Effects of the Cyclin-Dependent Kinase 10 (CDK10) on the Tamoxifen Sensitivity of Keloid Samples

Abstract
:1. Introduction
2. Results and Discussion
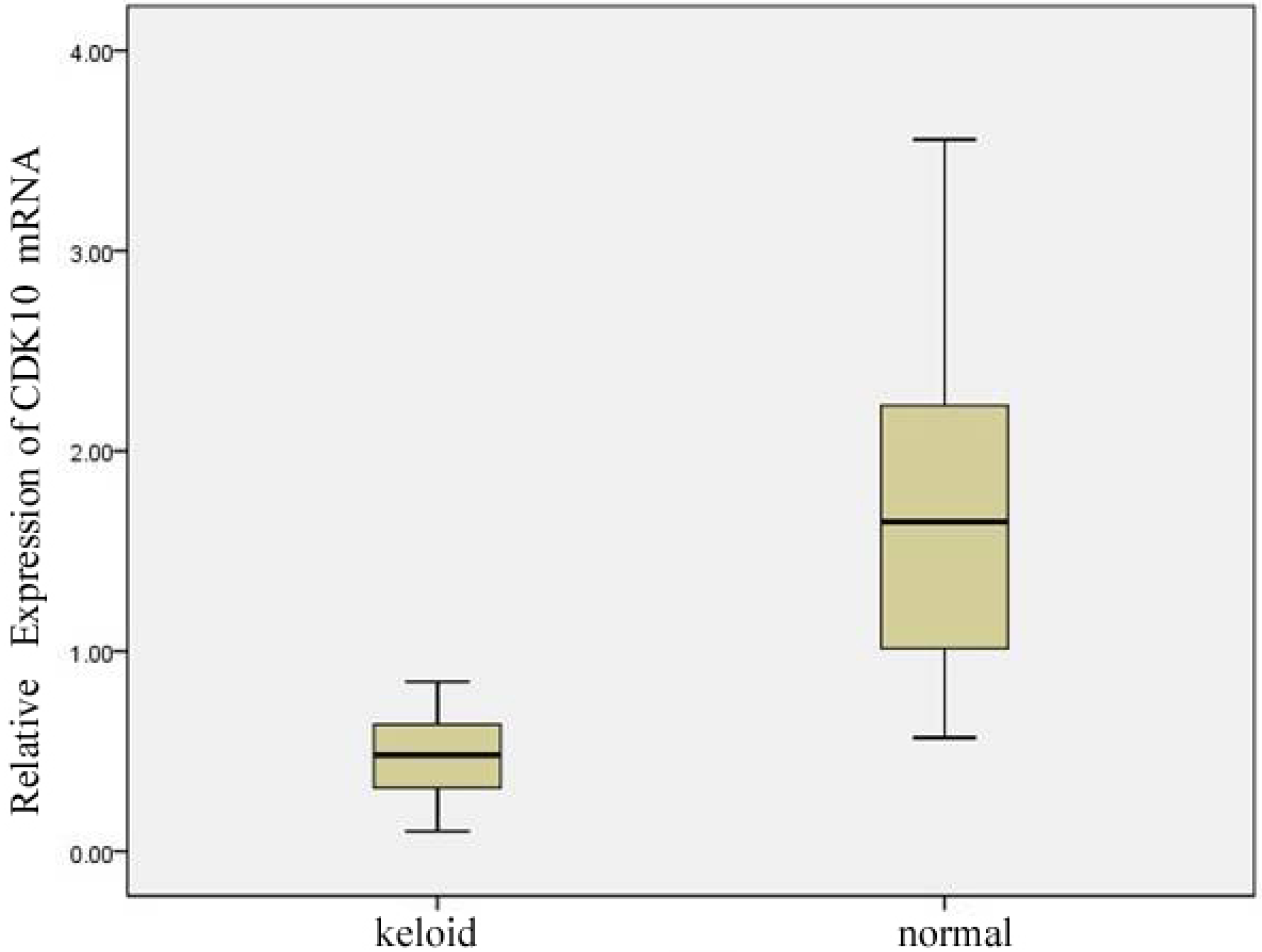
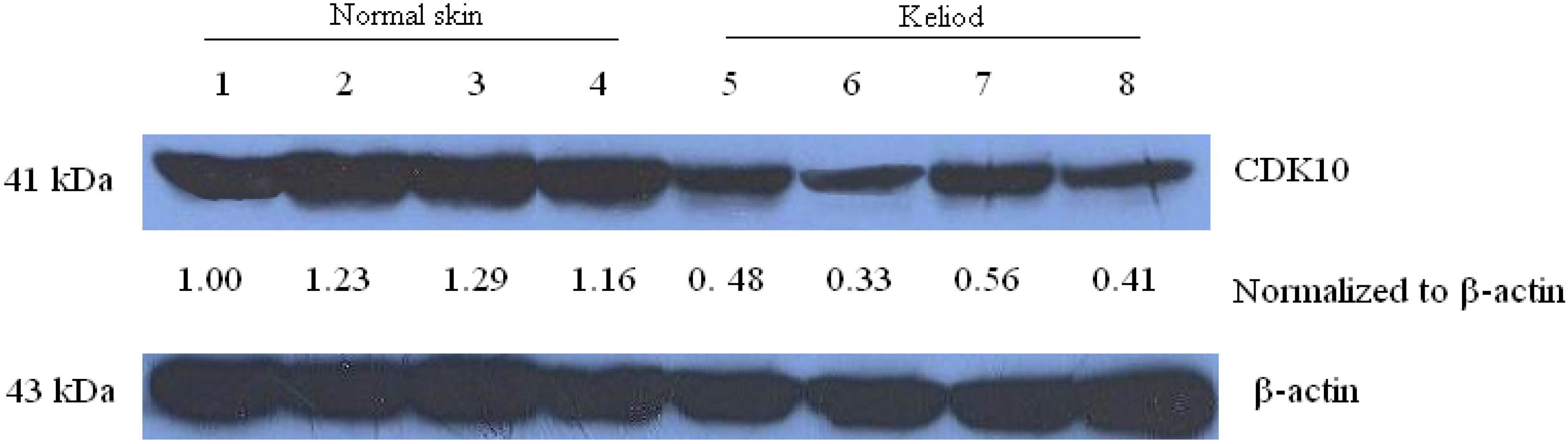
2.1. Expression of CDK10 in Keloid and Normal Skin Samples by Quantitative Real-time PCR and Western Blot Assay
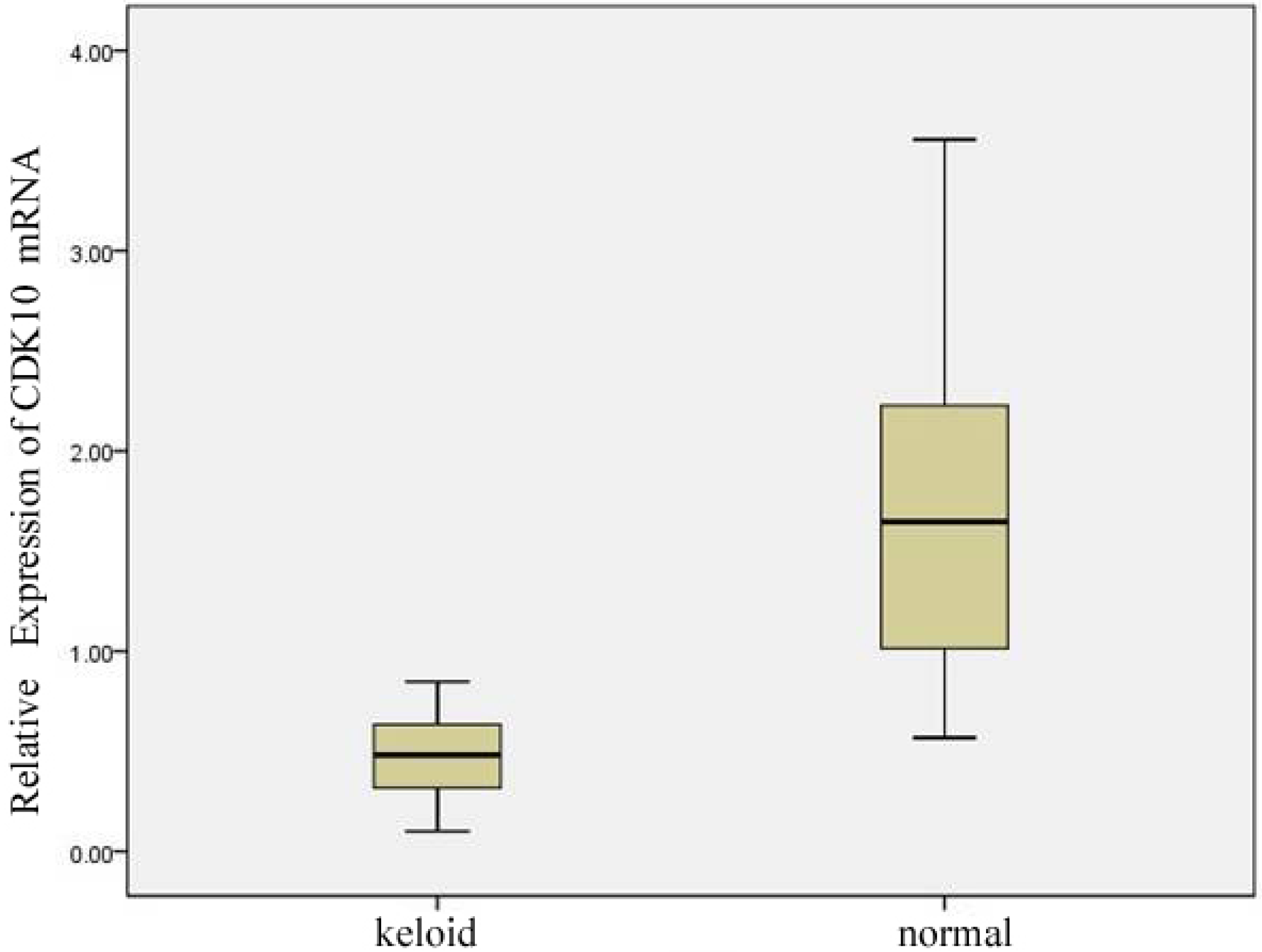
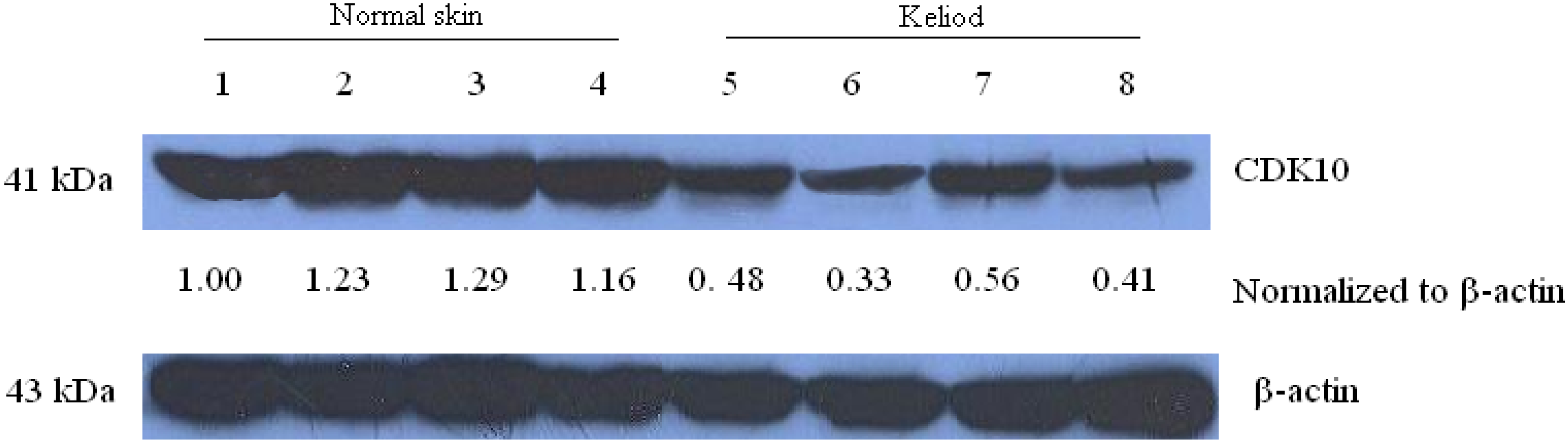
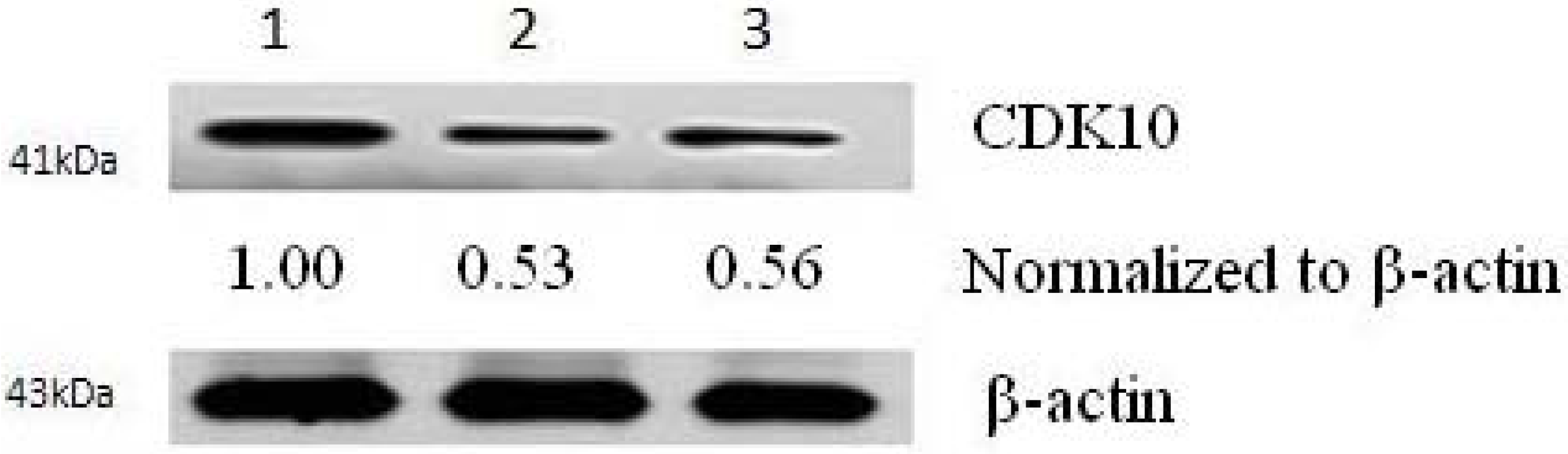
2.2. Results of Transfection
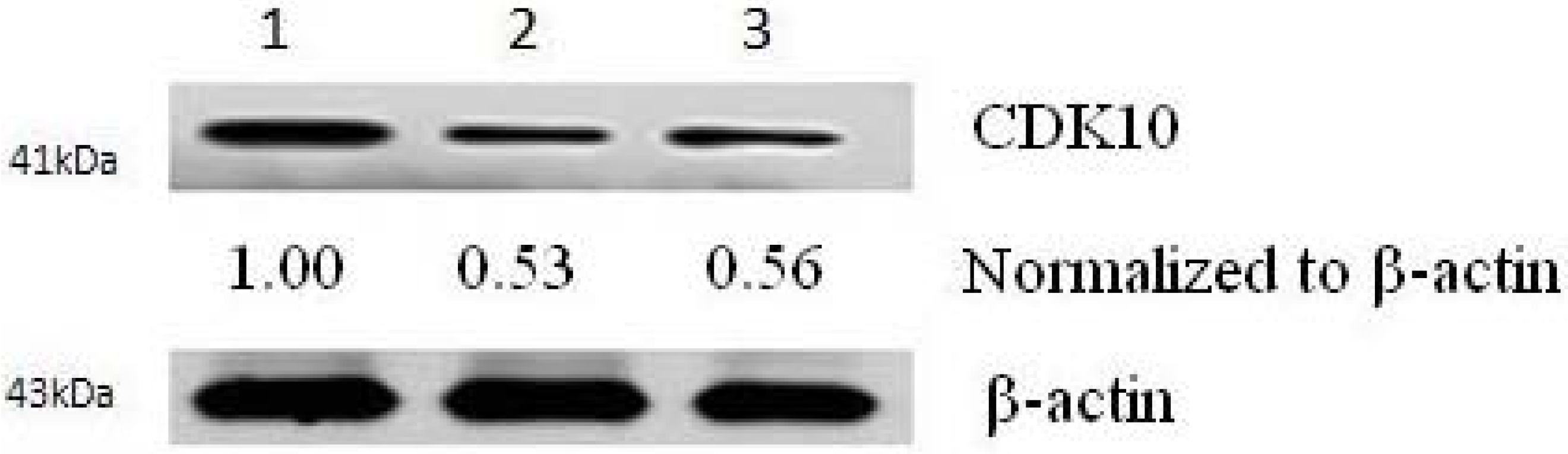
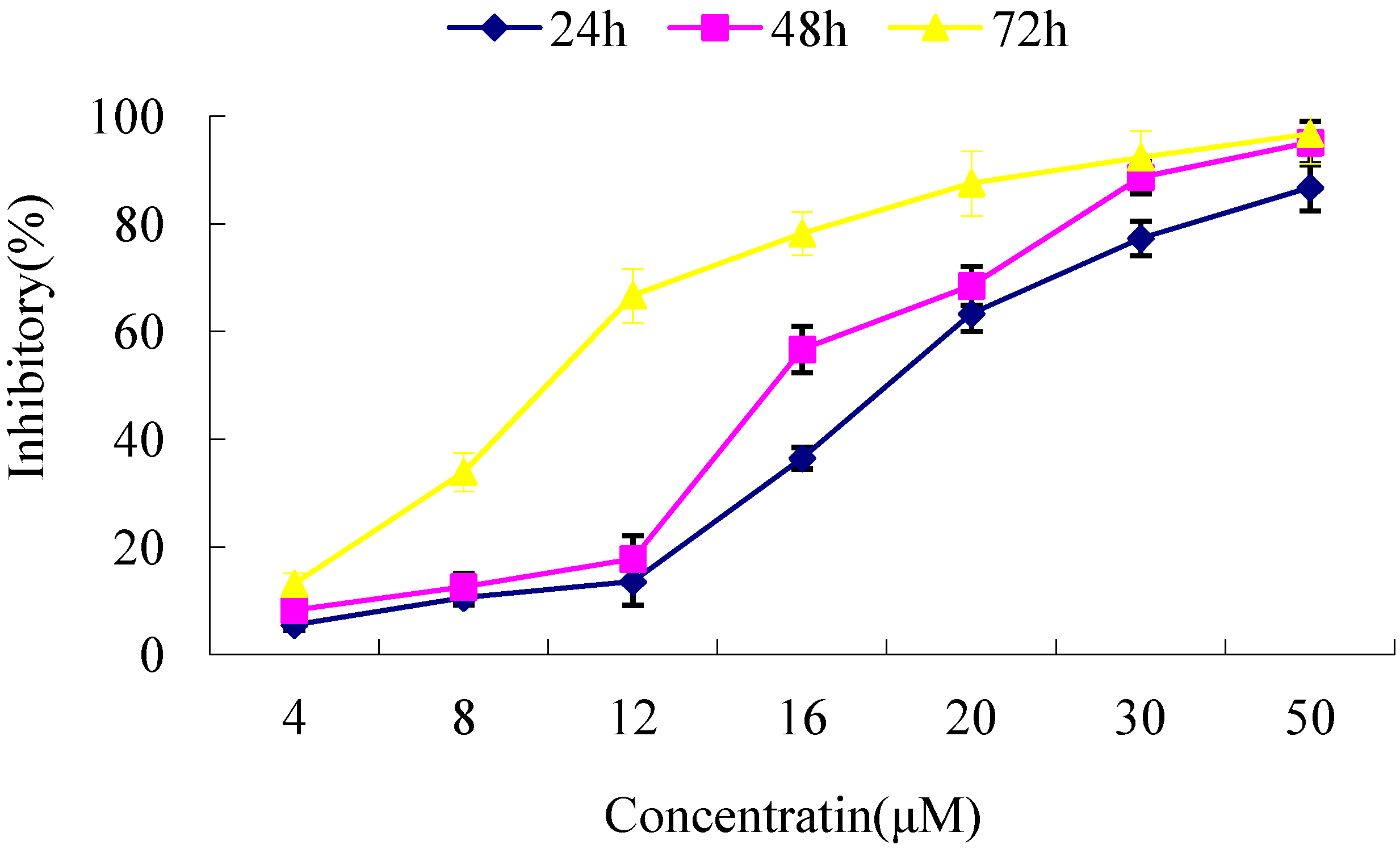
2.3. Cytotoxicity Assays


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell lines | IC50 (µM) |
|---|---|
| keloid fibroblast | 17.61 |
| pCMV6-CDK10-transfected cells | 9.41 * |
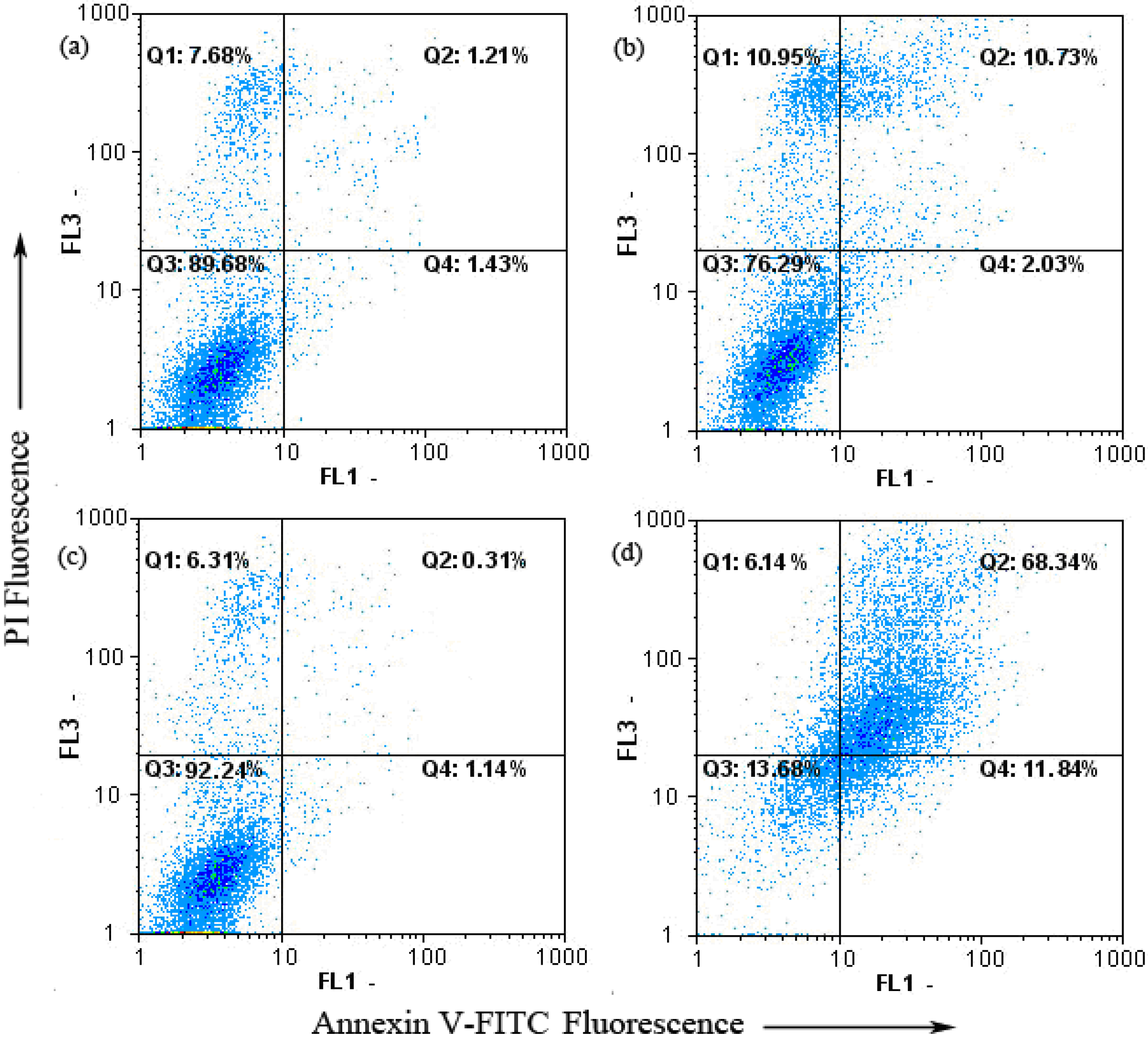
2.4. Keloid Fibroblast Cell Apoptosis as Detected by Annexin V-FITC/PI
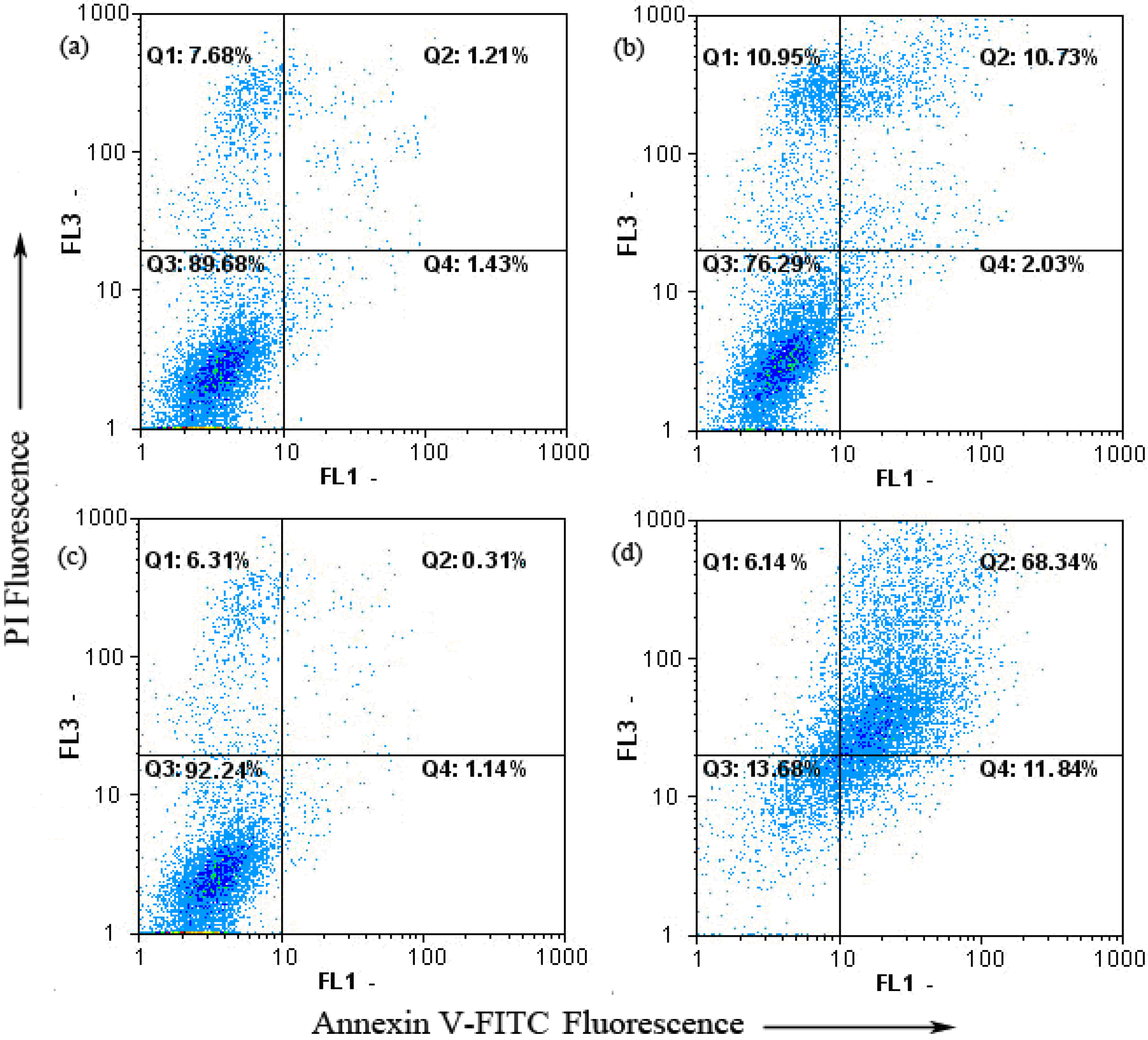
2.5. Bax and Bcl-2 Expression of Keloid Fibroblast as Detected by Western Blot

3. Discussion
4. Experimental Section
4.1. Patients and Treatment
4.2. RNA Extraction and Quantitative Real-time PCR
4.3. Western Blot Analysis
4.4. Transient Transfection
4.5. Cytotoxicity Assay
4.6. Flow Cytometric Analysis of Cell Apoptosis
4.7. Statistical Analysis
4. Conclusions
Acknowledgements
- Sample Availability: Not available.
References and Notes
- Vincent, A.S.; Phan, T.T.; Mukhopadhyay, A.; Lim, H.Y.; Halliwell, B.; Wong, K.P. Human skin keloid fibroblasts display bioenergetics of cancer cells. J. Invest. Dermatol. 2008, 128, 702–709. [Google Scholar] [CrossRef]
- Nurul Syazana, M.S.; Sukari Halim, A.; Hua Gan, S.; Shamsuddin, S. Antiproliferative effect of methanolic extraction of tualang honey on human keloid fibroblasts. BMC Complem. Altern. Med. 2011, 11, 82. [Google Scholar]
- Mikulec, A.A.; Hanasono, M.M.; Lum, J.; Kadleck, J.M.; Kita, M.; Koch, R.J. Effect of tamoxifen on transforming growth factor beta1 production by keloid and fetal fibroblasts. Arch. Facial Plast. Surg. 2001, 3, 111–114. [Google Scholar] [CrossRef]
- Kiyotani, K.; Mushiroda, T.; Nakamura, Y.; Zembutsu, H. Pharmacogenomics of tamoxifen: Roles of drug metabolizing enzymes and transporters. Drug Metab. Pharmacokinet. 2011. Epub ahead of print. [Google Scholar]
- Zhang, G.J.; Kimijima, I.; Onda, M.; Kanno, M.; Sato, H.; Watanabe, T.; Tsuchiya, A.; Abe, R.; Takenoshita, S. Tamoxifen-induced apoptosis in breast cancer cells relates to down-regulation of bcl-2, but not bax and bcl-X(L), without alteration of p53 protein levels. Clin. Cancer Res. 1999, 5, 2971–2977. [Google Scholar]
- Cameron, D.A.; Keen, J.C.; Dixon, J.M.; Bellamy, C.; Hanby, A.; Anderson, T.J.; Miller, W.R. Effective tamoxifen therapy of breast cancer involves both antiproliferative and pro-apoptotic changes. Eur. J. Cancer 2000, 36, 845–851. [Google Scholar]
- Adam, L.; Crepin, M.; Israel, L. Tumor growth inhibition, apoptosis, and Bcl-2 down-regulation of MCF-7 ras tumors by sodium phenylacetate and tamoxifen combination. Cancer Res. 1997, 57, 1023–1029. [Google Scholar]
- Liu, Z.H.; He, Y.P.; Qin, H. The growth-inhibition effect of tamoxifen in the combination chemotherapeutics on the human cholangiocarcinoma cell line QBC939. Mol. Biol. Rep. 2010, 37, 2693–2701. [Google Scholar] [CrossRef]
- Mancoll, J.S.; Macauley, R.L.; Phillips, L.G. The inhibitory effect of tamoxifen on keloid fibroblasts. Surg. Forum. 1996, 47, 718–720. [Google Scholar]
- Hardman, M.J.; Emmerson, E.; Campbell, L. Ashcroft GS Selective estrogen receptor modulators accelerate cutaneous wound healing in ovariectomized female mice. Endocrinology 2008, 149, 551–557. [Google Scholar]
- Levenyhal, D.; Furr, M.; Reiter, D. Treatment of keloids and hypertrophic scars: A meta-analysis and review of the literature. Arch. Facial Plast. Surg. 2006, 8, 362–368. [Google Scholar] [CrossRef]
- Blackwell, K.L.; Haroon, Z.A.; Shan, S.; Saito, W.; Broadwater, G.; Greenberg, C.S.; Dewhirst, M.W. Tamoxifen inhibits angiogenesis in estrogen receptor-negative animal models. Clin. Cancer Res. 2000, 6, 4359–4364. [Google Scholar]
- Kinzbrunner, B.; Ritter, S.; Domingo, J.; Rosenthal, C.J. Remission of rapidly growing desmoids tumors after tamoxifen therapy. Cancer 1993, 52, 2201–2204. [Google Scholar]
- Kuhn, M.A.; Wang, X.; Payne, W.G.; Ko, F.; Robson, M.C. Tamoxifen decreases fibroblast function and downregulates TGF(beta2) in dupuytren’s affected palmar fascia. J. Surg. Res. 2002, 103, 146–152. [Google Scholar] [CrossRef]
- Mancoll, J.S.; Zhao, J.; McCauley, R.L.; Phillips, L.G. The inhibitory effect of tamoxifen on keloid fibroblast. Surg. Forum 1996, 47, 718–720. [Google Scholar]
- Ruffy, M.B.; Kunnavatana, S.S.; Koch, R.J. Effects of tamoxifen on normal human dermal fibroblasts. Arch. Facial Plast. Surg. 2006, 8, 329–332. [Google Scholar] [CrossRef]
- Payne, W.G.; Ko, F.; Anspaugh, S.; Wheeler, C.K.; Wright, T.E.; Robson, M.C. Down-regulating causes of fibrosis with tamoxifen: A possible cellular/molecular approach to treat rhinophyma. Ann. Plast. Surg. 2006, 56, 301–305. [Google Scholar]
- Leventhal, D.; Furr, M.; Reiter, D. Treatment of keloids and hypertrophic scars: A meta-analysis and review of the literature. Arch. Facial Plast. Surg. 2006, 8, 362–368. [Google Scholar]
- Ekhdm, S.V.; Reed, S.I. Regulation of G1 cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 2000, 12, 676–684. [Google Scholar]
- Kaufmann, S.H.; Hengartner, M.O. Programmed cell death: Alive and well in the new millennium. Trends Cell Biol. 2001, 11, 526–534. [Google Scholar]
- Petros, A.M.; Olejniczak, E.T.; Fesik, S.W. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 2004, 1644, 83–94. [Google Scholar]
- Li, S.; MacLachlan, T.K.; De Luca, A.; Claudio, P.P.; Condorelli, G.; Giordano, A. The cdc-2-related kinase, PISSLRE, is essential for cell growth and acts in G2 phase of the cell cycle. Cancer Res. 1995, 55, 3992–3995. [Google Scholar]
- Grana, X.; Fau-Claudio, P.P.; De Luca, A.; Sang, N.; Giordano, A. PISSLRE, a human novel CDC2-related protein kinase. Oncogene 1994, 9, 2097–2103. [Google Scholar]
- Singhal, S.; Fau-Amin, K.M.; Kruklitis, R.; De Long, P.; Friscia, M.E.; Litzky, L.A.; Putt, M.E.; Kaiser, L.R.; Albelda, S.M. Alterations in cell cycle genes in early stage lung adenocarcinoma identified by expression profiling. Cancer Biol. Ther. 2003, 2, 291–298. [Google Scholar]
- Husson, H.; Carideo, E.G.; Neuberg, D.; Schultze, J.; Munoz, O.; Marks, P.W.; Donovan, J.W.; Chillemi, A.C.; O’Connell, P.; Freedman, A.S. Gene expression profiling of follicular lymphoma and normal germinal center B cells using cDNA arrays. Blood 2002, 99, 282–289. [Google Scholar] [CrossRef]
- Leman, E.S.; Magheli, A.; Yong, K.M.; Netto, G.; Hinz, S.; Getzenberg, R.H. Identification of nuclear structural protein alterations associated with seminomas. J. Cell Biochem. 2009, 108, 1274–1279. [Google Scholar]
- Iorns, E.; Fau-Turner, N.C.; Elliott, R.; Syed, N.; Garrone, O.; Gasco, M.; Tutt, A.N.; Crook, T.; Lord, C.J.; Ashworth, A. Identification of CDK10 as an important determinant of resistance to endocrine. Cancer Cell 2008, 13, 91–104. [Google Scholar] [CrossRef]
- Kasten, M.; Giordano, A. Cdk10, a Cdc2-related kinase, associates with the Ets2 transcription factor and modulate its transactivation activity. Oncogene 2001, 20, 1832–1838. [Google Scholar] [CrossRef]
- Lavoien, J.N.; L’Allemain, G.; Brunet, A.; Muller, R.; Pouyssegur, J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar]
- Wilcken, N.R.; Prall, O.W.; Musgrove, E.A.; Sutherland, R.L. Inducible over expression of cyclin D1 in breast cancer cells reverses the growth-inhibitory effects of ant estrogens. Clin. Cancer Res. 1997, 3, 849–854. [Google Scholar]
- Zhong, L.T.; Sarafian, T.; Kane, D.J.; Charles, A.C.; Mah, S.P.; Edwards, R.H.; Bredesen, D.E. Bcl-2 inhibits death of central neural cells induced by multiple agents. Proc. Natl. Acad. Sci.USA 1993, 90, 4533–4537. [Google Scholar]
- Wolter, K.G.; Hsu, Y.T.; Smith, C.L.; Nechushtan, A.; Xi, X.G.; Youle, R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997, 139, 1281–1292. [Google Scholar]
- Bruce-Keller, A.J.; Begley, J.G.; Fu, W.; Butterfield, D.A.; Bredesen, D.E.; Hutchins, J.B.; Hensley, K.; Mattson, M.P. Bcl-2 protects isolated plasma and mitochondrial membranes against lipid peroxidation induced by hydrogen peroxide and amyloid beta-peptide. J. Neurochem. 1998, 70, 31–39. [Google Scholar]
- Tsujimoto, Y. Role of Bcl-2 family proteins in apoptosis: Apoptosomes or mitochondria. Genes Cells 1998, 11, 697–707. [Google Scholar] [CrossRef]
- Adams, M.; Cory, S. Bcl-2-regulated apoptosis: Mechanism and therapeutic potential. Curr. Opin. Immunol. 2007, 19, 488–496. [Google Scholar]
- Reed, J.C. Regulation of apoptosis by Bcl-2 family proteins and its role in cancer and chemoresistance. Curr. Opin. Oncol. 1995, 7, 541–546. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Thati, B.; Noble, A.; Creaven, B.S.; Walsh, M.; McCann, M.; Devereux, M.; Kavanagh, K.; Egan, D.A. Role of cell cycle events and apoptosis in mediating the anti-cancer activity of a silver (I) complex of 4-hydroxy-3-nitro-coumarin-bis (phenanthroline) in human malignant cancer cells. Eur. J. Pharmacol. 2009, 602, 203–214. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Y.; Xiao, Z.; Yang, D.; Ren, L.; Liu, G.; Yang, L. Effects of the Cyclin-Dependent Kinase 10 (CDK10) on the Tamoxifen Sensitivity of Keloid Samples. Molecules 2012, 17, 1307-1318. https://doi.org/10.3390/molecules17021307
Liu Y, Xiao Z, Yang D, Ren L, Liu G, Yang L. Effects of the Cyclin-Dependent Kinase 10 (CDK10) on the Tamoxifen Sensitivity of Keloid Samples. Molecules. 2012; 17(2):1307-1318. https://doi.org/10.3390/molecules17021307
Chicago/Turabian StyleLiu, Ying, Zhibo Xiao, Daping Yang, Lihong Ren, Guofeng Liu, and Lin Yang. 2012. "Effects of the Cyclin-Dependent Kinase 10 (CDK10) on the Tamoxifen Sensitivity of Keloid Samples" Molecules 17, no. 2: 1307-1318. https://doi.org/10.3390/molecules17021307
APA StyleLiu, Y., Xiao, Z., Yang, D., Ren, L., Liu, G., & Yang, L. (2012). Effects of the Cyclin-Dependent Kinase 10 (CDK10) on the Tamoxifen Sensitivity of Keloid Samples. Molecules, 17(2), 1307-1318. https://doi.org/10.3390/molecules17021307