Salvianolic Acid A Inhibits PDGF-BB Induced Vascular Smooth Muscle Cell Migration and Proliferation While Does Not Constrain Endothelial Cell Proliferation and Nitric Oxide Biosynthesis

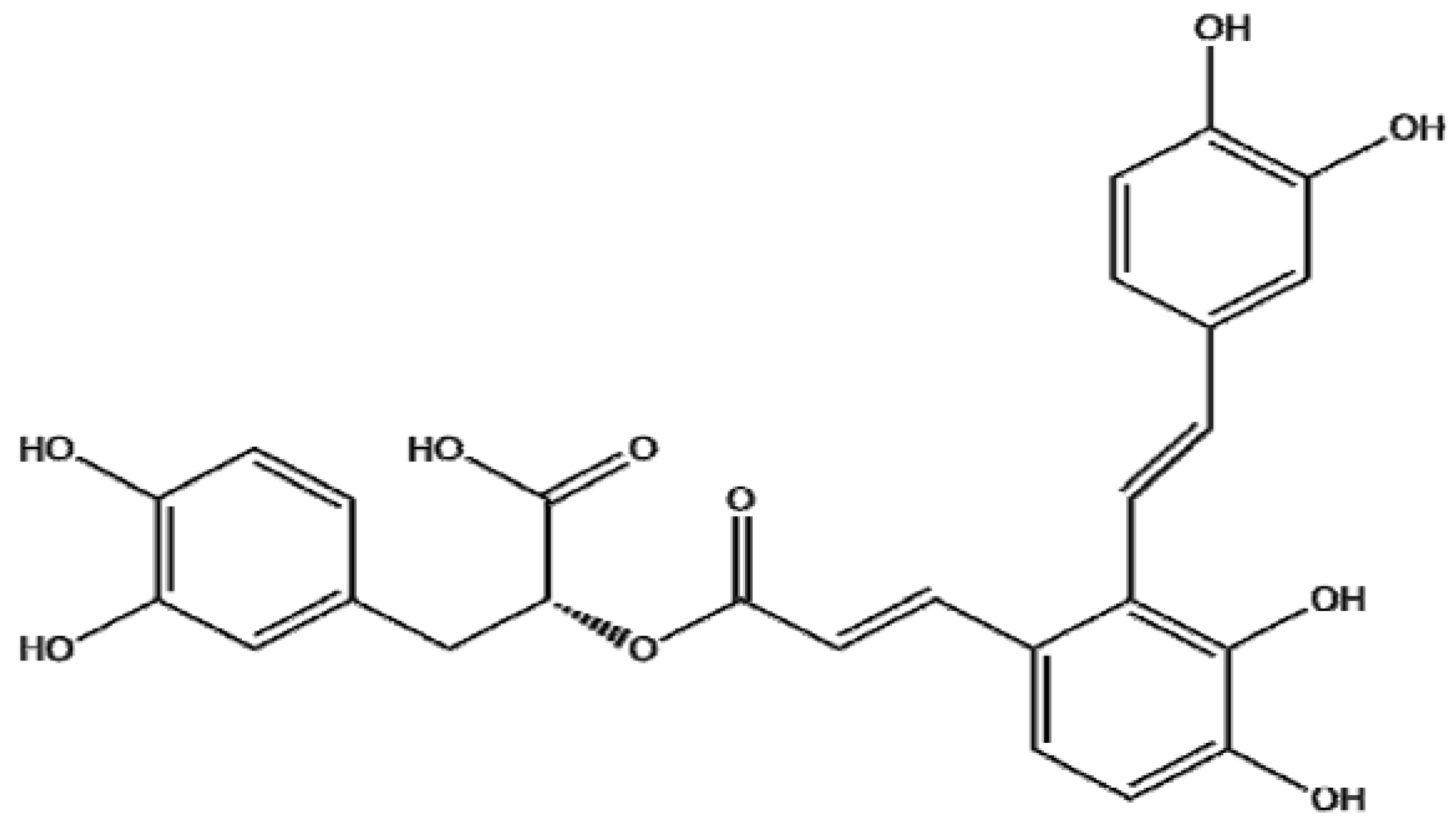{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
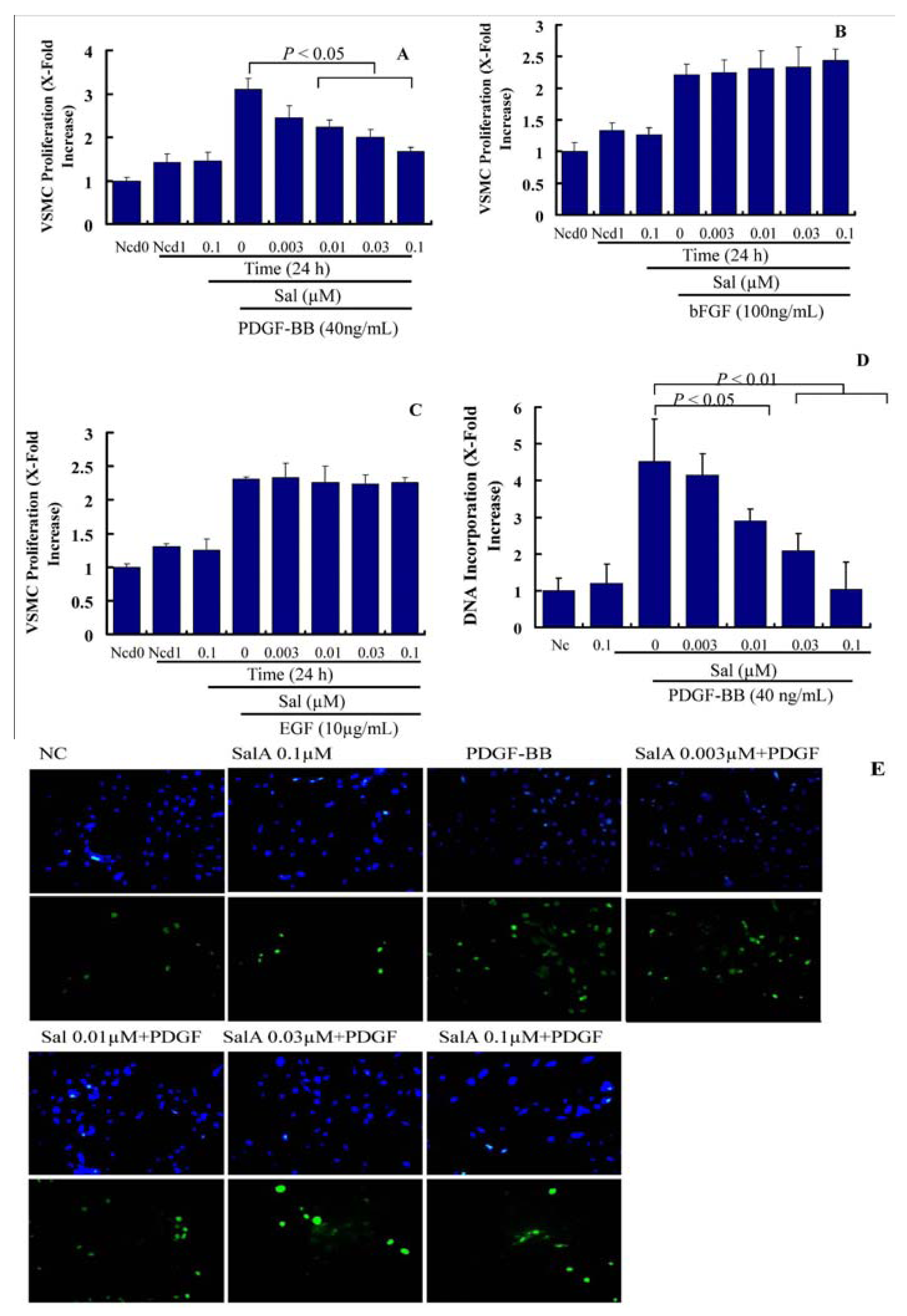
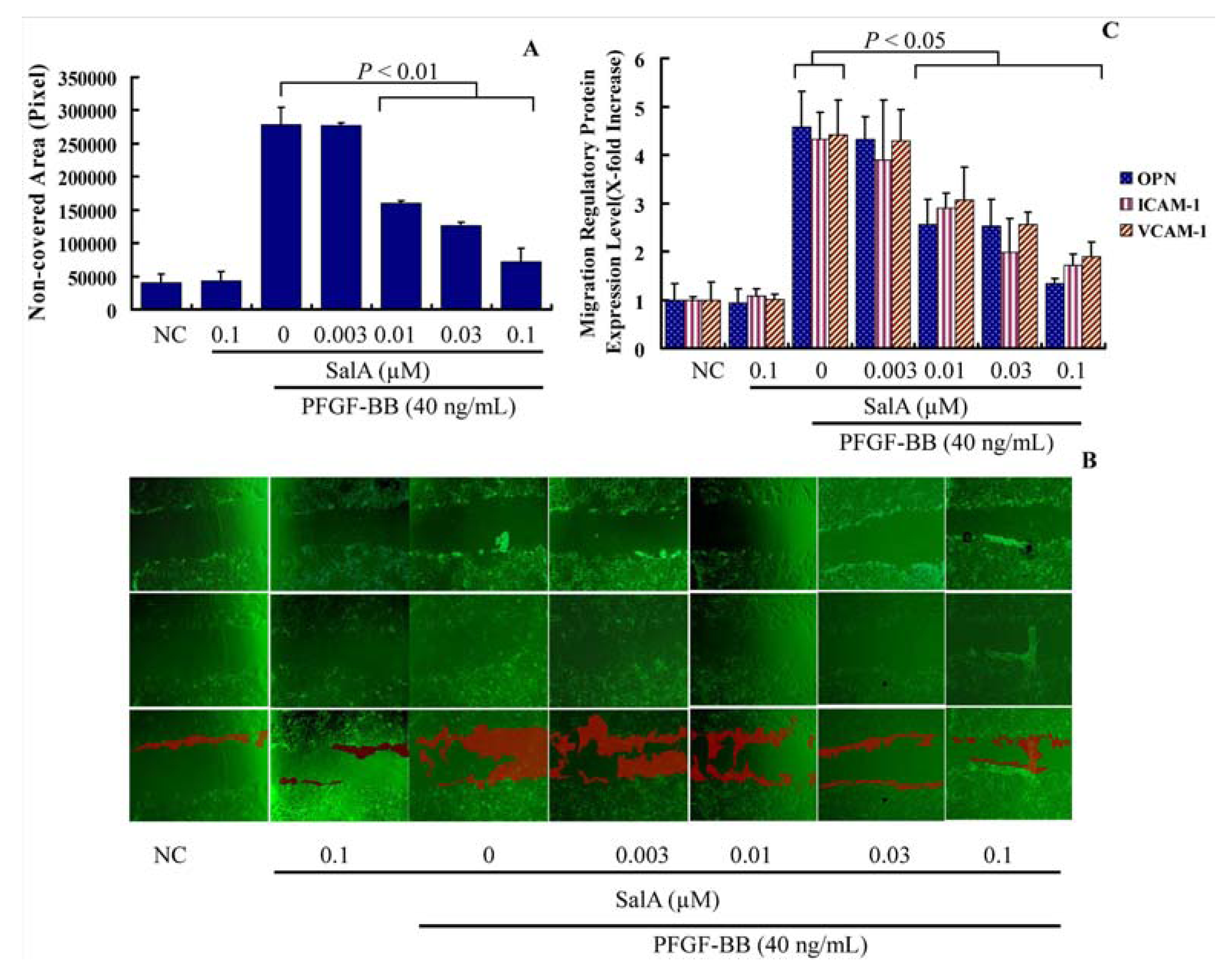
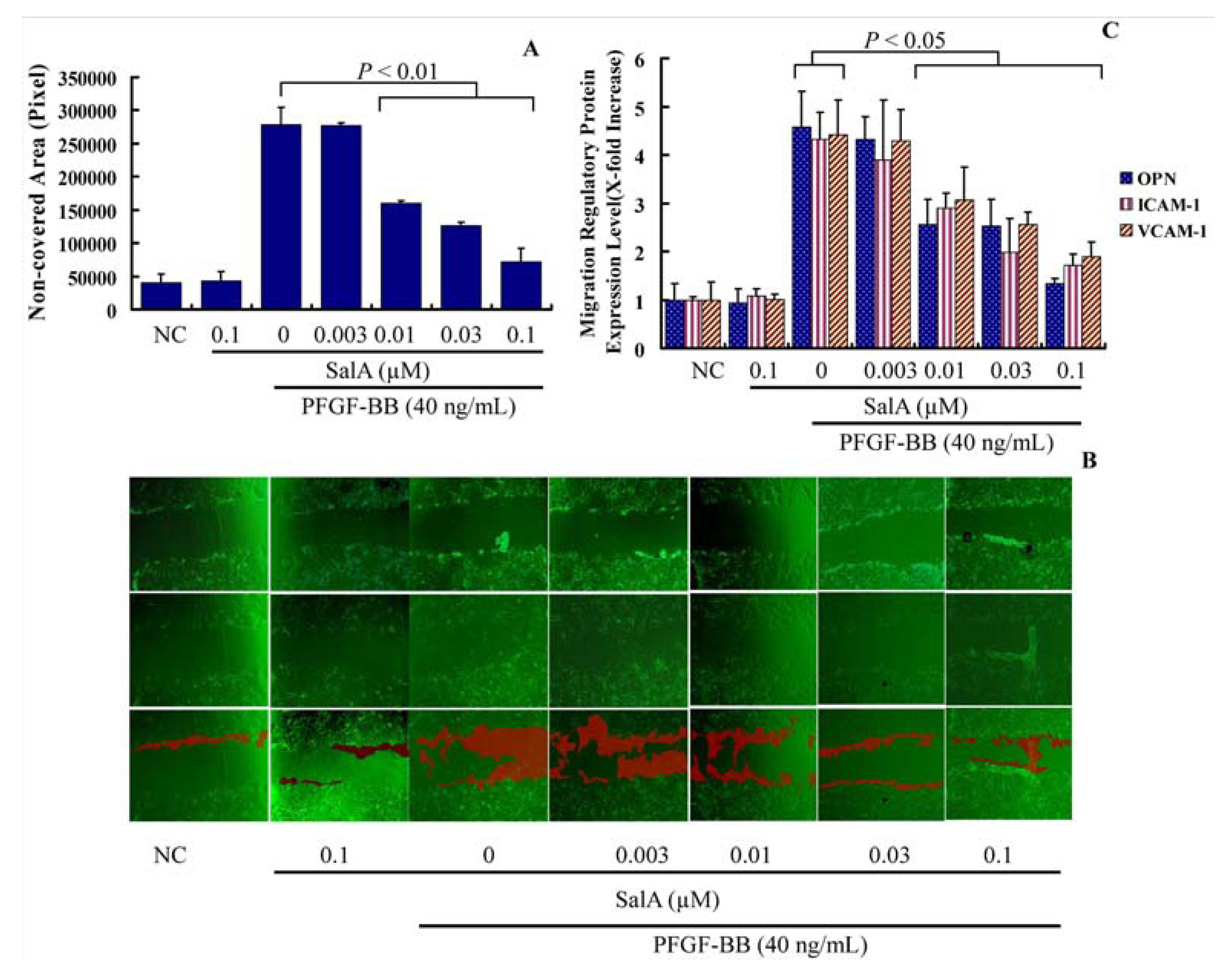
2.1. Sal A Inhibits PDGF-BB Induced VSMC Proliferation and Migration
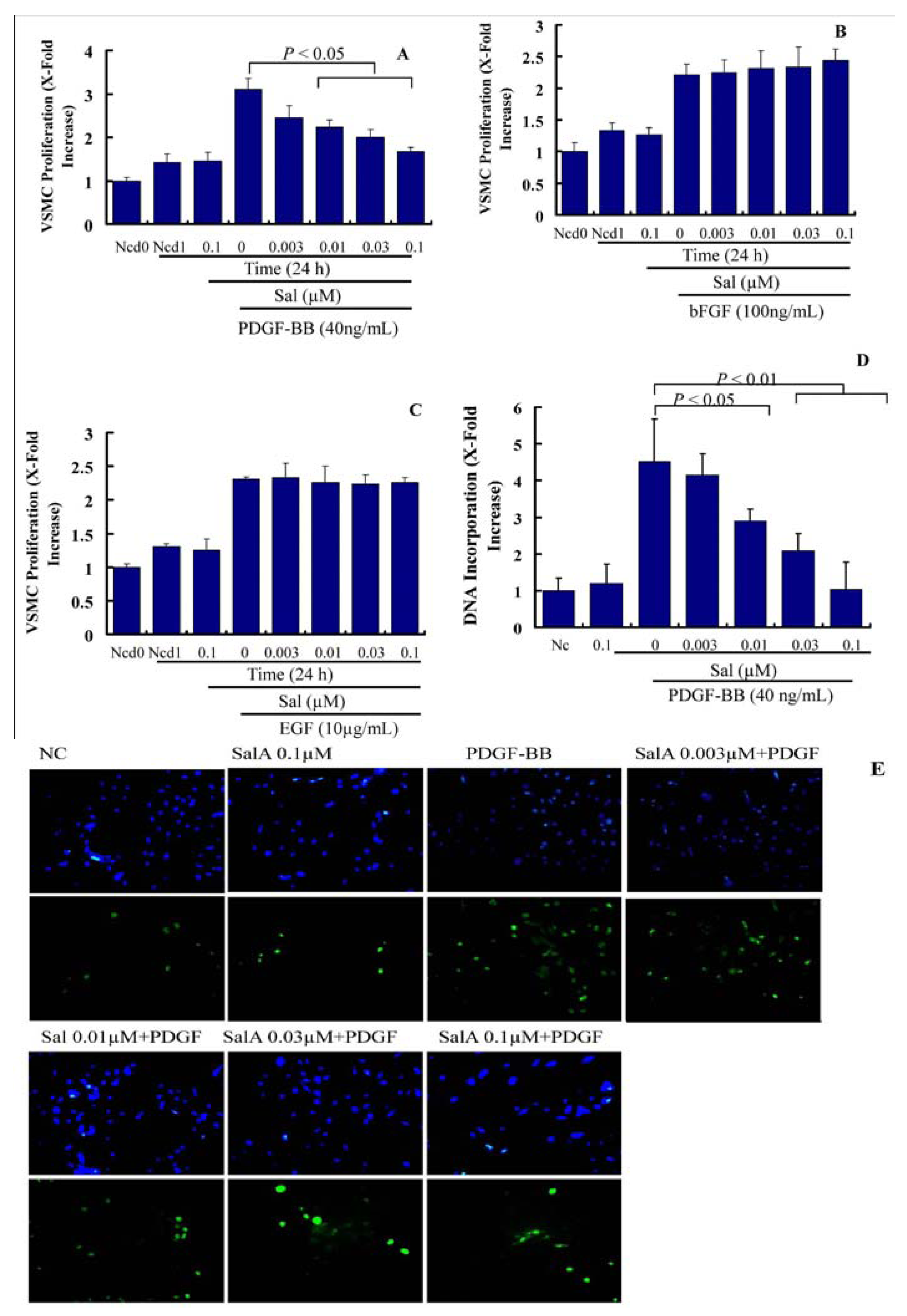
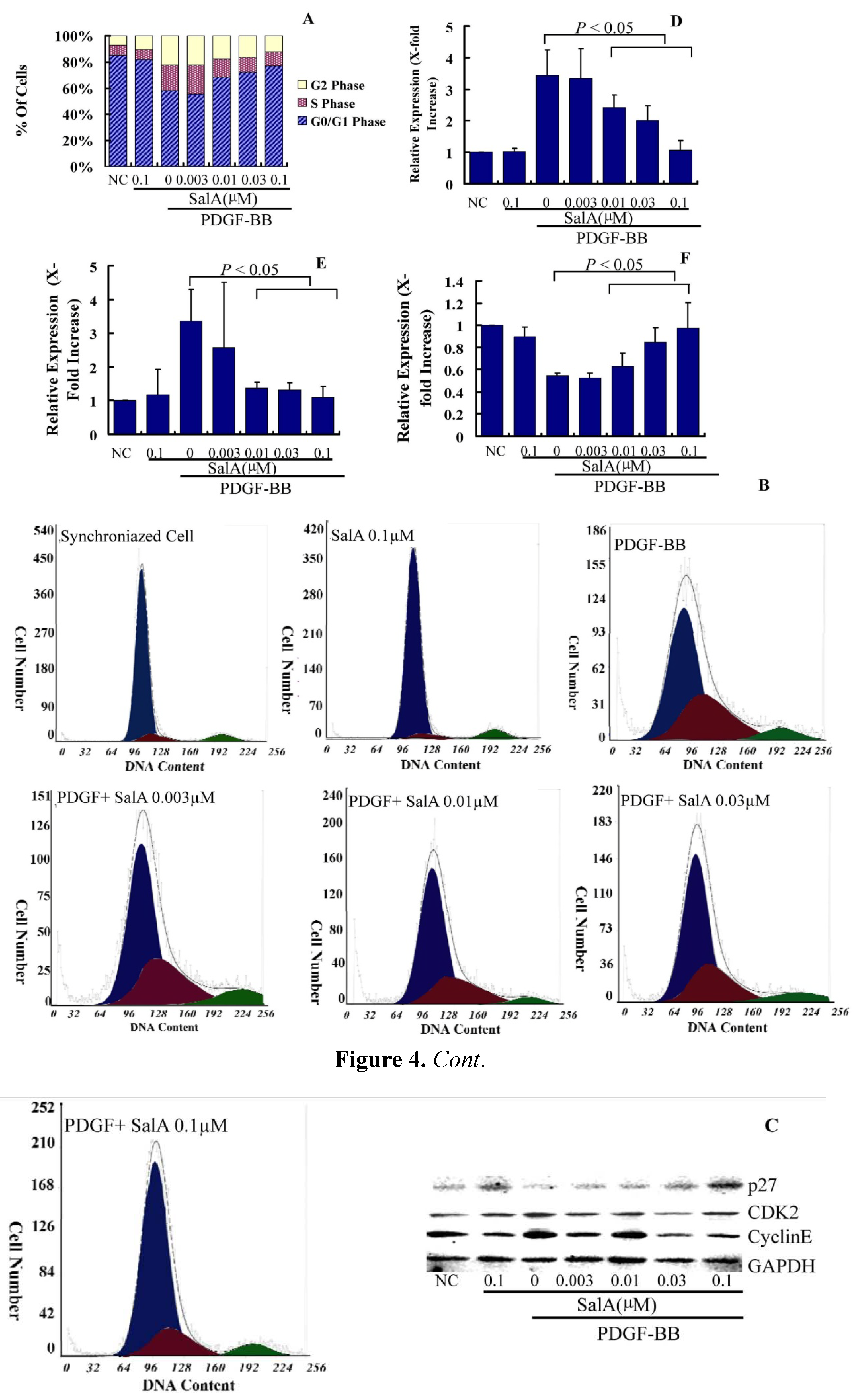
2.2. The Inhibition of on VSMC Proliferation by SalA is Associated with Cell Cycle Arrest
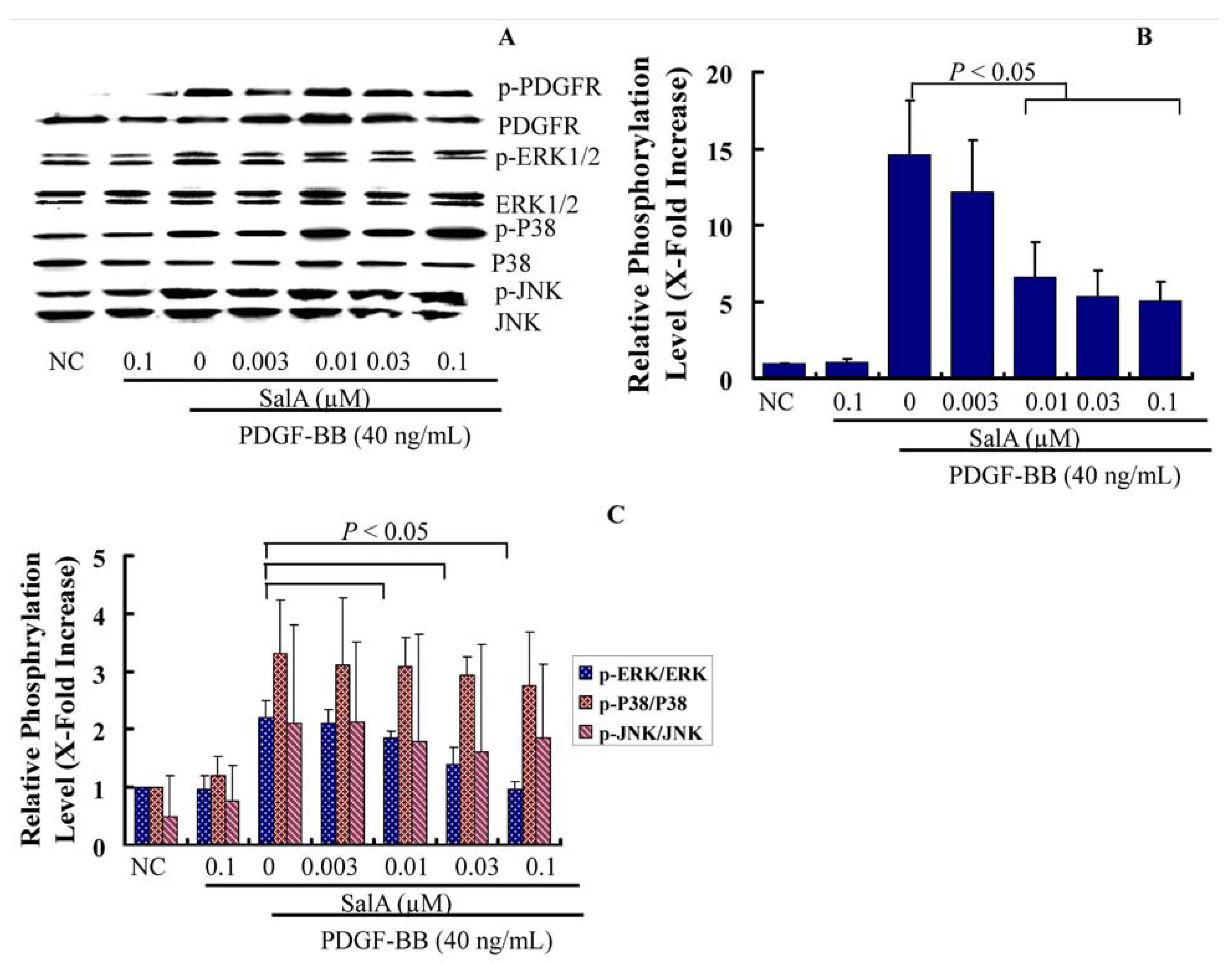
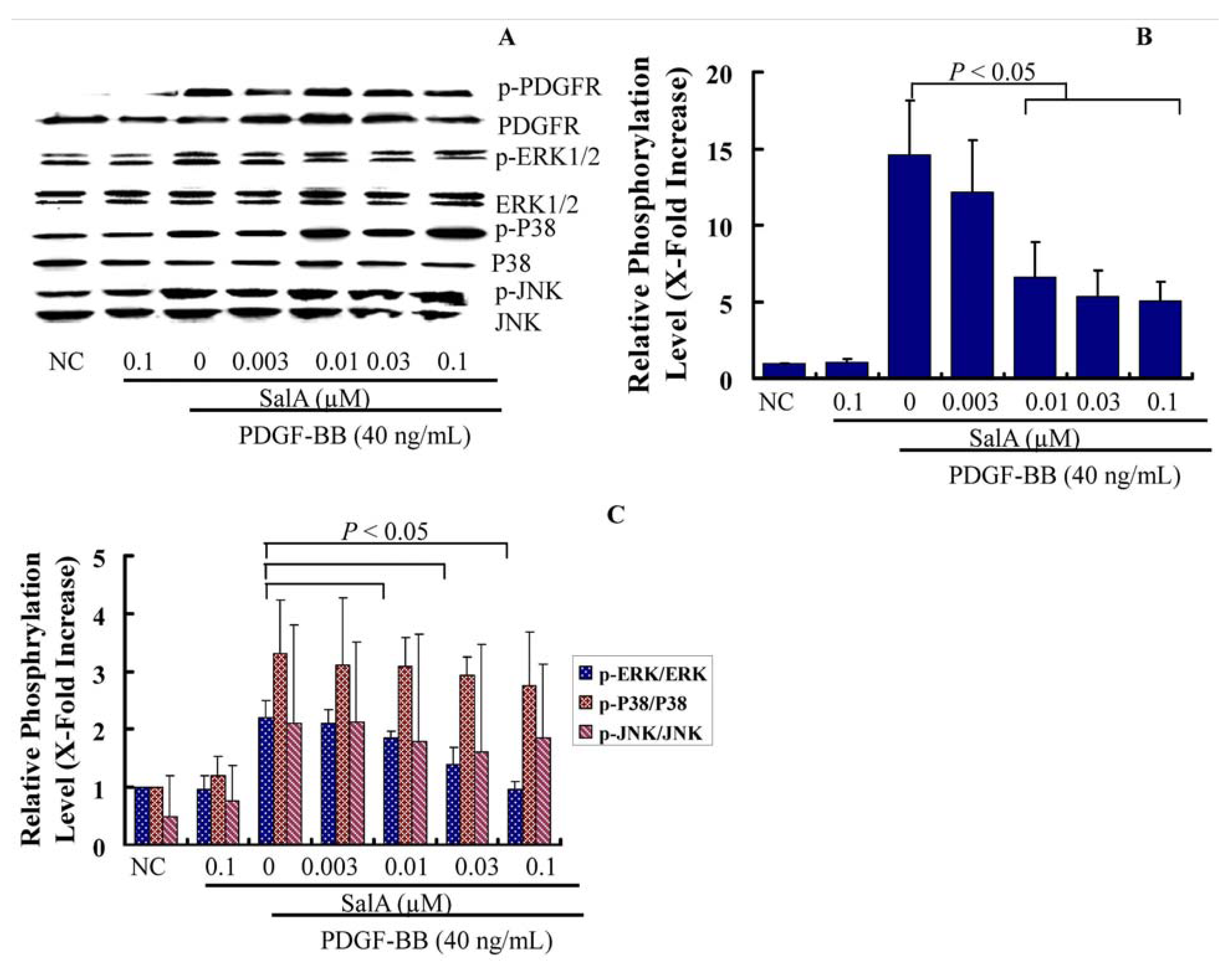
2.3. SalA Inhibits the PDGFRβ-ERK1/2 Signaling Cascade Activated by PDGF-BB in VSMCs
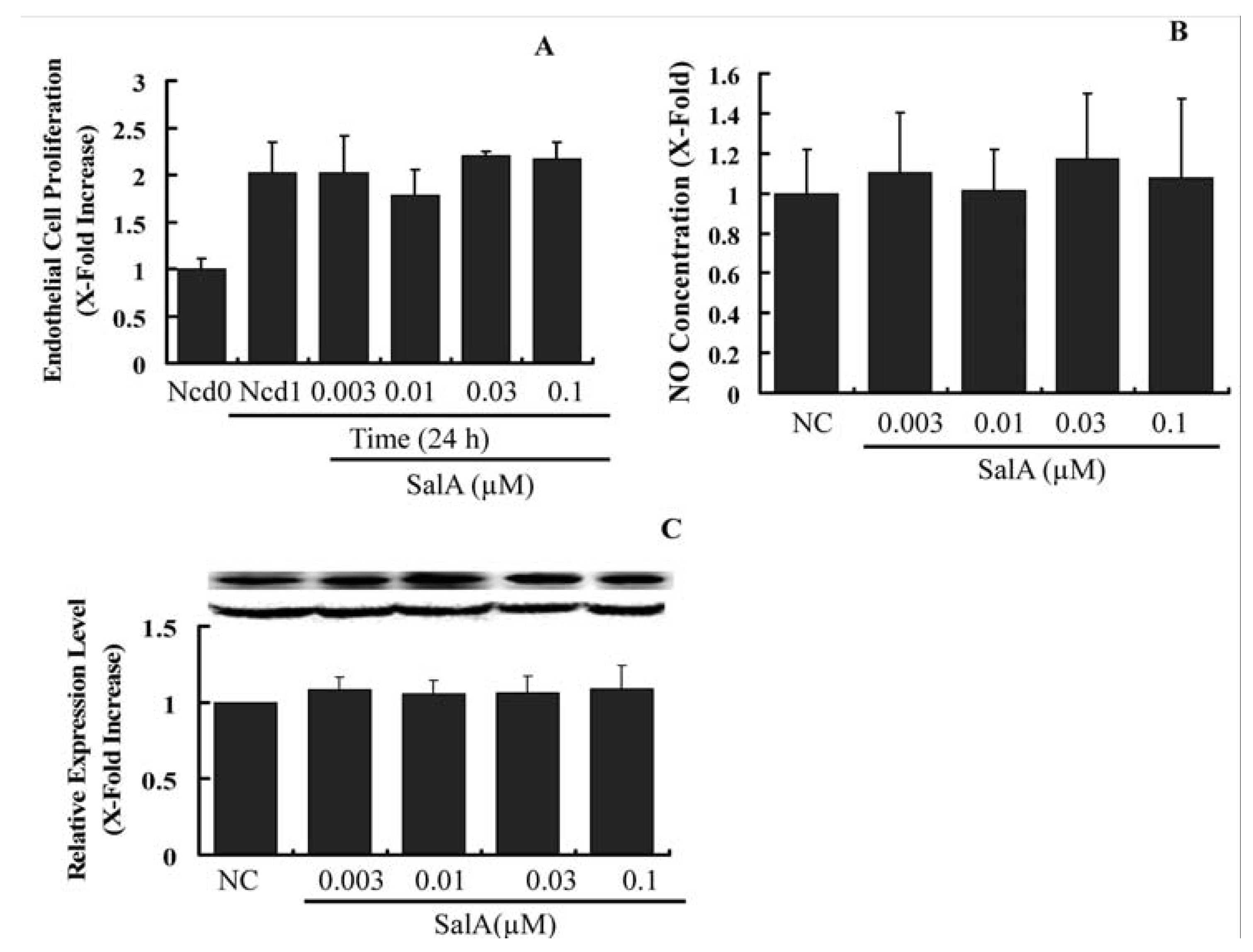
2.4. Sal A Does Not Constrain Endothelial Cell Proliferation and NO Biosynthesis
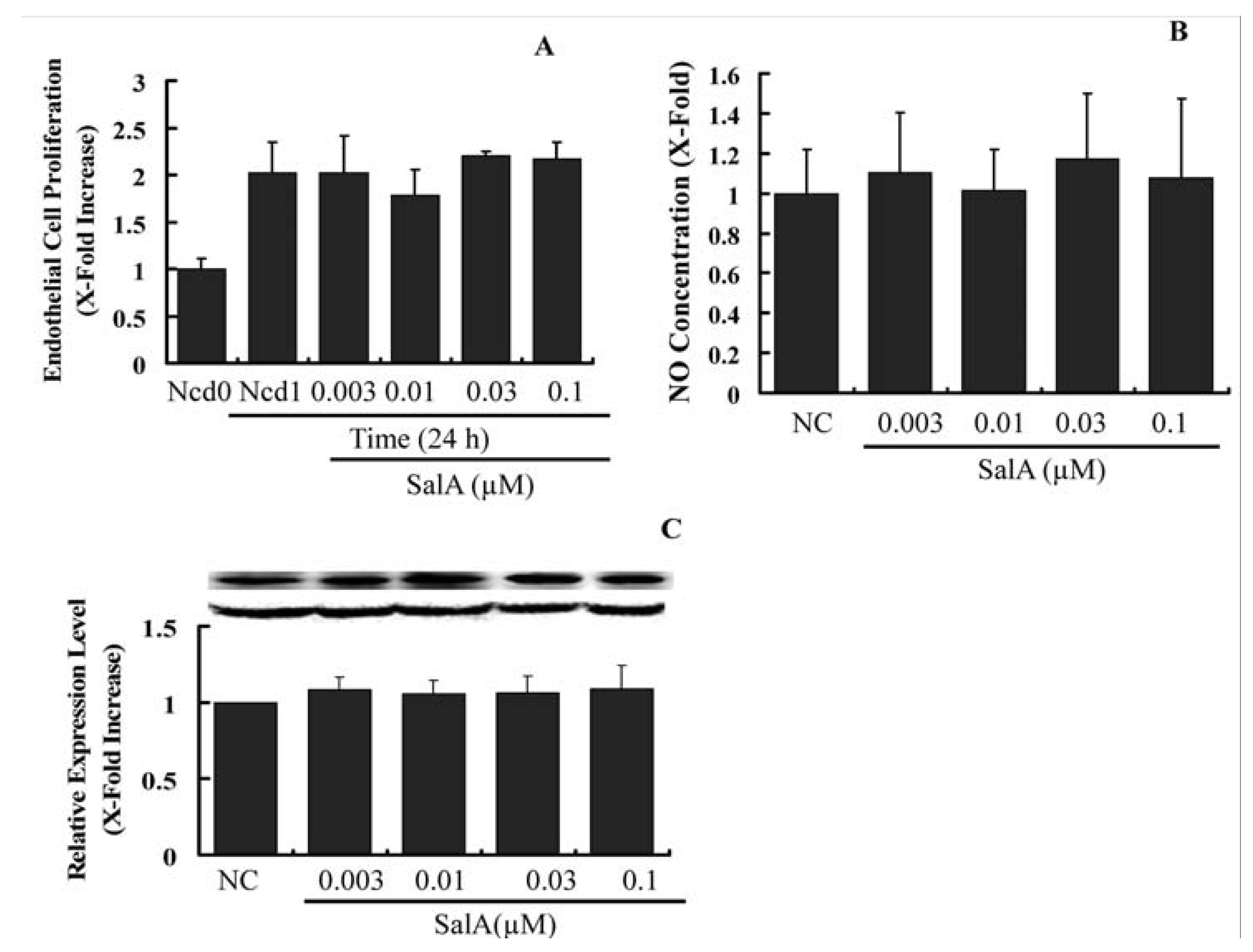
3. Experimental
3.1. Cell Culture
3.2. Antibodies and Major Reagents
3.3. Proliferation Assays
3.3.1. Crystal Violet Staining
3.3.2. BrdU Incorporation Assay
3.4. Wound-Healing Assay
3.5. Western Blot Analysis
3.6. Cell Cycle Progression Analysis
3.7. Assessment of NO, ICAM-1, VCAM-1 and OPN Production
3.8. Statistics
4. Conclusions
Acknowledgements
References and Notes
- Berk, B.C. Vascular smooth muscle growth: Autocrine growth mechanisms. Physiol. Rev. 2001, 81, 999–1030. [Google Scholar]
- Levitzki, A. PDGF receptor kinase inhibitors for the treatment of restenosis. Cardiovasc. Res. 2005, 65, 581–586. [Google Scholar] [CrossRef]
- Zhan, Y.; Kim, S.; Izumi, Y.; Izumiya, Y.; Nakao, T.; Miyazaki, H.; Iwao, H. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 795–801. [Google Scholar]
- Zheng, B.; Han, M.; Bernier, M.; Zhang, X.H.; Meng, F.; Miao, S.B.; He, M.; Zhao, X.M.; Wen, J.K. Krüppel-like factor 4 inhibits proliferation by platelet-derived growth factor receptor β-mediated, not by retinoic acid receptor α-mediated, phosphatidylinositol 3-kinase and ERK signaling in vascular smooth muscle cells. J. Biol. Chem. 2009, 284, 22773–22785. [Google Scholar]
- Chao, J.I.; Su, W.C.; Liu, H.F. Baicalein induces cancer cell death and proliferation retardation by the inhibition of CDC2 kinase and survivin associated with opposite role of p38 mitogenactivated protein kinase and AKT. Mol. Cancer Ther. 2007, 6, 3039–3048. [Google Scholar]
- Ho, J.H.; Hong, C.Y. Salvianolic acids: Small compounds with multiple mechanisms for cardiovascular protection. J. Biomed. Sci. 2011, 18, 30. [Google Scholar] [CrossRef]
- Pan, C.H.; Chen, C.W.; Sheu, M.J.; Wu, C.H. Salvianolic acid B inhibits SDF-1α-stimulated cell proliferation and migration of vascular smooth muscle cells by suppressing CXCR4 receptor. Vascul. Pharmacol. 2012, 56, 98–105. [Google Scholar]
- Yang, X.Y.; Sun, L.; Xu, P.; Gong, L.L.; Qiang, G.F.; Zhang, L.; Du, G.H. Effects of salvianolic scid A on plantar microcirculation and peripheral nerve function in diabetic rats. Eur. J. Pharmacol. 2011, 665, 40–46. [Google Scholar] [CrossRef]
- Jiang, M.; Wang, X.Y.; Zhou, W.Y.; Li, J.; Wang, J.; Guo, L.P. Cerebral protection of salvianolic acid A by the inhibition of granulocyte adherence. Am. J. Chin. Med. 2011, 39, 111–120. [Google Scholar] [CrossRef]
- Pan, H.; Li, D.; Fang, F.; Chen, D.; Qi, L.; Zhang, R.; Xu, T.; Sun, H. Salvianolic acid a demonstrates cardioprotective effects in rat hearts and cardiomyocytes after ischemia/reperfusion injury. J. Cardiovasc. Pharmacol. 2011, 58, 535–542. [Google Scholar]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar]
- Yang, X.; Thomas, D.P.; Zhang, X.; Culver, B.W.; Alexander, B.M.; Murdoch, W.J.; Rao, M.N.; Tulis, D.A.; Ren, J.; Sreejayan, N. Curcumin inhibits platelet-derived growth factor-stimulated vascular smooth muscle cell function and injury-induced neointima formation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 85–90. [Google Scholar] [CrossRef]
- Lamy, S.; Beaulieu, E.; Labbé, D.; Bédard, V.; Moghrabi, A.; Barrette, S.; Gingras, D.; Béliveau, R. Delphinidin, a dietary anthocyanidin, inhibits platelet-derived growth factor ligand/receptor (PDGF/PDGFR) signaling. Carcinogenesis 2008, 29, 1033–1041. [Google Scholar]
- Godichaud, S.; Si-Tayeb, K.; Augé, N.; Desmoulière, A.; Balabaud, C.; Payrastre, B.; Nègre-Salvayre, A.; Rosenbaum, J. The grape-derived polyphenol resveratrol differentially affects epidermal and platelet-derived growth factor signaling in human liver myofibroblasts. Int. J. Biochem. Cell Biol. 2006, 38, 629–637. [Google Scholar] [CrossRef]
- Chen, A.; Zhang, L. The antioxidant (-)-epigallocatechin-3-gallate inhibits rat hepatic stellate cell proliferation in vitro by blocking the tyrosine phosphorylation and reducing the gene expression of platelet-derived growth factor-beta receptor. J. Biol. Chem. 2003, 278, 23381–23389. [Google Scholar] [CrossRef]
- Ahn, H.Y.; Hadizadeh, K.R.; Seul, C.; Yun, Y.P.; Vetter, H.; Sachinidis, A. Epigallocathechin-3 gallate selectively inhibits the PDGFBB-induced intracellular signaling transduction pathway in vascular smooth muscle cells and inhibits transformation of sis-transfected NIH 3T3 fibroblasts and human glioblastoma cells (A172). Mol. Biol. Cell 1999, 10, 1093–1104. [Google Scholar]
- Egozi, D.; Shapira, M.; Paor, G.; Ben-Izhak, O.; Skorecki, K.; Hershko, D.D. Regulation of the cell cycle inhibitor p27 and its ubiquitin ligase Skp2 in differentiation of human embryonic stem cells. FASEB J. 2007, 21, 2807–2817. [Google Scholar] [CrossRef]
- Claesson-Welsh, L. Platelet-derived growth factor receptor signals. J. Biol. Chem. 1994, 269, 32023–32026. [Google Scholar]
- Hung, Y.C.; Wang, P.W.; Pan, T.L.; Bazylak, G.; Leu, Y.L. Proteomic screening of antioxidant effects exhibited by radix Salvia miltiorrhiza aqueous extract in cultured rat aortic smooth muscle cells under homocysteine treatment. J. Ethnopharmacol. 2009, 124, 463–474. [Google Scholar] [CrossRef]
- Lai, K.; Wang, H.; Lee, W.S.; Jain, M.K.; Lee, M.E.; Haber, E. Mitogenactivated protein kinase phosphatase-1 in rat arterial smooth muscle cell proliferation. J. Clin. Invest. 1996, 98, 1560–1567. [Google Scholar]
- Pyles, J.M.; March, K.L.; Franklin, M.; Mehdi, K.; Wilensky, R.L.; Adam, L.P. Activation of MAP kinase in vivo follows balloon over stretch injury of porcine coronary and carotid arteries. Circ. Res. 1997, 81, 904–910. [Google Scholar] [CrossRef]
- Izumi, Y.; Kim, S.; Namba, M.; Yasumoto, H.; Miyazaki, H.; Hoshiga, M.; Kaneda, Y.; Morishita, R.; Zhan, Y.; Iwao, H. Gene transfer of dominant-negative mutants of extracellular signal-regulated kinase and c-Jun NH2-terminal kinase prevents neointimal formation in balloon-injured rat artery. Circ. Res. 2001, 88, 1120–1126. [Google Scholar]
- Picard, P.; Smith, P.J.; Monge, J.C.; Stewart, D.J. Expression of endothelial factors after arterial injury in the rat. J. Cardiovasc. Pharmacol. 1998, 31, S323–S327. [Google Scholar] [CrossRef]
- Versari, D.; Lerman, L.O.; Lerman, A. The importance of reendothelialization after arterial injury. Curr. Pharm. Des. 2007, 13, 1811–1824. [Google Scholar] [CrossRef]
- Institute of Laboratory Animal Resources (USA), Guide for the Care and Use of Laboratory Animals; National Academy Press: Washington, DC, USA, 1996.
- Sun, L.; Zhang, T.; Yu, X.; Xin, W.; Lan, X.; Zhang, D.; Huang, C.; Du, G. Asymmetric dimethylarginine confers the communication between endothelial and smooth muscle cells and leads to VSMC migration through p38 and ERK1/2 signaling cascade. FEBS Lett. 2011, 585, 2727–2734. [Google Scholar] [CrossRef]
- Rothmeier, A.S.; Ischenko, I.; Joore, J.; Garczarczyk, D.; Fürst, R.; Bruns, C.J.; Vollmar, A.M.; Zahler, S. Investigation of the marine compound spongistatin 1 links the inhibition of PKC alpha translocation to nonmitotic effects of tubulin antagonism in angiogenesis. FASEB J. 2009, 23, 1127–1137. [Google Scholar] [CrossRef]
- Fürst, R.; Zirrgiebel, U.; Totzke, F.; Zahler, S.; Vollmar, A.M.; Koch, E. The Crataegus extract WS 1,442 inhibits balloon catheter-induced intimal hyperplasia in the rat carotid artery by directly influencing PDGFR-beta. Atherosclerosis 2010, 211, 409–417. [Google Scholar] [CrossRef]
- Zargham, R.; Thibault, G. α8β1 Integrin expression in the rat carotid artery: Involvement in smooth muscle cell migration and neointima formation. Cardiovasc. Res. 2005, 65, 813–822. [Google Scholar] [CrossRef]
- Park, E.S.; Yoo, J.M.; Lim, Y.; Tudev, M.; Yoo, H.S.; Hong, J.T.; Yun, Y.P. Inhibitory effects of docetaxel on platelet-derived growth factor (PDGF)-BB-induced proliferation of vascular smooth muscle cells through blocking PDGF-receptor β phosphorylation. J. Pharmacol. Sci. 2011, 116, 204–213. [Google Scholar]
- Sample Availability: Samples of the Salvianolic Acid A are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, L.; Zhao, R.; Zhang, L.; Zhang, T.; Xin, W.; Lan, X.; Huang, C.; Du, G. Salvianolic Acid A Inhibits PDGF-BB Induced Vascular Smooth Muscle Cell Migration and Proliferation While Does Not Constrain Endothelial Cell Proliferation and Nitric Oxide Biosynthesis. Molecules 2012, 17, 3333-3347. https://doi.org/10.3390/molecules17033333
Sun L, Zhao R, Zhang L, Zhang T, Xin W, Lan X, Huang C, Du G. Salvianolic Acid A Inhibits PDGF-BB Induced Vascular Smooth Muscle Cell Migration and Proliferation While Does Not Constrain Endothelial Cell Proliferation and Nitric Oxide Biosynthesis. Molecules. 2012; 17(3):3333-3347. https://doi.org/10.3390/molecules17033333
Chicago/Turabian StyleSun, Lan, Rui Zhao, Li Zhang, Tiantai Zhang, Wenyu Xin, Xi Lan, Chao Huang, and Guanhua Du. 2012. "Salvianolic Acid A Inhibits PDGF-BB Induced Vascular Smooth Muscle Cell Migration and Proliferation While Does Not Constrain Endothelial Cell Proliferation and Nitric Oxide Biosynthesis" Molecules 17, no. 3: 3333-3347. https://doi.org/10.3390/molecules17033333
APA StyleSun, L., Zhao, R., Zhang, L., Zhang, T., Xin, W., Lan, X., Huang, C., & Du, G. (2012). Salvianolic Acid A Inhibits PDGF-BB Induced Vascular Smooth Muscle Cell Migration and Proliferation While Does Not Constrain Endothelial Cell Proliferation and Nitric Oxide Biosynthesis. Molecules, 17(3), 3333-3347. https://doi.org/10.3390/molecules17033333