Enantioselective Evans-Tishchenko Reduction of b-Hydroxyketone Catalyzed by Lithium Binaphtholate

Abstract
:1. Introduction
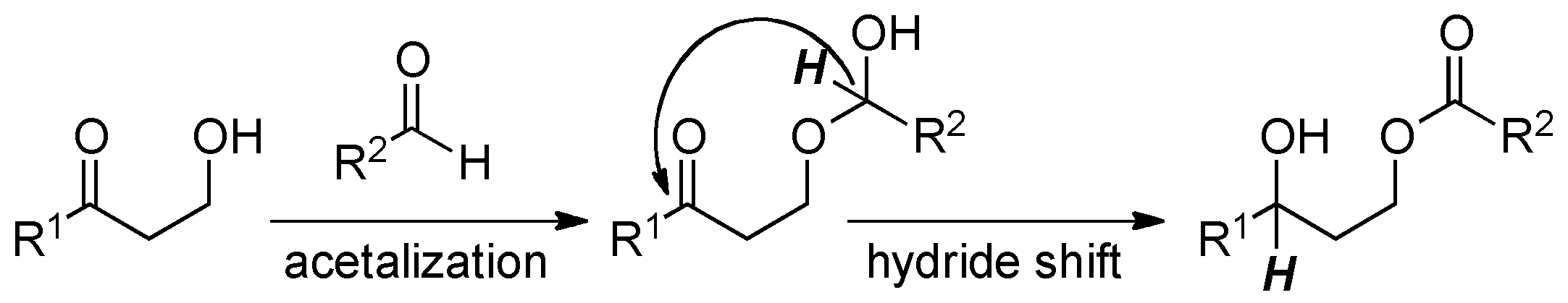
2. Results and Discussion
2.1. Optimization of Reaction Conditions


| Entry | Catalyst | Conditions | Solvent | Yield, % a | ee, % b |
|---|---|---|---|---|---|
| 1 | 1a (R = H) | rt, 24 h | THF | 36 | 2 |
| 2 | 1b (R = Me) | rt, 24 h | THF | 91 | 79 |
| 3 | 1c (R = Br) | rt, 24 h | THF | 76 | 83 |
| 4 | 1d (R = Ph) | rt, 0.5 h | THF | 82 | 96 |
| 5 | 1e (R = 4-MeC6H4) | rt, 0.5 h | THF | 80 | 89 |
| 6 | 1f (R = 3,5-Me2C6H3) | rt, 0.5 h | THF | 84 | 20 |
| 7 | 1d (R = Ph) | rt, 0.5 h | Et2O | 73 | 56 |
| 8 | 1d (R = Ph) | rt, 0.5 h | toluene | 63 | 63 |
| 9 | 1d (R = Ph) | −40 °C, 0.5 h | THF | 87 | 99 |
| 10 | 1d (R = Ph) | −78 °C, 48 h | THF | 56 | 99 |
2.2. Evans-Tishchenko Reduction of Various Achiral β-Hydroxy ketones

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 2 | 3 | Conditions | 4 | Yield of 4 (5) % a | ee of 4 (5) % b |
|---|---|---|---|---|---|---|
| 1 | 2a | 3a | −40 °C, 0.5 h | 4aa | 87 (0) | 99 |
| 2 | 2a | 3b | 0 °C, 1 h | 4ab | 93 (5) | 90 (90) |
| 3 | 2b | 3a | −40 °C, 0.5 h | 4ba | 96 (0) | 98 |
| 4 | 2b | 3b | 0 °C, 4 h | 4bb | 82 (13) | 83 |
| 5 | 2c | 3a | rt, 4 h | 4ca | 64 (31) | 93 (93) |
| 6 | 2d | 3a | −40 °C, 6 h | 4da | 91 c [4da:5da = 2:1] | 99 c |

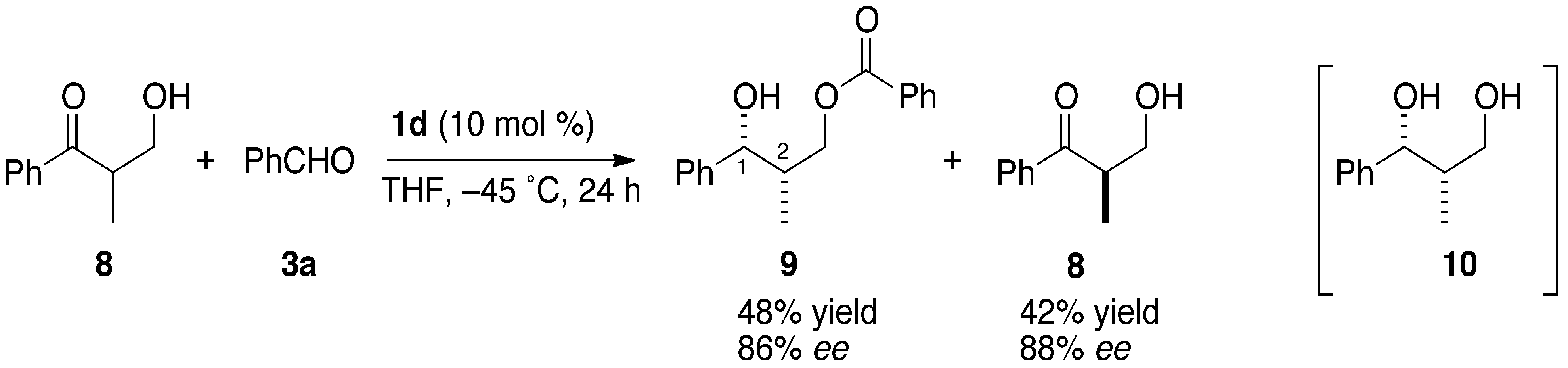
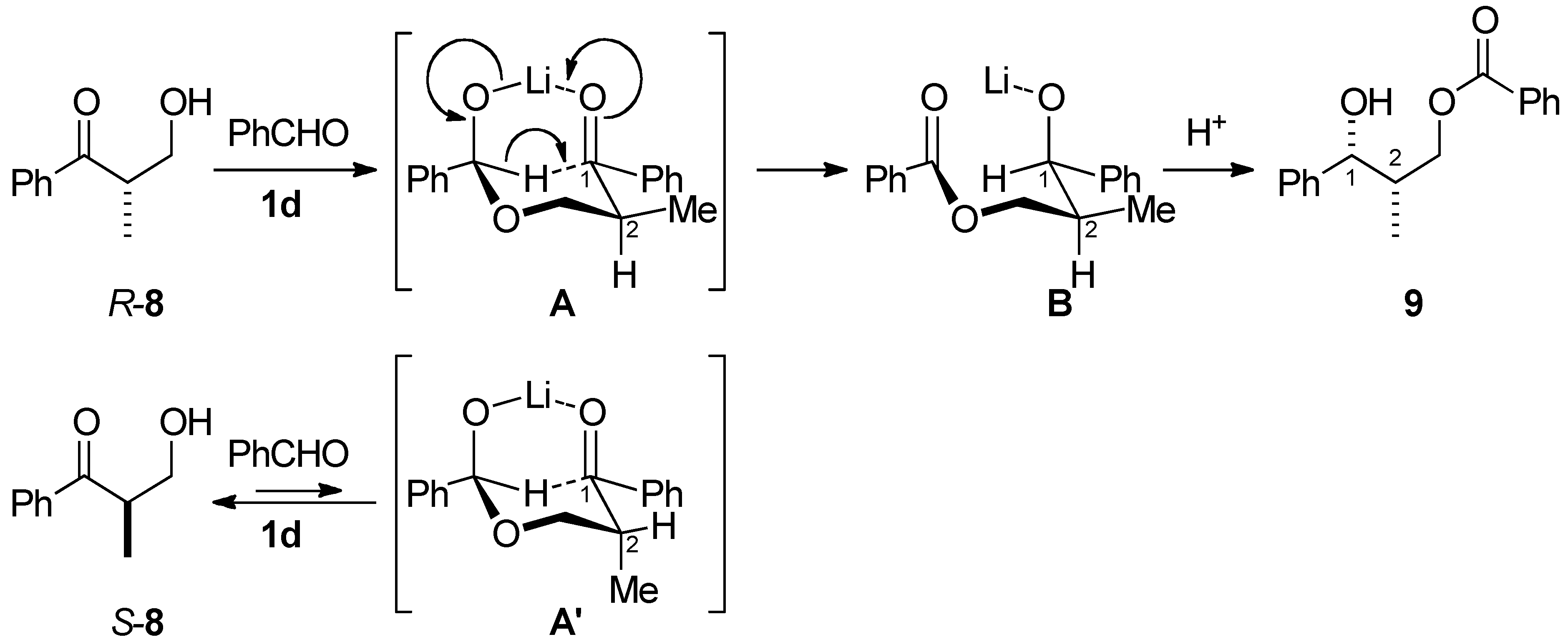
2.3. Evans-Tishchenko Reduction of Chiral β-Hydroxypropiophenone
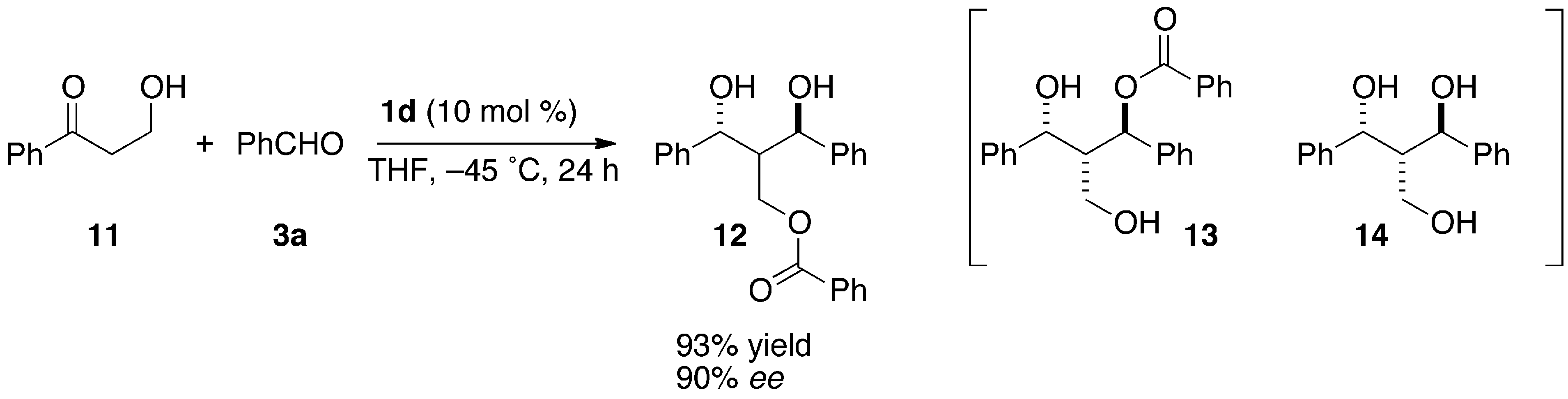
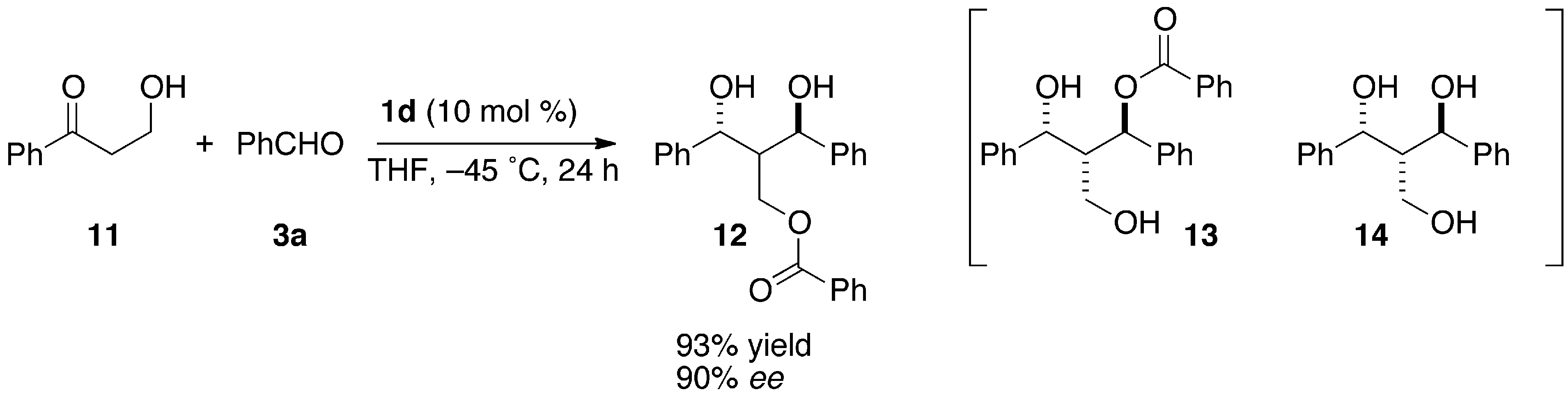
2.4. Reaction of An α-Unsubstituted-β-Hydroxypropiophenone
3. Experimental
3.1. General
3.2. Enantioselective Evans-Tishchenko Reduction of β-Hydroxyketones
3.2.1. (S)-2,2-Dimethyl-1-phenyl-1,3-propanediol 3-O-benzoate (4aa) [38]
Determination of the absolute configuration of 4aa.
3.2.2. (S)-2,2-Dimethyl-1-phenyl-1,3-propanediol 3-O-pivaloate (4ab)
3.2.3. (S)-2,2-Dimethyl-1-phenyl-1,3-propanediol 1-O-pivaloate (5ab)
3.2.4. (−)-2,2-Pentamethylene-1-phenyl-1,3-propanediol 3-O-benzoate (4ba)
3.2.5. (+)-2,2-Pentamethylene-1-phenyl-1,3-propanediol 3-O-pivaloate (4bb)
3.2.6. (+)-2,2-Pentamethylene-1-phenyl-1,3-propanediol 1-O-pivaloate (5bb)
3.2.7. (S)-2,2,4-Trimethyl-1,3-pentanediol 1-O-benzoate (4ca)
3.2.8. (S)-2,2,4-Trimethyl-1,3-pentanediol 3-O-benzoate (5ca)
3.2.9. (+)-2,2-Dimethyl-1,3-butanediol dibenzoate (7da)
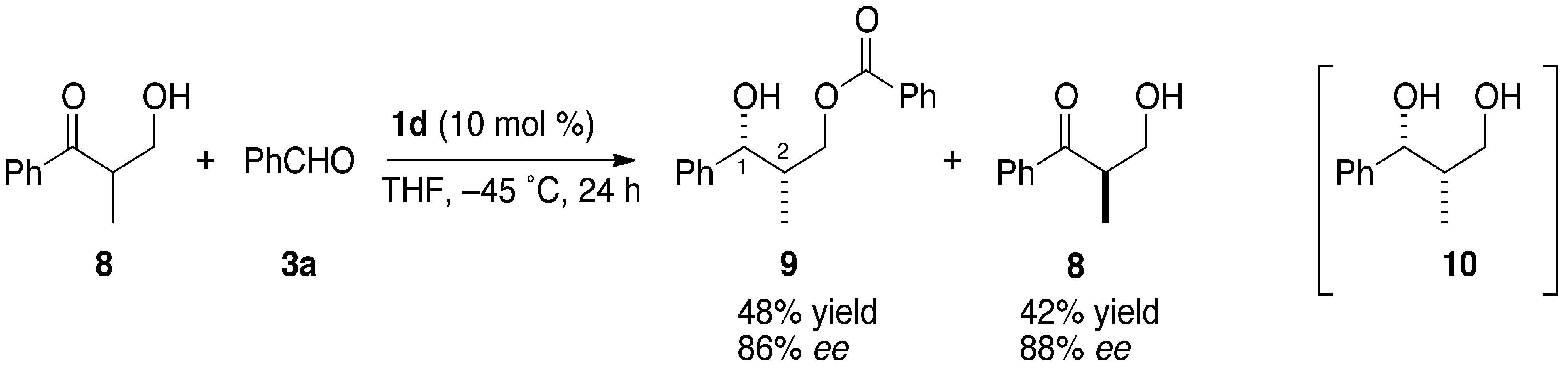
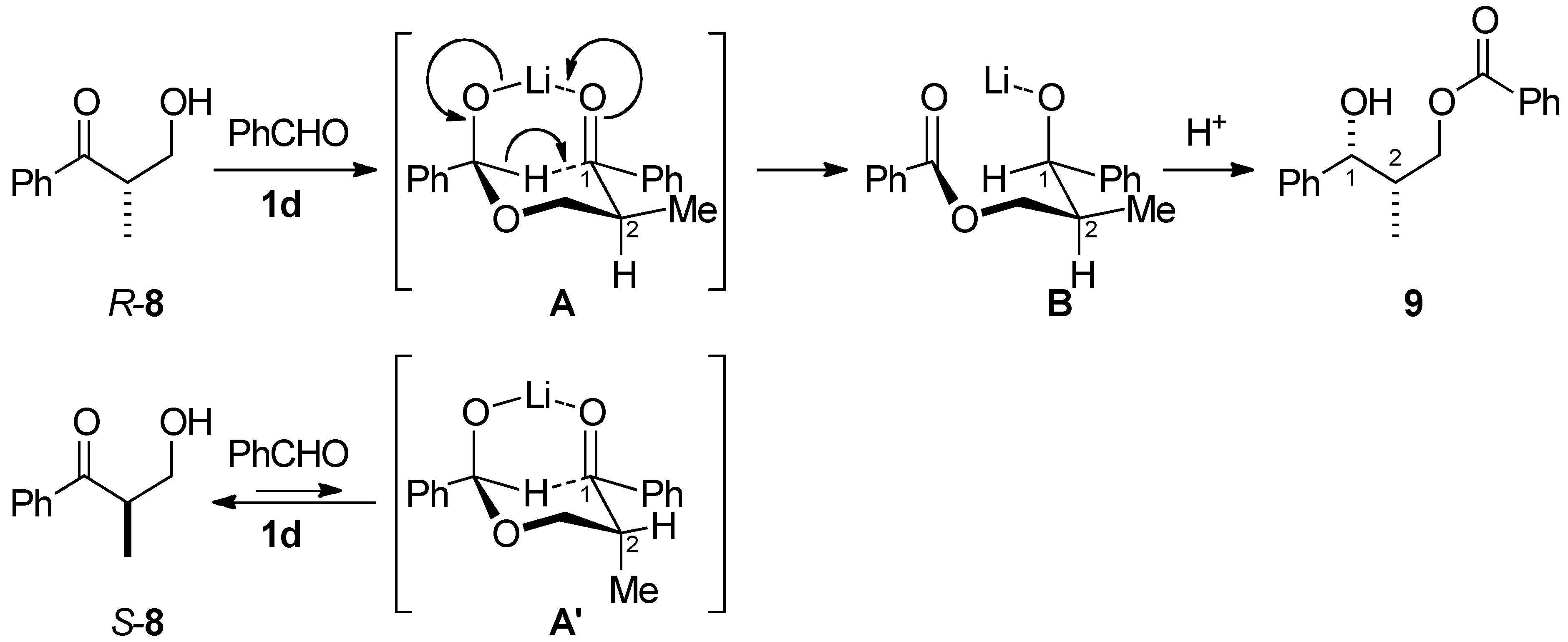
3.3. Kinetic Optical Resolution of 3-Hydroxy-2-methyl-1-phenylpropan-1-one (8)
3.3.1. (1R,2S)-2-Methyl-1-phenyl-1,3-propanediol 3-O-benzoate (9)
3.3.2. (S)-3-Hydroxy-2-methyl-1-phenylpropan-1-one (8)
3.4. The Aldol-Tishchenko Reaction of An α-Unsubstituted-β-Hydroxypropiophenone
(1R,3R)-2-Benzoyloxymethyl-1,3-diphenyl-1,3-propanediol (12)
4. Conclusions
References
- Oishi, T.; Nakata, T. New aspects of stereoselective synthesis of 1,3-polyols. Synthesis 1990, 635–645. [Google Scholar] [CrossRef]
- Bode, S.E.; Wolberg, M.; Müller, M. Stereoselective synthesis of 1,3-diols. Synthesis 2006, 557–588. [Google Scholar]
- Evans, D.A.; Hoveyda, A.H. Samarium-catalyzed intramolecular Tishchenko reduction of β-hydroxy ketones. A stereoselective approach to the synthesis of differentiated anti 1,3-diol monoesters. J. Am. Chem. Soc. 1990, 112, 6447–6449. [Google Scholar] [CrossRef]
- Umekawa, Y.; Sakaguchi, S.; Nishiyama, Y.; Ishii, Y. Stereoselective reduction of β-hydroxy ketones with aldehydes via Tishchenko reactions catalyzed by zirconocene complexes. J. Org. Chem. 1997, 62, 3409–3412. [Google Scholar] [CrossRef]
- Gillespie, K.M.; Munslow, I.J.; Scott, P. Stereoselective catalytic Tishchenko reduction of β-hydroxyketones using scandium triflate. Tetrahedron Lett. 1999, 40, 9371–9374. [Google Scholar] [CrossRef]
- Romo, D.; Meyer, S.D.; Johnson, D.D.; Schreiber, S.L. Total synthesis of (−)-rapamycin using an Evans-Tishchenko fragment coupling. J. Am. Chem. Soc. 1993, 115, 7906–7907. [Google Scholar] [CrossRef]
- Schöning, K.-U.; Hayashi, R.K.; Powell, D.R.; Kirschning, A. Synthetic studies toward ansatrienines: Application of the Evans-Tishchenko reaction to chiral enones. Tetrahedron: Asymmetry 1999, 10, 817–820. [Google Scholar] [CrossRef]
- Shotwell, J.B.; Krygowski, E.S.; Hines, J.; Koh, B.; Huntsman, E.W.D.; Choi, H.W.; Schneekloth, J.S.; Wood, J.L.; Crews, C.M. Total synthesis of luminacin D. Org. Lett. 2002, 4, 3087–3089. [Google Scholar]
- Jiang, Y.; Hong, J.; Burke, S.D. Stereoselective total synthesis of antitumor macrolide (+)-rhizoxin D. Org. Lett. 2004, 6, 1445–1448. [Google Scholar] [CrossRef]
- Aird, J.I.; Hulme, A.N.; White, J.W. An Evans-Tishchenko-ring-closing metathesis approach to medium-ring lactones. Org. Lett. 2007, 9, 631–634. [Google Scholar] [CrossRef]
- Smith, A.B., III; Lee, D. Total synthesis of (+)-tedanolide. J. Am. Chem. Soc. 2007, 129, 10957–10962. [Google Scholar] [CrossRef]
- Youngsaye, W.; Lowe, J.T.; Pohlki, F.; Ralifo, P.; Panek, J.S. Total synthesis and stereochemical reassignment of (+)-neopeltolide. Angew. Chem. Int. Ed. 2007, 46, 9211–9214. [Google Scholar] [CrossRef]
- Slavov, N.; Cvengros, J.; Neudörfl, J.-M.; Schmalz, H.-G. Total synthesis of the marine antibiotics pestalone and its surprisingly facile conversion into pestalalactone and pestalachloride A. Angew. Chem. Int. Ed. 2010, 49, 7588–7591. [Google Scholar] [CrossRef]
- Ichibakase, T.; Nakajima, M. Direct enantioselective aldol-Tishchenko reaction catalyzed by chiral lithium diphenylbinaphtholate. Org. Lett. 2011, 13, 1579–1581. [Google Scholar] [CrossRef]
- Mahrwald, R. The aldol-Tishchenko reaction: A tool in stereoselective synthesis. Curr. Org. Chem. 2003, 7, 1713–1723. [Google Scholar] [CrossRef]
- Mlynarski, J. Direct asymmetric aldol-Tishchenko reaction. Eur. J. Org. Chem. 2006, 4779–4786. [Google Scholar] [CrossRef]
- Mascarenhas, C.M.; Miller, S.P.; White, P.S.; Morken, J.P. First catalytic asymmetric aldol-Tishchenko reaction-insight into the catalyst structure and reaction mechanism. Angew. Chem. Int. Ed. 2001, 40, 601–603. [Google Scholar] [CrossRef]
- Schneider, C.; Hansch, M. First catalytic, enantioselective aldol-Tishchenko reaction with ketone aldols as enol equivalents. Synlett 2003, 837–840. [Google Scholar] [CrossRef]
- Gnanadesikan, V.; Horiuchi, Y.; Ohshima, T.; Shibasaki, M. Direct catalytic asymmetric aldol-Tishchenko reaction. J. Am. Chem. Soc. 2004, 126, 7782–7783. [Google Scholar] [CrossRef]
- Mlynarski, J.; Mitura, M. The first example of a catalytic asymmetric aldol-Tishchenko reaction of aldehydes and aliphatic ketones. Tetrahedron Lett. 2004, 45, 7549–7552. [Google Scholar] [CrossRef]
- Rohr, K.; Herre, R.; Mahrwald, R. Enantioselective direct aldol-Tishchenko reaction: Access to chiral stereopentads. Org. Lett. 2005, 7, 4499–4501. [Google Scholar] [CrossRef]
- Schiffers, R.; Kagan, H.B. Asymmetric catalytic reduction of ketones with hypervalent trialkoxysilanes. Synlett 1997, 1175–1178. [Google Scholar] [CrossRef]
- Holmes, I.P.; Kagan, H.B. The asymmetric addition of trimethylsilylcyanide to aldehydes catalysed by anionic chiral nucleophiles. Tetrahedron Lett. 2000, 41, 7453–7456. [Google Scholar] [CrossRef]
- Hatano, M.; Ikeno, T.; Miyamoto, T.; Ishihara, K. Chiral lithium binaphtholate aqua complex as a highly effective asymmetric catalyst for cyanohydrin synthesis. J. Am. Chem. Soc. 2005, 127, 10776–10777. [Google Scholar] [CrossRef]
- Hatano, M.; Ikeno, T.; Matsumura, T.; Torii, S.; Ishihara, K. Chiral lithium salts of phosphoric acids as Lewis acid-base conjugate catalysts for the enantioselective cyanosilylation of ketones. Adv. Synth. Catal. 2008, 350, 1776–1780. [Google Scholar] [CrossRef]
- Hatano, M.; Horibe, T.; Ishihara, K. Chiral lithium(I) binaphtholate salts for the enantioselective direct Mannich-type reaction with a change of syn/anti and absolute stereochemistry. J. Am. Chem. Soc. 2010, 132, 56–57. [Google Scholar] [CrossRef]
- Nakajima, M.; Orito, Y.; Ishizuka, T.; Hashimoto, S. The enantioselective aldol reaction of trimethoxysilyl enol ether catalyzed by lithium binaphtholate. Org. Lett. 2004, 6, 3763–3765. [Google Scholar] [CrossRef]
- Ichibakase, T.; Orito, Y.; Nakajima, M. Enantioselective construction of quaternary asymmetric carbon centers using an aldol reaction of trimethoxysilyl enol ethers catalyzed by lithium binaphtholate. Tetrahedron Lett. 2008, 49, 4427–4429. [Google Scholar] [CrossRef]
- Tanaka, K.; Ueda, T.; Ichibakase, T.; Nakajima, M. Enantioselective alkynylation of ketones with trimethoxysilylalkynes using lithium binaphtholate as a catalyst. Tetrahedron Lett. 2010, 51, 2168–2169. [Google Scholar] [CrossRef]
- Tanaka, K.; Kukita, K.; Ichibakase, T.; Kotani, S.; Nakajima, M. Lithium acetylides as alkynylating reagents for the enantioselective alkynylation of ketones catalyzed by lithium binaphtholate. Chem. Commun. 2011, 47, 5614–5616. [Google Scholar]
- Yakura, T.; Yoshimoto, Y.; Ishida, C.; Mabuchi, S. Synthesis of an immunomodulator (+)-conagenin and its analogs. Tetrahedron 2007, 63, 4429–4438. [Google Scholar] [CrossRef]
- Steif, F.; Wibbeling, B.; Meyer, O.; Hoppe, D. Enantio- and diastereoselective synthesis of (protected) 2-formyl- and 2-(hydroxymethyl)-1-phenylalkane-1,3-diols from chiral 2-methoxy-3-tosyl-1,3-oxazolidines by subsequent asymmetric formylation and aldolization. Synthesis 2000, 743–753. [Google Scholar]
- Sulmon, P.; De Kimpe, N.; Schamp, N. Preparation of α,α-dialkyl-β-haloketones. Org. Prep. Proc. Int. 1989, 21, 91–104. [Google Scholar] [CrossRef]
- Donnelly, J.A.; Hoey, J.G.; O’Donnell, R. Rearrangement of an oxetan-3-one and related alcohols by Grignard reagents. J. Chem. Soc. Perkin Trans. 1 1974, 1218–1220. [Google Scholar]
- Curzu, M.M.; Pinna, G.A. Hydroxymethylation of propiophenones in aqueous medium: A new route to 1-aryl-2-methyl-2-propen-1-ones. Synthesis 1984, 339–342. [Google Scholar]
- O’Neil, G.W.; Miller, M.M.; Carter, K.P. Direct conversion of β-hydroxyketones to cyclic disiloxanes. Org. Lett. 2010, 12, 5350–5353. [Google Scholar] [CrossRef]
- Wu, T.R.; Shen, L.; Chong, J.M. Asymmetric allylboration of aldehydes and ketones using 3,3‘-disubstitutedbinaphthol-modified boronates. Org. Lett. 2004, 6, 2701–2704. [Google Scholar] [CrossRef]
- Markert, M.; Mahrwald, R. LiClO4-amine mediated direct aldol process. Synthesis 2004, 1429–1433. [Google Scholar]
- Kotani, S.; Shimoda, Y.; Sugiura, M.; Nakajima, M. Novel enantioselective direct aldol-type reaction promoted by a chiral phosphine oxide as an organocatalyst. Tetrahedron Lett. 2009, 50, 4602–4605. [Google Scholar] [CrossRef]
- Seifert, A.; Scheffler, U.; Markert, M.; Mahrwald, R. Asymmetric acid-catalyzed Meerwein-Ponndorf-Verley-aldol reactions of enolizable aldehydes. Org. Lett. 2010, 12, 1660–1663. [Google Scholar] [CrossRef]
- Kokubo, N.; Ogawa, C.; Kobayashi, S. Lewis acid catalysis in water with a hydrophilic substrate: Scandium-catalyzed hydroxymethylation with aqueous formaldehyde in water. Angew. Chem. Int. Ed. 2008, 47, 6909–6911. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ichibakase, T.; Nakatsu, M.; Nakajima, M. Enantioselective Evans-Tishchenko Reduction of b-Hydroxyketone Catalyzed by Lithium Binaphtholate. Molecules 2011, 16, 5008-5019. https://doi.org/10.3390/molecules16065008
Ichibakase T, Nakatsu M, Nakajima M. Enantioselective Evans-Tishchenko Reduction of b-Hydroxyketone Catalyzed by Lithium Binaphtholate. Molecules. 2011; 16(6):5008-5019. https://doi.org/10.3390/molecules16065008
Chicago/Turabian StyleIchibakase, Tomonori, Masato Nakatsu, and Makoto Nakajima. 2011. "Enantioselective Evans-Tishchenko Reduction of b-Hydroxyketone Catalyzed by Lithium Binaphtholate" Molecules 16, no. 6: 5008-5019. https://doi.org/10.3390/molecules16065008
APA StyleIchibakase, T., Nakatsu, M., & Nakajima, M. (2011). Enantioselective Evans-Tishchenko Reduction of b-Hydroxyketone Catalyzed by Lithium Binaphtholate. Molecules, 16(6), 5008-5019. https://doi.org/10.3390/molecules16065008