Isolation of a Bis-Iodurated Tetra-THF as a Trace Product from the Oxidation of Squalene with RuO4 and Its Double Ring Expansion to a Novel bis-THF-bis-THP Compound

 ,
, 

 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
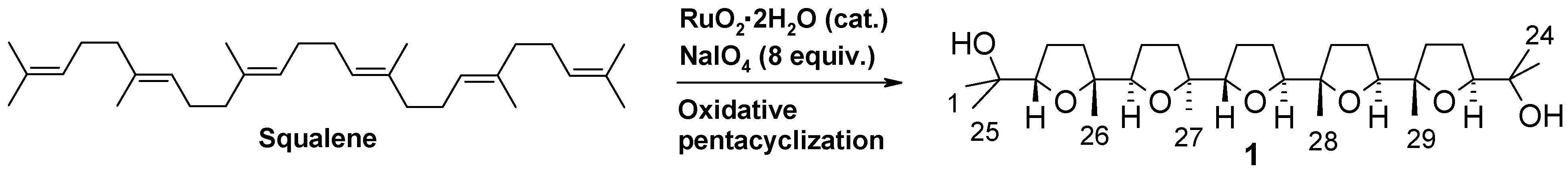{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
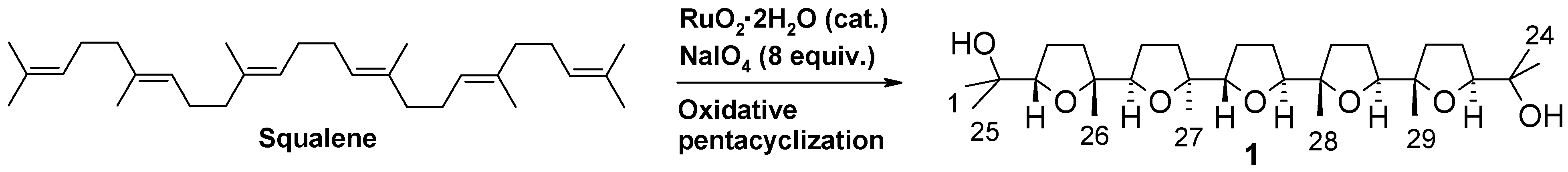
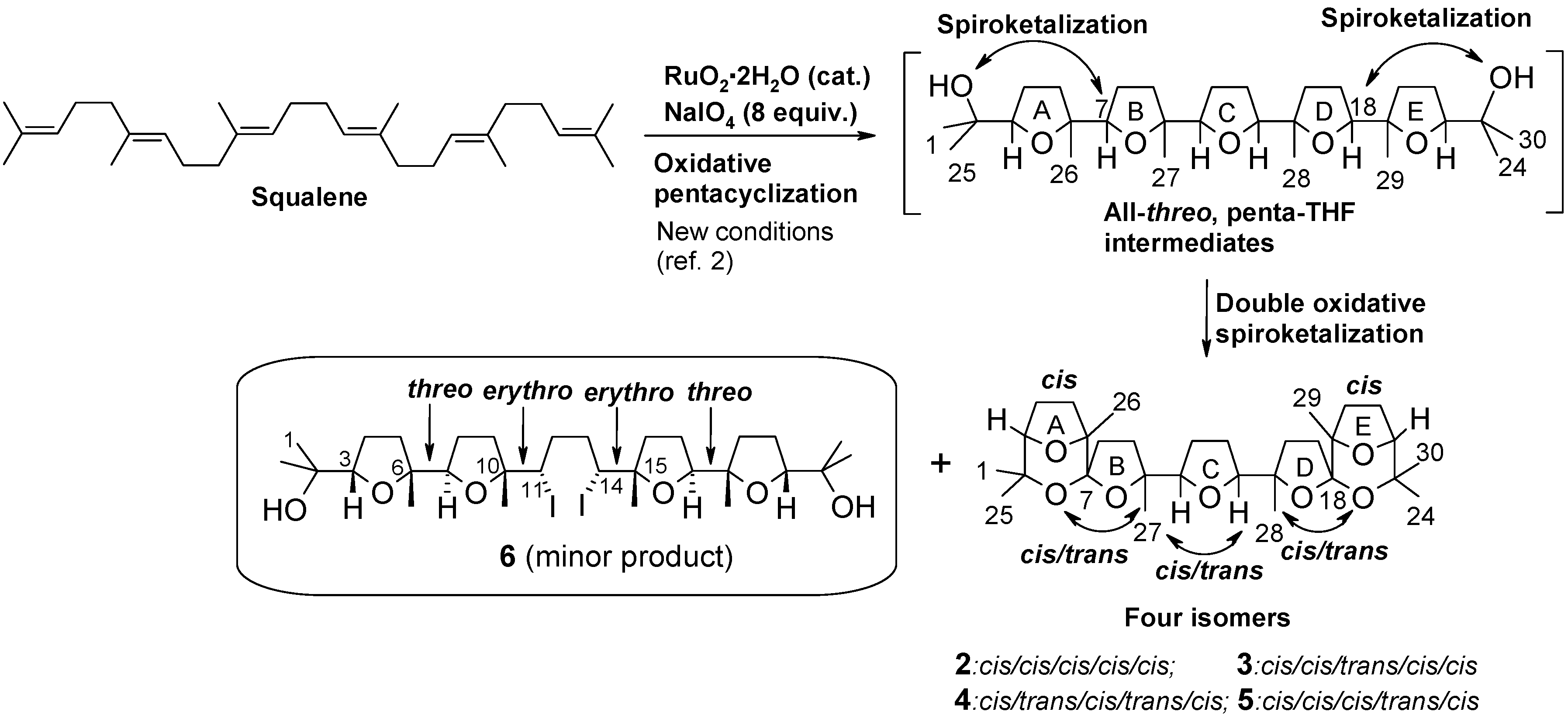
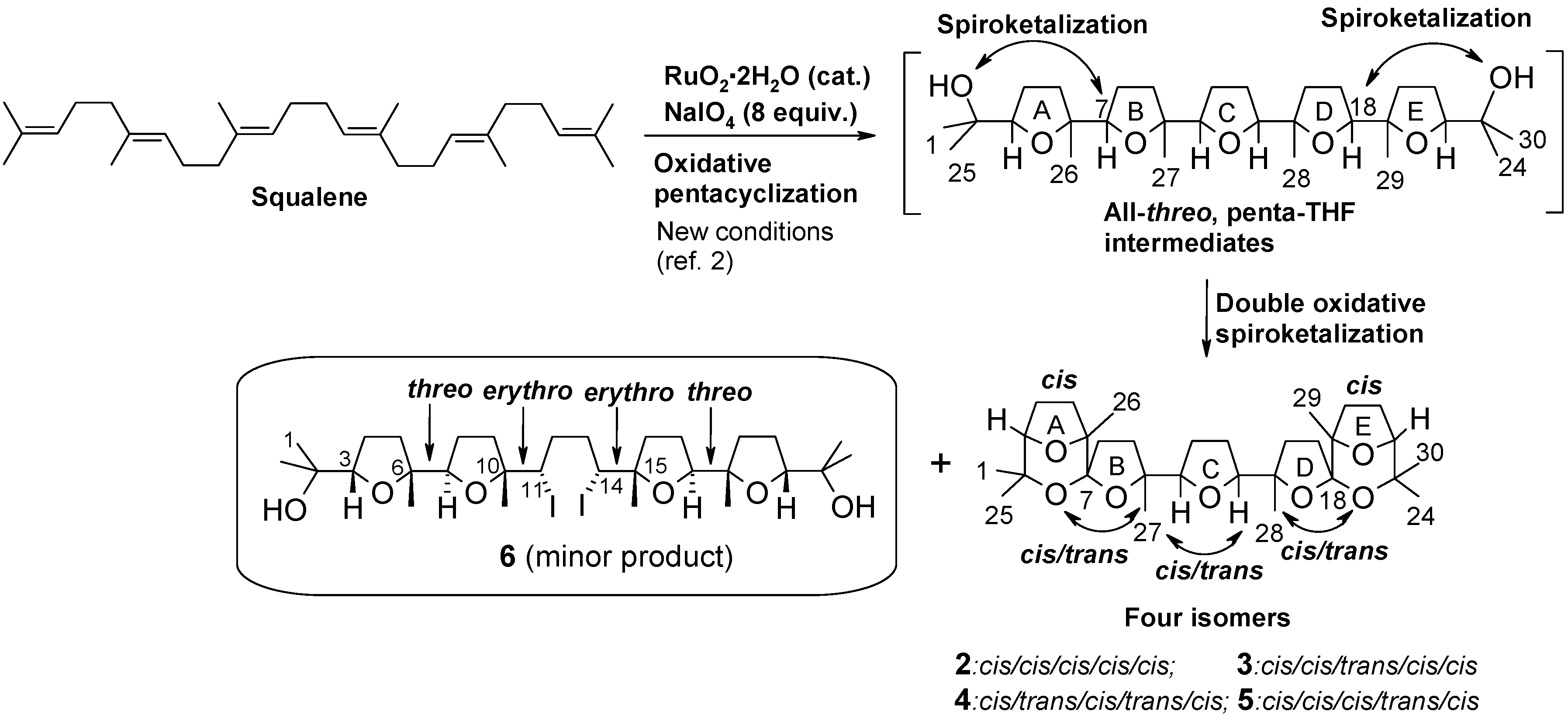
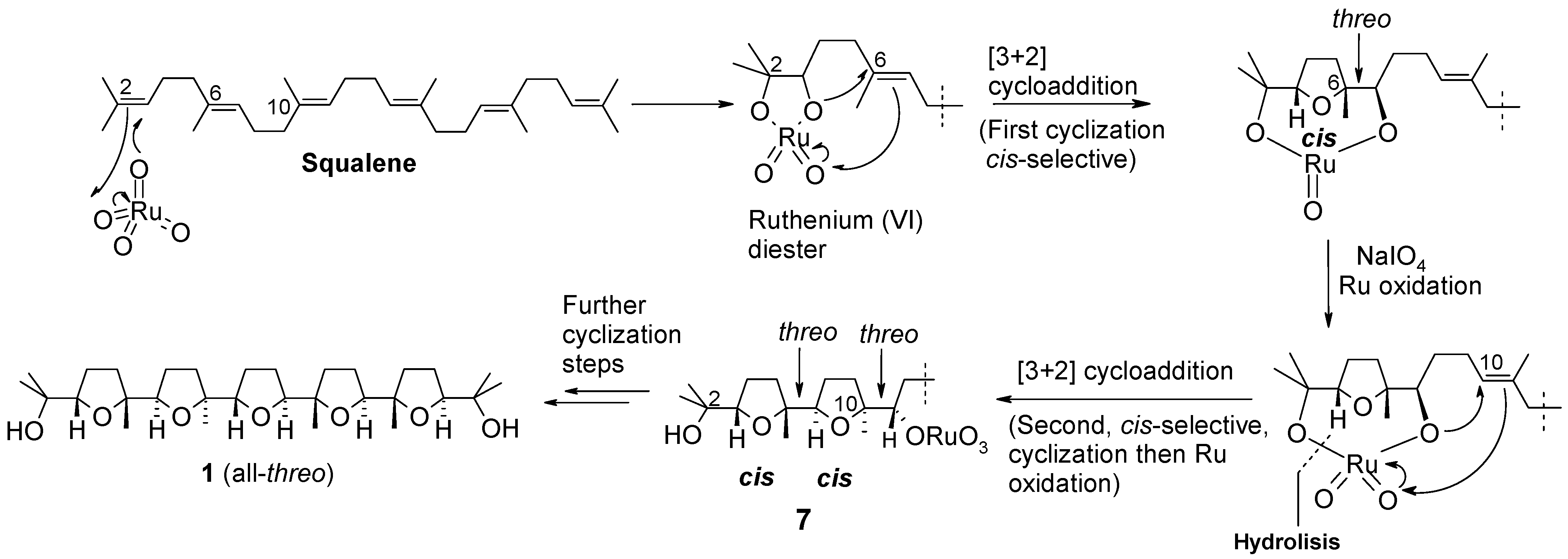
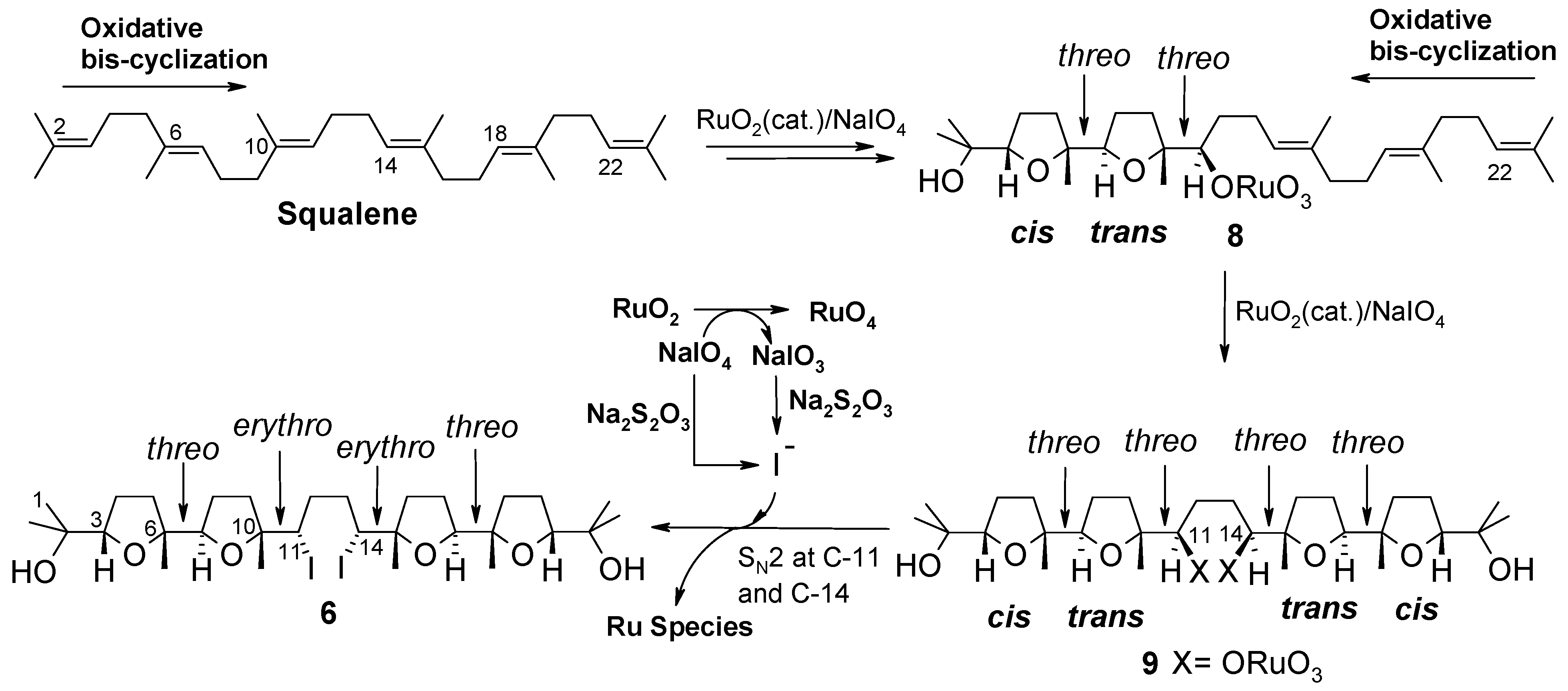
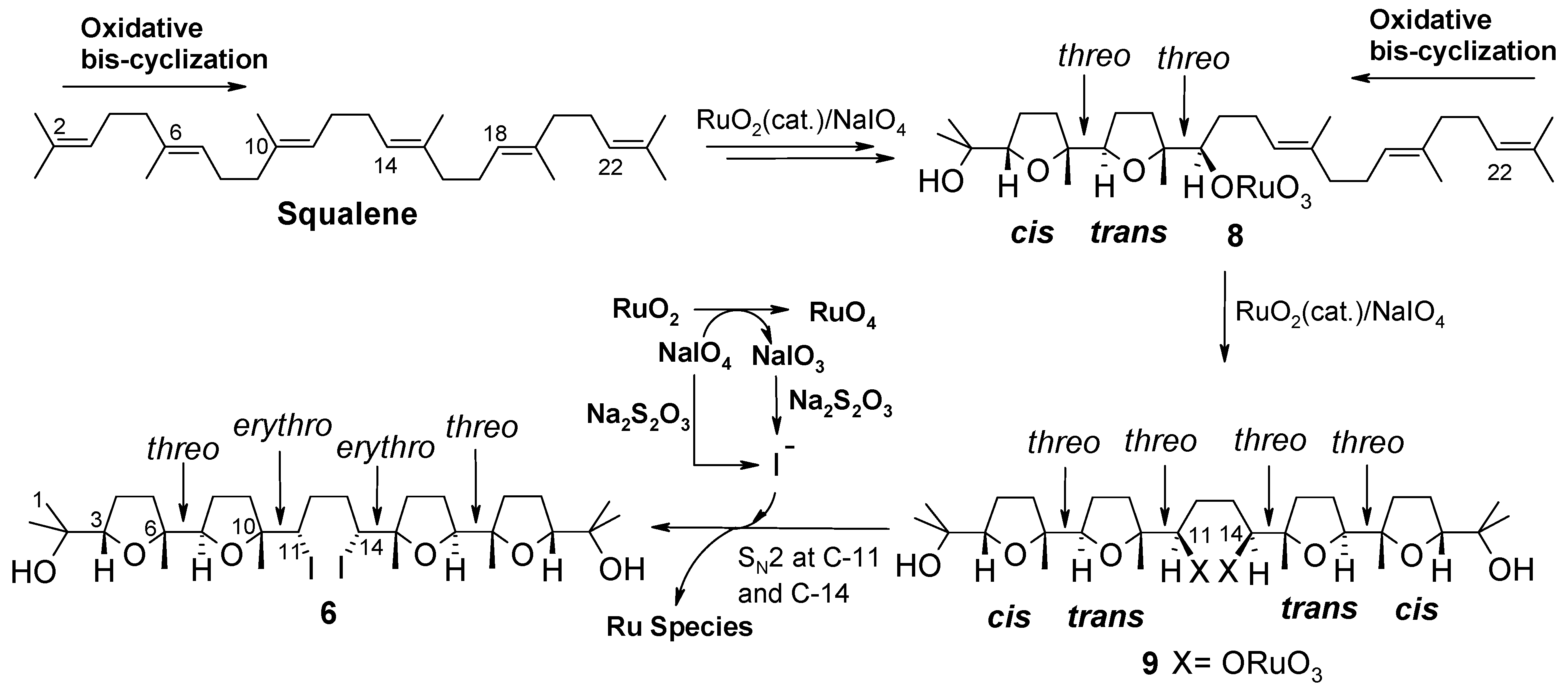
2. Results and Discussion
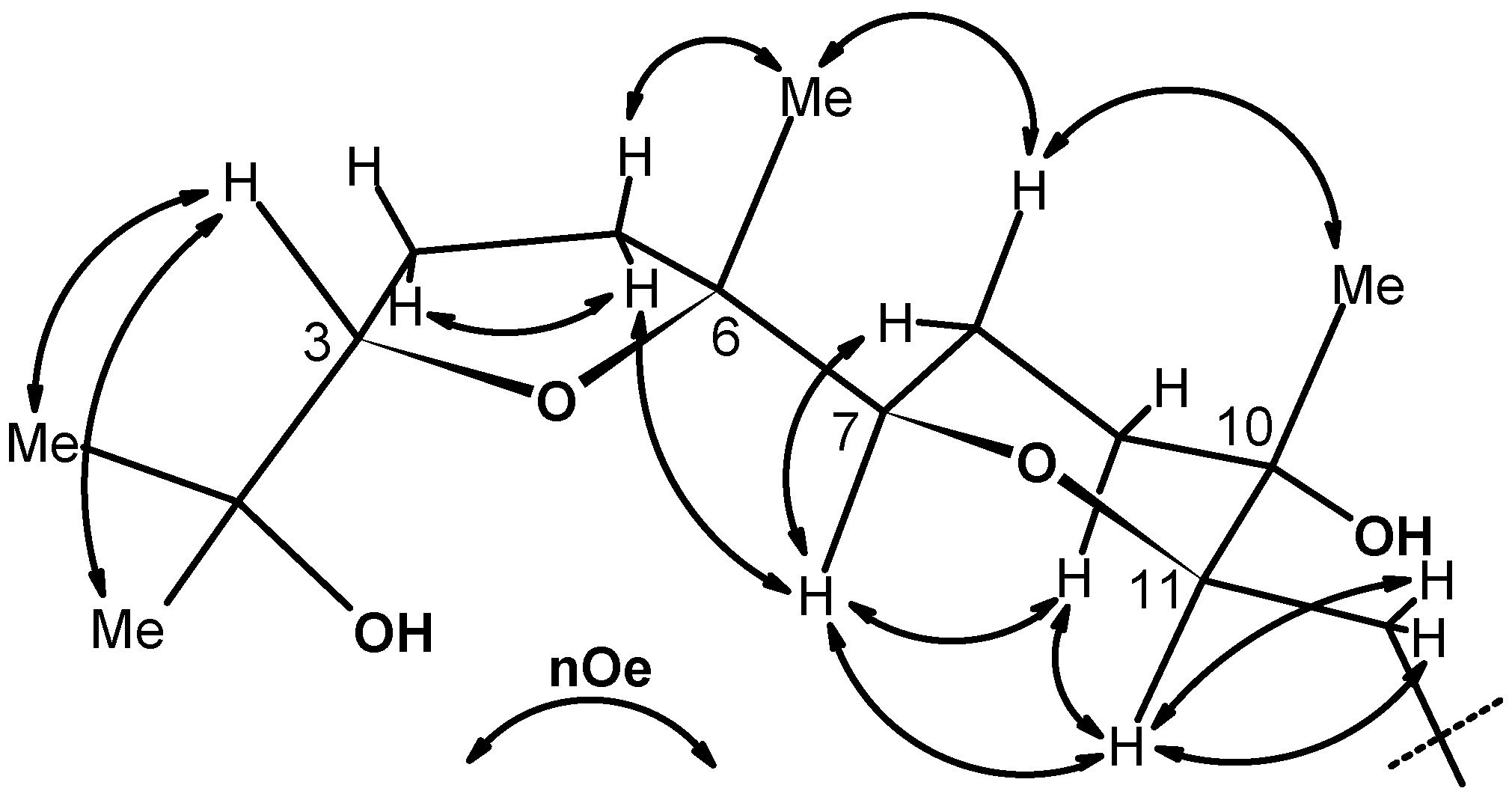
2.1. Chemistry
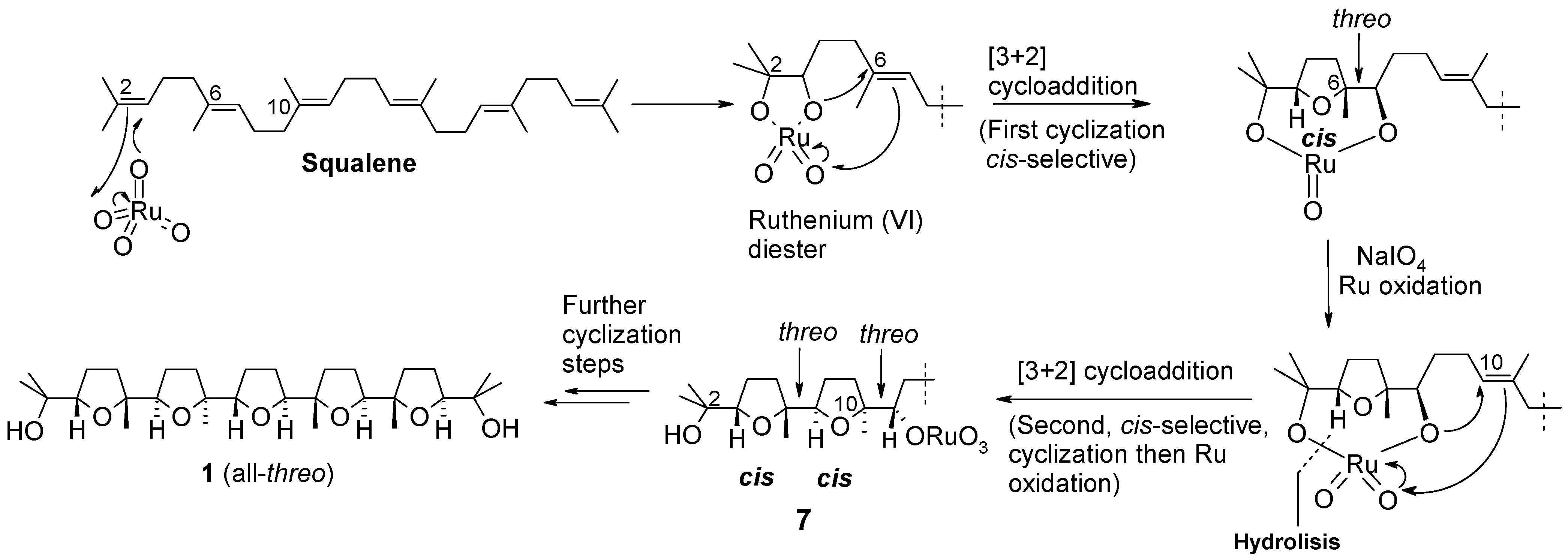
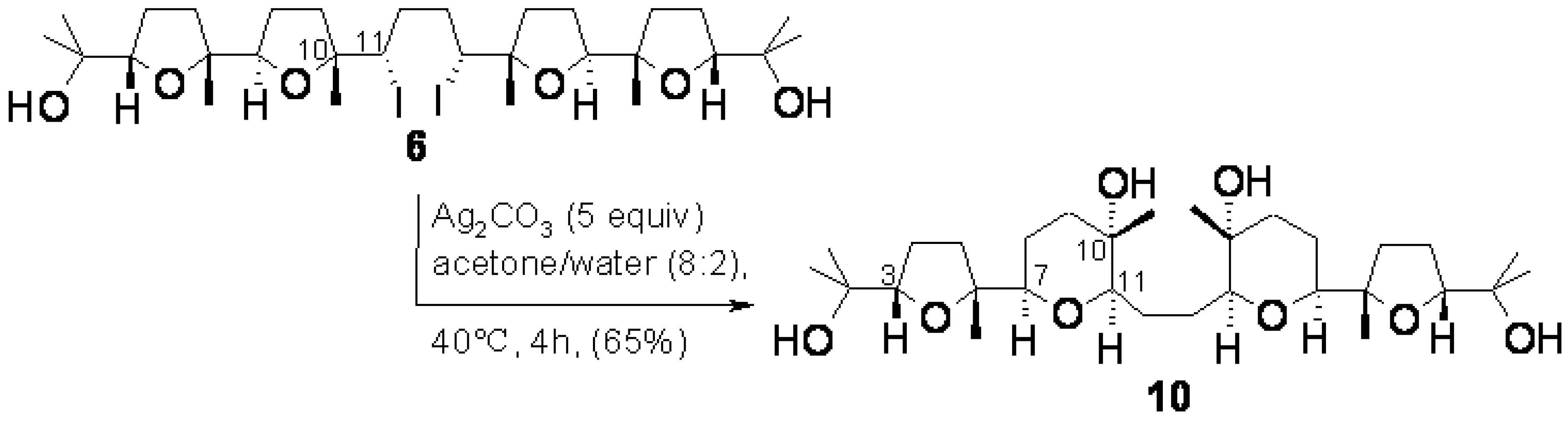
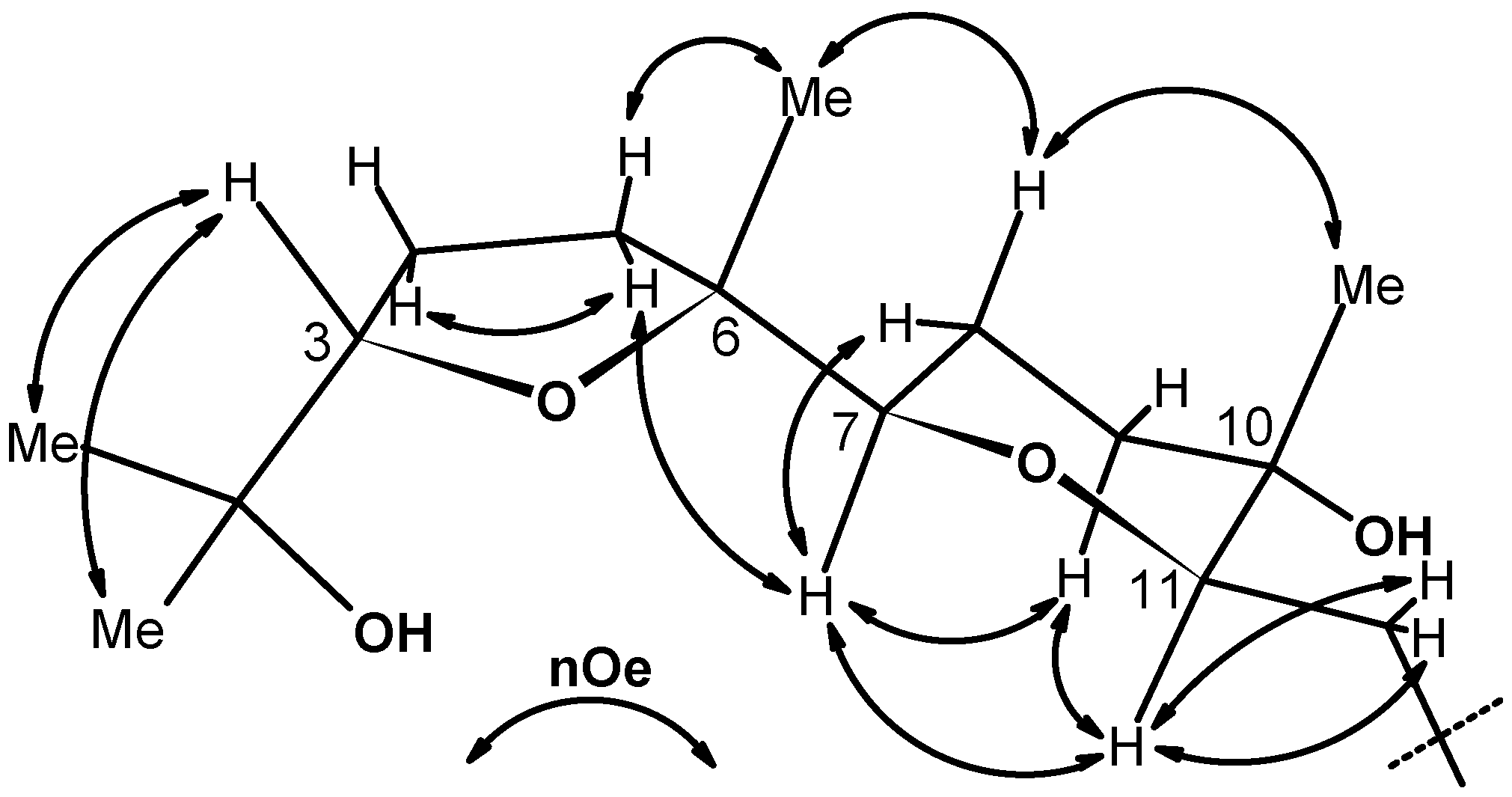
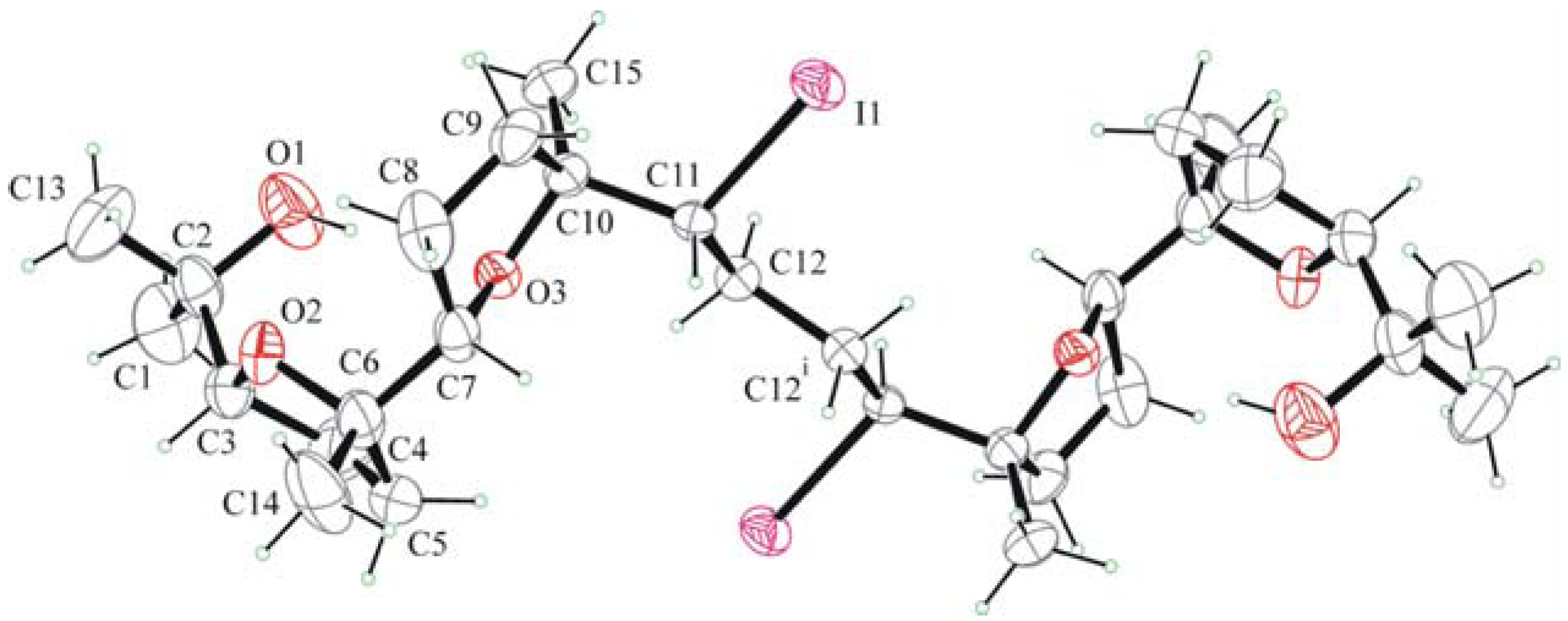
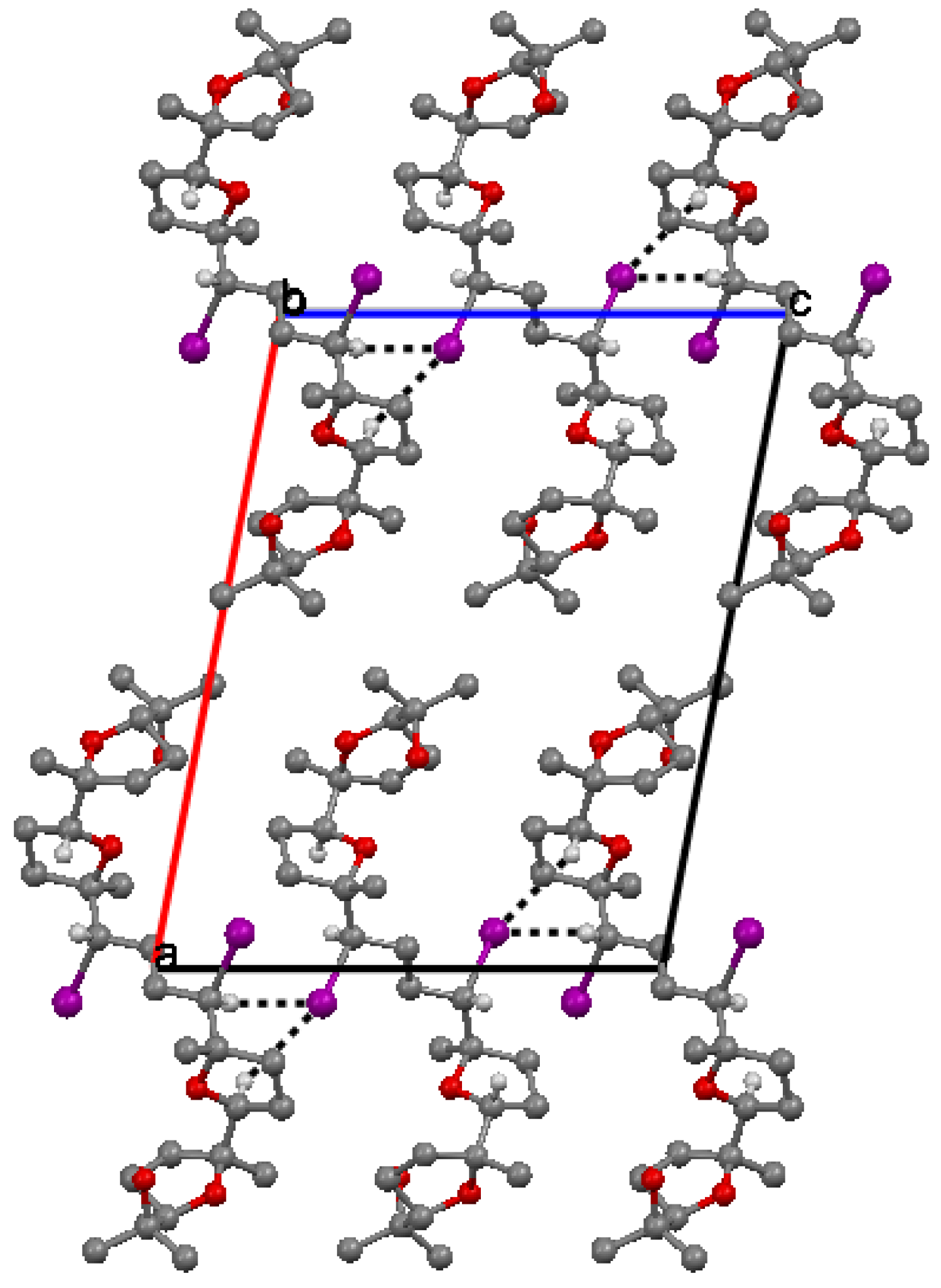
2.2. X-ray crystallography.
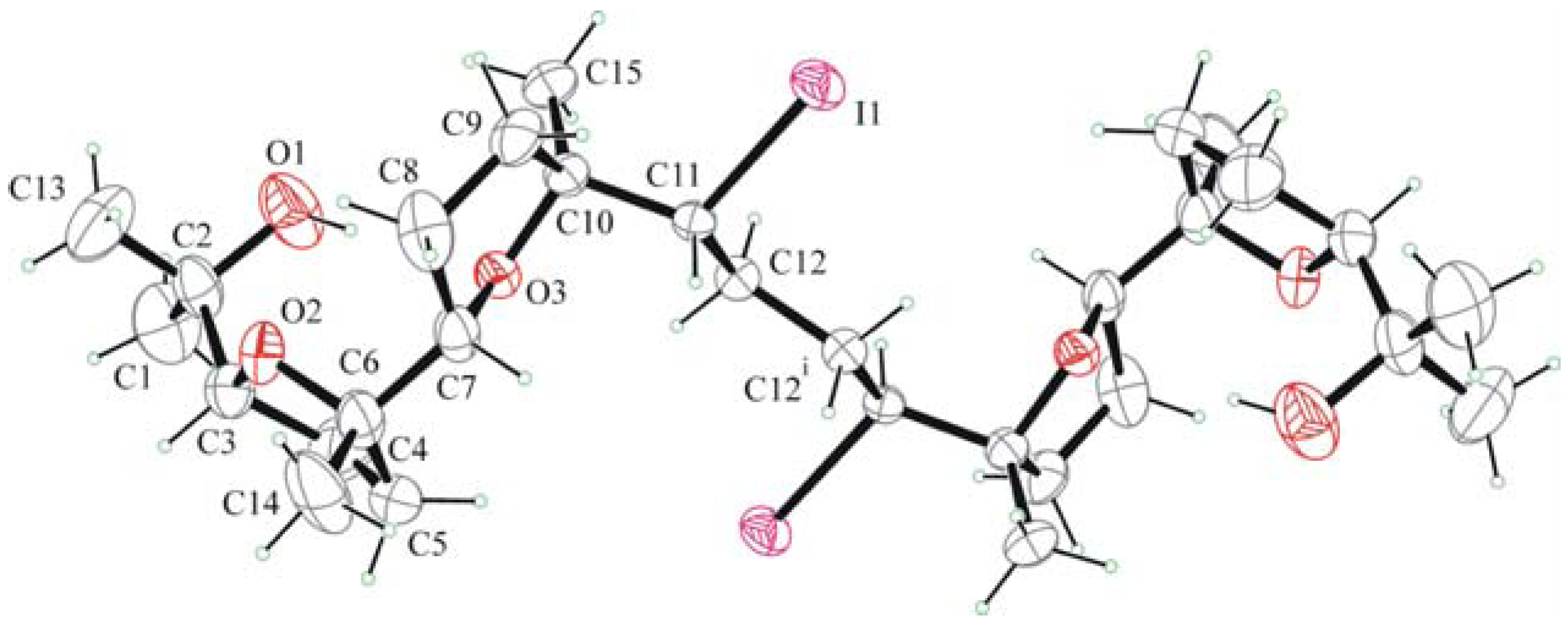
 . Along a molecules are stacked in layers through van der Waals contacts.
. Along a molecules are stacked in layers through van der Waals contacts.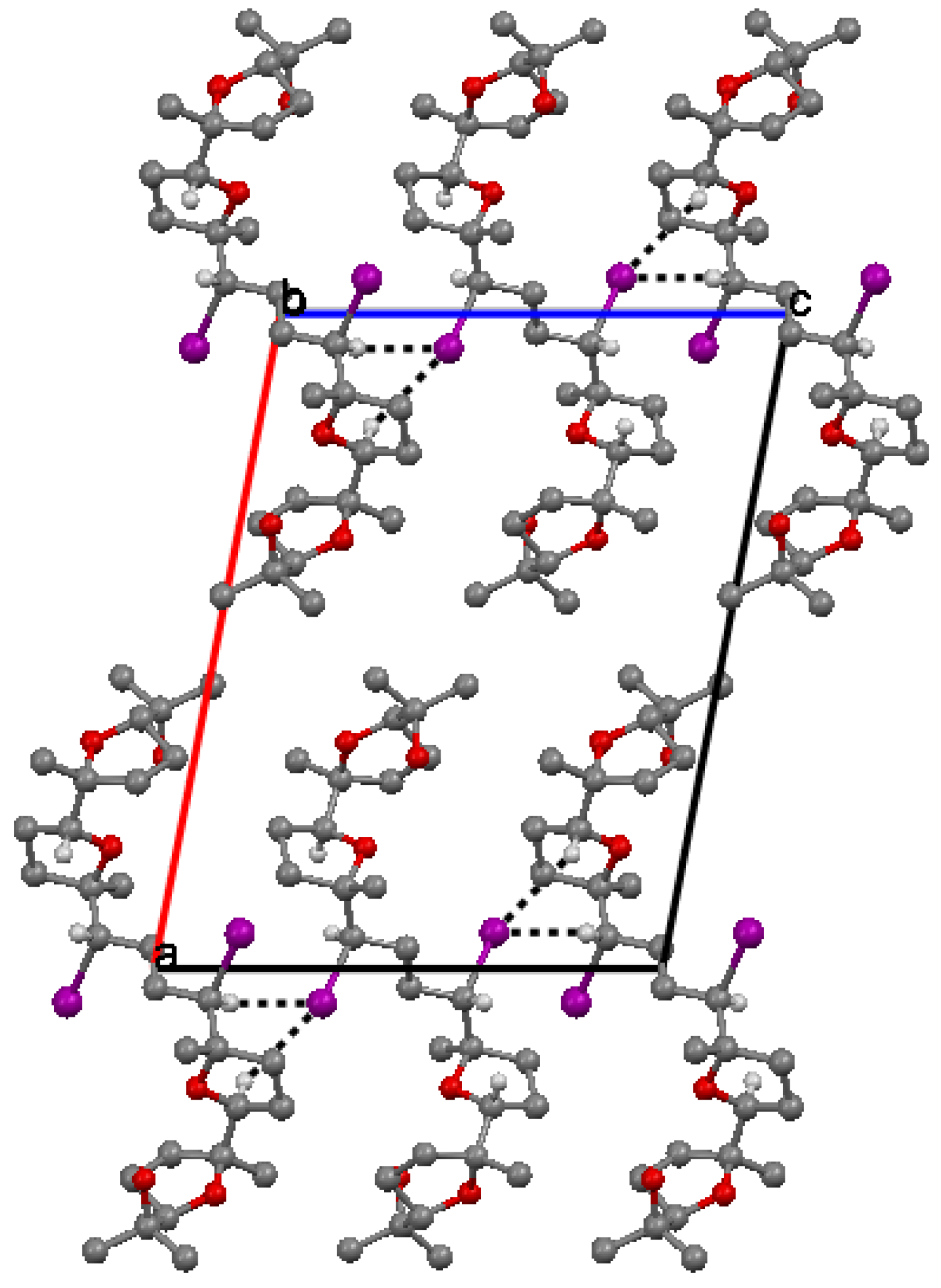
3. Experimental
3.1. General
3.2. Synthesis
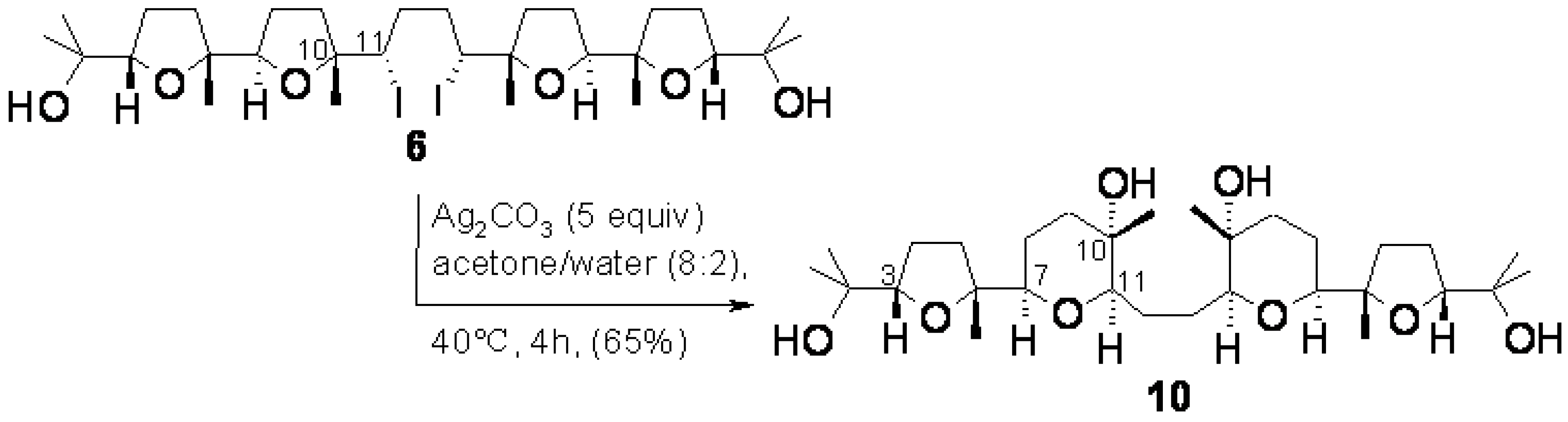
3.3. Ring expansion of 6 to 10
3.4. X-Ray Crystallography
4. Conclusion
Acknowledgments
References
- Bifulco, G.; Caserta, T.; Gomez-Paloma, L.; Piccialli, V. RuO4-promoted syn-oxidative polycyclization of isoprenoid polyenes: a new stereoselective cascade process. Tetrahedron Lett. 2002, 43, 9265–9269, corrigendum Tetrahedron Lett. 2003, 44, 3429. [Google Scholar] [CrossRef]
- Centore, R.; Tuzi, A.; Zaccaria, S.; Piccialli, V. Synthesis, stereostructure and H-bonding patterns of a tris-THF compound. J. Chem. Crystallogr. [CrossRef]
- Piccialli, V.; Oliviero, G.; Borbone, N.; Tuzi, A.; Centore, R.; Hemminki, A.; Ugolini, M.; Cerullo, V. Discovery of a new PCC-mediated stereoselective oxidative spiroketalization process. An access to a new type of poly-THF spiroketal compound displaying anticancer activity. Org. Biomol. Chem. 2009, 7, 3036–3039. [Google Scholar] [CrossRef]
- Piccialli, V.; Borbone, N.; Oliviero, G. RuO4-catalyzed oxidative polycyclization of the CS-symmetric isoprenoid polyene digeranyl. An unexpected stereochemical outcome. Tetrahedron 2008, 64, 11185–11192. [Google Scholar] [CrossRef]
- Piccialli, V. Oxidative cyclization of dienes and polyenes mediated by transition-metal-oxo species. Synthesis 2007, 17, 2585–2607. [Google Scholar] [CrossRef]
- Piccialli, V.; Caserta, T.; Caruso, L.; Gomez-Paloma, L.; Bifulco, G. RuO4-mediated oxidative polycyclization of linear polyenes. A new approach to the synthesis of the bis-THF diol core of antitumour cis-cis adjacent bis-THF annonaceous acetogenins. Tetrahedron 2006, 62, 10989–11007. [Google Scholar] [CrossRef]
- Caserta, T.; Piccialli, V.; Gomez-Paloma, L.; Bifulco, G. RuO4-catalyzed oxidative polycyclization of squalene. Determination of the configuration of the penta-tetrahydrofuranyl diol product. Tetrahedron 2005, 61, 927–939. [Google Scholar] [CrossRef]
- Bifulco, G.; Caserta, T.; Gomez-Paloma, L.; Piccialli, V. RuO4-promoted oxidative polycyclization of isoprenoid polyenes. A further insight into the stereochemistry of the process. Tetrahedron Lett. 2003, 44, 5499–5503. [Google Scholar]
- Piccialli, V.; Zaccaria, S.; Borbone, N.; Oliviero, G.; D'Errico, S.; Hemminki, A.; Cerullo, V.; Romano, V.; Tuzi, A.; Centore, R. Discovery of a novel one-step RuO4-catalysed tandem oxidative polycyclization/double spiroketalization process. Access to a new type of polyether bis-spiroketal compound displaying antitumour activity. Tetrahedron 2010, 66, 9370–9378. [Google Scholar]
- Carlsen, P.H.J.; Katsuki, T.; Martin, V.S.; Sharpless, K.B. A greatly improved procedure for ruthenium tetroxide catalyzed oxidations of organic compounds. J. Org. Chem. 1981, 46, 3936–3938. [Google Scholar] [CrossRef]
- Piccialli, V.; Cavallo, N. Improved RuO4-catalysed oxidative cyclisation of geraniol-type 1,5-dienes to cis-2,5-bis(hydroxymethyl)tetrahydrofuranyldiols. Tetrahedron Lett. 2001, 42, 4695–4699. [Google Scholar] [CrossRef]
- Albarella, L.; Musumeci, D.; Sica, D. Reactions of 1,5-dienes with ruthenium tetraoxide: Stereoselective synthesis of tetrahydrofurandiols. Eur. J. Org. Chem. 2001, 5, 997–1003. [Google Scholar]
- Roth, S.; Göhler, S.; Cheng, H.; Stark, C.B.W. A highly efficient procedure for ruthenium tetroxide catalyzed oxidative cyclizations of 1,5-dienes. Eur. J. Org. Chem. 2005, 19, 4109–4118. [Google Scholar]
- Göhler, S.; Cheng, H.; Stark, C.B.W. Catalytic diastereo- and positionselective oxidative mono-cyclization of 1,5,9-trienes and polyenes. Org. Biomol. Chem. 2007, 5, 1605–1614. [Google Scholar] [CrossRef]
- Göhler, S.; Roth, S.; Cheng, H.; Göksel, H.; Rupp, A.; Haustedt, L.O.; Stark, C.B.W. Multigram synthesis of diastereomerically pure tetrahydrofuran-diols. Synthesis 2007, 17, 2751–2754. [Google Scholar]
- de Champdoré, M.; Lasalvia, M.; Piccialli, V. OsO4-catalyzed oxidative cyclization of geranyl and neryl acetate to cis-2,5-bis(hydroxymethyl)tetrahydrofurans. Tetrahedron Lett. 1998, 39, 9781–9784. [Google Scholar]
- Donohoe, T.J.; Winter, J.J.G.; Helliwell, M.; Stemp, G. Hydrogen bonding control in the oxidative cyclisation of 1,5-dienes. Tetrahedron Lett. 2001, 42, 971–974. [Google Scholar] [CrossRef]
- Donohoe, T.J; Butterworth, S. A general oxidative cyclization of 1,5-dienes using catalytic osmium tetroxide. Angew. Chem. Int. Ed. 2003, 42, 948–951. [Google Scholar] [CrossRef]
- Klein, E.; Rojahn, W. Oxidation of olefins by potassium permanganate. Oxygen-labeling experiments and mechanism of the oxidation of 1,5-hexadiene. Evidence for a manganese intermediate with coordination number greater than four. Tetrahedron 1965, 21, 2353–2358. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Crossley, M.J.; Lehtonen, E.-M.M. Stereospecificity of oxidative cycloaddition reactions of 1,5-dienes. J. Chem. Soc., Chem. Commun. 1979, 918–920. [Google Scholar]
- Walba, D.M.; Wand, M.D.; Wilkes, M.C. Stereochemistry of the permanganate oxidation of 1,5-dienes. J. Am. Chem. Soc. 1979, 101, 4396–4397. [Google Scholar] [CrossRef]
- Walba, D.M.; Edwards, P.D. Total synthesis of ionophores the monensin BC-rings via permanganate promoted stereospecific oxidative cyclization. Tetrahedron Lett. 1980, 21, 3531–3534. [Google Scholar] [CrossRef]
- Spino, C.; Weiler, L. A stereoselective synthesis of the tetrahydrofuran unit in ionomycin. Tetrahedron Lett. 1987, 28, 731–734. [Google Scholar] [CrossRef]
- Walba, D.M.; Przybyla, C.A.; Walker, C.B. J. Total synthesis of ionophores. 6. Asymmetric induction in the permanganate-promoted oxidative cyclization of 1,5-dienes. J. Am. Chem. Soc. 1990, 112, 5624–5625. [Google Scholar]
- Kocienskyi, P.J.; Brown, R.C.D.; Pommier, A.; Procter, M.; Schmidt, B. Synthesis of salinomycin. J. Chem. Soc., Perkin Trans.1 1998, 9–40. [Google Scholar]
- Brown, R.C.D.; Hughes, R.M.; Keily, J.; Kenney, A. Diastereoselective synthesis of tetrahydrofuran-containing fragments by the permanganate oxidation of 1,5,9-trienes. Chem. Commun. 2000, 1735–1736. [Google Scholar]
- Brown, R.C.D.; Keily, J. F. Asymmetric permanganate-promoted oxidative cyclization of 1,5-dienes by using chiral phase-transfer catalysis. Angew. Chem. Int. Ed. 2001, 40, 4496–4498. [Google Scholar] [CrossRef]
- Towne, T.B.; McDonald, F.E. Syn-oxidative polycyclizations of hydroxypolyenes: highly stereoselective and potentially biomimetic syntheses of all-trans-polytetrahydrofurans. J. Am. Chem. Soc. 1997, 119, 6022–6028. [Google Scholar] [CrossRef]
- Morimoto, Y.; Iwai, T. Highly diastereoselective cyclizations of bishomoallylic tertiary alcohols promoted by rhenium(VII) oxide. Critical steric versus chelation effects in alkoxyrhenium intermediates. J. Am. Chem. Soc. 1998, 120, 1633–1634. [Google Scholar] [CrossRef]
- Sinha, S.C.; Keinan, E.; Sinha, S.C. Rules of Stereoselectivity in Tandem Oxidative Polycyclization Reaction with Rhenium(VII) Oxides. J. Am. Chem. Soc. 1998, 120, 9076–9077. [Google Scholar] [CrossRef]
- Keinan, E.; Sinha, S.C. Oxidative polycyclizations with rhenium(VII) oxides. Pure Appl. Chem. 2002, 74, 93–105. [Google Scholar] [CrossRef]
- Piancatelli, G.; Scettri, A.; D’Auria, M. Pyridinium chlorochromate: a versatile oxidant in organic synthesis. Synthesis 1982, 245–258. [Google Scholar]
- Piccialli, V.; Zaccaria, S.; Oliviero, G.; D’Errico, S.; D’Atri, V.; Borbone, N. Pyridinium chlorochromate-mediated oxidation of mono- and poly-tetrahydrofurans. Disclosure of novel oxidative pathways. Tetrahedron 2011. submitted. [Google Scholar]
- Brimble, M.A.; Edmonds, M.K. Synthesis of bis-2,5-linked tetrahydrofurans via iodoetherification. Tetrahedron 1995, 51, 9995–10012. [Google Scholar] [CrossRef]
- Nakata, T.; Nomura, S.; Matsukura, H.; Masamichi, M. Stereoselective synthesis of the C- and CD-ring systems of hemibrevetoxin B. Tetrahedron Lett. 1996, 37, 217–220. [Google Scholar] [CrossRef]
- Dickie, D.A.; Abeysekera, D.; McKenzie, I.D.; Jenkins, H.A.; Clyburne, J.A.C. Crystallographic studies on substituted m-terphenyls: identification of weak [CH3··I] interactions. Cryst. Eng. 2003, 6, 79–86. [Google Scholar] [CrossRef]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Crystallogr. 1995, A51, 33–38. [Google Scholar]
- Altomare, A.; Burla, M.C.; Cavalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, G.G.; Polidori, G.; Spagna, R. SIR97: a new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Nardelli, M. PARST95 – an update to PARST: a system of Fortran routines for calculating molecular structure parameters from the results of crystal structure analyses. J. Appl. Crystallogr. 1995, 28, 659–659. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0 – new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for Windows- a version of ORTEP-III with a graphical user interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar]
- Sample Availability: Samples of the compounds 6 and 10 are available from the authors
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Piccialli, V.; Zaccaria, S.; Centore, R.; Tuzi, A.; Borbone, N.; Oliviero, G. Isolation of a Bis-Iodurated Tetra-THF as a Trace Product from the Oxidation of Squalene with RuO4 and Its Double Ring Expansion to a Novel bis-THF-bis-THP Compound. Molecules 2011, 16, 5362-5373. https://doi.org/10.3390/molecules16075362
Piccialli V, Zaccaria S, Centore R, Tuzi A, Borbone N, Oliviero G. Isolation of a Bis-Iodurated Tetra-THF as a Trace Product from the Oxidation of Squalene with RuO4 and Its Double Ring Expansion to a Novel bis-THF-bis-THP Compound. Molecules. 2011; 16(7):5362-5373. https://doi.org/10.3390/molecules16075362
Chicago/Turabian StylePiccialli, Vincenzo, Sabrina Zaccaria, Roberto Centore, Angela Tuzi, Nicola Borbone, and Giorgia Oliviero. 2011. "Isolation of a Bis-Iodurated Tetra-THF as a Trace Product from the Oxidation of Squalene with RuO4 and Its Double Ring Expansion to a Novel bis-THF-bis-THP Compound" Molecules 16, no. 7: 5362-5373. https://doi.org/10.3390/molecules16075362
APA StylePiccialli, V., Zaccaria, S., Centore, R., Tuzi, A., Borbone, N., & Oliviero, G. (2011). Isolation of a Bis-Iodurated Tetra-THF as a Trace Product from the Oxidation of Squalene with RuO4 and Its Double Ring Expansion to a Novel bis-THF-bis-THP Compound. Molecules, 16(7), 5362-5373. https://doi.org/10.3390/molecules16075362