The Rotational Barrier in Ethane: A Molecular Orbital Study

1. Introduction
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B3G | B3+G | B3++G | MPG | MP+G | MP++G | MP2G | MP2+G | MP2++G | |
|---|---|---|---|---|---|---|---|---|---|
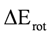 | 2.803 | 2.732 | 2.736 | 2.752 | 2.673 | 2.683 | 3.025 | 2.966 | 2.981 |
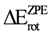 | 2.541 | 2.466 | 2.472 | 2.504 | 2.422 | 2.435 | 2.912 | 2.799 | 2.761 |
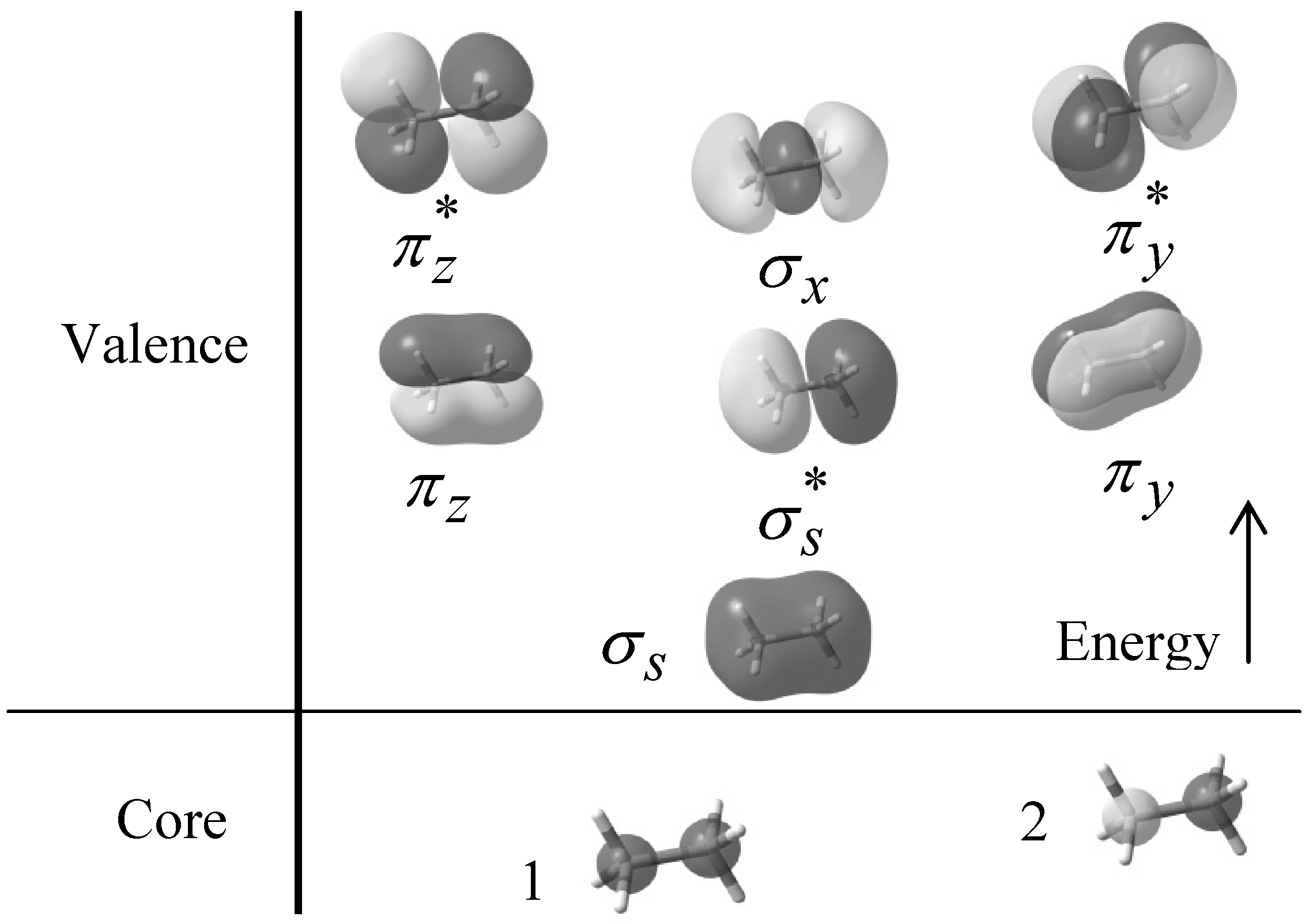
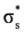 , πv, πz, σx,
, πv, πz, σx, 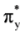 , and
, and 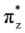 respectively. Figure 1 also shows the core orbital´s and the
respectively. Figure 1 also shows the core orbital´s and the  character of the degenerate πv, πz, , and molecular orbitals, and the σ character of the σs, and σx molecular orbitals at B3LYP/6-31+G(d, p) level of theory.
character of the degenerate πv, πz, , and molecular orbitals, and the σ character of the σs, and σx molecular orbitals at B3LYP/6-31+G(d, p) level of theory. 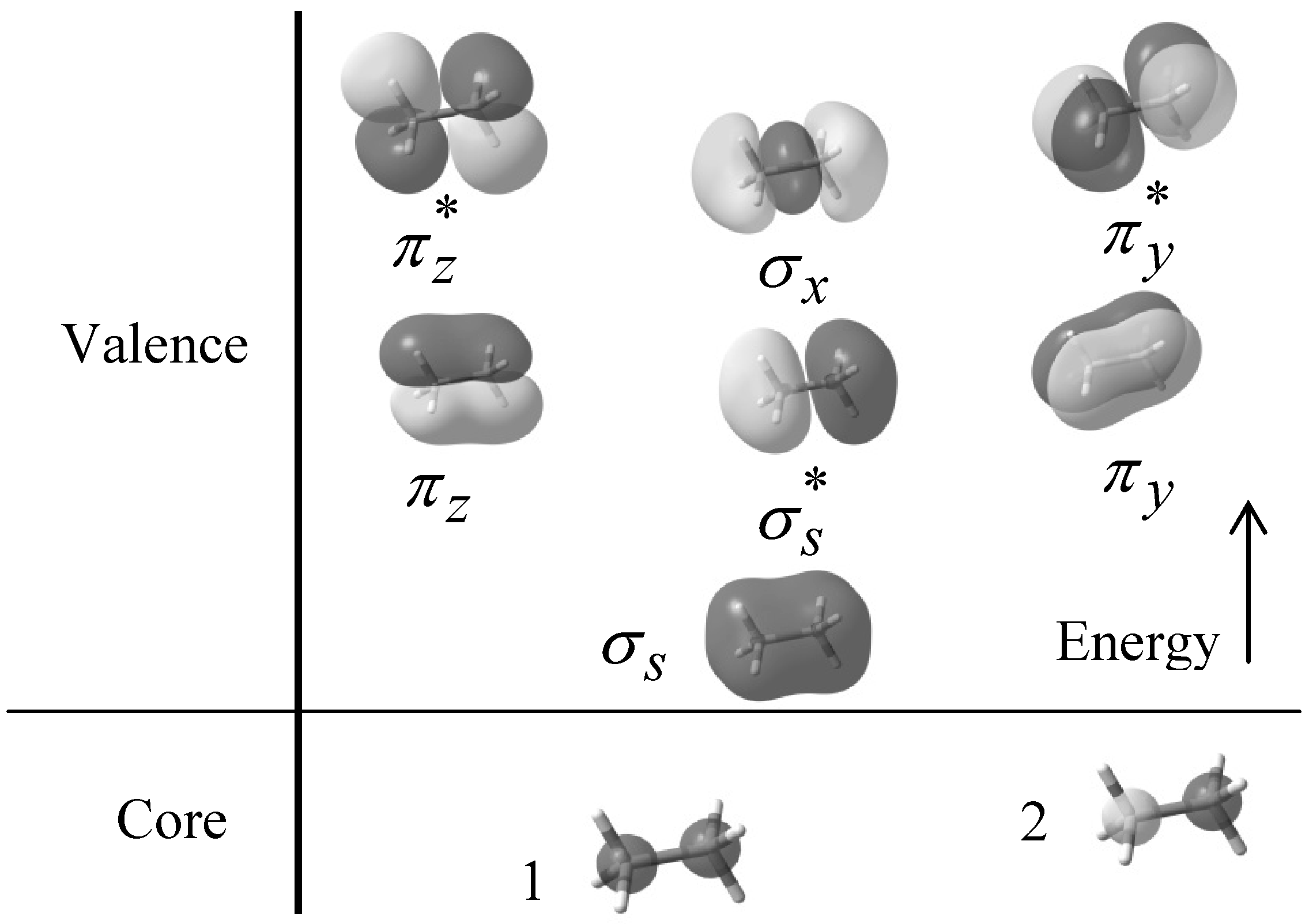
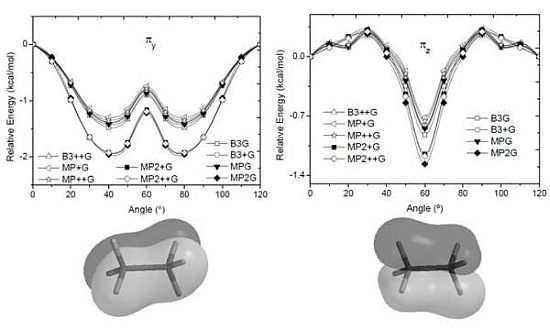
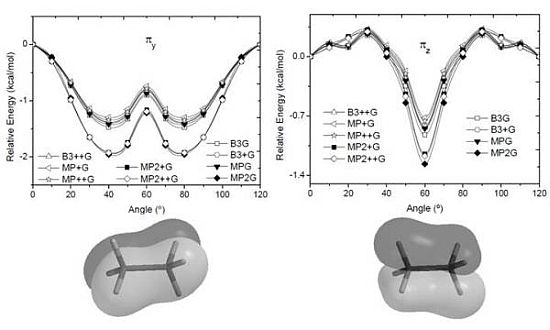
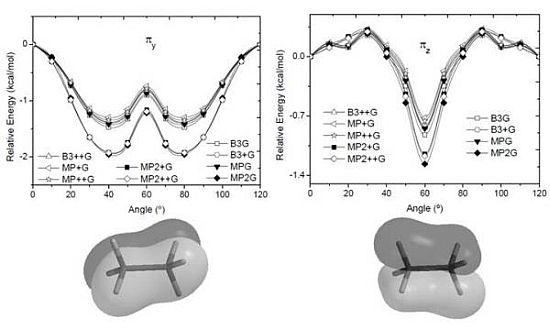
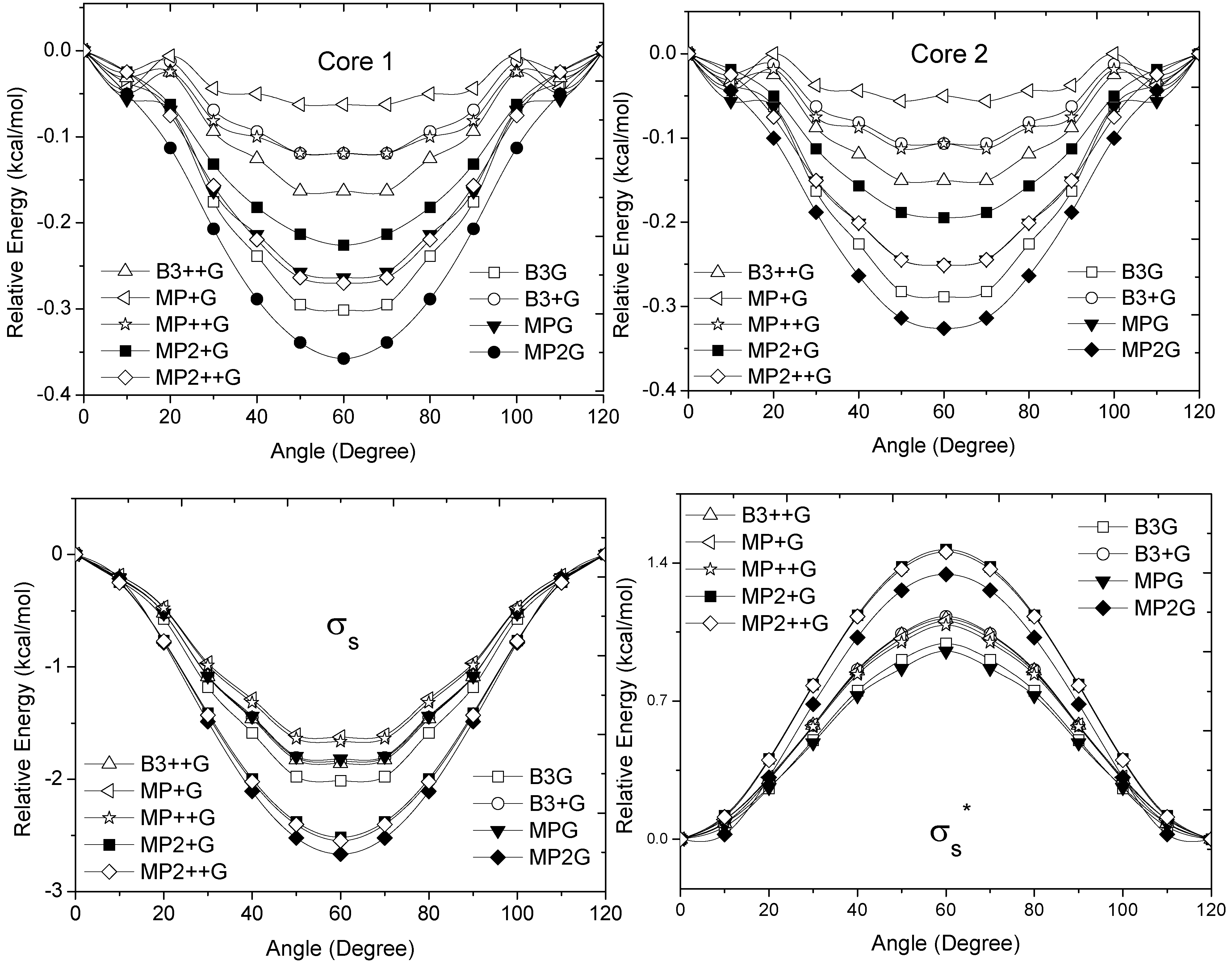
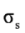 and orbitals present mainly C(1s) and C(2p) character and are main contributors to the C–C bond strength. All the bonding orbitals exhibited a minimum energy at staggered conformation (Figure 2). It is possible to observe that the energy changes between eclipsed and staggered conformations fluctuate from 2.667 kcal·mol−1 (HF/6-31G(d,p)) down to 1.626 kcal·mol−1 (mPWB95/6-31+G(d,p)). and occupy MOs of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP and mPWB95 functionals, MP2 correspond to HF theory and the symbols G, +G, and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.
and occupy MOs of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP and mPWB95 functionals, MP2 correspond to HF theory and the symbols G, +G, and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.
and orbitals present mainly C(1s) and C(2p) character and are main contributors to the C–C bond strength. All the bonding orbitals exhibited a minimum energy at staggered conformation (Figure 2). It is possible to observe that the energy changes between eclipsed and staggered conformations fluctuate from 2.667 kcal·mol−1 (HF/6-31G(d,p)) down to 1.626 kcal·mol−1 (mPWB95/6-31+G(d,p)). and occupy MOs of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP and mPWB95 functionals, MP2 correspond to HF theory and the symbols G, +G, and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.
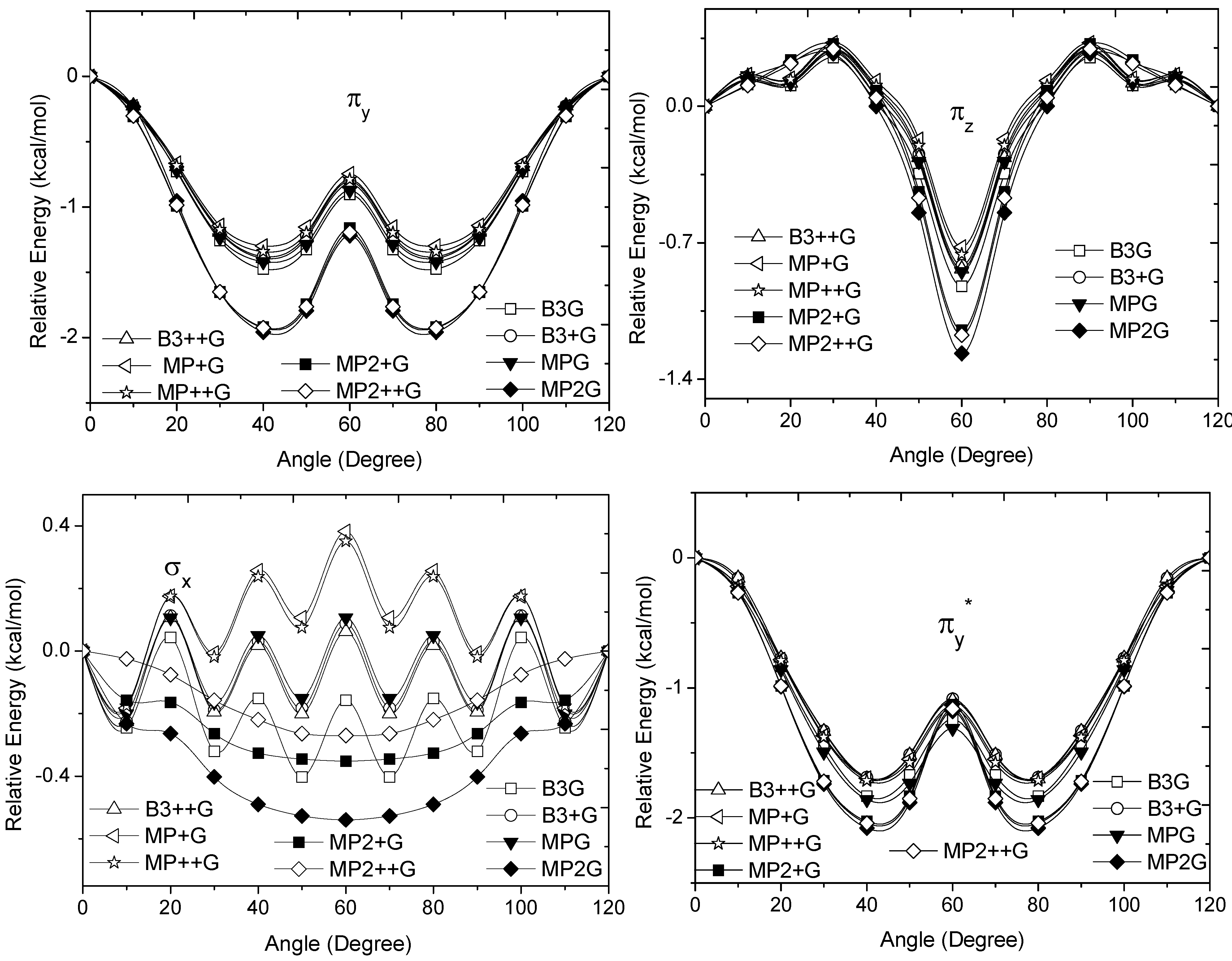
and occupy MOs of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP and mPWB95 functionals, MP2 correspond to HF theory and the symbols G, +G, and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively. 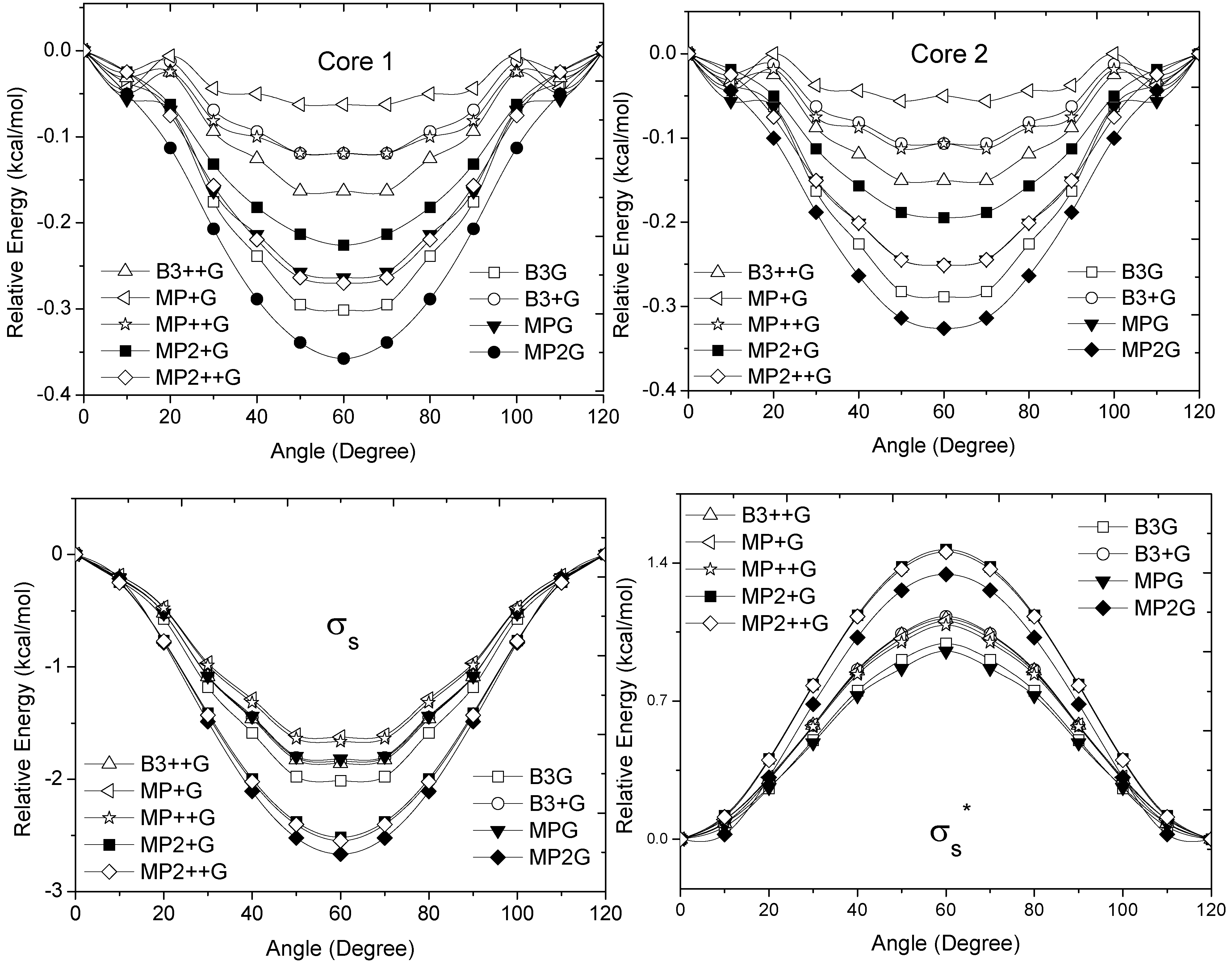 orbital bonding for the rotational barrier. Interestingly, the antibonding molecular orbital presented a minimum energy at the eclipsed conformation and a maximum at the staggered conformation with energies that change from −1.4683 kcal·mol−1 [HF/6-31G(d,p)] down to −0.9538 kcal·mol−1 [mPWB95/6-31+G(d,p)]. The negative sign of the value reveals that the molecular orbital had a minimum energy at φ = 0°, which stabilized the eclipsed conformer. and sets have a large H(1s) character and C(2p) character. These MOs are mainly associated to the vicinal hyperconjugative delocalization interactions between the methyl groups. The energy values, calculated for the change from staggered to eclipsed conformation in the orbitals πz, πv, and , vary from 0.7216 [mPWB95/6-31G(d,p)], 0.7467 [mPWB95/6-31+G(d,p)], 1.1044 [mPWB95/6-31+G(d,p)] and 1.1546 kcal·mol−1 [mPWB95/6-31+G(d,p)] to 1.267 [HF/6-31G(d,p)], 1.794 [HF/6-31G(d,p)], 1.2989 [B3LYP/6-31G(d,p)] and 1.3114 kcal·mol−1 [B3LYP/6-31+G(d,p)], respectively (Figure 3 and Figure 4). occupy MOs of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP, and mPWB95 functionals, MP2 correspond to the HF theory and the symbols G, +G and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.
occupy MOs of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP, and mPWB95 functionals, MP2 correspond to the HF theory and the symbols G, +G and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.
orbital bonding for the rotational barrier. Interestingly, the antibonding molecular orbital presented a minimum energy at the eclipsed conformation and a maximum at the staggered conformation with energies that change from −1.4683 kcal·mol−1 [HF/6-31G(d,p)] down to −0.9538 kcal·mol−1 [mPWB95/6-31+G(d,p)]. The negative sign of the value reveals that the molecular orbital had a minimum energy at φ = 0°, which stabilized the eclipsed conformer. and sets have a large H(1s) character and C(2p) character. These MOs are mainly associated to the vicinal hyperconjugative delocalization interactions between the methyl groups. The energy values, calculated for the change from staggered to eclipsed conformation in the orbitals πz, πv, and , vary from 0.7216 [mPWB95/6-31G(d,p)], 0.7467 [mPWB95/6-31+G(d,p)], 1.1044 [mPWB95/6-31+G(d,p)] and 1.1546 kcal·mol−1 [mPWB95/6-31+G(d,p)] to 1.267 [HF/6-31G(d,p)], 1.794 [HF/6-31G(d,p)], 1.2989 [B3LYP/6-31G(d,p)] and 1.3114 kcal·mol−1 [B3LYP/6-31+G(d,p)], respectively (Figure 3 and Figure 4). occupy MOs of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP, and mPWB95 functionals, MP2 correspond to the HF theory and the symbols G, +G and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.
occupy MOs of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP, and mPWB95 functionals, MP2 correspond to the HF theory and the symbols G, +G and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.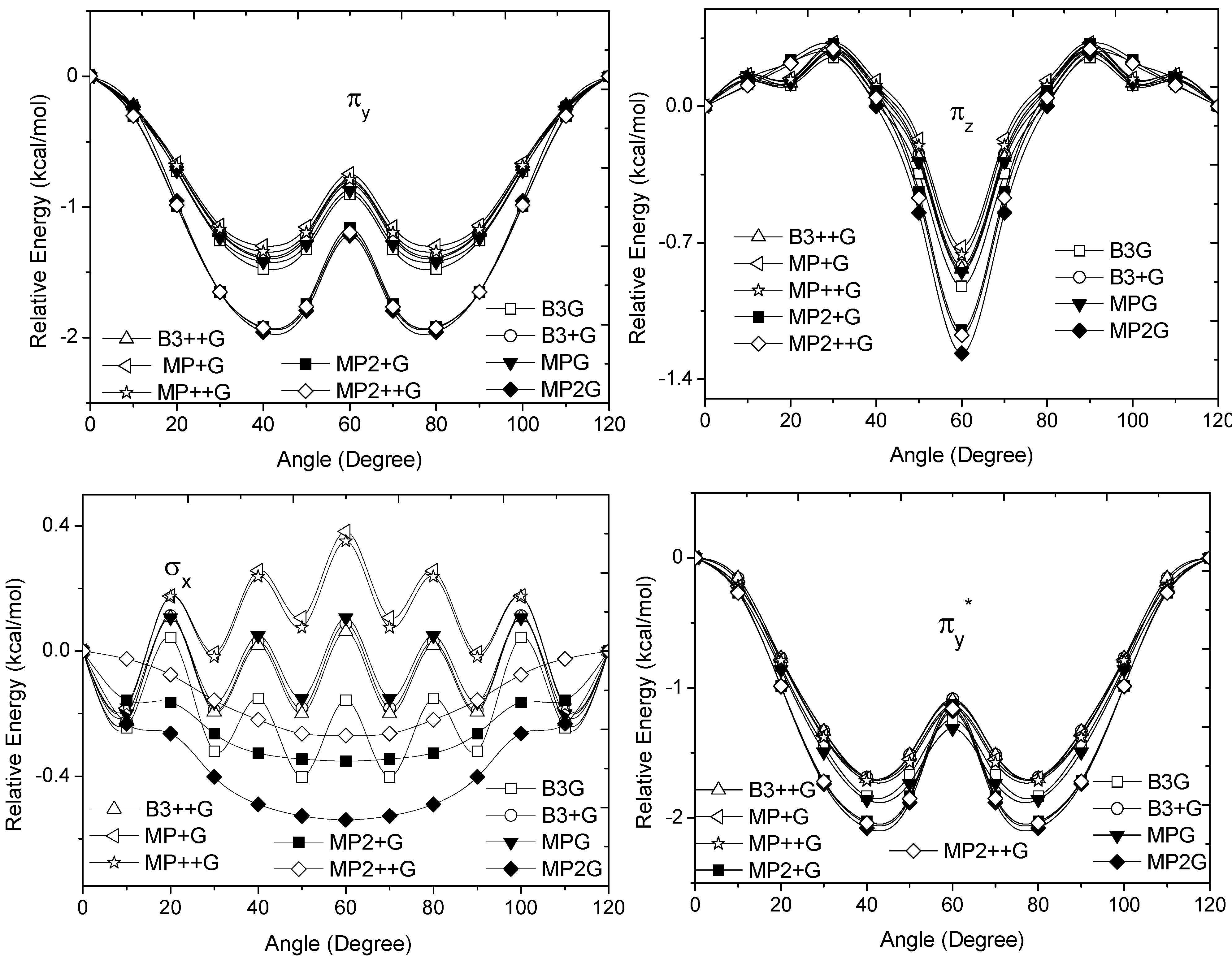 occupied MO of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP and mPWB95 functionals, MP2 correspond to the HF theory and the symbols G, +G, and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.
occupied MO of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP and mPWB95 functionals, MP2 correspond to the HF theory and the symbols G, +G, and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.
occupied MO of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP and mPWB95 functionals, MP2 correspond to the HF theory and the symbols G, +G, and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.
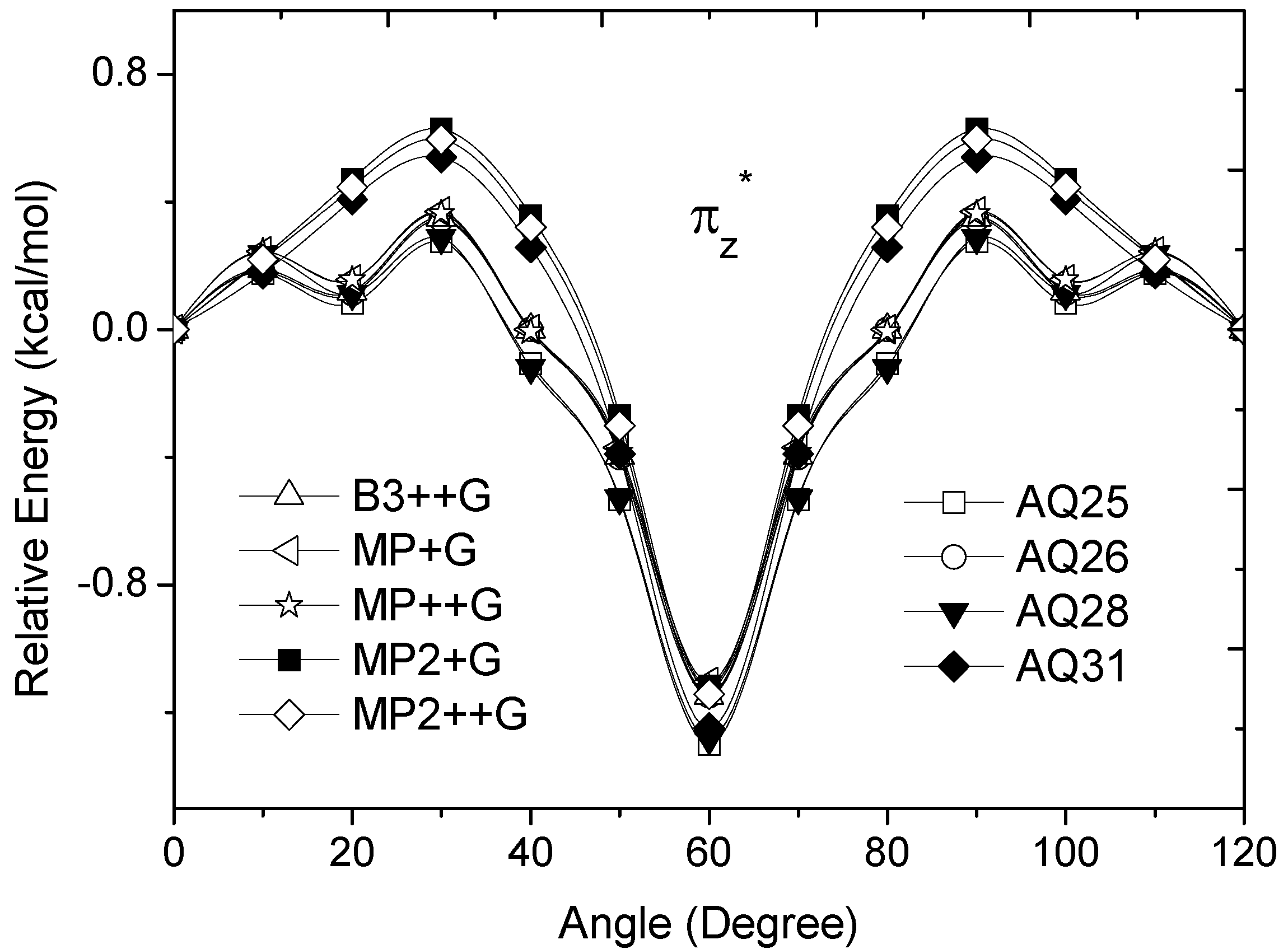
occupied MO of ethane as a function of the rotational angle, calculated with the entire theoretical model studied in this work. Labels B3, MP correspond to the B3LYP and mPWB95 functionals, MP2 correspond to the HF theory and the symbols G, +G, and ++G represent the basis set 6-31G(d, p), 6-31+G(d, p), and 6-31++G(d, p), respectively.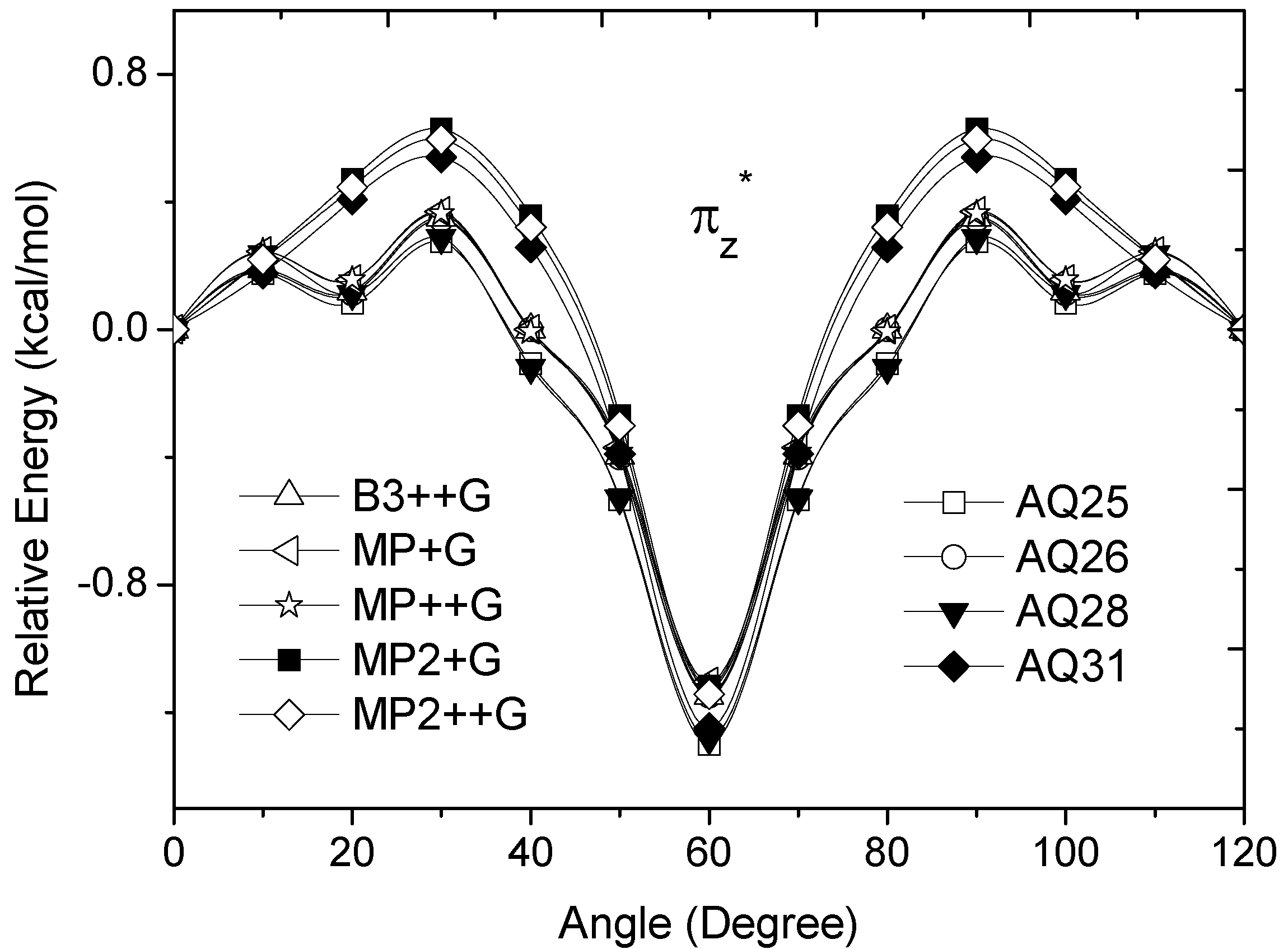 orbitals showed a smaller local maximum energy at staggered rather than at eclipsed conformation. Additionally, the energy presented a symmetric double minimum structure close to φ = 40° and φ = 80°. These results demonstrate that the energy changes in the πv and MOs destabilize the staggered conformer. MOs showed an asymmetric double minimum structure at φ = 0° (eclipsed conformation; local minimum) and φ = 60° (staggered conformation; global minimum) in addition to two maxima close to φ = 20° and φ = 100°. Thus, the energy change in the πz and MOs stabilizes both conformations. It is important to remark, however, that calculations based on DFT provided two energy minima close to φ = 40 and φ = 100°, which were not found when the HF model was used. MOs. The total electronic energy of the molecular orbital
orbitals showed a smaller local maximum energy at staggered rather than at eclipsed conformation. Additionally, the energy presented a symmetric double minimum structure close to φ = 40° and φ = 80°. These results demonstrate that the energy changes in the πv and MOs destabilize the staggered conformer. MOs showed an asymmetric double minimum structure at φ = 0° (eclipsed conformation; local minimum) and φ = 60° (staggered conformation; global minimum) in addition to two maxima close to φ = 20° and φ = 100°. Thus, the energy change in the πz and MOs stabilizes both conformations. It is important to remark, however, that calculations based on DFT provided two energy minima close to φ = 40 and φ = 100°, which were not found when the HF model was used. MOs. The total electronic energy of the molecular orbital 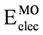 was calculated as a function of φ, considering two electrons for each MO. In all cases, the minimum and maximum values of both and
was calculated as a function of φ, considering two electrons for each MO. In all cases, the minimum and maximum values of both and 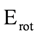 coincided. The overall net effect of the in conformational preference of the ethane can be estimated if the difference in energy between the in the eclipsed conformation and in staggered conformation (
coincided. The overall net effect of the in conformational preference of the ethane can be estimated if the difference in energy between the in the eclipsed conformation and in staggered conformation ( 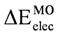 ) is compared to . We can see from the Table 2 that the was higher than the for all methods and basis set, in spite of the difference found between the different models in the behavior of the MO as a function of φ. This indicates that the energy difference between and as a function of φ produced an inversion of the minimum. Accordingly if is subtracted, the preference of for the staggered conformer is lost, and the eclipsed conformer becomes more stable. Additionally, we can note that the calculations using the HF model predict a higher than those performed with DFT, while the use of the functional mPWB95 of DFT allowed predict a lower value of (Table 2). Finally, the inclusion of diffuse functions in all calculations resulted in a decrease of .
) is compared to . We can see from the Table 2 that the was higher than the for all methods and basis set, in spite of the difference found between the different models in the behavior of the MO as a function of φ. This indicates that the energy difference between and as a function of φ produced an inversion of the minimum. Accordingly if is subtracted, the preference of for the staggered conformer is lost, and the eclipsed conformer becomes more stable. Additionally, we can note that the calculations using the HF model predict a higher than those performed with DFT, while the use of the functional mPWB95 of DFT allowed predict a lower value of (Table 2). Finally, the inclusion of diffuse functions in all calculations resulted in a decrease of .| B3G | B3+G | B3++G | MPG | MP+G | MP++G | MP2G | MP2+G | MP2++G | |
|---|---|---|---|---|---|---|---|---|---|
| | 2.803 | 2.732 | 2.736 | 2.752 | 2.673 | 2.683 | 3.025 | 2.966 | 2.981 |
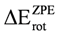 | 2.541 | 2.466 | 2.472 | 2.504 | 2.422 | 2.435 | 2.912 | 2.799 | 2.761 |
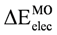 | 12.27 | 9.400 | 9.79 | 11.170 | 7.919 | 8.559 | 14.92 | 12.75 | 13.29 |
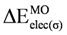 | 3.539 | 1.694 | 1.983 | 2.548 | 0.502 | 0.904 | 5.095 | 3.640 | 3.953 |
 | 8.735 | 7.706 | 7.806 | 8.622 | 7.417 | 7.656 | 9.827 | 9.111 | 9.337 |
 ) and π (
) and π (  ) contributions to for all used models is shown in Table 2. It was found that when the σ contribution is deleted, the conformational preference is reversed. A similar behavior was observed for the total π energy contribution to . It is important to note that the π energy contribution is higher than the σ contribution.
) contributions to for all used models is shown in Table 2. It was found that when the σ contribution is deleted, the conformational preference is reversed. A similar behavior was observed for the total π energy contribution to . It is important to note that the π energy contribution is higher than the σ contribution.3. Experimental
4. Conclusions
orbitals showed a smaller local maximum energy at staggered than at eclipsed conformation. In addition, the energy change of the πz and MO’s stabilizes the eclipsed and the staggered conformations. The DFT methods predict two energy minima close to φ = 40 and φ = 100°. The πz and orbitals stabilize both conformations. For the σx MO, the DFT energy changes contribute to stabilize the staggered conformation and shows irregular behavior. In addition, we found that for all models if is subtracted from the total energy of the ethane, the conformational preference in ethane is the eclipsed conformer.Acknowledgments
- Sample Availability: Not available.
References and Notes
- Kemp, J.D.; Pitzer, K.S. Hindered rotation of the methyl groups in ethane. J. Chem. Phys. 1936, 4, 749. [Google Scholar]
- Volhard, K.P.C.; Schore, N.E. Organic Chemistry Structure and Function, 5th ed; W.H. Freeman and Company: New York, NY, USA, 2007; pp. 79–83. [Google Scholar]
- Solomons, T.W.G. Organic Chemistry, 5th ed; John Wiley & Sons, Inc.: New York, NY, USA, 1992; p. 59. [Google Scholar]
- March, J.; Smith, M.B. March’s Advanced Organic Chemistry Reactions, Mechanisms and Structure, 6th ed; John Wiley & Sons, Inc.: New Jersey, NJ, USA, 2007; pp. 197–198. [Google Scholar]
- Brunck, T.K.; Weinhold, F. Quantum-mechanical studies on the origin of barriers to internal rotation about single bonds. J. Am. Chem. Soc. 1979, 101, 1700–1709. [Google Scholar]
- Pitzer, R.M. The barrier to internal rotation in ethane. Acc. Chem. Res. 1983, 16, 207–210. [Google Scholar]
- Bader, R.F.W.; Cheeseman, J.R.; Laidig, K.E.; Wiberg, K.B.; Breneman, C. Origin of rotation and inversion barriers. J. Am. Chem. Soc. 1990, 112, 6530–6536. [Google Scholar]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of internal rotation barriers and related phenomena. Isr. J. Chem. 1991, 31, 277–285. [Google Scholar]
- Mo, Y.; Wu, W.; Song, L.; Lin, M.; Zhang, Q.; Gao, J. The magnitude of hyperconjugation in ethane: A perspective from ab initio valence bond theory. Angew. Chem. Int. Ed. Engl. 2004, 43, 1986–1990. [Google Scholar]
- Bickelhaupt, F.M.; Baerends, E.J. The case for steric repulsion causing the staggered conformation of ethane. Angew. Chem. Int. Ed. Engl. 2003, 42, 4183–4188. [Google Scholar]
- Asturiol, D.; Salvador, P.; Mayer, I. Dissecting the hindered rotation of ethane. ChemPhysChem 2009, 10, 1987–1992. [Google Scholar]
- 12 Mo, Y.R.; Gao, J.L. Heoretical analysis of the rotational barrier of ethane. Acc. Chem. Res. 2007, 40, 113–119. [Google Scholar]
- Baerends, E.J. The rotation barrier in ethane. Nachr. Chem. 2004, 52, 581. [Google Scholar]
- Liu, S.; Govind, N. Toward understanding the nature of internal rotation barriers with a new energy partition scheme: Ethane and n-butane. J. Phys. Chem. A 2008, 112, 6690–6699. [Google Scholar]
- Pophristic, V.; Goodman, L. Hyperconjugation not steric repulsion leads to the staggered structure of ethane. Nature 2001, 411, 565–568. [Google Scholar]
- England, W.; Gordon, M.S. Localized charge distributions. I. General theory, energy partitioning, and the internal rotation barrier in ethane. J. Am. Chem. Soc. 1971, 93, 4649–4657. [Google Scholar] [CrossRef]
- Lowe, J.P. A butane analogue, 3-hexyne, is eclipsed. Science 1973, 179, 527–532. [Google Scholar]
- Goodman, L.; Gu, H. Flexing analysis of steric exchange repulsion accompanying ethane internal rotation. J. Chem. Phys. 1998, 109, 72–78. [Google Scholar]
- Weinhold, M.F. A new twist on molecular shape. Nature 2001, 411, 539–541. [Google Scholar]
- Goodman, L.; Gu, H.; Pophristic, V. Flexing analysis of ethane internal rotation energetic. J. Chem. Phys. 1999, 110, 4268–4275. [Google Scholar]
- Mulliken, R.S. Intensities of electronic transitions in molecular spectra. IV. Cyclic dienes and hyperconjugation. J. Chem. Phys. 1939, 7, 339–352. [Google Scholar] [CrossRef]
- Epiotis, N.D.; Cherry, W.R.; Shaik, S.; Yates, R.L.; Bernardi, E. Topics in Current Chemistry: Structural Theory of Organic Chemistry; Springer-Verlag: Berlin, Germany, 1997. [Google Scholar]
- Liu, S. Steric effect: A quantitative description from density functional theory. J. Chem. Phys. 2007, 126, 244103–244107. [Google Scholar]
- Liu, S. On the relationship between densities of Shannon entropy and Fisher information for atoms and molecules. J. Chem. Phys. 2007, 126, 191107–191109. [Google Scholar]
- Liu, S.; Govind, N.; Pedersen, L.G. Exploring the origin of the internal rotational barrier for molecules with one rotatable dihedral angle. J. Chem. Phys. 2008, 129, 094104–094106. [Google Scholar]
- Torrent-Sucarrat, M.; Liu, S.; de Proft, F. Steric effect: Partitioning in atomic and functional group contributions. J. Phys. Chem. A 2009, 113, 3698–3702. [Google Scholar]
- Esquivel, R.O.; Liu, S.; Angulo, J.C.; Dehesa, J.S.; Antolín, J.; Molina-Espíritu, M. Fisher information and steric effect: Study of the internal rotation barrier of ethane. J. Phys. Chem. A 2011, 115, 4406–4415. [Google Scholar]
- Weisskopf, V.F. Of atoms, mountains, and stars: A study in qualitative physics. Science 1975, 187, 605–612. [Google Scholar]
- Weinhold, F. Rebuttal to the Bickelhaupt-Baerends case for steric repulsion causing the staggered conformation of ethane. Angew. Chem. Int. Ed. Engl. 2003, 42, 4188–4194. [Google Scholar]
- Luken, W.L.; Beratan, D.N. Localized orbitals and the fermi hole. Theor. Chim. Acta 1982, 61, 265–281. [Google Scholar]
- Luken, W.L.; Culberson, J.C. Localized orbitals based on the fermi hole. Theor. Chim. Acta 1984, 66, 279–293. [Google Scholar]
- Luken, W.L. Properties of the fermi hole in molecules. Croat. Chem. Acta 1984, 57, 1283. [Google Scholar]
- Ludena, E.V.; Ugalde, J.M.; Lopez, X.; Fernandez-Rico, J.; Ramirez, G. A reinterpretation of the nature of the fermi hole. J. Chem. Phys. 2004, 120, 540–547. [Google Scholar]
- Cooper, D.L.; Ponec, R.A. One-electron approximation to domain averaged fermi hole analysis. Phys. Chem. Chem. Phys. 2008, 10, 1319–1329. [Google Scholar]
- Weinhold, F. Natural Bond Orbital Methods. In The Encyclopedia of Computational Chemistry; Schleyer, P.V.R., Ed.; John Wiley & Sons: Chichester, UK, 1998; pp. 1792–1811. [Google Scholar]
- Mo, Y. Geometrical optimization for strictly localized structures. J. Chem. Phys. 2003, 119, 1300–1306. [Google Scholar]
- Durant, J.L. Evaluation of transition state properties by density functional theory. Chem. Phys. Lett. 1996, 256, 595–602. [Google Scholar]
- Lynch, B.J.; Fast, P.L.; Harris, M.; Truhlar, D.G. Adiabatic connection for kinetics. J. Phys. Chem. A 2000, 104, 4811–4815. [Google Scholar]
- Zhao, Y.; Pu, J.; Lynch, B.J.; Truhlar, D.G. Tests of second-generation and third-generation density functionals for thermochemical kinetics. Phys. Chem. Chem. Phys. 2004, 6, 673–676. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Design of density functionals that are broadly accurate for thermochemistry, thermochemical kinetics, and nonbonded interactions. J. Phys. Chem. A 2005, 109, 5656–5667. [Google Scholar]
- Fǎrcaşiu, D.; Lukinskas, P.; Pamidighantam, S.V. Bridged and open carbocation structures as a function of the correlation level in ab initio calculations: The 4-methyl-2-pentyl cation. J. Phys. Chem. A 2002, 106, 11672–11675. [Google Scholar]
- Gimarc, B.M. Qualitative molecular orbital study of ethane and diborane. J. Am. Chem. Soc. 1973, 95, 1417–1421. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision A.1. Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. J. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Quijano-Quiñones, R.F.; Quesadas-Rojas, M.; Cuevas, G.; Mena-Rejón, G.J. The Rotational Barrier in Ethane: A Molecular Orbital Study. Molecules 2012, 17, 4661-4671. https://doi.org/10.3390/molecules17044661
Quijano-Quiñones RF, Quesadas-Rojas M, Cuevas G, Mena-Rejón GJ. The Rotational Barrier in Ethane: A Molecular Orbital Study. Molecules. 2012; 17(4):4661-4671. https://doi.org/10.3390/molecules17044661
Chicago/Turabian StyleQuijano-Quiñones, Ramiro F., Mariana Quesadas-Rojas, Gabriel Cuevas, and Gonzalo J. Mena-Rejón. 2012. "The Rotational Barrier in Ethane: A Molecular Orbital Study" Molecules 17, no. 4: 4661-4671. https://doi.org/10.3390/molecules17044661
APA StyleQuijano-Quiñones, R. F., Quesadas-Rojas, M., Cuevas, G., & Mena-Rejón, G. J. (2012). The Rotational Barrier in Ethane: A Molecular Orbital Study. Molecules, 17(4), 4661-4671. https://doi.org/10.3390/molecules17044661