New Sorafenib Derivatives: Synthesis, Antiproliferative Activity Against Tumour Cell Lines and Antimetabolic Evaluation

Abstract
:1. Introduction
2. Results and Discussion
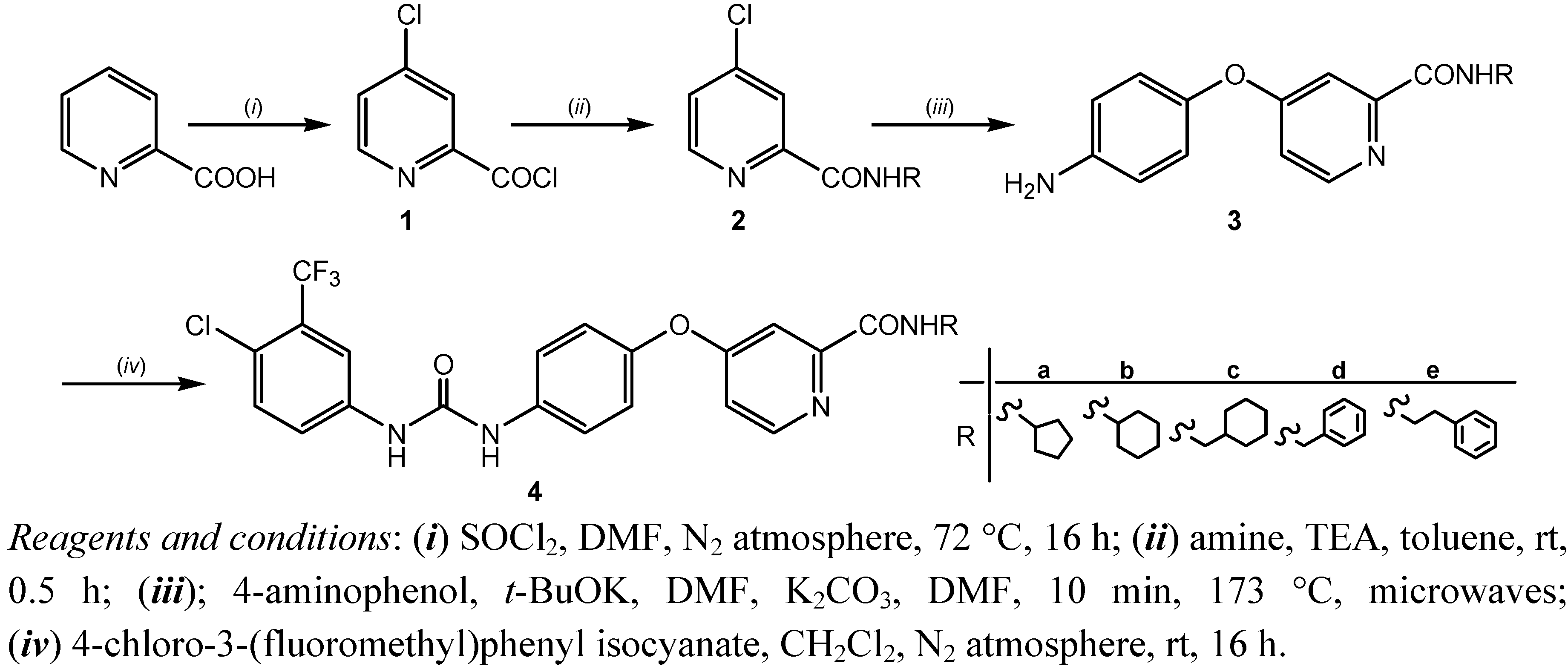
2.1. Chemistry
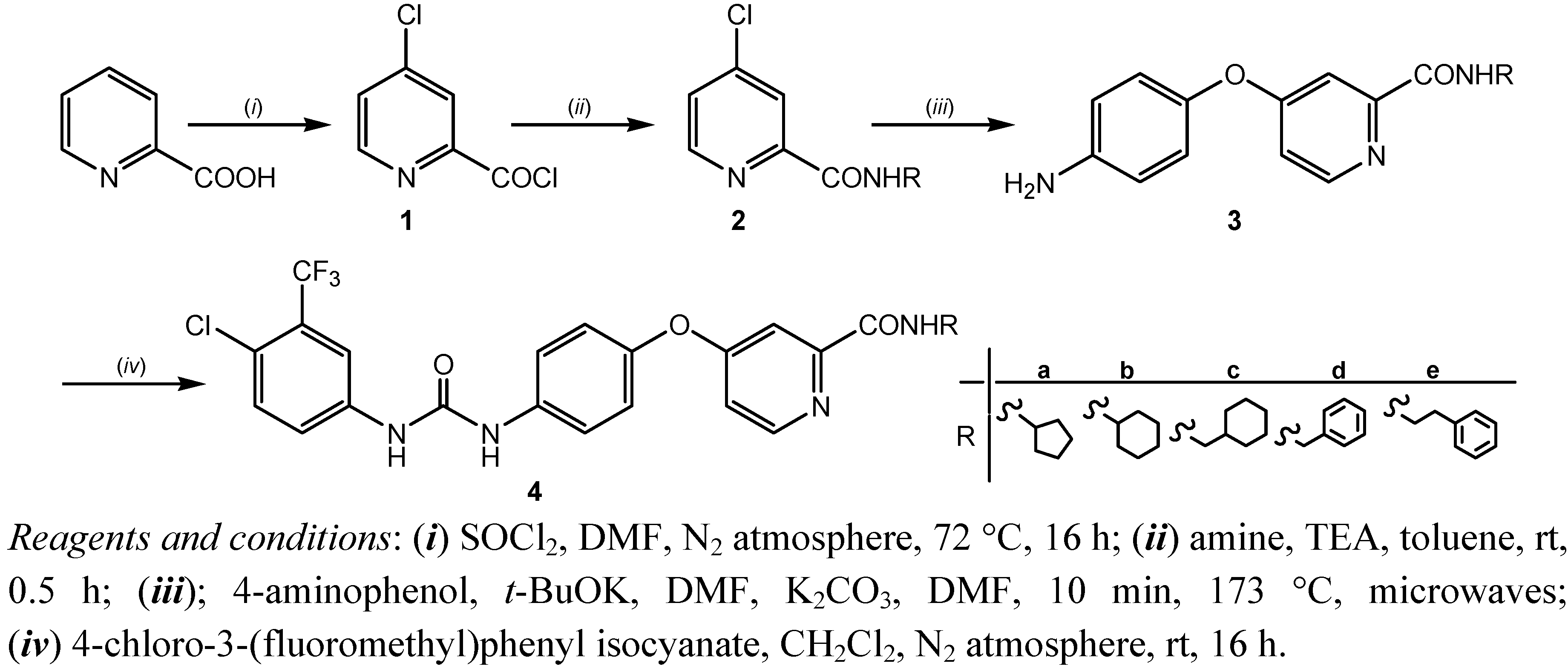

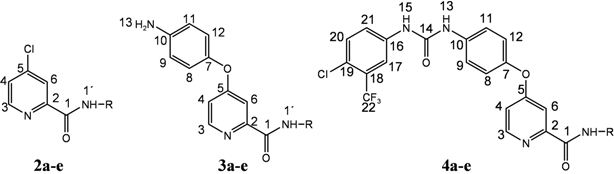
{kind=link}
{kind=link}
| Compd. | R | 1H and 13C-NMR spectra/δ ppm, J/Hz |
|---|---|---|
| 2a |  | 8.62 (d, 1H, 3, J = 5.4), 8.58 (d, 1H, 1', J = 7.6), 8.02 (d, 1H, 6, J = 1.8), 7.75 (dd, 1H, 4, J = 2.1, J = 3.2), 4.31–4.19 (m, 1H, 2'), 1.94–1.53 (m, 8H, 3'–6') |
| 161.26 (1), 151.02 (2), 149.02 (3), 143.61 (5), 125.36 (6), 120.98 (4), 49.82 (2'), 31.12 (3', 6'), 22.62 (4', 5') | ||
| 2b | 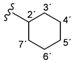 | 8.61 (d, 1H, 3, J = 5.2), 8.48 (d, 1H, 1', J = 8.1), 8.01 (d, 1H, 6, J = 1.9), 7.74 (dd, 1H, 4, J = 1.9, J = 3.2), 3.80–3.74 (m, 1H, 2'), 1.79–1.16 (m, 10H, 3'–7') |
| 161.54 (1), 151.90 (2), 149.91 (3), 144.53 (5), 126.28 (6), 121.90 (4), 48.13 (2'), 32.03 (3', 7'), 25.03 (5'), 24.73 (4', 6') | ||
| 2c | 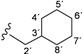 | 8.82 (t, 1H, 1', J = 8.8), 8.62 (d, 1H, 3, J = 8.6), 8.02 (d, 1H, 6, J = 1.8), 7.76 (dd, 1H, 4, J = 2.1, J = 3.1), 3.16 (t, 2H, 2', J = 6.6), 1.68–0.97 (m, 11H, 3'–8') |
| 163.08 (1), 152.37 (2), 150.46 (3), 145.00 (5), 126.76 (6), 122.39 (4), 45.52 (2'), 30.86 (4', 8'), 26.50 (6'), 25.81 (5', 7') | ||
| 2d | 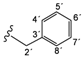 | 9.40 (t, 1H, 1', J = 6.2), 8.62 (d, 1H, 3, J = 5.5), 8.01 (d, 1H, 6, J = 2.0), 7.77 (dd, 1H, 4, J = 2.1, J = 3.3), 7.33–7.21 (m, 5H, arom. 4'–8'), 4.50 (d, 2H, 2', J = 6.1) |
| 163.26 (1), 152.23 (2), 150.57 (3), 145.04 (5), 139.76 (3'), 128.72 (4', 8'), 127.83 (5', 7'), 126.94 (6'), 125.36 (6), 120.98 (4), 42.98 (2') | ||
| 2e | 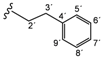 | 8.88 (t, 1H, 1', J = 5.6), 8.61 (d, 1H, 3, J = 5.0), 8.01 (d, 1H, 6, J = 1.8), 7.73 (dd, 1H, 4, J = 2.1, J = 3.2), 3.55–3.52 (q, 2H, 2', J = 7.0), 2.86 (t, 2H, 3', J = 7.5) |
| 162.50 (1), 151.73 (2), 149.98 (3), 144.52 (5), 139.24 (4'), 128.54 (5', 9'), 128.31 (6', 8'), 126.33 (6), 126.08 (7'), 121.86 (4), 40.44 (2'), 34.97 (3') | ||
| 3a | 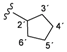 | 8.46 (d, 1H, 3, J = 2.5), 8.44 (d, 1H, 1', J = 8.5), 7.34 (d, 1H, 6, J = 2.4), 7.08 (dd, 1H, 4, J = 2.6, J = 2.9), 6.85 (d, 2H, 8, 12, J = 8.9), 6.66 (d, 2H, 9, 11, J = 8.9), 5.16 (s, 2H, 13), 4.22–4.15 (m, 1H, 2'), 1.99–1.53 (m, 8H, 3'–6') |
| 167.30 (5), 163.31 (1), 152.74 (2), 150.51 (3), 147.32 (7), 143.37 (10), 121.99 (8, 12), 115.36 (9, 11), 114.23 (6), 108.86 (4), 51.04 (2'), 32.56 (3', 6'), 23.95 (4', 5') | ||
| 3b | 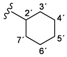 | 8.45 (d, 1H, 3, J = 5.7), 8.37 (d, 1H, 1', J = 8.5), 7.34 (d, 1H, 6, J = 2.4), 7.08 (dd, 1H, 4, J = 2.6, J = 2.9), 6.85 (d, 2H, 8, 12, J = 8.9), 6.65 (d, 2H, 9, 11, J = 8.9), 5.17 (s, 2H, 13), 3.77–3.68 (m, 1H, 2'), 1.78–1.04 (m, 10H, 3'–7') |
| 167.31 (5), 162.70 (1), 152.77 (2), 150.52 (3), 147.30 (7), 143.38 (10), 121.98 (8, 12), 115.37 (9, 11), 114.26 (6), 108.92 (4), 48.38 (2'), 32.58 (3', 7'), 25.54 (5'), 25.18 (4', 6') | ||
| 3c | 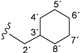 | 8.65 (t, 1H, 1', J = 6.2), 8.45 (d, 1H, 3, J = 5.6), 7.34 (d, 1H, 6, J = 2.3), 7.07 (dd, 1H, 4, J = 2.6, J = 3.1), 6.86 (d, 2H, 8, 12, J = 8.6), 6.65 (d, 2H, 9, 11, J = 8.9), 5.15 (s, 2H, 13), 3.12 (t, 2H, 2', J = 6.6), 1.66–0.84 (m, 11H, 3'–8') |
| 167.31 (5), 163.76 (1), 152.78 (2), 150.54 (3), 147.32 (7), 143.37 (10), 122.00 (8, 12), 115.36 (9, 11), 114.19 (6), 108.90 (4), 45.39 (2'), 37.91 (3'), 30.86 (4', 8'), 26.50 (6'), 25.81 (5', 7') | ||
| 3d | 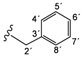 | 9.27 (t, 1H, 1', J = 6.4), 8.47 (d, 1H, 3, J = 5.6), 7.38 (d, 1H, 6, J = 2.4), 7.31–7.21 (m, 5H, 4'–8'), 7.08 (dd, 1H, 4, J = 2.7, J = 2.9), 6.85 (d, 2H, 8, 12, J = 8.8), 6.65 (d, 2H, 9, 11, J = 8.8), 5.16 (s, 2H, 13), 4.46 (d, 2H, 2', J = 6.4) |
| 167.32 (5), 163.96 (1), 152.63 (2), 150.66 (3), 147.33 (7), 143.36 (10), 139.89 (3'), 128.70 (4', 8'), 127.81 (5', 7'), 127.22 (6'), 122.01 (8, 12), 115.38 (9, 11), 114.35 (6), 109.09 (4), 42.91 (2') | ||
| 3e | 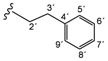 | 8.78 (t, 1H, 1', J = 6.0), 8.44 (d, 1H, 3, J = 5.5), 7.35 (d, 1H, 6, J = 2.6), 7.30–7.18 (m, 5H, 5'–9'), 7.08 (dd, 1H, 4, J = 2.6, J = 3.1), 6.86 (d, 2H, 8, 12, J = 8.7), 6.65 (d, 2H, 9, 11, J = 8.7), 5.17 (s, 2H, 13), 3.52 (q, 2H, 2', J = 7.0), 2.84 (t, 2H, 3', J = 7.5) |
| 166.80 (5), 163.23 (1), 152.14 (2), 150.09 (3), 146.84 (7), 142.83 (10), 139.29 (4'), 128.53 (5', 9'), 128.31 (6', 8'), 126.06 (7'), 121.51 (8, 12), 114.86 (9, 11), 113.73 (6), 108.42 (4), 39.55 (2'), 24.06 (3') | ||
| 4a | 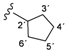 | 9.20 (s, 1H, 15), 8.98 (s, 1H, 13), 8.50 (d, 1H, 3, J = 5.4), 8.47 (d, 1H, 1', J = 6.7), 8.17 (s, 1H, 17), 7.69–7.58 (m, 4H, 20, 21, 8, 12), 7.38 (d, 1H, 6, J = 2.6), 7.18–7.14 (m, 3H, 4, 9, 11), 4.23–4.16 (m, 1H, 2'), 1.99–1.53 (m, 8H, 3'–6') |
| 166.47 (5), 163.21 (1), 152.94 (14), 152.92 (2), 150.75 (3), 148.36 (7), 139.80 (10), 137.52 (16), 132.44 (22), 124.00 (19), 123.58 (20), 122.54 (18), 121.86 (8, 12), 120.99 (9, 11), 117.36 (17), 117.28 (21), 114.59 (6), 109.20 (4), 51.07 (2'), 32.55 (3', 6'), 23.96 (4', 5') | ||
| 4b | 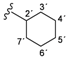 | 9.45 (s, 1H, 15), 9.19 (s, 1H, 13), 8.50 (d, 1H, 3, J = 5.6), 8.40 (d, 1H, 1', J = 8.6), 8.11 (s, 1H, 17), 7.68–7.58 (m, 4H, 20, 21, 8, 12), 7.39 (d, 1H, 6, J = 2.4), 7.18–7.14 (m, 3H, 4, 9, 11), 3.75–3.72 (m, 1H, 2'), 1.78–1.56 (m, 10H, 3'–7') |
| 166.49 (5), 162.61 (1), 152.99 (14), 152.93 (2), 150.77 (3), 148.31 (7), 139.85 (10), 137.56 (16), 132.47 (22), 123.93 (19), 123.45 (20), 122.46 (18), 121.87 (8, 12), 120.84 (9, 11), 117.21 (17), 117.13 (21), 114.59 (6), 109.31 (4), 48.42 (2'), 32.57 (3', 7'), 25.53 (5'), 25.18 (4',6') | ||
| 4c | 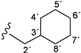 | 9.20 (s, 1H, 15), 8.97 (s, 1H, 13), 8.69 (t, 1H, 1', J = 6.1), 8.51 (d, 1H, 3, J = 5.5), 8.12 (s, 1H, 17), 7.69–7.58 (m, 4H, 20, 21, 8, 12), 7.38 (d, 1H, 6, J = 2.5), 7.18–7.14 (m, 3H, 4, 9, 11), 3.13 (t, 2H, 2', J = 6.4), 1.66–0.78 (m, 11H, 3'–8') |
| 166.49 (5), 163.66 (1), 152.94 (14), 152.92 (2), 150.78 (3), 148.35 (7), 139.80 (10), 137.52 (16), 132.45 (22), 124.04 (19), 123.58 (20), 122.58 (18), 121.90 (8, 12), 120.99 (9, 11), 117.36 (17), 117.29 (21), 114.57 (6), 109.22 (4), 45.41 (2'), 37.90 (3'), 30.86 (4', 8'), 26.50 (6'), 25.81 (5', 7') | ||
| 4d | 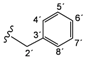 | 9.49 (s, 1H, 15), 9.31 (t, 1H, 1', J = 6.3), 9.22 (s, 1H, 13), 8.54 (d, 1H, 3, J = 5.6), 8.12 (s, 1H, 17), 7.65–7.58 (m, 4H, 20, 21, 8, 12), 7.42 (d, 1H, 6, J = 2.4), 7.31–7.16 (m, 8H, 4, 9, 11, 4'–8'), 4.46 (d, 2H, 2', J = 6.3) |
| 166.50 (5), 163.85 (1), 153.00 (14), 152.80 (2), 150.91 (3), 148.27 (7), 139.86 (10), 137.60 (16), 132.48 (22), 128.70 (4', 8'), 127.83 (5', 7'), 127.23 (6'), 126.66 (3'), 123.99 (19), 123.43 (20), 122.57 (18), 121.91 (8, 12), 120.83 (9, 11), 117.18 (17), 117.11 (21), 114.67 (6), 109.45 (4), 42.92 (2') | ||
| 4e | 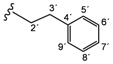 | 9.20 (s, 1H, 15), 8.98 (s, 1H, 13), 8.81 (t, 1H, 1', J = 5.8), 8.50 (d, 1H, 3, J = 5.6), 8.12 (s, 1H, 17), 7.68–7.58 (m, 4H, 20, 21, 8, 12), 7.39 (d, 1H, 6, J = 2.3), 7.31–7.14 (m, 8H, 4, 9, 11, 4'–8'), 3.51 (q, 2H, 2', J = 6.9), 2.84 (t, 2H, 3', J = 7.4) |
| 166.48 (5), 163.62 (1), 152.94 (14), 152.81 (2), 150.83 (3), 148.33 (7), 139.79 (10), 137.53 (16), 132.46 (22), 129.03 (5', 9'), 128.81 (6', 8'), 126.73 (4'), 126.57 (7'), 125.11 (19), 123.60 (20), 122.58 (18), 121.90 (8, 12), 121.01 (9, 11), 117.60 (17, 21), 114.60 (6), 109.24 (4), 40.83 (2'), 35.50 (3') |
2.2. Biological Evaluation
2.2.1. Cytostatic Activity
| Compd. | log P (Clog P) b | Tumour cell growth/IC50 (µmol·L−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HCT 116 | SW 620 | MCF-7 | H 460 | L1210 | CEM | HeLa | HEL | ||
| 2a | 1.95 (2.72) | >100 | >100 | >100 | ≥100 | >100 | >100 | >100 | >100 |
| 2b | 2.37 (3.28) | >100 | >100 | ≥100 | 35 ± 12 | >100 | >100 | >100 | >100 |
| 2c | 2.80 (3.90) | ≥100 | ≥100 | ≥100 | >100 | >100 | >100 | >100 | >100 |
| 2d | 2.55 (3.22) | ≥100 | 71 ± 28 | ≥100 | >100 | ≥100 | ≥100 | >100 | >100 |
| 2e | 2.83 (3.35) | ≥100 | >100 | 57 ± 46 | >100 | >100 | >100 | >100 | >100 |
| 3a | 2.13 (2.80) | 54 ± 21 | ≥100 | 53 ± 40 | >100 | 121 ± 50.5 | ≥100 | 77 ± 3.4 | >100 |
| 3b | 2.54 (3.36) | 37 ± 1 | ≥100 | ≥100 | 65 ± 33 | 234 ± 4.5 | >100 | >100 | >100 |
| 3c | 2.97 (3.98) | ≥100 | ≥100 | ≥100 | >100 | 163 ± 147 | >100 | >100 | >100 |
| 3d | 2.73 (3.30) | 29 ± 4 | 37 ± 6 | 29 ± 13 | 42 ± 9 | 66 ± 3.1 | 94 ± 2.2 | 94 ± 1.9 | >100 |
| 3e | 3.01 (3.43) | 25 ± 2 | 20 ± 7 | 34 ± 17 | 20 ± 0.0 | 63 ± 8.9 | 42 ± 3.0 | 39 ± 3.0 | >100 |
| 4a | 4.89 (6.93) | 3.0 ± 0.5 | 2.0 ± 0.4 | 4.0 ± 1.0 | 1.0 ± 0.0 | 4.2 ± 0.8 | 4.2 ± 0.6 | 3.1 ± 0.6 | 2.8 ± 0.0 |
| 4b | 5.31 (7.49) | 3.0 ± 0.8 | 3.0 ± 0.2 | 2.0 ± 0.6 | 2.0 ± 0.0 | 3.2 ± 1.7 | 3.6 ± 0.9 | 3.0 ± 0.2 | 3.2 ± 0.2 |
| 4c | 5.73 (8.11) | 3.0 ± 1.0 | 3.0 ± 0.4 | 2.0 ± 0.1 | 2.0 ± 0.2 | 1.8 ± 0.3 | 2.0 ± 0.5 | 2.7 ± 1.8 | 2.9 ± 0.0 |
| 4d | 5.49 (7.42) | 2.0 ± 0.8 | 2.0 ± 0.08 | 2.0 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.4 | 4.3 ± 0.4 | 2.6 ± 0.6 | 3.1 ± 0.2 |
| 4e | 5.77 (7.55) | 3.0 ± 0.7 | 2.0 ± 0.4 | 2.0 ± 0.1 | 2.0 ± 0.2 | 2.2 ± 0.2 | 2.5 ± 0.9 | 1.7 ± 0.2 | 4.6 ± 0.36 |
| Sorafenib | 3.76 (5.46) | 3.0 ± 0.8 | 6.0 ± 1.0 | 3.0 ± 0.7 | 3.0 ± 0.8 | 4.1 ± 0.0 | 3.4 ± 0.5 | 3.7 ± 0.6 | 7.5 ± 0.7 |
2.2.2. Antimetabolic Activity
| CEM | IC50 (µmol·L−1) | ||
|---|---|---|---|
| [CH3-3H]dThd | [5-3H]Urd | [4,5-3H]Leu | |
| 2a | >100 | >100 | >100 |
| 2c | >100 | >100 | >100 |
| 2e | >100 | >100 | >100 |
| 3a | >100 | >100 | >100 |
| 3c | >100 | 92.1 ± 43.0 | 70.6 ± 27.6 |
| 3e | >100 | 49.9 ± 17.9 | >100 |
| 4a | 12.5 ± 6.7 | 7.7 ± 2.1 | 6.1 ± 0.7 |
| 4c | 20.1 ± 10.9 | 12.2 ± 8.4 | 27.4 ± 12.7 |
| 4e | 12.7 ± 9.0 | 6.6 ± 1.8 | 11.8 ± 7.7 |
| Sorafenib | 8.6 ± 1.1 | 7.1 ± 0.2 | 12.0 ± 7.0 |
2.2.3. Uptake and Efflux of Tested Compounds in CaCo-2 Cell Cultures
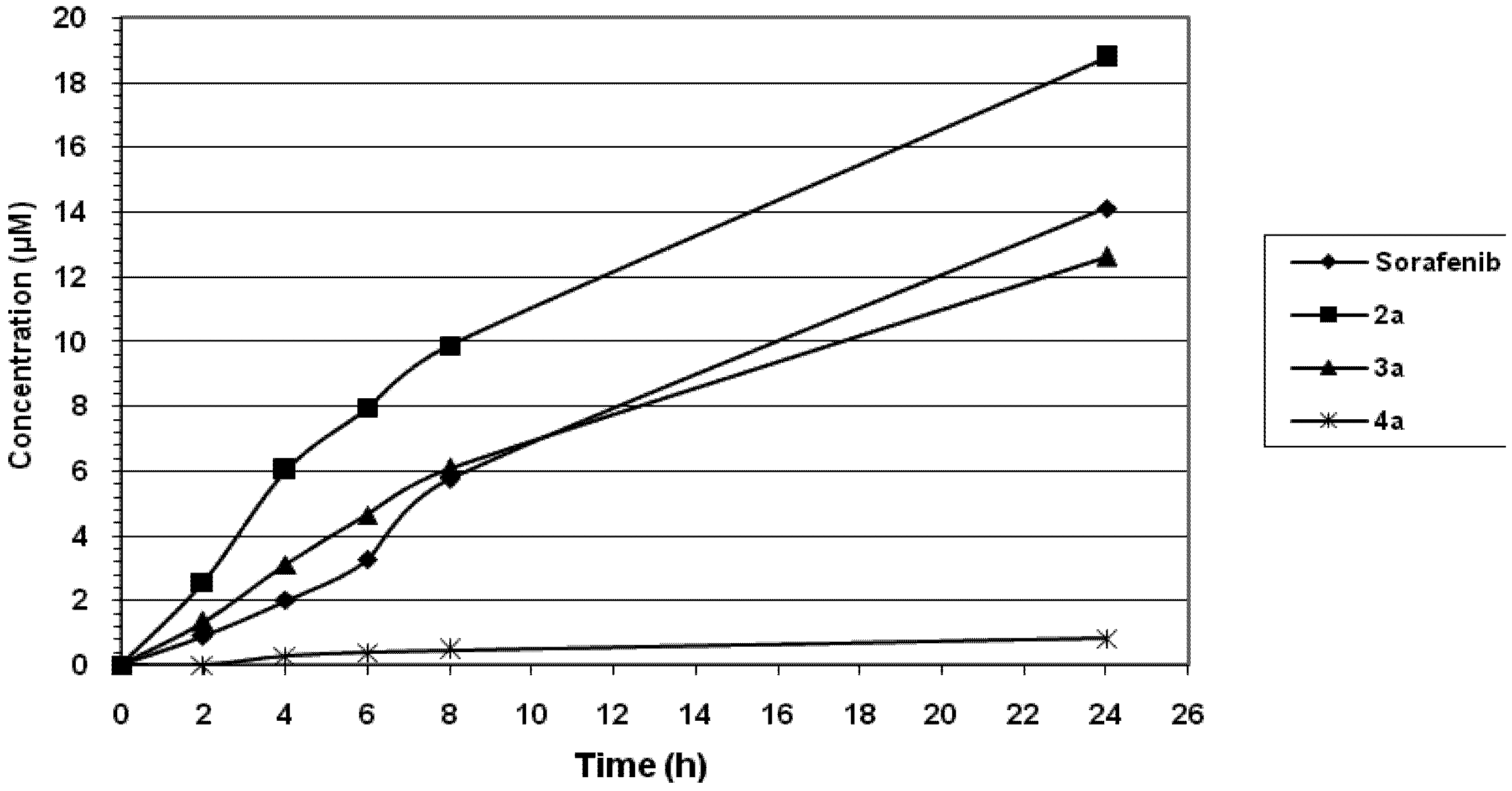
3. Experimental Section
3.1. Chemistry
3.1.1. General
3.1.2. General Method for the Synthesis of 4-chloropyridine-2-carboxamides 2a–e
3.1.3. General Method for the Synthesis of 4-(4-aminophenoxy)-pyridine-2-carboxamides 3a–e
3.1.4.General Procedure for the Synthesis of 4-[4-[[4-Chloro-3-(trifluoromethyl)phenyl]carbamoyl-amino]phenoxy]-pyridine-2-carboxamides4 a–e
3.2. Biological Evaluation
3.2.1. Cytostatic Activity
3.2.2. Antimetabolic Activity and Drug Influx/Efflux Assay in CaCo-2 Cell Cultures
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nature Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Keating, G.M.; Santoro, A. Sorafenib: A review of its use in advanced hepatocellular carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef]
- European Medicines Agency Home Page. Available online: http://www.ema.europa.eu/ (accessed on 5 July 2011).
- Gandey, A. Sorafenib new first-line option for advanced liver cancer. Available online: http://www.medscape.com/viewarticle/558023/ (accessed on 5 July 2011).
- Llovet, J.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Mekapati, S.B.; Kurup, A. QSAR and ADME. Bioorg. Med. Chem. 2004, 12, 3391–3400. [Google Scholar] [CrossRef]
- Bankston, D.; Dumas, J.; Natero, R.; Riedl, B.; Monahan, M.K.; Sibley, R. A scaleable synthesis of BAY 43-9006: A potent raf kinase inhibitor for the treatment of cancer. Org. Proc. Res. Dev 2002, 6, 777–781. [Google Scholar] [CrossRef]
- Keir, S.T.; Maris, J.M.; Lock, R.; Kolb, E.A.; Gorlick, R.; Carol, H.; Morton, C.L.; Reynolds, C.P.; Kang, M.H.; Watkins, A.; et al. Initial testing (stage 1) of the multi-target kinase inhibitor sorafenib by the pediatric preclinical testing program. Pediatr. Blood Cancer 2010, 55, 1126–1133. [Google Scholar] [CrossRef]
- Biobyte Corp, C-QSAR Database; Biobyte Corp: Claremont CA, California 91711, USA.
- Tzioumaki, N.; Manta, S.; Tsoukala, E.; Vande Voorde, J.; Liekens, S.; Komiotis, D.; Balzarini, J. Synthesis and biological evaluation of unsaturated keto and exomethylene D-arabinopyranonucleoside analogs: Novel 5-fluorouracil analogs that target thymidylate synthase. Eur. J. Med. Chem. 2011, 46, 993–1005. [Google Scholar] [CrossRef]
- Diez-Torrubia, A.; Balzarini, J.; Andrei, G.; Snoeck, R.; De Meester, I.; Camarasa, M.J.; Velázquez, S. Dipeptidyl peptidase IV dependent water-soluble prodrugs of highly lipophilic bicyclic nucleoside analogues. J. Med. Chem. 2011, 54, 1927–1942. [Google Scholar] [CrossRef]
- Rajić, Z.; Hadjipavlou-Litina, D.; Pontiki, E.; Kralj, M.; Šuman, L.; Zorc, B. The novel ketoprofen amides—Synthesis and biological evaluation as antioxidants, lipoxygenase inhibitors and cytostatic agents. Chem. Biol. Drug. Des. 2010, 75, 641–652. [Google Scholar] [CrossRef]
- Boyd, M.R.; Kenneth, D.P. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Babić, Ž.; Crkvenčić, M.; Rajić, Z.; Mikecin, A.-M.; Kralj, M.; Balzarini, J.; Petrova, M.; Vanderleyden, J.; Zorc, B. New Sorafenib Derivatives: Synthesis, Antiproliferative Activity Against Tumour Cell Lines and Antimetabolic Evaluation. Molecules 2012, 17, 1124-1137. https://doi.org/10.3390/molecules17011124
Babić Ž, Crkvenčić M, Rajić Z, Mikecin A-M, Kralj M, Balzarini J, Petrova M, Vanderleyden J, Zorc B. New Sorafenib Derivatives: Synthesis, Antiproliferative Activity Against Tumour Cell Lines and Antimetabolic Evaluation. Molecules. 2012; 17(1):1124-1137. https://doi.org/10.3390/molecules17011124
Chicago/Turabian StyleBabić, Željka, Maja Crkvenčić, Zrinka Rajić, Ana-Matea Mikecin, Marijeta Kralj, Jan Balzarini, Mariya Petrova, Jos Vanderleyden, and Branka Zorc. 2012. "New Sorafenib Derivatives: Synthesis, Antiproliferative Activity Against Tumour Cell Lines and Antimetabolic Evaluation" Molecules 17, no. 1: 1124-1137. https://doi.org/10.3390/molecules17011124
APA StyleBabić, Ž., Crkvenčić, M., Rajić, Z., Mikecin, A.-M., Kralj, M., Balzarini, J., Petrova, M., Vanderleyden, J., & Zorc, B. (2012). New Sorafenib Derivatives: Synthesis, Antiproliferative Activity Against Tumour Cell Lines and Antimetabolic Evaluation. Molecules, 17(1), 1124-1137. https://doi.org/10.3390/molecules17011124