Fast Method for Synthesis of Alkyl and Aryl-N-Methylnitrones

Abstract
:1. Introduction
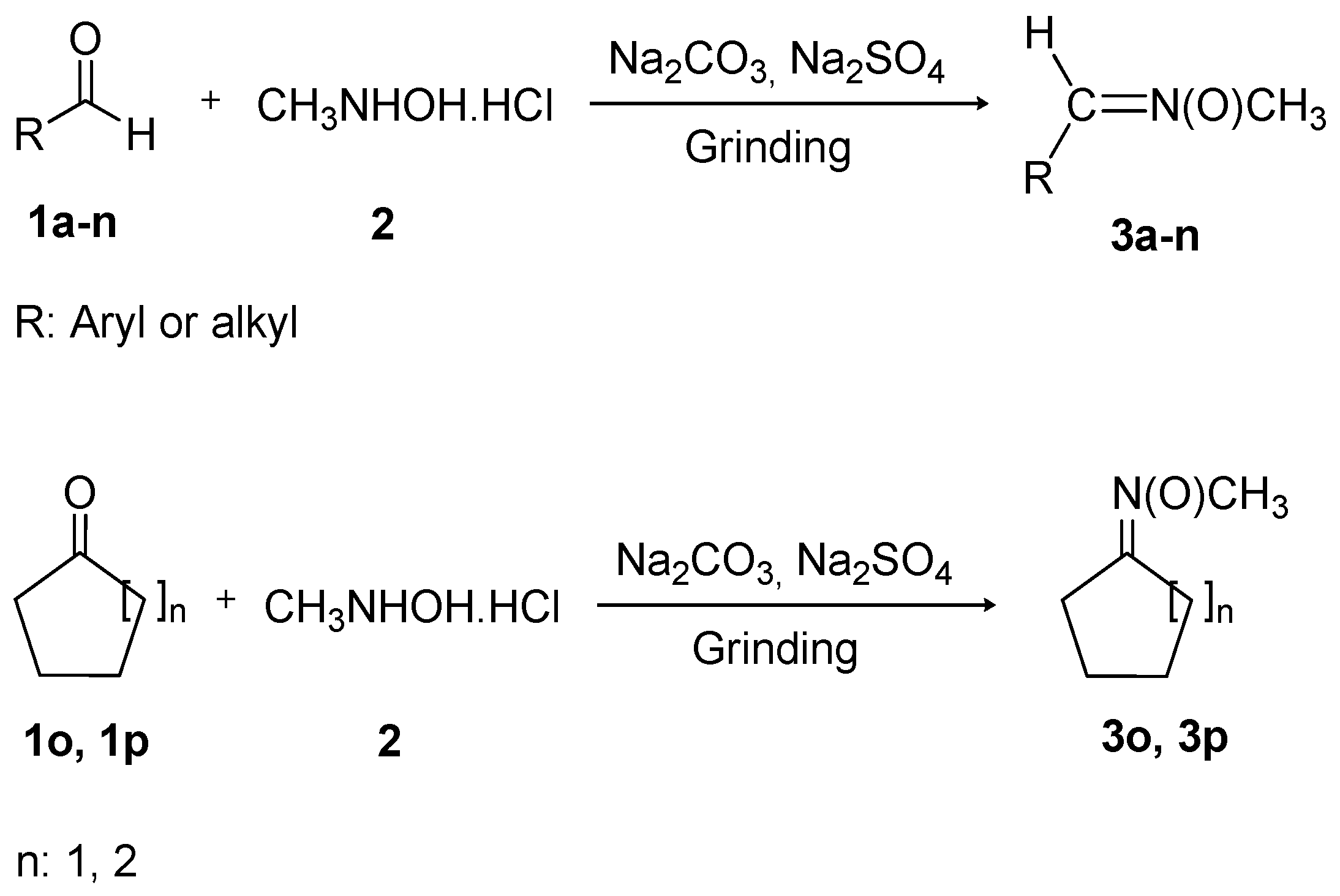
2. Results and Discussion
3. Experimental
3.1. General Method for the Synthesis of Compounds 3a-l
3.2. General Method for the Synthesis of Compounds 3m-p
4. Conclusions
Acknowledgment
References
- Black, D.S.C.; Crozier, R.F.; Davis, V.C. 1,3-Dipolar cycloaddition reactions of nitrones. Synthesis 1975, 1975, 205–221. [Google Scholar] [CrossRef]
- Dondas, H.A.; Cummins, J.E.; Grigg, R.; Thornton, P.M. X=Y–ZH Systems as potential 1,3-dipoles. Part 53: Sequential nucleophilic ring opening-1,3-dipolar cycloaddition reactions of Z-oxime anions with aziridines and dipolarophiles. Tetrahedron 2001, 57, 7951–7961. [Google Scholar] [CrossRef]
- Alibes, R.; Blanco, P.; de March, P.; Figueredo, M.; Font, J.; Alvarez-Larena, A.; Piniella, J.F. Synthesis and 1,3-dipolar cycloadditions of a new enantiopure cyclic nitrone. Tetrahedron Lett. 2002, 44, 523–525. [Google Scholar] [CrossRef]
- Kumar, K.R.R.; Mallesha, H.; Rangappa, K.S. Synthesis of novel isoxazolidine derivatives and studies for their antifungal properties. Eur. J. Med. Chem. 2003, 38, 613–619. [Google Scholar] [CrossRef]
- Gothelf, K.V.; Jorgenson, K.A. Asymmetric 1,3-Dipolar cycloaddition reactions. Chem. Rev. 1998, 98, 863–909. [Google Scholar]
- Broggini, G.; Zecchi, G. Pyrrolizidine and indolizidine syntheses involving 1,3-Dipolar cycloadditions. Synthesis 1999, 1999, 905–917. [Google Scholar] [CrossRef]
- Mulzer, J. Organic Synthesis Highlights; Verlag Chemie: Weinheim, Germany, 1991; pp. 3–4. [Google Scholar]
- Huisgen, R.; Grashey, R.; Sauer, J. The Chemistry of Alkenes; Wiley Interscience: London, UK, 1964; pp. 806–807. [Google Scholar]
- Tania, R.A.; Gareth, W.V.C.; Colin, L.R. Benign approaches for the synthesis of bis-imine schiff bases. Green Chem. 2006, 8, 50–53. [Google Scholar]
- Kumar, S.; Sharma, P.; Kapoor, K.K.; Hundal, M.S. An efficient, catalyst- and solvent-free, four component, and one-pot synthesis of polyhydroquinolines on grinding. Tetrahedron 2008, 64, 536–542. [Google Scholar] [CrossRef]
- Longhi, K.; Moreira, D.N.; Marzari, M.R.B.; Floss, V.M.; Bonacorso, H.G.; Zanatta, N.; Martins, M.A.P. An efficient solvent-free synthesis of NH-pyrazoles from β-dimethylaminovinylketones and hydrazine on grinding. Tetrahedron Lett. 2010, 51, 3193–3196. [Google Scholar] [CrossRef]
- Heravi, M.M.; Poormohammad, N.; Beheshtiha, Y.S.; Baghernejad, B. Efficient synthesis of 2,4-Disubstituted thiazoles under grinding. Synth. Commun. 2011, 41, 579–582. [Google Scholar] [CrossRef]
- Andrade, M.M.; Barros, M.T.; Pinto, R.C. Exploiting microwave-assisted neat procedures: Synthesis of N-aryl and N-alkylnitrones and their cycloaddition en route for isoxazolidines. Tetrahedron 2008, 64, 10521–10530. [Google Scholar] [CrossRef]
- Bigdeli, M.A.; Nikje, M.M.A. An efficient and rapid chemoselelective synthesis of α-aryl-N-methylnitrones in dry media. Monatsh. Chem. 2001, 132, 1547–1549. [Google Scholar] [CrossRef]
- Nikje, M.M.A.; Bigdeli, M.A. A microwave assistedoxidation of benzaldehydes under solvent-free condition usingoxone/wet-alumina. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 1445–1448. [Google Scholar] [CrossRef]
- Colacino, E.; Nun, P.; Colacino, F.M.; Martinez, J.; Lamaty, F. Solvent-free synthesis of nitrones in a ball-mill. Tetrahedron 2008, 64, 5569–5576. [Google Scholar] [CrossRef]
- Colonna, S.; Piront, V.; Carrea, G.; Pasta, P.; Zambianchi, F. Oxidation of secondary amines by molecular oxygen and cyclohexanone monooxygenase. Tetrahedron 2004, 60, 569–575. [Google Scholar] [CrossRef]
- Coutouli, A.E.; Malamidou, X.E.; Stampelos, X.N.; Alexopoulou, I.N. Formation and reduction reactions of 3-indol-3-yl-isoxazolidines. Tetrahedron 1997, 53, 707–718. [Google Scholar] [CrossRef]
- Cummins, H.C.; Coates, R.M. α-Oxygenation of aldehydes and cyclic ketones by acylation & arrangement of nitrones. J. Org. Chem. 1983, 48, 2070–2076. [Google Scholar]
Sample Availability: Samples of the substituted N-methyl nitrones 3a-p are available from the authors. |

{kind=link}
| Substrate | Entry | Reaction Time (min) | Products | mp (or bp) (°C) | mp (or bp) (°C) Lit. | Yield (%) |
|---|---|---|---|---|---|---|
| 1a | 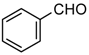 | 4 | 3a | 82–84 | 82–84 [14] | 94 |
| 1b |  | 5 | 3b | 204–205 | 210–212 [13] | 93 |
| 1c |  | 5 | 3c | 129–130 | 129–130 [17] | 95 |
| 1d | 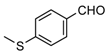 | 5 | 3d | 102–103 | 103–104 [17] | 98 |
| 1e | 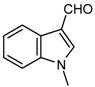 | 7 | 3e | 148–149 | 148–150 [18] | 98 |
| 1f | 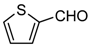 | 4 | 3f | 118–119 | 118–119 [17] | 96 |
| 1g | 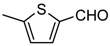 | 5 | 3g | 90–91 † | - | 96 |
| 1h | 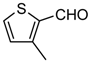 | 8 | 3h | 135–136 ‡ | - | 98 |
| 1i |  | 6 | 3i | 193–195 † | - | 91 |
| 1j | 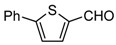 | 6 | 3j | 113–114 † | - | 93 |
| 1k | 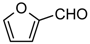 | 3 | 3k | 89–90 | 89–90 [14] | 94 |
| 1l |  | 5 | 3l | oil † | - | 92 |
| 1m |  | 10 | 3m | oil ‡ | - | 62 |
| 1n | 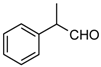 | 10 | 3n | oil ‡ | - | 76 |
| 1o |  | 10 | 3o | (72 °C, 0.1 torr) | (70 °C, 0.1 torr) [19] | 71 |
| 1p | 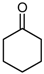 | 10 | 3p | (88 °C, 0.1 torr) | (80 °C, 0.1 torr) [19] | 68 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yavuz, S.; Ozkan, H.; Colak, N.; Yildirir, Y. Fast Method for Synthesis of Alkyl and Aryl-N-Methylnitrones. Molecules 2011, 16, 6677-6683. https://doi.org/10.3390/molecules16086677
Yavuz S, Ozkan H, Colak N, Yildirir Y. Fast Method for Synthesis of Alkyl and Aryl-N-Methylnitrones. Molecules. 2011; 16(8):6677-6683. https://doi.org/10.3390/molecules16086677
Chicago/Turabian StyleYavuz, Serkan, Hamdi Ozkan, Naki Colak, and Yilmaz Yildirir. 2011. "Fast Method for Synthesis of Alkyl and Aryl-N-Methylnitrones" Molecules 16, no. 8: 6677-6683. https://doi.org/10.3390/molecules16086677
APA StyleYavuz, S., Ozkan, H., Colak, N., & Yildirir, Y. (2011). Fast Method for Synthesis of Alkyl and Aryl-N-Methylnitrones. Molecules, 16(8), 6677-6683. https://doi.org/10.3390/molecules16086677