The Pharmacological Properties and Therapeutic Use of Apomorphine

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
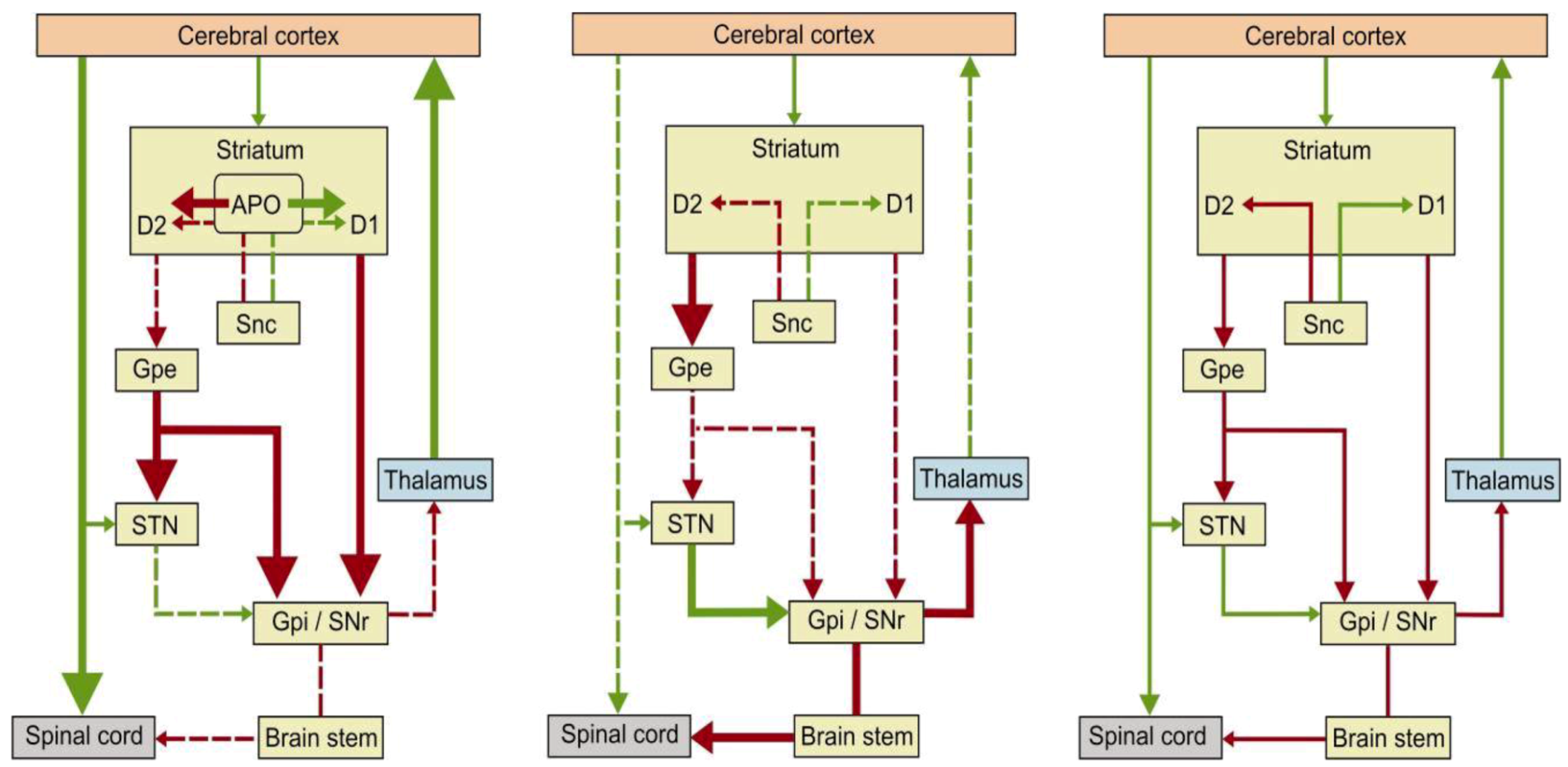
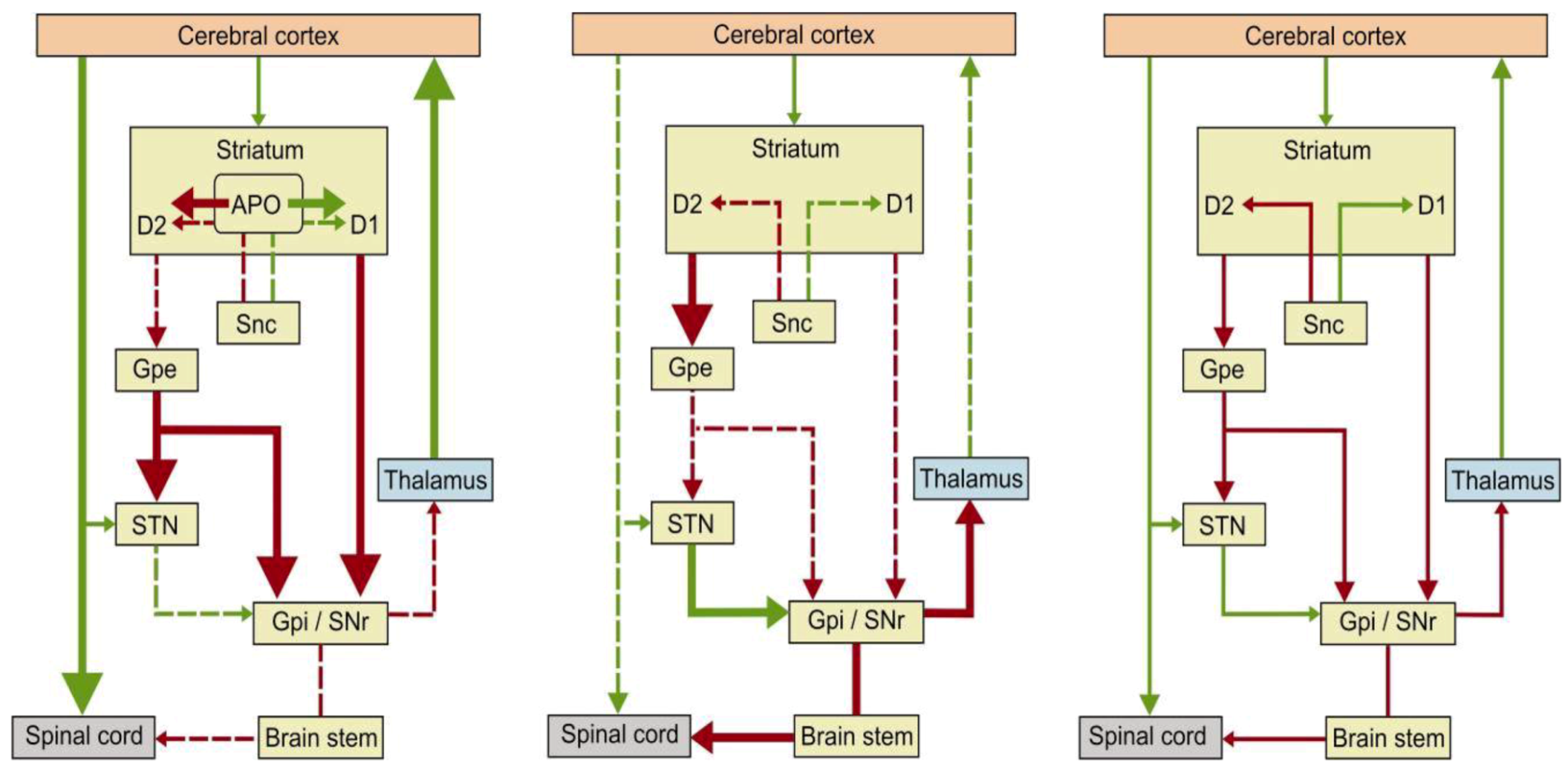
2. Apomorphine Treatment in Parkinson’s Disease
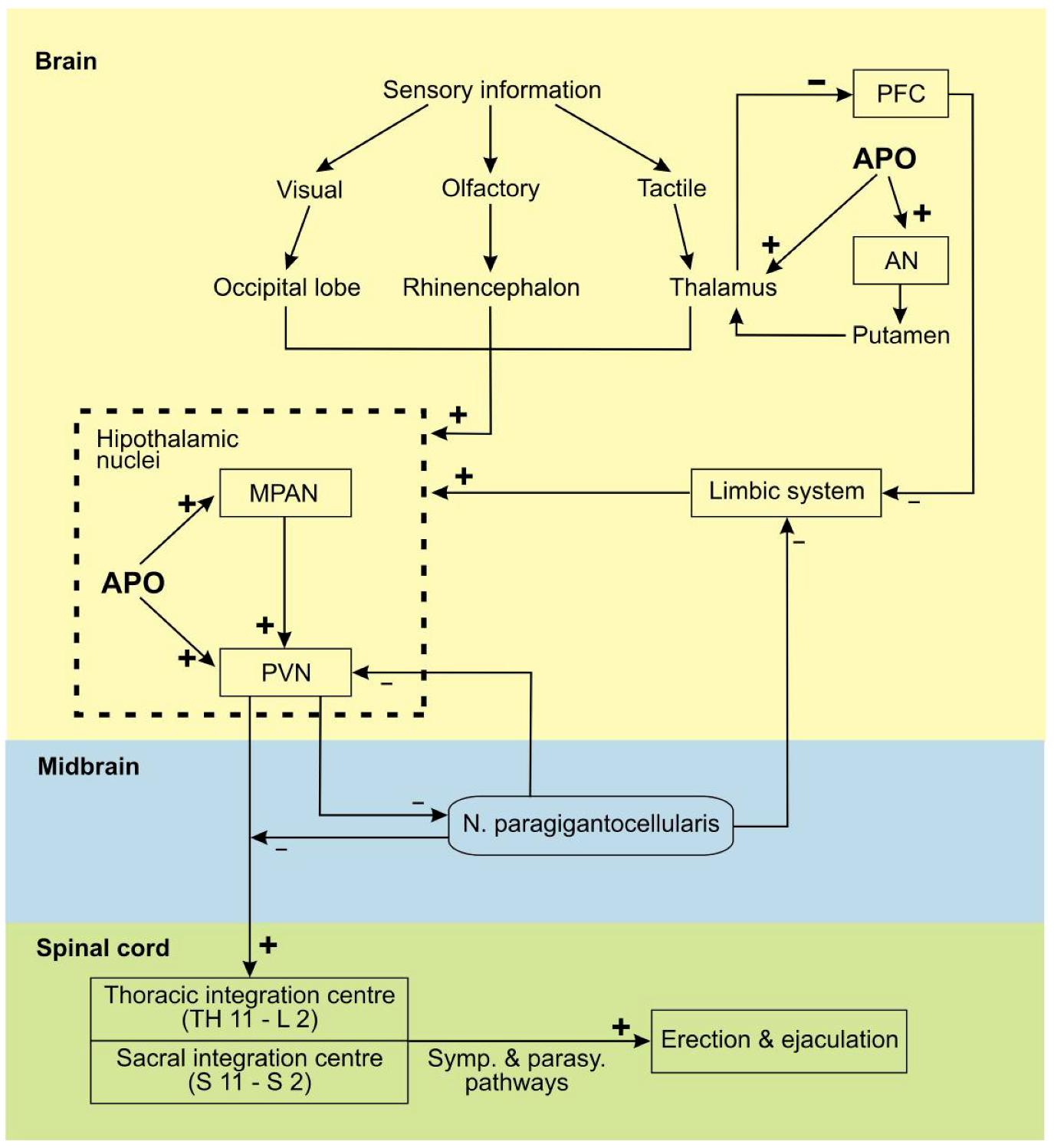
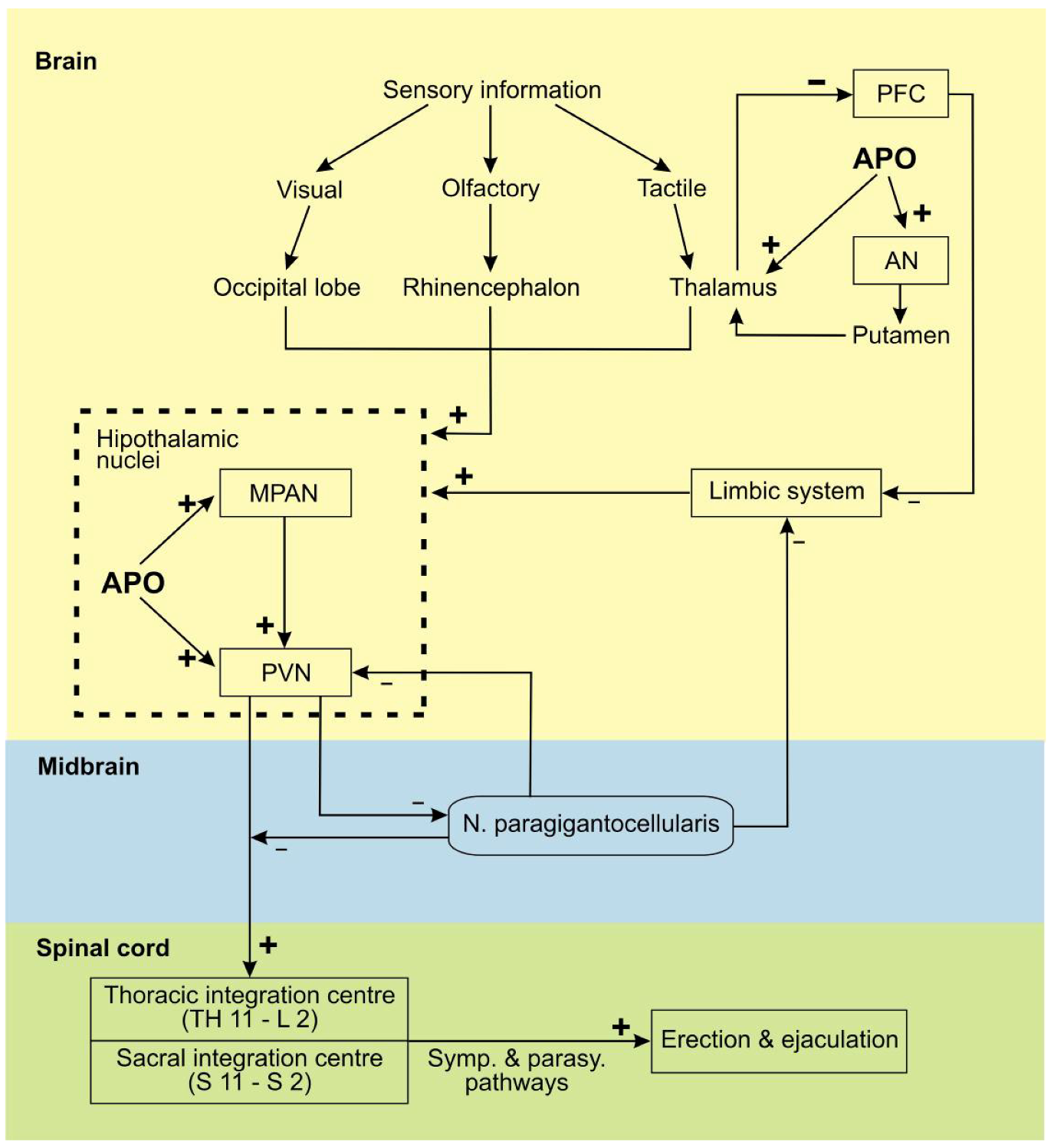
3. Apomorphine Treatment in Erectile Dysfunction
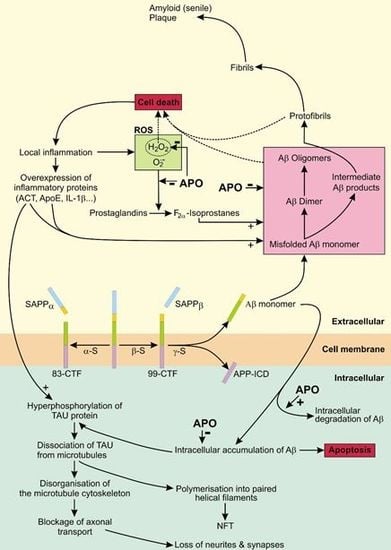
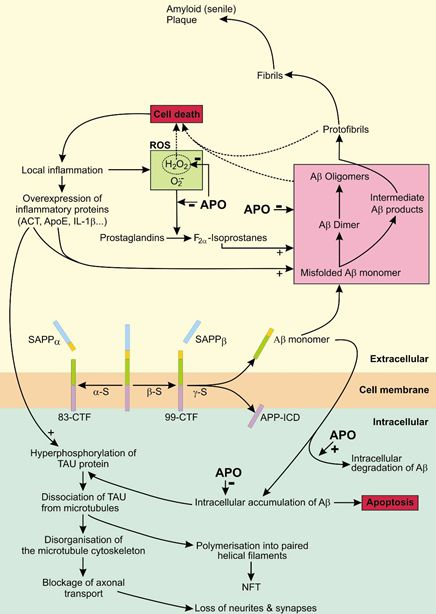
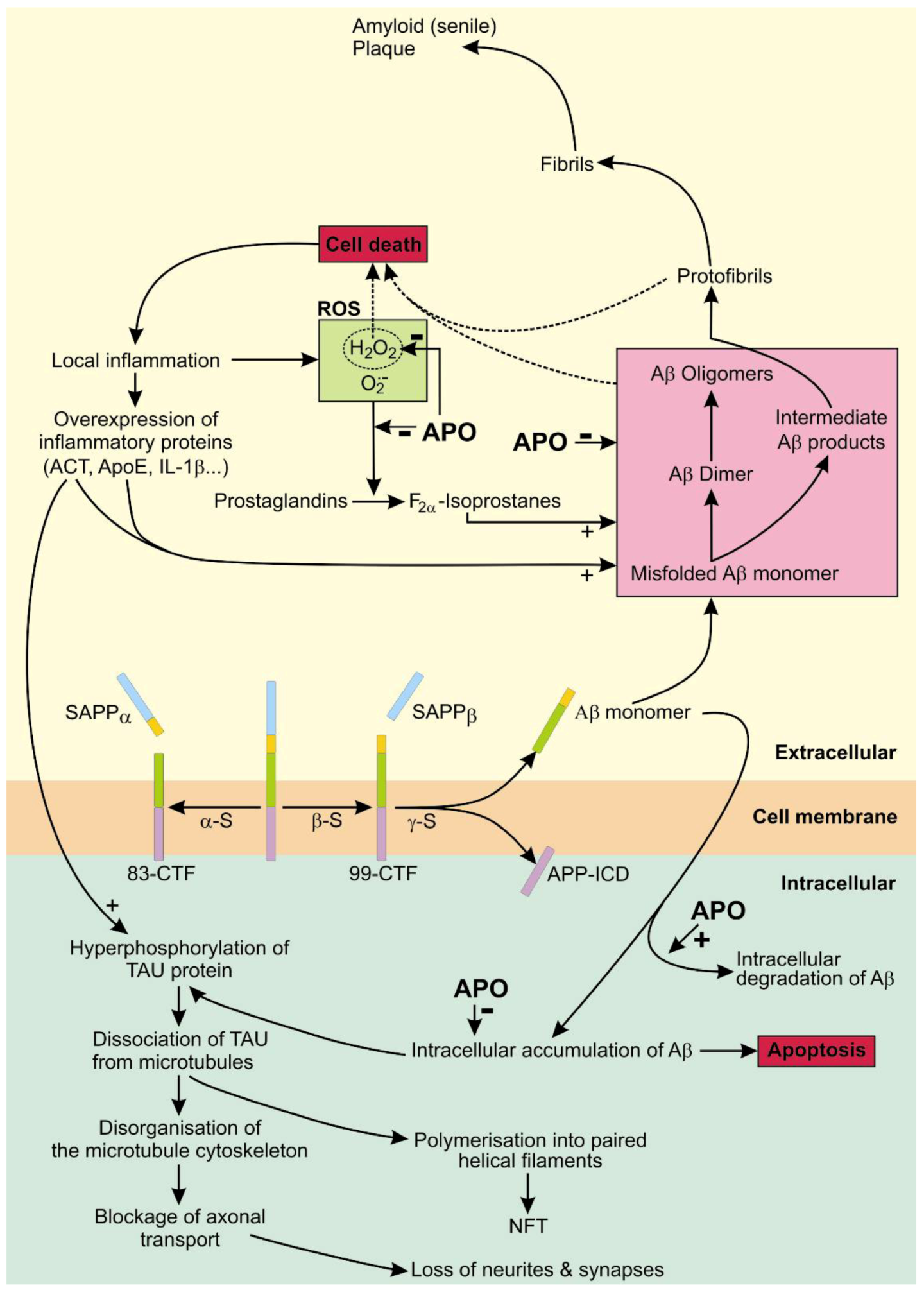
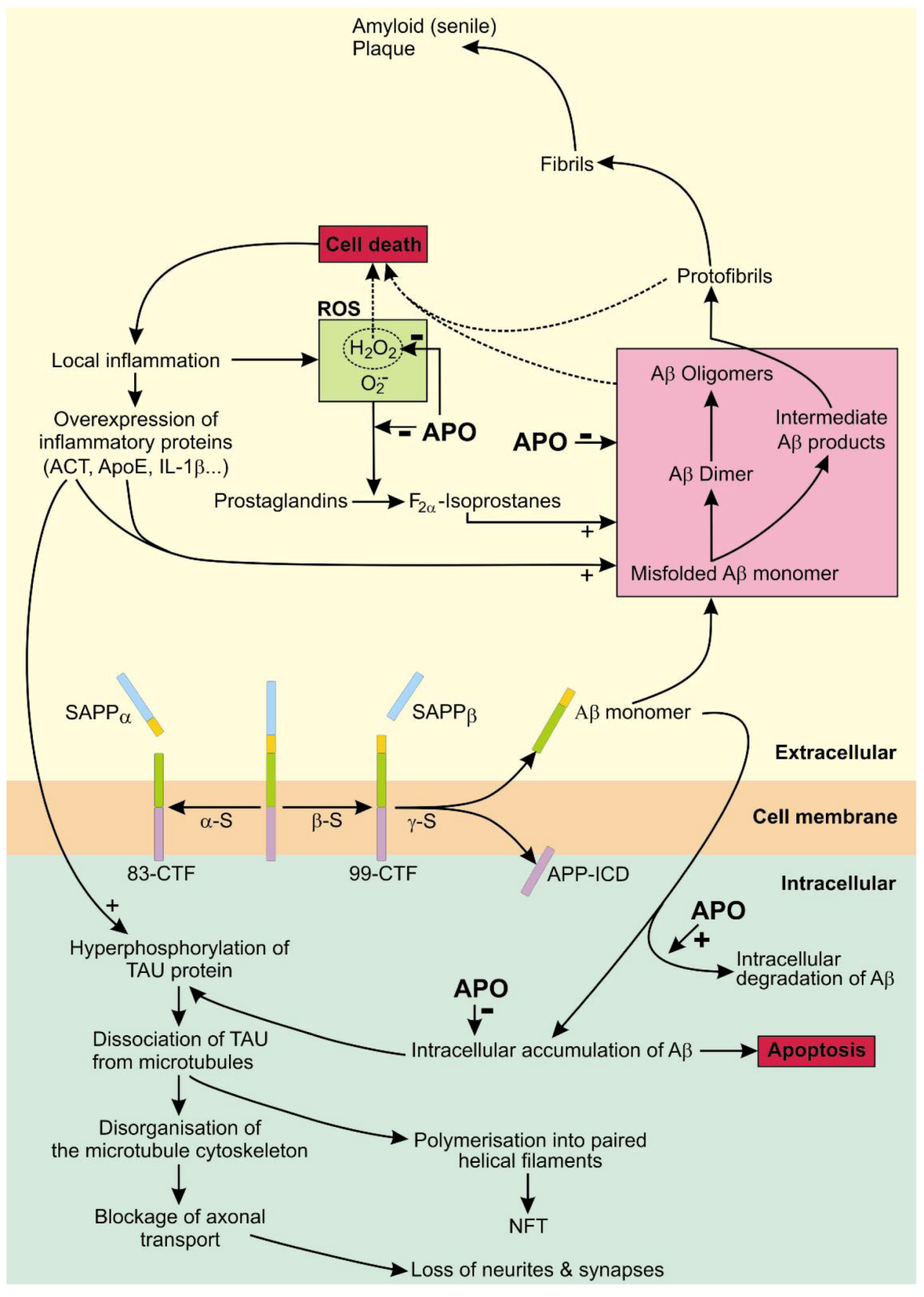
4. Apomorphine Treatment in Alzheimer’s Disease
4.1. The Role of APP in AD
4.2. The Role of Tau in AD
4.3. APO As a Potential Treatment of AD
5. Conclusions
Acknowledgements
Conflict of Interest
- Sample Availability: Not available.
References and Notes
- LeWitt, P.A. Subcutaneously administered apomorphine: Pharmacokinetics and metabolism. Neurology 2004, 62 (Suppl. 4), S8–S11. [Google Scholar] [CrossRef]
- Muguet, D.; Broussolle, E.; Chazot, G. Apomorphine in patients with Parkinson’s disease. Biomed. Pharmacother. 1995, 49, 197–209. [Google Scholar] [CrossRef]
- Millan, M.J.; Maiofiss, L.; Cussac, D.; Audinot, V.; Boutin, J.A.; Newman-Tancredi, A. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J. Pharmacol. Exp. Ther. 2002, 303, 791–804. [Google Scholar] [CrossRef]
- Nutt, J.G.; Gancher, S.T.; Woodward, W.R. Does an inhibitory action of levodopa contribute to motor fluctuations? Neurology 1988, 38, 1553–1557. [Google Scholar] [CrossRef]
- Gassen, M.; Glinka, Y.; Pinchasi, B.; Youdim, M.B. Apomorphine is a highly potent free radical scavenger in rat brain mitochondrial fraction. Eur. J. Pharmacol. 1996, 308, 219–225. [Google Scholar] [CrossRef]
- Grunblatt, E.; Mandel, S.; Gassen, M.; Youdim, M.B. Potent neuroprotective and antioxidant activity of apomorphine in MPTP and 6-hydroxydopamine induced neurotoxicity. J. Neural. Transm. Suppl. 1999, 55, 57–70. [Google Scholar]
- Li, A.; Guo, H.; Luo, X.; Sheng, J.; Yang, S.; Yin, Y.; Zhou, J. Apomorphine-induced activation of dopamine receptors modulates FGF-2 expression in astrocytic cultures and promotes survival of dopaminergic neurons. FASEB J. 2006, 20, 1263–1265. [Google Scholar] [CrossRef]
- Gancher, S.T.; Woodward, W.R.; Boucher, B.; Nutt, J.G. Peripheral pharmacokinetics of apomorphine in humans. Ann. Neurol. 1989, 26, 232–238. [Google Scholar] [CrossRef]
- Apomorphine-Compound Summary (CID 6005) Apomorphine PubChem. Available online: http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=6005 (accessed on 28 February 2012).
- Deleu, D.; Hanssens, Y.; Northway, M.G. Subcutaneous apomorphine: An evidence-based review of its use in Parkinson’s disease. Drugs Aging 2004, 21, 687–709. [Google Scholar] [CrossRef]
- Holford, N.H.; Sheiner, L.B. Understanding the dose-effect relationship: Clinical application of pharmacokinetic-pharmacodynamic models. Clin. Pharmacokinet. 1981, 6, 429–453. [Google Scholar] [CrossRef]
- Neef, C.; van Laar, T. Pharmacokinetic-pharmacodynamic relationships of apomorphine in patients with Parkinson’s disease. Clin. Pharmacokinet. 1999, 37, 257–271. [Google Scholar] [CrossRef]
- Neumeyer, J.L.; Lal, S.; Baldessarini, R.J. Historical Highlights of the Chemistry, Pharmacology, and Early Clinical Uses of Apomorphine. In Basic Pharmacology; Gessa, G.L., Corsini, G.U., Eds.; Raven Press: New York, NY, USA, 1981; Volume 1, pp. 1–17. [Google Scholar]
- Okun, M.S.; Foote, K.D. Parkinson’s disease DBS: What, when, who and why? The time has come to tailor DBS targets. Expert Rev. Neurother. 2010, 10, 1847–1857. [Google Scholar] [CrossRef]
- Scherkl, R.; Hashem, A.; Frey, H.H. Apomorphine-induced emesis in the dog-routes of administration, efficacy and synergism by naloxone. J. Vet. Pharmacol. Ther. 1990, 13, 154–158. [Google Scholar] [CrossRef]
- Sydow, O. Parkinson’s disease: Recent development in therapies for advanced disease with a focus on deep brain stimulation (DBS) and duodenal levodopa infusion. FEBS J. 2008, 275, 1370–1376. [Google Scholar] [CrossRef]
- Tsuji, A. Small molecular drug transfer across the blood-brain barrier via carrier-mediated transport systems. NeuroRx 2005, 2, 54–62. [Google Scholar] [CrossRef]
- Stefani, A.; Bassi, A.; Mazzone, P.; Pierantozzi, M.; Gattoni, G.; Altibrandi, M.G.; Giacomini, P.; Peppe, A.; Bernardi, G.; Stanzione, P. Subdyskinetic apomorphine responses in globus pallidus and subthalamus of parkinsonian patients: Lack of clear evidence for the ‘indirect pathway’. Clin. Neurophysiol. 2002, 113, 91–100. [Google Scholar] [CrossRef]
- Merello, M.; Lees, A.J.; Balej, J.; Cammarota, A.; Leiguarda, R. GPi firing rate modification during beginning-of-dose motor deterioration following acute administration of apomorphine. Mov. Disord. 1999, 14, 481–483. [Google Scholar] [CrossRef]
- Marsden, C.D.; Parkes, J.D.; Quinn, N. Fluctuations and Disability in Parkinson’s Disease: Clinical Aspects. In Movement Disorders; Marsden, C.D., Fahn, S., Eds.; Butterworth Scientific: London, UK, 1982; pp. 96–122. [Google Scholar]
- Ahlskog, J.E.; Muenter, M.D. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov. Disord. 2001, 16, 448–458. [Google Scholar] [CrossRef]
- Carron, R.; Fraix, V.; Maineri, C.; Seigneuret, E.; Piallat, B.; Krack, P.; Pollak, P.; Benabid, A.L.; Chabardès, S. High frequency deep brain stimulation of the subthalamic nucleus versus continuous subcutaneous apomorphine infusion therapy: A review. J. Neural. Transm. 2011, 118, 915–924. [Google Scholar] [CrossRef]
- Antonini, A.; Isaias, I.U.; Rodolfi, G.; Landi, A.; Natuzzi, F.; Siri, C.; Pezzoli, G. A 5-year prospective assessment of advanced Parkinson disease patients treated with subcutaneous apomorphine infusion or deep brain stimulation. J. Neurol. 2011, 258, 579–585. [Google Scholar] [CrossRef]
- de Gaspari, D.; Siri, C.; Landi, A.; Cilia, R.; Bonetti, A.; Natuzzi, F.; Morgante, L.; Mariani, C.B.; Sganzerla, E.; Pezzoli, G.; et al. Clinical and neuropsychological follow up at 12 months in patients with complicated Parkinson’s disease treated with subcutaneous apomorphine infusion or deep brain stimulation of the subthalamic nucleus. J. Neurol. Neurosurg. Psychiatr. 2006, 77, 450–453. [Google Scholar] [CrossRef]
- Rudzinska, M.; Szczudlik, A. Apomorphine in off state—Clinical experience. Neurol. Neurochir. Pol. 2007, 41, S40–S48. [Google Scholar]
- Gancher, S.T.; Nutt, J.G.; Woodward, W.R. Time course of tolerance to apomorphine in parkinsonism. Clin. Pharmacol. Ther. 1992, 52, 504–510. [Google Scholar] [CrossRef]
- Grandas, F.; Gancher, S.; Lera, G.; Rodriguez, M.; Woodward, W.R.; Nutt, J.; Obeso, J.A. Time interval between repeated injections conditions the duration of motor improvement to apomorphine in Parkinson’s disease. Neurology 1992, 42, 1287–1290. [Google Scholar] [CrossRef]
- Gonzales-Rios, F.; Vlaiculescu, A.; Ben Natan, L.; Protais, P.; Costentin, J. Dissociated effects of apomorphine on various nociceptive responses in mice. J. Neural. Transm. 1986, 67, 87–103. [Google Scholar] [CrossRef]
- Bonuccelli, U.; Piccini, P.; Del Dotto, P.; Rossi, G.; Corsini, G.U.; Muratorio, A. Naloxone partly counteracts apomorphine side effects. Clin. Neuropharmacol. 1991, 14, 442–449. [Google Scholar] [CrossRef]
- Poewe, W.; Wenning, G.K. Apomorphine: An underutilized therapy for Parkinson’s disease. Mov. Disord. 2000, 15, 789–794. [Google Scholar] [CrossRef]
- Clarke, C.E.; Worth, P.; Grosset, D.; Stewart, D. Systematic review of apomorphine infusion, levodopa infusion and deep brain stimulation in advanced Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 728–741. [Google Scholar] [CrossRef]
- Corsini, G.U.; Del Zompo, M.; Gessa, G.L.; Mangoni, A. Therapeutic efficacy of apomorphine combined with an extracerebral inhibitor of dopamine receptors in Parkinson’s disease. Lancet 1979, 1, 954–956. [Google Scholar]
- Hughes, A.J.; Kleedorfer, B.B.; Turjanski, N.; Fernandez, W.; Lees, A.J.; Stern, G.M. Subcutaneous apomorphine in Parkinson’s disease: Response to chronic administration for up to five years. Mov. Disord. 1992, 8, 165–170. [Google Scholar]
- Deuschl, G.; Schade-Brittinger, C.; Krack, P.; Volkmann, J.; Schafer, H.; Botzel, K.; Daniels, C.; Deutschländer, A.; Dillmann, U.; Eisner, W.; et al. A randomized trial of deep-brain stimulation for Parkinson’s disease. N. Engl. J. Med. 2006, 355, 896–908. [Google Scholar] [CrossRef]
- NIH Consensus Conference. Impotence. NIH consensus development panel on impotence. JAMA 1993, 270, 83–90. [Google Scholar]
- Lasker, G.F.; Maley, J.H.; Kadowitz, P.J. A review of the pathophysiology and novel treatments for erectile dysfunction. Adv. Pharmacol. Sci. 2010. [Google Scholar] [CrossRef]
- Fedele, D.; Coscelli, C.; Cucinotta, D.; Forti, G.; Santeusanio, F.; Viaggi, S.; Fiori, G.; Velonà, T.; Lavezzari, M. Incidence of erectile dysfunction in Italian men with diabetes. J. Urol. 2001, 166, 1368–1371. [Google Scholar] [CrossRef]
- Martin-Morales, A.; Sanchez-Cruz, J.J.; Saenz de Tejada, I.; Rodriguez-Vela, L.; Jimenez-Cruz, J.F.; Burgos-Rodriguez, R. Prevalence and independent risk factors for erectile dysfunction in Spain: Results of the Epidemiologia de la Disfuncion Erectil Masculina Study. J. Urol. 2001, 166, 569–574. [Google Scholar] [CrossRef]
- Montorsi, P.; Ravagnani, P.M.; Galli, S.; Rotatori, F.; Briganti, A.; Salonia, A. The artery size hypothesis: A macrovascular link between erectile dysfunction and coronary artery disease. Am. J. Cardiol. 2005, 6, 19–23. [Google Scholar]
- Carson, C.C., III. Erectile dysfunction: Diagnosis and management with newer oral agents. Proc. (Bayl. Univ. Med. Cent.) 2000, 13, 356–360. [Google Scholar]
- Shaul, P.W. Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J. Physiol. 2003, 15, 21–33. [Google Scholar] [CrossRef]
- Stief, C.G. Central mechanisms of erectile dysfunction: What a clinician may want to know. Int. J. Impot. Res. 2003, 15 (Suppl. 2), S3–S6. [Google Scholar] [CrossRef]
- Gratzke, C.; Angulo, J.; Chitaley, K.; Dai, Y.T.; Kim, N.N.; Paick, J.S.; Simonsen, U.; Uckert, S.; Wespes, E.; Andersson, K.E.; et al. Anatomy, physiology, and pathophysiology of erectile dysfunction. J. Sex. Med. 2010, 7, 445–475. [Google Scholar] [CrossRef]
- Dean, R.C.; Lue, T.F. Physiology of penile erection and pathophysiology of erectile dysfunction. Urol. Clin. N. Am. 2005, 32, 379–395. [Google Scholar] [CrossRef]
- Heaton, J.P.; Morales, A.; Adams, M.A.; Johnston, B.; el-Rashidy, R. Recovery of erectile function by the oral administration of apomorphine. Urology 1995, 45, 200–206. [Google Scholar] [CrossRef]
- Von Keitz, A.T.; Ströberg, P.; Bukofzer, S.; Mallard, N.; Hibberd, M. A European multicentre study to evaluate the tolerability of apomorphine sublingual administered in a forced dose-escalation regimen in patients with erectile dysfunction. BJU Int. 2002, 89, 409–415. [Google Scholar] [CrossRef]
- Maclennan, K.M.; Boshier, A.; Wilton, L.V.; Shakir, S.A. Examination of the safety and use of apomorphine prescribed in general practice in England as a treatment for erectile dysfunction. BJU Int. 2006, 98, 125–131. [Google Scholar] [CrossRef]
- Afif-Abdo, J.; Teloken, C.; Damião, R.; Koff, W.; Wroclawski, E.; Yamasaki, R.; Torres, L.O.; Sabaneeff, J.; Faria, G.; Pompeo, A.C.; et al. Comparative cross-over study of sildenafil and apomorphine for treating erectile dysfunction. BJU Int. 2008, 102, 829–834. [Google Scholar] [CrossRef]
- Montorsi, F.; Perani, D.; Anchisi, D.; Salonia, A.; Scifo, P.; Rigiroli, P.; Zanoni, M.; Heaton, J.P.; Rigatti, P.; Fazio, F. Apomorphine-induced brain modulation during sexual stimulation: A new look at central phenomena related to erectile dysfunction. Int. J. Impot. Res. 2003, 15, 203–209. [Google Scholar] [CrossRef]
- Caruso, S.; Agnello, C.; Intelisano, G.; Farina, M.; Di Mari, L.; Cianci, A. Placebo-controlled study on efficacy and safety of daily apomorphine SL intake in premenopausal women affected by hypoactive sexual desire disorder and sexual arousal disorder. Urology 2004, 63, 955–959. [Google Scholar] [CrossRef]
- Sarazin, M.; de Souza, L.C.; Lehéricy, S.; Dubois, B. Clinical and research diagnostic criteria for alzheimer’s disease. Neuroimaging Clin. N. Am. 2012, 22, 23–32. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Liu, X.; Bandy, D.; Yu, M.; Lee, W.; Ayutyanont, N.; Keppler, J.; Reeder, S.A.; Langbaum, J.B.; et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 6820–6825. [Google Scholar]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Mölsä, P.K.; Marttila, R.J.; Rinne, U.K. Long-term survival and predictors of mortality in Alzheimer’s disease and multi-infarct dementia. Acta Neurol. Scand. 1995, 91, 159–164. [Google Scholar]
- Mölsä, P.K.; Marttila, R.J.; Rinne, U.K. Survival and cause of death in Alzheimer’s disease and multi-infarct dementia. Acta Neurol. Scand. 1986, 74, 103–107. [Google Scholar]
- Wimo, A.; Jönsson, L.; Gustavsson, A.; McDaid, D.; Ersek, K.; Georges, J.; Gulácsi, L.; Karpati, K.; Kenigsberg, P.; Valtonen, H. The economic impact of dementia in Europe in 2008-cost estimates from the Eurocode project. Int. J. Geriatr. Psychiatry 2011, 26, 825–832. [Google Scholar] [CrossRef]
- Zahs, K.R.; Ashe, K.H. “Too much good news”—are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer’s disease? Trends Neurosci. 2010, 33, 381–389. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Slunt, H.H.; Gonzales, V.; Savonenko, A.V.; Wen, J.C.; Jenkins, N.A.; Copeland, N.G.; Younkin, L.H; Lester, H.A.; Younkin, S.G.; et al. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS. Med. 2005, 2, e355. [Google Scholar]
- Gandy, S.; Heppner, F.L. Breaking up (amyloid) is hard to do. PLoS. Med. 2005, 2, e417. [Google Scholar] [CrossRef]
- Gouras, G.K.; Tampellini, D.; Takahashi, R.H.; Capetillo-Zarate, E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 119, 523–541. [Google Scholar] [CrossRef]
- Lee, J.; Retamal, C.; Cuitino, L.; Caruano-Yzermans, A.; Shin, J.E.; van Kerkhof, P.; Marzolo, M.P.; Bu, G. Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J. Biol. Chem. 2008, 283, 11501–11508. [Google Scholar]
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar]
- Kinoshita, A.; Fukumoto, H.; Shah, T.; Whelan, C.M.; Irizarry, M.C.; Hyman, B.T. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell. Sci. 2003, 116, 3339–3346. [Google Scholar] [CrossRef]
- Fukumori, A.; Okochi, M.; Tagami, S.; Jiang, J.; Itoh, N.; Nakayama, T.; Yanagida, K.; Ishizuka-Katsura, Y.; Morihara, T.; Kamino, K.; et al. Presenilin-dependent gamma-secretase on plasma membrane and endosomes is functionally distinct. Biochemistry 2006, 45, 4907–4914. [Google Scholar]
- Parvathy, S.; Hussain, I.; Karran, E.H.; Turner, A.J.; Hooper, N.M. Cleavage of Alzheimer’s amyloid precursor protein by alpha-secretase occurs at the surface of neuronal cells. Biochemistry 1999, 38, 9728–9734. [Google Scholar] [CrossRef]
- Gandy, S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J. Clin. Invest. 2005, 115, 1121–1129. [Google Scholar]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 2007, 282, 10311–10324. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Levy, M.; Dickson, D.W.; Potter, H. Alpha1-antichymotrypsin, an inflammatory protein overexpressed in Alzheimer’s disease brain, induces tau phosphorylation in neurons. Brain 2006, 129, 3020–3034. [Google Scholar] [CrossRef]
- Mrak, R.E.; Griffin, W.S. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging 2001, 22, 903–908. [Google Scholar] [CrossRef]
- Yao, Y.; Chinnici, C.; Tang, H.; Trojanowski, J.Q.; Lee, V.M.; Praticò, D. Brain inflammation and oxidative stress in a transgenic mouse model of Alzheimer-like brain amyloidosis. J. Neuroinflamm. 2004, 1, 21. [Google Scholar] [CrossRef]
- Yang, A.J.; Chandswangbhuvana, D.; Shu, T.; Henschen, A.; Glabe, C.G. Intracellular accumulation of insoluble, newly synthesized abetan-42 in amyloid precursor protein-transfected cells that have been treated with Abeta1-42. J. Biol. Chem. 1999, 274, 20650–20656. [Google Scholar]
- Wild-Bode, C.; Yamazaki, T.; Capell, A.; Leimer, U.; Steiner, H.; Ihara, Y.; Haass, C. Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42. J. Biol. Chem. 1997, 272, 16085–16088. [Google Scholar]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Ohyagi, Y.; Asahara, H.; Chui, D.H.; Tsuruta, Y.; Sakae, N.; Miyoshi, K.; Yamada, T.; Kikuchi, H.; Taniwaki, T.; Murai, H.; et al. Intracellular Ab42 activates p53 promoter: A pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 2005, 19, 255–257. [Google Scholar]
- Ohyagi, Y. Intracellular amyloid b-protein as a therapeutic target for treating Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 555–561. [Google Scholar] [CrossRef]
- Ma, L.; Ohyagi, Y.; Miyoshi, K.; Sakae, N.; Motomura, K.; Taniwaki, T.; Furuya, H.; Takeda, K.; Tabira, T.; Kira, J. Increase in p53 protein levels by presenilin 1 gene mutations and its inhibition by secretase inhibitors. J. Alzheimers Dis. 2009, 16, 565–575. [Google Scholar]
- Capetillo-Zarate, E.; Gracia, L.; Yu, F.; Banfelder, J.R.; Lin, M.T.; Tampellini, D.; Gouras, G.K. High-resolution 3D reconstruction reveals intra-synaptic amyloid fibrils. Am. J. Pathol. 2011, 179, 2551–2558. [Google Scholar] [CrossRef]
- Glabe, C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer's disease. J. Mol. Neurosci. 2001, 17, 137–145. [Google Scholar] [CrossRef]
- Gouras, G.K.; Almeida, C.G.; Takahashi, R.H. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol. Aging 2005, 26, 1235–1244. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral betaamyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar]
- Steele, J.W.; Gandy, S. Apomorphine and Alzheimer Aβ: Roles for regulated αcleavage, autophagy, and antioxidation? Ann. Neurol. 2011, 69, 221–225. [Google Scholar] [CrossRef]
- Shin, R.W.; Iwaki, T.; Kitamoto, T.; Tateishi, J. Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab. Invest. 1991, 64, 693–702. [Google Scholar]
- Mawal-Dewan, M.; Henley, J.; van de Voorde, A.; Trojanowski, J.Q.; Lee, V.M. The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J. Biol. Chem. 1994, 269, 30981–30987. [Google Scholar]
- Taniguchi, T.; Kawamata, T.; Mukai, H.; Hasegawa, H.; Isagawa, T.; Yasuda, M.; Hashimoto, T.; Terashima, A.; Nakai, M.; Mori, H.; et al. Phosphorylation of tau is regulated by PKN. J. Biol. Chem. 2001, 276, 10025–10031. [Google Scholar]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 2000, 156, 15–20. [Google Scholar] [CrossRef]
- Oddo, S.; Billings, L.; Kesslak, J.P.; Cribbs, D.H.; LaFerla, F.M. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004, 43, 321–332. [Google Scholar] [CrossRef]
- Milton, V.J.; Sweeney, S.T. Oxidative stress in synapse development and function. Dev. Neurobiol. 2012, 72, 100–110. [Google Scholar] [CrossRef]
- Gomez-Ramos, A.; Diaz-Nido, J.; Smith, M.A.; Perry, G.; Avila, J. Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. J. Neurosci. Res. 2003, 71, 863–870. [Google Scholar] [CrossRef]
- Tamagno, E.; Gugliemotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the γ- and β-secretase cleavages of the β-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Giliberto, L.; Tamagno, E.; Tabaton, M. Oxidative stress mediates the pathogenic effect of different Alzheimer’s disease risk factors. Front. Aging Neurosci. 2010, 2. [Google Scholar] [CrossRef]
- Himeno, E.; Ohyagi, Y.; Ma, L.; Nakamura, N.; Miyoshi, K.; Sakae, N.; Motomura, K.; Soejima, N.; Yamasaki, R.; Hashimoto, T.; et al. Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation. Ann. Neurol. 2011, 69, 248–256. [Google Scholar] [CrossRef]
- Ma, L.; Ohyagi, Y.; Nakamura, N.; Iinuma, K.M.; Miyoshi, K.; Himeno, E.; Soejima, N.; Yanagihara, Y.T.; Sakae, N.; Yamasaki, R.; et al. Activation of glutathione peroxidase and inhibition of p53-related apoptosis by apomorphine. J. Alzheimers. Dis. 2011, 27, 225–237. [Google Scholar]
- Ishige, K.; Chen, Q.; Sagara, Y.; Schubert, D. The activation of dopamine D4 receptors inhibits oxidative stress-induced nerve cell death. J. Neurosci. 2001, 21, 6069–6076. [Google Scholar]
- Tampellini, D.; Rahman, N.; Gallo, E.F.; Huang, Z.; Dumont, M.; Capetillo-Zarate, E.; Ma, T.; Zheng, R.; Lu, B.; Nanus, D.M.; et al. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J. Neurosci. 2009, 29, 9704–9713. [Google Scholar]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar]
- Hara, H.; Ohta, M.; Adachi, T. Apomorphine protects against 6-hydroxydopamine- induced neuronal cell death through activation of the Nrf2-ARE pathway. J. Neurosci. Res. 2006, 84, 860–866. [Google Scholar] [CrossRef]
- Boutaud, O.; Ou, J.J.; Chaurand, P.; Caprioli, R.M.; Montine, T.J.; Oates, J.A. Prostaglandin H2 (PGH2) accelerates formation of amyloid beta1–42 oligomers. J. Neurochem. 2002, 82, 1003–1006. [Google Scholar] [CrossRef]
- Pratico, D.; Sung, S. Lipid peroxidation and oxidative imbalance: Early functional events in Alzheimer’s disease. J. Alzheimers Dis. 2004, 6, 171–175. [Google Scholar]
- Lashuel, H.A.; Hartley, D.M.; Balakhaneh, D.; Aggarwal, A.; Teichberg, S.; Callaway, D.J. New class of inhibitors of amyloid-beta fibril formation. Implications for the mechanism of pathogenesis in Alzheimer's disease. J. Biol. Chem. 2002, 277, 42881–42890. [Google Scholar]
- Linazasoro, G. The apomorphine test in gait disorders associated with parkinsonism. Clin. Neuropharmacol. 1996, 19, 171–176. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ribarič, S. The Pharmacological Properties and Therapeutic Use of Apomorphine. Molecules 2012, 17, 5289-5309. https://doi.org/10.3390/molecules17055289
Ribarič S. The Pharmacological Properties and Therapeutic Use of Apomorphine. Molecules. 2012; 17(5):5289-5309. https://doi.org/10.3390/molecules17055289
Chicago/Turabian StyleRibarič, Samo. 2012. "The Pharmacological Properties and Therapeutic Use of Apomorphine" Molecules 17, no. 5: 5289-5309. https://doi.org/10.3390/molecules17055289
APA StyleRibarič, S. (2012). The Pharmacological Properties and Therapeutic Use of Apomorphine. Molecules, 17(5), 5289-5309. https://doi.org/10.3390/molecules17055289