Synthesis and in Vitro Antioxidant Activity Evaluation of 3-Carboxycoumarin Derivatives and QSAR Study of Their DPPH• Radical Scavenging Activity

 ,
, 
Abstract
:
1. Introduction
2. Experimental Methods and Results
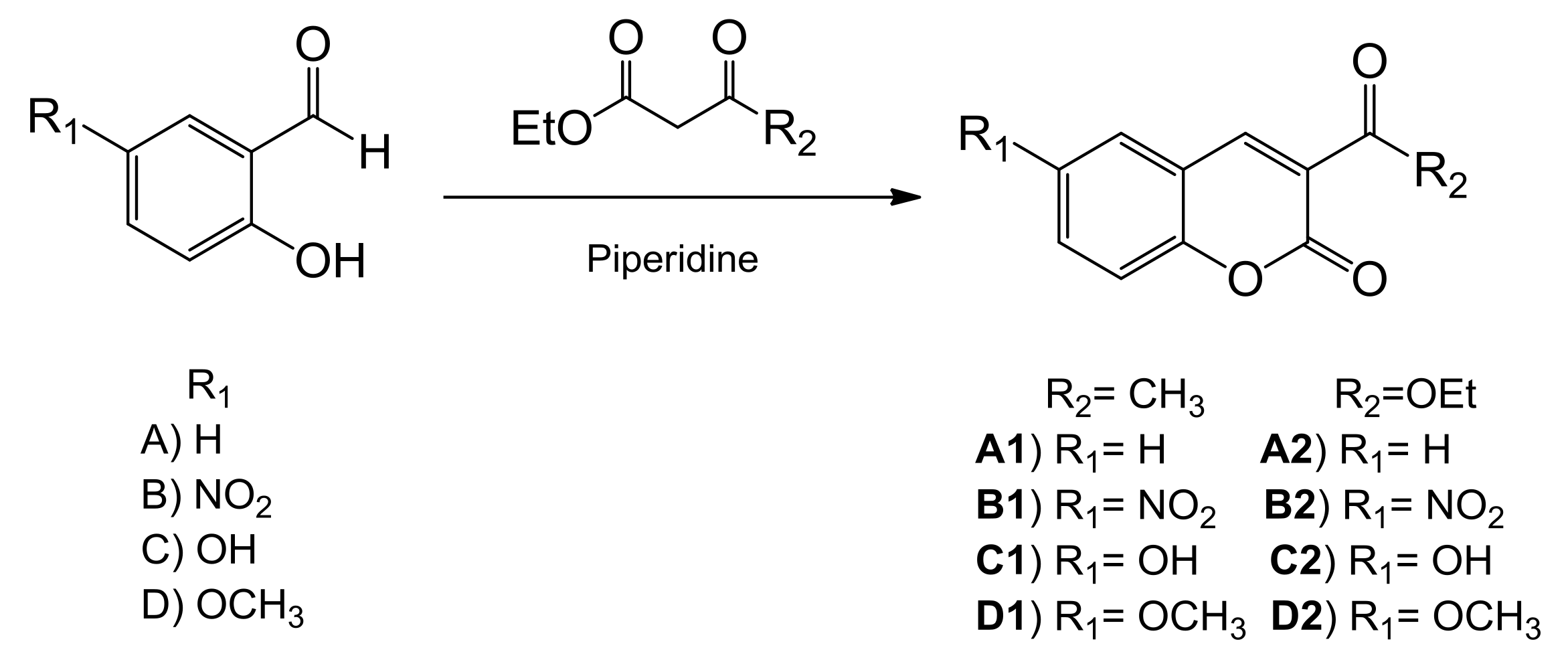
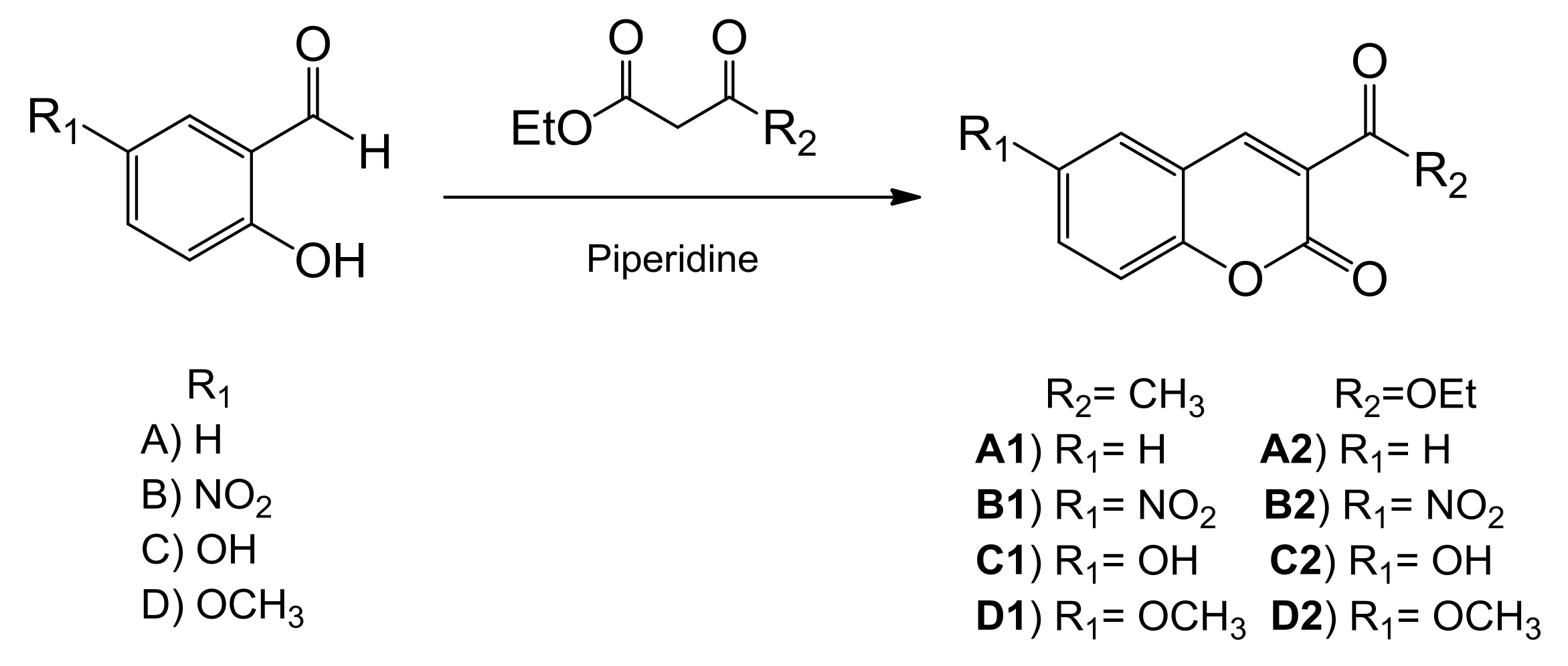
2.1. Synthesis and Characterization
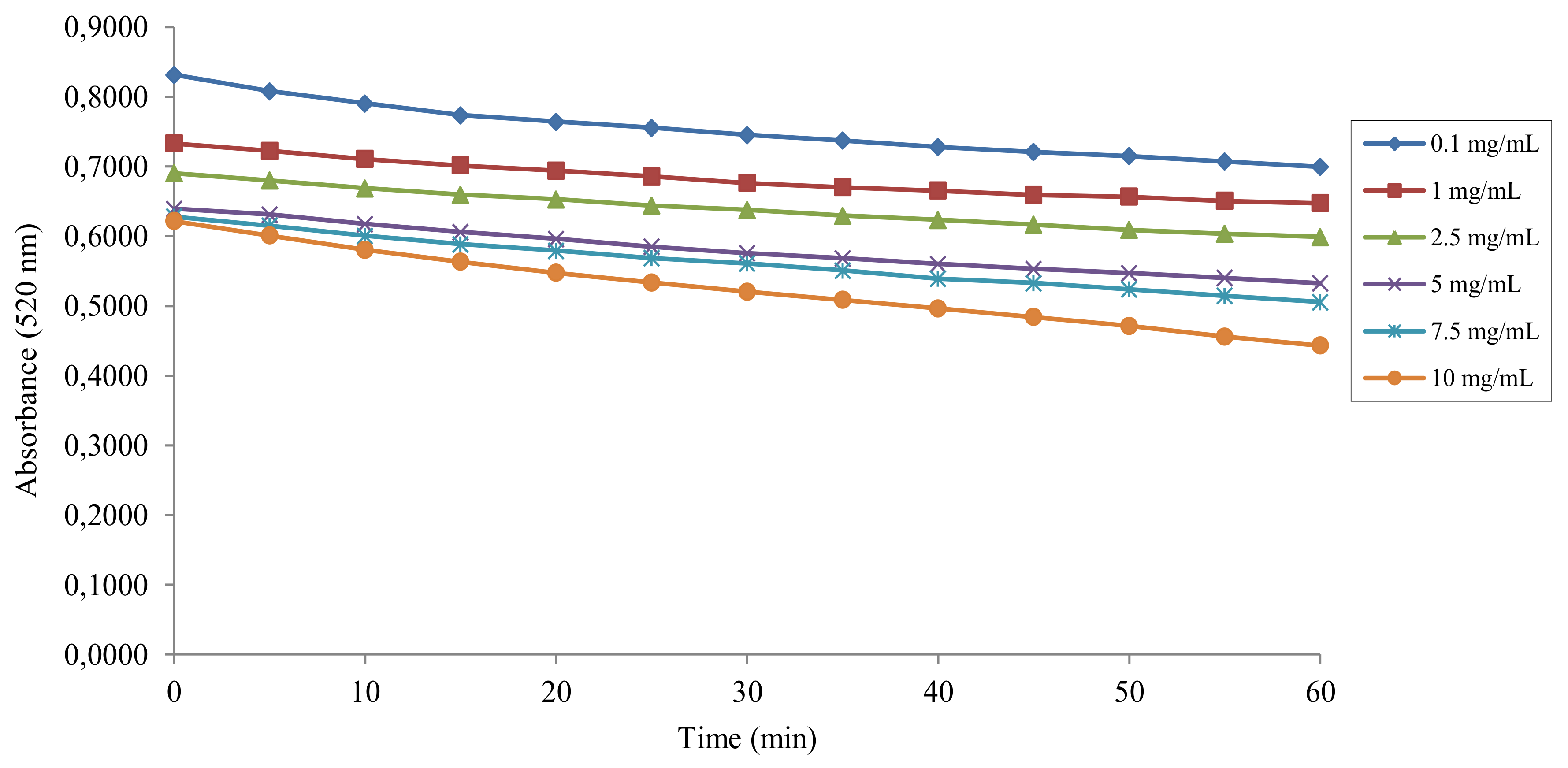
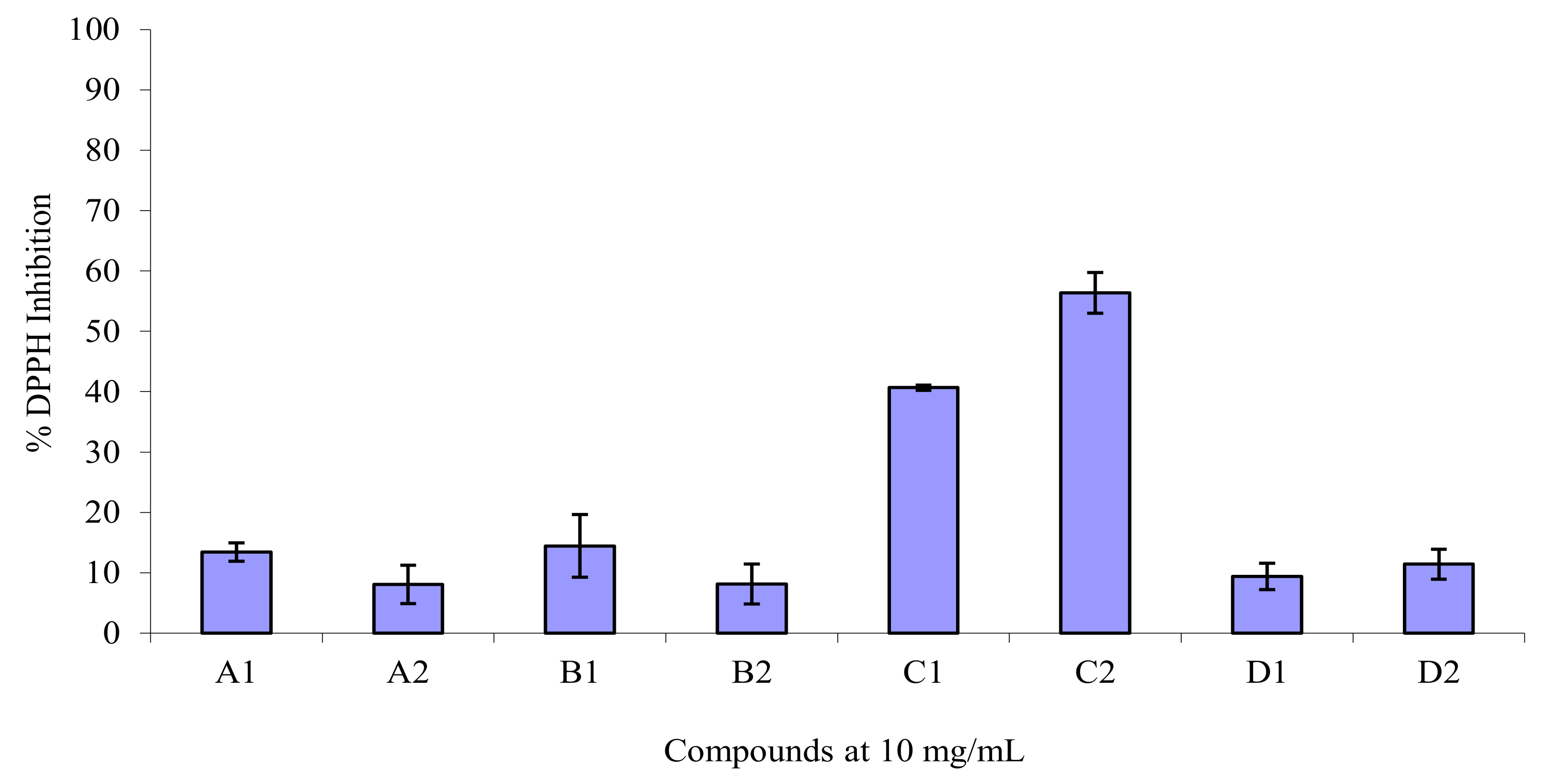
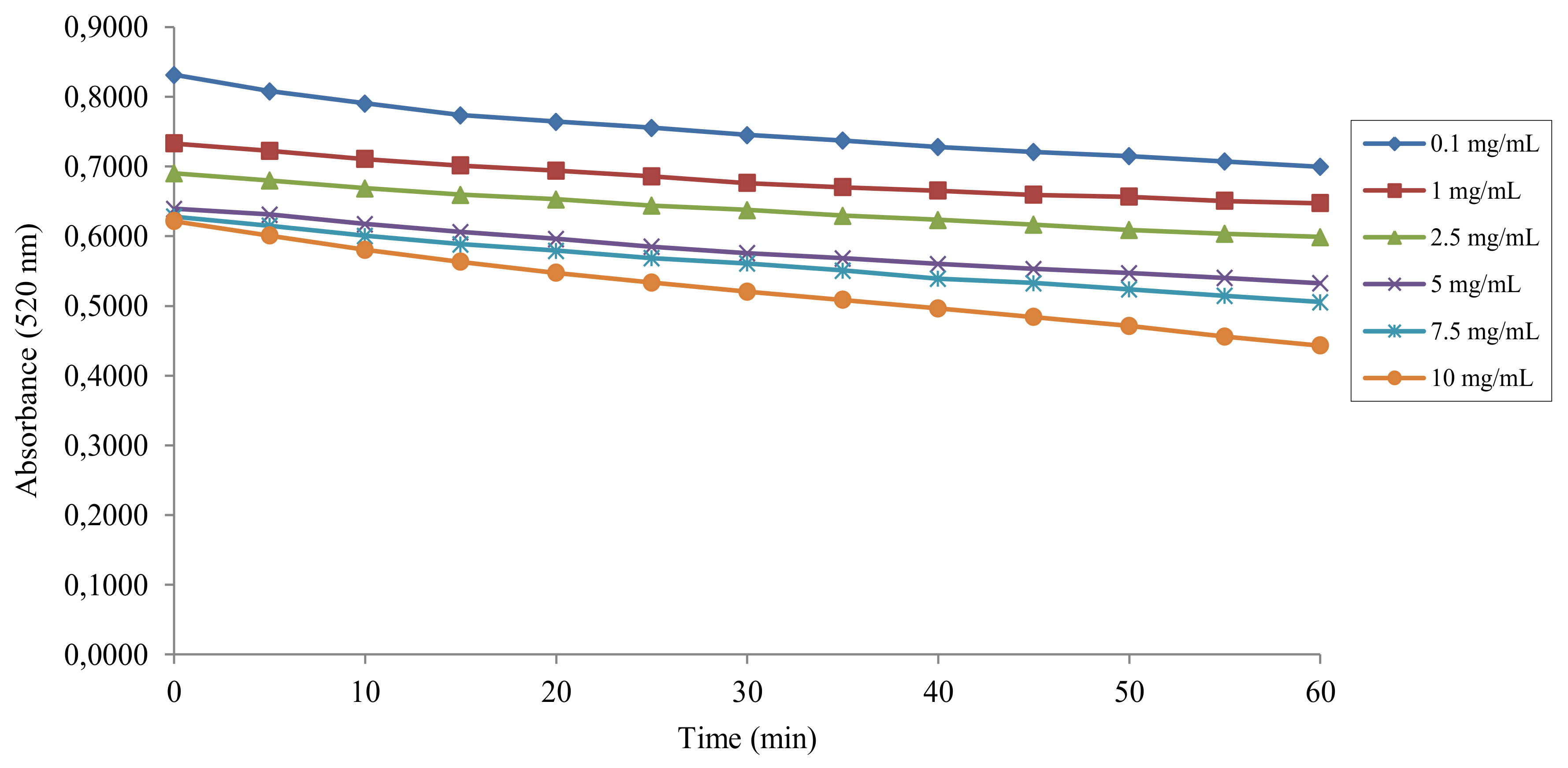
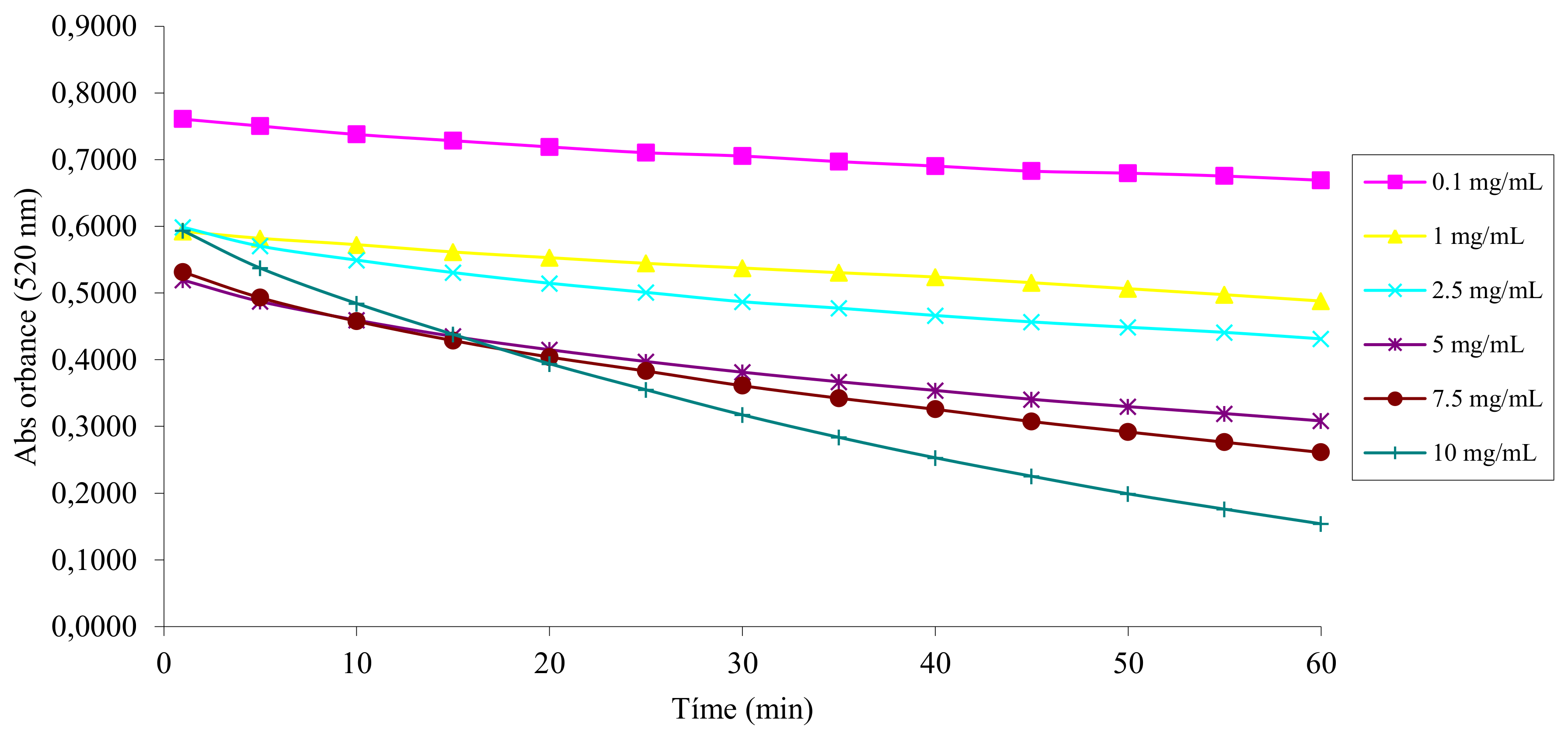
2.2. DPPH• Radical Scavenging Activity
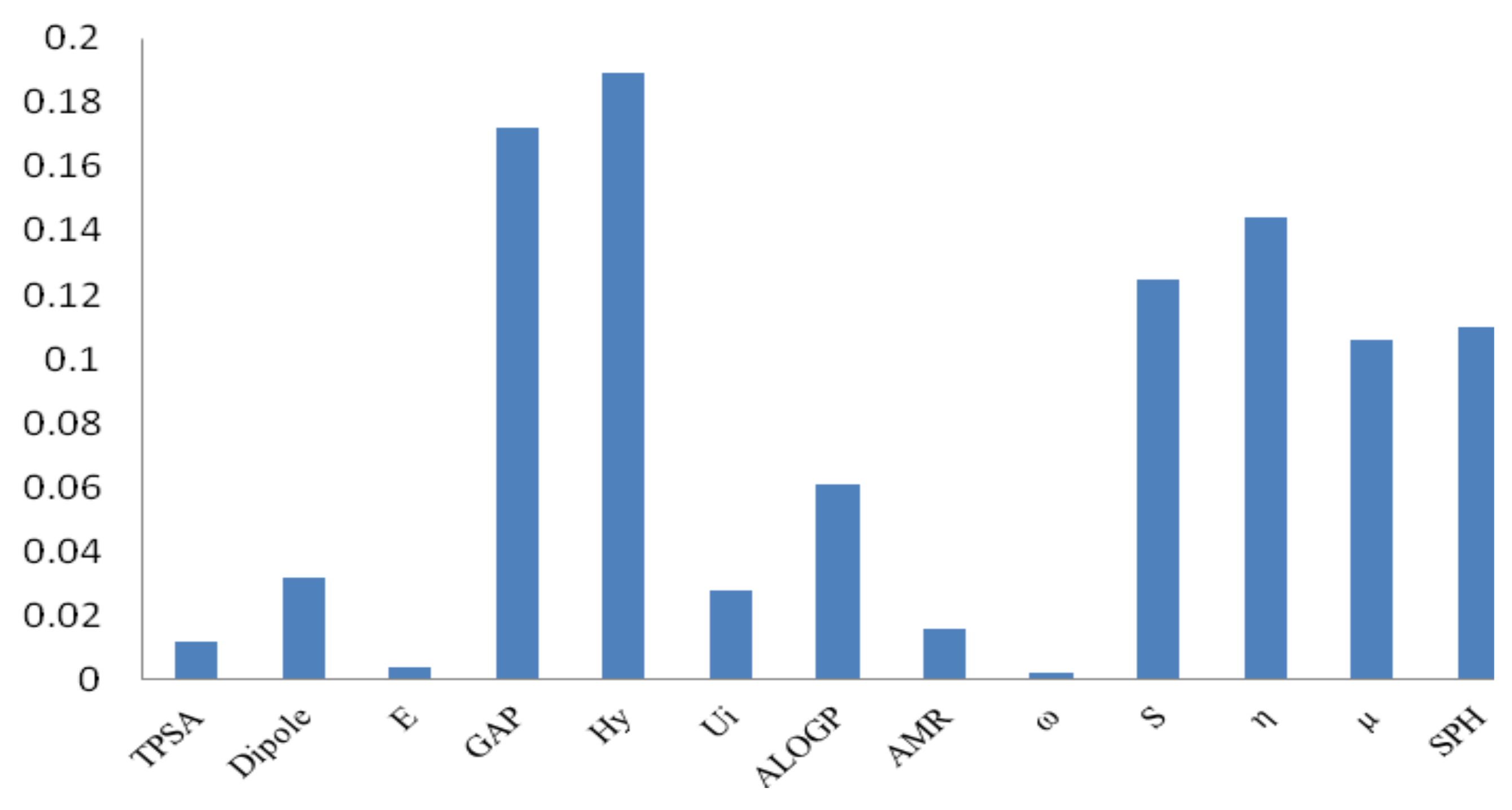
3. Computational Details and Results










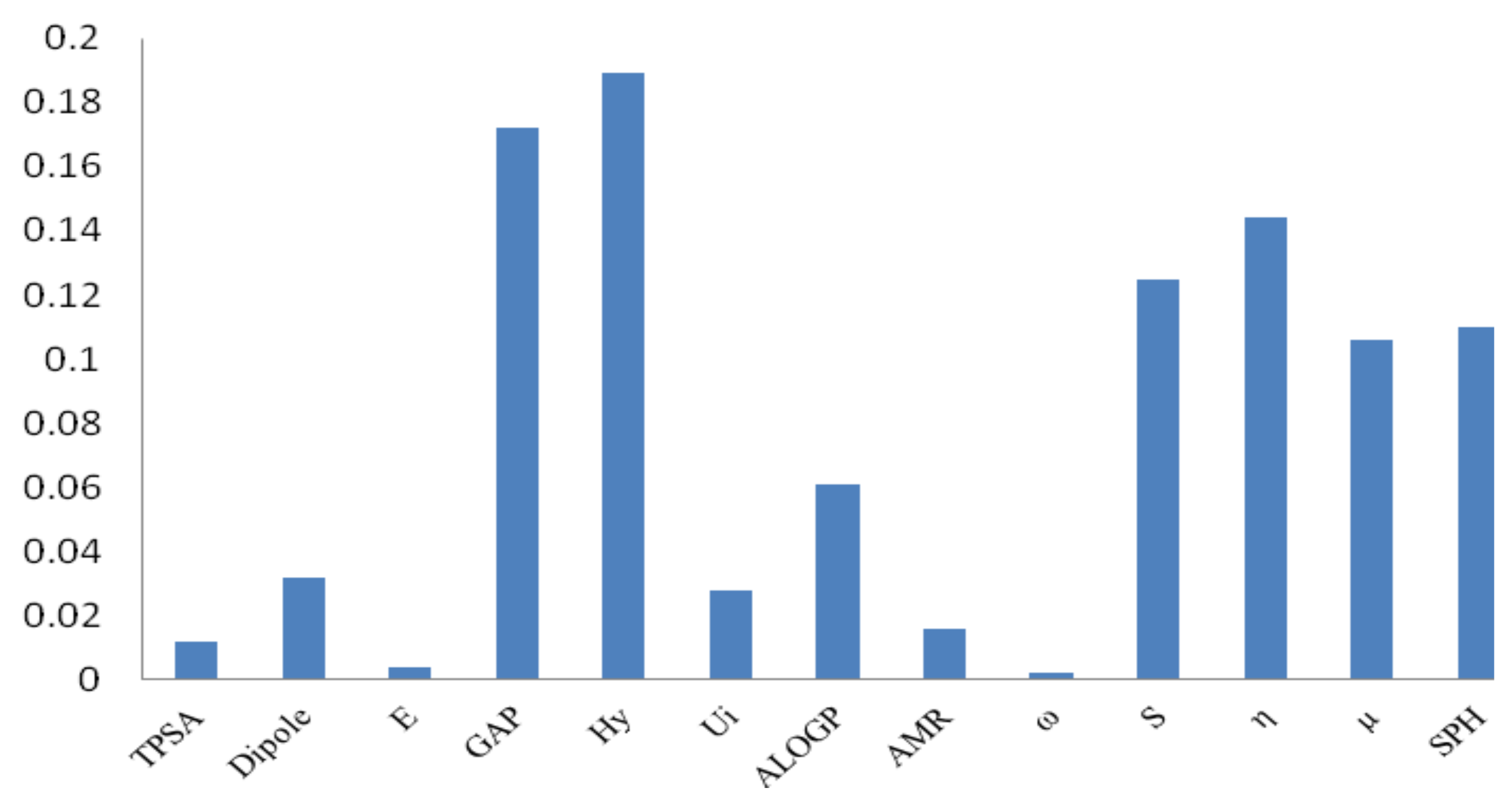
3.1. Genetic Algorithms (GA)




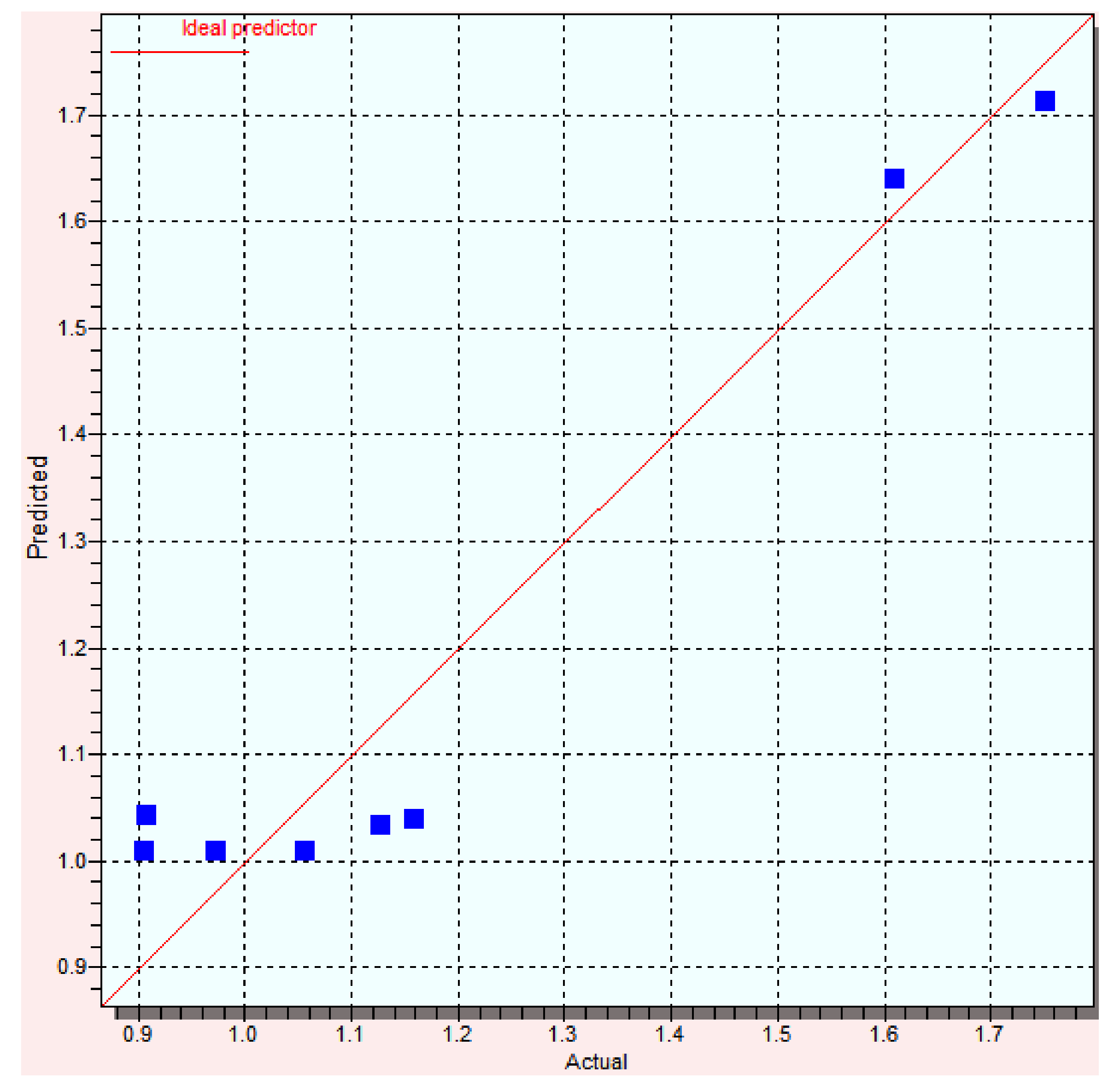
3.2. Backpropagation Neural Network
4. Experimental
4.1. General
General Procedure for the Synthesis of Coumarin Derivatives
4.2. Antiradical Activity Measurement with the DPPH• Assay
5. Conclusions
Acknowledgments
References
- Kontogiorgis, C.; Handjipavlou, D. Biological evaluation of several coumarin derivatives designed as possible anti-inflammatory/antioxidant agents. J. Enzym. Inhib. 2006, 18, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, A.; Karali, N. Synthesis, characterization and primary antituberculosis activity evaluation of 4-(3-coumarinyl)-3-benzyl-4-thiazolin-2-one benzilidenehydrazones. Turk. J. Chem. 2003, 27, 545–551. [Google Scholar]
- Kostova, I. Synthetic and Natural Coumarins as Cytotoxic Agents. Curr. Med. Chem. 2005, 5, 29–46. [Google Scholar] [CrossRef]
- Kusanur, R.A.; Ghate, M.; Kulkarni, M.V. Synthesis of spiro[indolo-1,5-benzodiazepines] from 3-acetyl coumarins for use as possible antianxiety agents. J. Chem. Sci. 2004, 116, 265–270. [Google Scholar] [CrossRef]
- Kempen, I.; Papapostolou, D.; Thierry, N.; Pochet, L.; Counerotte, S.; Masereel, B.; Foidart, J.M.; Reboud-Ravaux, M.; Noe, A.; Pirotte, B. 3-bromophenyl 6-acetoxymethyl-2-oxo-2H-1-benzopyran-3-carboxylate inhibits cancer cell invasion in vitro and tumor growth in vivo. Br. J. Cancer 2003, 88, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Lacy, A.; O’Kennedy, R. Studies on Coumarins and Coumarin-Related Compounds to Determine their Therapeutic Role in the Treatment of Cancer. Curr. Pharm. Design. 2004, 10, 3797–3811. [Google Scholar] [CrossRef]
- Kontogiorgis, C.; Handjipavlou, D. Synthesis and antiinflamatory activity of coumarin derivatives. J. Med. Chem. 2005, 48, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.C.; Tsai, S.H.; Chen, C.-S.; Chang, Y.-C.; Lee, C.-M.; Lai, Z.-Y.; Lin, C.-M. Structure-activity relationship of coumarin derivatives on xanthine oxidase-inhibiting and free radical-scavenging activities. Biochem. Pharm. 2008, 75, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Berhanu, W.M.; Pillai, G.G.; Oliferenko, A.A.; Katritzky, A.R. Quantitative structure–activity/property relationships: The ubiquitous links between cause and effect. ChemPlusChem 2012, 77, 507–517. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Kuanar, M.; Slavov, S.; Hall, C.D.; Karelson, M.; Kahn, I.; Dobchev, D.A. Quantitative correlation of physical and chemical properties with chemical structure: Utility for prediction. Chem. Rev. 2010, 110, 5714. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Epa, V.C.; Burden, F.R.; Winkler, D.A. Quantitative structure–property relationship modeling of diverse materials properties. Chem. Rev. 2012, 112, 2889–2919. [Google Scholar] [CrossRef] [PubMed]
- Topliss, J.G.; Costello, R.J. Chance correlations in structure-activity studies using multiple regression analysis. J. Med. Chem. 1972, 15, 1066–1068. [Google Scholar] [CrossRef] [PubMed]
- Puzyn, T.; Leszczynski, J.; Cronin, M.T.D. Recent Advances in QSAR Studies; Springer: New York, NY, USA, 2010; pp. 30–41. [Google Scholar]
- Parr, R.G.; Yang, W. Chemical potential derivatives. In Density-Functional Theory of Atoms and Molecules, 1st ed.; Oxford University Press: New York, NY, USA, 1989; pp. 87–95. [Google Scholar]
- Parthasarathi, R.; Subramanian, V.; Royb, D.R.; Chattarajb, P.K. Electrophilicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 2004, 12, 5533–5543. [Google Scholar] [CrossRef] [PubMed]
- Pasha, F.A.; Cho, S.J.; Beg, Y.; Tripathi, Y.B. Quantum chemical QSAR study of flavones and their radical-scavenging activity. Med. Chem. Res. 2008, 16, 408–417. [Google Scholar] [CrossRef]
- Ray, S.; Sengupta, C.; Roy, K. QSAR modeling of antiradical and antioxidant activities of flavonoids using electrotopological state (E-State) atom parameters. Cen. Eur. J. Chem. 2007, 5, 1094–1113. [Google Scholar] [CrossRef]
- Xue, Y.; Zheng, Y.; An, L.; Zhang, L.; Qian, Y.; Yu, D.; Gong, X. A theoretical study of the structure–radical scavenging activity of hydroxychalcones. Comp. Theor. Chem. 2012, 982, 74–83. [Google Scholar] [CrossRef]
- Sarkar, A.; Middya, T.R.; Jana, A.D. A QSAR study of radical scavenging antioxidant activity of a series of flavonoids using DFT based quantum chemical descriptors—The importance of group frontier electron density. J. Mol. Model. 2011, 18, 2621–2631. [Google Scholar] [CrossRef] [PubMed]
- Asadollahi, T.; Dadfarnia, S.; Mohammad, A.; Shabani, H.; Ghasemi, J.B.; Sarkhosh, M. QSAR Models for CXCR2 Receptor Antagonists Based on the Genetic Algorithm for Data Preprocessing Prior to Application of the PLS Linear Regression Method and Design of the New Compounds Using In Silico Virtual Screening. Molecules 2011, 16, 1928–1955. [Google Scholar] [CrossRef] [PubMed]
- Terfloth, L.; Johann, G. Neural networks and genetic algorithms in drug design. Drug Discov. Today 2001, 6, 102–108. [Google Scholar] [CrossRef]
- Nevillers, J. Strengths and weaknesses of the backpropagation neural network in QSAR and QSPR studies. In Neural Networks in QSAR and Drug Design, 1st ed.; Nevillers, J., Ed.; Academic Press Inc.: San Diego, CA, USA, 1996; pp. 1–20. [Google Scholar]
- Gasteiger, J.; Zupan, J. Neural Networks in Chemistry. Angew. Chem. Int. Ed. 1993, 32, 503–527. [Google Scholar] [CrossRef]
- Bonsignore, L.; Cottiglia, F.; Maccioni, A.; Secci, D. Shynthesis of coumarin-3-O-acylisoureas by dicyclohexylcarboiimide. J. Heterocycl. Chem. 1995, 32, 573–577. [Google Scholar] [CrossRef]
- Martínez-Martínez, F.J.; Padilla-Martínez, I.I.; Trujillo-Ferrara, J. 1H and 13C NMR assignments of 2-oxo-2H-1-benzopyran-3-acyl and 3-amide derivatives. Magn. Res. Chem. 2001, 39, 765–776. [Google Scholar] [CrossRef]
- García-Báez, E.V.; Martínez-Martínez, F.J.; Höpfl, H.; Padilla-Martínez, I.I. π-Stacking interactions and C—H···X (X = O, aryl) hydrogen bonding as directing features of the supramolecular self-association in 3-carboxy and 3-amido coumarin derivatives. Cryst. Growth Des. 2003, 3, 35–45. [Google Scholar] [CrossRef]
- Morales, F.J.; Jiménez-Pérez, S. Free radical scavenging capacity of Maillard reaction products as related to color and fluorescence. Food Chem. 2001, 72, 119–125. [Google Scholar] [CrossRef]
- Molyneux, P. The use of the stable free radical diphenylpicrylhydrazyl (DPPH) for estimating antioxidant activity. Songklanakarin J. Sci. Technol. 2004, 26, 211–219. [Google Scholar]
- Hsiu-Chen, L.; Shin-Hui, T.; Chien-Shu, C.; Yuan-Ching, C.; Chi-Ming, L.; Zhi-Yang, L.; Chun-Mao, L. Structure–activity relationship of coumarin derivatives on xanthine oxidase-inhibiting and free radical-scavenging activities. Bioch. Pharm. 2008, 75, 1416–1425. [Google Scholar]
- Hehre, W. A Guide to Molecular Mechanics and Quantum Chemical Calculations; Wavefunction Inc.: Irvine, CA, USA, 2003. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 2004, 10, 209–220. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- DRAGON for Windows, Version 5. Software for Molecular Descriptor Calculations. Talete srl: Milano, Italy, 2006.
- Thomsen, R.; Christensen, M. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.D.; Barlow, T.W.; Richards, W.G. Reduced Dimensional Representations of Molecular Structure. J. Chem. Inf. Comput. Sci. 1997, 37, 939–942. [Google Scholar] [CrossRef]
- Todeschini, R.; Vighi, M.; Finizio, A.; Gramatica, P. 3D-modelling and prediction by WHIM descriptors. Part 8. Toxicity and physico-chemical properties of environmental priority chemicals by 2D-TI and 3D-WHIM descriptors. SAR. QSAR. Environ. Res. 1997, 7, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K; Crippen, G.M. Atomic Physicochemical Parameters for Three-Dimensional-Structure-Directed Quantitative Structure-Activity Relationships. 2. Modeling Dispersive and Hydrophobic Interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Roy, D.R. Update 1 of: Electrophilicity Index. Chem. Rev. 2007, 107, PR46–PR74. [Google Scholar] [CrossRef]
- NeuroShell Predictor Reference Manual, Ward Systms Group, Inc.: Frederick, MD, USA, September 2008.
- Goldberg, D.E. Genetic Algorithms in Search, Optimization, and Machine Learning; Addison-Wesley: Boston, MA, USA, 1989. [Google Scholar]
- Roy, K.; Mitra, I.; Kar, S.; Ojha, P.K.; Das, R.N.; Kabir, H. Comparative studies on some metrics for external validation of QSPR models. J. Chem. Inf. Model. 2012, 52, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Ojha, P.K.; Mitra, I.; Das, R.N.; Roy, K. Further exploring rm2 metrics for validation of QSPR models. Chemom. Intell. Lab. Syst. 2011, 107, 194–205. [Google Scholar] [CrossRef]
- Mladenović, M.; Mihailović, M.; Bogojević, D.; Matić, S.; Nićiforović, N. In Vitro Antioxidant Activity of Selected 4-Hydroxy-chromene- 2-one Derivatives—SAR, QSAR and DFT Studies. Int. J. Mol. Sci. 2011, 12, 2822–2841. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds A1–D2 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R2 | R1 | % DPPH• inhibition | Log Yexp |
|---|---|---|---|---|
| A1 | COOCH3 | H | 13.42 | 1.13 |
| B1 | COOCH3 | NO2 | 14.43 | 1.16 |
| C1 | COOCH3 | OH | 40.67 | 1.61 |
| D1 | COOCH3 | OCH3 | 9.39 | 0.97 |
| A2 | COOEt | H | 8.06 | 0.91 |
| B2 | COOEt | NO2 | 8.10 | 0.91 |
| C2 | COOEt | OH | 56.39 | 0.75 |
| D2 | COOEt | OCH3 | 11.41 | 1.06 |
| PROGRAM | DESCRIPTOR | TYPE | DESCRIPTION |
|---|---|---|---|
| DRAGON | SPH | Geometrical | Spherosity |
| DRAGON | Ui | Molecular Properties | Unsaturation index |
| DRAGON | Hy | Molecular Properties | Hydrophilic factor |
| DRAGON | AMR | Molecular Properties | Molar refractivity |
| DRAGON | ALOGP | Molecular Properties | Ghose-Crippen-Viswanadhan octanol-water partition coefficient |
| DRAGON | TPSA | Molecular Properties | Topological Polar Surface Area |
| GAUSSIAN | E | Quantum-Chemical | Total Energy |
| GAUSSIAN | Dipole | Quantum-Chemical | Dipole Moment |
| GAUSSIAN | η | Quantum-Chemical | Hardness |
| GAUSSIAN | ω | Quantum-Chemical | Electrophilicity |
| GAUSSIAN | µ | Quantum-Chemical | Chemical potential |
| GAUSSIAN | S | Quantum-Chemical | Softness |
| GAUSSIAN | Gap HOMO-LUMO | Quantum-Chemical | Energy difference LUMO-HOMO |
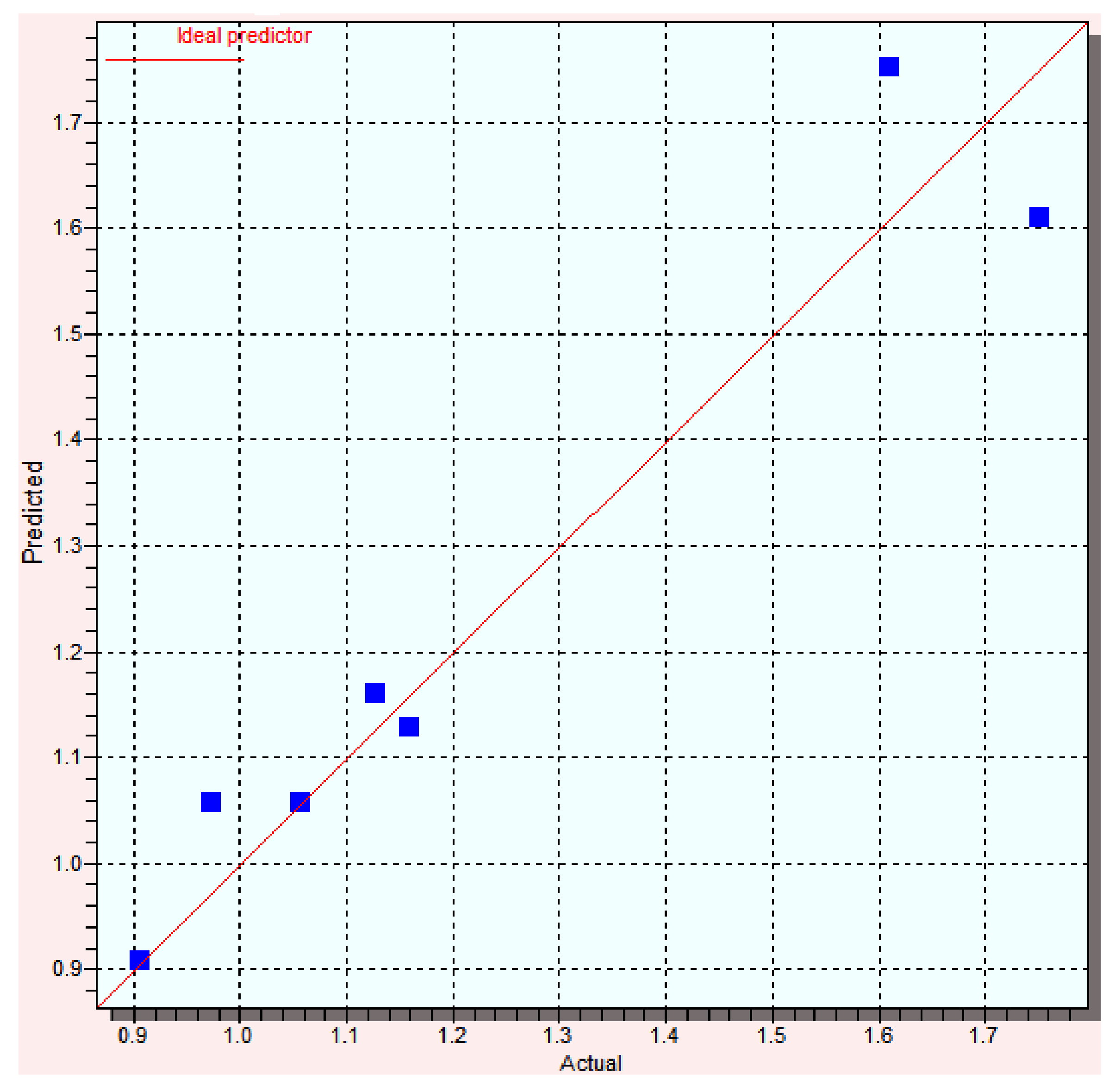
| Compound | Log Yexp | Log Ypred | Error | % Error |
|---|---|---|---|---|
| A1 | 1.13 | 1.16 | −0.03 | 2.65 |
| B1 | 1.16 | 1.13 | 0.03 | 2.58 |
| C1 | 1.61 | 1.75 | −0.14 | 8.69 |
| D1 | 0.97 | 1.05 | −0.08 | 8.24 |
| A2 | 0.91 | 0.91 | 0.00 | 0.00 |
| B2 | 0.91 | 0.91 | 0.00 | 0.00 |
| C2 | 1.75 | 1.61 | 0.14 | 8.00 |
| D2 | 1.06 | 1.06 | 0.00 | 0.00 |
| Compound | Hy |
|---|---|
| A1 | −0.766 |
| B1 | −0.634 |
| C1 | −0.200 |
| D1 | −0.734 |
| A2 | −0.734 |
| B2 | −0.621 |
| C2 | −0.198 |
| D2 | −0.709 |
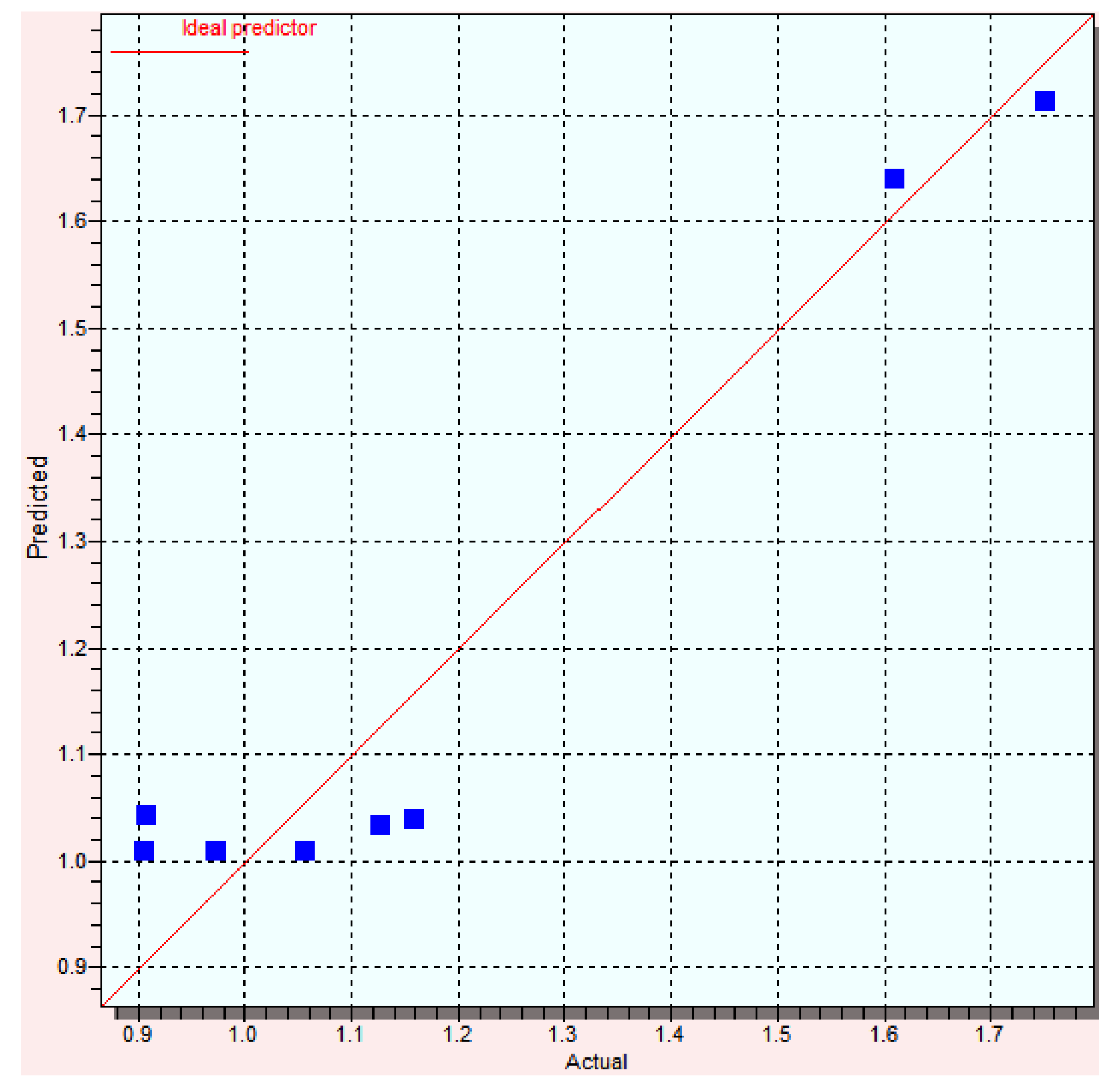
| Compound | Log Yexp | Log Ypred | Error | % Error |
|---|---|---|---|---|
| 1 | 1.13 | 1.03 | 0.1 | 8.85 |
| B1 | 1.16 | 1.04 | 0.12 | 10.34 |
| C1 | 1.61 | 1.64 | −0.03 | 1.86 |
| D1 | 0.97 | 1.01 | −0.04 | 4.12 |
| A2 | 0.91 | 1.01 | −0.1 | 10.99 |
| B2 | 0.91 | 1.04 | −0.13 | 14.29 |
| C2 | 1.75 | 1.71 | 0.04 | 2.29 |
| D2 | 1.06 | 1.01 | 0.05 | 4.72 |
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martínez-Martínez, F.J.; Razo-Hernández, R.S.; Peraza-Campos, A.L.; Villanueva-García, M.; Sumaya-Martínez, M.T.; Cano, D.J.; Gómez-Sandoval, Z. Synthesis and in Vitro Antioxidant Activity Evaluation of 3-Carboxycoumarin Derivatives and QSAR Study of Their DPPH• Radical Scavenging Activity. Molecules 2012, 17, 14882-14898. https://doi.org/10.3390/molecules171214882
Martínez-Martínez FJ, Razo-Hernández RS, Peraza-Campos AL, Villanueva-García M, Sumaya-Martínez MT, Cano DJ, Gómez-Sandoval Z. Synthesis and in Vitro Antioxidant Activity Evaluation of 3-Carboxycoumarin Derivatives and QSAR Study of Their DPPH• Radical Scavenging Activity. Molecules. 2012; 17(12):14882-14898. https://doi.org/10.3390/molecules171214882
Chicago/Turabian StyleMartínez-Martínez, Francisco J., Rodrigo Said Razo-Hernández, Ana Lilia Peraza-Campos, Manuel Villanueva-García, Maria Teresa Sumaya-Martínez, Daniel Jaramillo Cano, and Zeferino Gómez-Sandoval. 2012. "Synthesis and in Vitro Antioxidant Activity Evaluation of 3-Carboxycoumarin Derivatives and QSAR Study of Their DPPH• Radical Scavenging Activity" Molecules 17, no. 12: 14882-14898. https://doi.org/10.3390/molecules171214882
APA StyleMartínez-Martínez, F. J., Razo-Hernández, R. S., Peraza-Campos, A. L., Villanueva-García, M., Sumaya-Martínez, M. T., Cano, D. J., & Gómez-Sandoval, Z. (2012). Synthesis and in Vitro Antioxidant Activity Evaluation of 3-Carboxycoumarin Derivatives and QSAR Study of Their DPPH• Radical Scavenging Activity. Molecules, 17(12), 14882-14898. https://doi.org/10.3390/molecules171214882