A New and Efficient Method for the Synthesis of 3,4-Disubstituted Pyrrolidine-2,5-diones

Abstract
:1. Introduction
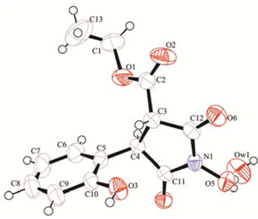
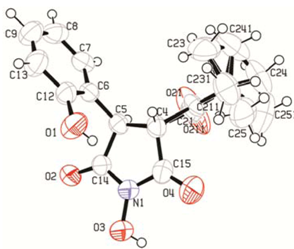
2. Results and Discussion
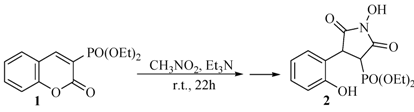
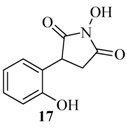
2.1. Rearrangement Reaction of Coumarins with Electron Withdrawing Substituent in the Third Position
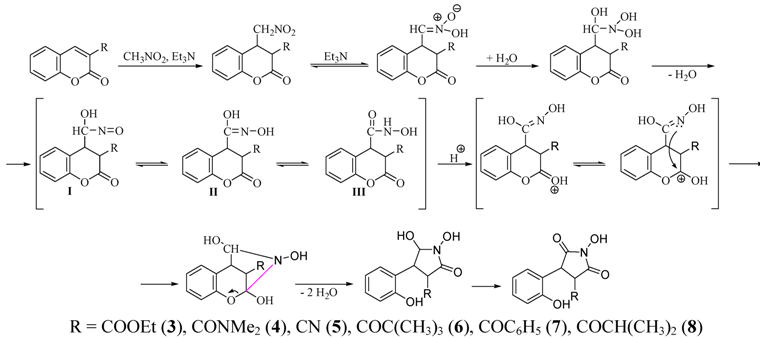

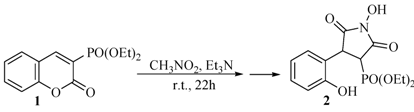{kind=link}
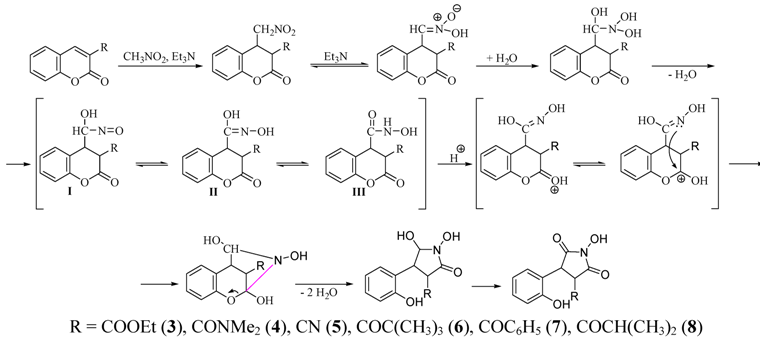{kind=link}
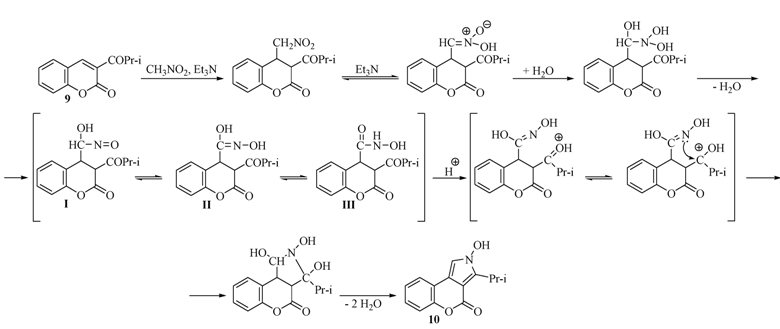{kind=link}
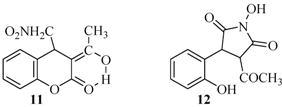{kind=link}
{kind=link}
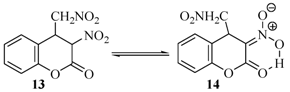
{kind=link}
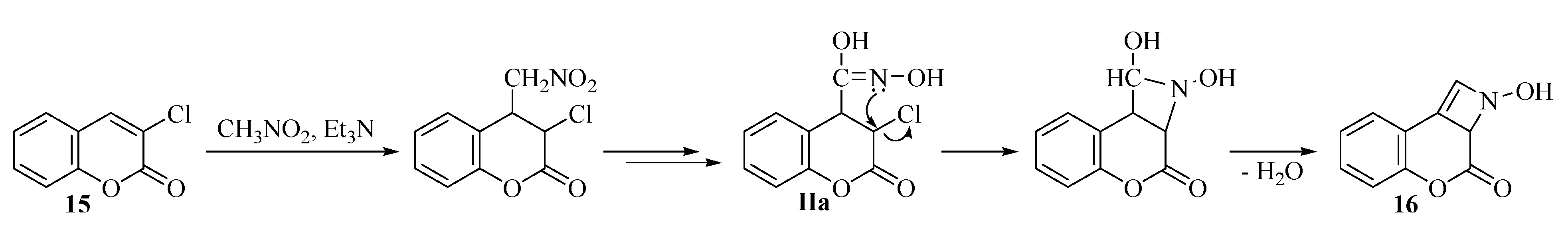
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Time, h | Yield, % | |
|---|---|---|---|
| 3 | 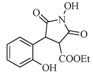 | 22 | 70% |
| 4 | 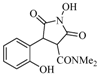 | 18 | 69% |
| 5 | 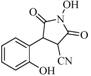 | 68 | 34% |
| 6 | 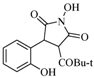 | 18 | 62% |
| 7 | 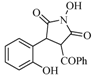 | 115 | 52% |
| 8 | 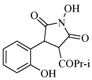 | 120 | 49% |
| 10 | 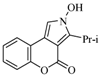 | 22% | |
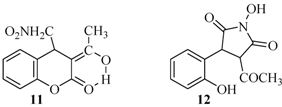
| 11 |  | 46 | 75% |
| 12 | 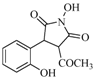 | 11% | |
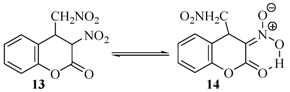
| 13 | 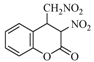 | 20 | 90% |
| 16 | 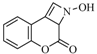 | 480 | 55% |
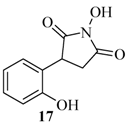
| 17 | 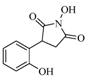 | 120 | 21% |
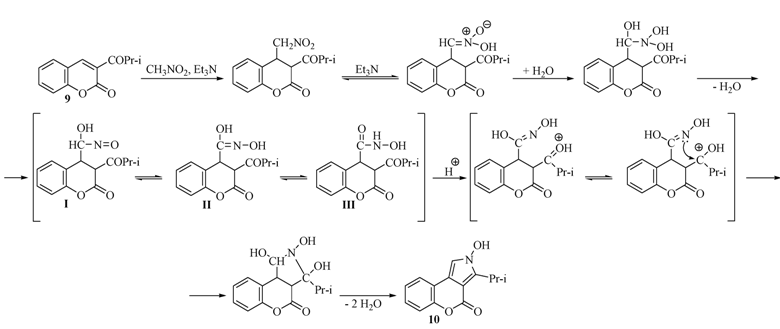
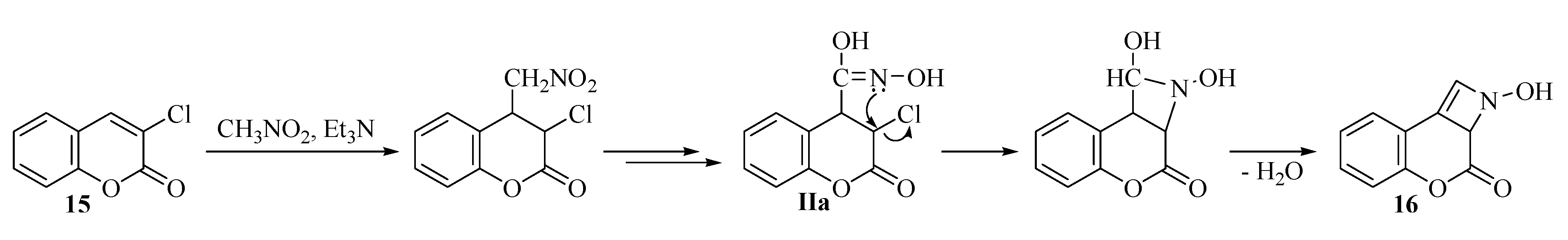
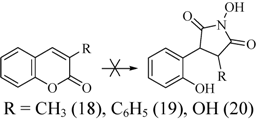
2.2. Rearrangement Reactions of Coumarin without a Substituent in the 3-Position
2.3. Rearrangement Reactions of Coumarins with Electron Donating Substituents in the 3-Position
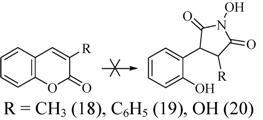

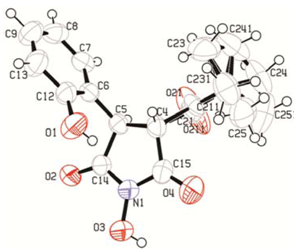
3. Experimental
3.1. General
3.2. General Procedure
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds 3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 16 are available from the authors.
References and Notes
- Chien, S.-C.; Chen, M.-L. Anti-inflammatory activities of new succinic and maleic derivatives from the fruiting body of antrodia camphorata. J. Agric. Food Chem. 2008, 56, 7017–7022. [Google Scholar] [CrossRef]
- Banerjee, P.S.; Sharma, P.K. New antiepileptic agents: Structure-activity relationships. Med. Chem. Res. 2011. [Google Scholar] [CrossRef]
- Obniska, J.; Kolaczkowski, M.; Bojarski, A.J.; Duszynska, B. Synthesis, anticonvulsant activity and 5-HT1A, 5-HT2A receptor affinity of new N-[(4-arylpiperazin-1-yl)-alkyl] derivatives of 2-azaspiro[4.4]nonane and [4.5]decane-1,3-dione. Eur. J. Med. Chem. 2006, 41, 874–881. [Google Scholar]
- Groutas, W.C.; Brubaker, M.J.; Chong, L.S.; Venkataraman, R.; Huang, H.; Epp, J.B.; Kuang, R.; Hoidal, J.R. Design, synthesis and biological evaluation of succinimide derivatives as potential mechanism-based inhibitors of human leukocyte elastase, cathepsin G and proteinase 3. Bioorg.Med. Chem. 1995, 3, 375–381. [Google Scholar] [CrossRef]
- Metobo, S.E.; Jin, H.; Tsiang, M.; Kim, C.U. Design, synthesis and biological evaluation of novel tricyclic HIV-1 integrase inhibitors by modification of its pyridine ring. Bioorg.Med. Chem. Lett. 2006, 16, 3985–3988. [Google Scholar] [CrossRef]
- Pawlowski, M.; Chlon, G.; Obniska, J.; Zejc, A.; Charakchieva-Minol, S.; Mokrosz, M. Synthesis, 5-HT1A and 5-HT2A receptor affinity of new 1-phenylpiperazinylpropyl derivatives of purine-2,6- and pyrrolidine-2,5-diones. IlFarmaco 2000, 55, 461–468. [Google Scholar]
- Thaqi, A.; Scott, J.L.; Gilbert, J.; Sakoff, J.A.; McCluskey, A. Synthesis and biological activity of D-5,6-norcantharimides: Importance of the 5,6-bridge. Eur. J. Med. Chem. 2010, 45, 1717–1723. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.-M.; ElTahir, K.E.H.; Asiri, Y.A. Synthesis, anti-inflammatory activity and COX-1/COX-2 inhibition of novel substituted cyclic imides. Part 1: Molecular docking study. Eur. J. Med. Chem. 2011, 46, 1648–1655. [Google Scholar]
- Vershueren, W.G.; Dierynck, I.; Amssoms, K.I.E.; Hu, L.; Boonants, P.M.J.G.; Pille, G.M.E.; Daeyaert, F.F.D.; Hertogs, K.; Surleraux, D.L.N.G.; Wigerinck, P.B.T.P. Design and Optimization of Tricyclic Phtalimide Analogues as Novel Inhibitors of HIV-1 Integrase. J. Med. Chem. 2005, 48, 1930–1940. [Google Scholar]
- Jindal, D.P.; Bedi, V.; Jit, B.; Karkra, N.; Guleria, S.; Bansal, R.; Palusczak, A.; Hartmann, R.W. Synthesis and study of some new N-substituted imide derivatives as potential anticancer agents. IlFarmaco 2005, 60, 283–290. [Google Scholar]
- Zajdel, P.; Subra, G.; Bojarska, A.J.; Duszynska, B.; Tatarczynska, E.; Nikiforuk, A.; Choinacka-Wojcik, E.; Pawlowski, M.; Martinez, J. Novel class of arylpiperazines containing N-acylated amino acids: Their synthesis, 5-HT1A, 5-HT2A receptor affinity, and in vivo pharmacological evaluati. Bioorg.Med. Chem. 2007, 15, 2907–2919. [Google Scholar] [CrossRef]
- Curtin, M.L.; Garland, R.B.; Heyman, H.R.; Frey, R.R.; Michaelides, M.R.; Li, J.; Pease, L.J.; Glaser, K.B.; Marcotte, P.A.; Davidsen, S.K. Succinimide hydroxamic acids as potent Inhibitors of histone deacetylase (HDAC). Bioorg.Med. Chem. Lett. 2002, 12, 2919–2923. [Google Scholar]
- Zajdel, P.; Krol, J.; Grychowska, K.; Pawlowski, M.; Subra, G.; Nomezine, G.; Martinez, J.; Satala, G.; Bojarski, A.; Zhou, Z.; O’Donnell, M.J.; Scott, W. Solid-phase synthesis of arylpiperazine derivatives and implementation of the distributed drug discovery (D3) project in the search for CNS agents. Molecules 2011, 16, 4104–4121. [Google Scholar]
- Katritzky, A.R.; Nair, S.K.; Witek, R.M.; Hutchins, S.M. Synthesis of 3,3-diarylpyrrolidines from diaryl ketones. ARKIVOC 2003, V, 9–18. [Google Scholar]
- Gribble, G.W.; Fu, L. Reductive acylation of 2- and 3-nitropyrroles-Efficient syntheses of pyrrolylamides and pyrrolylimides. Tetrahedron Lett. 2007, 48, 9155–9158. [Google Scholar] [CrossRef]
- Reddy, P.Y.; Kondo, S.; Toru, T.; Ueno, Y. Lewis Acid and Hexamethyldisilazane-Promoted Efficient Synthesis of N-Alkyl- and N-Arylimide Derivatives. J. Org. Chem. 1997, 62, 2652–2654. [Google Scholar] [CrossRef]
- Houghten, R.A.; Nefzi, A.; Alvarez-Gutierrez, J.M. Solid phase synthesis of 1,3-disubstituted succinimides. Tetrahedron Lett. 2000, 41, 609–612. [Google Scholar] [CrossRef]
- Vo-Hoang, Y.; Gasse, C.; Vidal, M.; Garbay, C.; Galons, H. Efficient synthesis of N-benzyl-3-aminopyrrolidine-2,5-dione and N-benzyl-3-aminopyrrolidin-2-one. Tetrahedron Lett. 2004, 45, 3603–3605. [Google Scholar] [CrossRef]
- Zajdel, P.; Subra, G.; Verdie, P.; Bojarski, A.J.; Duszynska, B.; Basista, K.; Obniska, J.; Martinez, J.; Pawlowski, M. The influence of an ethylene spacer on the 5-HT1A and 5-HT2A receptor affinity of arylpiperazine derivatives of amides with N-acylated amino acids and 3-differently substituted pyrrolidine-2,5-diones. Eur. J. Med. Chem. 2009, 44, 800–808. [Google Scholar] [CrossRef]
- Furukawa, I.; Abe, T.; Fujisawa, H.; Ohta, T. Novel synthesis of N-alkyl-3,4-disubstituted pyrrolidine-2,5-diones: condensation of a-oxoketene O,N-acetals and maleic anhydride. Tetrahedron 1997, 53, 17643–17652. [Google Scholar] [CrossRef]
- Cheng, M.; De, B.; Wahl, C.T.; Almstead, N.G.; Natchus, M.G.; Pikul, S. Oxazolidinone to succinamide: a novel rearrangement reaction. Tetrahedron Lett. 1999, 40, 5975–5977. [Google Scholar] [CrossRef]
- Brace, N.O.; Mull, S.G. New succinamic acids, -g-lactones, and -succinimides from (3-perfluoroalkyl-2-iodoalkyl)succinic acid anhydrides and amines Highly surface active amphiphile. J. Fluorine Chem. 2006, 127, 108–125. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Cabrero, G.; Ruiz, M.P. Organocatalytic ring expansion of beta-lactams to gamma-lactams through a novel N1-C4 bond cleavage. Direct synthesis of enantiopure succinimide derivatives. Org. Lett. 2005, 7, 3981–3984. [Google Scholar] [CrossRef]
- Ilieva, E.D.; Petkova, N.I.; Nikolova, R.D. Ring opening reactions of 3-phosphonocoumarin under Michael reaction conditions. Phosphorus Sulfur Silicon Relat.Elem. 2012, 187, 39–50. [Google Scholar] [CrossRef]
- Perucca, E. The new generation of antiepileptic drugs: advantages and disadvantages. Br. J. Clin. Pharmacol. 1996, 42, 531–543. [Google Scholar]
- 26. Unverferth, K.; Engel, J.; Hofgen, A; Rostock, A.; Gunther, R.; Lankau, H.J.; Menzer, M.; Rolfs, A.; Liebscher, J.; Muller, B.; Hofmann, H.J. Synthesis, anticonvulsant activity, and structure-activity relationships of sodium channel blocking 3-aminopyrroles. J. Med. Chem. 1998, 41, 63–67. [Google Scholar]
- Wong, M.G.; Defina, J.A.; Andrews, P.R. Conformational Analysis of Clinically Active Anticonvulsant Drugs. J. Med. Chem. 1986, 29, 562–572. [Google Scholar] [CrossRef]
- Bruno-Blanch, L.; Galvez, J.; Garcia-Domenach, R. Topological virtual screening: A way to find new anticonvulsant drugs from chemical diversity. Bioorg.Med. Chem. Lett. 2003, 13, 2749–2754. [Google Scholar] [CrossRef]
- Simeonov, M.; Spassov, S.; Bojilova, A.; Ivanov, C.; Radeglia, R. Conformation and tautomeric equilibria of 3-acyl-3,4-dihydrocoumarins: a 1H and 13C-NMR study. J. Mol. Struct. 1985, 127, 127–133. [Google Scholar] [CrossRef]
- Nikolova, R.D.; Bojilova, A.G.; Rodios, N.A. A new and efficient method for conjugate addition of trialkylphosphites to 3-acylsubstitute coumarins. Tetrahedron 2004, 60, 10335–10342. [Google Scholar] [CrossRef]
- Bojilova, A.; Nikolova, R.; Ivanov, C.; Rodios, N.A.; Terzis, A.; Raptopoulou, C.P. A comparative study of the interaction of salicylaldehydes with phosphonoacetates under Knoevenagel reaction conditions. Synthesis of 1,2-benzoxaphosphorines and their dimers. Tetrahedron 1996, 52, 12597–12612. [Google Scholar]
- Knoevenagel, E. Condensationen Zwischen Malonester und Aldehyden unter dem Einfluss von Ammoniak und Organischen Aminen. Chem. Ber. 1898, 31, 2585–2595. [Google Scholar] [CrossRef]
- Czerney, P.; Hartmann, H. Zur darstellung von 3-Cyancumarinen. J. Prakt. Chem. 1981, 323, 691–693. [Google Scholar] [CrossRef]
- Baker, W.; Howes, C.S. Stereochemistry of arylidenecyanoacetic acids and arylarylidene acrtonitriles. J. Chem. Soc. 1953, 119–124. [Google Scholar] [CrossRef]
- Knoevenagel, E.; Arnot, R. Condensationen von Salicylaldehyd mit Cyanessigester, Benzoylessigester und Acetylaceton. Chem. Ber. 1904, 37, 4496–4502. [Google Scholar] [CrossRef]
- Knoevenagel, E. Ueber eine Darstellungsweise der Alkkyliden-Acetessigester. Chem. Ber. 1898, 31, 730–737. [Google Scholar] [CrossRef]
- Rodios, N.; Bojilova, A.; Terzis, A.; Raptopoulou, C.P. Reaction of 3-nitro- and 3-diethylphosphonocoumarin with phenacyl bromide. X-ray molecular structure of 3-nitro-3,4-phenacylidenecoumsrin. J. Heterocycl. Chem. 1994, 31, 1129–1133. [Google Scholar]
- Perkin, W.H. Uber die Künstliche Bildung des Cumarins und seiner Homologen. Annalen 1868, 147, 229–241. [Google Scholar]
- Bogdal, D. Coumarins: Fast synthesis by Knoevenagel condensation under microwave irradiation. J. Chem. Res. (S) 1998, 468–469. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ilieva, E.D.; Petkova, N.I.; Nikolova, R.D. A New and Efficient Method for the Synthesis of 3,4-Disubstituted Pyrrolidine-2,5-diones. Molecules 2012, 17, 4936-4949. https://doi.org/10.3390/molecules17054936
Ilieva ED, Petkova NI, Nikolova RD. A New and Efficient Method for the Synthesis of 3,4-Disubstituted Pyrrolidine-2,5-diones. Molecules. 2012; 17(5):4936-4949. https://doi.org/10.3390/molecules17054936
Chicago/Turabian StyleIlieva, Eleonora D., Nevena I. Petkova, and Rositca D. Nikolova. 2012. "A New and Efficient Method for the Synthesis of 3,4-Disubstituted Pyrrolidine-2,5-diones" Molecules 17, no. 5: 4936-4949. https://doi.org/10.3390/molecules17054936
APA StyleIlieva, E. D., Petkova, N. I., & Nikolova, R. D. (2012). A New and Efficient Method for the Synthesis of 3,4-Disubstituted Pyrrolidine-2,5-diones. Molecules, 17(5), 4936-4949. https://doi.org/10.3390/molecules17054936