Opportunities Offered by Chiral η6-Arene/N-Arylsulfonyl-diamine-RuII Catalysts in the Asymmetric Transfer Hydrogenation of Ketones and Imines

Abstract
:1. Introduction
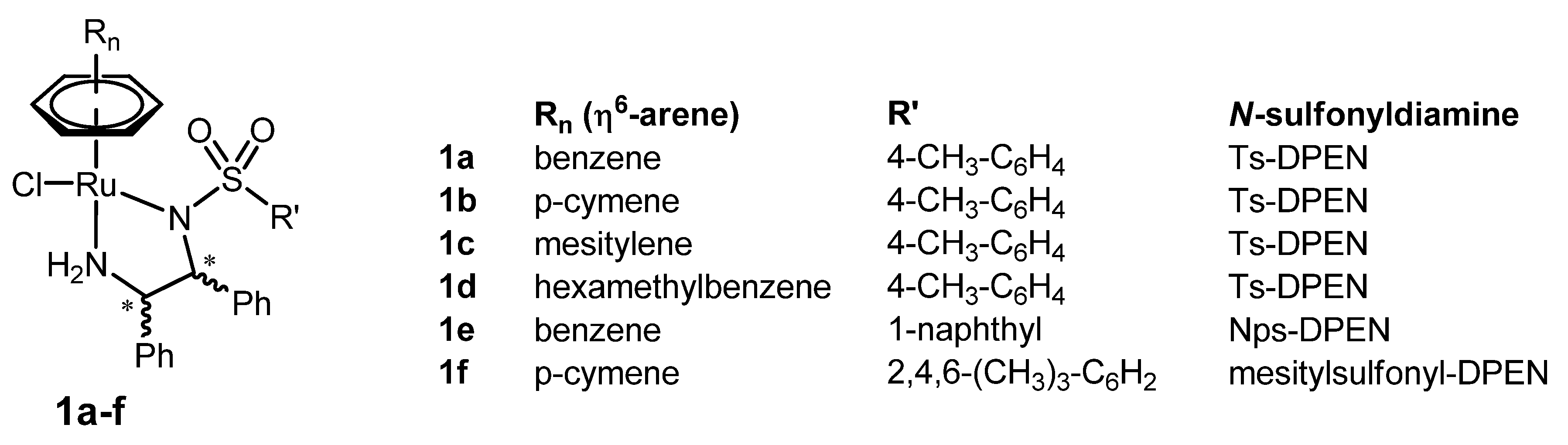
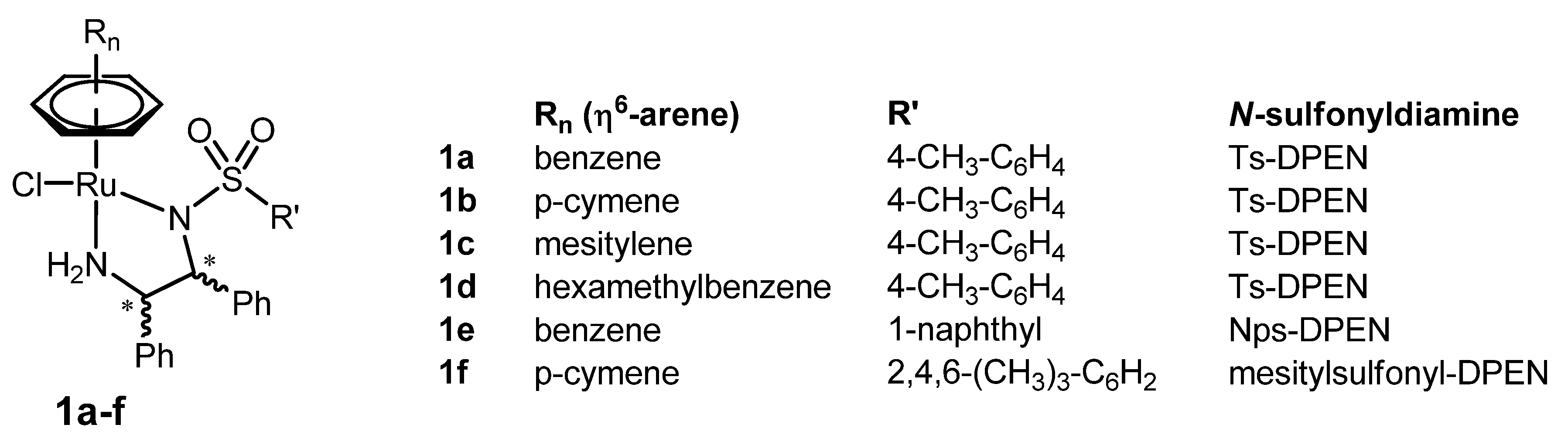
2. Original η6-Arene/N-sulfonyldiamine-RuII Catalysts
2.1. The Pioneering Works on η6-Arene/N-sulfonyldiamine-RuII Catalysts
2.2. ATH Catalyzed by 1 in Water
2.3. Attempts to Immobilize Unmodified Complexes 1
2.4. ATH Catalyzed by 1 in Ionic Liquids
3. Modifications of the η6-Arene/N-sulfonyldiamine-RuII Complexes
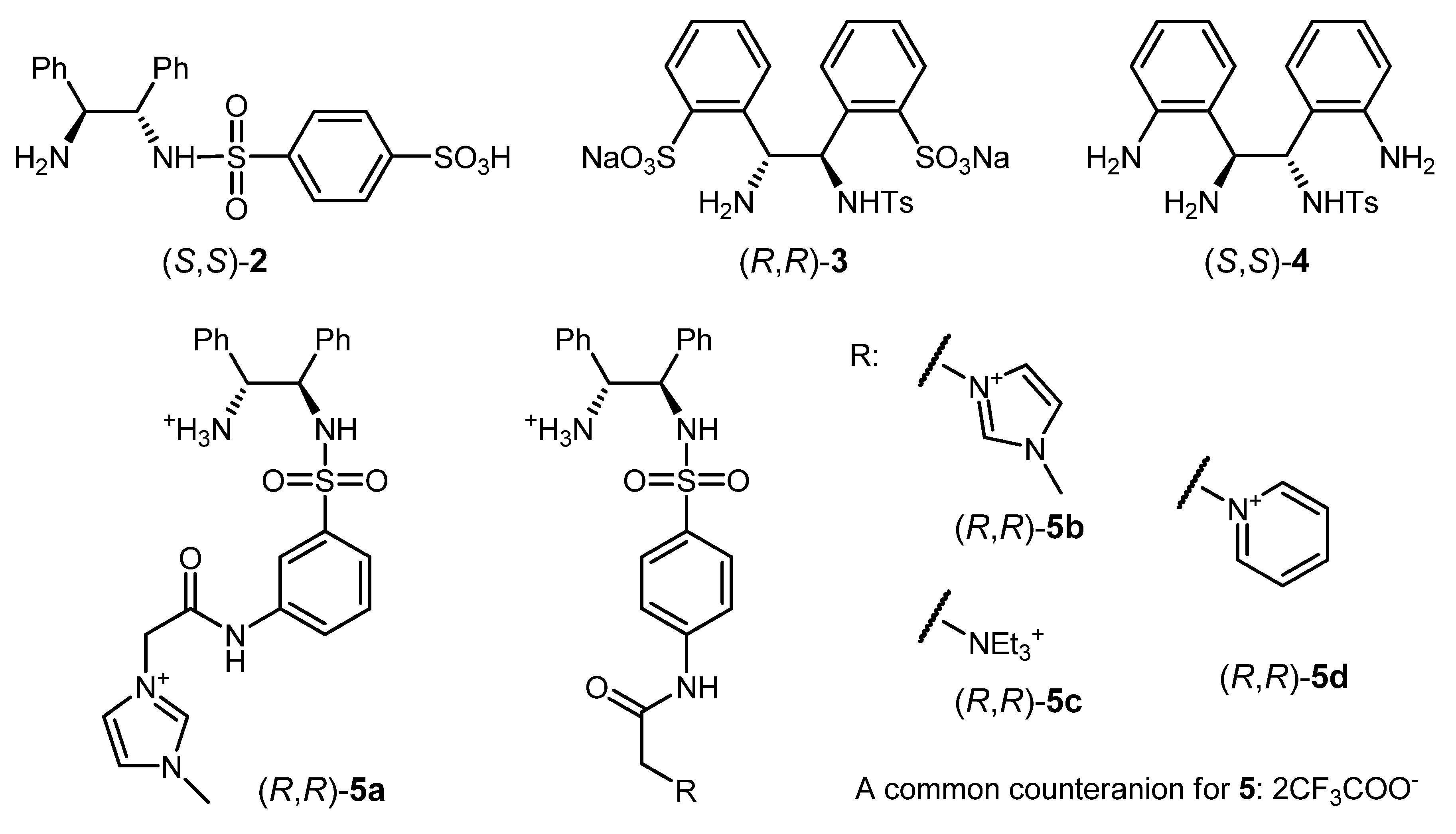
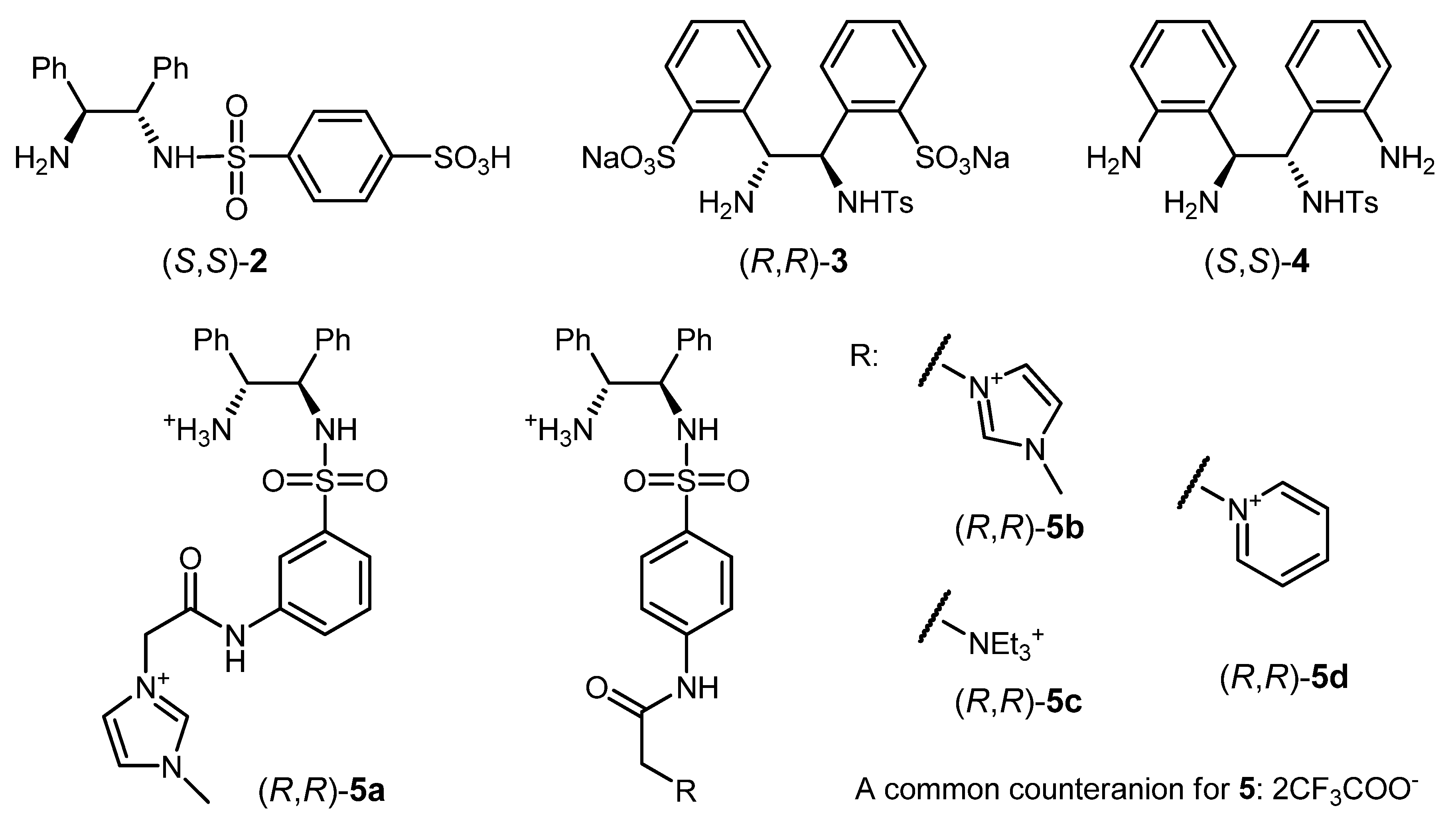
3.1. Modifications of 1 Facilitating ATH in Aqueous Media
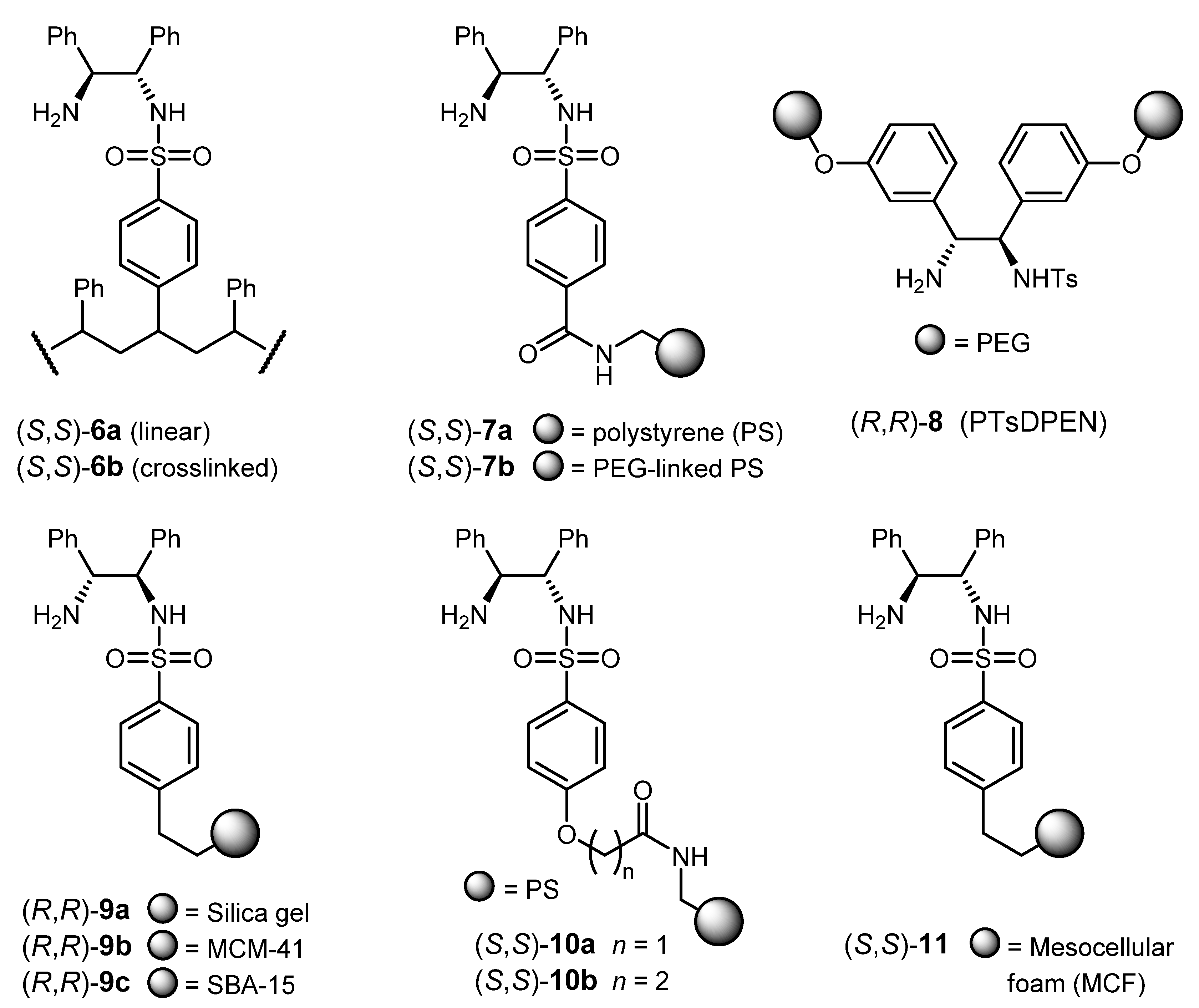
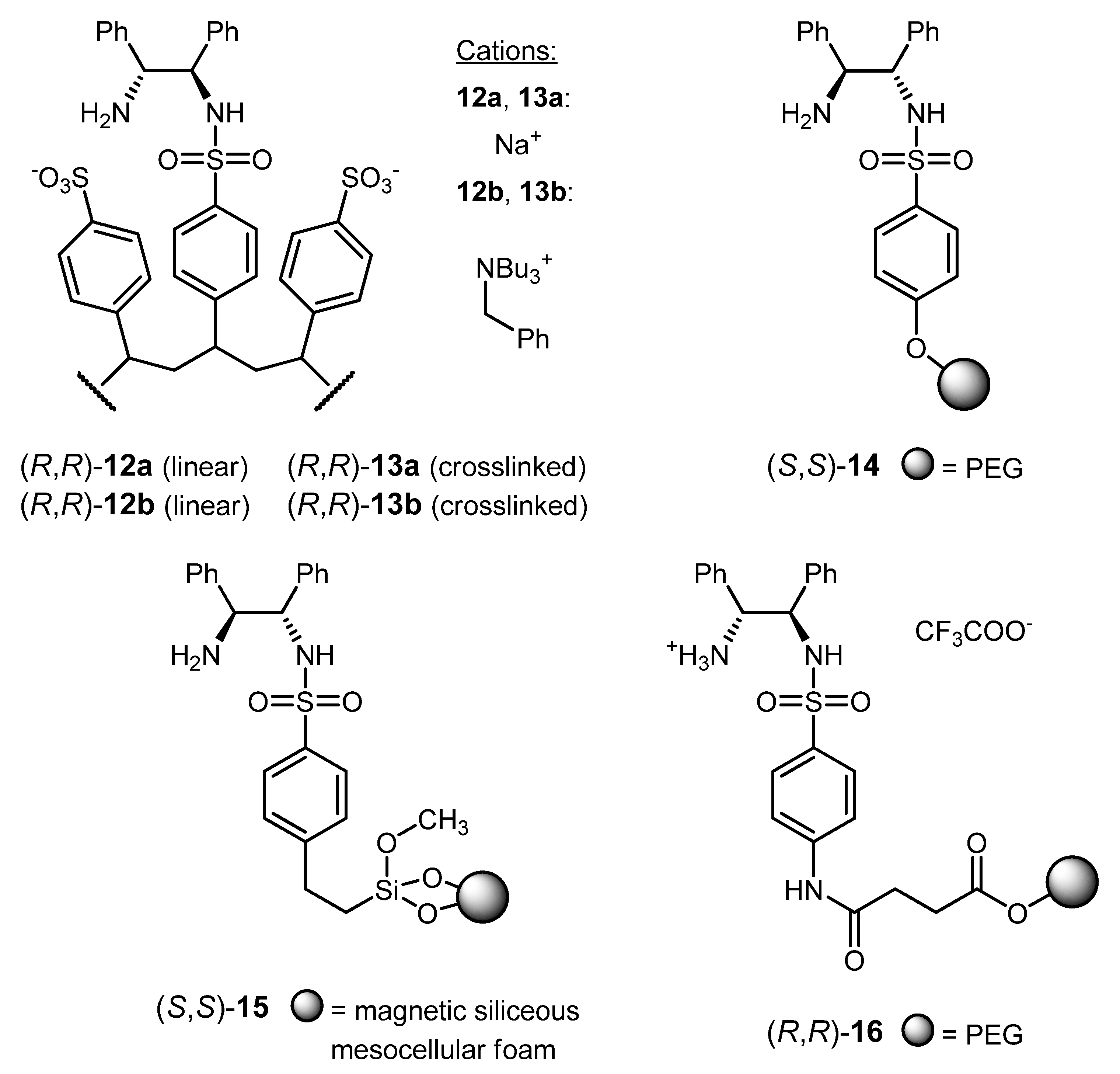
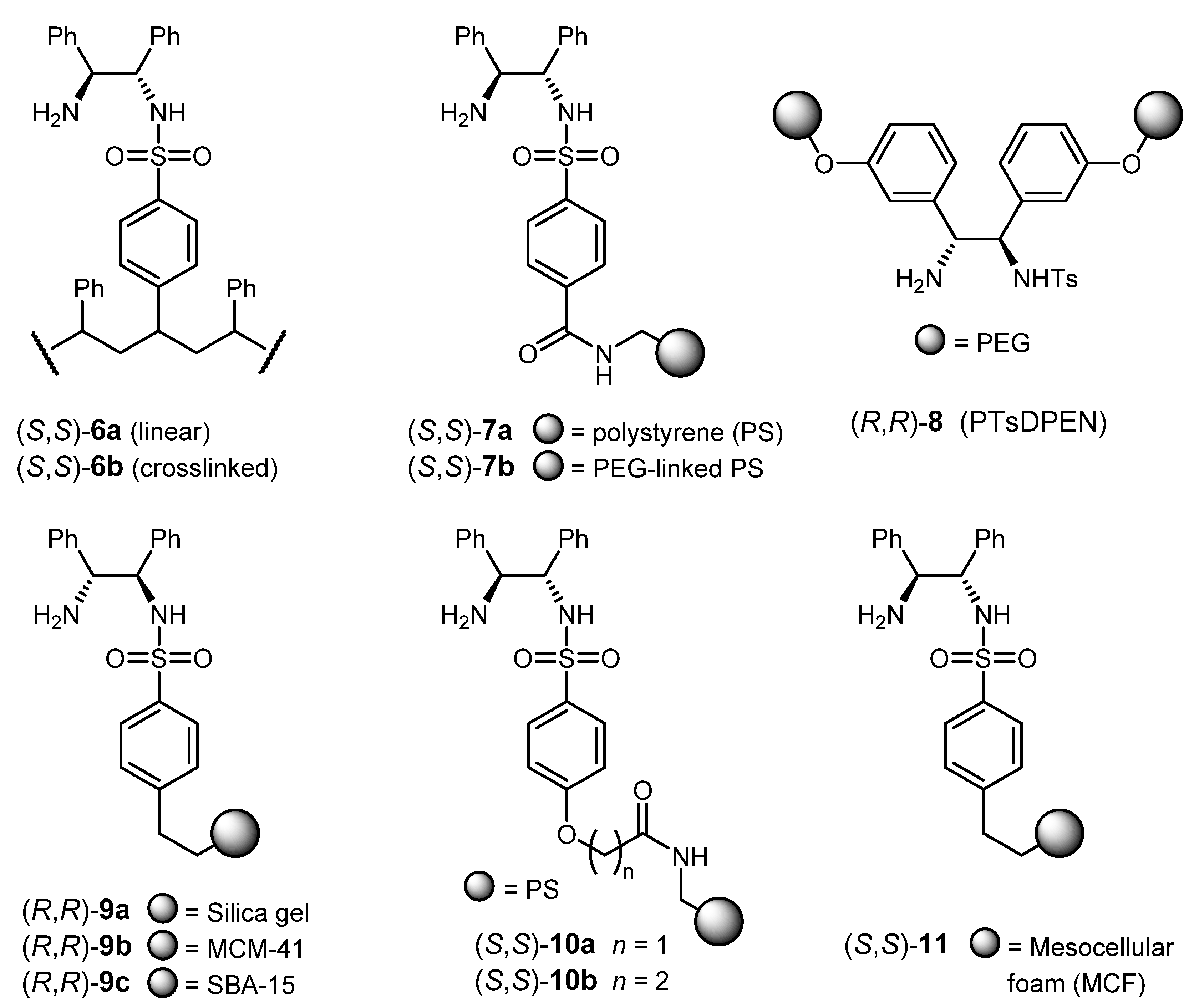
3.2. ATH Conducted on Immobilized Catalysts Derived from 1
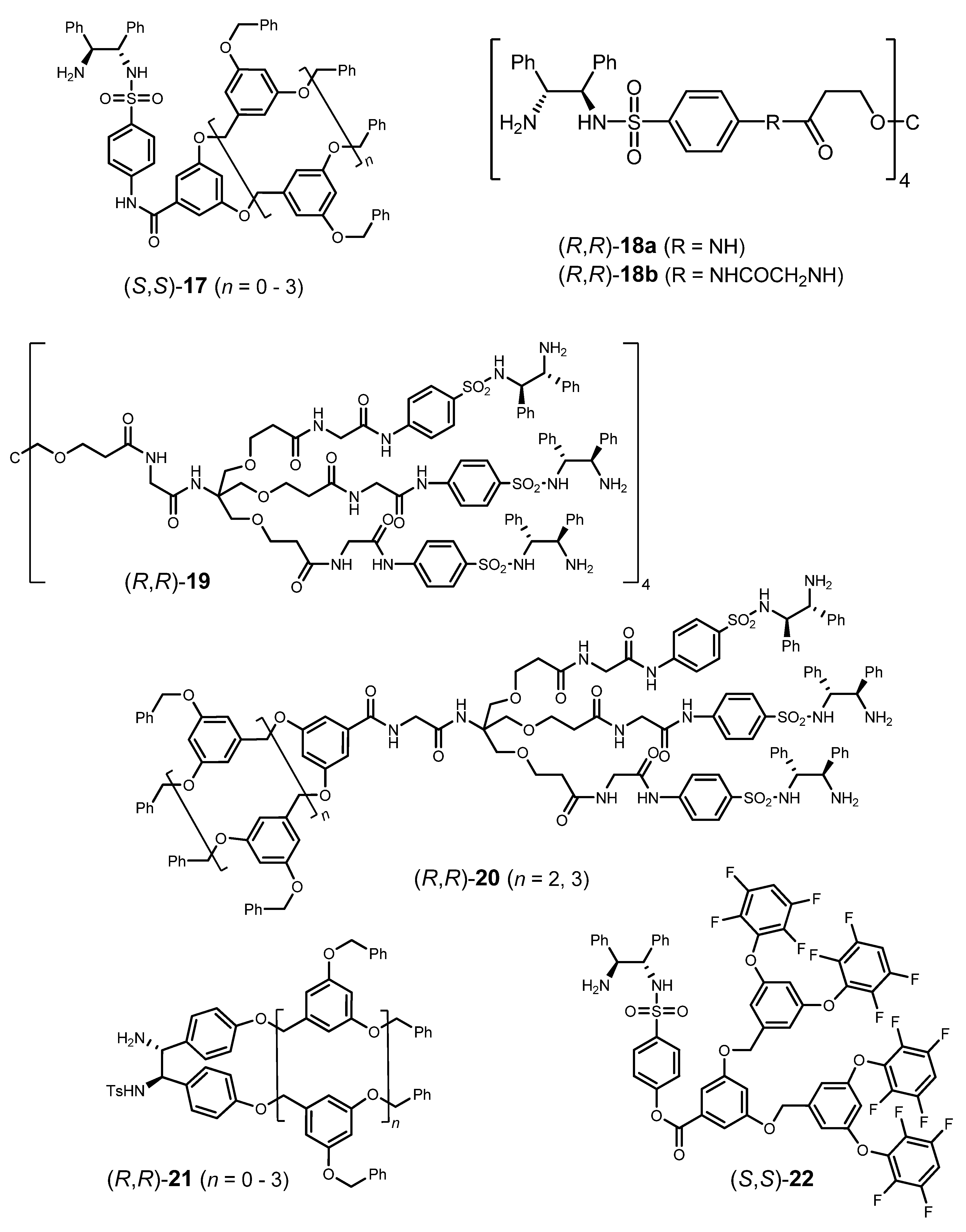
3.3. ATH Conducted on Dendrimeric Catalysts Derived from 1
3.4. ATH Conducted in Ionic Liquids Using Modified Catalysts 1
3.5. Biomimetic Modifications of 1
4. Mechanistic Considerations
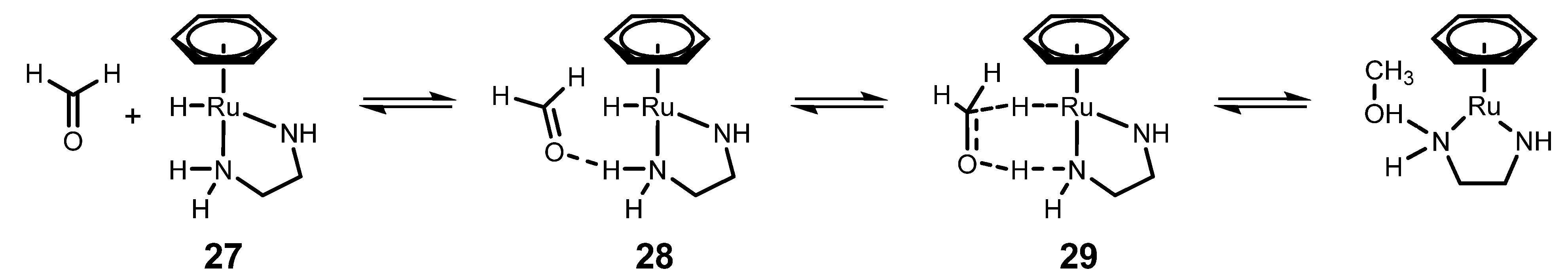
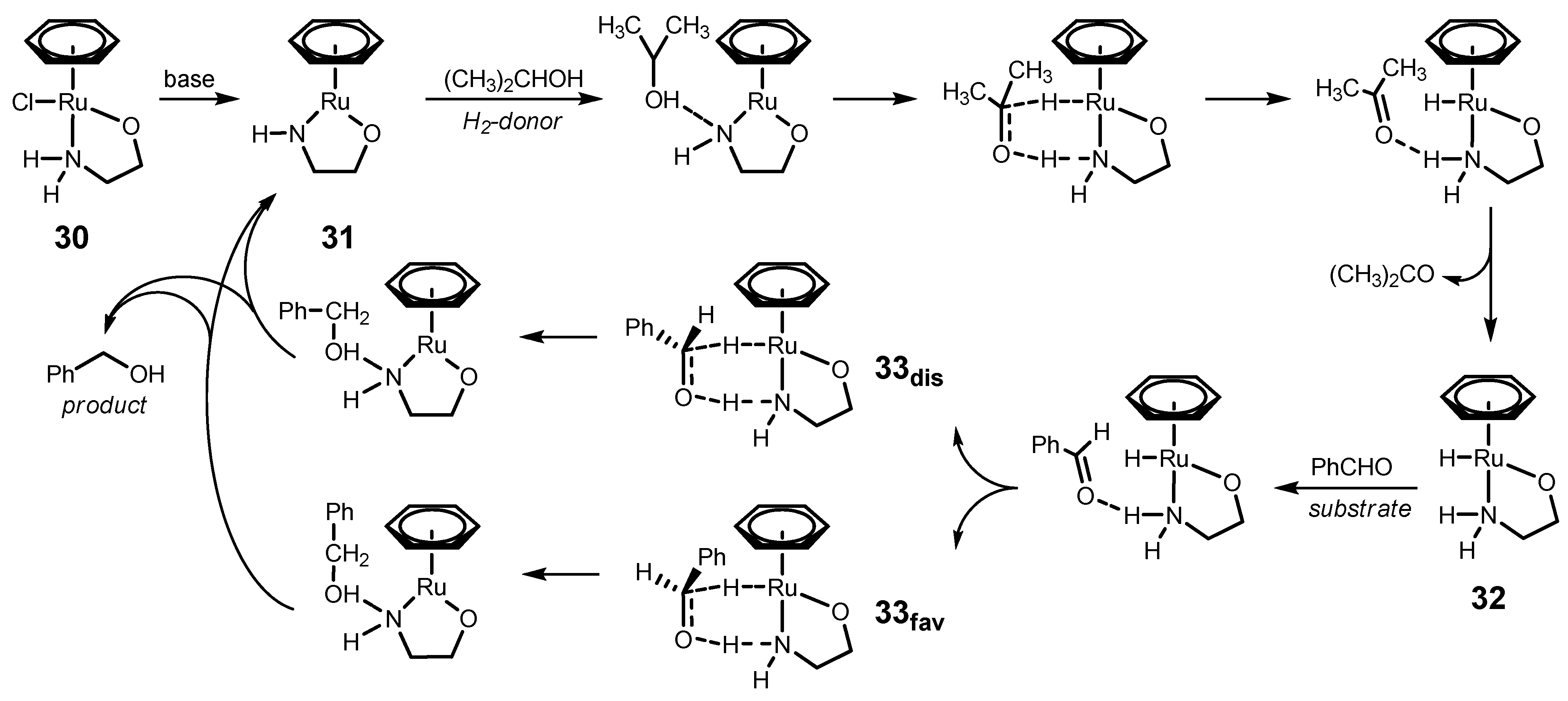
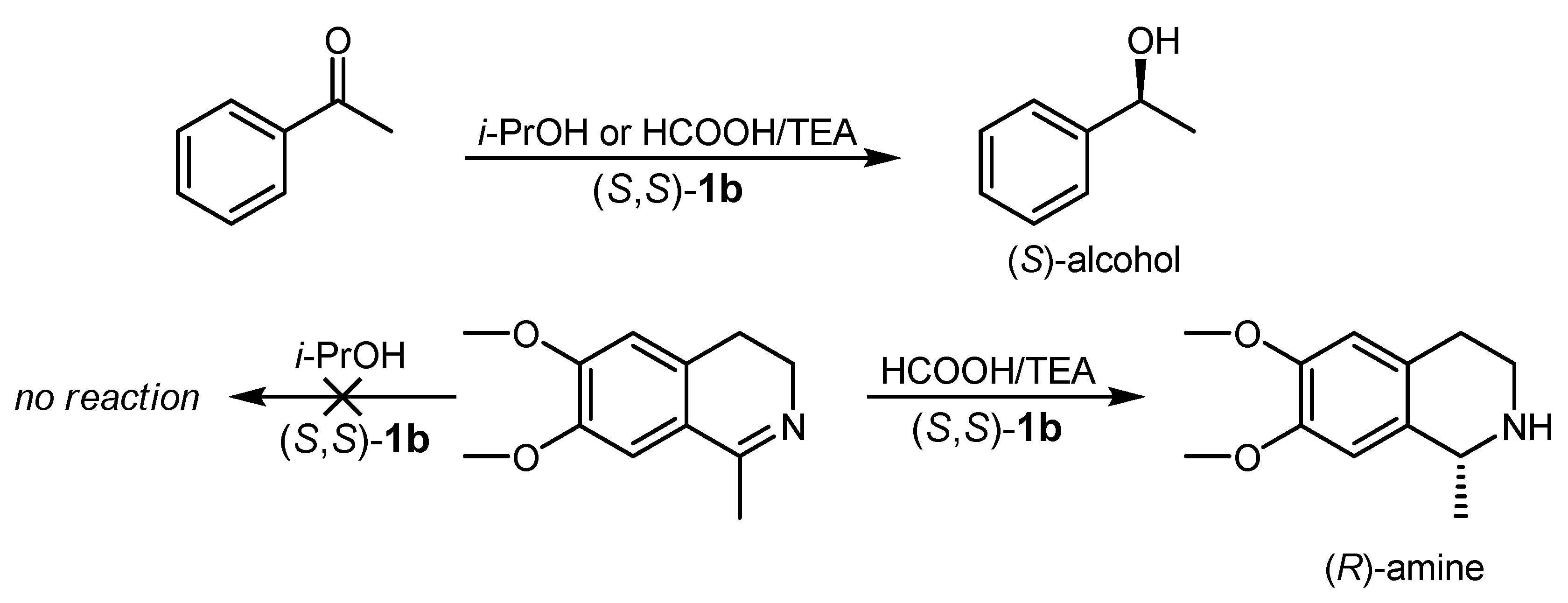
4.1. ATH of Ketones Catalyzed by 1 in the Presence of Isopropanol and a Strong Base
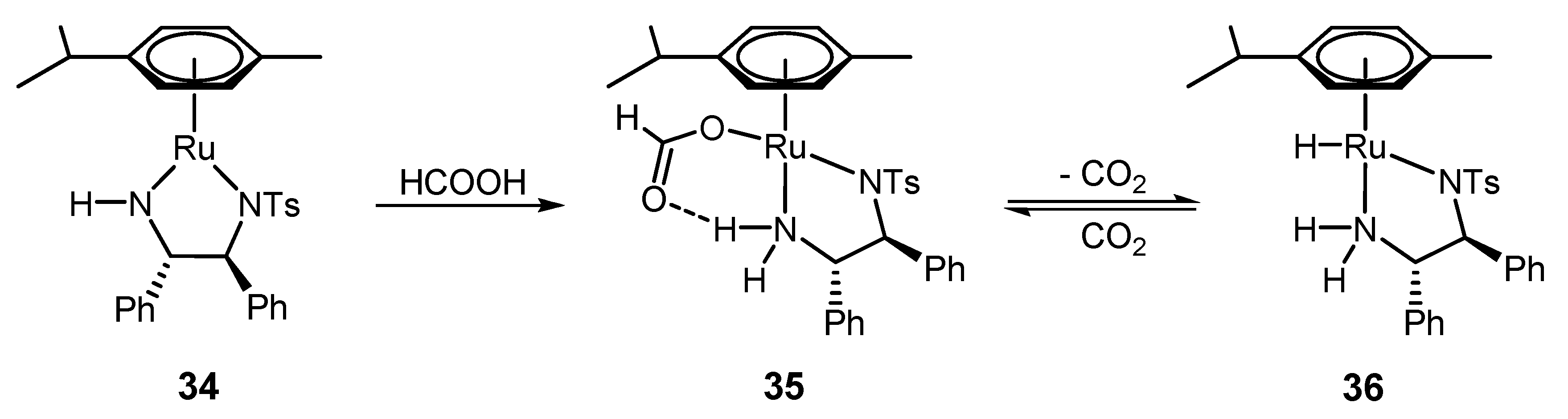
4.2. ATH Catalyzed by 1 in the Presence of the HCOOH/Triethylamine Azeotrope
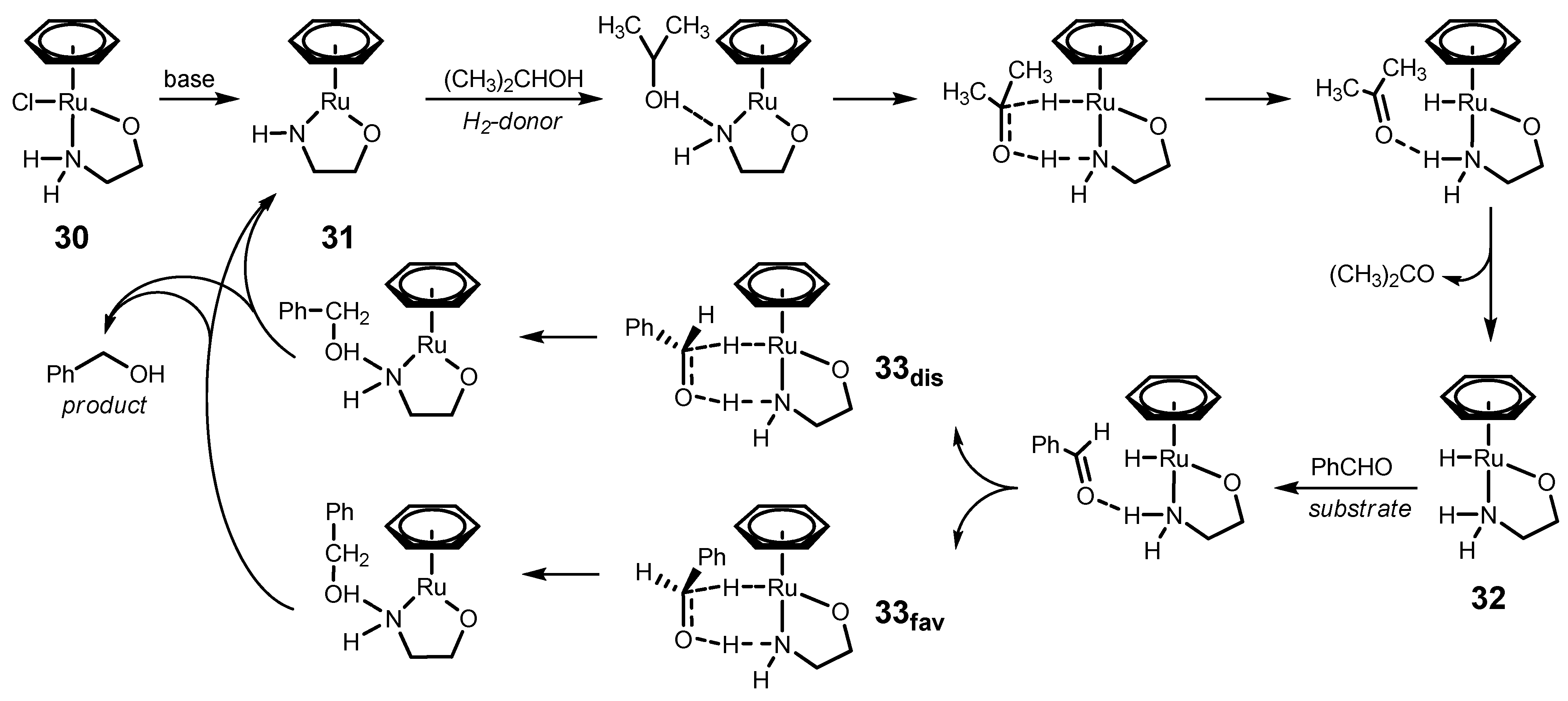
4.3. Mechanism of ATH Catalyzed by 1 in Water
4.4. ATH of Imines Catalyzed by 1 in the Presence of the HCOOH/Triethylamine Azeotrope
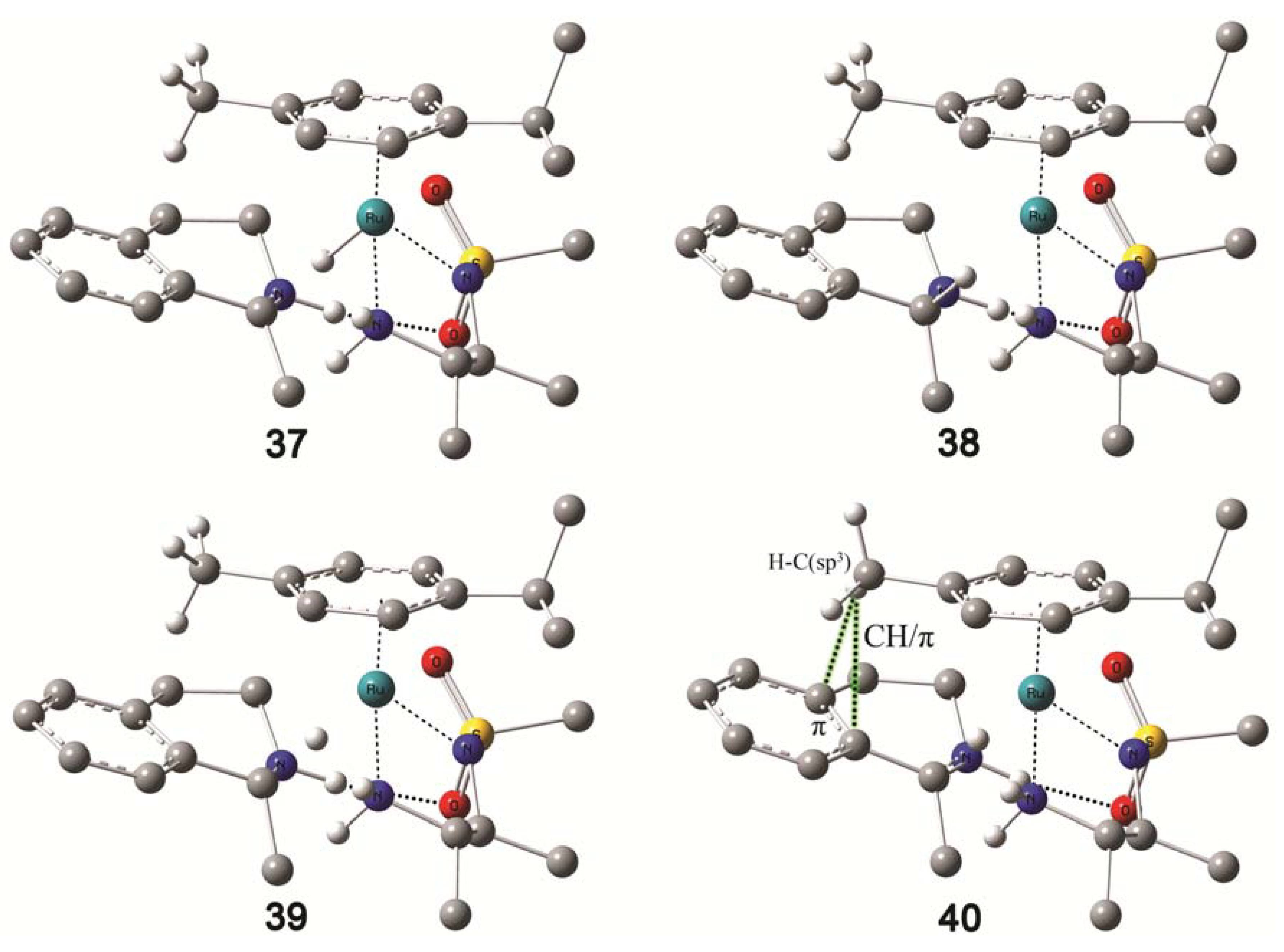
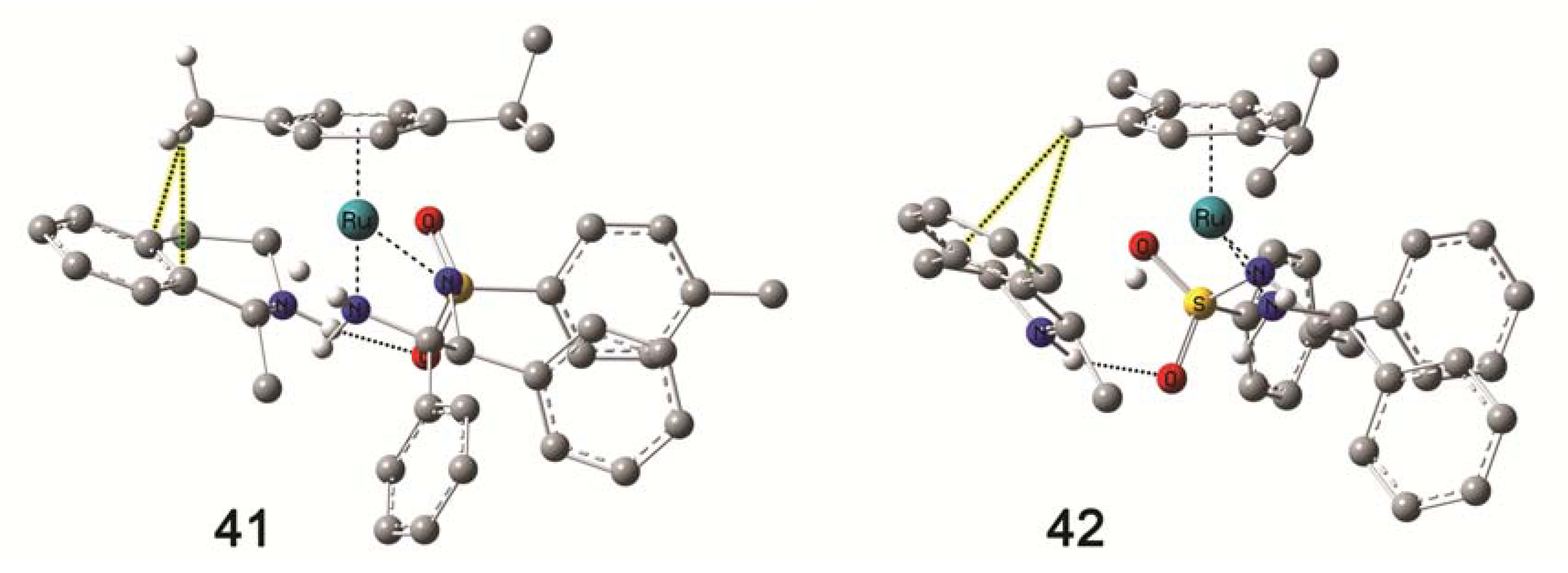
4.5. Molecular Modelling
5. Conclusions
Acknowledgements
References and Notes
- Davies, N.M.; Teng, X.W. Importance of chirality in drug therapy and pharmacy practice: Implications for psychiatry. Adv. Pharm. 2003, I, 242–252. [Google Scholar]
- Arnum, P.V. Single-enantiomer drugs drive advances in asymmetric synthesis. Pharm. Technol. 2006, 30, 58–67. [Google Scholar]
- Lin, G.-Q.; Li, Y.-M.; Chan, A.S.C. Principles and Applications of Asymmetric Synthesis; John Wiley & Sons, Inc.: New York, NY, USA, 2001. [Google Scholar]
- Liu, W. Resolutions at Large Scale: Case Studies. In Handbook of Chiral Chemicals, 2nd ed.; Ager, D., Ed.; CRC Taylor & Francis Group: Boca Raton, FL, USA, 2006; pp. 75–95. [Google Scholar]
- Li, Z.J.; Grant, D.J.W. Relationship between physical properties and crystal structures of chiral drugs. J. Pharm. Sci. 1997, 86, 1073–1078. [Google Scholar] [PubMed]
- Subramanian, G. Chiral Separation Techniques: A Practical Approach, 3rd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2006, 35, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Drauz, K.; Waldmann, H. Enzyme Catalysis in Organic Synthesis, 2nd ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2002. [Google Scholar]
- Noyori, R. Asymmetric Catalysis In Organic Synthesis; John Wiley & Sons, Inc.: New York, NY, USA, 1994. [Google Scholar]
- Blaser, H.-U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Selective hydrogenation for fine chemicals: Recent trends and new developments. Adv. Synth. Catal. 2003, 345, 103–151. [Google Scholar] [CrossRef]
- Ojima, I. Catalytic Asymmetric Synthesis, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Noyori, R. Asymmetric catalysis: Science and opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric hydrogenations (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 1998–2007. [Google Scholar] [CrossRef]
- Knowles, W.S.; Noyori, R. Pioneering perspectives on asymmetric hydrogenation. Acc. Chem. Res. 2007, 40, 1238–1239. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.P.; Kagan, H.B. The asymmetric synthesis of hydratopic acid and amino-acids by homogeneous catalytic hydrogenation. Chem. Commun. 1971, 481. [Google Scholar] [CrossRef]
- Kagan, H.B.; Dang, T.-P. Asymmetric catalytic reduction with transition metal complexes. I. A catalytic system of Rhodium(I) with (–)-2,3-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane, a new chiral diphosphine. J. Am. Chem. Soc. 1972, 94, 6429–6433. [Google Scholar] [CrossRef]
- Knowles, W.S.; Sabacky, M.J.; Vineyard, B.D. Catalytic asymmetric hydrogenation. J. Chem. Soc. Chem. Commun. 1972, 10–11. [Google Scholar] [CrossRef]
- Börner, A. Phosphorus Ligands in Asymmetric Catalysis, Synthesis and Applications; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Tang, W.; Zhang, X. New chiral phosphorous ligands for enantioselective hydrogenation. Chem. Rev. 2003, 103, 3029–3069. [Google Scholar] [CrossRef] [PubMed]
- Helmchen, G.; Pfaltz, A. Phosphinooxazolines – A new class of versatile, modular P,N-ligands for asymmetric catalysis. Acc. Chem. Res. 2000, 33, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Murata, K.; Okano, K.; Miyagi, M.; Iwane, H.; Noyori, R.; Ikariya, T. A practical stereoselective synthesis of chiral hydrobenzoins via asymmetric transfer hydrogenation of benzils. Org. Lett. 1999, 1, 1119–1121. [Google Scholar] [CrossRef]
- Vedejs, E.; Trapencieris, P.; Suna, E. Substituted isoquinolines by noyori transfer hydrogenation: Enantioselective synthesis of chiral diamines containing an aniline subunit. J. Org. Chem. 1999, 64, 6724–6729. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Murata, K.; Ikariya, T. Stereoselective synthesis of optically active a-hydroxy ketones and anti-1,2-diols via asymmetric transfer hydrogenation of unsymmetrically substituted 1,2-diketones. Org. Lett. 2000, 2, 3833–3836. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Muratsugu, S.; Ikariya, T. Stereoselective synthesis of optically active pyridyl alcohols via asymmetric transfer hydrogenation of pyridyl ketones. Tetrah. Lett. 2000, 41, 9277–9280. [Google Scholar] [CrossRef]
- Yamada, I.; Noyori, R. Asymmetric transfer hydrogenation of benzaldehydes. Org. Lett. 2000, 2, 3425–3427. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Murata, K.; Ikariya, T. Practical synthesis of optically active amino alcohols via asymmetric transfer hydrogenation of functionalized aromatic ketones. J. Org. Chem. 2002, 67, 1712–1715. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Chen, Y.-C.; Cui, X.; Wang, Q.-W.; Zhu, J.; Deng, J.-G. Transfer hydrogenation of activated C=C bonds catalyzed by ruthenium amido complexes: Reaction scope, limitation and enantioselectivity. J. Org. Chem. 2005, 70, 3584–3591. [Google Scholar] [CrossRef] [PubMed]
- Samano, V.; Ray, J.A.; Thompson, J.B.; Mook, R.A.; Jung, D.K.; Koble, C.S.; Martin, M.T.; Bigham, E.C.; Regitz, C.S.; Feldman, P.L.; et al. Synthesis of ultra-short-acting neuromuscular blocker GW 0430: A remarkably stereo- and regioselective synthesis of mixed tetrahydroisoquinolinium chlorofumarates. Org. Lett. 1999, 1, 1993–1996. [Google Scholar] [CrossRef] [PubMed]
- Meuzelaar, G.J.; van Vliet, M.C.A.; Maat, L.; Sheldon, R.A. Improvements in the total synthesis of morphine. Eur. J. Org. Chem. 1999, 1999, 2315–2321. [Google Scholar] [CrossRef]
- Miyagi, M.; Takehara, J.; Collet, S.; Okano, K. Practical synthesis of (S)-1-(3-trifluoromethylphenyl)ethanol via Ruthenium(II)-catalyzed asymmetric transfer hydrogenation. Org. Proc. Res. Dev. 2000, 4, 346–348. [Google Scholar] [CrossRef]
- Tietze, L.F.; Zhou, Y.; Töpken, E. Synthesis of simple enantiopure tetrahydro-β-carbolines and tetrahydroisoquinolines. Eur. J. Org. Chem. 2000, 2000, 2247–2252. [Google Scholar] [CrossRef]
- Szawkalo, J.; Czarnocki, Z. Enantioselective synthesis of some tetracyclic isoquinoline alkaloids by asymmetric transfer hydrogenation catalysed by a chiral ruthenium complex. Monatsh. Chem. 2005, 136, 1619–1627. [Google Scholar] [CrossRef]
- Roszkowski, P.; Wojtasiewicz, K.; Leniewski, A.; Maurin, J.K.; Lis, T.; Czarnocki, Z. Enantioselective synthesis of 1-substituted tetrahydro-b-carboline derivatives via the asymmetric transfer hydrogenation. J. Mol. Catal., A Chem. 2005, 232, 143–149. [Google Scholar] [CrossRef]
- Roszkowski, P.; Maurin, J.K.; Czarnocki, Z. Enantioselective synthesis of (R)-(–)-praziquantel (PZQ). Tetrahedron Asymmetry 2006, 17, 1415–1419. [Google Scholar] [CrossRef]
- Szawkalo, J.; Czarnocki, S.J.; Zawadzka, A.; Wojtasiewicz, K.; Leniewski, A.; Maurin, J.K.; Czarnocki, Z.; Drabowicz, J. Enantioselective synthesis of some tetrahydroisoquinoline and tetrahydro-b-carboline alkaloids. Tetrahedron Asymmetry 2007, 18, 406–413. [Google Scholar] [CrossRef]
- Cheng, J.-J.; Yang, Y.-S. Enantioselective total synthesis of (−)-(S)-stepholidine. J. Org. Chem. 2009, 74, 9225–9228. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.J.; Wills, M. Asymmetric transfer hydrogenation of C=O and C=N bonds. Tetrahedron Asymmetry 1999, 10, 2045–2061. [Google Scholar] [CrossRef]
- Wills, M.; Palmer, M.; Smith, A.; Kenny, J.; Walsgrove, T. Recent developments in the area of asymmetric transfer hydrogenation. Molecules 2000, 5, 4–18. [Google Scholar] [CrossRef]
- Everaere, K.; Mortreux, A.; Carpentier, J.-F. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of carbonyl compounds with 2-propanol and ephedrine-type ligands. Adv. Synth. Catal. 2003, 345, 67–77. [Google Scholar] [CrossRef]
- Wang, C.; Wu, X.; Xiao, J. Broader, greener, and more efficient: Recent advances in asymmetric transfer hydrogenation. Chem. Asian J. 2008, 3, 1750–1770. [Google Scholar] [CrossRef] [PubMed]
- Nugent, T.C.; El-Shazly, M. Chiral amine synthesis – recent developments and trends for enamide reduction, reductive amination, and imine reduction. Adv. Synth. Catal. 2010, 352, 753–819. [Google Scholar] [CrossRef]
- Fleury-Brégeot, N.; de la Fuente, V.; Castillón, S.; Claver, C. Highlights of transition metal-catalyzed asymmetric hydrogenation of imines. ChemCatChem 2010, 2, 1346–1371. [Google Scholar] [CrossRef]
- Bäckvall, J.-E. Transition metal hydrides as active intermediates in hydrogen transfer reactions. J. Organomet. Chem. 2002, 652, 105–111. [Google Scholar] [CrossRef]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
- Samec, J.S.M.; Backvall, J.-E.; Andersson, P.G.; Brandt, P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006, 35, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Fabrello, A.; Bachelier, A.; Urrutigoïty, M.; Kalck, P. Mechanistic analysis of the transition metal-catalyzed hydrogenation of imines and functionalized enamines. Coord. Chem. Rev. 2010, 254, 273–287. [Google Scholar] [CrossRef]
- Sandoval, C.A.; Ohkuma, T.; Utsumi, N.; Tsutsumi, K.; Murata, K.; Noyori, R. Mechanism of asymmetric hydrogenation of acetophenone catalyzed by chiral η6-Arene-N-tosylethylenediamine-ruthenium(II) complexes. Chem. Asian J. 2006, 1, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, C.A.; Bie, F.; Matsuoka, A.; Yamaguchi, Y.; Naka, H.; Li, Y.; Kato, K.; Utsumi, N.; Tsutsumi, K.; Ohkuma, T.; et al. Chiral η6-Arene/N-tosylethylenediamine-ruthenium(II) complexes: Solution behavior and catalytic activity for asymmetric hydrogenation. Chem. Asian J. 2010, 5, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Takehara, J.; Hashiguchi, S.; Fujii, A.; Inoue, S.-i.; Ikariya, T.; Noyori, R. Amino alcohol effects on the ruthenium(II)-catalysed asymmetric transfer hydrogenation of ketones in propan-2-ol. Chem. Commun. 1996, 233–234. [Google Scholar] [CrossRef]
- Püntener, K.; Schwink, L.; Knochel, P. New efficient catalysts for enantioselective transfer hydrogenations. Tetrahedron Lett. 1996, 37, 8165–8168. [Google Scholar] [CrossRef]
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of aromatic ketones catalyzed by chiral Ruthenium(II) complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of imines. J. Am. Chem. Soc. 1996, 118, 4916–4917. [Google Scholar] [CrossRef]
- Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid-triethylamine mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar] [CrossRef]
- Matsumura, K.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of a,b-acetylenic ketones. J. Am. Chem. Soc. 1997, 119, 8738–8739. [Google Scholar] [CrossRef]
- Gladiali, S.; Alberico, E. Transferhydrogenations. In Transition Metals for Organic Synthesis, 2nd ed.; Beller, M., Bolm, C., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004; Volume 2, pp. 145–166. [Google Scholar]
- Chen, Y.-C.; Wu, T.-F.; Deng, J.-G.; Liu, H.; Cui, X.; Zhu, J.; Jiang, Y.-Z.; Choi, M.C.K.; Chan, A.S.C. Multiple dendritic catalysts for asymmetric transfer hydrogenation. J. Org. Chem. 2002, 67, 5301–5306. [Google Scholar] [CrossRef] [PubMed]
- Dwars, T.; Oehme, G. Complex-catalyzed hydrogenation reactions in aqueous media. Adv. Synth. Catal. 2002, 344, 239–260. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; Hems, W.; King, F.; Xiao, J. Accelerated asymmetric transfer hydrogenation of aromatic ketones in water. Org. Biomol. Chem. 2004, 2, 1818–1821. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, X.; King, F.; Xiao, J. Insight into and practical application of pH-controlled asymmetric transfer hydrogenation of aromatic ketones in water. Angew. Chem. Int. Ed. 2005, 44, 3407–3411. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Yamakawa, M.; Hashiguchi, S. Metal-ligand bifunctional catalysis: A nonclassical mechanism for asymmetric hydrogen transfer between alcohols and carbonyl compounds. J. Org. Chem. 2001, 66, 7931–7944. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, H.; Cun, L.; Zhu, J.; Deng, J.; Jiang, Y. Asymmetric transfer hydrogenation of ketones catalyzed by hydrophobic metal-amido complexes in aqueous micelles and vesicles. J. Org. Chem. 2005, 70, 9424–9429. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-F.; Fan, Q.-H.; Huang, Y.-Y.; Wu, L.; He, Y.-M.; Tang, W.-J.; Gu, L.-Q.; Chan, A.S.C. Mixture of poly(ethylene glycol) and water as environmentally friendly media for efficient enantioselective transfer hydrogenation and catalyst recycling. J. Mol. Catal., A Chem. 2007, 275, 47–53. [Google Scholar] [CrossRef]
- Wang, W.; Li, Z.; Mu, W.; Su, L.; Wang, Q. Highly efficient asymmetric transfer hydrogenation of ketones in emulsions. Catal. Commun. 2010, 11, 480–483. [Google Scholar] [CrossRef]
- Haack, K.-J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. The catalyst precursor, catalyst, and intermediate in the Ru(II)-promoted asymmetric hydrogen transfer between alcohols and ketones. Angew. Chem. Int. Ed. Engl. 1997, 36, 285–288. [Google Scholar] [CrossRef]
- de Smet, K.; Pleysier, A.; Vankelecom, I.F.J.; Jacobs, P.A. Recycling of homogeneous hydrogenation catalysts by dialysis coupled catalysis. Chem. Eur. J. 2003, 9, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Šiklová, H.; Leitmannová, E.; Kačer, P.; Červený, L. Immobilization of Ru-TsDPEN catalyst on functionalized MCM-41. React. Kinet. Catal. Lett. 2007, 92, 129–136. [Google Scholar] [CrossRef]
- Yang, H.; Li, J.; Yang, J.; Liu, Z.; Yang, Q.; Li, C. Asymmetric reactions on chiral catalysts entrapped within a mesoporous cage. Chem. Commun. 2007, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, L.; Zhong, L.; Yang, Q.; Li, C. Enhanced cooperative activation effect in the hydrolytic kinetic resolution of epoxides on [Co(salen)] catalysts confined in nanocages. Angew. Chem. Int. Ed. 2007, 46, 6861–6865. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, L.; Wang, P.; Yang, Q.; Li, C. The enantioselective cyanosilylation of aldehydes on a chiral VO(Salen) complex encapsulated in SBA-16. Green Chem. 2009, 11, 257–264. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, L.; Su, W.; Yang, Q.; Li, C. Asymmetric ring-opening of epoxides on chiral Co(Salen) catalyst synthesized in SBA-16 through the "ship in a bottle" strategy. J. Catal. 2007, 248, 204–212. [Google Scholar] [CrossRef]
- Kawasaki, I.; Tsunoda, K.; Tsuji, T.; Yamaguchi, T.; Shibuta, H.; Uchida, N.; Yamashita, M.; Ohta, S. A recyclable catalyst for asymmetric transfer hydrogenation with a formic acid-triethylamine mixture in ionic liquid. Chem. Commun. 2005, 2134–2136. [Google Scholar] [CrossRef] [PubMed]
- Joerger, J.-M.; Paris, J.-M.; Vaultier, M. [Ru(arene)(diamine)] catalysts in ionic liquids: Recyclable catalytic systems for transfer hydrogenation. ARKIVOC 2006, 152–160. [Google Scholar]
- Hut’ka, M.; Toma, Š. Hydrogen transfer reduction of different ketones in ionic liquids. Chem. Mon. 2008, 139, 793–798. [Google Scholar] [CrossRef]
- Václavík, J.; Kačer, P.; Červený, L. Rational Design of Chiral Ruthenium Complexes for Asymmetric Hydrogenations. In Homogeneous Catalysts: Types, Reactions and Applications; Poehler, A.C., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2011; in press. [Google Scholar]
- Hayes, A.; Clarkson, G.; Wills, M. The importance of 1,2-anti-disubstitution in monotosylated diamine ligands for ruthenium(II)-catalysed asymmetric transfer hydrogenation. Tetrahedron Asymmetry 2004, 15, 2079–2084. [Google Scholar] [CrossRef]
- Yamakawa, M.; Yamada, I.; Noyori, R. Angew. Chem. Int. Ed. 2001, 40, 2818–2821. [CrossRef]
- Soleimannejad, J.; Sisson, A.; White, C. Functionalized-arene ruthenium half-sandwich compounds as enantioselective hydrogen transfer catalysts. crystal structures of [RuCl{TsNCH(R)CH(R)NH2}(h6-C6H5OCH2CH2OH)] (R=H or Ph). Inorg. Chim. Acta 2003, 352, 121–128. [Google Scholar] [CrossRef]
- Matsunaga, H.; Ishizuka, T.; Kunieda, T. Highly efficient asymmetric transfer hydrogenation of ketones catalyzed by chiral ‘roofed’ cis-diamine-Ru(II) complex. Tetrahedron Lett. 2005, 46, 3645–3648. [Google Scholar] [CrossRef]
- Li, X.; Blacker, J.; Houson, I.; Wu, X.; Xiao, J. An efficient Ir(III) catalyst for the asymmetric transfer hydrogenation of ketones in neat water. Synlett 2006, 2006, 1155,1160. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Y.; Li, X.; Chan, A.S.C. Structure effect of TsDPEN derivatives on enantioselectivity of asymmetric transfer hydrogenation. J. Organomet. Chem. 2008, 693, 2177–2180. [Google Scholar] [CrossRef]
- Martins, J.E.D.; Clarkson, G.J.; Wills, M. Ru(II) complexes of N-alkylated TsDPEN ligands in asymmetric transfer hydrogenation of ketones and imines. Organic Lett. 2009, 11, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.E.D.; Contreras Redondo, M.A.; Wills, M. Applications of N’-alkylated derivatives of TsDPEN in the asymmetric transfer hydrogenation of C=O and C=N bonds. Tetrahedron Asymmetry 2010, 21, 2258–2264. [Google Scholar] [CrossRef]
- Hannedouche, J.; Clarkson, G.J.; Wills, M. A new class of “tethered” Ruthenium(II) catalyst for asymmetric transfer hydrogenation reactions. J. Am. Chem. Soc. 2004, 126, 986–987. [Google Scholar] [CrossRef] [PubMed]
- Cheung, F.K.; Hayes, A.M.; Hannedouche, J.; Yim, A.S.Y.; Wills, M. “Tethered” Ru(II) catalysts for asymmetric transfer hydrogenation of ketones. J. Org. Chem. 2005, 70, 3188–3197. [Google Scholar] [CrossRef] [PubMed]
- Cheung, F.K.; Clarke, A.J.; Clarkson, G.J.; Fox, D.J.; Graham, M.A.; Lin, C.; Criville, A.L.; Wills, M. Kinetic and structural studies on ‘tethered’ Ru(II) arene ketone reduction catalysts. Dalton Trans. 2010, 39, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.J.; Hayes, A.M.; Wills, M. The “reverse-tethered” Ruthenium (II) catalyst for asymmetric transfer hydrogenation: Further applications. J. Org. Chem. 2006, 71, 7035–7044. [Google Scholar] [CrossRef] [PubMed]
- Kačer, P.; Kuzma, M.; Leitmannová, E.; Červený, L. Ruthenium complexes for asymmetric transfer hydrogenation. In Organometallic Compounds: Preparation, Structure and Properties; Chin, H.F., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2009; pp. 361–387. [Google Scholar]
- Wu, X.; Xiao, J. Aqueous-phase asymmetric transfer hydrogenation of ketones – a greener approach to chiral alcohols. Chem. Commun. 2007, 2449–2466. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, C.; Xiao, J. Asymmetric transfer hydrogenation in water with platinum group metal catalysts. Platinum Met. Rev. 2010, 54, 3–19. [Google Scholar] [CrossRef]
- Bubert, C.; Blacker, J.; Brown, S.M.; Crosby, J.; Fitzjohn, S.; Muxworthy, J.P.; Thorpe, T.; Williams, J.M.J. Synthesis of water-soluble aminosulfonamide ligands and their application in enantioselective transfer hydrogenation. Tetrahedron Lett. 2001, 42, 4037–4039. [Google Scholar] [CrossRef]
- Thorpe, T.; Blacker, J.; Brown, S.M.; Bubert, C.; Crosby, J.; Fitzjohn, S.; Muxworthy, J.P.; Williams, J.M.J. Efficient rhodium and iridium-catalysed asymmetric transfer hydrogenation using water-soluble aminosulfonamide ligands. Tetrahedron Lett. 2001, 42, 4041–4043. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, H.; Chen, L.; Cui, X.; Zhu, J.; Deng, J. Asymmetric transfer hydrogenation of prochiral ketones in aqueous media with new water-soluble chiral vicinal diamine as ligand. Org. Lett. 2003, 5, 2103–2106. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, F.; Ma, Y.; Cui, X.; Cun, L.; Zhu, J.; Deng, J.; Yu, B. Asymmetric transfer hydrogenation of imines and iminiums catalyzed by water-soluble catalyst in water. Chem. Commun. 2006, 1766–1768. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, J.; Wang, F.; Liao, J.; Zhang, H.; Lian, C.; Zhu, J.; Deng, J. Asymmetric transfer hydrogenation of ketones and imines with novel water-soluble chiral diamine as ligand in neat water. Green Chem. 2007, 9, 23–25. [Google Scholar] [CrossRef]
- Zhou, Z.; Ma, Q.; Sun, Y.; Zhang, A.; Li, L. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of aromatic ketones in water using novel water-soluble chiral monosulfonamide ligands. Heteroatom Chem. 2010, 21, 505–514. [Google Scholar] [CrossRef]
- Cross, D.J.; Kenny, J.A.; Houson, I.; Campbell, L.; Walsgrove, T.; Wills, M. Tetrahedron Asymmetry, 2001; 12, 1801–1806.
- Zhou, Z.; Sun, Y. Water-soluble chiral aminosulfonamides as ligands for ruthenium(II)-catalyzed asymmetric transfer hydrogenation. Catal. Commun. 2009, 10, 1685–1688. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, Y.; Zhang, A. Asymmetric transfer hydrogenation of prochiral ketones catalyzed by aminosulfonamide-ruthenium complexes in ionic liquid. Cent. Eur. J. Chem. 2011, 9, 175–179. [Google Scholar] [CrossRef]
- Ter Halle, R.; Schulz, E.; Lemaire, M. Heterogeneous enantioselective catalytic reduction of ketones. Synlett 1997, 1257–1258. [Google Scholar] [CrossRef]
- Bayston, D.J.; Travers, C.B.; Polywka, M.E.C. Synthesis and evaluation of a chiral heterogeneous transfer hydrogenation catalyst. Tetrahedron Asymmetry 1998, 9, 2015–2018. [Google Scholar] [CrossRef]
- Li, X.; Chen, W.; Hems, W.; King, F.; Xiao, J. Asymmetric transfer hydrogenation of ketones with a polymer-supported chiral diamine. Tetrahedron Lett. 2004, 45, 951–953. [Google Scholar] [CrossRef]
- Li, X.; Wu, X.; Chen, W.; Hancock, F.E.; King, F.; Xiao, J. Asymmetric transfer hydrogenation in Wwater with a supported noyori-ikariya catalyst. Org. Lett. 2004, 6, 3321–3324. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.N.; Gu, P.M.; Wang, F.; Tu, Y.Q. Efficient heterogeneous asymmetric transfer hydrogenation of ketones using highly eecyclable and accessible silica-immobilized Ru-TsDPEN catalysts. Org. Lett. 2004, 6, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.N.; Deng, J.G.; Tu, Y.Q.; Wang, S.H. Highly efficient and recyclable heterogeneous asymmetric transfer hydrogenation of ketones in water. Chem. Commun. 2004, 2070–2071. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-N.; Gu, P.-M.; Deng, J.-G.; Tu, Y.-Q.; Ma, Y.-P. Efficient heterogeneous asymmetric transfer hydrogenation catalyzed by recyclable silica-supported Ruthenium complexes. Eur. J. Org. Chem. 2005, 2005, 3221–3227. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Li, F.; Wang, Q.; Tao, F. Preparation of polymer-supported Ru-TsDPEN catalysts and use for enantioselective synthesis of (S)-fluoxetine. Org. Biomol. Chem. 2005, 3, 2513–2518. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ying, J.Y. Asymmetric transfer hydrogenation over Ru-TsDPEN catalysts supported on siliceous mesocellular foam. Chem. Commun. 2007, 1825–1827. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, Y.; Haraguchi, N.; Itsuno, S. Design of novel polymer-supported chiral catalyst for asymmetric transfer hydrogenation in water. Tetrahedron Lett. 2006, 47, 3239–3243. [Google Scholar] [CrossRef]
- Haraguchi, N.; Tsuru, K.; Arakawa, Y.; Itsuno, S. Asymmetric transfer hydrogenation of imines catalyzed by a polymer-immobilized chiral catalyst. Org. Biomol. Chem. 2009, 7, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Wu, Y.; Li, X.; Chan, A.S.C. Asymmetric transfer hydrogenation of ketones with a polyethylene glycol bound Ru catalyst in water. Tetrahedron Asymmetry 2008, 19, 832–837. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Han, D.; Gao, Q.; Li, C. Asymmetric transfer hydrogenation using recoverable ruthenium catalyst immobilized into magnetic mesoporous silica. J. Mol. Catal., A Chem. 2009, 298, 31–35. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, Y. Synthesis of polyethylene glycol supported chiral monosulfonamide and its application in asymmetric transfer hydrogenation of prochiral ketones. React. Kinet. Mech. Catal. 2010, 99, 391–396. [Google Scholar] [CrossRef]
- Arakawa, Y.; Chiba, A.; Haraguchi, N.; Itsuno, S. Asymmetric transfer hydrogenation of aromatic ketones in water using a polymer-supported chiral catalyst containing a hydrophilic pendant group. Adv. Synth. Catal. 2008, 350, 2295–2304. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, D.; Jia, X.; Huang, L.; Li, X.; Chan, A.S.C. A convenient synthesis of (R)-salmeterol via Rh-catalyzed asymmetric transfer hydrogenation. Tetrahedron Asymmetry 2008, 19, 1824–1828. [Google Scholar] [CrossRef]
- Huang, L.; Liu, J.; Shan, W.; Liu, B.; Shi, A.; Li, X. The asymmetric synthesis of (R,R)-formoterol via transfer hydrogenation with polyethylene glycol bound Rh catalyst in PEG2000 and water. Chirality 2010, 22, 206–211. [Google Scholar] [PubMed]
- Zhu, Y.; Stubbs, L.P.; Ho, F.; Liu, R.; Ship, C.P.; Maguire, J.A.; Hosmane, N.S. Magnetic nanocomposites: A new perspective in catalysis. ChemCatChem 2010, 2, 365–374. [Google Scholar] [CrossRef]
- Schätz, A.; Reiser, O.; Stark, W.J. Nanoparticles as semi-heterogeneous catalyst supports. Chem. Eur. J. 2010, 16, 8950–8967. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, K.V.S.; Glorius, F. Superparamagnetic nanoparticles for asymmetric catalysis-a perfect match. Catal. Sci. Technol. 2011, 1, 13–22. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Luque, R.; Fihri, A.; Zhu, H.; Bouhrara, M.; Basset, J.-M. Magnetically Recoverable Nanocatalysts. Chem. Rev. 2011, in press. [Google Scholar] [CrossRef] [PubMed]
- Kassube, J.; Gade, L. Stereoselective Dendrimer Catalysis. In Dendrimer Catalysis; Gade, L., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 20, pp. 61–96. [Google Scholar]
- Méry, D.; Astruc, D. Dendritic catalysis: Major concepts and recent progress. Coord. Chem. Rev. 2006, 250, 1965–1979. [Google Scholar] [CrossRef]
- de Jesús, E.; Flores, J.C. Dendrimers: Solutions for catalyst separation and recycling – a review. Ind. Eng. Chem. Res. 2008, 47, 7968–7981. [Google Scholar] [CrossRef]
- Dijkstra, H.P.; van Klink, G.P.M.; van Koten, G. The use of ultra- and nanofiltration techniques in homogeneous catalyst recycling. Acc. Chem. Res. 2002, 35, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Wu, T.-F.; Deng, J.-G.; Liu, H.; Jiang, Y.-Z.; Choi, M.C.K.; Chan, A.S.C. Dendritic catalysts for asymmetric transferhydrogenation. Chem. Commun. 2001, 1488–1489. [Google Scholar] [CrossRef]
- Hawker, C.J.; Frechet, J.M.J. Preparation of polymers with controlled molecular architecture. A new convergent approach to dendritic macromolecules. J. Am. Chem. Soc. 1990, 112, 7638–7647. [Google Scholar] [CrossRef]
- Newkome, G.R.; Lin, X. Symmetrical, four-directional, poly(ether-amide) cascade polymers. Macromolecules 1991, 24, 1443–1444. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Wu, T.-F.; Jiang, L.; Deng, J.-G.; Liu, H.; Zhu, J.; Jiang, Y.-Z. Synthesis of dendritic catalysts and application in asymmetric transfer hydrogenation. J. Org. Chem. 2005, 70, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cui, X.; Cun, L.; Zhu, J.; Deng, J. Tunable dendritic ligands of chiral 1,2-diamine and their application in asymmetric transfer hydrogenation. Tetrahedron Asymmetry 2005, 16, 2525–2530. [Google Scholar] [CrossRef]
- Gaikwad, A.V.; Boffa, V.; Ten Elshof, J.E.; Rothenberg, G. Cat-in-a-cup: Facile separation of large homogeneous catalysts. Angew. Chem. Int. Ed. 2008, 47, 5407–5410. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Q. A fluorinated dendritic TsDPEN-Ru(II) catalyst for asymmetric transfer hydrogenation of prochiral ketones in aqueous media. Chem. Commun. 2010, 46, 4616–4618. [Google Scholar] [CrossRef] [PubMed]
- Geldbach, T.J.; Dyson, P.J. A versatile ruthenium precursor for biphasic catalysis and its application in ionic liquid biphasic transfer hydrogenation: Conventional vs. task-specific catalysts. J. Am. Chem. Soc. 2004, 126, 8114–8115. [Google Scholar] [CrossRef] [PubMed]
- Letondor, C.; Ward, T.R. Artificial metalloenzymes for enantioselective catalysis: Recent advances. ChemBioChem 2006, 7, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Steinreiber, J.; Ward, T.R. Artificial metalloenzymes as selective catalysts in aqueous media. Coord. Chem. Rev. 2008, 252, 751–766. [Google Scholar] [CrossRef]
- Rosati, F.; Roelfes, G. Artificial metalloenzymes. ChemCatChem 2010, 2, 916–927. [Google Scholar] [CrossRef]
- Letondor, C.; Humbert, N.; Ward, T.R. Artificial metalloenzymes based on biotin-avidin technology for the enantioselective reduction of ketones by transfer hydrogenation. Proc. Natl. Acad. Sci. USA 2005, 102, 4683–4687. [Google Scholar] [CrossRef] [PubMed]
- Letondor, C.; Pordea, A.; Humbert, N.; Ivanova, A.; Mazurek, S.; Novic, M.; Ward, T.R. Artificial transfer hydrogenases based on the biotin-(strept)avidin technology: Fine tuning the selectivity by saturation mutagenesis of the host protein. J. Am. Chem. Soc. 2006, 128, 8320–8328. [Google Scholar] [CrossRef] [PubMed]
- Creus, M.; Pordea, A.; Rossel, T.; Sardo, A.; Letondor, C.; Ivanova, A.; LeTrong, I.; Stenkamp, R.E.; Ward, T.R. X-Ray structure and designed evolution of an artificial transfer hydrogenase. Angew. Chem. Int. Ed. 2008, 47, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.R. Artificial metalloenzymes based on the biotin-avidin technology: Enantioselective catalysis and beyond. Acc. Chem. Res. 2010, 44, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Dürrenberger, M.; Heinisch, T.; Wilson, Y.M.; Rossel, T.; Nogueira, E.; Knörr, L.; Mutschler, A.; Kersten, K.; Zimbron, M.J.; Pierron, J.; et al. Artificial transfer hydrogenases for the enantioselective reduction of cyclic imines. Angew. Chem. Int. Ed. 2011, 50, 3026–3029. [Google Scholar] [CrossRef] [PubMed]
- Polborn, K.; Severin, K. Biomimetic catalysis with immobilised organometallic Ruthenium complexes: Substrate- and regioselective transfer hydrogenation of ketones. Chem. Eur. J. 2000, 6, 4604–4611. [Google Scholar] [CrossRef]
- Locatelli, F.; Gamez, P.; Lemaire, M. Molecular imprinting of polymerised catalytic complexes in asymmetric catalysis. J. Mol. Catal., A Chem. 1998, 135, 89–98. [Google Scholar] [CrossRef]
- Weng, Z.; Muratsugu, S.; Ishiguro, N.; Ohkoshi, S.-i.; Tada, M. Preparation of surface molecularly imprinted Ru-complex catalysts for asymmetric transfer hydrogenation in water media. Dalton Trans. 2011, 40, 2338–2347. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, M.; Ito, H.; Noyori, R. The metal-ligand bifunctional catalysis: A theoretical study on the Ruthenium(II)-catalyzed hydrogen transfer between alcohols and carbonyl compounds. J. Am. Chem. Soc. 2000, 122, 1466–1478. [Google Scholar] [CrossRef]
- Ikariya, T.; Blacker, A.J. Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res. 2007, 40, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Ikariya, T. Mechanistic aspects of formation of chiral Ruthenium hydride complexes from 16-electron ruthenium amide complexes and formic acid: Facile reversible decarboxylation and carboxylation. Adv. Synth. Catal. 2004, 346, 37–41. [Google Scholar] [CrossRef]
- Kuzma, M.; Pelantová, H.; Sedmera, P.; Kačer, P.; Šiklová, H.; Červený, L. Study on Influence of Amines on Behavior of the Noyoris Catalysts Under Transfer Hydrogenation Conditions Studied by NMR. In International Symposium on Relations between Homogeneous and Heterogeneous Catalysis XIII (ISHHC XIII); Somorjai, G., Ed.; University of California: Berkeley, CA, USA, 2007. [Google Scholar]
- Wu, X.; Liu, J.; di Tommaso, D.; Iggo, J.A.; Catlow, C.R.A.; Bacsa, J.; Xiao, J. A multilateral mechanistic study into asymmetric transfer hydrogenation in water. Chem. Eur. J. 2008, 14, 7699–7715. [Google Scholar] [CrossRef] [PubMed]
- Åberg, J.B.; Samec, J.S.M.; Backvall, J.-E. Mechanistic investigation on the hydrogenation of imines by [p-(Me2CH)C6H4Me]RuH(NH2CHPhCHPhNSO2C6H4-p-CH). Experimental support for an ionic pathway. Chem. Commun. 2006, 2771–2773. [Google Scholar] [CrossRef]
- Balcells, D.; Maseras, F. Computational approaches to asymmetric synthesis. New J. Chem. 2007, 31, 333–343. [Google Scholar] [CrossRef]
- Václavík, J.; Kuzma, M.; Přech, J.; Kačer, P. Asymmetric transfer hydrogenation of imines and ketones using chiral Ru(II)Cl(η6-p-cymene)[(S,S)-N-TsDPEN] as a catalyst: A computational study. Organometallics 2011. submitted. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Substrate | Product | S/C [a]; Time; Temp.; Conversion (%) | % ee (config) [b] | Ref. [c] |
|---|---|---|---|---|---|---|
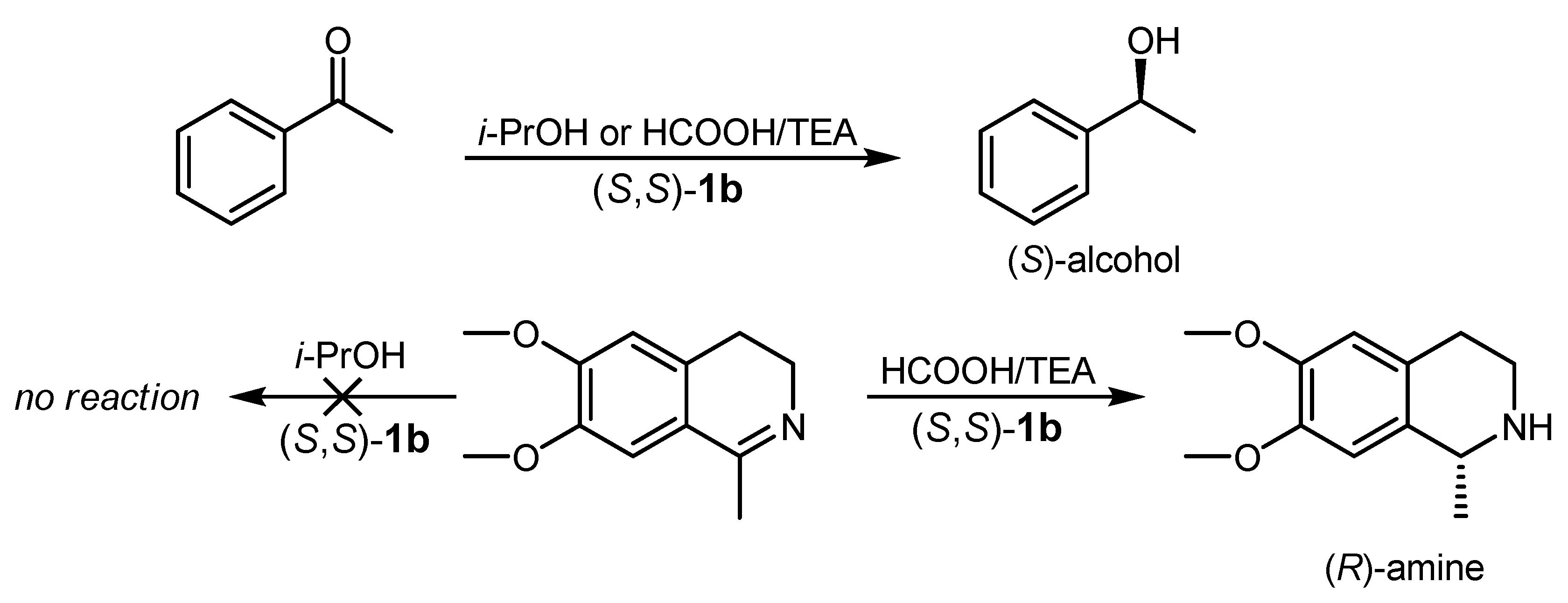
| 1 | (R,R)-1a | 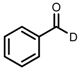 | 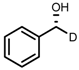 | 200; 0.5 h; 28 °C; 100 | 97 (R) | [26] |
| 2 | (S,S)-1b |  | 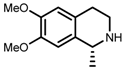 | 200; 3 h; 28 °C; 99 | 95 (R) | [53] |
| 3 | (R,R)-1b [d] | 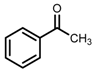 | 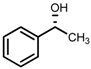 | 100; 20 h; 28 °C; > 99 | 97.7 (R) | [57] |
| 4 | (S,S)-1c | 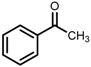 |  | 200; 20 h; 28 °C; 99 | 98 (S) | [54] |
| 5 | (S,S)-1c [d] | 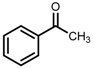 | 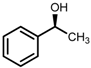 | 200; 15 h; r.t.; 99 | 97 (S) | [52] |
| 6 | (R,R)-1d | 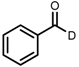 | 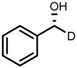 | 200; 7 h; 28 °C; 99 | 96 (R) | [26] |
| 7 | (S,S)-1e | 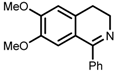 | 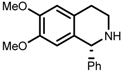 | 200; 8 h; 28 °C; 99 | 84 (R) | [53] |
| 8 | (R,R)-1f | 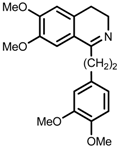 |  | 200; 12 h; 28 °C; 99 | 92 (S) | [53] |
| Entry | Catalyst config. | Solution | Time; Temp.; Conversion (%) | % ee (config) [b] | Ref. [c] |
|---|---|---|---|---|---|
| 1 | (R,R) | H2O/HCOONa | 2 h; 40 °C; >99 | 94 % (R) | [59] |
| 2 | (R,R) | HCOOH/Et3N | 12 h; 40°C; 98 | 97 % (R) | [59] |
| 3 | (R,R) | H2O/HCOONa/CTAB [d] | 4 h; 28 °C; >99 | 95 % (R) | [62] |
| 4 | (S,S) | H2O/HCOONa/PEG | 15 h; 40 °C; >99 | 96 % (S) | [63] |
| Entry | Catalyst config. | Substrate | Solution | S/C [a]; Time; Temp.; Conversion (%) | % ee (config) [b] | Ref. [c] |
|---|---|---|---|---|---|---|
| 1 | (S,S) [d] | DHIQ [e] | HCOOH/Et3N; acetonitrile | 200; 7 h; r.t.; 99 | 89% (R) | [67] |
| 2 | (S,S) [f] | acetophenone | H2O/HCOONa; Et4N+Br | 100; 5 h; 30 °C; >99 | 93% (S) | [68] |
| Entry | Ligand | Substrate | Solution | Time; Temp.; Conversion (%) | % ee (config) [b] | Ref. [c] |
|---|---|---|---|---|---|---|
| 1 | (S,S)-2 | acetophenone | H2O/i-PrOH/t-BuOK | 48 h; 22 °C; 96 | 94 (S) | [91] |
| 2 | (R,R)-3 | acetophenone | H2O/HCOONa; SDS [d] | 24 h; 40 °C; >99 | 95 (R) | [93] |
| 3 | (R,R)-3 | α-bromo-acetophenone | H2O/CH2Cl2/HCOONa; SDS | 24 h; 28 °C; 87 (isol. yield) | 94 | [93] |
| 4 | (R,R)-3 | DHIQ [e] | H2O/HCOONa; CTAB [f] | 10 h; 28 °C; 97 (isol. yield) | 95 (S) | [94] |
| 5 | (S,S)-4 | acetophenone | H2O/HCOONa | 0.5 h; 28 °C; 33 | 95 (S) | [95] |
| 6 | (R,R)-5c | acetophenone | H2O/HCOONa | 2 h; 40 °C; 100 | 94 (R) | [96] |
| 7 | (R,R)-5d | acetophenone | H2O/HCOONa | 2 h; 40 °C; 100 | 92 (R) | [96] |
| Entry | Ligand | Substrate | Solution | S/C [b]; Time; Temp.; Conversion (%) | % ee (config) [c] | Ref. [d] |
|---|---|---|---|---|---|---|
| 1 | (S,S)-6b | acetophenone | i-PrOH/KOH | 20; 48 h; – ; 23 | 84 (S) | [100] |
| 2 | (S,S)-7a | acetophenone | HCOOH/Et3N/ CH2Cl2 | 100; 18 h; 30 °C; 71 | > 99 (S) | [101] |
| 3 | (S,S)-7b | acetophenone | HCOOH/Et3N | 100; 28 h; 30 °C; 95 | 96.7 (S) | [101] |
| 4 | (R,R)-8 | acetophenone | HCOOH/Et3N | 100; 20 h; 50 °C; 95 | 94 (R) | [102] |
| 5 | (R,R)-8 | acetophenone | H2O/HCOONa | 100; 1 h; 40 °C; 99 | 92 (R) | [103] |
| 6 | (R,R)-9a | acetophenone | HCOOH/Et3N | 100; 6 h; 40 °C; >99 | 97 (R) | [104] |
| 7 | (R,R)-9b | acetophenone | H2O/HCOONa; TBAB [e] | 100; 2 h; 40 °C; >99 | 96 (R) | [105] |
| 8 | (S,S)-10b | 2-cyano-acetophenone | HCOOH/Et3N | 100; 17 h; 30 °C; 98 (isol. yield) | 97 (S) | [107] |
| 9 | (S,S)-11 | DHIQ [f] | HCOOH/Et3N/CH2Cl2 | 100; 12 h; r.t.; 100 (isol. yield) | 91 (R) | [108] |
| 10 | (R,R)-12b | acetophenone | H2O/HCOONa | 100; 3 h; 40 °C; 100 | 98 (R) | [109] |
| 11 | (R,R)-13b | acetophenone | H2O/HCOONa | 100; 3 h; 40 °C; 100 | 98 (R) | [109] |
| 12 | (R,R)-13a | DHIQ [f] | H2O/HCOONa; CTAC [g] | 100; 24 h; r.t.; 69 | 94 (S) | [110] |
| 13 | (S,S)-14 | acetophenone | H2O/HCOONa | 100; 2 h; r.t.; >99 | 96 (S) | [111] |
| 14 | (S,S)-15 | DHIQ [f] | HCOOH/Et3N/CH2Cl2 | 100; 1.5 h; 40 °C; 99 | 94 (R) | [112] |
| 15 | (R,R)-16 | acetophenone | H2O/HCOONa | 100; 9 h; 40 °C; 100 | 97 (R) | [113] |
| Entry | Ligand | Solution | Time; Temp.; Conversion (%) | % ee (config.) [b] | Ref. [c] | |
|---|---|---|---|---|---|---|
| 1 | (S,S)-17 [d] | HCOOH/Et3N | 20 h; 28 °C; >98 | 96.5 (S) | [125] | |
| 2 | (R,R)-18a | HCOOH/Et3N | 20 h; 28 °C; 99 | 97.6 (R) | [57] | |
| 3 | (R,R)-19 | HCOOH/Et3N | 20 h; 28 °C; 97 | 97.1 (R) | [57] | |
| 4 | (R,R)-20 [e] | HCOOH/Et3N | 20 h; 28 °C; 97 | 97.1 (R) | [128] | |
| 5 | (R,R)-21 [f] | HCOOH/Et3N/CH2Cl2 | 20 h; 28 °C; 97 | 96.1 (R) | [129] | |
| 6 | (S,S)-17 [e] | i-PrOH/i-PrOK | 48 h; 25 °C; 65 | 95 (S) | [130] | |
| 7 | (S,S)-22 | H2O/HCOONa; TBAI [g] | 4 h; 40 °C; > 99 | 97 (S) | [131] | |
| Entry | Ligand | Solution | S/C [a]; Time; Temp.; Conversion (%) | % ee (config.) [b] | Ref. [c] |
|---|---|---|---|---|---|
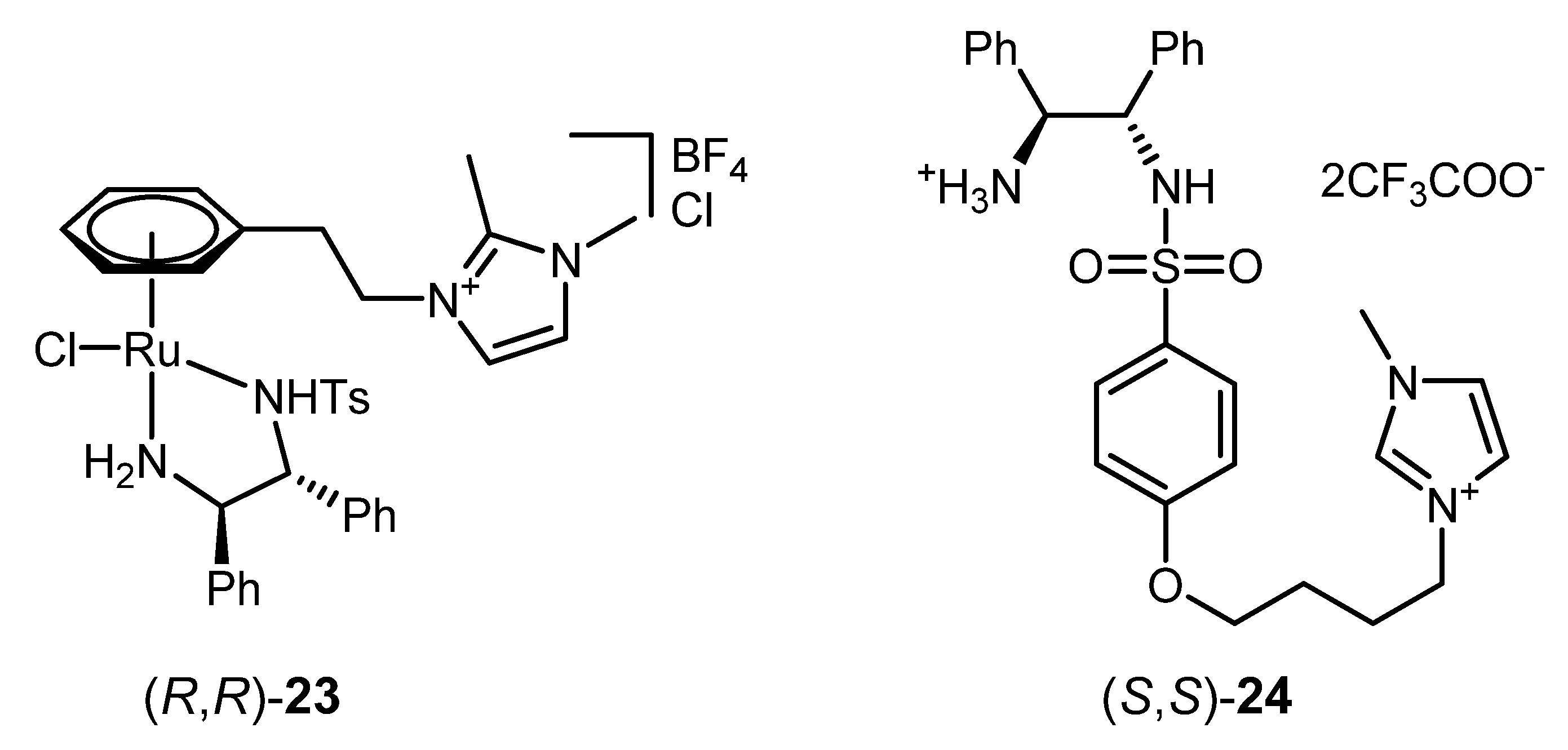
| 1 | (R,R)-23 [d] | HCOOH/Et3N/[C4C1C1Im]PF6 [e] | 200; 24 h; 35 °C; > 99 | > 99 | [132] |
| 2 | (S,S)-24 [f] | HCOOH/Et3N/[bmim][PF6] [g] | 100; 24 h; r.t.; 98 | 92 (S) | [72] |
| 3 | (R,R)-5a [h] | HCOOH/Et3N/[bmim][PF6] [g] | 100; 8 h; 40 °C; 100 | 97 (R) | [99] |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Václavík, J.; Kačer, P.; Kuzma, M.; Červený, L. Opportunities Offered by Chiral η6-Arene/N-Arylsulfonyl-diamine-RuII Catalysts in the Asymmetric Transfer Hydrogenation of Ketones and Imines. Molecules 2011, 16, 5460-5495. https://doi.org/10.3390/molecules16075460
Václavík J, Kačer P, Kuzma M, Červený L. Opportunities Offered by Chiral η6-Arene/N-Arylsulfonyl-diamine-RuII Catalysts in the Asymmetric Transfer Hydrogenation of Ketones and Imines. Molecules. 2011; 16(7):5460-5495. https://doi.org/10.3390/molecules16075460
Chicago/Turabian StyleVáclavík, Jiří, Petr Kačer, Marek Kuzma, and Libor Červený. 2011. "Opportunities Offered by Chiral η6-Arene/N-Arylsulfonyl-diamine-RuII Catalysts in the Asymmetric Transfer Hydrogenation of Ketones and Imines" Molecules 16, no. 7: 5460-5495. https://doi.org/10.3390/molecules16075460
APA StyleVáclavík, J., Kačer, P., Kuzma, M., & Červený, L. (2011). Opportunities Offered by Chiral η6-Arene/N-Arylsulfonyl-diamine-RuII Catalysts in the Asymmetric Transfer Hydrogenation of Ketones and Imines. Molecules, 16(7), 5460-5495. https://doi.org/10.3390/molecules16075460