H, J1,2 = 8.0 Hz, H-1a), 4.83 (d, 1 H, Jgem = 10.2 Hz, OCH2), 4.76 (2 d, 2 H, Jgem = 12.0 Hz, OCH2), 4.47 (d, 1 H, Jgem = 12.2 Hz, OCH2), 4.32 (dd, 1 H, H-9b), 4.19 (dd, 1 H, J6,7 = 1.7 Hz, H-6b), 4.10 (dd, 1 H, H-9'b), 4.07 (near d, 1 H, H-4a), 3.94–3.89 (m, 2 H, H-2a, H-5a), 3.81–3.75 (m, 7 H, H-6a, 2 OMe), 3.65–3.61 (m, 2 H, H-6'a, H-5b), 3.57 (dd, 1 H, H-3a), 2.67–2.63 (m, 2 H, H-3beq, OH), 2.12–1.99 (m, 12 H, 4 Ac), 1.91 (t, 1 H, H-3bax); 13C-NMR (100 MHz, CDCl3) δ 193.2, 191.4, 170.7, 170.3, 170.1, 169.9, 167.9, 155.2, 154.0, 151.7, 138.4, 137.9, 129.0, 128.4, 128.3, 128.2, 128.1, 127.8, 127.7, 127.6, 125.3, 118.6, 114.4, 102.9, 98.7, 95.3, 80.6, 78.7, 77.2, 75.3, 74.5, 72.6, 72.3, 72.2, 68.6, 68.4, 67.5, 66.0, 63.1, 62.3, 55.6, 53.0, 51.6, 37.5, 29.7, 29.5, 21.4, 21.0, 20.8, 20.7. m/z (MALDI): found [M+Na]+ 1094.32, C48H56Cl3NO20calcd for [M+Na]+ 1094.32.
4-Methoxyphenyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzyl-β-D-galactopyranoside (8). To a solution of 6 (3.77 g, 3.51 μmol) in AcOH/CH2Cl2 (3:2, 70 mL) was added Zn/Cu couple (18.9 g) at r.t. The reaction mixture was heated to 40 °C and was stirred for 45 min at the same temperature as the reaction was monitored by TLC (4:1 toluene–EtOAc). The precipitate was filtered through Celite and the filtrate was co-evaporated with toluene. The obtained residue was exposed to high vacuum for 6 h. The crude residue was dissolved in pyridine (35 mL) and acetic anhydride (1.32 μL, 14.0 mmol), DMAP (4.3 mg, 35.1 μmol) were then added to the mixture at 0 °C. After stirring for 3 d at r.t as the reaction was monitored by TLC (2:1 toluene–EtOAc), the reaction mixture was evaporated. The residue was diluted with CHCl3, washed with 2 M HCl, H2O, satd aq NaHCO3 and brine, dried over Na2SO4, and concentrated. The obtained residue was purified by silica gel column chromatography (4:1 toluene–EtOAc) to give 8 (3.35 g, 97%). [α]D −11.3° (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.37–7.24 (m, 10 H, 2 Ph), 6.97 (2 d, 4 H, Ar), 5.95 (d, 1 H, JNH,5 = 6.3 Hz, NH), 5.62 (d, 1 H, J3,4 = 2.3 Hz, H-4a), 5.41 (m, 1 H, H-8b), 5.32 (d, 1 H, H-7b), 4.96–4.81 (m, 5 H, H-1a, H-4b, 3OCH2), 4.68 (d, 1 H, OCH2), 4.34 (d, 1 H, H-9b), 4.14–4.05 (m, 3 H, H-5b, H-6b, H-9'b), 3.90–3.69 (m, 11 H, H-2a, H-3a, H-5a, H-6a, H-6'a, 2 OMe), 2.62 (m, 1 H, Jgem = 11.5 Hz, H-3beq), 2.16–1.87 (m, 19 H, 6 Ac, H-3bax); 13C-NMR (100 MHz, CDCl3) δ 170.4, 170.2, 170.0, 169.8, 169.7, 169.5, 167.7, 154.9, 151.2, 138.2, 137.5, 128.7, 128.0, 127.9, 127.7, 127.3, 127.2, 118.0, 114.1, 102.2, 98.4, 79.1, 78.3, 75.0, 72.4, 71.8, 71.3, 68.7, 68.3, 67.0, 65.9, 62.7, 62.3, 55.2, 52.5, 48.6, 37.5, 22.7, 20.7, 20.5, 20.4, 20.4, 20.3. m/z (MALDI): found [M+Na]+ 1004.17, C49H59NO20 calcd for [M+Na]+ 1004.35.
4-Methoxyphenyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-β-D-galactopyranoside (10). To a solution of 8 (3.34 g, 3.41 μmol) in 1,4-dioxane (34 mL) was added Pd(OH)2/C (3.34 g). After stirring for 45 min at r.t. under a hydrogen atmosphere as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the mixture was filtered through Celite. The filtrate was concentrated and the obtained crude residue was roughly purified by silica gel column chromatography. The obtained product was exposed to high vacuum for 24 h. The residue was then dissolved in pyridine (34 mL). Benzoic anhydride (3.09 g, 13.6 mmol) and DMAP (20.8 mg, 0.171 μmol) were added to the mixture at 0 °C. After stirring for 9 h at r.t. as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the reaction was quenched by the addition of MeOH at 0 °C. The mixture was co-evaporated with toluene and the residue was then diluted with CHCl3, and washed with 2 M HCl, H2O, satd aq NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (1:1 toluene–EtOAc) to give 10 (3.22 g, 94%). [α]D +40.3° (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 8.00–7.37 (m, 10 H, 2 Ph), 7.06–6.76 (2 d, Ar), 5.91 (near t, 1 H, J1,2 = 7.8 Hz, J2,3 = 10.6 Hz, H-2a), 5.78 (d, 1 H, J3,4 = 3.4 Hz, H-4a), 5.60 (dd, 1 H, H-3a), 5.50 (m, 1 H, J7,8 = 6.8 Hz, H-8b), 5.33–5.27 (m, 2 H, H-1a, H-7b), 5.15 (d, 1 H, NH), 4.87 (m, 1 H, J3eq,4 = 4.6 Hz, H-4b), 4.41 (dd, 1 H, Jgem = 12.4 Hz, H-9), 4.30 (near t, 1 H, J5,6 = 6.9 Hz, H-5a), 4.17–4.02 (m, 3 H, H-5b, H-6b, H-9'b), 3.89–3.73 (m, 7 H, 2 OMe, H-6a), 3.58 (near t, 1 H, Jgem = 10.1 Hz, H-6'a), 2.55 (dd, 1 H, Jgem = 12.4 Hz, H-3beq), 2.25–1.89 (m, 19 H, 6 Ac, H-3bax); 13C-NMR (100 MHz, CDCl3) δ 170.5, 170.4, 170.0, 169.7, 169.5, 167.7, 165.1, 165.0, 155.1, 151.0, 133.0, 129.4, 129.3, 129.1, 128.8, 128.1, 128.1, 118.2, 114.1, 100.1, 98.9, 72.5, 71.7, 71.6, 69.4, 68.6, 67.9, 67.0, 62.9, 55.2, 52.7, 48.7, 37.6, 22.8, 20.8, 20.5, 20.4, 20.3. m/z (MALDI): found [M+Na]+ 1032.21, C49H55NO22 calcd for [M+Na]+ 1032.31.
(Methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-D-galactopyranosyl trichloroacetimidate (12). To a solution of 10 (3.22 g, 3.19 mmol) in MeCN/PhMe/H2O (32 mL, 6:5:3) was added diammonium cerium (IV) nitrate (CAN; 17.5 g, 31.9 mmol) at r.t. The mixture was stirred for 2 h at r.t., as the proceeding of the reaction was monitored by TLC (10:1 CHCl3–MeOH). The reaction mixture was diluted with CHCl3 and washed with H2O, satd aq NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4 and concentrated. The resulting residue was purified by silica gel column chromatography (70:1 CHCl3–MeOH) to give 11. The obtained hemiacetal compound 11 (2.27 g, 2.51 mmol) was dissolved in CH2Cl2 (50 mL). To the mixture was added CCl3CN (2.5 mL, 25.1 mmol), DBU (449 μL, 3.01 mmol) at 0 °C. After stirring for 6 h at 0 °C as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the reaction mixture was evaporated. The crude residue was purified by silica gel column chromatography (70:1 CHCl3–MeOH) to give 12 (2.10 g, 63% over 2 steps). 12α: 1H-NMR (500 MHz, CDCl3) δ 8.61 (s, 1 H, C=NH), 7.95–7.28 (m, 10 H, 2 Ph), 6.80 (d, 1 H, J1,2 = 3.4 Hz, H-1a), 5.90–5.82 (m, 2 H, H-2a, H-3a), 5.79 (m, 1 H, J3,4 = 2.8 Hz, H-4a), 5.38–5.30 (m, 3 H, H-7b, H-8b, NHb), 4.87–4.86 (m, 1 H, H-4b), 4.51 (br t, 1 H, H-5a), 4.28 (dd, 1 H, Jgem = 12.6 Hz, J8,9 = 2.9 Hz, H-9b), 4.11 (dd, 1 H, J8,9´ = 5.9 Hz, H-9'b), 4.06–3.97 (m, 3 H, H-6a, H-5b, H-6b), 3.78 (s, 3 H, COOMe), 3.42 (dd, 1 H, J5,6' = 2.6 Hz, Jgem = 10.6 Hz, H-6'a), 3.55 (dd, 1 H, Jgem = 11.6 Hz, J3eq,4 = 3.6 Hz, H-3beq), 2.35–1.87 (m, 19 H, 6 Ac, H-3bax).
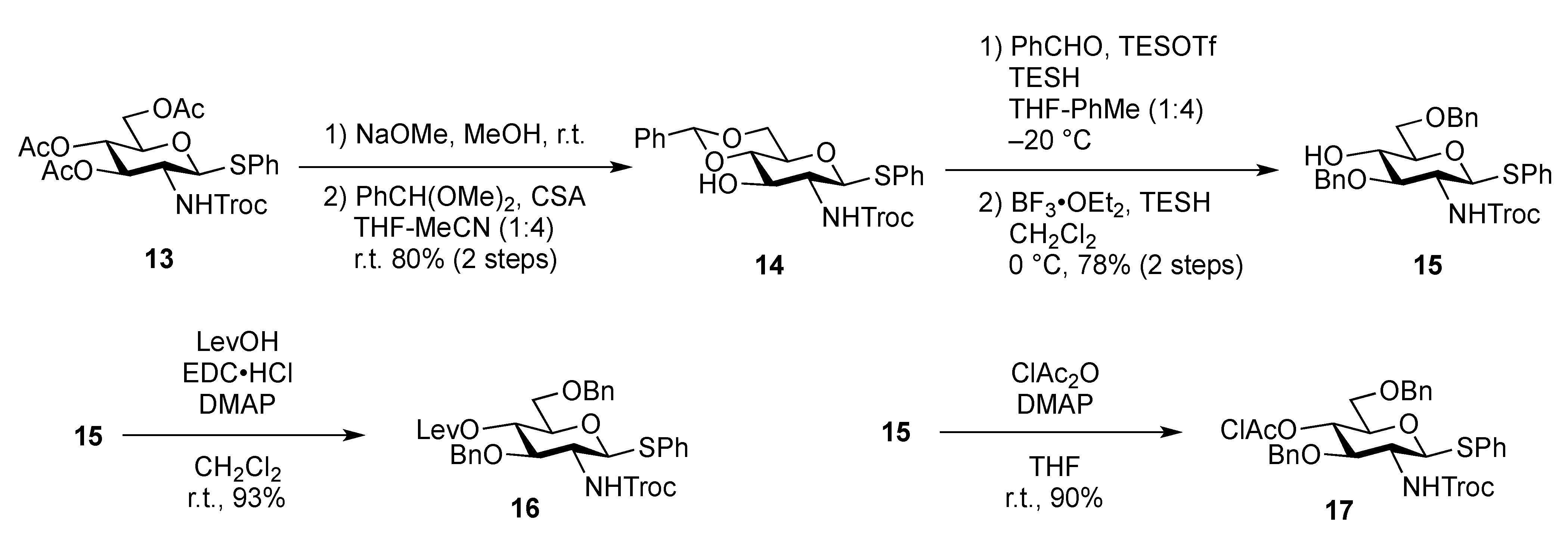
Phenyl 4,6-O-benzylidene-2-deoxy-1-thio-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranoside (
14). To a solution of
13 (8.43 g, 14.7 mmol) in MeOH (84 mL) was added NaOMe (28% solution in MeOH, 39.7 mg, 0.735 mmol) at 0 °C. After stirring for 2.5 h at room temperature as the reaction was monitored by TLC (10:1 CHCl
3–MeOH), the reaction was neutralized with Dowex (H
+) resin. The resin was filtered through cotton and the filtrate was then evaporated. The residue was exposed to high vacuum for 12 h. The obtained crude mixture was then dissolved in THF/MeCN (1:4, 135 mL). To the mixture were added benzaldehyde dimethyl acetal (4.39 mL, 29.4 mmol) and (±)-camphor-10-sulfonic acid (CSA) (512 mg, 2.21 mmol) at 0 °C. After stirring for 1 h at room temperature as the reaction was monitored by TLC (20:1 CHCl
3–MeOH), the reaction was quenched by the addition of triethylamine. The reaction mixture was concentrated and then subjected to crystallization from hot acetone/
n-hexane to give
14 (6.27 g, 80%) as a white crystal. The physical data of
14 was identical to those reported in the literature [
27].
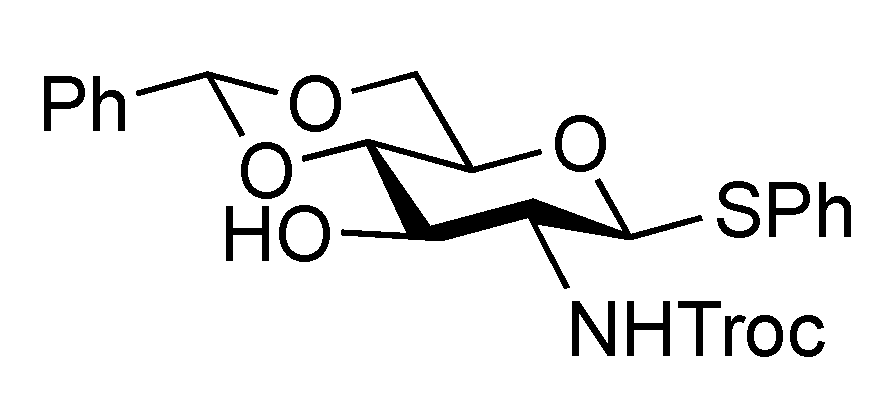
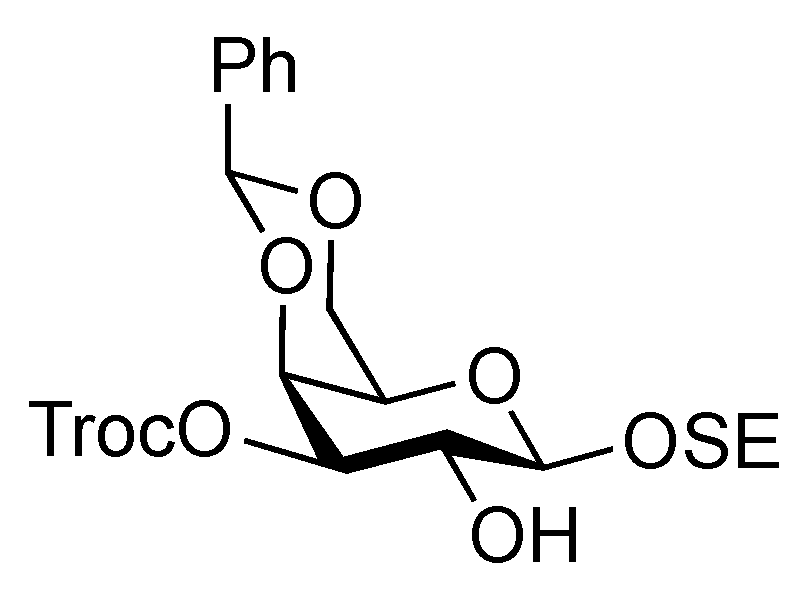
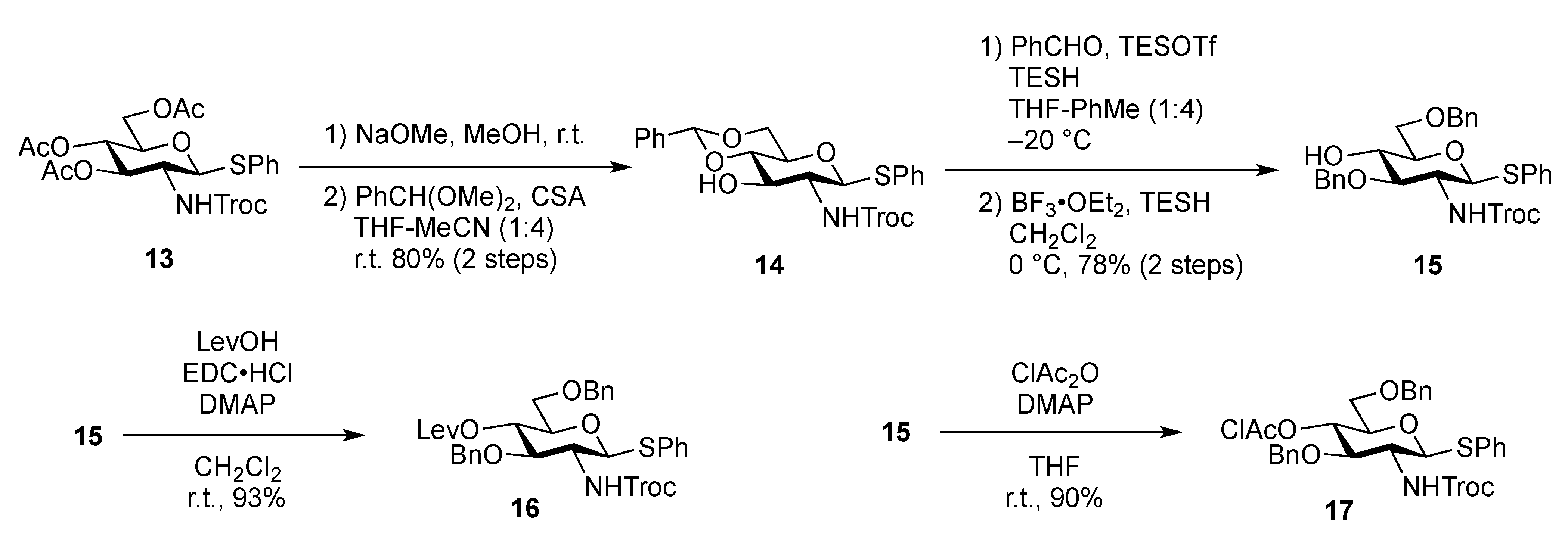
Phenyl 3,6-di-O-benzyl-2-deoxy-1-thio-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranoside (15). To a solution of 14 (4.89 g, 9.15 mmol) in THF/toluene (1:4, 9.0 mL) was added TESOTf (4.1 mL, 18.3 mmol) at −20 °C. After stirring for 45 min at −20 °C, benzaldehyde (4.7 mL, 45.7 mmol) and triethylsilane (2.2 mL, 13.7 mmol) were added to the mixture. After stirring for 2 h at −20 °C as the reaction was monitored by TLC (1:4 EtOAc–toluene), the reaction was quenched by satd aq Na2CO3. Dilution of the mixture with EtOAc provided a solution, which was then washed with satd aq Na2CO3 and brine. The organic layer was subsequently dried over Na2SO4 and concentrated. The obtained residue was exposed to high vacuum for 24 h. The resulting residue was dissolved in CH2Cl2 (183 mL) and cooled to 0 °C. BF3·OEt2 (4.7 mL, 18.3 mmol) and triethylsilane (14.6 mL, 91.5 mmol) were added to the solution at 0 °C and the mixture was then stirred for 1 h at 0 °C as the reaction was monitored by TLC (1:4 EtOAc–toluene). The reaction was quenched by the addition of satd aq Na2CO3 at 0 °C and then diluted with CHCl3, and washed with satd aq Na2CO3 and brine. The organic layer was subsequently dried over Na2SO4 and concentrated. The resulting residue was purified by silica gel column chromatography (200:1 CHCl3–MeOH) to give 15 (4.50 g, 78%). [α]D −24.2° (c 0.3, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.49–7.21 (m, 15 H, 3 Ph), 5.17 (d, 1 H, J2,NH = 8.2 Hz, NH), 4.91 (d, 1 H, , J1,2 = 10.1 Hz, H-1), 4.75 (s, 4 H, 2 OCH2), 4.56 (2 d, 2 H, OCH2), 3.77–3.67 (m, 4 H, H-3, H-5, H-6, H-6'), 3.52–3.42 (m, 2 H, H-2, H-4), 2.85 (s, 1 H, OH); 13C-NMR (100 MHz, CDCl3) δ 153.8, 137.9, 137.7, 132.7, 132.3, 128.9, 128.5, 128.4, 128.1, 128.0, 127.8, 127.7, 95.4, 86.0, 81.9, 78.0, 74.5, 74.4, 73.7, 72.6, 70.4, 56.0. m/z (MALDI): found [M+Na]+ 648.05, C29H30Cl3NO6S calcd for [M+Na]+ 648.08.
Phenyl 3,6-di-O-benzyl -2-deoxy-4-O-levulinoyl-1-thio-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranoside (16). To a solution of 15 (200 mg, 0.314 mmol) in CH2Cl2 (3.1 mL) were added levulinic acid (55 mg, 0.472 mmol), EDC·HCl (90 mg, 0.472 mmol), and DMAP (4.2 mg, 3.44 μmol). After stirring for 4 h at r.t. as the reaction was monitored by TLC (2:3 EtOAc–n-hexane), the reaction mixture was diluted with CHCl3, and washed with 2 M HCl and brine. The organic layer was subsequently dried over Na2SO4, and concentrated, and the obtained residue was purified by silica gel column chromatography (1:4 EtOAc–n-hexane) to give 16 (215 mg, 93%). [α]D +3.7° (c 0.9, CHCl3); 1H-NMR (600 MHz, acetone-d6) δ 7.53–7.24 (m, 16 H, 3 Ph, NH), 5.06 (d, 1 H, J1,2 = 10.3 Hz, H-1), 5.03 (t, 1 H, J3,4 = 9.6 Hz, J4,5 = 9.7 Hz, H-4), 4.83 (2 d, 2 H, Jgem = 12.4 Hz, OCH2), 4.72 (2 d, 2 H, Jgem = 11.0 Hz, OCH2), 4.52 (2 d, 2 H, Jgem = 11.7 Hz, OCH2), 3.99 (t, 1 H, J2,3 = 9.7 Hz, H-3), 3.79–3.75 (m, 2 H, H-2, H-5), 3.65 (dd, 1 H, Jgem = 11.0 Hz, J5,6 = 2.8 Hz, H-6), 3.65 (dd, 1 H, J5,6' = 6.9 Hz, H-6'), 2.73–2.40 (m, 4 H, CH2CH2C(=O)O), 2.09 (s, 3 H, C(=O)CH3); 13C-NMR (150 MHz, CDCl3) δ 166.1, 153.7, 137.6, 137.5, 132.6, 132.1, 129.0, 128.5, 128.3, 128.0, 128.0, 127.9, 127.7, 95.4, 85.2, 79.0, 76.8, 74.5, 74.4, 73.6, 73.2, 69.5, 56.7, 40.5. m/z (MALDI): found [M+Na]+ 746.09, C34H36Cl3NO8S calcd for [M+Na]+ 746.11.
Phenyl 3,6-di-O-benzyl-4-O-chloroacetyl-2-deoxy-1-thio-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranoside (17). To a solution of 15 (200 mg, 0.314 mmol) in THF (3.1 mL) were added monochloroacetic anhydride (81 mg, 0.472 mmol) and DMAP (0.5 mg, 4.09 μmol) at 0 °C. After stirring for 7 h at r.t. as the reaction was monitored by TLC (2:3 EtOAc–n-hexane), THF was evaporated and the residue was then diluted with CHCl3, and washed with 2 M HCl, H2O, satd aq NaHCO3, and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography (1:8 EtOAc–n-hexane) to give 17 (200 mg, 90%). [α]D −2.6° (c 1.0, CHCl3); 1H-NMR (600 MHz, CDCl3) δ 7.51–7.21 (m, 15 H, 3 Ph), 5.37 (d, 1 H, JNH,2 = 8.2 Hz, NH), 5.12 (d, 1 H, J1,2 = 10.3 Hz, H-1), 5.07 (near t, 1 H, J3,4 = 8.9 Hz, J4,5 = 9.6 Hz, H-4), 4.75 (2 d, 2 H, Jgem = 11.8 Hz, OCH2), 4.61 (2 d, 2 H, Jgem =11.7 Hz, OCH2), 4.48 (m, 2 H, Jgem = 11.7 Hz, OCH2), 4.11 (near t, 1 H, J2,3 = 9.6 Hz, J3,4 = 8.9 Hz, H-3), 3.68–3.55 (m, 5 H, H-5, H-6, H-6', CH2Cl), 3.36 (br dd, 1 H, J2,3 = 9.6 Hz, H-2); 13C-NMR (125 MHz, CDCl3) δ 166.1, 153.7, 137.7, 137.6, 132.8, 132.0, 129.1, 128.5, 128.4, 128.2, 128.0, 127.9, 127.8, 95.4, 85.2, 79.0, 77.6, 74.7, 74.5, 73.7, 73.4, 69.7, 56.9, 40.5. m/z (MALDI): found [M+Na]+ 724.05, C31H31Cl4NO7S calcd for [M+Na]+ 724.05.
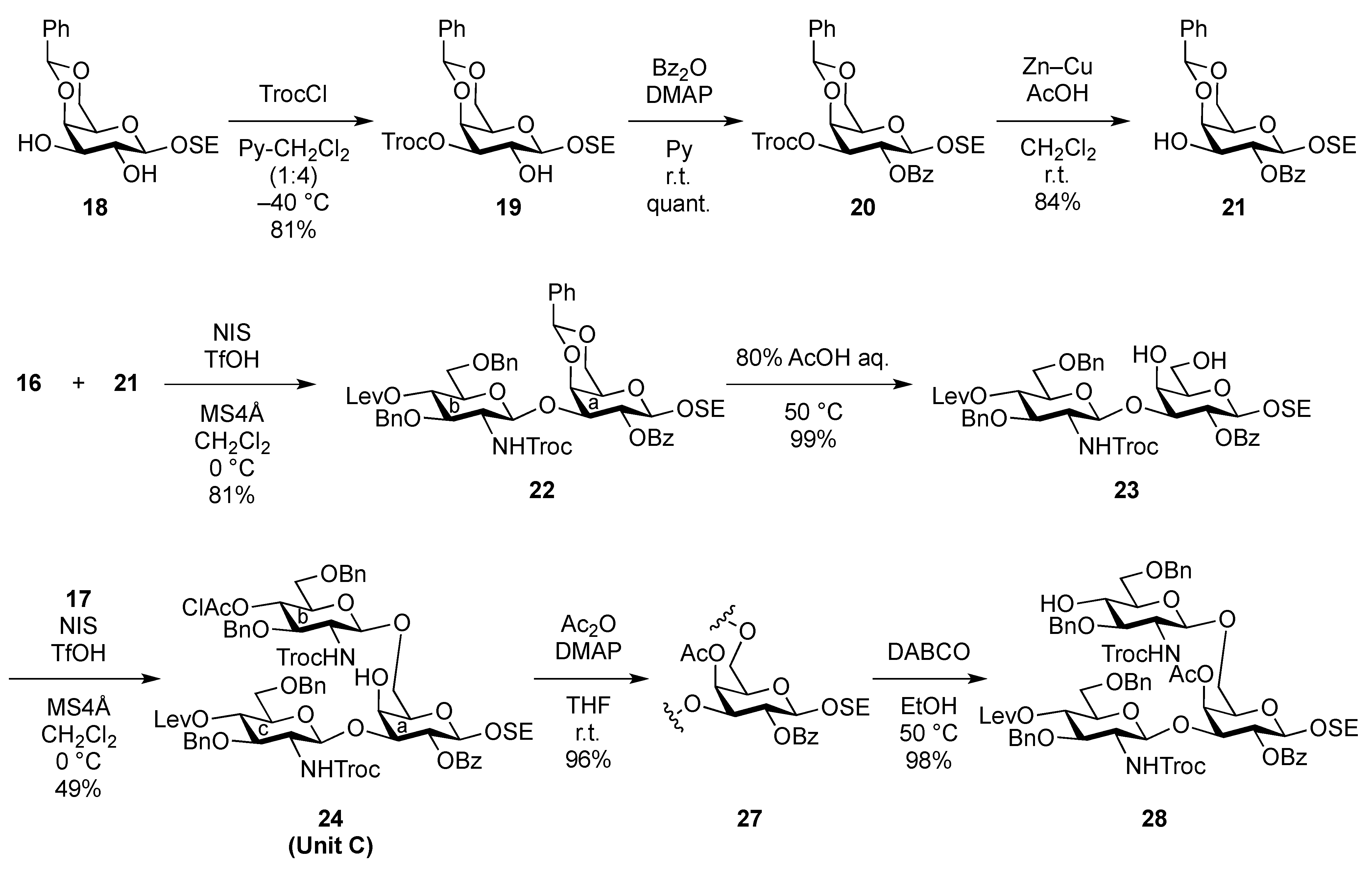
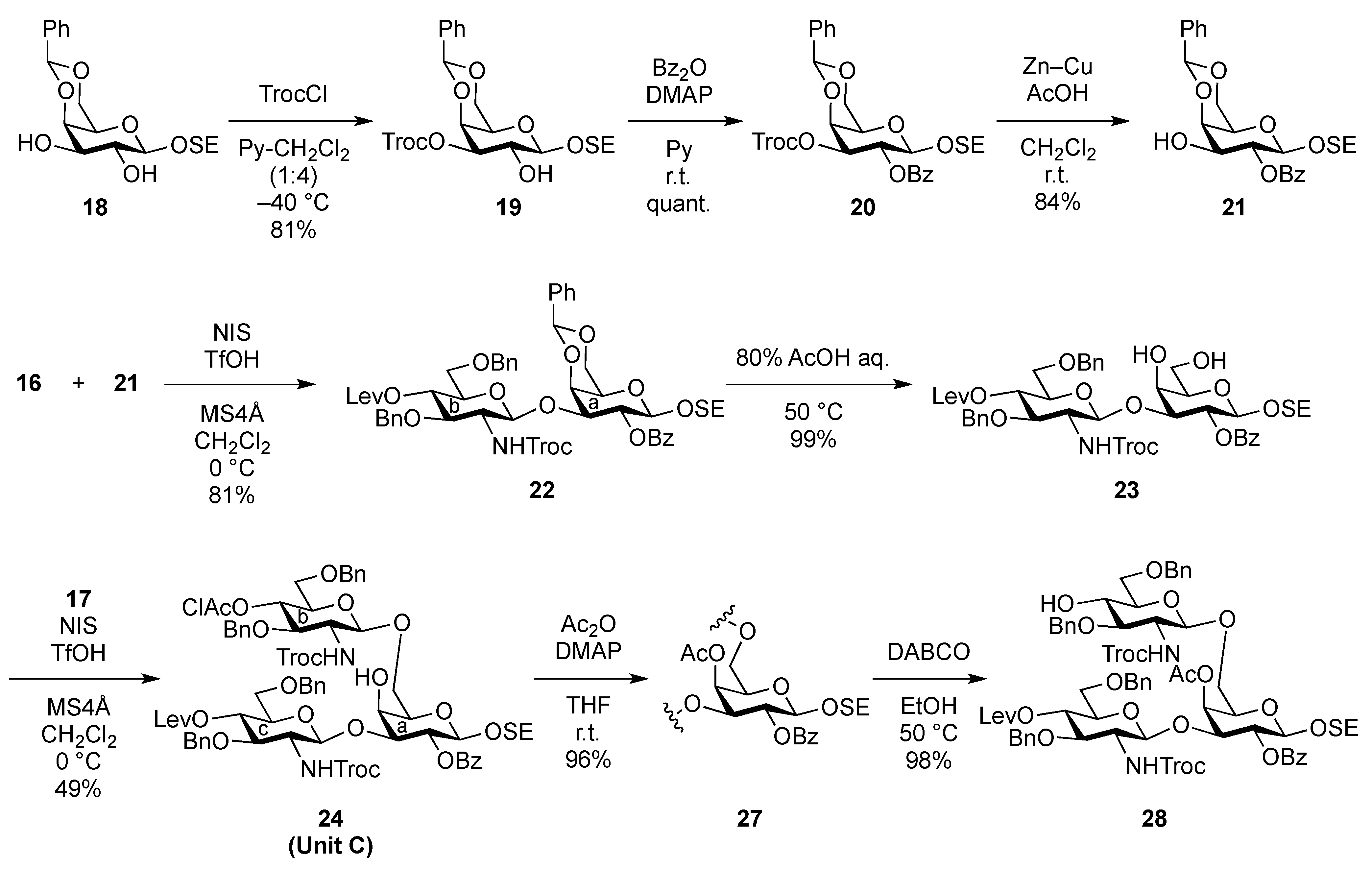
2-(Trimethylsilyl)ethyl 4,6-O-benzylidene-3-O-(2,2,2-trichloroethoxycarbonyl)-β-D-galactopyranoside (19). To a solution of 18 (49.5 mg, 0.134 mmol) in pyridine/CH2Cl2 (1:4, 1.3 mL) was added trichloroethyl chloroformate (20 μL, 0.148 mmol) at −40 °C. After stirring for 3 h at the same temperature as the reaction was monitored by TLC (30:1 CHCl3–MeOH), solvents were removed by co-evaporation with toluene, and then the residue was diluted with CHCl3, washed with 2 M HCl, H2O, satd aq NaHCO3, and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (1:5 EtOAc–n-hexane) to give 19 (59.1 mg, 81%). [α]D +41.7° (c 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.48–7.30 (m, 5 H, Ph), 5.49 (s, 1 H, PhCH<), 4.82–4.72 (m, 3 H, H-3, OCH2CCl3), 4.45 (d, 1 H, J3,4 = 3.7 Hz, H-4), 4.34 (d, 1 H, J1,2 = 7.7 Hz, H-1), 4.33 (d, 1 H, Jgem = 13.8 Hz, H-6), 4.08–4.01 (m, 3 H, H-2, H-6', OCH2CH2SiMe3), 3.60–3.53 (m, 1 H, OCH2CH2SiMe3), 3.48 (s, 1 H, H-5), 2.51 (br s, 1 H, OH), 1.03–0.97 (m, 2 H, OCH2CH2SiMe3), 0.00 (s, 9 H, OCH2CH2SiMe3); 13C-NMR (100 MHz, CDCl3) δ 153.6, 137.3, 129.0, 128.1, 126.2, 102.3, 100.9, 94.2, 78.2, 77.2, 76.9, 73.0, 69.0, 68.5, 67.5, 66.2, 18.1, −1.4. m/z (MALDI): found [M+Na]+ 565.11, C16H16Cl3O7 calcd for [M+Na]+ 565.06.
2-(Trimethylsilyl)ethyl 2-O-benzoyl-4,6-O-benzylidene-3-O-(2,2,2-trichloroethoxycarbonyl)-β-D-galactopyranoside (20). To a solution of 19 (2.39 g, 4.39 mmol) in THF (1.3 mL) were added benzoic anhydride (1.49 g, 6.59 mmol) and DMAP (268 mg, 2.20 μmol) at 0 °C. After stirring for 8.5 h at r.t. as the reaction was monitored by TLC (30:1 CHCl3–MeOH), THF was evaporated and the residue was then diluted with CHCl3, washed with 2 M HCl, H2O, satd aq NaHCO3, and brine. The organic layer was subsequently dried over Na2SO4, concentrated. The residue was purified by silica gel column chromatography (20:1 toluene–EtOAc) to give 20 (2.90 g, quant.). [α]D +38.8° (c 1.2, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 8.11–7.43 (m, 10 H, 2 Ph), 5.79–7.75 (near t, 1 H, J2,3 = 10.6 Hz, J1,2 = 7.8 Hz, H-2), 5.64 (s, 1 H, PhCH<), 5.22 (dd, 1 H, J3,4 = 3.7 Hz, H-3), 4.81–4.78 (2 d, 2 H, OCH2CCl3, H-1), 4.68 (d, 1 H, Jgem = 11.9 Hz, OCH2CCl3), 4.58 (d, 1 H, H-4), 4.67–4.50 (dd, 1 H, Jgem = 12.4 Hz, J5,6 = 1.8 Hz, H-6), 4.23–4.19 (dd, 1 H, J5,6´ = 1.8 Hz, H-6'), 4.15–4.08 (m, 1 H, OCH2CH2SiMe3), 3.68–3.62 (m, 2 H, H-5, OCH2CH2SiMe3), 1.03–0.91 (m, 2 H, OCH2CH2SiMe3), 0.00 (s, 9 H, OCH2CH2SiMe3); 13C-NMR (100 MHz, CDCl3) δ 164.8, 153.7, 137.2, 133.1, 129.8, 129.7, 129.1, 128.3, 128.2, 126.4, 101.1, 100.5, 94.1, 76.8, 76.6, 73.4, 69.2, 68.9, 67.0, 66.2, 17.9, −1.5. m/z (MALDI): found [M+Na]+ 669.09, C28H33Cl3O9Si calcd for [M+Na]+ 669.09.
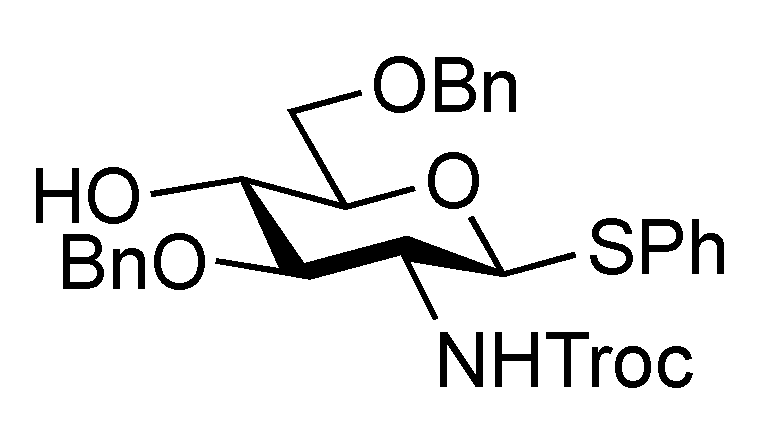
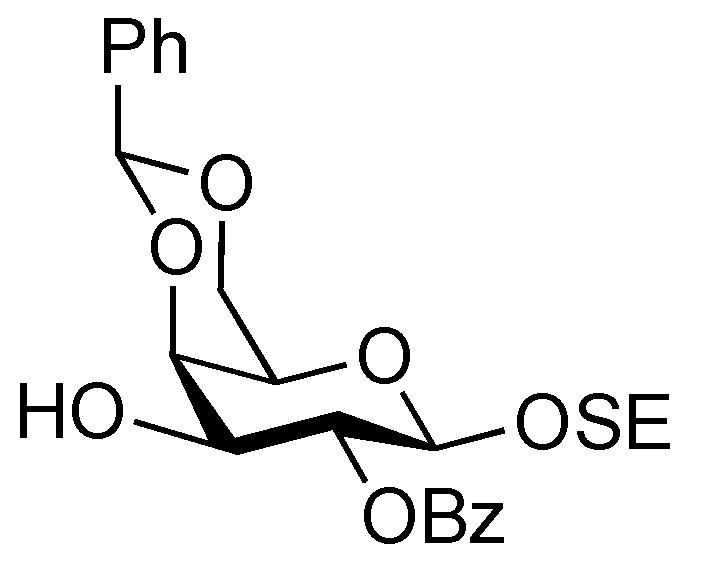
2-(Trimethylsilyl)ethyl 2-O-benzoyl-4,6-O-benzylidene-β-D-galactopyranoside (21). To a solution of 20 (2.18 g, 3.36 μmol) in AcOH/CH2Cl2 (3:2, 33.6 mL) was added Zn/Cu couple (6.00 g) at r.t. The reaction mixture was stirred for 1 h at r.t. as the reaction was monitored by TLC (30:1 CHCl3–MeOH). The precipitate was filtered through Celite and the filtrate was co-evaporated with toluene. The residue was diluted with CHCl3 and washed with satd aq NaHCO3 and brine, dried over Na2SO4, and concentrated. The obtained residue was purified by silica gel column chromatography (4:1 EtOAc–n-hexane) to give 21 (1.33 g, 84%). [α]D −3.8° (c 1.1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 8.15–7.45 (m, 10 H, 2 Ph), 5.65 (s, 1 H, PhCH<), 5.43 (near t, 1 H, J2,3 = 10.3 Hz, J1,2 = 8.3 Hz, H-2), 4.68 (d, 1 H, J1,2 = 8.3 Hz, H-1), 4.45 (d, 1 H, Jgem = 12.3 Hz, H-6), 4.33 (d, 1 H, J3,4 = 4.2 Hz, H-4), 4.18 (dd, 1 H, Jgem = 12.4 Hz, H-6'), 4.12 (m, 1 H, OCH2CH2SiMe3), 3.97 (m, 1 H, J3,OH = 11.0 Hz, J2,3 = 10.3 Hz, H-3), 3.68–3.60 (m, 2 H, H-5, OCH2CH2SiMe3), 2.72 (d, 1 H, OH), 0.98 (m, 2 H, CH2CH2SiMe3), 0.00 (s, 3 H, CH2CH2SiMe3); 13C-NMR (100 MHz, CDCl3) δ 166.2, 137.4, 132.9, 130.0, 129.8, 129.2, 128.2, 126.4, 101.4, 100.3, 75.6, 72.9, 71.9, 69.0, 67.0, 66.5, 17.9, −1.5. m/z (MALDI): found [M+Na]+ 495.12, C25H32O7Si calcd for [M+Na]+ 495.18.
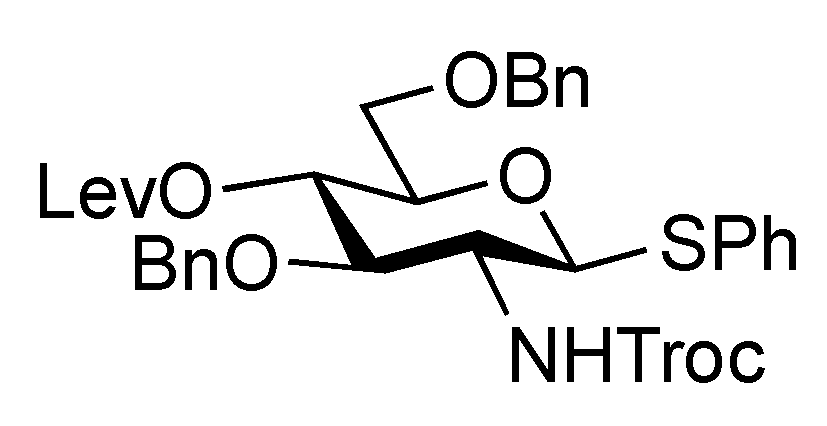
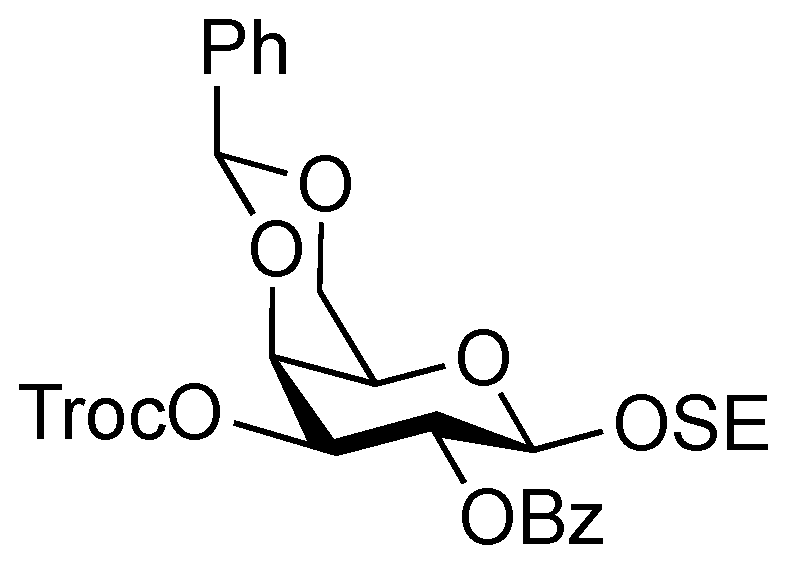
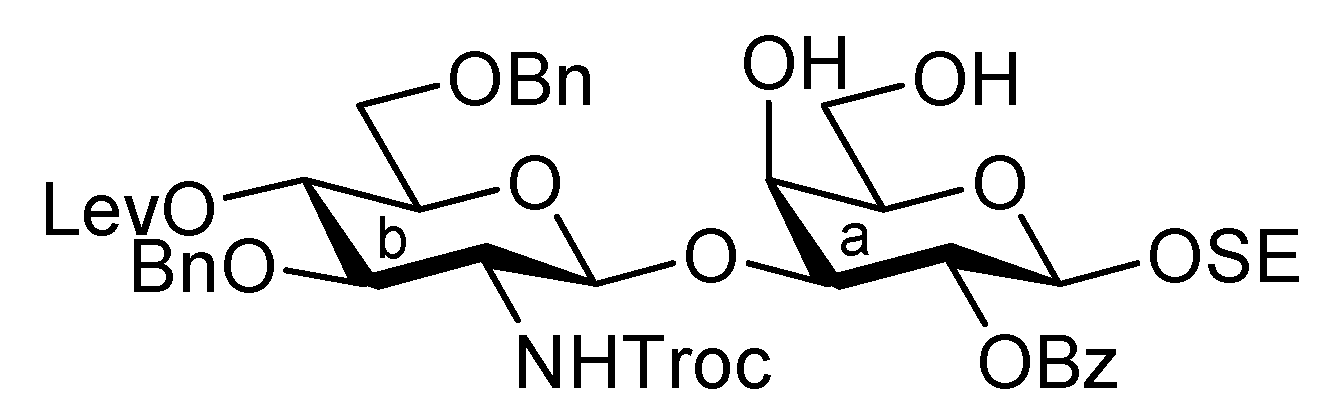
2-(Trimethylsilyl)ethyl 3,6-di-O-benzyl-2-deoxy-4-O-levulinoyl-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranosyl-(1→3)-2-O-benzoyl-4,6-O-benzylidene-β-D-galactopyranoside (22). To a mixture of 16 (230 mg, 0.317 mmol) and 21 (100 mg, 0.212 mmol) in CH2Cl2 (3.2 mL) was added 4 Å molecular sieves AW-300 (400 mg) at r.t. After stirring for 1 h, the mixture was cooled to 0 °C. NIS (143 mg, 0.636 mmol) and TfOH (2.8 μL, 31.7 μmol) were then added to the mixture at 0 °C. After stirring for 2 h at the same temperature as the reaction was monitored by TLC (1:4 EtOAc–toluene), the reaction was quenched by the addition of triethylamine. The solution was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd aq Na2S2O3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (1:4 EtOAc–toluene, then 200:1 CHCl3–MeOH) to give 22 (186 mg, 81%). [α]D +17.4° (c 0.8, CHCl3); 1H-NMR (600 MHz, CDCl3) δ 8.07–7.13 (m, 20 H, 4 Ph), 5.55 (near t, 1 H, J2,3 = 10.3 Hz, J1,2 = 7.6 Hz, H-2a), 5.49 (s, 1 H, PhCH<), 5.26 (d, 1 H, JNH,2 = 6.2 Hz, NH), 5.08 (d, 1 H, J1,2 = 7.6 Hz, H-1b), 4.92 (near t, 1 H, J3,4 = 9.9 Hz, J4,5 = 9.4 Hz, H-4b), 4.53 (d, 1 H, H-1a), 4.55–4.51 (m, 2 H, 2 OCH2), 4.46–4.41 (m, 3 H, 3 OCH2), 4.40 (d, 1 H, J3,4 = 3.4 Hz, H-4a), 4.26 (d, 1 H, Jgem = 12.4 Hz, H-6'a), 4.15 (t, 1 H, J3,4 = 9.9 Hz, H-3b), 4.00–3.95 (m, 2 H, H-3a, OCH2CH2SiMe3), 3.83 (d, 1 H, Jgem = 12.4 Hz, H-6a), 3.66 (br m, 1 H, H-5b), 3.55 (m, 3 H, H-6b, H-6'b, OCH2CH2SiMe3), 3.35 (s, 1 H, H-5a), 3.28 (br d, 1 H, OCH2), 3.17 (br dd, 1 H, H-2b), 2.61–2.36 (m, 4 H, CH2CH2C(=O)O), 2.10 (s, 3 H, C(=O)CH3), 0.86–0.76 (m, 2 H, CH2CH2SiMe3), −0.12 (m, 9 H, CH2CH2SiMe3); 13C-NMR (125 MHz, CDCl3) δ 206.2, 171.6, 164.9, 153.4, 138.1, 138.0, 137.8, 133.0, 130.2, 129.9, 128.9, 128.4, 128.3, 128.3, 128.1, 127.7, 127.6, 127.6, 127.5, 126.5, 101.1, 100.8, 100.2, 95.4, 78.7, 77.5, 76.2, 74.2, 73.4, 73.3, 73.2, 71.6, 70.5, 69.9, 68.8, 66.7, 66.6, 58.2, 37.7, 29.7, 27.8, 17.9, −1.6. m/z (MALDI): found [M+Na]+ 1108.51, C53H62Cl3NO15Si calcd for [M+Na]+ 1108.29.
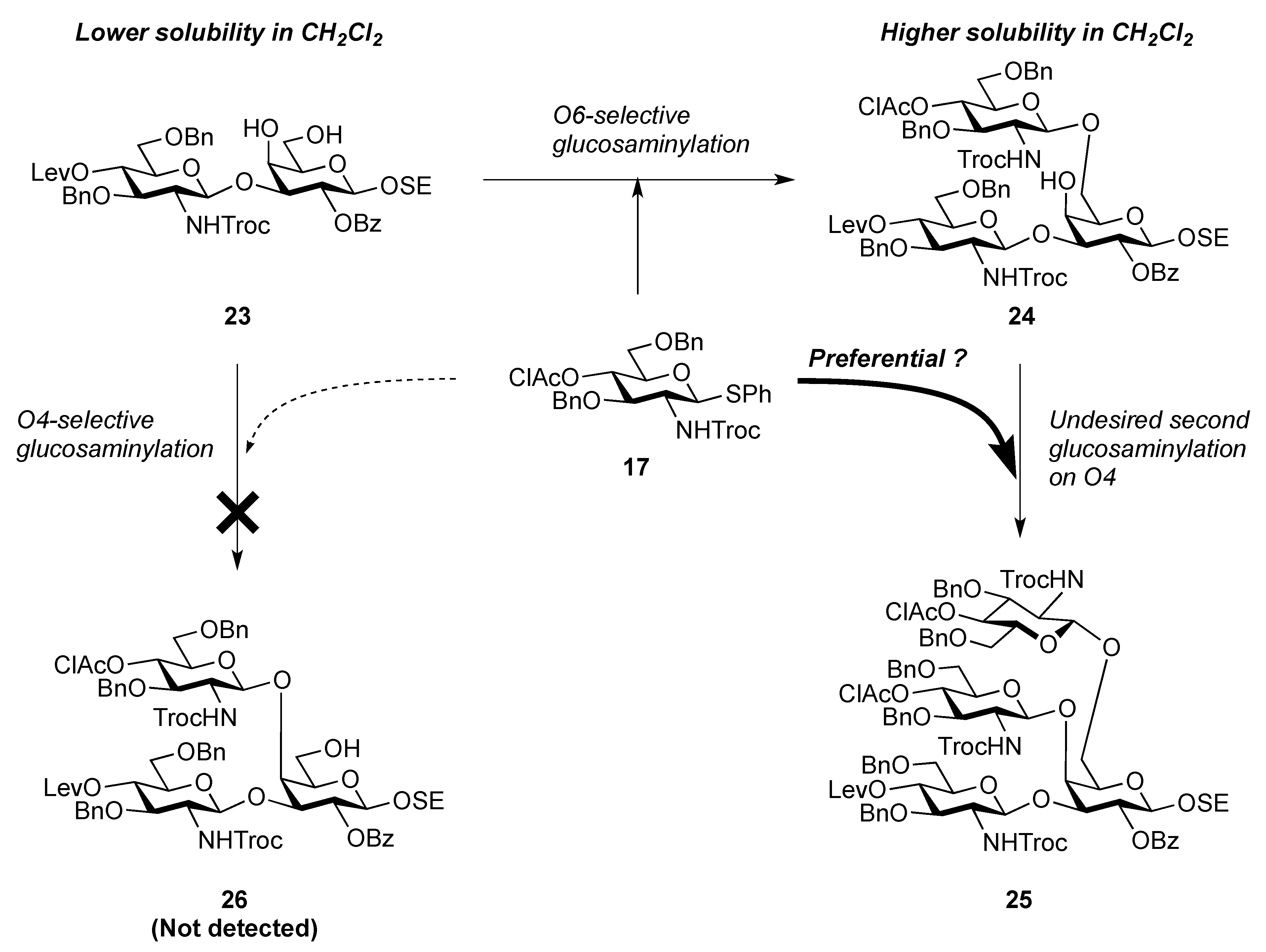
2-(Trimethylsilyl)ethyl 3,6-di-O-benzyl-2-deoxy-4-O-levulinoyl-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranosyl-(1→3)-2-O-benzoyl-β-D-galactopyranoside (23). Compound 22 (42 mg, 38.3 μmol) was dissolved in 80% AcOH aq (1.5 mL) and the solution was stirred for 4 h at 50 °C as the reaction was monitored by TLC (15:1 CHCl3–MeOH). After the reaction mixture was co-evaporated with toluene, the crude residue was purified by silica gel column chromatography (50:1 CHCl3–MeOH) to give 23 (38 mg, 99%). [α]D −1.43° (c 0.7, CHCl3); 1H-NMR (400 MHz, acetone-d6) δ 8.34–7.32 (m, 15 H, 3 Ph), 7.03 (d, 1 H, JNH,2 = 9.2 Hz, NH), 5.60 (near t, 1 H, J2,3 = 9.7 Hz, J1,2 = 8.0 Hz, H-2a), 5.13 (d, 1 H, J1,2 = 8.6 Hz, H-1b), 5.09 (near t, J3,4 = 9.9 Hz, J4,5 = 8.3 Hz, H-4b), 4.77–4.64 (m, 5 H, H-1a, 4 OCH2), 4.48 (d, 1 H, J3,4 = 3.4 Hz, H-4a), 4.51 (d, 1 H, OCH2), 4.23 (dd, 1 H, H-3a), 4.13–4.08 (m, 2 H, H-3b, OCH2), 3.96–3.63 (m, 9 H, 2 OCH2, H-2b, H-5b, H-6b, H-6'b, H-5a, H-6a, H-6'a), 2.98 (s, 2 H, 2 OH), 2.80–2.49 (m, 4 H, CH2CH2C(=O)O), 2.20 (s, 3 H, C(=O)CH3), 1.01–0.83 (m, 2 H, CH2CH2SiMe3), 0.00 (s, 9 H, CH2CH2SiMe3); 13C-NMR (100 MHz, acetone-d6) δ 206.5, 172.5, 165.6, 154.8, 139.5, 133.7, 131.7, 130.7, 129.2, 129.0, 128.8, 128.4, 128.3, 128.2, 128.0, 102.7, 101.7, 96.8, 82.4, 80.2, 75.9, 74.6, 74.1, 74.0, 73.9, 72.1, 72.0, 70.4, 69.5, 67.0, 62.6, 58.3, 38.1, 29.6, 28.6, 18.6, −1.4. m/z (MALDI): found [M+Na]+ 1020.26, C46H58Cl3NO15Si calcd for [M+Na]+ 1020.25.
2-(Trimethylsilyl)ethyl 3,6-di-O-benzyl-2-deoxy-4-O-levulinoyl-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranosyl-(1→3)-[3,6-di-O-benzyl-4-O-chloroacetyl-2-deoxy-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranosyl-(1→6)]-2-O-benzoyl-β-D-galactopyranoside (24). To a mixture of 17 (694 mg, 0.987 mmol) and 23 (658 mg, 0.658 mmol) in CH2Cl2 (6.6 mL) was added 4 Å molecular sieves (1.40 g) at r.t. After stirring for 1 h, the mixture was cooled to 0 °C. NIS (296 mg, 1.32 mmol) and TfOH (8.7 μL, 98.7 μmol) were then added to the mixture at 0 °C. After stirring for 2 h at r.t. as the reaction was monitored by TLC (30:1 CHCl3–MeOH), the reaction was quenched by the addition of triethylamine. The solution was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd aq Na2S2O3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography (100:1 CHCl3–MeOH) to give 24 (511 mg, 49%). [α]D −19.4° (c 1.8, CHCl3); 1H-NMR (400 MHz, DMSO-d6) δ 8.20–7.32 (m, 27 H, 5 Ph, NHb, NHc), 5.38 (t, 1 H, J2,3 = 8.3 Hz, J1,2 = 8.7 Hz, H-2a), 5.14 (t, 1 H, J4,5 = 9.6 Hz, H-4c), 5.06 (d, 1 H, Jgem = 12.4 Hz, OCH2), 5.01 (t, 1 H, J3,4 = 9.6 Hz, J4,5 = 9.1 Hz, H-4b), 4.90 (d, 1 H, J1,2 = 8.2 Hz, H-1c), 4.75 (d, 1 H, J1,2 = 8.3 Hz, H-1b), 4.66 (d, 1 H, H-1a), 4.91–4.61 (m, 10 H, OH, 9 OCH2), 4.49 (2 d, 2 H, OCH2), 4.24 (br s, 1 H, H-4a), 4.04–3.52 (m, 18 H, H-3a, H-5a, H-6a, H-6'a, H-2b, H-3b, H-5b, H-6b, H-6'b, H-2c, H-3c, H-5c, H-6c, H-6'c, OCH2CH2SiMe3, CH2Cl), 2.83–2.80 (m, 2 H, CH2CH2C(=O)O), 2.56 (m, 2 H, CH2CH2C(=O)O), 2.26 (s, 3 H, C(=O)CH3), 0.93–0.78 (m, 2 H, CH2CH2SiMe3), 0.00 (s, 9 H, CH2CH2SiMe3); 13C-NMR (100 MHz, DMSO-d6) δ 206.8, 171.5, 166.5, 164.7, 154.4, 153.8, 138.4, 138.3, 138.2, 133.2, 130.3, 129.6, 128.6, 128.4, 128.3, 128.2, 127.7, 127.7, 127.6, 101.9, 101.1, 100.2, 96.4, 96.0, 80.7, 79.4, 78.9, 73.7, 73.6, 73.3, 72.8, 72.7, 72.6, 72.5, 72.2, 72.0, 70.9, 70.7, 69.6, 69.0, 68.7, 68.6, 66.1, 57.0, 41.1, 37.4, 29.7, 27.8, 17.6, −1.3. m/z (MALDI): found [M+Na]+ 1611.27, C71H83Cl7N2O22Si calcd for [M+Na]+ 1611.29.
2-(Trimethylsilyl)ethyl 3,6-di-O-benzyl-2-deoxy-4-O-levulinoyl-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranosyl-(1→3)-[3,6-di-O-benzyl-4-O-chloroacetyl-2-deoxy-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranosyl-(1→6)]-4-O-acetyl-2-O-benzoyl-β-D-galactopyranoside (27). To a solution of 24 (759 mg, 0.476 mmol) in THF (4.8 mL) were added acetic anhydride (94 μL, 0.956 mmol) and DMAP (5.8 mg, 47.6 μmol) at 0 °C. The reaction mixture was stirred for 1.5 h at r.t. as the reaction was monitored by TLC (15:1 CHCl3–MeOH). After THF was evaporated, the obtained crude residue was purified by silica gel column chromatography (100:1 CHCl3–MeOH) to give 27 (745 mg, 96%). [α]D −5.5° (c 1.8, CHCl3); 1H-NMR (400 MHz, DMSO-d6) δ 8.20–7.32 (m, 27 H, 5 Ph, NHb, NHc), 5.58 (br s, 1 H, H-4a), 5.34 (near t, 1 H, J2,3 = 9.7 Hz, J1,2 = 8.2 Hz, H-2a), 5.16–5.07 (m, 3 H, H-4c, H-4b, OCH2), 4.86–4.61 (m, 10 H, H-1b, H-1c, H-1a, 7 OCH2), 4.58–4.39 (m, 3 H, 3 OCH2), 4.31 (br dd, 1 H, H-3a), 4.18 (br dd, 1 H, H-6a), 4.06–3.48 (m, 17 H, H-5a, H-6'a, H-2b, H-3b, H-5b, H-6b, H-6'b, H-2c, H-3c, H-5c, H-6c, H-6'c, OCH2, OCH2CH2SiMe3, CH2Cl), 2.80 (m, 2 H, CH2CH2C(=O)O), 2.54 (m, 2 H, CH2CH2C(=O)O), 2.25–2.22 (2 s, 6 H, 2 C(=O)CH3), 0.96–0.79 (m, 2 H, CH2CH2SiMe3), 0.00 (s, 9 H, CH2CH2SiMe3); 13C-NMR (100 MHz, DMSO-d6) δ 206.7, 179.5, 171.3, 170.0, 166.5, 164.5, 154.4, 153.8, 138.5, 138.3, 138.3, 138.2, 133.3, 130.1, 129.6, 128.7, 128.4, 128.3, 128.3, 128.1, 127.7, 127.6, 127.6, 127.5, 127.2, 101.3, 100.9, 100.0, 96.3, 96.0, 79.3, 79.0, 77.6, 73.6, 73.1, 72.7, 72.4, 72.3, 72.0, 71.1, 70.3, 70.2, 69.0, 68.8, 68.4, 66.4, 57.0, 41.1, 37.4, 29.7, 27.8, 20.8, 17.6, −1.3. m/z (MALDI): found [M+Na]+ 1653.57, C73H85Cl7N2O23Si calcd for [M+Na]+ 1653.30.
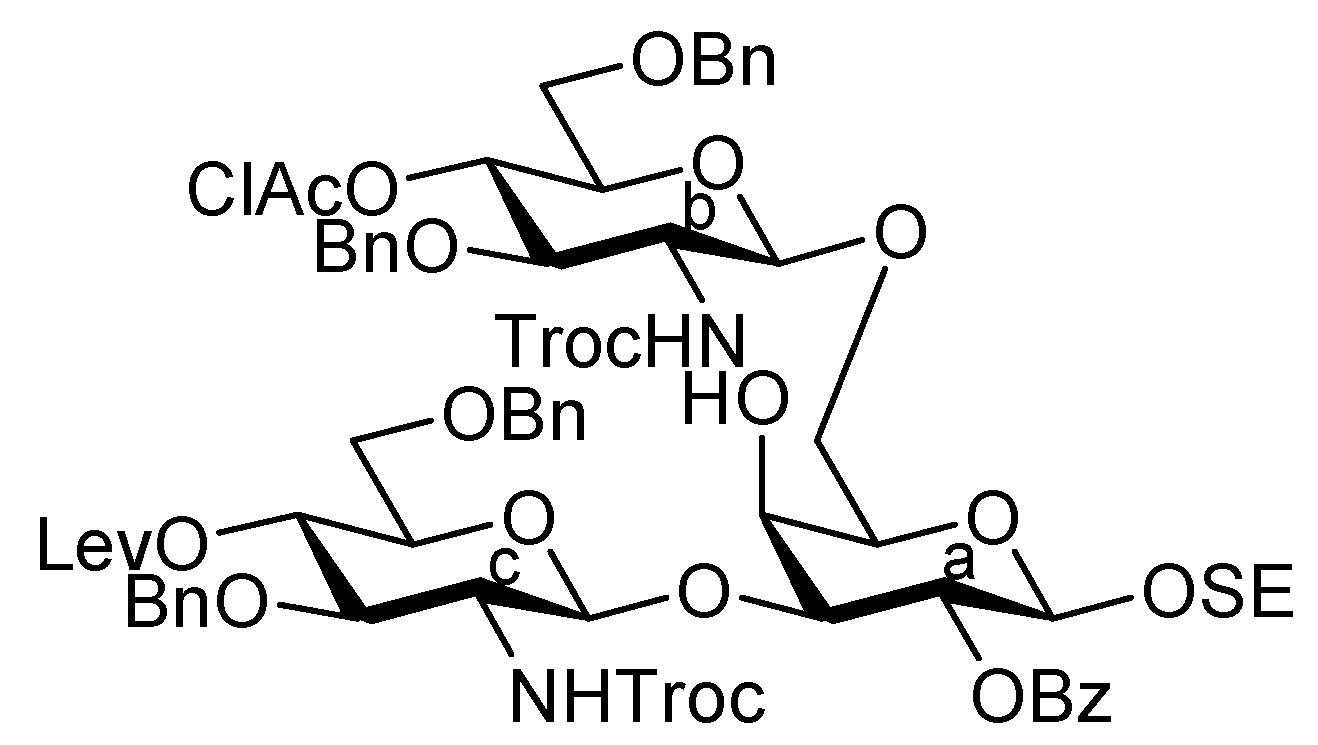
2-(Trimethylsilyl)ethyl 3,6-di-O-benzyl-2-deoxy-4-O-levulinoyl-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranosyl-(1→3)-[3,6-di-O-benzyl-2-deoxy-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranosyl-(1→6)]-4-O-acetyl-2-O-benzoyl-β-D-galactopyranoside (28). To a solution of 27 (712 mg, 0.435 mmol) in EtOH (4.4 mL) was added DABCO (732 mg, 6.52 mmol) at 0 °C. After stirring for 1 h at 50 °C as the reaction was monitored by TLC (30:1 CHCl3–MeOH), EtOH was evaporated. The obtained crude residue was purified by silica gel column chromatography (100:1 CHCl3–MeOH) to give 28 (665 mg, 98%). [α]D −1.8° (c 1.5, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 8.19–7.27 (m, 25 H, 5 Ph), 5.63 (d, 1 H, J3,4 = 3.4 Hz, H-4a), 5.52 (near t, 1 H, J2,3 = 10.3 Hz, J1,2 = 8.0 Hz, H-2a), 5.32 (br d, 1 H, NHb), 5.18 (d, 1 H, JNH,2 = 6.9 Hz, NHc), 5.07 (d, 1 H, J1,2 = 9.2 Hz, H-1c), 5.05 (near t, 1 H, J3,4 = 9.2 Hz, H-4c), 4.85 (d, 1 H, J1,2 = 8.6 Hz, H-1b), 4.91–4.80 (m, 3 H, 3 OCH2), 4.71 (d, 1 H, OCH2), 4.63 (d, 1 H, H-1a), 4.69–4.59 (m, 6 H, 6 OCH2), 4.50 (d, 1 H, OCH2), 4.16 (br t, 1 H, J2,3 = 10.3 Hz, H-3c), 4.09 (m, 1 H, OCH2CH2SiMe3), 4.07 (dd, 1 H, H-3a), 4.01 (dd, 1 H, J5,6 = 4.1 Hz, Jgem = 12.8 Hz, H-6a), 3.89–3.56 (m, 11 H, H-5a, H-6'a, H-3b, H-5b, H-6b, H-6'b, H-5c, H-6c, H-6'c, OCH2, OCH2CH2SiMe3), 3.44 (br dd, 1 H, H-2b), 3.23 (br dd, 1 H, H-2c), 2.96 (s, 1 H, OH), 2.72–2.37 (m, 4 H, CH2CH2C(=O)O), 2.22 (m, 6 H, 2 C(=O)CH3), 1.01–0.86 (m, 2 H, CH2CH2SiMe3), 0.00 (s, 9 H, CH2CH2SiMe3); 13C-NMR (100 MHz, CDCl3) δ 206.1, 171.4, 169.9, 164.8, 154.0, 153.3, 138.2, 138.1, 137.7, 137.5, 133.2, 129.9, 128.5, 128.4, 128.3, 128.0, 127.9, 127.8, 127.7, 127.6, 127.6, 100.7, 100.2, 95.6, 95.4, 80.5, 77.5, 74.3, 74.3, 73.9, 73.7, 73.4, 73.4, 73.2, 73.1, 71.5, 71.4, 70.5, 70.1, 69.6, 67.4, 57.9, 57.3, 37.6, 29.6, 27.8, 20.8, 17.9, −1.5. m/z (MALDI): found [M+Na]+ 1577.28, C71H84Cl6N2O22Si calcd for [M+Na]+ 1577.33.
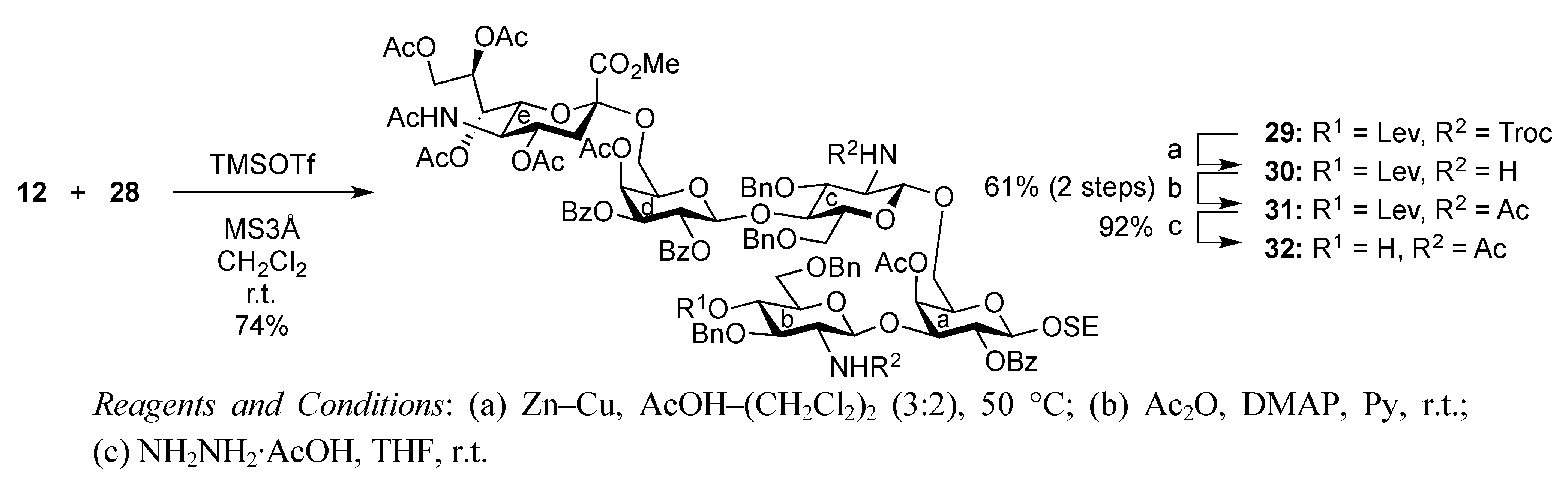
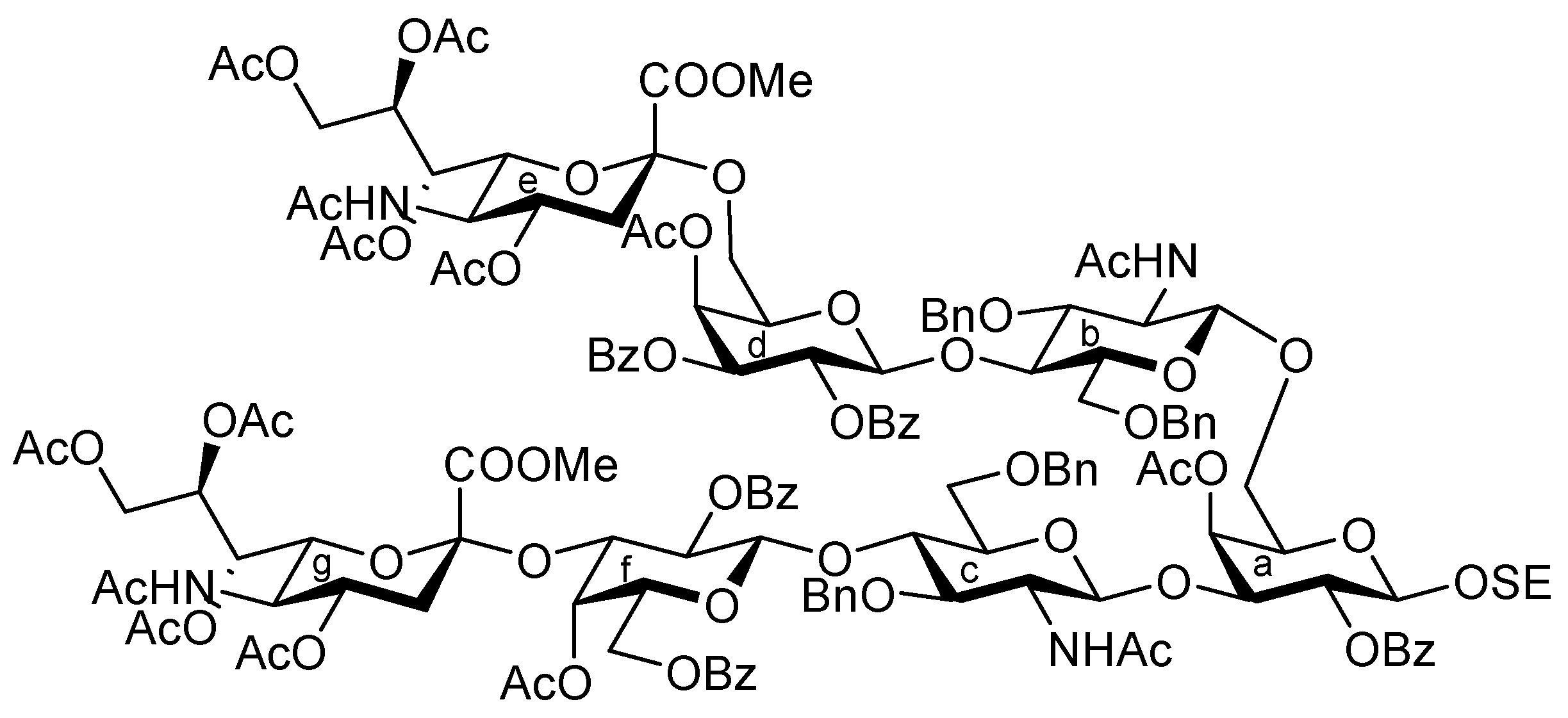
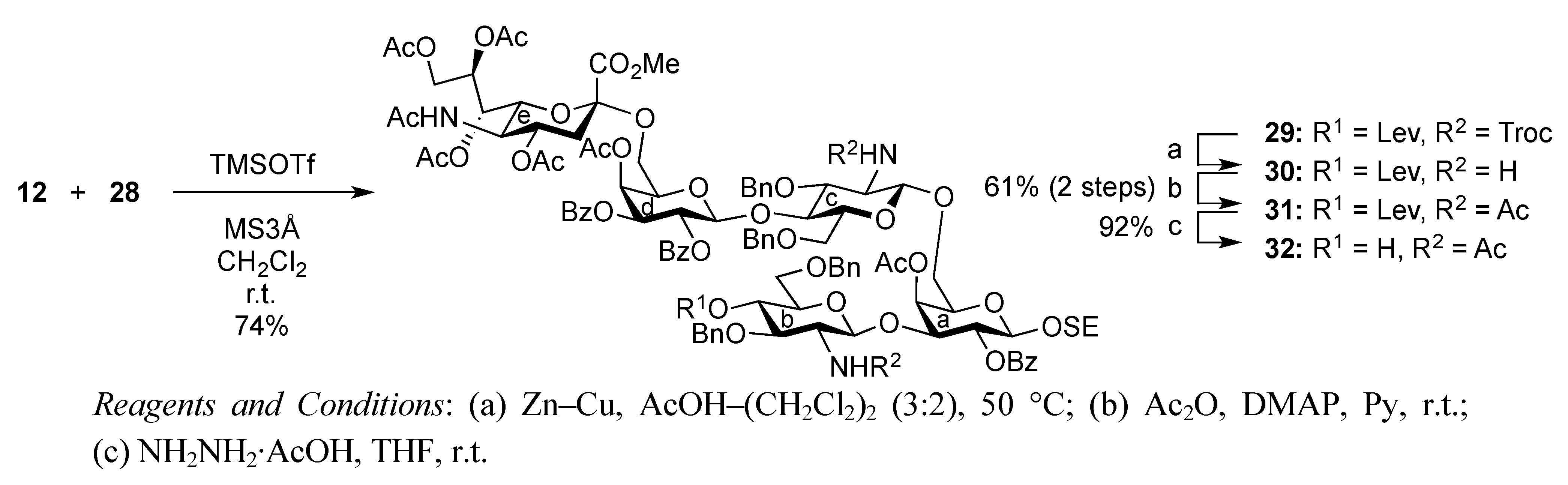
2-(Trimethylsilyl)ethyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-(2,2,2-trichloroethoxycarbamoyl)-β-D-glucopyranosyl-(1→6)-[3,6-di-O-benzyl-2-deoxy-4-O-levulinoyl-2-(2,2,2-trichloroethoxycarbamoyl)-α-D-glucopyranosyl-(1→3)]-4-O-acetyl-2-O-benzoyl-β-D-galactopyranoside (29). To a mixture of 12 (80 mg, 76.5 μmol) and 28 (70 mg, 45.0 μmol) in CH2Cl2 (0.9 mL) was added 3 Å molecular sieves (250 mg) at r.t. After stirring for 1 h, the mixture was cooled to 0 °C. TMSOTf (1.4 μL, 7.65 μmol) was then added to the mixture at 0 °C. After stirring for 25 h at r.t., TMSOTf (1.4 μL, 7.65 μmol) was added to the mixture. After the stirring was continued for 3 h at r.t. as the reaction was monitored by TLC (30:1 CHCl3–MeOH), the reaction was quenched by the addition of triethylamine. The mixture was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd aq NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (90:1 CHCl3–MeOH) followed by gel filtration column chromatography (LH-20) using MeOH as eluent, giving 29 (81 mg, 74%). [α]D −2.9° (c 1.4, CHCl3); 1H-NMR (600 MHz, CDCl3) δ 8.05–7.14 (m, 35 H, 7 Ph), 5.61 (d, 1 H, J3,4 = 3.3 Hz, H-4d), 5.59 (near t, 1 H, J1,2 = 8.3 Hz, H-2d), 5.45 (d, 1 H, J3,4 = 3.4 Hz, H-4a), 5.40–5.36 (m, 1 H, H-8e), 5.36 (t, 1 H, J1,2 = 8.2 Hz, H-2a), 5.34 (m, 1 H, H-7e), 5.30 (dd, 1 H, J2,3 = 10.2 Hz, H-3d), 5.23 (2 br d, 2 H, NHe, NHb), 5.02 (d, 1 H, J2,NH = 6.9 Hz, NHc), 5.12 (d, 1 H, Jgem = 11.0 Hz, OCH2), 4.94 (d, 1 H, H-1d), 4.94–4.86 (m, 2 H, H-1c, H-4c), 4.84 (m, 1 H, J3eq,4 = 4.2 Hz, H-4e), 4.73–4.68 (m, 3 H, 3 OCH2), 4.58–4.30 (m, 10 H, H-1a, H-1b, H-9e, 7 OCH2), 4.14–3.71 (m, 15 H, H-3a, H-6'a, H-3b, H-4b, H-3c, H-4c, H-5d, H-6d, H-6'd, H-5e, H-6e, OCH2, OCH3), 3.58–3.37 (m, 10 H, H-6a, H-2b, H-5b, H-6b, H-6'b, H-5c, H-6c, H-6'c, 2 OCH2), 3.28–3.26 (m, 1 H, H-5a), 3.08 (br dd, 1 H, H-2c), 2.60–2.51 (m, 3 H, H-3eeq, CH2CH2C(=O)O), 2.38–2.24 (m, 2 H, CH2CH2C(=O)O), 2.16–1.91 (m, 25 H, 8 C(=O)CH3, H-3eax), 0.85–0.69 (m, 2 H, CH2CH2SiMe3), −0.14 (s, 9 H, CH2CH2SiMe3); 13C-NMR (100 MHz, CDCl3) δ 206.0, 171.4, 170.9, 170.7, 170.3, 170.1, 169.8, 169.6, 167.8, 165.3, 165.2, 164.8, 153.9, 153.3, 138.6, 138.1, 138.0, 137.7, 133.3, 133.1, 129.8, 129.6, 129.5, 129.1, 128.5, 128.4, 128.4, 128.3, 128.3, 128.2, 127.9, 127.8, 127.7, 127.6, 127.6, 127.4, 100.6, 100.1, 99.8, 99.0, 95.4, 77.4, 76.8, 76.1, 74.4, 74.3, 73.9, 73.7, 73.3, 73.3, 73.1, 72.8, 71.7, 71.5, 71.4, 70.2, 70.1, 69.6, 68.7, 68.4, 68.1, 67.3, 62.4, 57.9, 57.0, 52.9, 49.4, 37.7, 37.6, 29.6, 27.8, 23.1, 21.0, 20.8, 20.5, 17.8, −1.5. m/z (MALDI): found [M+Na]+ 2463.09, C113H131Cl6N3O42Si calcd for [M+Na]+ 2462.60.
2-(Trimethylsilyl)ethyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranosyl-(1→6)-[2-acetamido-3,6-di-O-benzyl-2-deoxy-4-O-levulinoyl-β-D-glucopyranosyl-(1→3)]-4-O-acetyl-2-O-benzoyl-β-D-galactopyranoside (31). To a solution of 29 (220 mg, 89.9 μmol) in CH2Cl2/AcOH (2:3, 3.5 mL) was added Zn/Cu couple (2.20 g) at r.t. The reaction mixture was heated to 50 °C and was stirred for 2.5 h at the same temperature as the reaction was monitored by TLC (15:1 CHCl3–MeOH). The precipitate was filtered through Celite and the filtrate was co-evaporated with toluene. The obtained residue was exposed to high vacuum for 6 h. The crude residue was dissolved in pyridine (3.6 mL) and acetic anhydride (68 μL, 0.719 mmol), DMAP (2.5 mg, 47.6 μmol) were then added to the mixture at 0 °C. After stirring for 12 h at r.t as the reaction was monitored by TLC (15:1 CHCl3–MeOH), the reaction mixture was evaporated. The obtained residue was diluted with CHCl3 and washed with 2 M HCl, satd aq NaHCO3, and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (40:1 CHCl3–MeOH) to give 31 (120 mg, 61%). [α]D −1.9° (c 0.5, CHCl3); 1H-NMR (600 MHz, CDCl3) δ 8.02–7.13 (m, 35 H, 7 Ph), 5.93 (d, 1 H, J2,NH = 8.3 Hz, NHb), 5.63 (d, 1 H, J3,4 = 3.4 Hz, H-4d), 5.56 (near t, 1 H, J1,2 = 7.5 Hz, J2,3 = 8.2 Hz, H-2d), 5.41–5.31 (m, 5 H, H-2a, H-4a, H-3d, H-7e, H-8e), 5.26 (d, 1 H, J2,NH = 7.6 Hz, NHc), 5.13 (d, 1 H, J5,NH = 7.5 Hz, NHe), 4.95 (d, 1 H, J1,2 = 8.2 Hz, H-1c), 4.90–4.83 (m, 4 H, H-4c, H-1d, H-4e, OCH2), 4.70 (d, 1 H, Jgem = 11.7 Hz, OCH2), 4.58 (d, 1 H, J1,2 = 6.9 Hz, H-1c), 4.44 (d, 1 H, J1,2 = 8.3 Hz, H-1a), 4.54–4.31 (m, 7 H, H-9e, 3 OCH2), 4.13 (t, 1 H, J2,3 = 9.6 Hz, H-3c), 4.10–4.02 (m, 4 H, H-6a, H-4b, H-5e, H-9e), 3.97–3.75 (m, 9 H, H-3a, H-6'a, H-3b, H-6d, H-6e, OCH2CH2SiMe3, OCH3), 3.67–3.35 (m, 11 H, H-5a, H-2b, H-5b, H-6b, H-6'b, H-5c, H-6c, H-6'c, H-5d, H-6'd, OCH2CH2SiMe3), 3.07 (dd, 1 H, H-2c), 2.54–2.51 (m, 3 H, H-3eeq, CH2CH2C(=O)O), 2.35–2.27 (m, 2 H, CH2CH2C(=O)O), 2.15–1.89 (m, 28 H, 9 C(=O)CH3, H-3eax), 0.88–0.69 (m, 2 H, OCH2CH2SiMe3), −0.16 (s, 9 H, OCH2CH2SiMe3); 13C (125 MHz, CDCl3) δ 206.1, 171.5, 170.9, 170.7, 170.5, 170.3, 170.1, 169.9, 169.7, 167.8, 165.3, 165.1, 138.9, 138.2, 138.1, 133.4, 133.3, 133.2, 129.8, 129.7, 129.6, 129.1, 128.6, 128.5, 128.5, 128.4, 128.3, 127.9, 127.9, 127.8, 127.7, 127.6, 127.5, 127.4, 100.6, 100.2, 99.9, 99.7, 99.0, 78.0, 77.4, 74.6, 73.6, 73.4, 73.3, 73.2, 73.1, 72.7, 71.8, 71.8, 71.6, 71.5, 70.2, 70.1, 69.7, 68.8, 68.8, 68.1, 67.6, 67.4, 67.2, 62.7, 62.5, 57.6, 52.9, 49.5, 37.8, 37.7, 29.7, 27.9, 23.4, 23.2, 22.7, 21.0, 20.8, 20.7, 20.6, 17.8, −1.5. m/z (MALDI): found [M+Na]+ 2198.83, C111H133N3O40Si calcd for [M+Na]+ 2198.81.
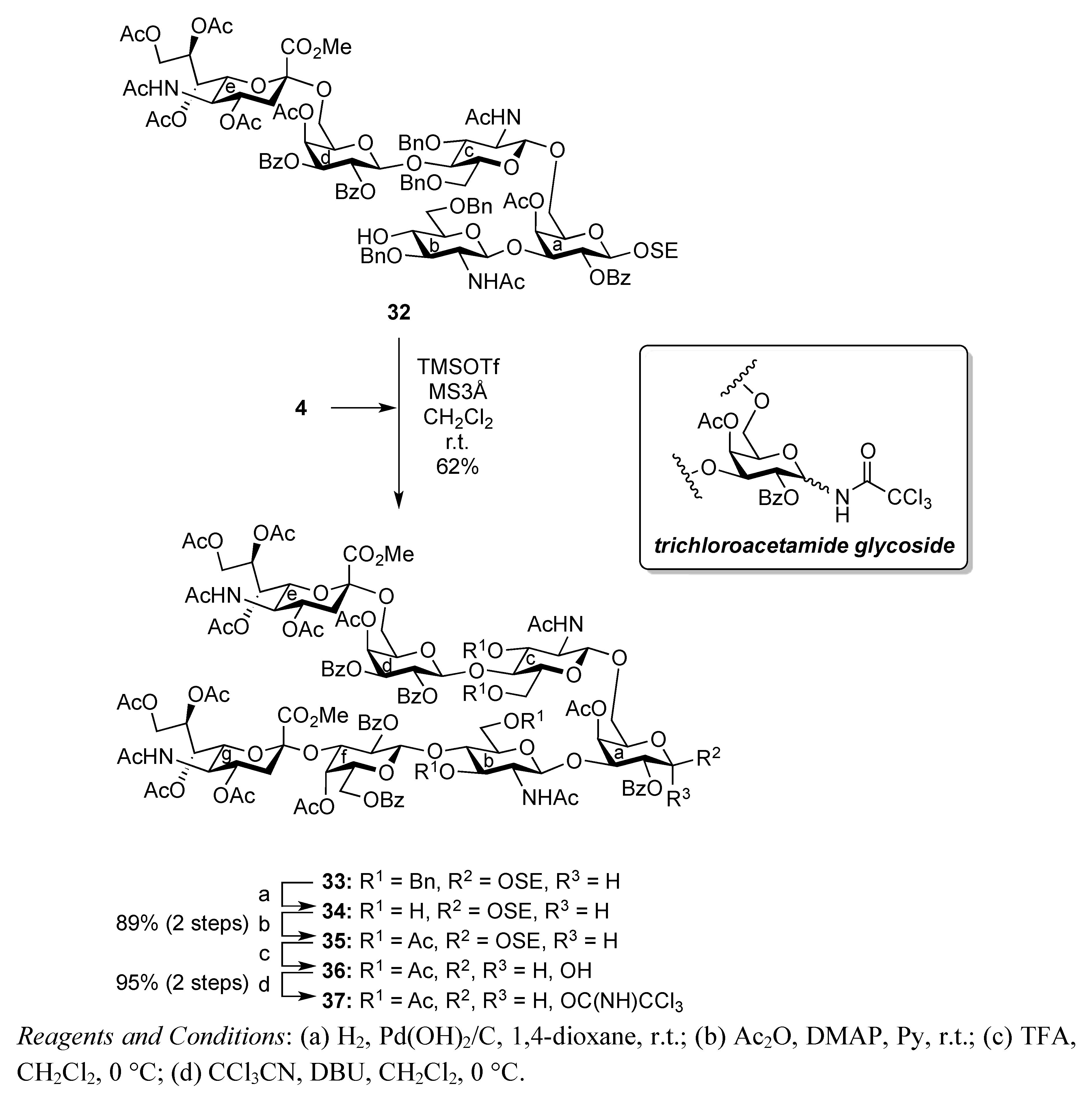
2-(Trimethylsilyl)ethyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranosyl-(1→6)-[2-acetamido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranosyl-(1→3)]-4-O-acetyl-2-O-benzoyl-β-D-galactopyranoside (32). To a solution of 31 (119 mg, 54.7 μmol) in THF (2.0 mL) was added hydrazine monoacetate (4.9 mg, 54.7 μmol) at 0 °C. After stirring for 1 h at r.t. as the reaction was monitored by TLC (30:1 CHCl3–MeOH), THF was evaporated. The residue was diluted with CHCl3 and washed with 2 M HCl, satd aq NaHCO3, and brine, dried over Na2SO4, and concentrated. The obtained residue was purified by silica gel column chromatography (40:1 CHCl3–MeOH) to give 32 (104 mg, 92%). [α]D −5.2° (c 1.9, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 8.02–7.20 (m, 35 H, 7 Ph), 6.01 (d, 1 H, JNH,2 = 8.6 Hz, NHb), 5.64 (d, 1 H, J3,4 = 3.5 Hz, H-4d), 5.58 (near t, 1 H, J1,2 = 8.0 Hz, J2,3 = 10.3 Hz, H-2d), 5.43–5.32 (m, 6 H, H-2a, H-4a, H-3d, H-7e, H-8e, NHc), 5.21 (d, 1 H, JNH,5 = 7.4 Hz, NHe), 4.89–4.81 (m, 3 H, H-1d, H-4e, OCH2), 4.82 (d, 1 H, J1,2 = 9.7 Hz, H-1c), 4.72–4.65 (2 d, 2 H, Jgem = 11.5 Hz, 2 OCH2), 4.60–4.45 (m, 5 H, H-1b, 4 OCH2), 4.44 (d, 1 H, J1,2 = 8.0 Hz, H-1a), 4.37–4.32 (m, 2 H, OCH2, H-9e), 4.12–4.04 (m, 4 H, H-6a, H-6d, H-6e, H-9'e), 3.98–3.62 (m, 14 H, H-3a, H-5a, H-6'a, H-2b, H-3b, H-6b, H-6'b, H-3c, H-5d, H-6'd, OCH3, OCH2CH2SiMe3), 3.54 (t, 1 H, J3,4 = J4,5 = 12.0 Hz, H-4c), 3.49–3.36 (m, 6 H, H-5c, H-6c, H-6'c, H-4b, H-5b, OCH2CH2SiMe3), 3.16 (dd, 1 H, H-2c), 2.96 (s, 1 H, OH), 2.53 (dd, 1 H, Jgem = 12.4 Hz, J3eq,4 = 4.6 Hz, H-3eeq), 2.18–1.87 (m, 25 H, 8 C(=O)CH3, H-3eax), 0.88–0.69 (m, 2 H, OCH2CH2SiMe3), −0.16 (s, 9 H, OCH2CH2SiMe3); 13C-NMR (125 MHz, CDCl3) δ 170.8, 170.6, 170.4, 170.2, 170.1, 169.9, 169.6, 169.5, 167.7, 165.6, 165.2, 165.0, 138.7, 138.4, 138.0, 137.7, 133.4, 133.2, 133.1, 129.7, 129.7, 129.6, 129.5, 129.0, 128.4, 128.4, 128.3, 128.3, 128.2, 127.8, 127.8, 127.7, 127.6, 127.5, 127.3, 100.4, 100.2, 100.1, 99.6, 98.9, 79.8, 77.9, 75.2, 74.5, 73.8, 73.5, 73.5, 73.2, 73.1, 73.0, 72.8, 72.6, 71.7, 71.6, 71.4, 70.7, 70.1, 70.0, 68.7, 68.1, 67.5, 67.3, 67.2, 62.5, 62.4, 56.4, 54.0, 52.8, 49.2, 37.7, 29.6, 23.3, 23.1, 22.8, 20.9, 20.7, 20.6, 20.5, 17.7, −1.6. m/z (MALDI): found [M+Na]+ 2100.96, C106H127N3O38Si calcd for [M+Na]+ 2100.78.
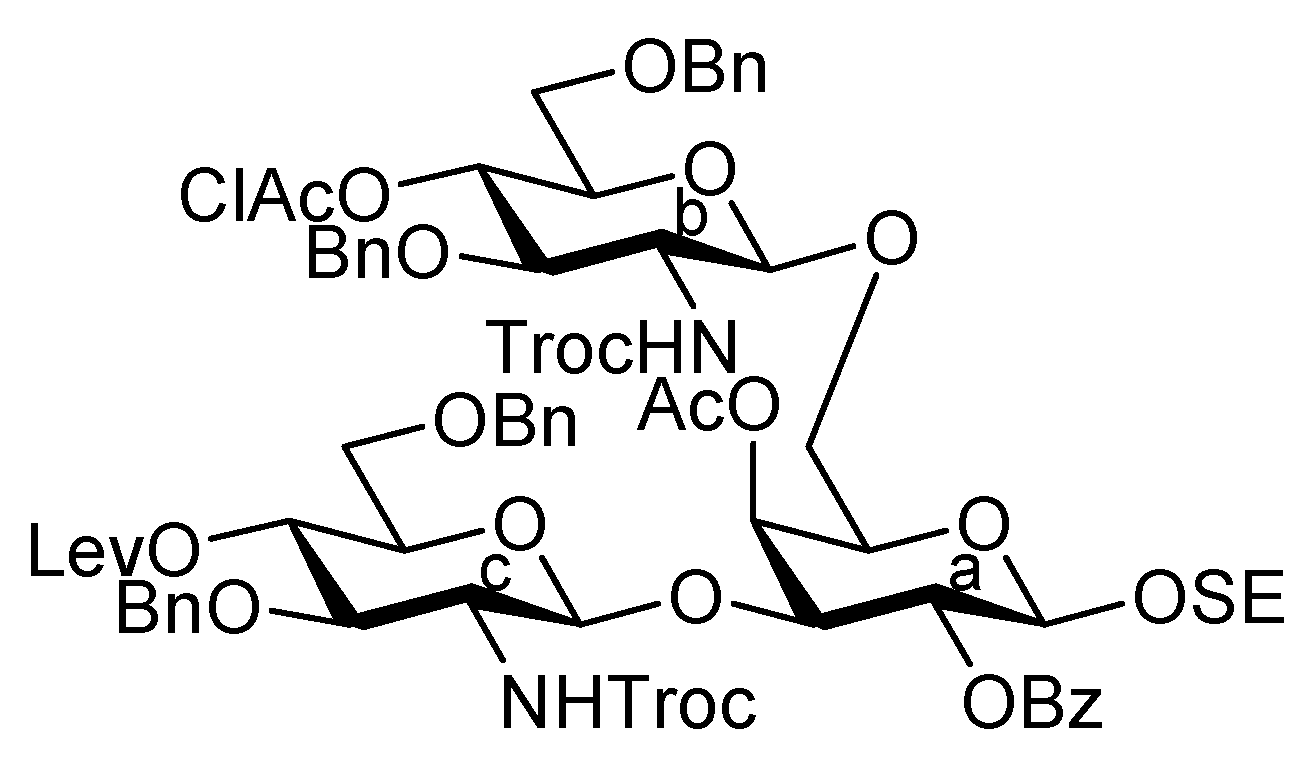
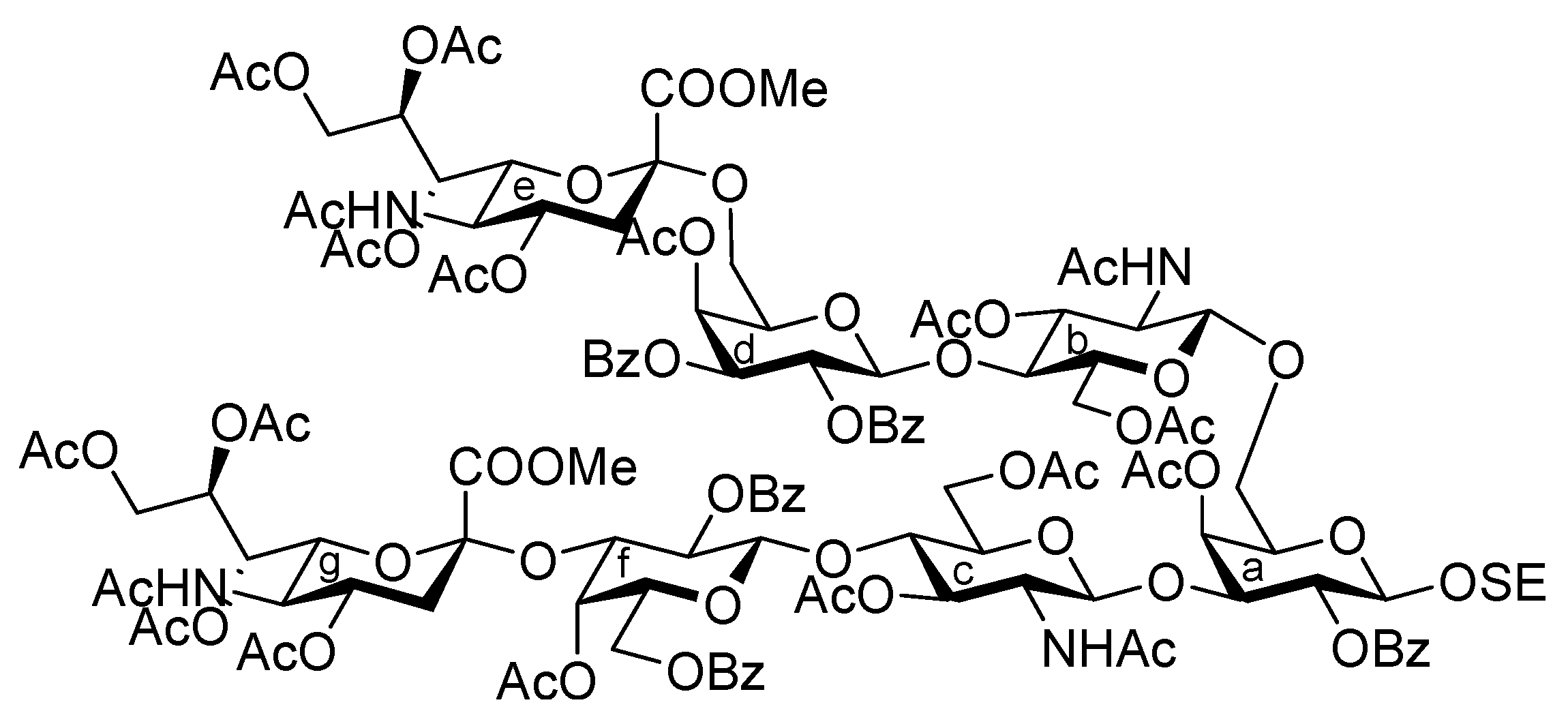
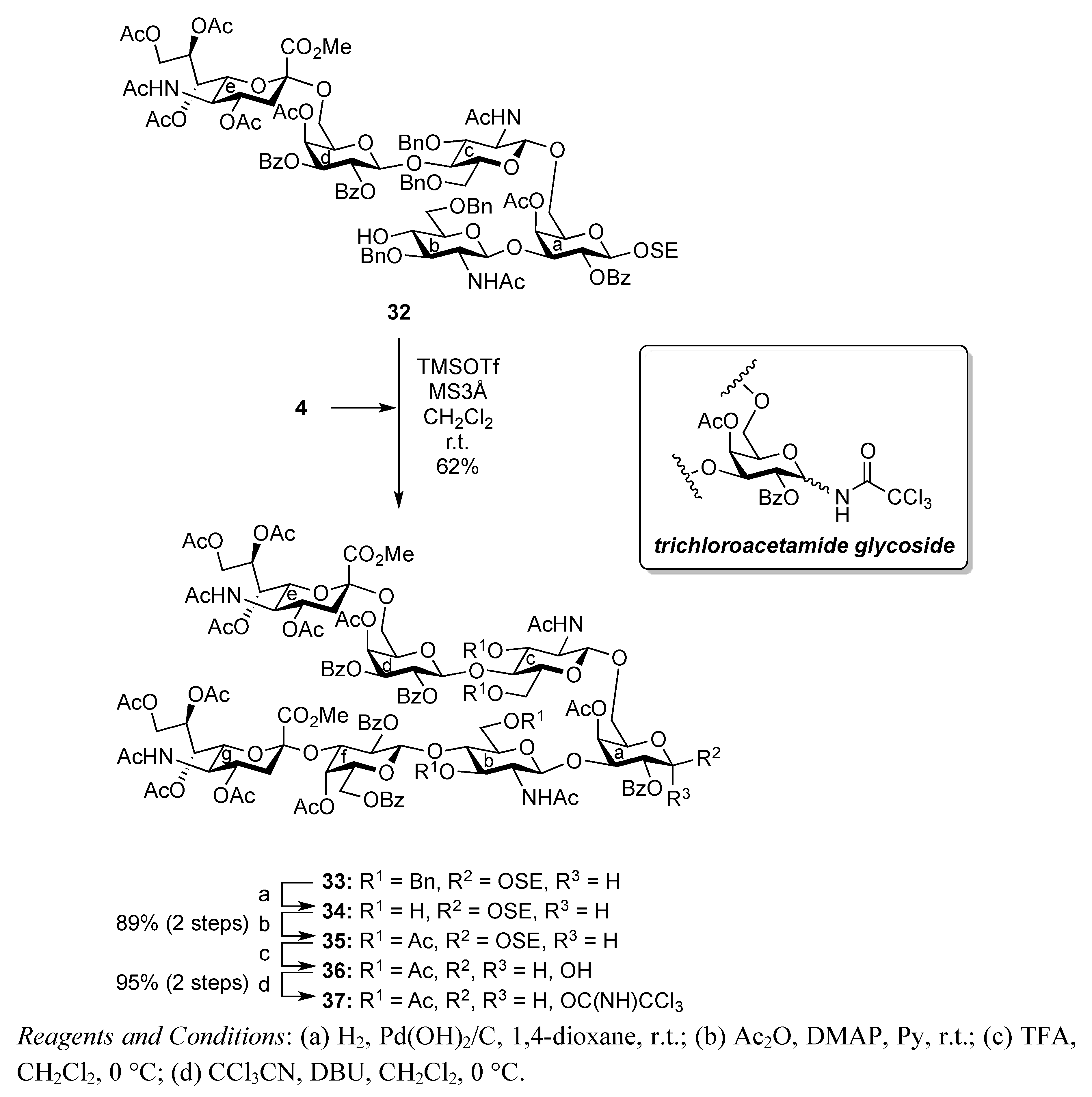
2-(Trimethylsilyl)ethyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→3)-4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranosyl-(1→3)-[(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranosyl-(1→6)]-4-O-acetyl-2-O-benzoyl-β-D-galactopyranoside (33). To a mixture of 4 (103 mg, 98.7 μmol) and 32 (93 mg, 44.5 μmol) in CH2Cl2 (1.8 mL) was added 3 Å molecular sieves (300 mg) at r.t. After stirring for 1 h, the mixture was cooled to 0 °C. TMSOTf (1.8 μL, 9.94 μmol) was then added to the mixture at 0 °C. The reaction was stirred for 2 h at r.t. as the reaction was monitored by TLC (15:1:1 CHCl3–MeOH–EtOAc). Another portion of TMSOTf (1.8 μL, 9.94 μmol) was added to the mixture at 0 °C. After the stirring was continued for 4 h at r.t, the reaction was quenched by the addition of triethylamine. The reaction mixture was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd aq NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (40:1 CHCl3–MeOH) to give 33 (82 mg, 62%). [α]D −5.1° (c 0.8, CHCl3); 1H-NMR (600 MHz, CDCl3) δ 8.22–7.12 (m, 45 H, 9 Ph), 5.92 (d, 1 H, NHb), 5.65 (m, 1 H, H-4g), 5.63 (d, 1 H, J3,4 = 3.4 Hz, H-4d), 5.55 (near t, 1 H, J1,2 = 8.2 Hz, J2,3 = 10.2 Hz, H-2d), 5.40 (m, 1 H, H-8e), 5.37 (dd, 1 H, H-3d), 5.33–5.13 (m, 7 H, H-2a, H-4a, H-7e, H-2f, H-7g, NHc, NHe), 5.06 (d, 1 H, J3,4 = 3.4 Hz, H-4f), 5.01 (d, 1 H, J1,2 = 7.5 Hz, H-1f), 5.00 (d, 1 H, J5,NH = 7.5 Hz, NHg), 4.87–4.82 (m, 5 H, H-1d, H-4e, H-4g, 2 OCH2), 4.75 (dd, 1 H, J2,3 = 10.2 Hz, H-3f), 4.71 (d, 1 H, Jgem = 11.6 Hz, OCH2), 4.68 (d, 1 H, J1,2 = 7.5 Hz, H-1c), 4.68–4.47 (m, 4 H, H-1b, 3 OCH2), 4.42 (d, 1 H, J1,2 = 10.7 Hz, H-1a), 4.28 (m, 4 H, H-9e, H-9g, 2 OCH2), 4.11–4.00 (m, 7 H, H-4b, H-4c, H-6c, H-6'c, H-5e, H-6e, H-9'e), 3.97–3.55 (m, 23 H, H-3a, H-5a, H-6a, H-6'a, H-2b, H-3b, H-6b, H-6'b, H-3c, H-6d, H-6'd, H-6f, H-6'f, H-5g, H-6g, H-9'g, 2 OCH3, OCH2CH2SiMe3), 3.49–3.40 (m, 3 H, H-5b, H-5c, OCH2CH2SiMe3), 3.31–3.28 (m, 2 H, H-5c, H-5f), 3.14 (br dd, 1 H, H-2c), 2.53 (dd, 1 H, Jgem = 11.3 Hz, J3eq,4 = 4.8 Hz, H-3geq), 2.49 (m, 1 H, Jgem = 11.7 Hz, J3eq,4 = 4.8 Hz, H-3eeq), 2.15–1.74 (m, 43 H, 14 Ac, H-3eax), 1.70 (t, 1 H, H-3gax), 1.51 (s, 3 H, Ac), 0.88–0.68 (m, 2 H, OCH2CH2SiMe3), −0.17 (s, 9 H, OCH2CH2SiMe3); 13C-NMR (150 MHz, CDCl3) δ 170.9, 170.7, 170.7, 170.3, 170.2, 170.1, 170.1, 170.0, 169.8, 169.7, 169.6, 168.0, 167.8, 165.7, 165.5, 165.3, 165.2, 164.9, 138.8, 138.8, 138.7, 138.1, 133.4, 133.3, 133.2, 133.1, 133.0, 130.3, 129.9, 129.8, 129.7, 129.7, 129.6, 129.5, 129.0, 128.6, 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 129.6, 129.5, 129.0, 128.6, 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 127.8, 127.5, 127.4, 127.2, 127.2, 127.0, 100.5, 100.3, 99.6, 99.5, 99.0, 96.8, 78.1, 77.6, 77.5, 75.2, 75.0, 74.6, 74.5, 73.6, 73.3, 73.0, 72.7, 72.6, 71.8, 71.7, 71.4, 71.3, 70.4, 70.2, 70.1, 69.4, 68.7, 68.1, 68.0, 67.5, 67.3, 67.2, 67.1, 66.6, 62.4, 61.3, 53.0, 52.9, 49.4, 48.8, 37.8, 37.3, 29.7, 23.3, 23.2, 23.1, 22.6, 21.3, 21.0, 20.8, 20.8, 20.7, 20.6, 20.6, 20.3, 17.7, −1.5. m/z (MALDI): found [M+Na]+ 2986.28, C148H174N4O58Si calcd for [M+Na]+ 2986.05.
2-(Trimethylsilyl)ethyl (methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→3)-4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→3)-[(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→6)]-4-O-acetyl-2-O-benzoyl-β-D-galactopyranoside (35). To a solution of 33 (42.0 mg, 14.2 μmol) in 1,4-dioxane (0.6 mL) was added Pd(OH)2/C (210 mg). After stirring for 4 h at r.t. under a hydrogen atmosphere as the reaction was monitored by TLC (10:1 CHCl3–MeOH), the mixture was filtered through Celite. The filtrate was concentrated and the obtained crude residue was roughly purified by silica gel column chromatography (10:1 CHCl3–MeOH). The obtained product was exposed to high vacuum for 24 h. The residue was then dissolved in pyridine (1.4 mL). Acetic anhydride (11 μL, 0.114 mmol) and DMAP (1.0 mg, 8.18 μmol) were added to the mixture at 0 °C. After stirring for 72 h at r.t. as the reaction was monitored by TLC (10:1 CHCl3–MeOH), the reaction was quenched by the addition of MeOH at 0 °C. The mixture was co-evaporated with toluene and the residue was then diluted with CHCl3, and washed with 2 M HCl, H2O, satd aq NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (40:1 CHCl3–MeOH) to give 35 (35 mg, 89%). [α]D +5.1° (c 0.7, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 8.17–7.27 (m, 25 H, 5 Ph), 5.92 (d, 1 H, JNH,2 = 9.2 Hz, NHb), 5.68 (d, 1 H, J3,4 = 3.5 Hz, H-4d), 5.77 (m, 1 H, H-8g), 5.55 (near t, 2 H, J1,2 = 8.0 Hz, J2,3 = 10.3 Hz, H-2d), 5.44 (m, 1 H, H-3d), 5.38 (m, 1 H, H-8e), 5.32–5.16 (m, 7 H, H-2a, H-3a, H-3b, H-7e, H-2f, H-7g, NHe), 5.09 (d, 1 H, J3,4 = 2.8 Hz, H-4f), 4.96 (d, 1 H, JNH,5 = 9.8 Hz, NHg), 4.91–4.80 (m, 6 H, NHc, H-3c, H-1d, H-4e, H-1f, H-4g), 4.74–4.71 (dd, 1 H, J2,3 = 9.7 Hz, H-3f), 4.47 (d, 1 H, J1,2 = 8.0 Hz, H-1b), 4.43 (d, 1 H, J1,2 = 8.0 Hz, H-1a), 4.41–4.28 (m, 6 H, H-6a, H-1c, H-6d, H-9e, H-6f, H-9g), 4.17–3.93 (m, 10 H, H-6'a, H-6b, H-6'b, H-6'd, H-5e, H-6e, H-9'e, H-6'f, H-9'g, OCH2CH2SiMe3), 3.88–3.68 (m, 15 H, H-3a, H-2b, H-4b, H-5b, H-2c, H-4c, H-6c, H-6'c, H-5g, 2 OCH3), 3.55–3.53 (m, 2 H, H-5d, H-6g), 3.48–3.42 (m, 3 H, H-5a, H-5f, OCH2CH2SiMe3), 3.27–3.25 (m, 1 H, J5,4 = 9.7 Hz, H-5c), 2.55 (dd, 1 H, Jgem = 12.6 Hz, J3eq,4 = 4.6 Hz, H-3geq), 2.51 (dd, 1 H, Jgem = 12.6 Hz, J3eq,4 = 4.6 Hz, H-3eeq), 2.20–1.78 (m, 52 H, 17 Ac, H-3eax), 1.62 (t, 1 H, H-3gax), 1.54 (s, 6 H, 2 Ac), 0.88–0.72 (m, 2 H, OCH2CH2SiMe3), −0.15 (s, 9 H, OCH2CH2SiMe3); 13C-NMR (150 MHz, CDCl3) δ 170.9, 170.7, 170.7, 170.5, 170.4, 170.4, 170.2, 170.1, 170.1, 170.0, 169.8, 169.8, 169.7, 168.0, 167.7, 165.6, 165.3, 165.0, 164.9, 164.8, 133.5, 133.3, 133.2, 130.3, 129.8, 129.7, 129.5, 129.0, 128.9, 128.7, 128.5, 128.5, 128.4, 128.4, 101.2, 100.7, 100.6, 100.5, 100.4, 99.2, 96.8, 75.6, 74.6, 72.9, 72.7, 72.5, 72.4, 71.9, 71.7, 71.1, 70.9, 70.0, 69.8, 69.6, 69.4, 68.6, 67.8, 67.4, 67.1, 66.2, 62.9, 62.4, 62.2, 62.0, 61.0, 60.9, 53.9, 53.0, 49.4, 48.8, 37.8, 37.2, 23.2, 23.1, 22.7, 21.3, 21.0, 20.9, 20.8, 20.7, 20.6, 20.5, 20.4, 17.8, −1.5. m/z (MALDI): found [M+Na]+ 2793.65, C128H158N4O62Si calcd for [M+Na]+ 2793.90.
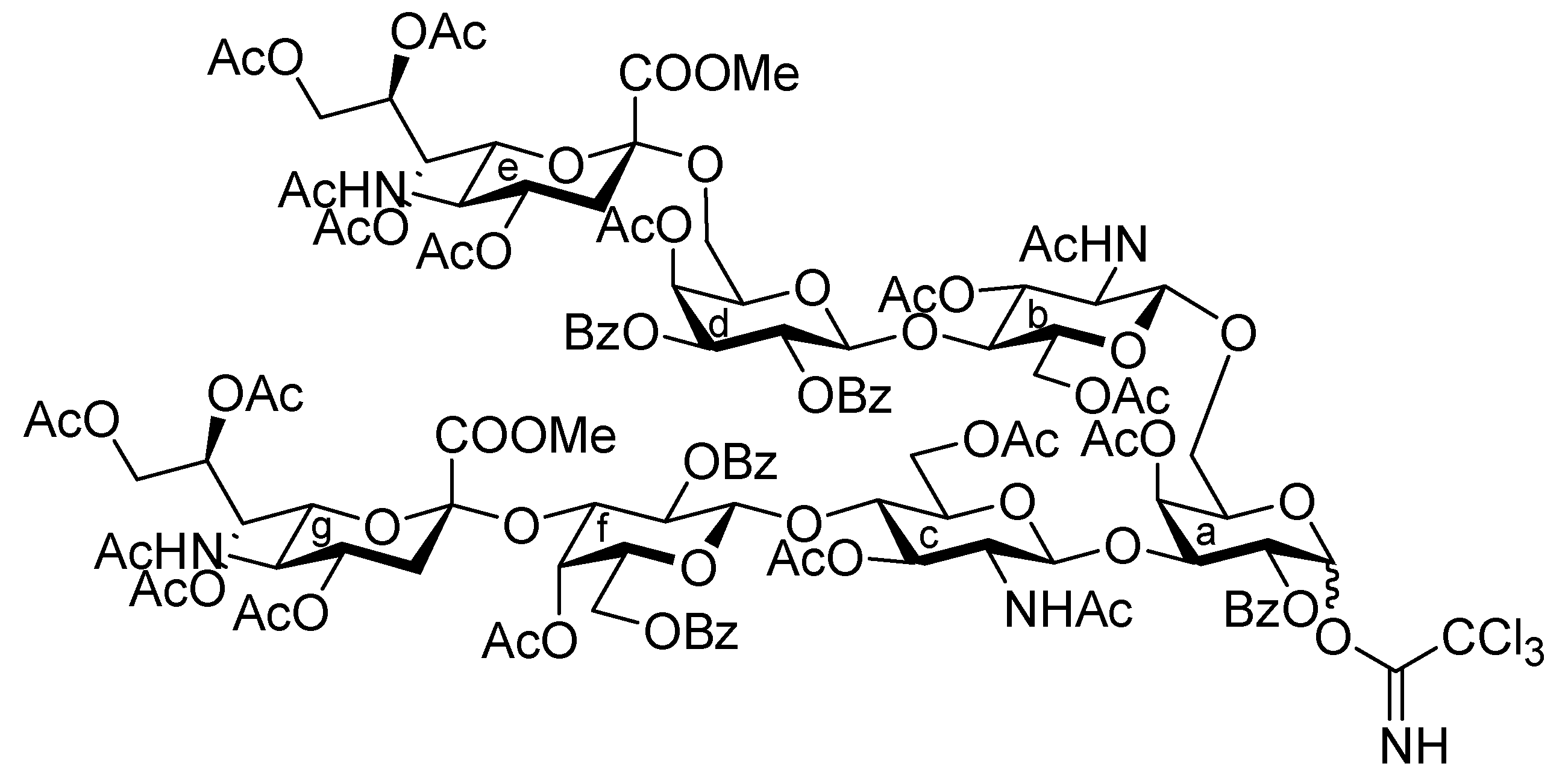
(Methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→3)-4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→3)-[(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→6)]-4-O-acetyl-2-O-benzoyl-D-galactopyranosyl trichloroacetimidate (37). To a solution of 35 (28 mg, 10.2 μmol) in CH2Cl2 (0.75 mL) was added TFA (0.25 mL) at 0 °C. After stirring for 2 h at 0 °C as the reaction was monitored by TLC (15:1:1 CHCl3–MeOH–EtOAc), the reaction mixture was co-evaporated with toluene and then roughly purified by silica gel column chromatography (20:1 CHCl3–MeOH). The obtained product was exposed to high vacuum for 24 h and then dissolved in CH2Cl2 (1.0 mL). CCl3CN (10.2 μL, 0.102 mmol) and DBU (1.8 μL, 12.2 μmol) were added to the mixture at 0 °C. After stirring for 45 min at 0 °C as the reaction was monitored by TLC (10:1 CHCl3–MeOH), the reaction mixture was evaporated. The obtained crude residue was purified by silica gel column chromatography (30:1 CHCl3–MeOH) to give 37 (27 mg, 95%, α:β = 3:1). 37α: 1H-NMR (600 MHz, CDCl3) δ 8.60 (s, 1 H, C(=NH)), 8.19–7.27 (m, 25 H, 5 Ph), 6.48 (d, 1 H, J1,2 = 4.1 Hz, H-1a), 5.81 (d, 1 H, JNH,2 = 9.0 Hz, NHc), 5.68 (d, 1 H, J3,4 = 3.5 Hz, H-4d), 5.60 (m, 1 H, H-8g), 5.55 (near t, 1 H, J1,2 = 8.3 Hz, J2,3 = 10.3 Hz, H-2d), 5.48 (dd, 1 H, J2,3 = 10.3 Hz, H-2a), 5.43–5.42 (m, 2 H, H-3a, H-4a), 5.38 (m, 1 H, H-8e), 5.32 (dd, 1 H, H-7e), 5.24 (m, 2 H, H-3a, H-7g), 5.20 (near t, 1 H, J1,2 = 8.3 Hz, J2,3 = 9.6 Hz, H-2f), 5.16 (t, 1 H, J2,3 = J3,4 = 9.0 Hz, H-3b), 5.13 (d, 1 H, J5,NH = 9.6 Hz, NHe), 4.92 (d, 1 H, J3,4 = 2.3 Hz, H-4f), 4.90–4.80 (m, 8 H, H-3c, H-1d, H-4e, H-1f, H-3d, H-4g, NHc, NHg), 4.73 (dd, 1 H, H-3f), 4.55 (d, 1 H, J1,2 = 7.6 Hz, H-1b), 4.45 (d, 1 H, J1,2 = 7.4 Hz, H-1c), 4.41–4.01 (m, 13 H, H-3a, H-6a, H-6'a, H-5e, H-6e, H-9e, H-9'e, H-6d, H-6'd, H-6f, H-6'f, H-9g, H-9'g), 3.88–3.70 (m, 15 H, H-2b, H-4b, H-6b, H-6'b, H-2c, H-4c, H-6c, H-6'c, H-5e, 2 OCH3), 3.55–3.36 (m, 5 H, H-5a, H-5b, H-5c, H-5d, H-5f), 2.55 (dd, 1 H, Jgem = 13.1 Hz, J3eq,4 = 4.8 Hz, H-3eeq ), 2.51 (dd, 1 H, Jgem = 11.6 Hz, J3eq,4 = 4.8 Hz, H-3geq), 2.20–1.78 (m, 52 H, 17 Ac, H-3eax), 1.63–1.39 (m, 7 H, 2 Ac, H-3gax).
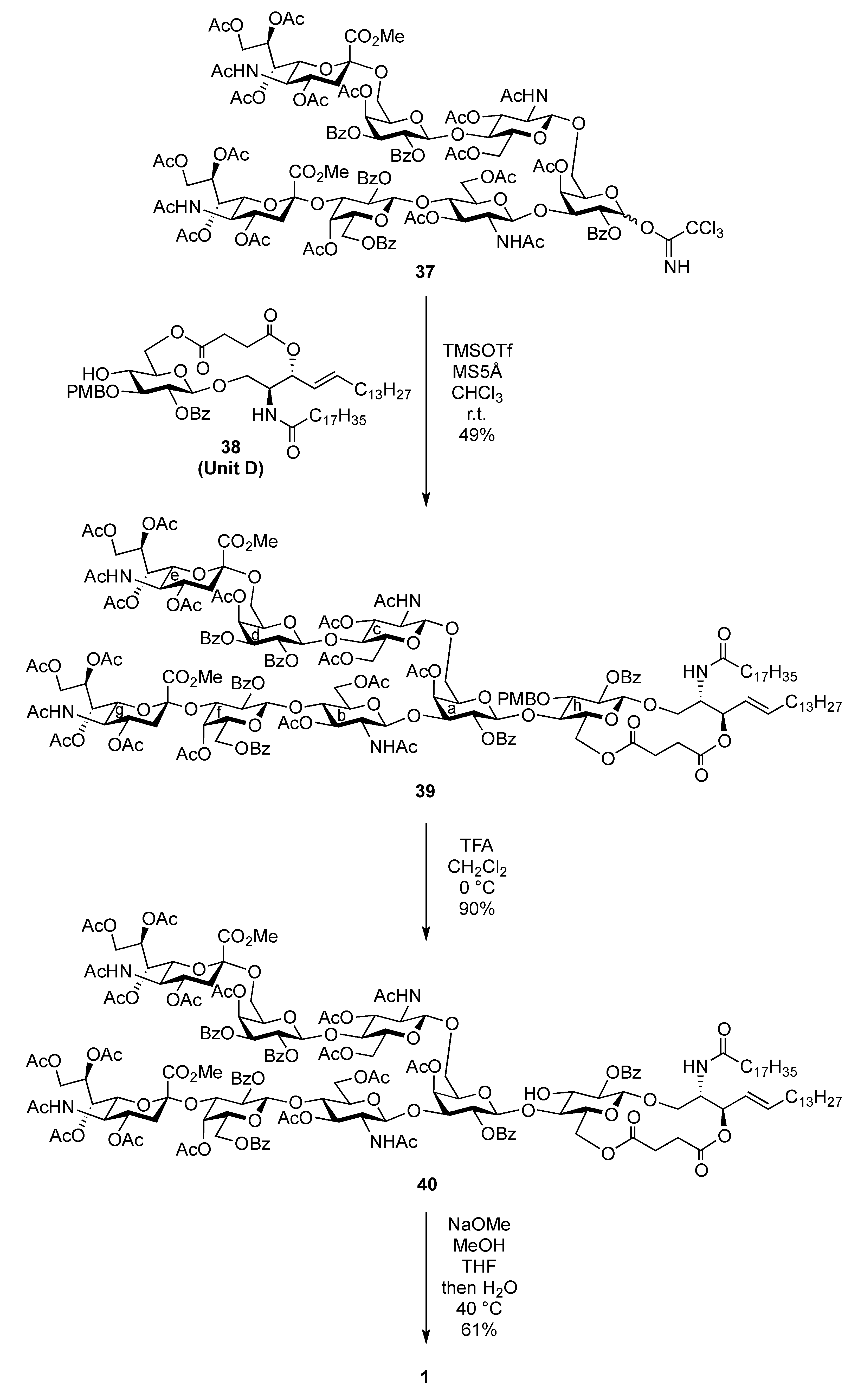
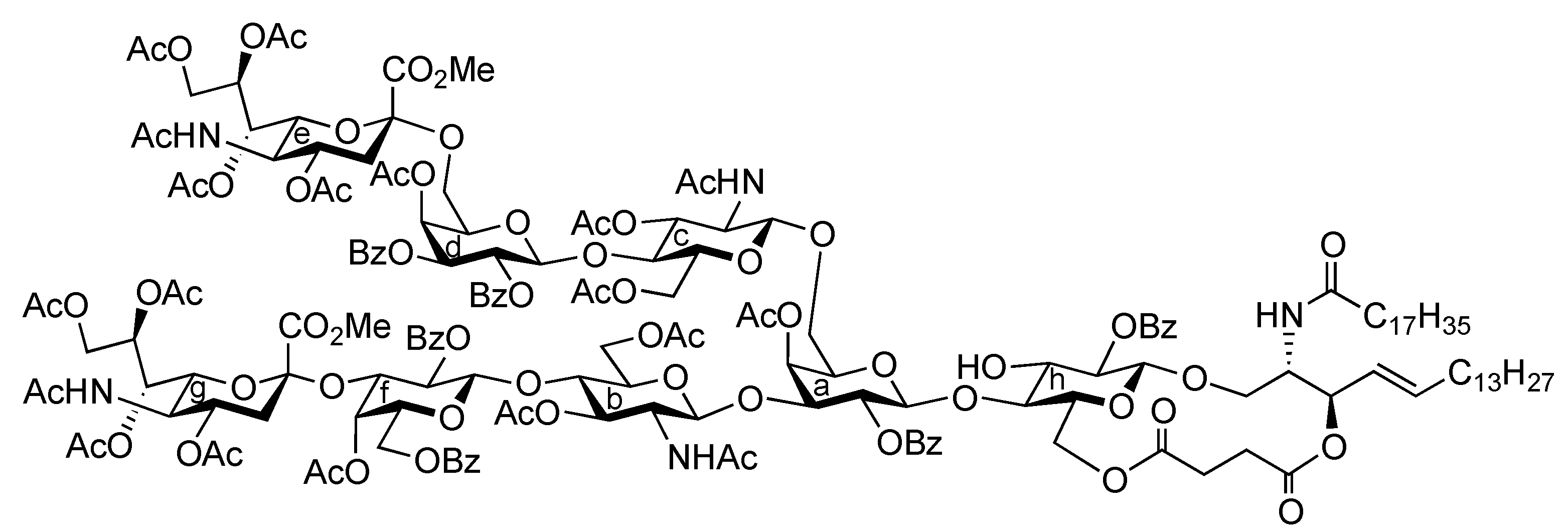
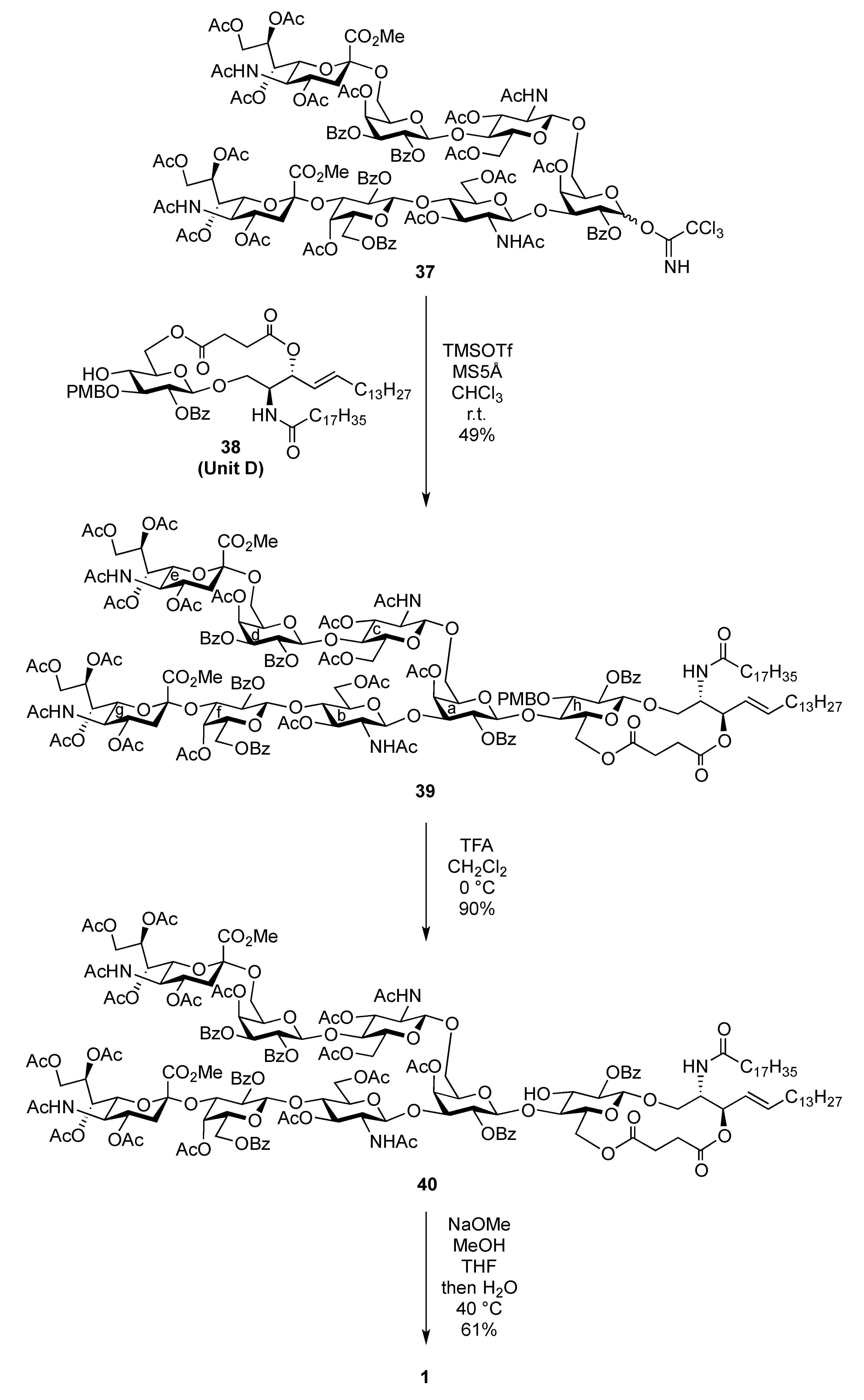
(2S,3R,4E)-1-O-({(Methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→3)-4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→3)-[(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→6)]-4-O-acetyl-2-O-benzoyl-β-D-galactopyranosyl}-(1→4)-2-O-benzoyl-3-O-p-methoxybenzyl-β-D-glucopyranosyl)-2-octadecanamido-3,6´-succinyl-octadec-4-ene-1,3-diol (39). To a mixture of 37 (38 mg, 13.4 μmol) and 38 (14 mg, 13.4 μmol) in CH2Cl2 (1.3 mL) was added 5 Å molecular sieves (100 mg) at r.t. After stirring for 1 h, the mixture was cooled to 0 °C. TMSOTf (0.3 μL, 1.34 μmol) was then added to the mixture at 0 °C. After stirring for 1 h at r.t. as the reaction was monitored by TLC (15:1:5 CHCl3–MeOH–EtOAc), the reaction was quenched by the addition of triethylamine. The reaction mixture was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd aq NaHCO3 and brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography (40:1:5 CHCl3–MeOH–EtOAc) to give 39 (24 mg, 49%). [α]D −10.3° (c 2.0, CHCl3); 1H-NMR (500 MHz, CD3CN) δ 8.13–7.39 (m, 30 H, 6 Ph), 7.04 (d, 2 H, Ar), 6.66 (d, 2 H, Ar), 6.14 (d, 1 H, J5,NH = 9.7 Hz, NHe), 6.09 (d, 1 H, J2,NH = 9.2 Hz, NHCer), 6.00 (d, 1 H, J5,NH = 9.7 Hz, NHg), 5.95 (d, 1 H, J2,NH = 9.1 Hz, NHb), 5.79 (d, 1 H, J2,NH = 9.8 Hz, NHc), 5.63 (dt, 1 H, J5,6 = 6.9 Hz, J = 14.9 Hz, J = 6.9 Hz, H-5Cer), 5.54 (d, 1 H, J3,4 = 3.5 Hz, H-4d), 5.47 (m, 3 H, H-3d, H-7e, H-8e), 5.37 (near t, 1 H, J1,2 = 7.5 Hz, J2,3 = 10.3 Hz, H-2d), 5.36 (m, 1 H, H-8g), 5.27 (d, 1 H, J3,4 = 3.5 Hz, H-4a), 5.25–5.17 (m, 4 H, H-2a, H-4f, H-7g, H-4Cer), 5.11 (br t, 1 H, J3,4 = J2,3 = 7.5 Hz, H-3Cer), 5.03 (near t, 1 H, J1,2 = 8.0 Hz, J2,3 = 10.3 Hz, H-2f), 4.94 (t, 1 H, J1,2 = J2,3 = 8.3 Hz, H-2h), 4.88 (t, 1 H, J2,3 = J3,4 = 9.3 Hz, H-3b), 4.76 (m, 8 H, H-1c, H-3c, H-1d, H-4e, H-1f, H-3f, H-4g, ArCH2), 4.68 (d, 1 H, J1,2 = 8.1 Hz, H-1a), 4.54 (d, 1 H, J1,2 = 8.5 Hz, H-1b), 4.47 (d, 1 H, H-1h), 4.46 (d, 1 H, Jgem = 10.9 Hz, ArCH2), 4.35 (dd, 1 H, J8,9 = 2.9 Hz, Jgem = 12.6 Hz, H-9e), 4.30–4.21 (m, 4 H, H-6b, H-6'b, H-6d, H-9g), 4.11–3.89 (m, 13 H, H-3a, H-6'a, H-6c, H-6'c, H-6'd, H-5e, H-6e, H-9'e, H-6f, H-9g, H-6h, H-6'h, H-2Cer), 3.78–3.53 (m, 21 H, H-5a, H-4b, H-2c, H-4c, H-5d, H-6'a, H-6'f, H-5g, H-6g, H-3h, H-4h, H-1Cer, 3 OCH3), 3.51 (m, 1 H, H-2b), 3.40–3.32 (m, 3 H, H-5b, H-5h, H-1'Cer), 3.18–3.12 (m, 2 H, H-5c, H-5f), 2.56 (dd, 1 H, J3eq,4 = 4.6 Hz, Jgem = 12.6 Hz, H-3eeq), 2.48–2.34 (m, 5 H, H-3geq, 2 C(=O)CH2), 2.21–1.61 (m, 62 H, H-3eax, H-6Cer, H-6'Cer, C(=O)CH2Cer, 19 Ac), 1.55–1.08 (m, 53 H, H-3gax, 26 –CH2–), 0.87 (t, 6 H, 2 –CH3Cer); 13C-NMR (150 MHz, CDCl3) δ 172.6, 171.0, 170.9, 170.8, 170.7, 170.6, 170.5, 170.4, 170.3, 170.2, 170.1, 170.0, 169.7, 169.7, 169.5, 168.0, 167.7, 165.6, 165.2, 165.2, 165.1, 165.0, 164.5, 159.1, 138.6, 133.8, 133.3, 133.1, 130.7, 130.3, 129.8, 129.7, 129.6, 129.5, 129.5, 129.5, 129.0, 128.9, 128.8, 128.6, 128.6, 128.5, 128.4, 128.3, 124.8, 113.9, 101.6, 101.3, 100.8, 100.6, 100.3, 99.7, 99.3, 96.8, 91.8, 80.8, 79.7, 76.3, 75.9, 74.7, 74.3, 73.2, 72.9, 72.7, 72.6, 72.5, 71.9, 71.8, 71.7, 71.7, 71.0, 70.9, 70.1, 69.8, 69.7, 69.4, 68.6, 67.8, 67.4, 67.1, 66.3, 63.2, 63.0, 62.6, 62.1, 61.9, 61.1, 60.9, 55.1, 53.6, 53.1, 53.0, 49.9, 49.4, 48.8, 37.9, 37.2, 37.1, 36.6, 32.7, 32.2, 31.9, 30.0, 29.7, 29.6, 29.5, 29.5, 29.3, 29.3, 29.2, 28.8, 27.1, 25.5, 23.6, 23.2, 23.1, 22.7, 21.3, 21.0, 20.9, 20.8, 20.7, 20.7, 20.6, 20.5, 20.5, 19.7, 14.1. m/z (MALDI): found [M+Na]+ 3709.15, C184H239N5O73 calcd for [M+Na]+ 3709.50.
(2S,3R,4E)-1-O-({(Methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→3)-4-O-acetyl-2,6-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→3)-[(methyl 5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonate)-(2→6)-4-O-acetyl-2,3-di-O-benzoyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→6)]-4-O-acetyl-2-O-benzoyl-β-D-galactopyranosyl}-(1→4)-2-O-benzoyl-β-D-glucopyranosyl)-2-octadecanamido-3,6'-succinyl-octadec-4-ene-1,3-diol (40). To a solution of 39 (9.0 mg, 2.47 μmol) in CH2Cl2 (0.60 mL) was added TFA (0.30 mL) at 0 °C. After stirring for 30 min at 0 °C as the reaction was monitored by TLC (10:1 CHCl3–MeOH), the reaction was quenched by the addition of satd aq NaHCO3. The mixture was diluted with CHCl3 and washed with brine. The organic layer was subsequently dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography (20:1 CHCl3–MeOH) to give 40 (8.0 mg, 91%). [α]D −6.3° (c 0.8, CHCl3); 1H-NMR (600 MHz, CDCl3) δ 8.17–7.27 (m, 30 H, 6 Ph), 6.68 (d, 1 H, J2,NH = 8.2 Hz, NHCer), 6.21 (d, 1 H, J2,NH = 9.0 Hz, NHb), 5.85 (near dt, 1 H, J5,6 = 6.9 Hz, J = 14.4 Hz, J = 7.5 Hz, H-5Cer), 5.70 (d, 1 H, J3,4 = 3.4 Hz, H-4d), 5.59 (m, 1 H, H-8g), 5.57 (near t, 1 H, J1,2 = 8.3 Hz, J2,3 = 10.3 Hz, H-2d), 5.45 (dd, 1 H, H-3d), 5.41–5.38 (m, 1 H, H-8e), 5.35–5.32 (m, 2 H, H-2a, H-7e), 5.27–5.17 (m, 7 H, H-4a, H-3b, H-2f, H-7g, NHe, H-3Cer, H-4Cer), 5.09 (d, 1 H, J3,4 = 2.7 Hz, H-4f), 5.06 (near t, 1 H, J1,2 = 4.8 Hz, J2,3 = 5.5 Hz, H-2h), 4.94 (d, 1 H, J5,NH =10.3 Hz, NHg), 4.91–4.81 (m, 5 H, H-3c, H-1d, H-4e, H-4g, NHc), 4.74–4.70 (m, 2 H, H-1a, H-3f), 4.62 (d, 1 H, H-1h), 4.45–4.34 (m, 6 H, H-1b, H-1c, H-9e, H-6f, H-9g, H-6h), 4.27–4.23 (m, 3 H, H-2cer, H-6a, H-6d), 4.17–3.98 (m, 11 H, H-6'a, H-6c, H-6'c, H-6d, H-5e, H-6e, H-9'e, H-9'g, H-5h, H-6h, H-6'h), 3.88–3.65 (m, 17 H, H-3a, H-2b, H-4b, H-6b, H-6'b, H-2c, H-4c, H-5g, H-3h, H-4h, H-1Cer, 2 OCH3), 3.60–3.53 (m, 3 H, H-5d, H-6g, H-1Cer), 3.50–3.44 (m, 2 H, H-5b, H-5f), 3.29–3.23 (m, 2 H, H-5a, H-5c), 2.56–2.39 (m, 6 H, H-3eeq, H-3geq, 2 C(=O)CH2), 2.20–1.78 (m, 56 H, 17 Ac, H-3eax, H-6Cer, H-6'Cer, C(=O)CH2Cer), 1.62 (t, 1 H, Jgem = J3ax,4 = 12.6 Hz, H-3gax), 1.54 (s, 3 H, Ac), 1.51–1.46 (m, 2 H, C(=O)CH2CH2), 1.42 (s, 3 H, Ac), 1.38–1.20 (m, 50 H, 25 –CH2–), 0.88 (m, 6 H, 2 –CH3Cer); 13C-NMR (150 MHz, CDCl3) δ 172.9, 171.0, 170.9, 170.8, 170.8, 170.7, 170.6, 170.5, 170.5, 170.3, 170.3, 170.3, 170.1, 170.1, 170.0, 169.9, 169.8, 169.7, 169.7, 168.0, 167.8, 165.7, 165.4, 165.3, 165.2, 165.0, 164.7, 138.5, 134.0, 133.5, 133.3, 133.2, 130.3, 129.8, 129.7, 129.7, 129.6, 129.5, 129.4, 129.0, 129.0, 128.7, 128.6, 128.5, 128.4, 128.4, 128.4, 125.0, 101.7, 101.2, 100.7, 99.8, 99.6, 99.3, 98.5, 96.8. 82.0, 77.5, 75.2, 74.5, 74.2, 73.8, 73.5, 72.9, 72.7, 72.6, 72.5, 72.0, 71.9, 71.7, 71.2, 71.0, 70.9, 70.7, 70.1, 69.9, 69.6, 69.4, 68.6, 67.8, 67.6, 67.4, 67.2, 67.1, 66.6, 66.3, 62.9, 69.4, 68.6, 67.8, 67.6, 67.4, 67.2, 67.1, 66.6, 66.3, 62.9, 62.5, 62.4, 62.0, 61.1, 60.9, 54.2, 53.9, 53.0, 53.0, 50.1, 49.5, 48.8, 37.8, 37.2, 36.4, 32.2, 31.9, 30.4, 29.7, 29.7, 29.7, 29.6, 29.5, 29.5, 29.3, 29.2, 28.8, 25.6, 23.6, 23.2, 23.1, 22.7, 22.5, 21.4, 21.1, 20.8, 20.8, 20.7, 20.6, 20.5, 20.4, 14.1. m/z (MALDI): found [M+Na]+ 3589.60, C176H231N5O72 calcd for [M+Na]+ 3589.45. HRMS (ESI): found [1/2M+Na]+ 1806.2182, C176H231N5O72 calcd for [1/2M+Na]+ 1806.2181.
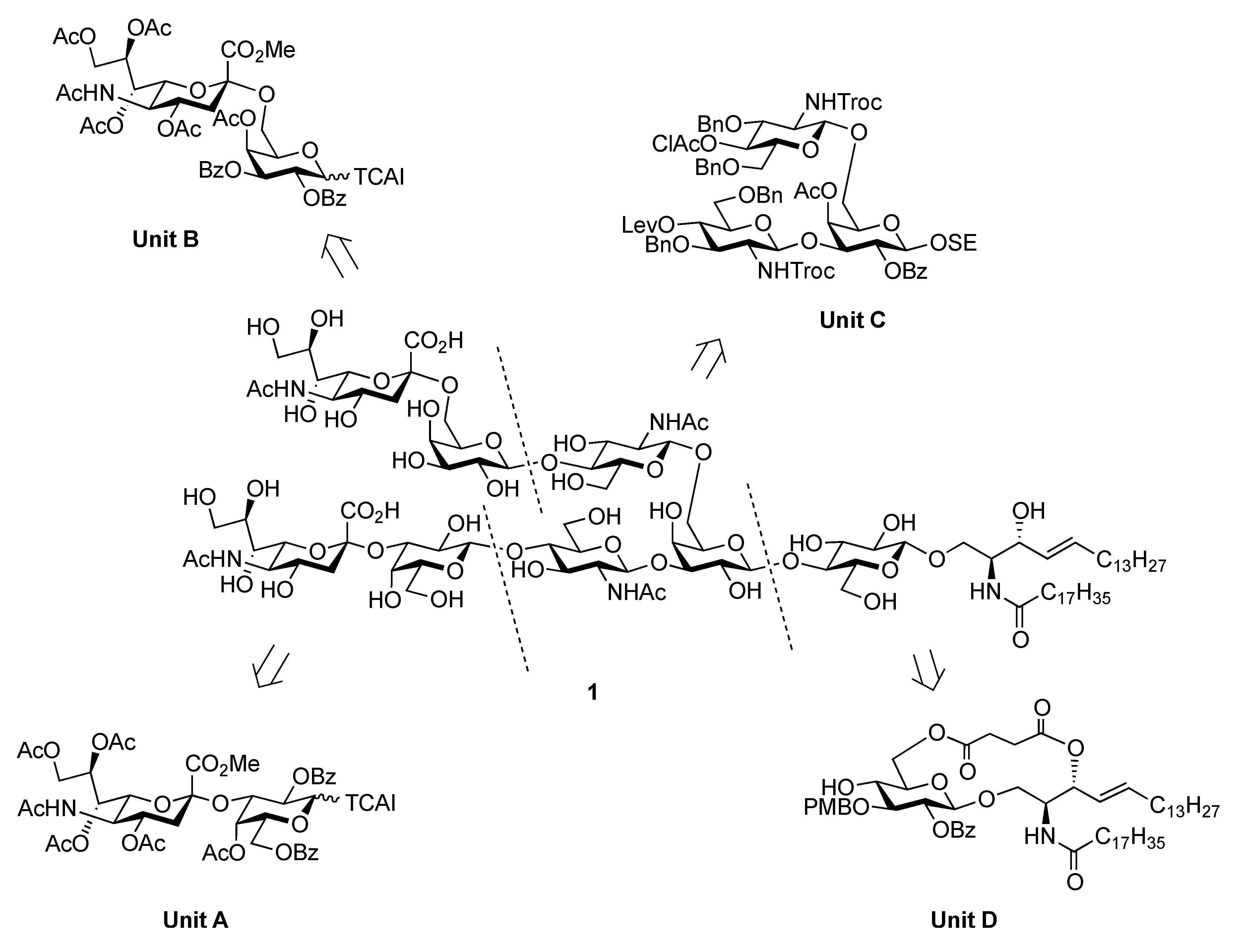
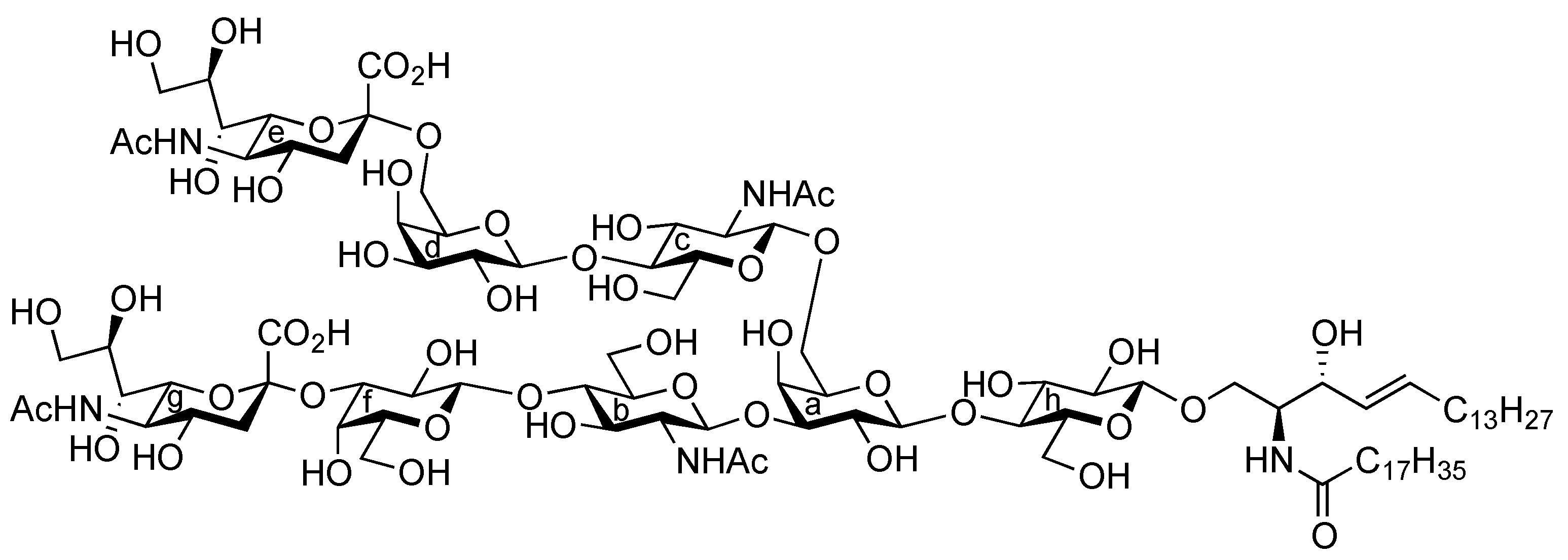
{3-O-[(5-Acetamido-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonic acid)-β-D-galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl]-6-O-[(5-acetamido-3,5-dideoxy-D-glycero-α-D-galacto-2-nonulopyranosylonic acid)-β-D-galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl]-β-D-galactopyranosyl}-(1→4)-β-D-glucopyranosyl-(1→1)-(2S,3R,4E)-2-octadecanamido-4-octadecene-1,3-diol (1). To a solution of 40 (8.0 mg, 2.24 μmol) in MeOH/THF (1:1, 1.0 mL) was added NaOMe (28% solution in MeOH, 12.1 μg, 0.672 μmol) at 0 °C. After stirring for 7 d at 40 °C as the reaction was monitored by TLC (6:4:1 CHCl3–MeOH–0.2% aq CaCl2) and MALDI-TOF MS, water (0.1 mL) was added to the reaction mixture. After stirring for 2 d at 40 °C, the reaction was neutralized with Dowex (H+) resin. The resin was filtered through cotton and the filtrate was then evaporated. The residue was purified by gel filtration column chromatography (LH-20) using MeOH as eluent followed by silica gel column chromatography (6:4:1 CHCl3–MeOH–H2O) to give 1 (3.0 mg, 61%). [α]D −78.8° (c 0.4, MeOH); 1H-NMR (600 MHz, 1:1 CD3OD–D2O) δ 5.73 (m, 1 H, H-5Cer), 5.46 (m, 1 H, H-4Cer), 2.86 (dd, 1 H, H-3geq), 2.78 (dd, 1 H, H-3eeq), 2.20 (t, 2 H, NHCOCH2), 2.04 (s, 14 H, H-6Cer, H-6'Cer, 4 NAc), 1.73 (m, 2 H, H-3gax, H-3eax), 1.58 (m, 2 H, NHCOCH2CH2), 1.30 (m, 50 H, 25 –CH2–), 0.92 (t, 6 H, 2 –CH3); 13C-NMR (125 MHz, 5:6:0.5 CD3OD–D2O–CDCl3) δ 174.2, 174.1, 173.7, 173.3, 134.1, 128.6, 102.9, 102.3, 102.0, 101.8, 99.9, 79.9, 79.0, 77.8, 76.5, 75.1, 74.6, 74.1, 74.0, 73.5, 73.1, 72.5, 72.2, 72.0, 71.7, 71.6, 71.1, 70.9, 70.6, 70.2, 69.3, 68.7, 68.2, 67.9, 67.8, 67.5, 67.3, 66.8, 62.4, 62.2, 60.4, 59.7, 54.6, 54.5, 52.4, 51.6, 51.5, 48.1, 46.7, 39.7, 39.4, 35.6, 31.6, 31.1, 31.0, 29.0, 28.8, 28.7, 28.6, 28.6, 28.5, 28.4, 25.3, 21.8, 21.6, 21.4, 21.2, 21.1, 12.9. HRMS (ESI): found [M+Na]− 2225.0943, C98H171N5O49 calcd for [M+Na]– 2225.0940.

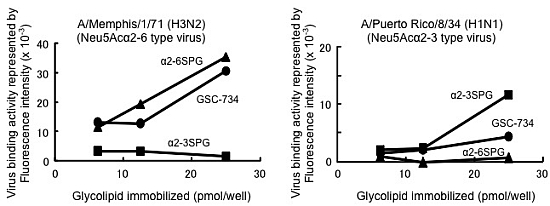
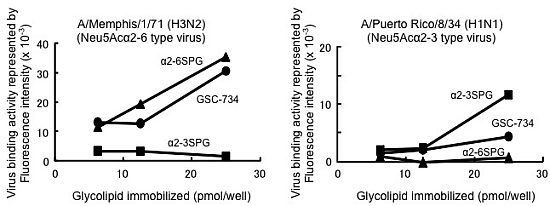
 ,
, 
{kind=link}
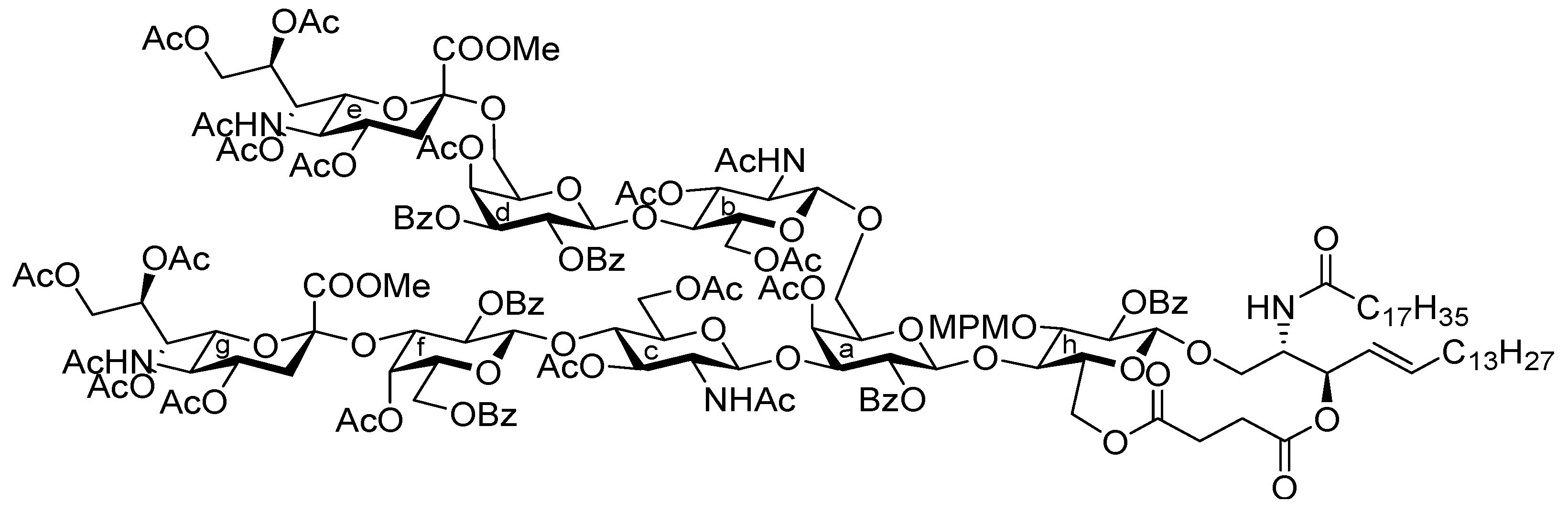
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}