Antioxidant, Lipoxygenase and Histone Deacetylase Inhibitory Activities of Acridocarpus orientalis From Al Ain and Oman

Abstract
:1. Introduction
2. Results and Discussion
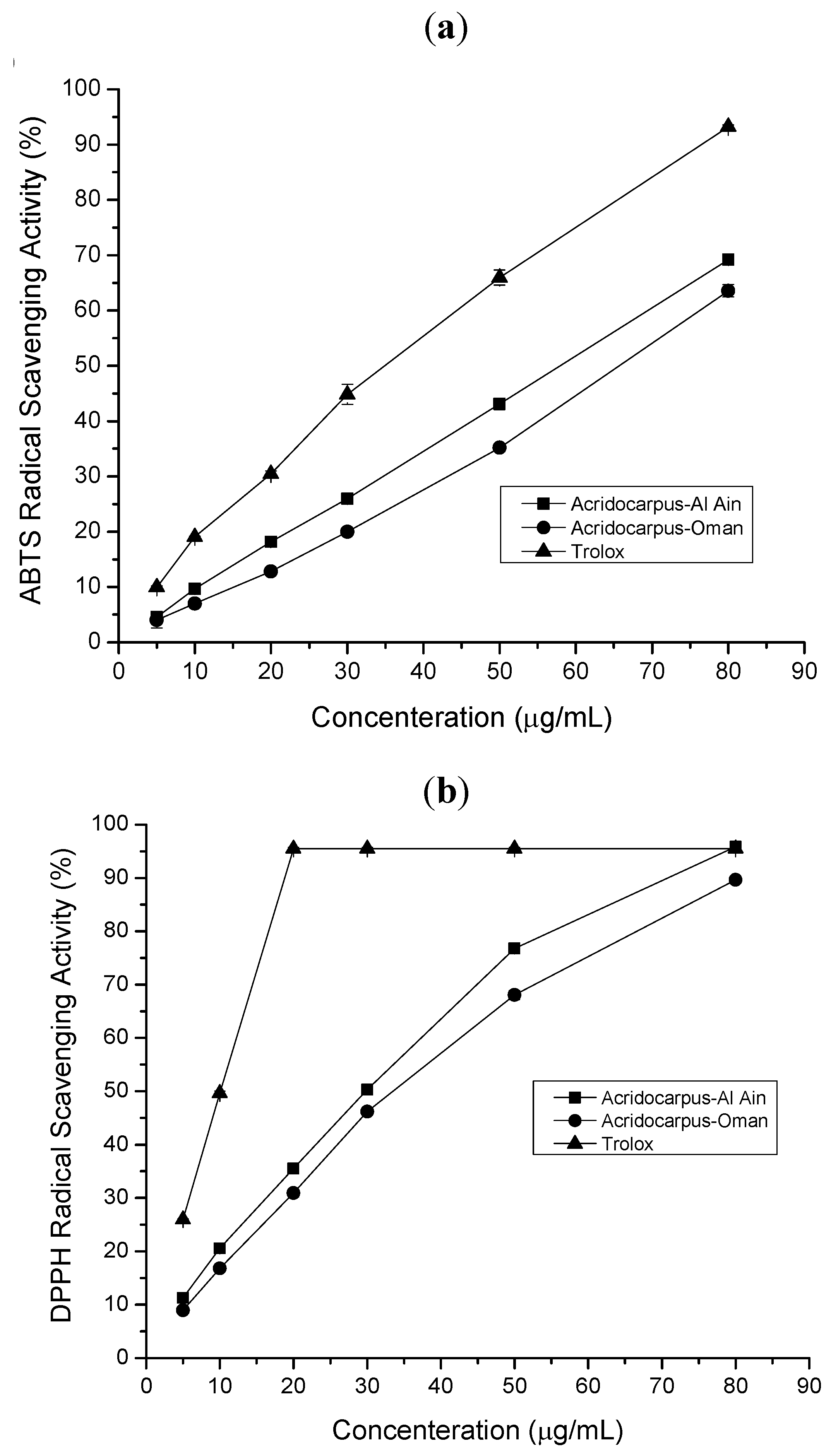
2.1. Antioxidant, Free Radical Scavenging Activity

{kind=link}
{kind=link}
{kind=link}
| Extract | FRAP Assay | ABTS Assay | DPPH Assay | β-Carotene Assay | ||
|---|---|---|---|---|---|---|
| TAC (mmol/g) | TAC (mmol/g) | IC50 (µg/mL) | TAC (mmol/g) | IC50 (µg/mL) | IC50 (µg/mL) | |
| Acridocarpus-Al Ain (a) | 1.10 ± 0.01 | 1.04 ± 0.02 | 58.06 ± 1.39 b,c | 1.14 ± 0.01 | 29.84 ± 0.59 b,c | 5.0 ± 0.04 b,c |
| Acridocarpus-Oman (b) | 0.96 ± 0.02 | 0.98 ± 0.01 | 72.32 ± 1.61 a,c | 1.04 ± 0.36 | 32.44 ± 0.34 a,c | 6.32 ± 0.04 a,c |
| Trolox (c) | 3.86 ± 0.09 a,b | 1.83 ± 0.45 | 33.58 ± 1.43 a,b | 3.99 ± 0.03 | 10.07 ± 0.09 a,b | 3.03 ± 0.04 a,b |
2.1.1. FRAP Assay
2.1.2. ABTS Radical Scavenging Assay
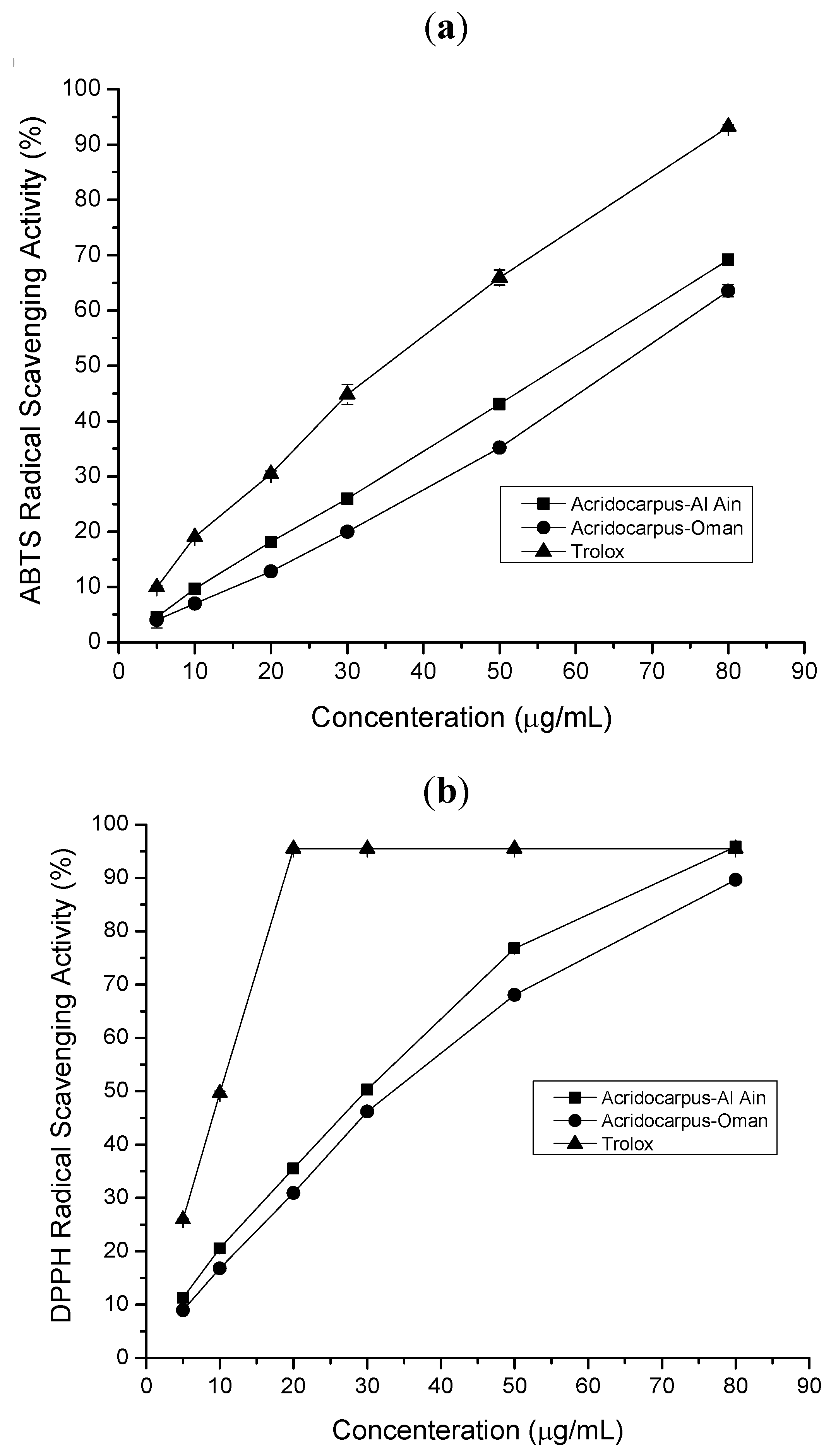
2.1.3. DPPH Radical Scavenging Assay
2.1.4. β-Carotene Bleaching Test
2.2. Total Phenolic Content
| Extract | Yield (%) | Total Phenolic Content * (mg/g) |
|---|---|---|
| Acridocarpus-Al Ain (a) | 30% | 184.24 ± 4.39 b |
| Acridocarpus-Oman (b) | 22% | 149.23 ± 2.84 a |
2.3. Correlation between Antioxidant Activity and Phenolic Contents
| Assay | Correlation (R2) | Significance |
|---|---|---|
| FRAP activity | 0.93 | p < 0.01 |
| ABTS• scavenging activity | 0.94 | p < 0.01 |
| DPPH• scavenging activity | 0.87 | p < 0.01 |
| β-Carotene bleaching inhibition | 0.99 | p < 0.001 |
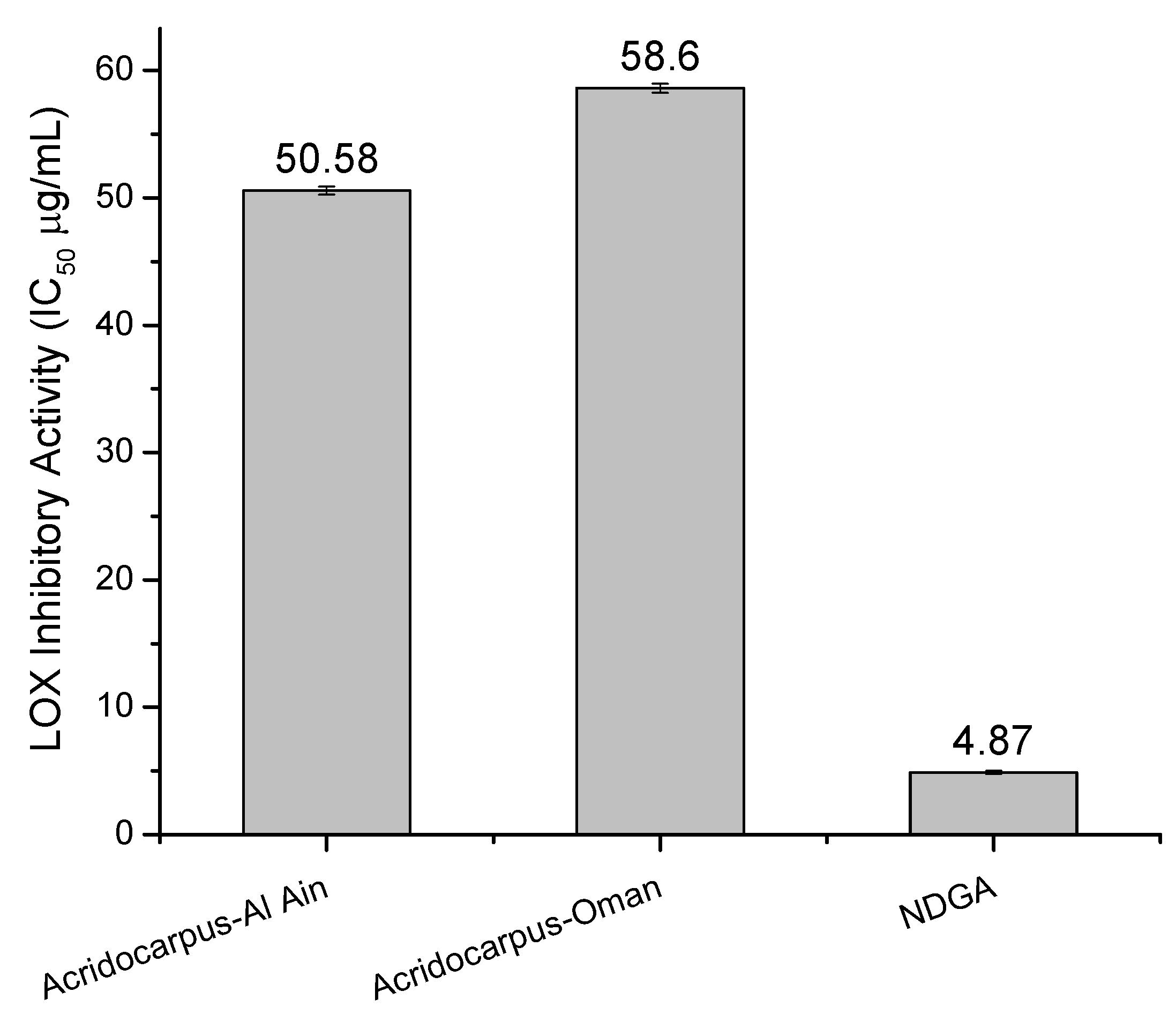
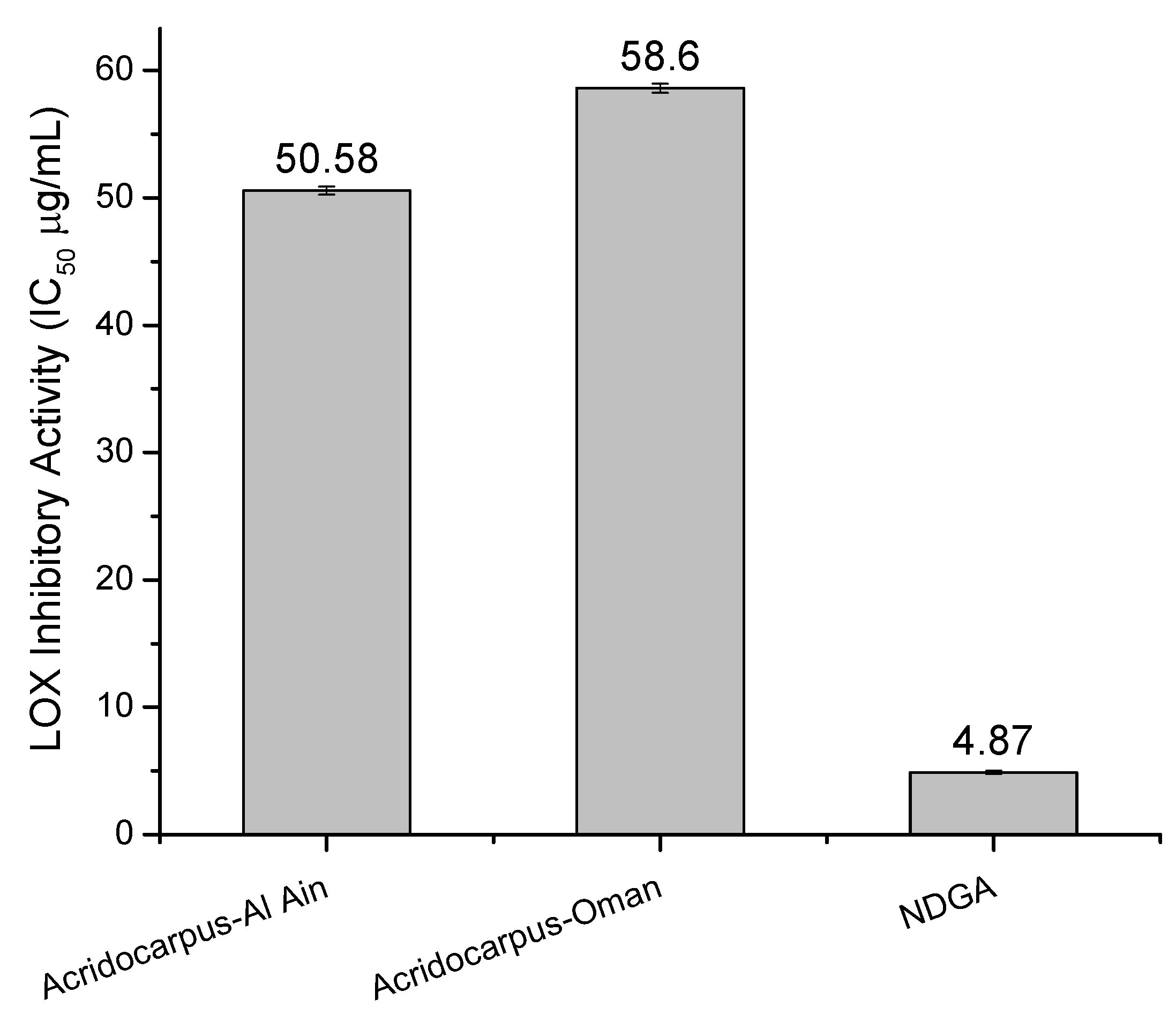
2.4. LOX Inhibitory Assay
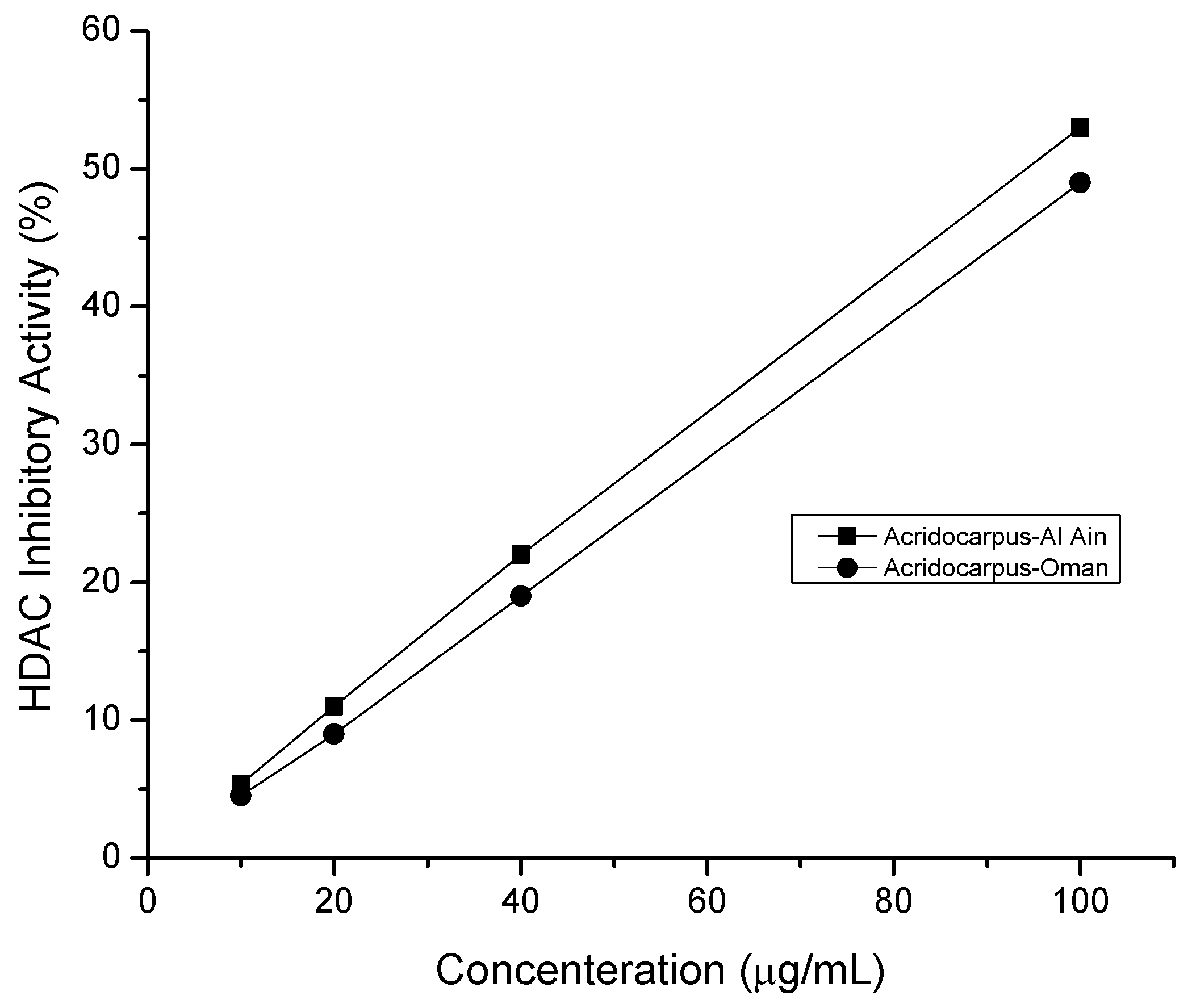
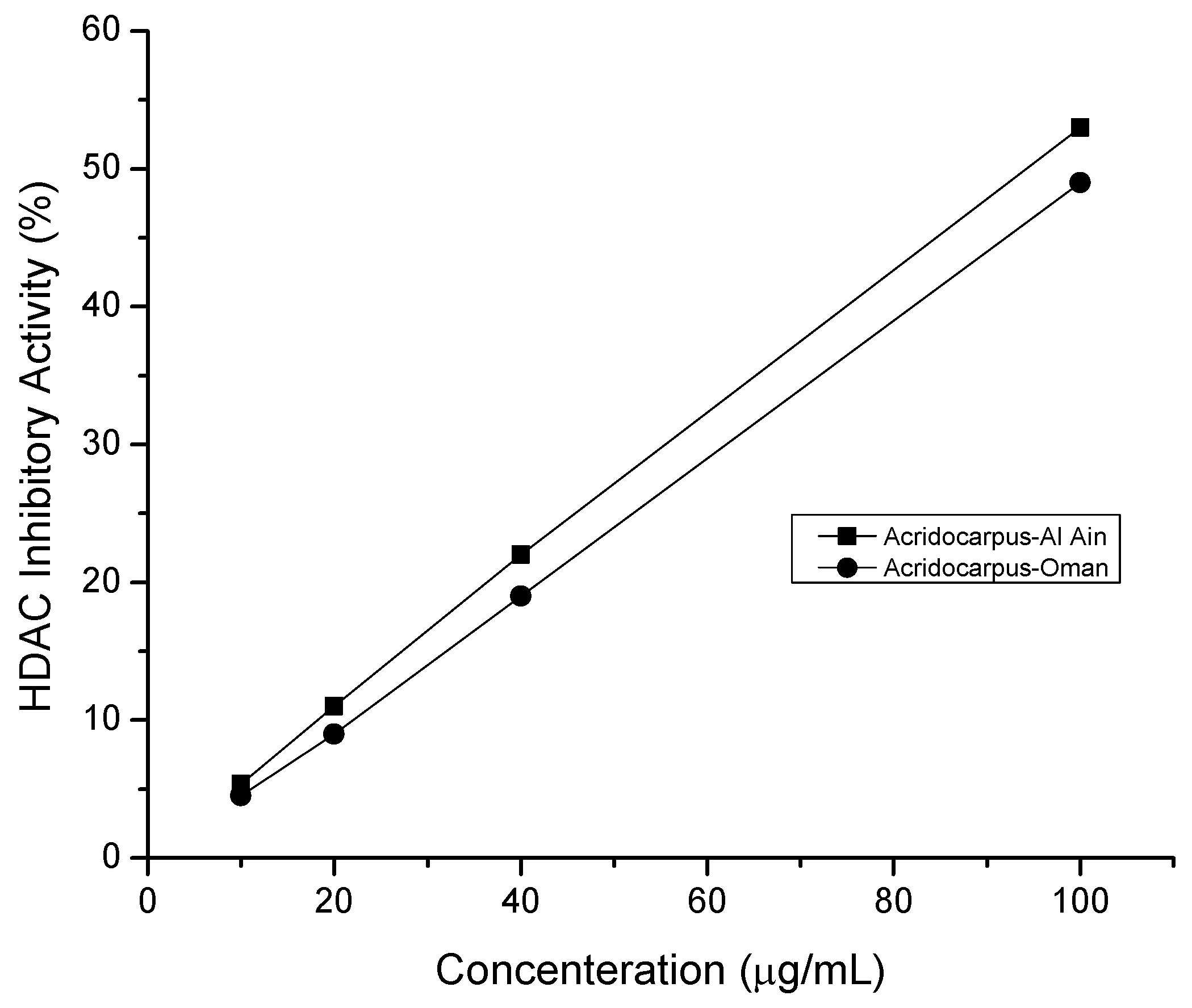
2.5. HDAC Inhibitory Assay
3. Experimental
3.1. Chemicals
3.2. Plant Materials
3.3. Preparation of the Plant Extracts
3.4. Total Phenolic Content Determination
3.5. Estimation of Total Antioxidant Activity
3.5.1. FRAP assay
3.5.2. ABTS Assay
3.5.3. DPPH• Radical Assay
3.5.4. β-Carotene Bleaching Assay
3.6. LOX Inhibitory Assay
3.7. HDAC Inhibition Activity Screening
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Williams, R.J.; Spencer, J.P.E.; Rice-Evans, C. Flavonoids: Antioxidants or signalling molecules? Free Radic. Biol. Med. 2004, 36, 838–849. [Google Scholar] [CrossRef]
- Soobrattee, M.A.; Neergheen, V.S.; Luximon-Ramma, A.; Aruoma, O.; Bahorun, T. Phenolics as potential antioxidant theraputic agents: Mechanism and actions. Mut. Res. 2005, 579, 200–213. [Google Scholar] [CrossRef]
- Mitjavila, M.T.; Moreno, J.J. The effects of polyphenols on oxidative stress and the arachidonic acid cascade: Implications for the prevention/treatment of high prevalence diseases. Biochem. Pharmacol. 2012, 84, 1113–1122. [Google Scholar] [CrossRef]
- Moreno, J.J. New aspects of the role of hydroxyeicosatetraenoic acids in cell growth and cancer development. Biochem. Pharmacol. 2009, 77, 1–10. [Google Scholar] [CrossRef]
- Dobrian, A.D.; Lieb, D.C.; Cole, B.K.; Taylor-Fishwick, D.A.; Chakrabarti, S.K.; Nadler, J.L. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog. Lipid. Res. 2011, 50, 115–131. [Google Scholar] [CrossRef]
- Pontiki, E.; Hadjipavlou-Litina, H. Histone deacetylase inhibitors (HDACIs). Structure-activity relationships: History and new QSAR perspectives. Med. Res. Rev. 2012. [Google Scholar] [CrossRef]
- Mercurio, C.; Minucci, S.; Pelicci, P.G. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol. Res. 2010, 62, 18–34. [Google Scholar] [CrossRef]
- Shah, M.H.; Binkley, P.; Chan, K.; Xiao, J.; Arbogast, D.; Collamore, M.; Farra, Y.; Young, D.; Grever, M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin. Cancer Res. 2006, 12, 3997–4003. [Google Scholar] [CrossRef]
- Walkinshaw, D.R.; Yang, X.J. Histone deacetylase inhibitors as novel anticancer therapeutics. Curr.Oncol. 2008, 15, 237–243. [Google Scholar]
- Kisksi, T.; Guenaoui, C.; Fawzi, N. Early growth stages of the rare Acridocarpus orientalis in the UAE-A First step towards conservation. Nat. Resour. 2012, 3, 1–5. [Google Scholar]
- Monthana, R.A.; Lindequist, U.; Gruenert, R.; Bednarski, P.J. Studies of the in vitro anticancer, antimicrobial and antioxidant potentials of selected Yemeni medicinal plants from the island Soqotra. BMC Complem. Altern. Med. 2009. [Google Scholar] [CrossRef]
- Hammiche, V.; Maiza, K. Traditional medicine in central sahara:Pharmacopoeia of Tassili N’ajjer. J. Ethnopharmacol. 2006, 105, 358–367. [Google Scholar] [CrossRef]
- Malebo, H.M.; Tanja, W.; Cal, M.; Swaleh, S.A.M.; Omolo, M.O.; Hassanali, A.; Séquin, U.; Hamburger, M.; Brun, R.; Ndiege, I.O. Antiplasmodial, anti-trypanosomal, anti-leishmanial and cytotoxicity activity of selected Tanzanian medicinal plants. Tanzan. J. Health Res. 2009, 11, 226–234. [Google Scholar]
- Khasawneh, M.A.; Elwy, H.M.; Hamza, A.A.; Fawzi, N.M.; Hassan, A.H. Antioxidant, anti-lipoxygenase and cytotoxic activity of Leptadenia pyrotechnica (Forssk.) decne polyphenolic constituents. Molecules 2011, 16, 7510–7521. [Google Scholar]
- Yang, J.; Liu, R.H.; Halim, L. Antioxidant and antiproliferative activities of common edible nut seeds. LWT-Food Sci. Technol. 2009, 42, 1–8. [Google Scholar]
- Lim, Y.Y.; Quah, E.P.L. Antioxidant properties of different cultivars of Portulaca oleracea. Food Chem. 2007, 103, 734–740. [Google Scholar] [CrossRef]
- Kosar, M.; Dorman, H.J.D.; Hiltunen, R. Effect of an acid treatment on the phytochemical and antioxidant characteristics of extracts from selected Lamiaceae species. Food Chem. 2005, 91, 525–533. [Google Scholar] [CrossRef]
- Conforti, F.; Sosa, S.; Marrelli, M.; Menichini, F.; Statti, G.A.; Uzunov, D.; Tubaro, A.; Menichini, F.; Loggia, R.L. In vivo anti-inflammatory and in vitro antioxidant activities of mediterranean dietary plants. J. Ethnopharmacol. 2008, 116, 144–151. [Google Scholar] [CrossRef]
- Conforti, F.; Sosa, S.; Marrelli, M.; Menichini, F.; Statti, G.A.; Uzunov, D.; Tubaro, A.; Menichini, F. The protective ability of mediterranean dietary plants against the oxidative damage: The role of radical oxygen species in inflammation and the polyphenol, flavonoid and sterol contents. Food Chem. 2009, 112, 587–594. [Google Scholar] [CrossRef]
- Wojdylo, A.; Oszmiański, J.; Czemerys, R. Antioxidant activity and phenolic compounds in 32 selected herbs. Food Chem. 2007, 105, 940–949. [Google Scholar] [CrossRef]
- Kumaraswamy, M.V.; Satish, S. Antioxidant and anti-lipoxygenase activity of Thespesia lampas Dalz and Gibs. Advan. Biol. Res. 2008, 2, 56–59. [Google Scholar]
- Bora-Tatar, G.; Dayangac-Erden, D.; Demir, A.S.; Dalkara, S.; Yelekci, K.; Erdem-Yurter, H. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorg. Med. Chem. 2009, 17, 5219–5228. [Google Scholar] [CrossRef]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.W.; Wuth, B.; Williams, D.E.; Ho, E.; Dashwood, R.H. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: Competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol. Cancer 2011, 10, 1–18. [Google Scholar] [CrossRef]
- Singleton, V.L.; Orthofer, R.; Lamuela-raventos, R.M. Analysis of total phenols and other oxidation substrates and antioxidants by means of folin-ciocalteu reagent. Meth. Enzymol. 1999, 299, 152–178. [Google Scholar] [CrossRef]
- Nenadis, N.; Lazaridou, O.; Tsimidou, M.Z. Use of reference compounds in antioxidant activity assessment. J. Agric. Food. Chem. 2007, 55, 5452–5460. [Google Scholar] [CrossRef]
- Wu, H. Affecting the activity of soybean lipoxygenase-1. J. mol. Graph. 1996, 14, 331–337. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ksiksi, T.; Hamza, A.A. Antioxidant, Lipoxygenase and Histone Deacetylase Inhibitory Activities of Acridocarpus orientalis From Al Ain and Oman. Molecules 2012, 17, 12521-12532. https://doi.org/10.3390/molecules171112521
Ksiksi T, Hamza AA. Antioxidant, Lipoxygenase and Histone Deacetylase Inhibitory Activities of Acridocarpus orientalis From Al Ain and Oman. Molecules. 2012; 17(11):12521-12532. https://doi.org/10.3390/molecules171112521
Chicago/Turabian StyleKsiksi, Taoufik, and Alaaeldin A. Hamza. 2012. "Antioxidant, Lipoxygenase and Histone Deacetylase Inhibitory Activities of Acridocarpus orientalis From Al Ain and Oman" Molecules 17, no. 11: 12521-12532. https://doi.org/10.3390/molecules171112521
APA StyleKsiksi, T., & Hamza, A. A. (2012). Antioxidant, Lipoxygenase and Histone Deacetylase Inhibitory Activities of Acridocarpus orientalis From Al Ain and Oman. Molecules, 17(11), 12521-12532. https://doi.org/10.3390/molecules171112521