Industrial Aplication of Catalytic Systems for n-Heptane Isomerization

Abstract
:1. Introduction

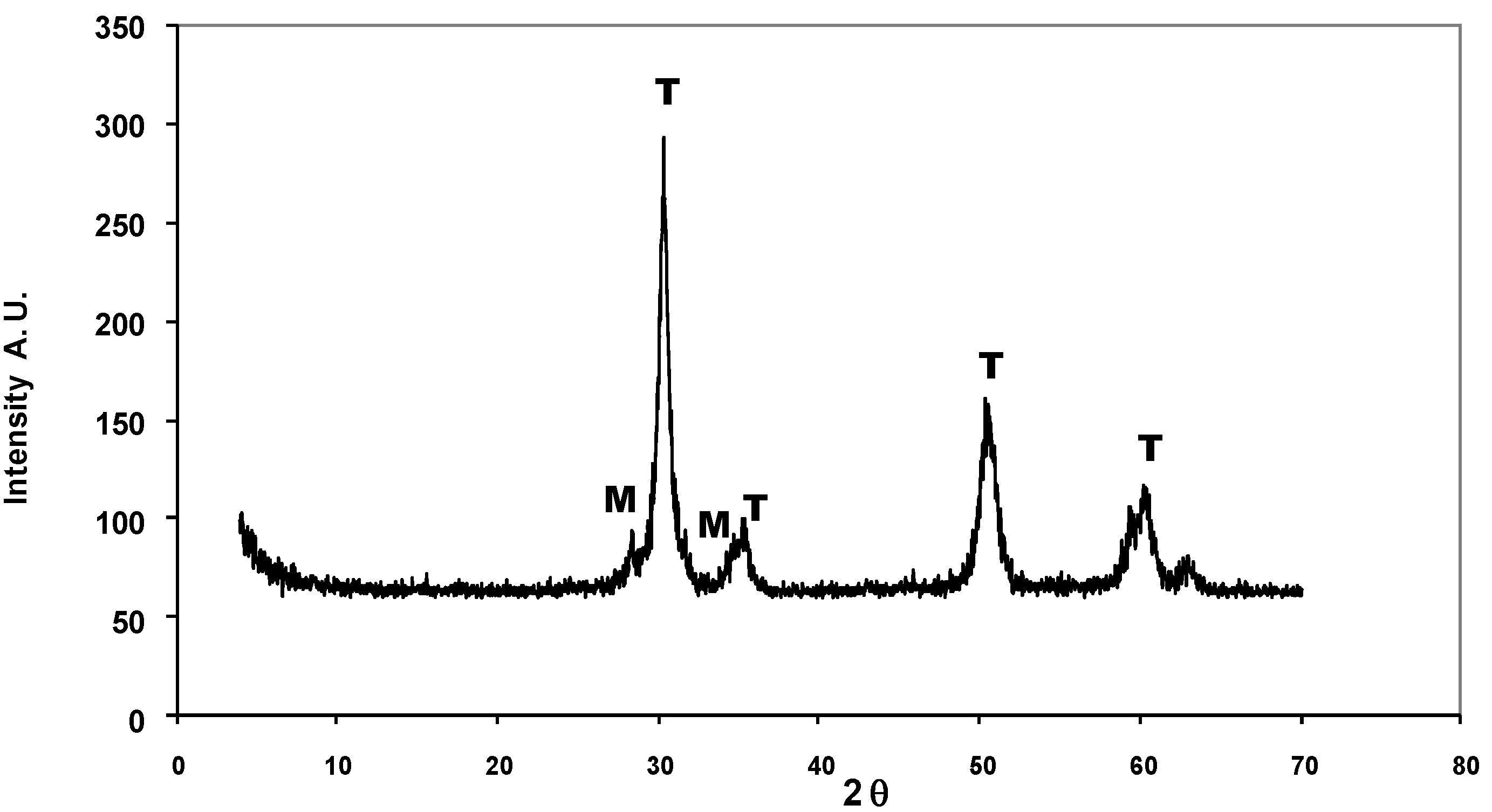{kind=link}
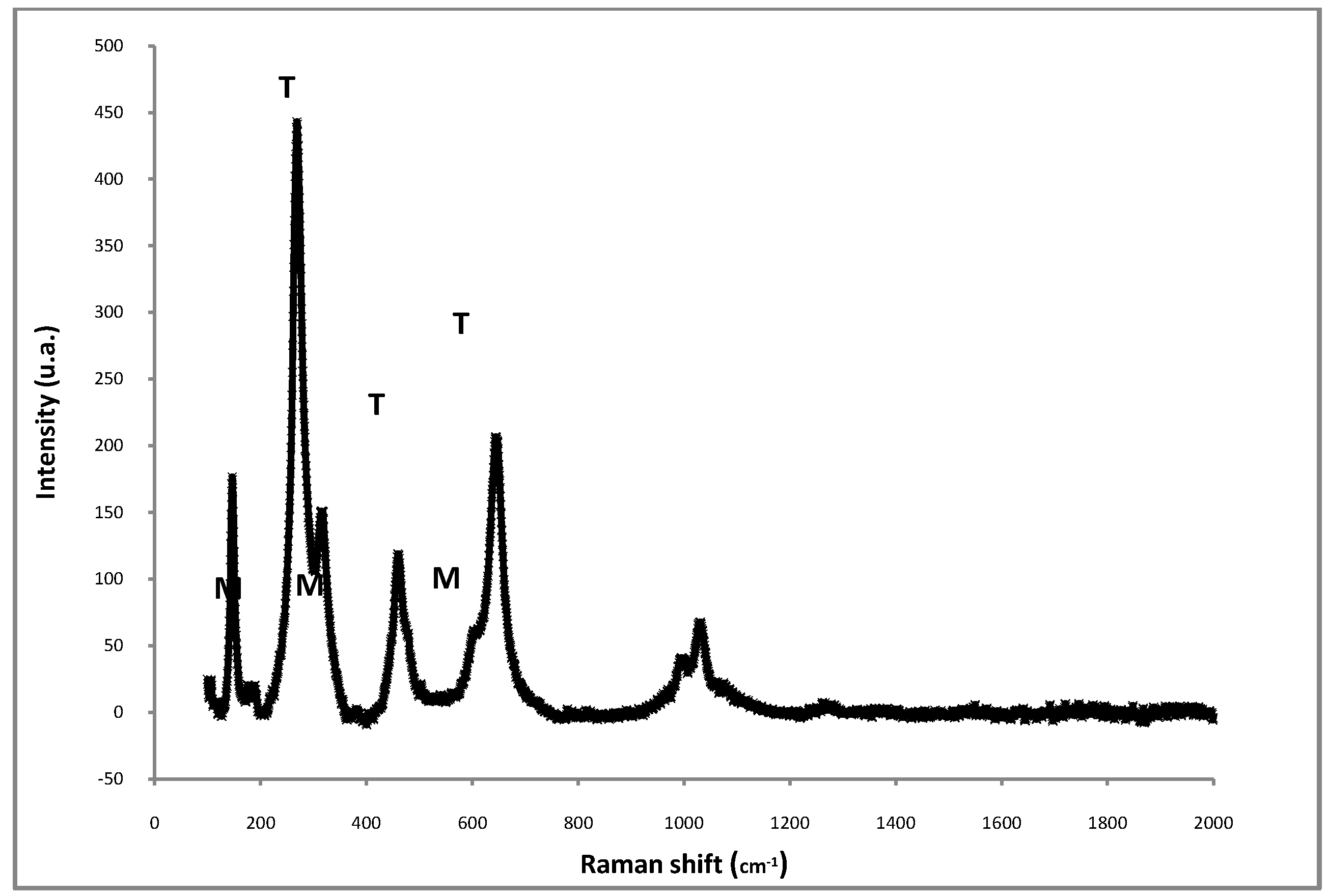{kind=link}
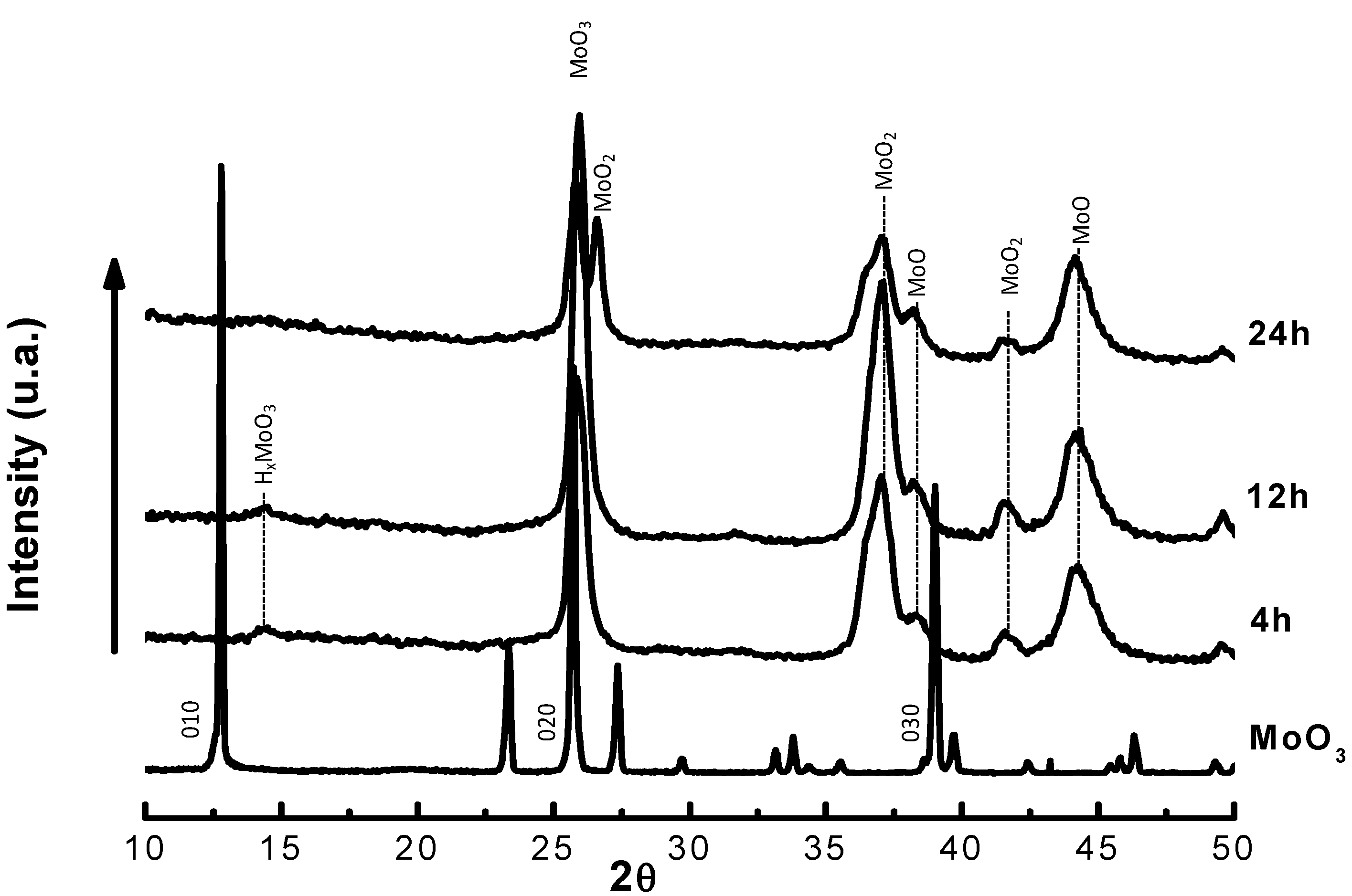{kind=link}
{kind=link}
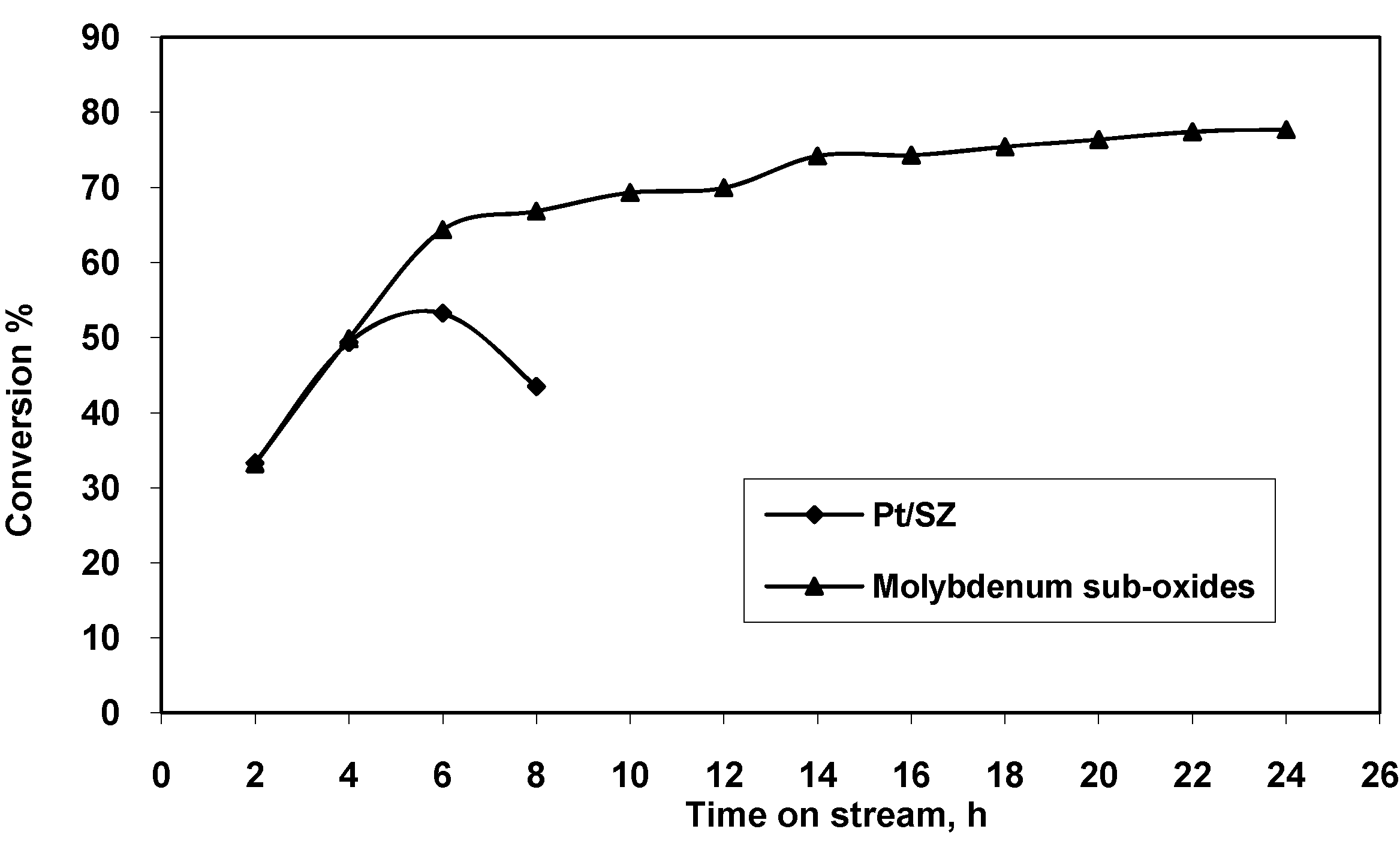{kind=link}
| Hydrocarbons | Ozone Production (g O3/g Hydrocarbon) |
|---|---|
| ALKANES | |
| n-pentane | 1.040 |
| n-hexane | 0.980 |
| n-heptane | 0.810 |
| n-octane | 0.610 |
| n-nonane | 0.540 |
| BRANCHED ALKANES | |
| 2-methylbutane | 1.380 |
| 2-methylhexane | 1.080 |
| 2-methylheptane | 0.960 |
| 2,4-dimethylhexane | 1.500 |
| 2,2,4-trimethylpentane | 1.600 |
| ALKENES | |
| 1-pentene | 6.220 |
| 1-octene | 5.290 |
| 3-octene | 5.290 |
| 4-methyl-1-pentene | 4.420 |
| AROMATICS | |
| Toluene | 2.730 |
| Ethylbenzene | 2.700 |
| m-xylene | 7.380 |
| p-xylene | 7.380 |
| C7 ALKANES | OCTANE NUMBERS | ||
|---|---|---|---|
| Research | Motor | Pump | |
| 2,2,3-trimethyl butane | 112.10 | 101.30 | 106.70 |
| 2,2-dimethyl pentane | 92.80 | 95.60 | 94.20 |
| 2,4-dimethyl pentane | 83.10 | 83.80 | 83.45 |
| 3,3-dimethyl pentane | 80.80 | 86.60 | 83.70 |
| 2,3-dimethyl pentane | 91.10 | 88.50 | 89.80 |
| 2-methyl hexane | 42.40 | 46.40 | 44.40 |
| 3-methyl hexane | 52.00 | 55.80 | 53.90 |
| 3-ethyl pentane | 65.00 | 69.30 | 67.15 |
| n-heptane | 0.00 | 0.00 | 0.00 |
- (a) The strong acidity of sulfated zirconia has been attributed to the electron-withdrawing anion groups, which lead to coordinatively unsaturated and electron defficient metal centers that behave as strong Lewis acid sites. Water vapor titrates such Lewis sites and converts them to Bronsted acids with very reactive protons [17].
- (b) Arata and Hino [19] proposed that water adsorption generates the Lewis and Bronsted acid sites responsible for the catalytic activity, which was confirmed by IR-spectroscopy using pyridine as the test molecule.
- (c) Yamaguchi [20], using IR-spectroscopy, showed that superacid centers are Lewis sites associated to the metallic cation. Acid strength of these sites is intensified by the inductive effect of the electrons of the double bond in the S=O structure.
- (d) Babou et al. [21] considered the acid sites of the sulfated zirconia as H2SO4 molecules supported on the zirconia surface, which can be reversibly hydrated. At high dehydration conditions an adsorbed SO3 species with a high Lewis acidity is obtained. In an intermediate hydration stage, the presence of H3O+ and HSO4− species promote a high protonic Bronsted acid strength. This reversible effect of water is important for catalytic applications because it modifies the system acidity.
2. Results and Discussion
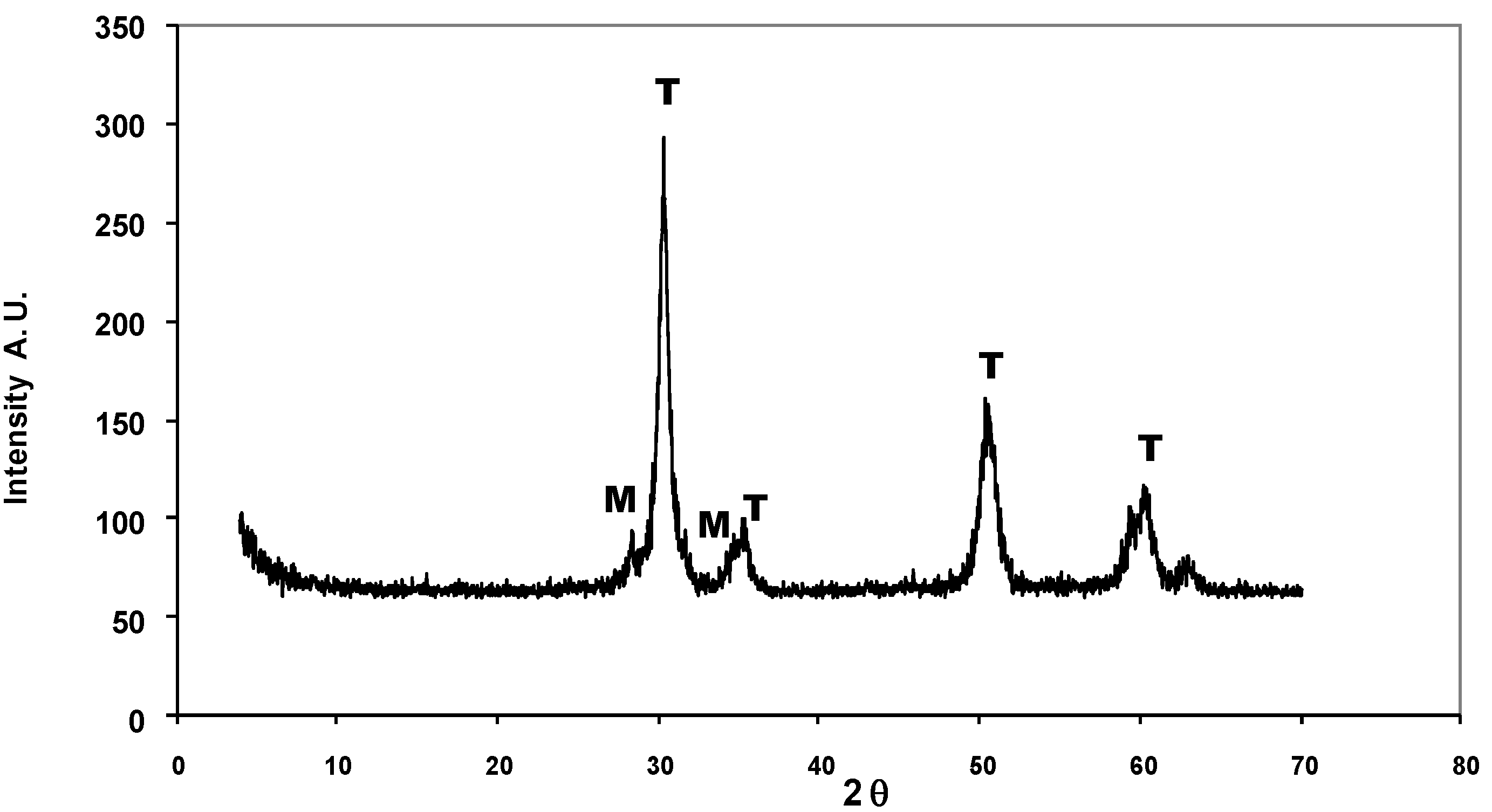
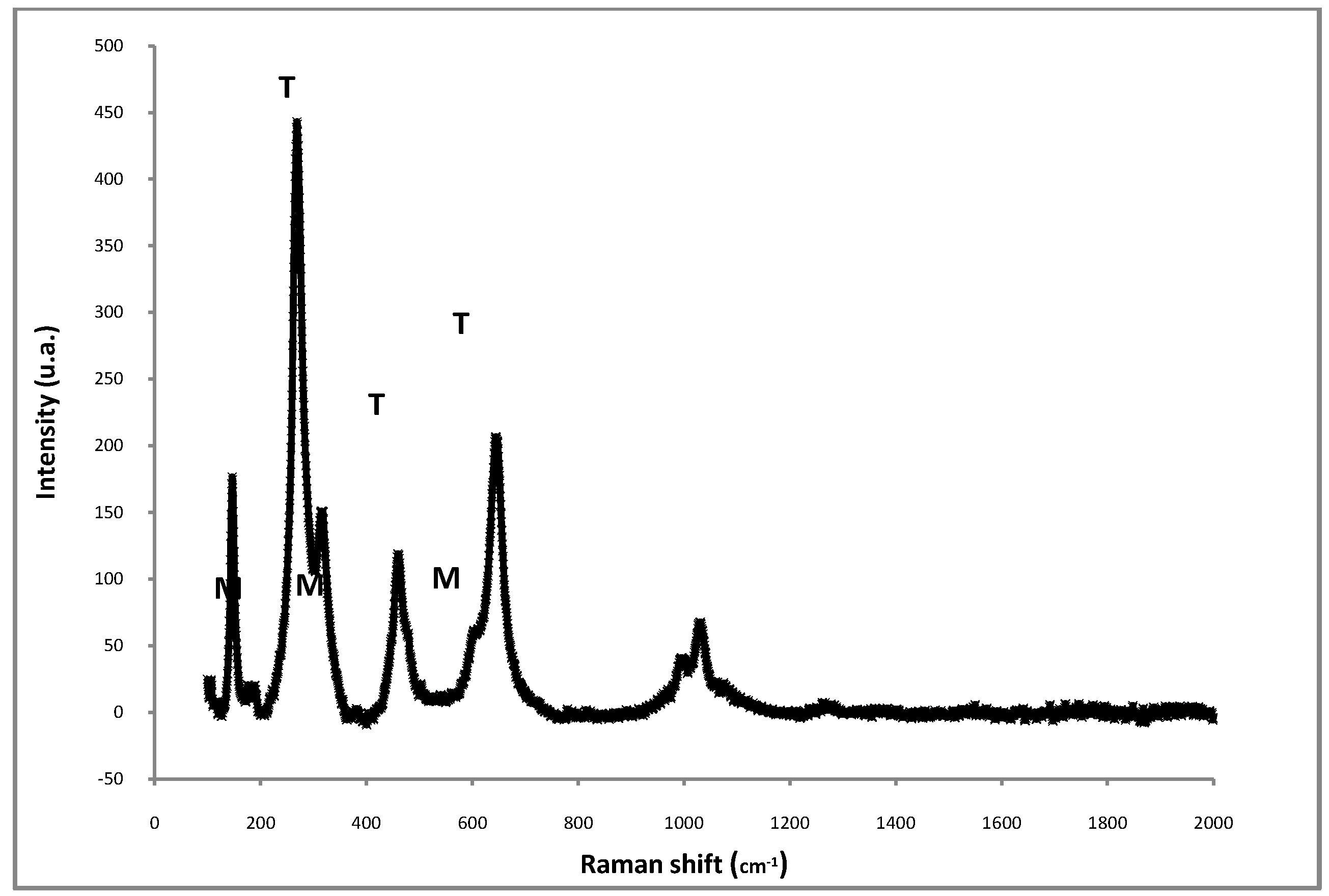
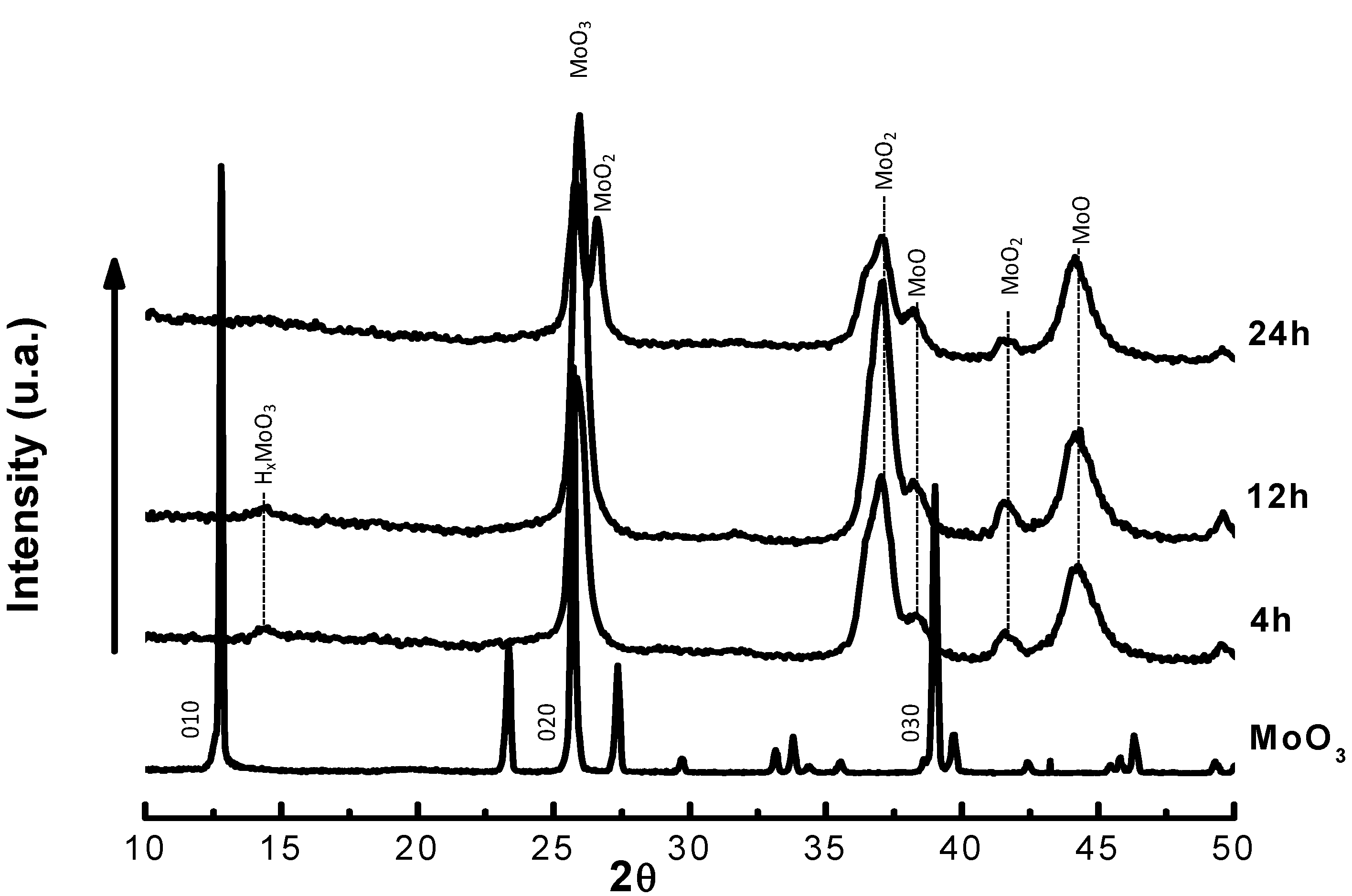
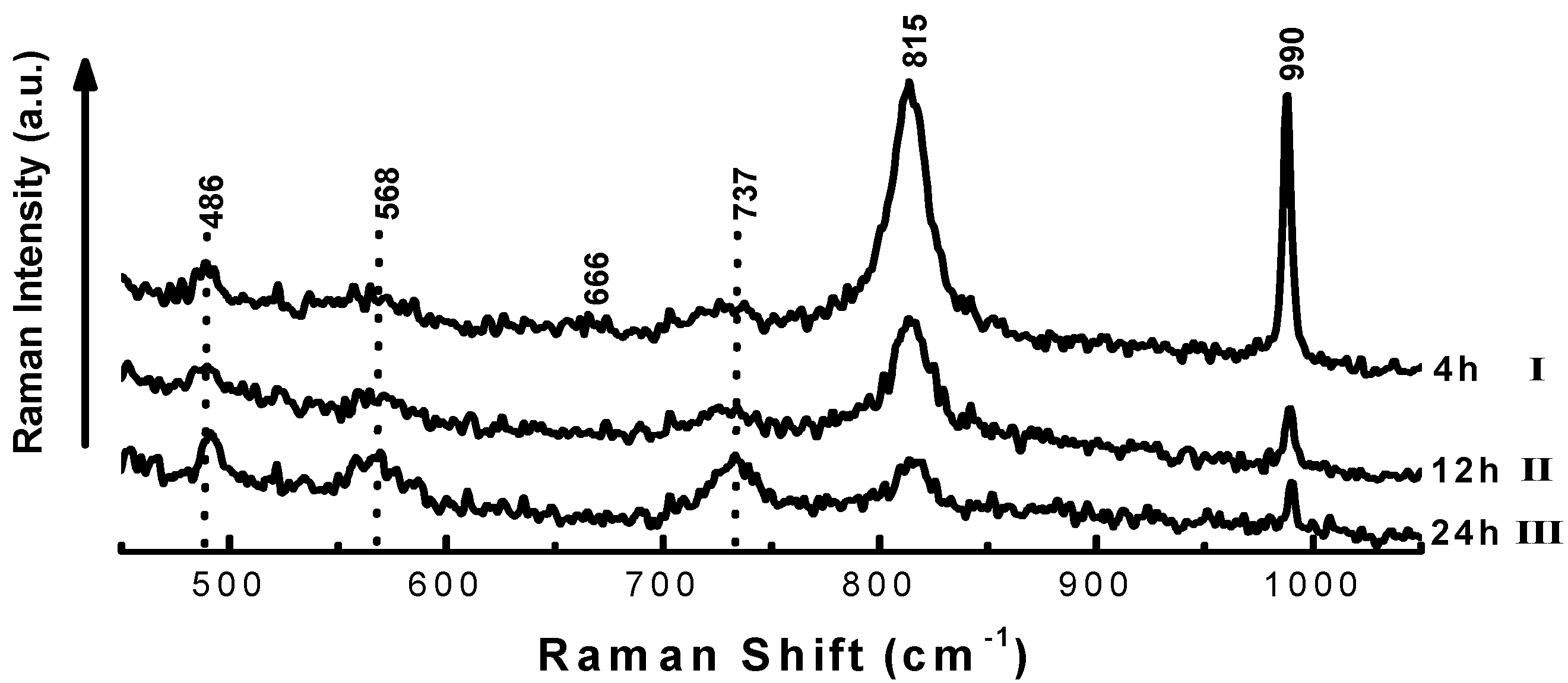
| Time on stream (h) | 2 | 4 | 6 | 8 |
|---|---|---|---|---|
| Conversion (wt %) | 33.33 | 49.35 | 53.23 | 43.47 |
| Selectivity to isomerization (wt %) | 73.96 | 70.49 | 69.96 | 77.57 |
| Selectivity to cracking (wt %) | 7.58 | 25.21 | 26.38 | 19.14 |
| Branched isomers (wt %) | 24.48 | 34.54 | 36.97 | 33.48 |
| Dimethylpentanes | 6.50 | 10.02 | 10.66 | 9.15 |
| Methylhexanes | 17.37 | 23.69 | 25.46 | 23.55 |
| 3-ethylpentane | 0.61 | 0.83 | 0.86 | 0.78 |
| Dimethylpentanes/ Methylhexanes | 0.37 | 0.42 | 0.41 | 0.39 |
| Cracked products (wt %) | ||||
| C1–C4 | 1.30 | 5.02 | 5.70 | 3.22 |
| C5–C6 | 1.20 | 7.32 | 8.23 | 5.04 |
| Time on stream (h) | 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 | 18 | 20 | 22 | 24 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conversion (wt %) | 33.21 | 49.84 | 64.36 | 66.85 | 69.31 | 70.24 | 74.21 | 74.43 | 75.43 | 76.39 | 77.45 | 78.01 |
| Selectivity to isomeriz. (wt %) | 89.94 | 92.82 | 92.11 | 92.12 | 90.68 | 92.33 | 89.79 | 89.43 | 87.93 | 86.44 | 85.34 | 87.45 |
| Selectivity to cracking (wt%) | 7.85 | 4.59 | 7.31 | 7.62 | 9.07 | 7.47 | 9.77 | 10.17 | 11.64 | 12.81 | 14.16 | 11.99 |
| Branched isomers (wt %) | 29.66 | 45.94 | 58.87 | 61.15 | 62.41 | 64.40 | 66.17 | 66.10 | 65.86 | 65.57 | 65.63 | 67.74 |
| Dimethylpentanes | 4.67 | 7.44 | 10.72 | 11.59 | 12.36 | 12.98 | 14.31 | 14.44 | 14.70 | 14.89 | 15.29 | 16.07 |
| Methylhexanes | 24.11 | 37.01 | 46.14 | 47.50 | 47.91 | 49.21 | 49.61 | 49.41 | 48.92 | 48.50 | 48.16 | 49.40 |
| 3-ethylpentane | 0.89 | 1.49 | 2.01 | 2.06 | 2.14 | 2.22 | 2.25 | 2.24 | 2.24 | 2.18 | 2.17 | 2.26 |
| DimethylC5/ MethylC6 | 0.194 | 0.201 | 0.232 | 0.244 | 0.258 | 0.264 | 0.288 | 0.292 | 0.300 | 0.307 | 0.317 | 0.325 |
| Cracked products (wt %) | ||||||||||||
| C1–C4 | 1.16 | 1.23 | 1.74 | 1.91 | 2.78 | 2.16 | 3.21 | 3.58 | 4.55 | 5.32 | 6.32 | 4.96 |
| C5–C6 | 1.43 | 1.04 | 2.93 | 3.15 | 3.46 | 3.05 | 3.99 | 3.94 | 4.17 | 4.40 | 4.57 | 4.33 |

3. Experimental
2.1. Sulfated Zirconia (SZ) Catalyst Preparation
2.2. Molybdenum Sub-Oxides Catalyst Preparation
2.3. Characterization
2.4. Catalysts Evaluation
2.4.1. Isomerization reaction with the Pt/SZ catalyst
2.4.2. Isomerization reaction with the molybdenum sub-oxides catalyst
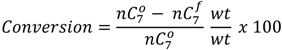
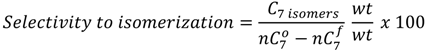
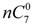 and
and 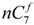 are the concentration of
are the concentration of 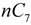 at the beginning of the test and at each sampling time respectively.
at the beginning of the test and at each sampling time respectively.4. Conclusions
References and Notes
- Ledoux, M.J.; Del Gallo, P.; Pham-Huu, C.; York, A.P.E. Molybdenum oxycarbide isomerization catalysts for cleaner fuel production. Catal. Today 1996, 27, 145–150. [Google Scholar]
- Blekkan, E.A.; Pham-Huu, C.; Ledoux, M.J.; Guille, J. Isomerization of n-heptane on an oxygen-modified molybdenum carbide catalyst. Ind. Eng. Chem. Res. 1994, 33, 1657–1664. [Google Scholar] [CrossRef]
- Ledoux, M.J.; Guille, J.; Pham-Huu, C.; Blekkan, E.A.; Peschiera, E. Process for the isomerisation of straight hydrocarbons containing at least 7 carbon atoms using catalysts with a base of molybdenum oxycarbide. U.S. Patent 5,576,466, 1996. [Google Scholar]
- Ledoux, M.J.; Meunier, F.; Heinrich, B.; Pham-Huu, C.; Harlin, M.E.; Krause, A.O.I. Part I. n-Butane dehydrogenation on unsupported carbon modified MoO3 (MoOxCy): Effect of stream on the catalyst stability. Appl. Catal. A 1999, 181, 157–170. [Google Scholar] [CrossRef]
- Delporte, P.; Meunier, F.; Pham-Huu, C.; Vennegues, P.; Ledoux, M.J.; Guille, J. Physical characterization of molybdenum oxycarbide catalyst: TEM, XRD and XPS. Catal. Today 1995, 23, 251–267. [Google Scholar]
- Delporte, P.; Pham-Huu, C.; Ledoux, M.J. Effect of the reaction temperature and hydrocarbon partial pressure on the activity of carbon-modified MoO3 for n-hexane isomerization. Appl. Catal. A 1997, 149, 151–180. [Google Scholar] [CrossRef]
- Bouchy, C.; Pham-Huu, C.; Heinrich, B.; Chaumont, C.; Ledoux, M.J. Microstructure and characterization of a highly selective catalyst for the isomerization of alkanes: A molybdenum oxycarbide. J. Catal. 2000, 190, 92–103. [Google Scholar] [CrossRef]
- Iglesia, E.; Baumgartner, J.E.; Ribeiro, F.H.; Boudart, M. Bifuntional reactions of alkanes on tungsten carbides modified by chemisorbed oxygen. J. Catal. 1991, 131, 523–544. [Google Scholar] [CrossRef]
- Brown, S.C.; Hargreaves, J.S.J.; Taylor, S.H. A study of “superacidic” MoO3/ZrO2 catalysts for methane oxidation. Catal. Lett. 1999, 57, 109–113. [Google Scholar] [CrossRef]
- Scheithauer, M.; Grasselli, R.K.; Knözinger, H. Genesis and structure of WOx/ZrO2 solid acid catalysts. Langmuir 1998, 14, 3019–3029. [Google Scholar] [CrossRef]
- Zalewski, D.J.; Saeed, A.; Doolin, P.K. Characterization of catalytically active sulfated zirconia. Catal. Today 1999, 53, 419–432. [Google Scholar] [CrossRef]
- Alemán-Vázquez, L.O.; Mariel-Reyes, P.R.; Cano-Domínguez, J.L. The effect of sulfates concentration in sulfated zirconia (SZ) catalysts on n-heptane isomerization. Pet.Sci. Tech. 2010, 28, 374–381. [Google Scholar] [CrossRef]
- Normair, C.J.; Goulding, P.A.; McAlpine, I. Role of anions in the surface area. Catal. Today 1994, 20, 313–317. [Google Scholar] [CrossRef]
- Sameer, V.; Wolf, E.E. A highly active and stable platinum-modified sulfated zirconia catalyst 1. Preparation and activity for n-pentane isomerization. Appl. Catal. A 2004, 264, 117–124. [Google Scholar] [CrossRef]
- Sayari, A.; Dicko, A. The state of platinum in Pt on sulfated zirconia superacid catalysts. J. Catal. 1994, 145, 561–564. [Google Scholar] [CrossRef]
- Grau, J.M.; Parera, J.M. Single and composite bifunctional catalysts of H-MOR or SO42−-ZrO2 for n-octane hydroisomerization-cracking. Influence of the porosity of the acid component. Appl. Catal. A: General 1997, 162, 17–27. [Google Scholar] [CrossRef]
- Iglesia, E.; Soled, S.L.; Kramer, G.M. Isomerization of alkanes on sulfated zirconia: Promotion by Pt and by adamantyl hydride transfer species. J. Catal. 1993, 144, 238–253. [Google Scholar] [CrossRef]
- Comelli, R.A.; Finelli, Z.R.; Vaudagna, S.R. Hydroisomerization of n-hexane on Pt/SO42−-ZrO2: Effect of total and hydrogen partial pressure. Catal. Lett. 1997, 45, 227–231. [Google Scholar] [CrossRef]
- Hino, M.; Kobayashi, S.; Arata, K. Reactions of butane and isobutene catalyzed by zirconium oxide treated with sulfate ion. J. Am. Chem. Soc. 1979, 101, 6439–6441. [Google Scholar] [CrossRef]
- Yamaguchi, T. Application of ZrO2 as a catalyst and a catalyst support. Catal. Today 1994, 20, 199–217. [Google Scholar] [CrossRef]
- Babou, F.; Coudurier, G.; Vedrine, J.C. Acidic properties of sulfated zirconia: An infrared spectroscopic study. J. Catal. 1995, 152, 341–349. [Google Scholar]
- Garin, F.; Andriamasinoro, D.; Abdulsamad, A.; Sommer, J. Conversion of butane over the solid superacid ZrO2/SO42− in the presence of hydrogen. J. Catal. 1991, 131, 199–203. [Google Scholar]
- Benaïssa, M.; Santiesteban, J.G.; Díaz, G.; Chang, C.D. Interaction of sulfate groups with the surface of zirconia: An HRTEM characterization study. J. Catal. 1996, 161, 694–703. [Google Scholar] [CrossRef]
- Jouanne, M.; Morhange, J.F.; Kanehisa, M.; Haro-Poniatowski, E.; Fuentes, G.A.; Torres, E.; Hernández-Tellez, E. Structural transformation in nanosized zirconium oxide. Phys. Rev. B 2001, 64, 154041–154047. [Google Scholar]
- Torres-García, E.; Peláiz-Barranco, A.; Vázquez-Ramos, C.; Fuentes, G.A. Thermal and structural characterization of the ZrO2−x(OH)2x to ZrO2 transition. J. Mater. Res. 2001, 16, 2209–2212. [Google Scholar] [CrossRef]
- Vera, C.R.; Pieck, C.L.; Shimizu, K.; Parera, J.M. Tetragonal structure anionic vacancies and catalytic activity of SO42−-ZrO2 catalysts for n-butane isomerization. Appl. Catal. A: General 2002, 230, 137–151. [Google Scholar] [CrossRef]
- Torres-García, E.; Rodríguez-Gattorno, G.; Ascencio-Gutierrez, J.A.; Alemán-Vázquez, L.O.; Cano-Domínguez, J.L.; Martínez-Hernández, A.; Santiago-Jacinto, P. New insights on molybdenum suboxide: Nature of carbons in isomerization reactions. J. Phys.Chem. B 2005, 109, 17518–17525. [Google Scholar]
- Rodríguez-Gattorno, G.; Martínez-Hernández, A.; Aleman-Vázquez, L.O.; Torres-García, E. Structural and thermal study of carbon-modified molybdenum sub-oxide catalysts. Appl. Catal. A: General 2007, 321, 117–124. [Google Scholar] [CrossRef]
- Resofszki, G.; Muhler, M.; Sprenger, S.; Wild, U.; Paál, Z. Electron spectroscopy of sulfated zirconia, its activity in n-hexane conversion and possible reasons of its deactivation. Appl. Catal. A: General 2003, 240, 71–81. [Google Scholar] [CrossRef]
- Ahmad, R.; Melsheimer, J.; Friederike, C.; Jentoft, F.C.; Schlögl, R. Isomerization of n-butane and of n-pentane in the presence of sulfated zirconia: Formation of surface deposits investigated by in situ UV-vis diffuse reflectance spectroscopy. J. Catal. 2003, 218, 365–374. [Google Scholar]
- Samples Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alemán-Vázquez, L.O.; Cano-Domínguez, J.L.; Torres-García, E.; Villagómez-Ibarra, J.R. Industrial Aplication of Catalytic Systems for n-Heptane Isomerization. Molecules 2011, 16, 5916-5927. https://doi.org/10.3390/molecules16075916
Alemán-Vázquez LO, Cano-Domínguez JL, Torres-García E, Villagómez-Ibarra JR. Industrial Aplication of Catalytic Systems for n-Heptane Isomerization. Molecules. 2011; 16(7):5916-5927. https://doi.org/10.3390/molecules16075916
Chicago/Turabian StyleAlemán-Vázquez, Laura Olivia, José Luis Cano-Domínguez, Enelio Torres-García, and José Roberto Villagómez-Ibarra. 2011. "Industrial Aplication of Catalytic Systems for n-Heptane Isomerization" Molecules 16, no. 7: 5916-5927. https://doi.org/10.3390/molecules16075916
APA StyleAlemán-Vázquez, L. O., Cano-Domínguez, J. L., Torres-García, E., & Villagómez-Ibarra, J. R. (2011). Industrial Aplication of Catalytic Systems for n-Heptane Isomerization. Molecules, 16(7), 5916-5927. https://doi.org/10.3390/molecules16075916