Synthetic Organic Electrochemistry in Ionic Liquids: The Viscosity Question

Abstract
:1. Introduction

2. Results and Discussion
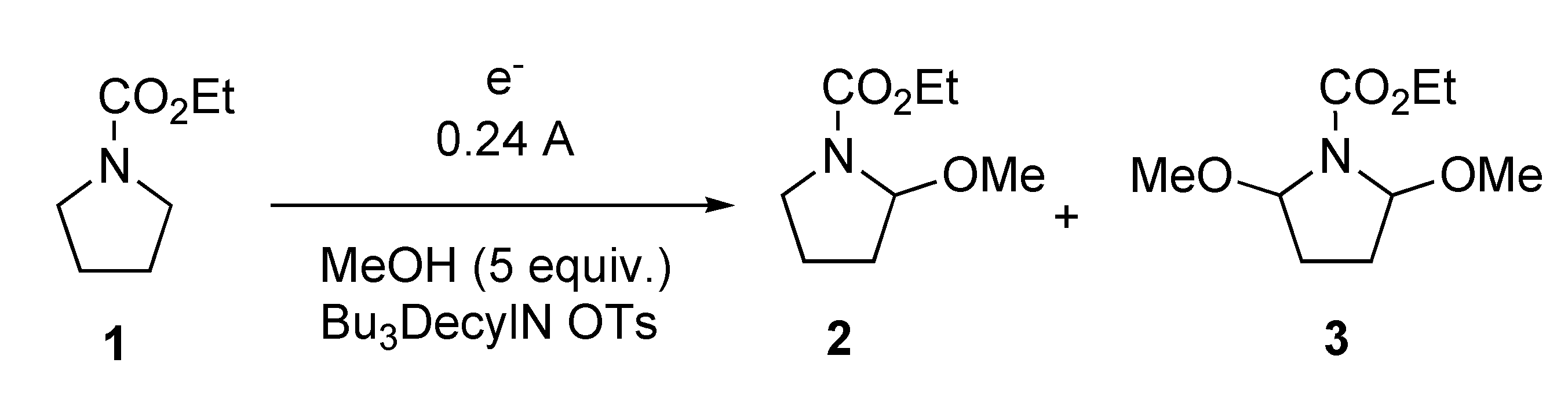

{kind=link}
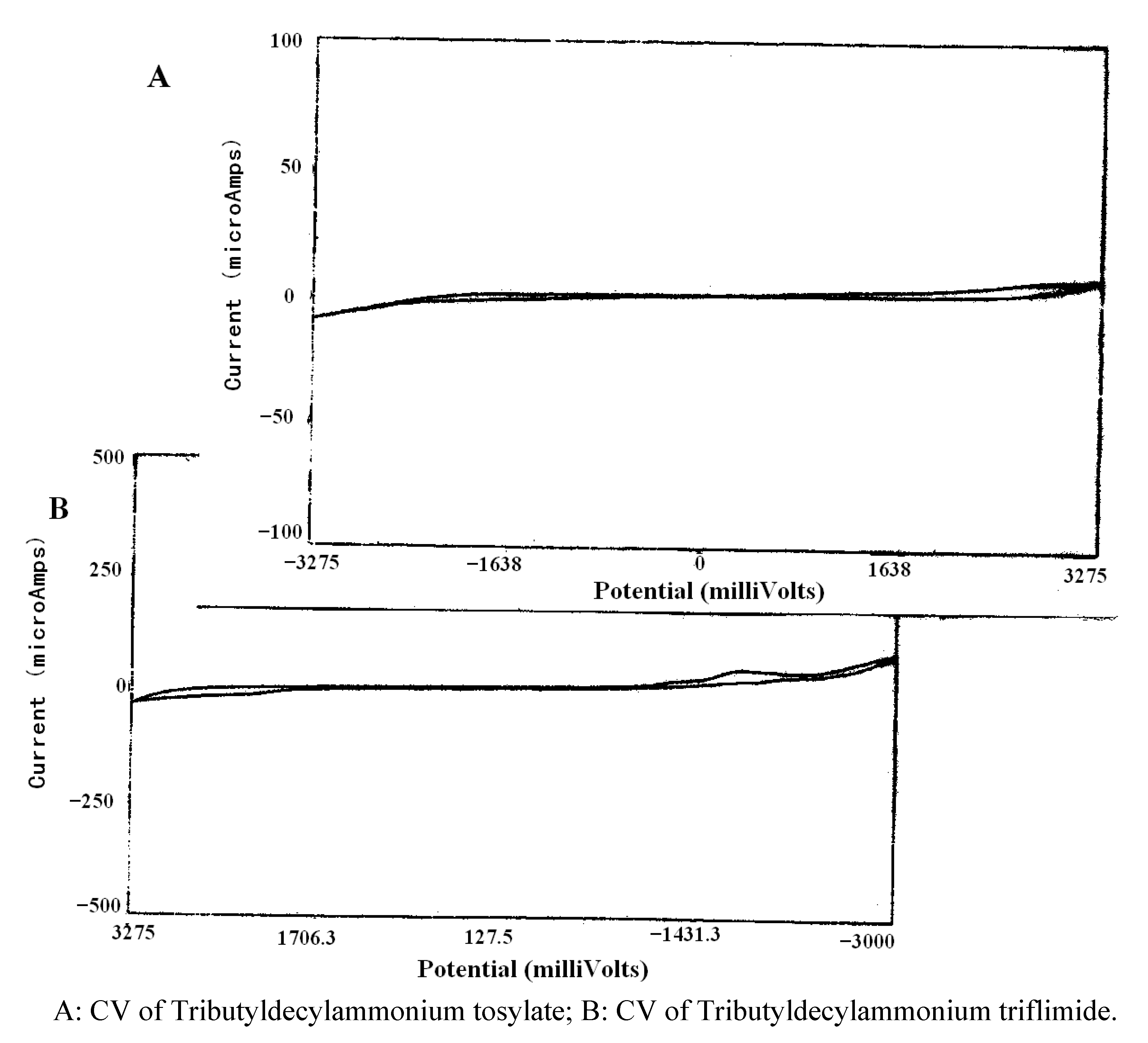
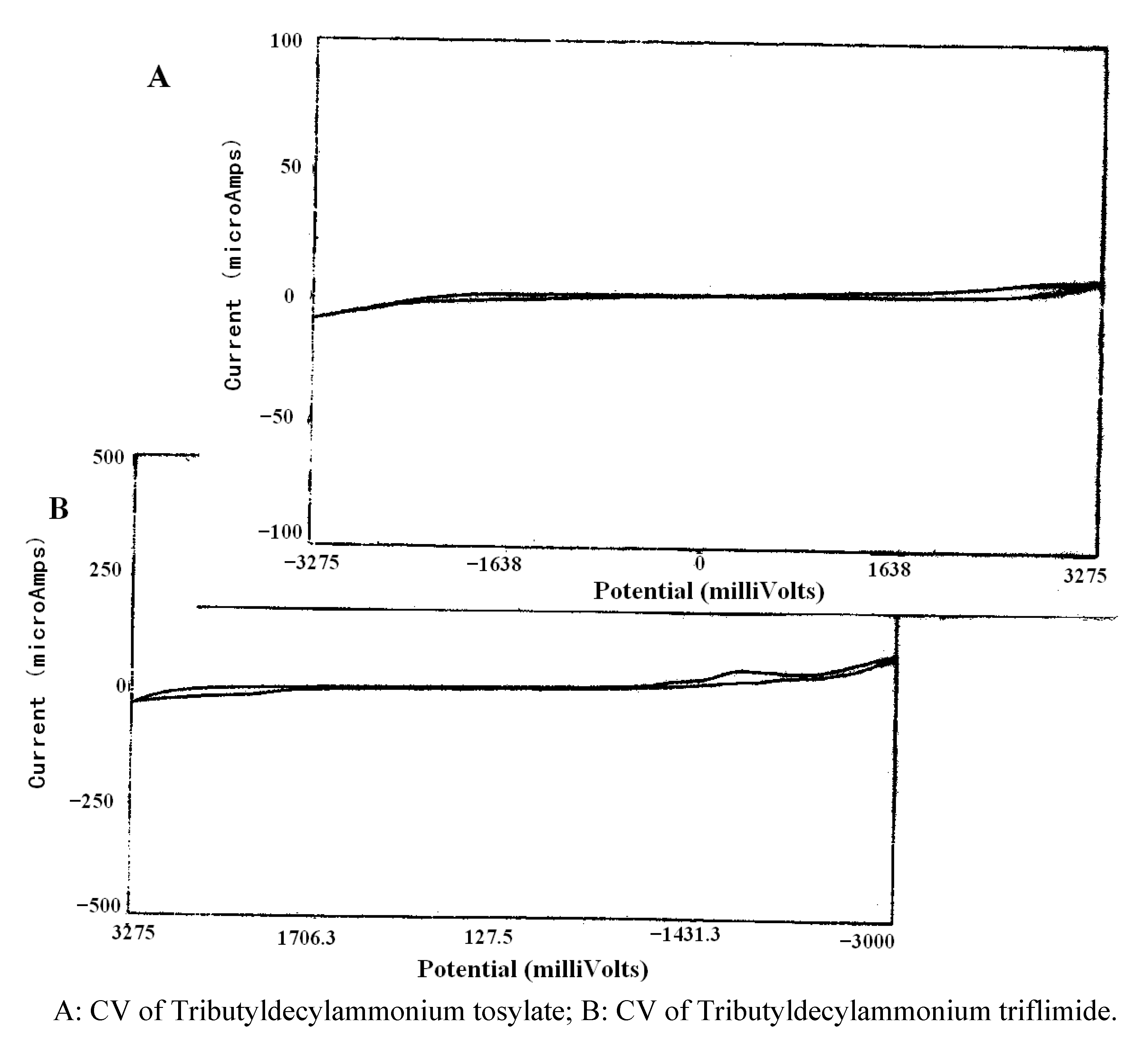{kind=link}
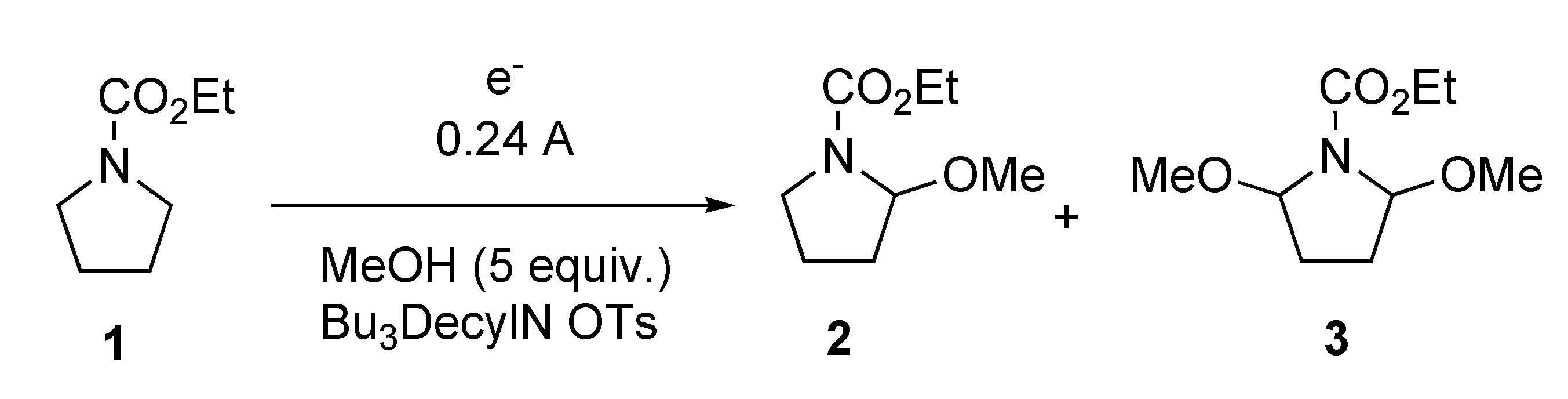{kind=link}
| Entry | Volume% MeOH | % Yield of 2 | State of RTIL | Viscosity (cP) |
|---|---|---|---|---|
| 1 | 33 | 0 | Dark black | 120 |
| 2 | 50 | 88 | Pale yellow | 15 |
| 3 | 67 | 90 | Clear | 7 |
| Entry | Volume% MeOH | % Yield of 2 | State of RTIL | Viscosity (cP) |
|---|---|---|---|---|
| 1 | 10 | 10 | Black | 150 |
| 2 | 20 | 45 | Dark brown | 50 |
| 3 | 33 | 89 | Pale yellow | 10 |
| 4 | 50 | 93 | Clear | 5 |
| 5 a | 33 | 89 | Pale yellow | |
| 6 b | 33 | 91 | Pale yellow | |
| 7 c | 33 | 93 | Pale yellow |
| Entry | Volume% MeOH | % Yield of 2 | State of RTIL | Viscosity (cP) |
|---|---|---|---|---|
| 1 | 5 | 0 | Black | 25 |
| 2 | 10 | 4 | Dark orange | 16 |
| 3 | 20 | 9 | Dark yellow | 10 |
| 4 | 25 | 83 | Clear | 6 |
| 5 a | 33 | 89 | Pale yellow | |
| 6 b | 33 | 91 | Pale yellow | |
| 7 c | 33 | 93 | Pale yellow |
| Entry | Carbamate | % Yield of Methoxylated Product |
|---|---|---|
| 1 |  | 95 |
| 2 | 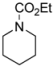 | 89 |
| 3 | 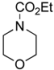 | 81 |
| 4 | N,N-diethyl | No reaction |
3. Experimental
3.1. Decyltributylammonium Tosylate
3.1.1. Decyl Tosylate
3.1.2. Decyltributylammonium tosylate
3.1.3. Decyltributylammonium bis(trifluoromethanesulfonyl)imide
3.1.4. 1-Ethyl-3-Methylimidazolium bis(trifluoromethanesulfonyl)imide [43]
3.2. General Procedure for Preparing Carbamates
N-Carboethoxypyrrolidine
3.3. Electrochemical Oxidation of N-Carboethoxypyrrolidine
The Control [45]
3.4. Using Ionic Liquid as Solvent/Electrolyte
3.5. Viscosity Measurements
4. Conclusions
Conflict of Interest
References and Notes
- Wilkes, J.S. A short history of ionic liquids—From molten salts to neoteric solvents. Green Chem. 2002, 10, 73–80. [Google Scholar]
- Hallett, J.P.; Welton, T. Room-temperature ionic liquids: Solvents for synthesis and catalysis. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar]
- Wasserscheid, P.; Welton, T. Ionic Liquids in Synthesis, 2nd ed; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Handy, S.T. Room temperature ionic liquids: Different classes and physical properties. Curr. Org. Chem. 2005, 9, 959–988. [Google Scholar]
- Wasserscheid, P.; Keim, W. Ionic liquids—New “solutions” for transition metal catalysis. Angew. Chem. Int. Ed. 2000, 39, 3772–3789. [Google Scholar]
- Parvulescu, V.I.; Hardacre, C. Catalysis in ionic liquids. 2007, 107, 2615–2665. [Google Scholar]
- Ennis, E.; Handy, S.T. A facile route to C2-substituted imidazolium ionic liquids. Molecules 2009, 14, 2235–2245. [Google Scholar]
- Handy, S.T. One-pot halogenation-Heck coupling reactions in ionic liquids. Synlett 2006, 3176–3178. [Google Scholar]
- Handy, S.T.; Okello, M. The 2-position of imidazolium ionic liquids: Substitution and exchange. J. Org. Chem. 2005, 70, 1915–1918. [Google Scholar]
- Handy, S.T.; Okello, M. Fructose-derived ionic liquids: Recyclable homogeneous supports. Tetrahedron Lett. 2003, 44, 8399–8402. [Google Scholar]
- Handy, S.T. Greener solvents: Room temperature ionic liquids from biorenewable sources. Chem. Eur. J. 2003, 9, 2938–2944. [Google Scholar]
- Handy, S.T.; Okello, M.; Dickenson, G. Solvents from biorenewable sources: Ionic liquids based on fructose. Org. Lett. 2003, 5, 2513–2515. [Google Scholar]
- Frontana-Uribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic electrosynthesis: A promising green methodology in organic chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar]
- Hapiot, P.; Lagrost, C. Electrochemical reactivity in room-temperature ionic liquids. Chem. Rev. 2008, 108, 2238–2264. [Google Scholar]
- Martiz, B.; Keyrouz, R.; Gmouh, S.; Vaultier, M.; Jouikov, V. Superoxide-stable ionic liquids: New and efficient media for electrosynthesis of functional siloxanes. Chem. Commun. 2004, 674–675. [Google Scholar]
- Damlin, P.; Kvarnström, C.; Ivaska, A. Electrochemical synthesis and in situ spectoelectrochemical characterization of poly(3,4-ethylenedioxythhiophene) (PEDOT) in room temperature ionic liquids. J. Electroanal. Chem. 2004, 570, 113–122. [Google Scholar]
- Sekiguchi, K.; Atobe, M.; Fuchigami, T. Electrooxidative polymerization of aromatic compounds in 1-ethyl-3-methylimidazolium trifluoromethanesulfonate room-temperature ionic liquid. J. Electroanal. Chem. 2003, 557, 1–7. [Google Scholar]
- Shen, Y.; Atobe, M.; Fuchigami, T. Electroorganic synthesis using a fluoride ion mediator under ultrasonic irradiation: Synthesis of oxindole and 3-oxotetrahydroisoquinoline derivatives. Org. Lett. 2004, 6, 2441–2444. [Google Scholar]
- Sawamura, T.; Inagi, S.; Fuchigami, T. Anodic fluorination and fluorodesulfurization in ionic liquid hydrogen fluoride salts with polyether additives. J. Electrochem. Soc. 2009, E26–E28. [Google Scholar]
- Hasegawa, M.; Ishii, H.; Cao, Y.; Fuchigami, T. Regioselective anodic monofluorination of ethers, lactones, carbonates, and esters using ionic liquid fluoride salts. J. Electrochem. Soc. 2006, D162–D166. [Google Scholar]
- Hasegawa, M.; Ishii, H.; Fuchigami, T. Selective anodic fluorination of phthalides in ionic liquids. Green Chem. 2003, 5, 512–515. [Google Scholar]
- Hasegawa, M.; Ishii, H.; Fuchigami, T. Electroorganic synthesis under solvent-free conditions. Highly regioselective anodic monofluorination of cyclic ethers, lactones, and a cyclic carbonate. Tetrahedron Lett. 2002, 43, 1503–1505. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fuchigami, T. Electroorganic reactions in ionic liquids 5 {1}. Anodic fluorodesulfurization of phthalide, ethylene carbonate, and glucopyranosides having arylthio groups. Electrochim. Acta 2004, 49, 3367–3372. [Google Scholar] [CrossRef]
- Barhdadi, R.; Comminges, C.; Doherty, A.P.; Nédélee, J.Y.; O’Toole, S.; Troupel, M. The electrochemistry of TEMPO-mediated oxidation of alcohols in ionic liquid. J. Appl. Electrochem. 2007, 37, 723–728. [Google Scholar]
- Herath, A.C.; Becker, J.Y. 2,2,6,6-Tetramethylpiperidine-1-oxyl (TEMPO)-mediated catalytic oxidation of benzyl alcohol in acetonitrile and ionic liquid 1-buthyl-3-methyl-imidazolium hexafluorophosphate [BMIM][PF6]: Kinetic analysis. Electrochim. Acta 2008, 53, 4324–4330. [Google Scholar]
- Ho, K.-P.; Wong, K.-Y.; Chan, T.H. Indirect catalytic epoxidation with hydrogen peroxide electrogenerated in ionic liquids. Tetrahedron 2006, 62, 6650–6658. [Google Scholar]
- Tang, M.C.-Y.; Wong, K.-Y.; Chan, T.H. Electrosynthesis of hydrogen peroxide in room temperature ionic liquids and in situ epoxidation of alkenes. Chem. Commun. 2005, 1345–1347. [Google Scholar]
- Gaillon, L.; Bedioui, F. First example of electroassisted biomimetic activation of molecular oxygen by a (salen)Mn epoxidation catalyst in a room-temperature ionic liquid. Chem. Commun. 2001, 1458–1459. [Google Scholar]
- Lagrost, C.; Hapiot, P.; Vaultier, M. The influence of room-temperature ionic liquids on the stereoselectivity and kinetics of the electrochemical pinacol coupling of acetophenone. Green Chem. 2005, 7, 468–474. [Google Scholar]
- Villagrán, C.; Banks, C.; Pitner, W.; Hardacre, C.; Compton, R.G. Electroreduction of N-methylphthalimide in room temperature ionic liquids under insonated and silent conditions. Ultrasonics Sonochemistry 2005, 12, 423–428. [Google Scholar]
- Mellah, M.; Zeitouny, J.; Gmouh, S.; Vaultier, M.; Jouikov, V. Oxidative self-coupling of aromatic compounds in ionic liquids. Electrochem. Commun. 2005, 7, 869–874. [Google Scholar]
- Mellah, M.; Gmouh, M.; Vaultier, M.; Jouikov, V. Electrocatalytic dimerisation of PhBr and PhCH2Br in [BMIM]+ NTf2− ionic liquid. Electrochem. Commun. 2005, 5, 591–593. [Google Scholar]
- Sweeny, B.K.; Peters, D.G. Cyclic Voltametric study of the catalytic behavior of nickel(I) salen electrogenerated at a glassy carbon electrode in an ionic liquid (1-butyl-3-methylimidazolium tetrafluoroborate, BMIM+ BF4−). Electrochem. Commun. 2001, 3, 712–715. [Google Scholar]
- Barhdadi, R.; Courtinard, C.; Nédélee, J.Y.; Troupel, M. Room-temperature ionic liquids as new solvents for organic electrosynthesis. The first examples of direct or nickel-catalysed electroreductive coupling involving organic halides. Chem. Commun. 2003, 1434–1435. [Google Scholar]
- Jabbar, M.A.; Shimakoshi, H.; Hisaeda, Y. Enhanced reactivity of hydrophobic vitamin B12 towards the dechlorination of DDT in ionic liquid. Chem. Commun. 2007, 1653–1655. [Google Scholar]
- Feroci, M.; Chiarotto, I.; Orsini, M.; Sotgiu, G.; Inesi, A. Reactivity of electrogenerated N-heterocyclic carbenes in room-temperature ionic liquids. Cyclization to 2-azetidinone ring via C-3/C-4 bond formation. Adv. Synth. Catal. 2008, 350, 1355–1359. [Google Scholar]
- Yang, H.; Gu, Y.; Deng, Y.; Shi, F. Electrochemical activation of carbon dioxide in ionic liquid: synthesis of cyclic carbonates at mild reaction conditions. Chem. Commun. 2002, 274–275. [Google Scholar]
- Zhang, L.; Niu, D.; Zhang, K.; Zhang, G.; Luo, Y.; Lu, J. Electrochemical activation of CO2 in ionic liquid (BMIMBF4): Synthesis of organic carbonates under mild conditions. Green Chem. 2008, 10, 202–206. [Google Scholar]
- Chiarotto, I.; Feeney, M.M.M.; Feroci, M.; Inesi, A. Electrogenerated N-heterocyclic carbene: N-Acylation of chiral oxaolidin-2-ones in ionic liquids. Electrochim. Acta 2009, 54, 1638–1644. [Google Scholar]
- Chiarotto, I.; Feroci, M.; Feeney, M.M.M.; Inesi, A. Study on the reactivity of aldehydes in electrolyzed ionic liquids: Benzoin Condensation—Volatile Organic Compounds (VOCs) vs. Room Temperature Ionic Liquids (RTILs). Adv. Synth. Catal. 2010, 352, 3287–3292. [Google Scholar]
- Kita, Y.; Haruta, J.; Tagawa, H.; Tamura, Y. Facile and efficient carboalkoxylation and carboaryloxylation of amines. J. Org. Chem. 1980, 45, 4519–4522. [Google Scholar]
- Seddon, K.R.; Stark, A.; Torres, M.-J. Influence of chloride, water, and organic solvents on the physical properties of ionic liquids. Pure Appl. Chem. 2000, 72, 2275–2287. [Google Scholar]
- Chu, Y.; Deng, H.; Cheng, J.-P. An Acidity Scale of 1,3-Dialkylimidazolium Salts in Dimethyl Sulfoxide Solution. J. Org. Chem. 2007, 72, 7790–7793. [Google Scholar]
- Ballini, R.; Fiorini, D.; Maggi, R.; Righi, P.; Sartori, G.; Sartorio, R. TBD-catalyzed solventless synthesis of symmetrically N,N'-substituted ureas from primary amines and diethyl carbonate. Green Chem. 2003, 5, 396–398. [Google Scholar]
- Shono, T.; Matsumura, Y.; Tsubata, K. Electroorganic chemistry. 46. A new carbon-carbon bond forming reaction at the α -position of amines utilizing anodic oxidation as a key step. J. Am. Chem. Soc. 1981, 103, 1172–1176. [Google Scholar]
- Torii, S.; Inokuchi, T.; Akahosi, F.; Kubota, M. Facile access to 6-methoxy-1,2,3,6-tetrahydro- and 4-hydroxy-1,2,3,4-tetrahydropyridines by electrochemical haloalkoxylation-dehydrohalogenation sequence as a key operation. Synthesis 1987, 3, 242–245. [Google Scholar]
- Nishitani, T.; Horikawa, H.; Iwasaki, T.; Matsumoto, K.; Inoue, I.; Miyoshi, M. Synthetic electroorganic chemistry. 14. Synthesis of 5-fluorouracil derivatives having N-acylazacycloalkanes and lactams. J. Org. Chem. 1982, 47, 1706–1712. [Google Scholar]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bornemann, S.; Handy, S.T. Synthetic Organic Electrochemistry in Ionic Liquids: The Viscosity Question. Molecules 2011, 16, 5963-5974. https://doi.org/10.3390/molecules16075963
Bornemann S, Handy ST. Synthetic Organic Electrochemistry in Ionic Liquids: The Viscosity Question. Molecules. 2011; 16(7):5963-5974. https://doi.org/10.3390/molecules16075963
Chicago/Turabian StyleBornemann, Steven, and Scott T. Handy. 2011. "Synthetic Organic Electrochemistry in Ionic Liquids: The Viscosity Question" Molecules 16, no. 7: 5963-5974. https://doi.org/10.3390/molecules16075963
APA StyleBornemann, S., & Handy, S. T. (2011). Synthetic Organic Electrochemistry in Ionic Liquids: The Viscosity Question. Molecules, 16(7), 5963-5974. https://doi.org/10.3390/molecules16075963