B-norsteroids from Hymenoscyphus pseudoalbidus

Abstract
:1. Introduction
2. Results and Discussion
2.1. Compound Isolation
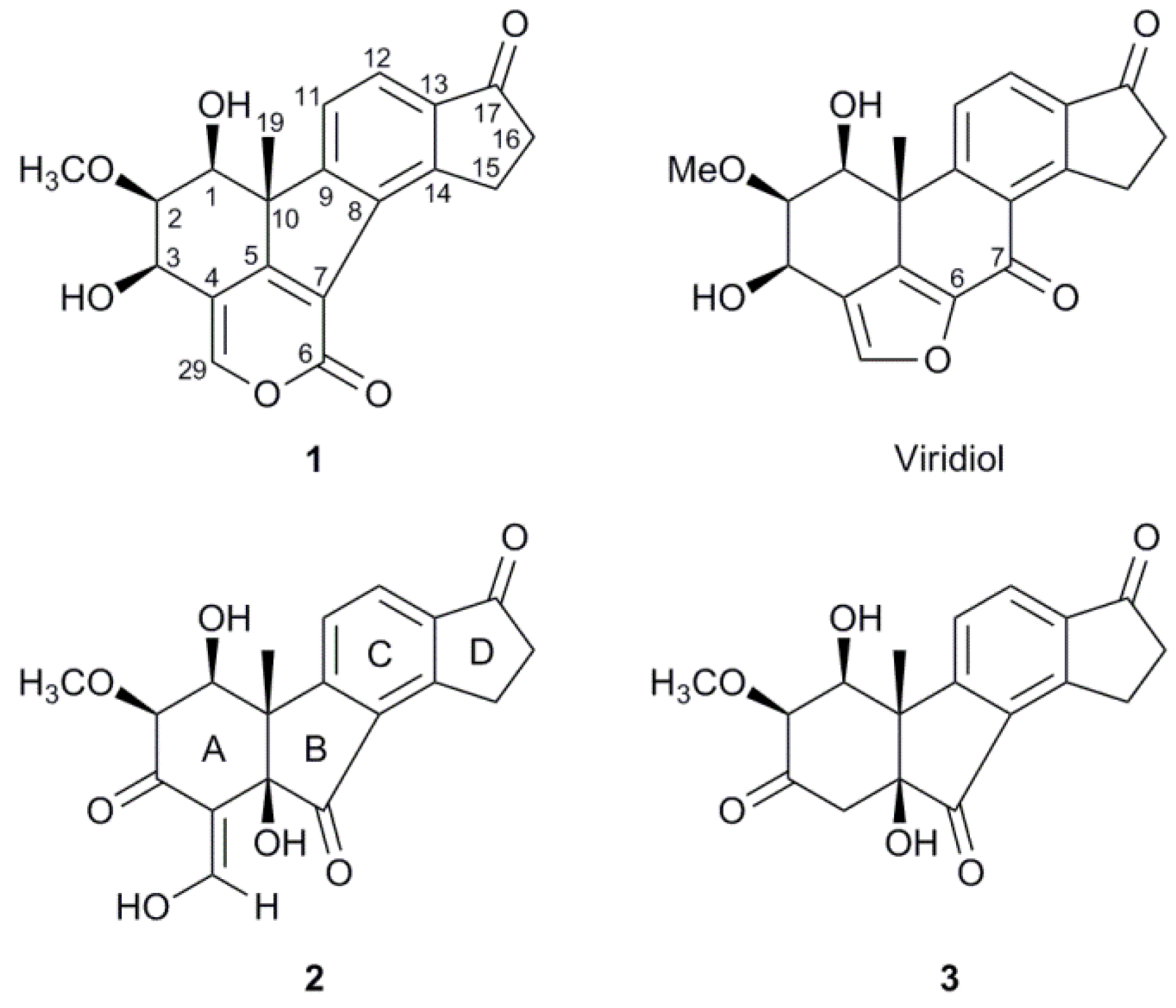
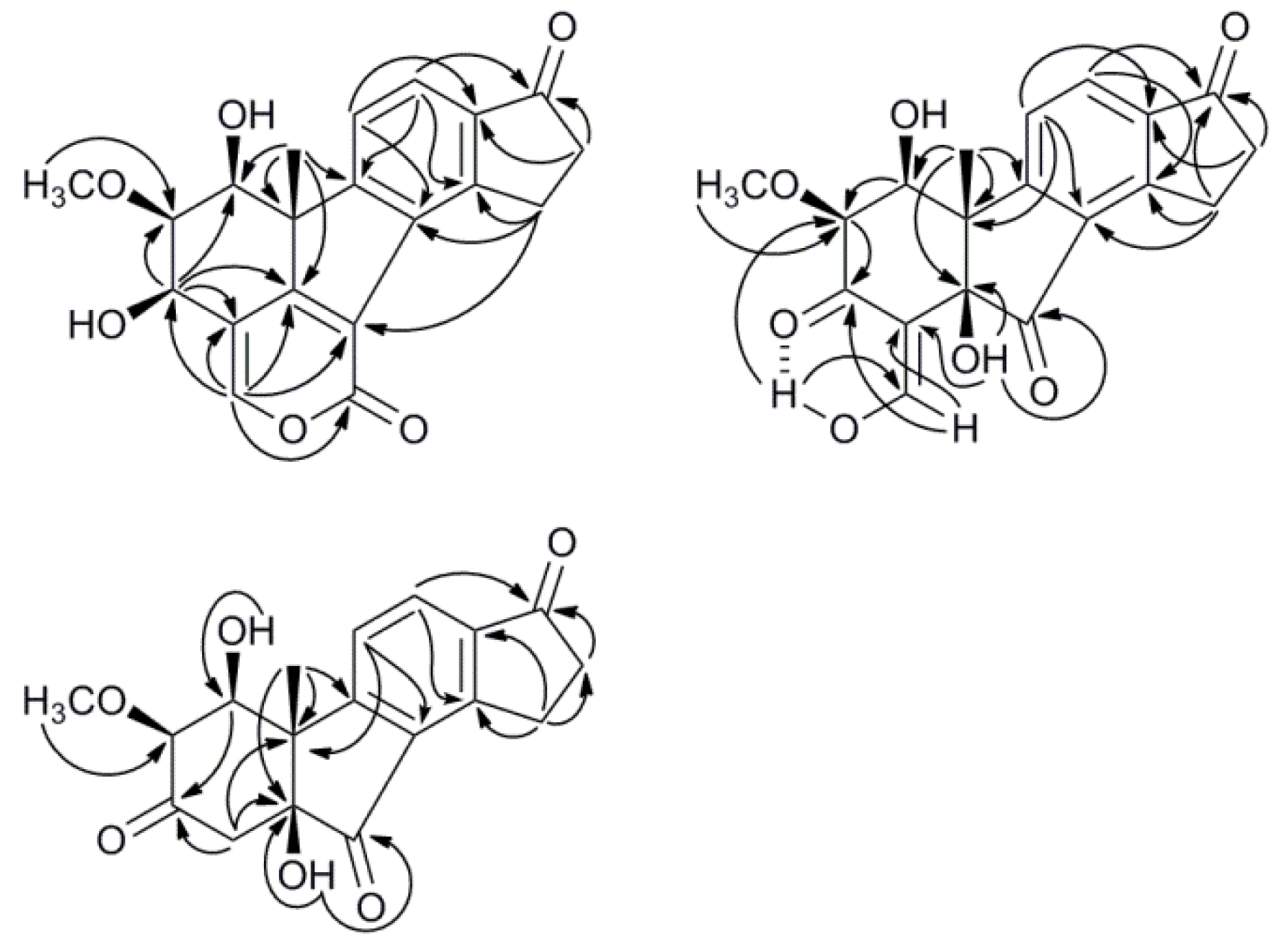
2.2. Structure Elucidation

| 1 | 2 | 3 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pos. | δC | Mult. | δH | Mult. | J (Hz) | δC | Mult. | δH | Mult. | J (Hz) | δC | Mult. | δH | Mult. | J (Hz) | ||
| 1 | 70.6 | CH | 3.60 | dd | 7.8, 7.5 | 71.9 | CH | 4.66 | d | 3.1 | 77.7 | CH | 4.23 | dd | 3.7, 1.9 | ||
| 2 | 78.3 | CH | 3.748 | dd | 7.5, 6.0 | 77.5 | CH | 3.56 | d | 3.1 | 83.8 | CH | 3.17 | d | 1.9 | ||
| 3 | 62.8 | CH | 4.99 | d | 6.0 | 184.4 | C | 203.8 | C | ||||||||
| 4 | 114.8 | C | 110.7 | C | 46.1 | CH2 | α 2.83 | d | 16.8 | ||||||||
| β 2.78 | d | 16.8 | |||||||||||||||
| 5 | 164.4 | C | 80.1 | C | 78.8 | C | |||||||||||
| 6 | 158.2 | C | |||||||||||||||
| 7 | 122.7 | C | 201.0 | C | 204.9 | C | |||||||||||
| 8 | 133.7 | C | 130.0 | C | 131.3 | C | |||||||||||
| 9 | 158.4 | C | 161.8 | C | 165.6 | C | |||||||||||
| 10 | 53.3 | C | 49.4 | C | 52.4 | C | |||||||||||
| 11 | 122.9 | CH | 7.72 | d | 7.8 | 122.8 | CH | 7.50 | d | 7.9 | 124.5 | CH | 7.72 | d | 8.0 | ||
| 12 | 123.0 | CH | 7.77 | d | 7.8 | 130.9 | CH | 8.06 | d | 7.9 | 130.5 | CH | 7.97 | d | 8.0 | ||
| 13 | 137.7 | C | 138.6 | C | 138.8 | C | |||||||||||
| 14 | 149.6 | C | 156.1 | C | 155.1 | C | |||||||||||
| 15 | 26.6 | CH2 | 3.73 | m | 25.1 | CH2 | 3.36 | ddd | 18.9, 7.1, 4.5 | 25.3 | CH2 | 3.36 | m | ||||
| 3.56 | ddd | 18.9, 7.7, 4.7 | |||||||||||||||
| 16 | 36.3 | CH2 | 2.74 | m | 36.4 | CH2 | 2.78 | m | 36.5 | CH2 | 2.65 | m | |||||
| 17 | 206.6 | C | 205.1 | C | 203.9 | C | |||||||||||
| 19 | 19.6 | CH3 | 1.57 | s | 25.3 | CH3 | 1.53 | s | 20.1 | CH3 | 1.52 | s | |||||
| 29 | 150.8 | CH | 7.69 | s | 188.1 | CH | 9.12 | d | 5.3 | ||||||||
| OCH3 | 60.9 | CH3 | 3.754 | s | 60.7 | CH3 | 3.68 | s | 58.6 | CH3 | 3.27 | s | |||||
| 1-OH | 3.35 | d | 7.8 | 2.70 | s | 5.05 | d | 3.7 | |||||||||
| 3-OH | 3.26 | s | |||||||||||||||
| 5-OH | 2.93 | s | 5.3 | 5.13 | s | ||||||||||||
| 29-OH | 15.32 | d | |||||||||||||||

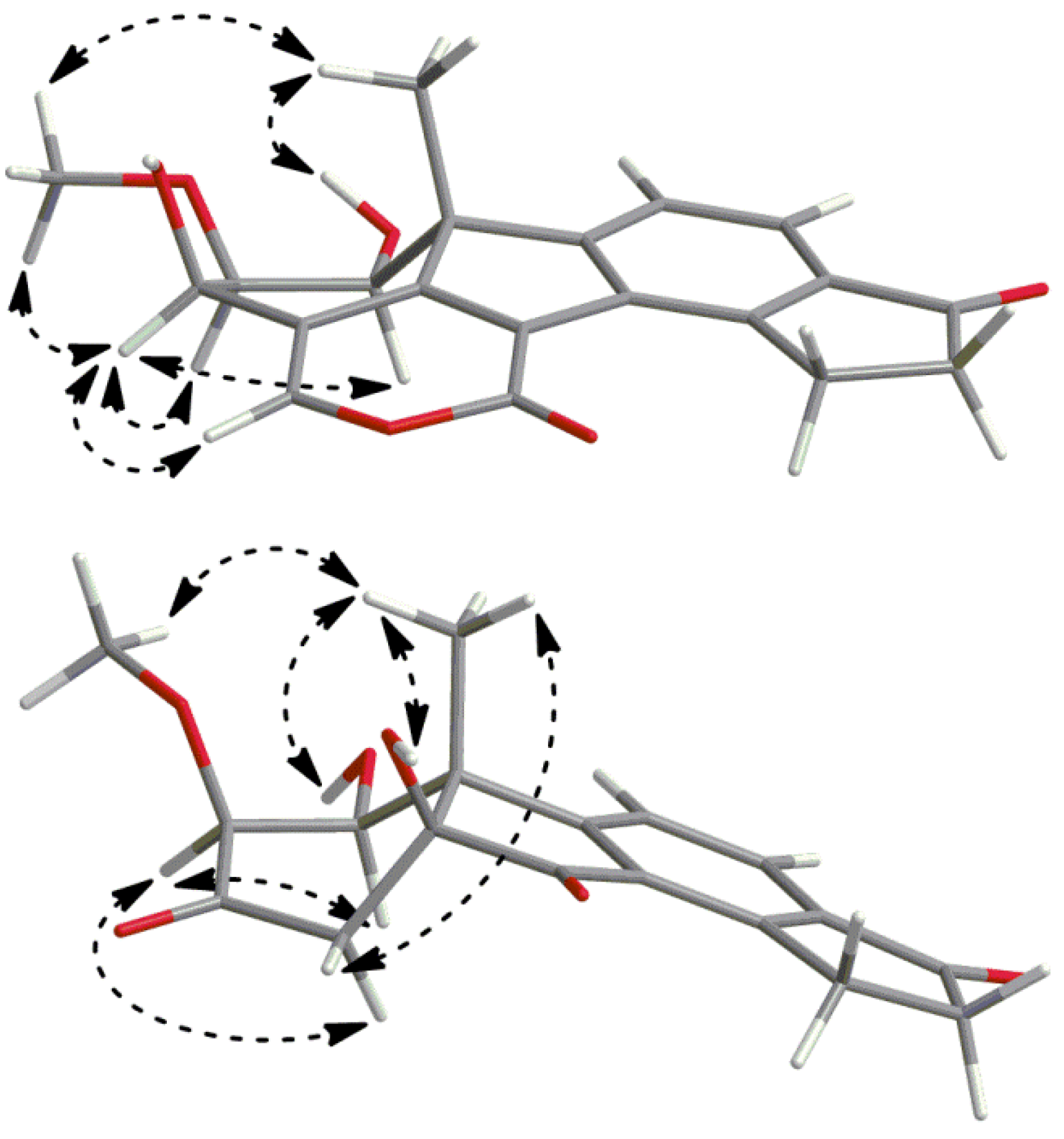
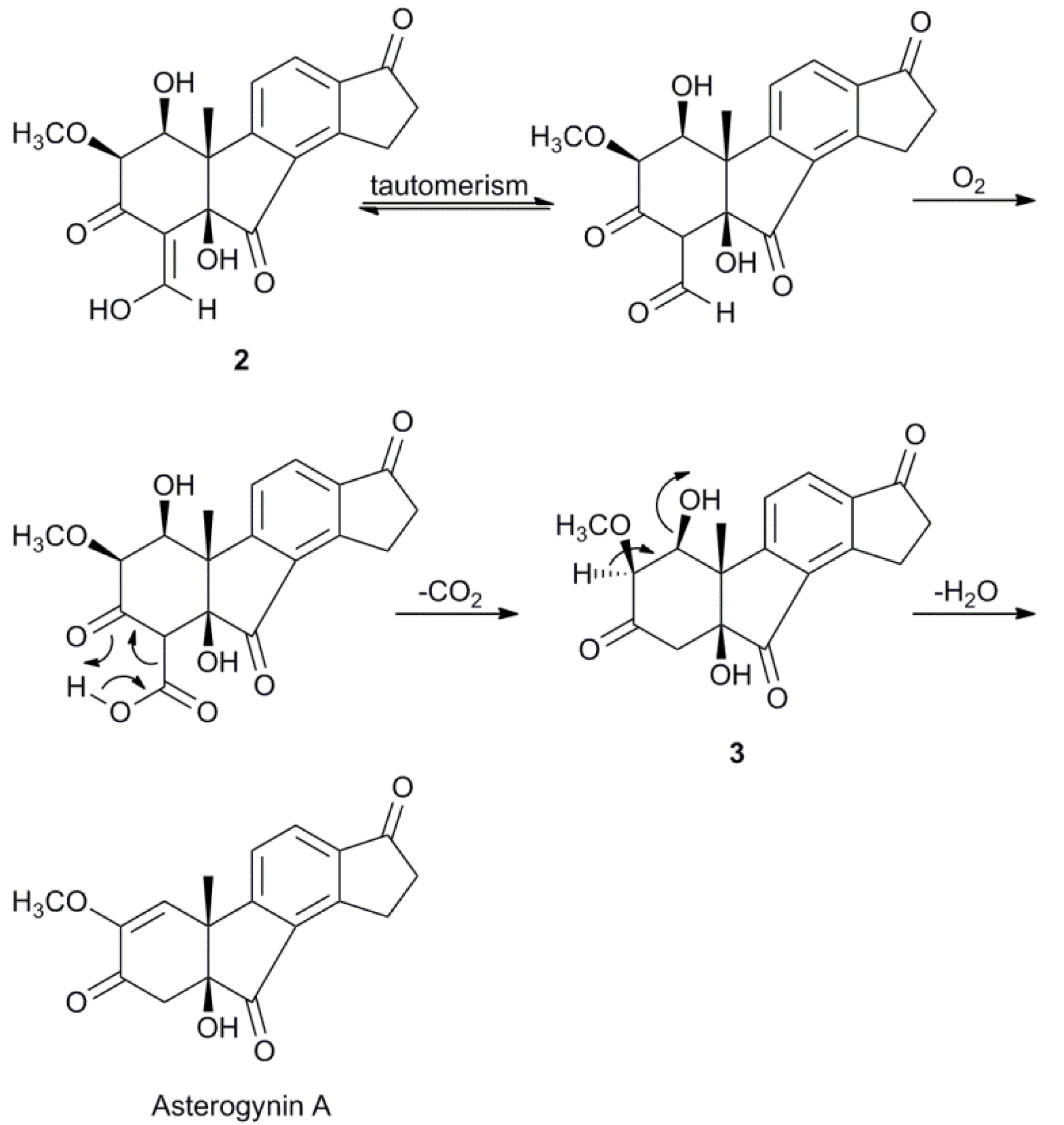
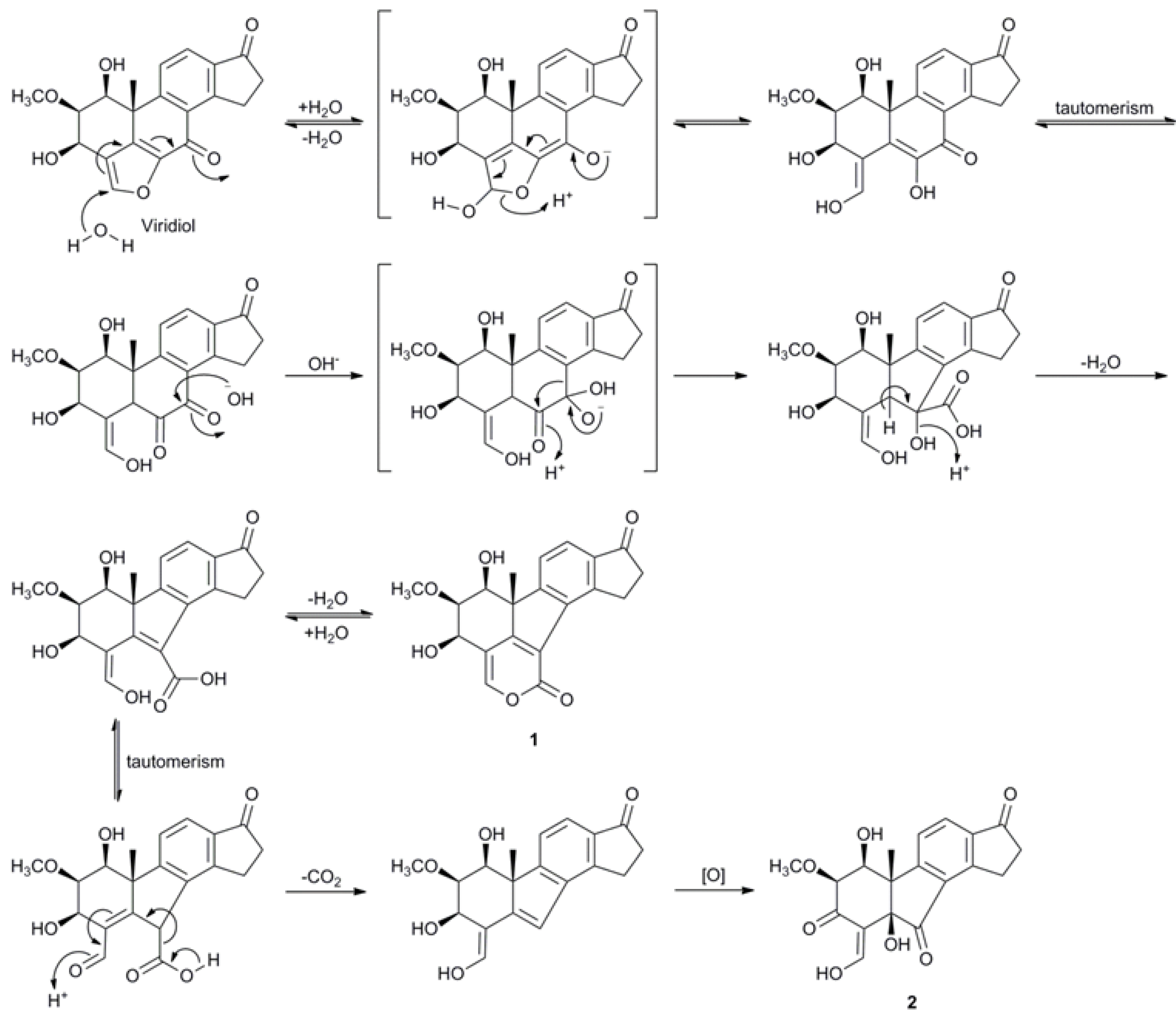
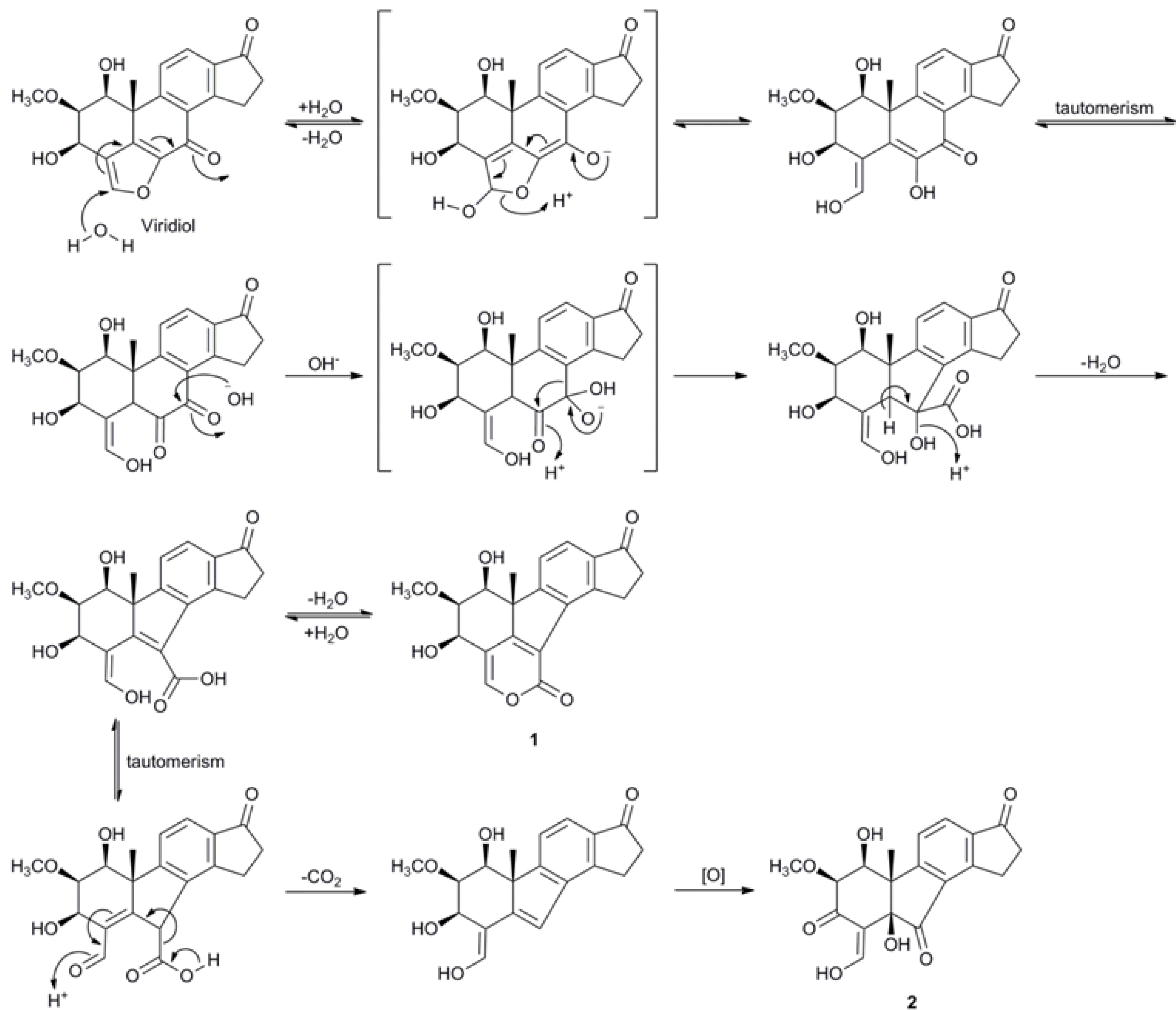
2.3. Biosynthetic Considerations
3. Experimental
3.1. General
3.2. Biological Material
3.3. Extraction and Isolation
3.4. Molecular Modeling
3.5. Compounds
 : + 94 (c 0.03, MeOH); UV (MeOH) λmax (log ε) 218 (3.84), 250 (4.08), 344 (3.67) nm; HRMS m/z 355.1172 [M+H]+ (calcd for C20H19O6, 355.1176); 1H-NMR data, see Table 1, 13C-NMR , see Table 1.
: + 94 (c 0.03, MeOH); UV (MeOH) λmax (log ε) 218 (3.84), 250 (4.08), 344 (3.67) nm; HRMS m/z 355.1172 [M+H]+ (calcd for C20H19O6, 355.1176); 1H-NMR data, see Table 1, 13C-NMR , see Table 1.
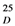
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Bakys, R.; Vasaitis, R.; Barklund, P.; Ihrmark, K.; Stenlid, J. Investigations concerning the role of Chalara fraxinea in declining Fraxinus excelsior. Plant Pathol. 2009, 58, 284–292. [Google Scholar] [CrossRef]
- Queloz, V.; Grunig, C.R.; Berndt, R.; Kowalski, T.; Sieber, T.N.; Holdenrieder, O. Cryptic speciation in Hymenoscyphus albidus. Forest Pathol. 2011, 41, 133–142. [Google Scholar] [CrossRef]
- Kowalski, T.; Holdenrieder, O. Pathogenicity of Chalara fraxinea. Forest Pathol. 2009, 39, 1–7. [Google Scholar] [CrossRef]
- Schumacher, J.; Kehr, R.; Leonhard, S. Mycological and histological investigations of Fraxinus excelsior nursery saplings naturally infected by Chalara fraxinea. Forest Pathol. 2010, 40, 419–429. [Google Scholar]
- Haselwandter, K.; Dobernigg, B.; Beck, W.; Jung, G.; Cansier, A.; Winkelmann, G. Isolation and identification of hydroxamate siderophores of ericoid myccorrhizal fungi. Biometals 1992, 5, 51–56. [Google Scholar] [CrossRef]
- Thines, E.; Anke, H.; Steglich, W.; Sterner, O. New botrydial sesquiterpenoids from Hymenoscyphus epiphyllus. Z. Naturforsch. (C) 1997, 52, 413–420. [Google Scholar]
- Biabani, M.A.F.; Laatsch, H. Advances in chemical studies on low-molecular weight metabolites of marine fungi. J. Prakt. Chem.-Chem. Ztg. 1998, 340, 589–607. [Google Scholar] [CrossRef]
- Andersson, P.F.; Johansson, S.B.K.; Stenlid, J.; Broberg, A. Isolation, identification and necrotic activity of viridiol from Chalara fraxinea, the fungus responsible for dieback of ash. For. Pathol. 2010, 40, 43–46. [Google Scholar] [CrossRef]
- Moffatt, J.S.; Bulock, J.D.; Yuen, T.H. Viridiol, a steroid-like product from Trichoderma viride. J. Chem. Soc.Chem. Comm. 1969. [Google Scholar] [CrossRef]
- Howell, C.R.; Stipanovic, R.D. Phytotoxicity to crop plants and herbicidal effects on weeds of viridiol produced by Gliocladium virens. Phytopathology 1984, 74, 1346–1349. [Google Scholar] [CrossRef]
- Brian, P.W.; McGowan, J.C. Viridin—A highly fungistatic substance produced by Trichoderma viride. Nature 1945, 156, 144–145. [Google Scholar]
- Shukla, Y.J.; Pawar, R.S.; Ding, Y.Q.; Li, X.C.; Ferreira, D.; Khan, I.A. Pregnane glycosides from Hoodia gordonii. Phytochemistry 2009, 70, 675–683. [Google Scholar]
- Lin, W.H.; Fang, J.M.; Cheng, Y.S. Diterpenoids and steroids from Taiwania cryptomerioides. Phytochemistry 1998, 48, 1391–1397. [Google Scholar]
- Wei, X.M.; Rodriguez, A.D.; Wang, Y.H.; Franzblau, S.G. Novel ring B abeo-sterols as growth inhibitors of Mycobacterium tuberculosis isolated from a Caribbean Sea sponge, Svenzea zeai. Tetrahedron Lett. 2007, 48, 8851–8854. [Google Scholar] [CrossRef]
- Miyamoto, T.; Kodama, K.; Aramaki, Y.; Higuchi, R.; Van Soest, R.W.M. Orostanal, a novel abeo-sterol inducing apoptosis in leukemia cell from a marine sponge, Stelletta hiwasaensis. Tetrahedron Lett. 2001, 42, 6349–6351. [Google Scholar] [CrossRef]
- Anke, T.; Werle, A.; Kappe, R.B.; Sterner, O. Laschiatrion, a new antifungal agent from a Favolaschia species (Basidiomycetes) active against human pathogens. J. Antibiot. 2004, 57, 496–501. [Google Scholar] [CrossRef]
- Cao, S.G.; Ross, L.; Tamayo, G.; Clardy, J. Asterogynins: Secondary Metabolites from a Costa Rican Endophytic Fungus. Org. Lett. 2010, 12, 4661–4663. [Google Scholar] [CrossRef]
- Hanson, J.R. The viridin family of steroidal antibiotics. Nat. Prod. Rep. 1995, 12, 381–384. [Google Scholar] [CrossRef]
- Sakuno, E.; Yabe, K.; Hamasaki, T.; Nakajima, H. A new inhibitor of 5'-hydroxyaverantin dehydrogenase, an enzyme involved in aflatoxin biosynthesis, from Trichoderma hamatum. J. Nat. Prod. 2000, 63, 1677–1678. [Google Scholar] [CrossRef]
- Wipf, P.; Kerekes, A.D. Structure reassignment of the fungal metabolite TAEMC161 as the phytotoxin viridiol. J. Nat. Prod. 2003, 66, 716–718. [Google Scholar] [CrossRef]
- Ernst, L.; Wray, V.; Chertkov, V.A.; Sergeyev, N.M. High-resolution proton-coupled 13C-NMR spectra of monosubstituted benzenes. Theoretical and empirical correlations of JCH. J. Magn. Reson. 1977, 25, 123–139. [Google Scholar]
- Allinger, N.L.; Tribble, M.T. Conformational analysis – LXXX. The hydrindanone ring system. Tetrahedron 1972, 28, 1191–1202. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in electronic structure theory: GAMESS a decade later. In Theory and applications of computational chemistry: The first forty years; Dykstra, C.E., Frenkling, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherland, 2005; pp. 1167–1189. [Google Scholar]
- Zheng, Y.C.; Almlöf, J. Density functionals without meshes and grids. Chem. Phys. Lett. 1993, 214, 397–401. [Google Scholar] [CrossRef]
- Glaesemann, K.R.; Gordon, M.S. Investigation of a grid-free density functional theory (DFT) approach. J. Chem. Phys. 1998, 108, 9959–9969. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Denisov, G.S. A review with comprehensive data on experimental indirect scalar NMR spin-spin coupling constants across hydrogen bonds. Magn. Reson. Chem. 2008, 46, 599–624. [Google Scholar]
- Fierman, M.; Nelson, A.; Khan, S.I.; Barfield, M.; O’Leary, D.J. Scalar coupling across the hydrogen bond in 1,3-and 1,4-diols. Org. Lett. 2000, 2, 2077–2080. [Google Scholar] [CrossRef]
- Jansma, A.; Zhang, Q.; Li, B.; Ding, Q.; Uno, T.; Bursulaya, B.; Liu, Y.; Furet, P.; Gray, N.S.; Geierstanger, B.H. Verification of a designed intramolecular hydrogen bond in a drug scaffold by nuclear magnetic resonance spectroscopy. J. Med. Chem. 2007, 50, 5875–5877. [Google Scholar]
- Chen, Y.H.; Luo, Y.G.; Ju, J.H.; Wendt-Pienkowski, E.; Rajski, S.R.; Shen, B. Identification of fredericamycin E from Streptomyces griseus: Insights into fredericamycin A biosynthesis highlighting carbaspirocycle formation. J. Nat. Prod. 2008, 71, 431–437. [Google Scholar] [CrossRef]
- Tischler, M.; Ayer, S.W.; Andersen, R.J.; Mitchell, J.F.; Clardy, J. Anthosterone A and anthosterone B, ring-A contracted steroids from the sponge Anthoracuata graceae. Can. J. Chem. 1988, 66, 1173–1178. [Google Scholar] [CrossRef]
- Sample Availability: Contact the corresponding author.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Andersson, P.F.; Bengtsson, S.; Stenlid, J.; Broberg, A. B-norsteroids from Hymenoscyphus pseudoalbidus. Molecules 2012, 17, 7769-7781. https://doi.org/10.3390/molecules17077769
Andersson PF, Bengtsson S, Stenlid J, Broberg A. B-norsteroids from Hymenoscyphus pseudoalbidus. Molecules. 2012; 17(7):7769-7781. https://doi.org/10.3390/molecules17077769
Chicago/Turabian StyleAndersson, Pierre F., Stina Bengtsson, Jan Stenlid, and Anders Broberg. 2012. "B-norsteroids from Hymenoscyphus pseudoalbidus" Molecules 17, no. 7: 7769-7781. https://doi.org/10.3390/molecules17077769
APA StyleAndersson, P. F., Bengtsson, S., Stenlid, J., & Broberg, A. (2012). B-norsteroids from Hymenoscyphus pseudoalbidus. Molecules, 17(7), 7769-7781. https://doi.org/10.3390/molecules17077769