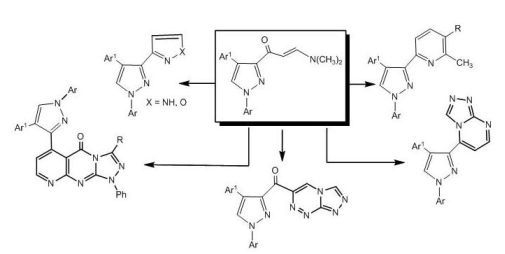
2. Results and Discussion
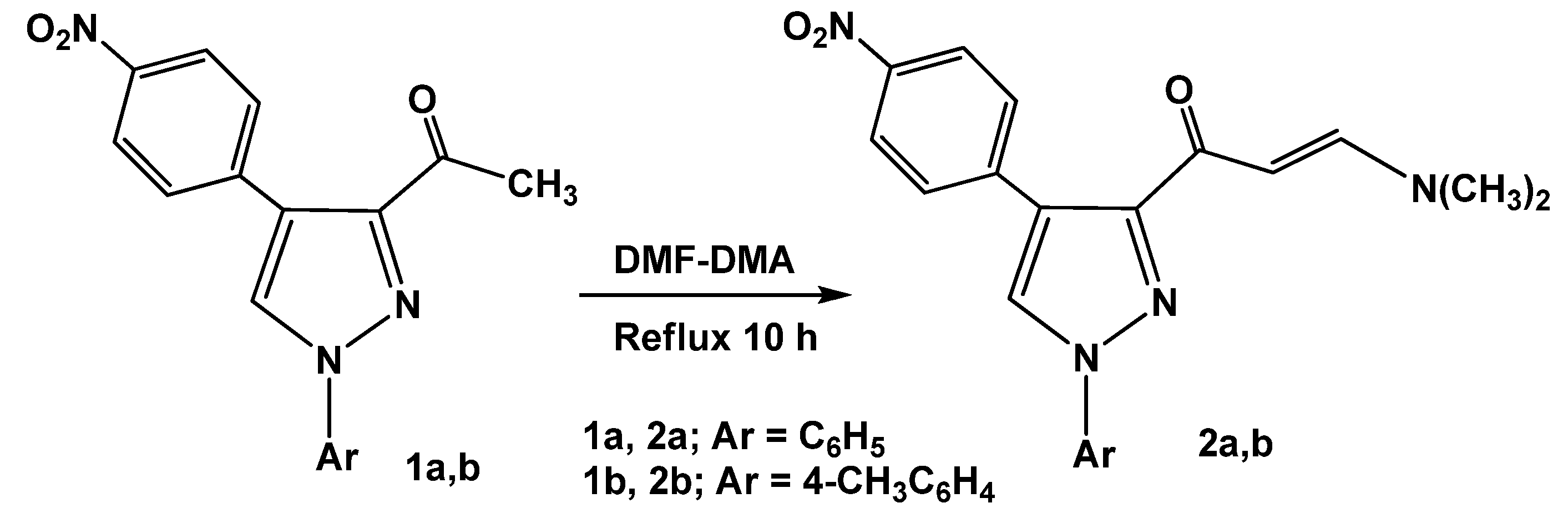
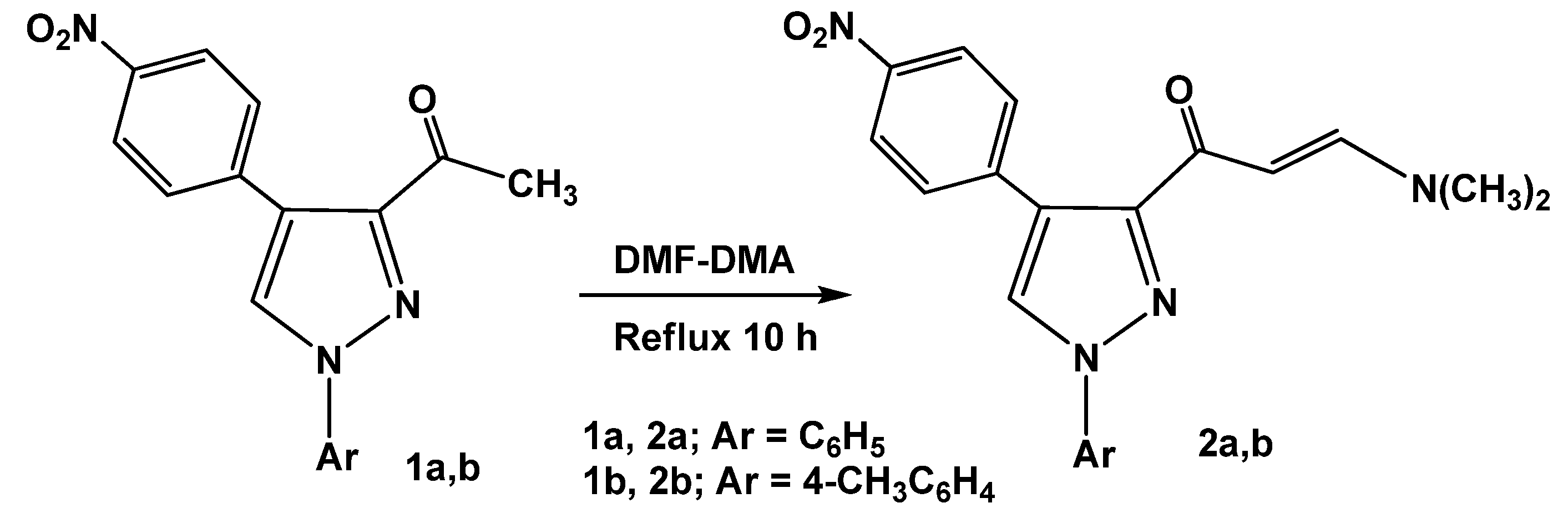
The synthetic route for preparation of the previously unreported 3-[
E-3-(
N,
N-dimethyl-amino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1
H-pyrazoles (
2a,b), involving condensation of 3-acetyl-4-(4-nitrophenyl)-1-aryl-1
H-pyrazoles (
1a,b) [
26] with dimethylformamide dimethylacetal (DMF-DMA) under reflux for 10 hours in the absence of solvent, is depicted in
Scheme 1.
Scheme 1.
Synthesis of 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles (2a,b).
Scheme 1.
Synthesis of 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles (2a,b).
The structures of
2a,b were confirmed by their spectral data (IR, MS and
1H-NMR) and elemental analyses. For example, the
1H-NMR spectrum revealed two doublet signals at
δ 5.88, 7.67 ppm with coupling constant
J = 13 Hz assignable to olefinic protons (CH=CH) in a
trans configuration [
26,
27] besides two singlet signals of the dimethylamino group at
δ 2.8, 3.1 ppm.
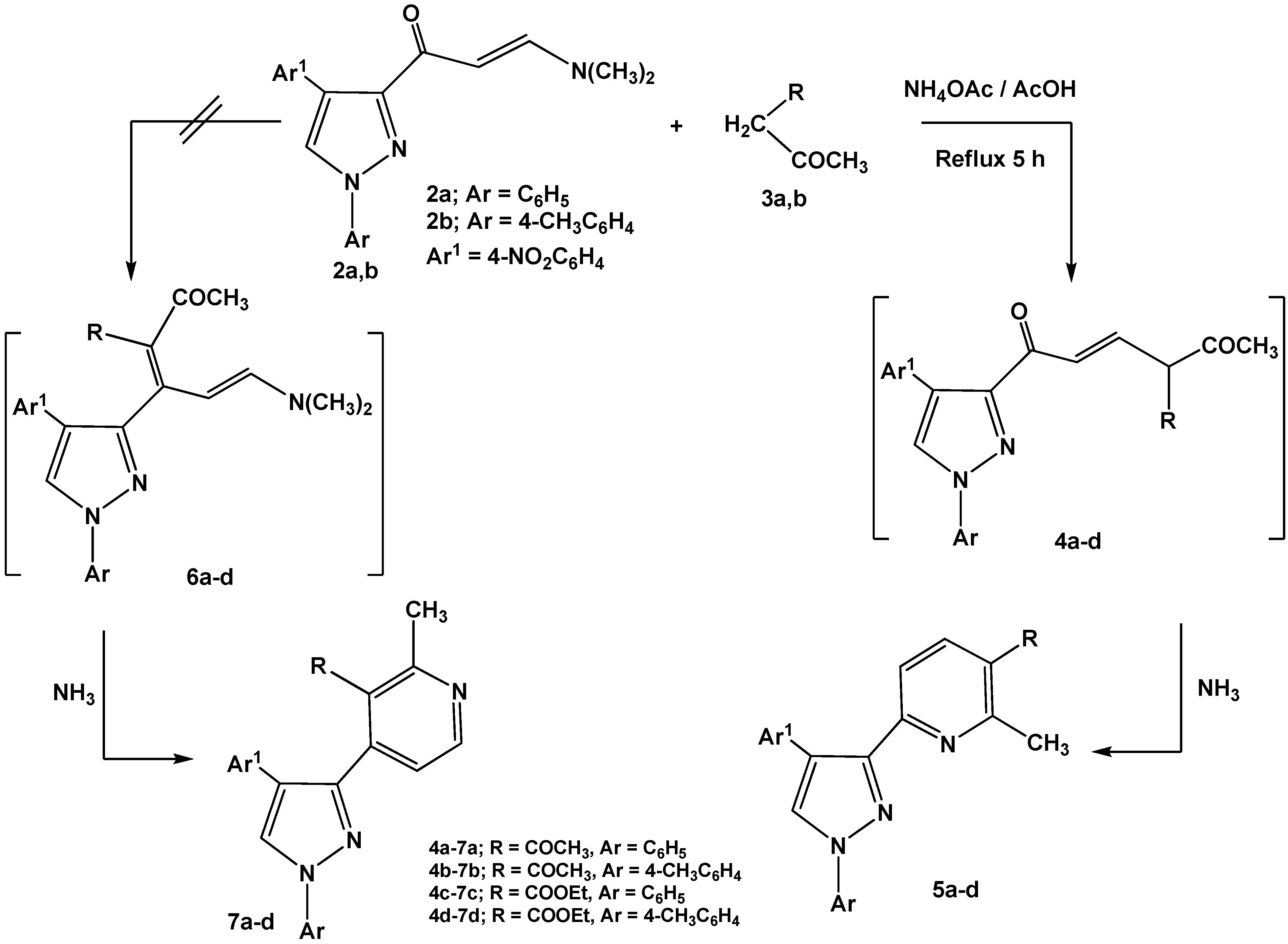
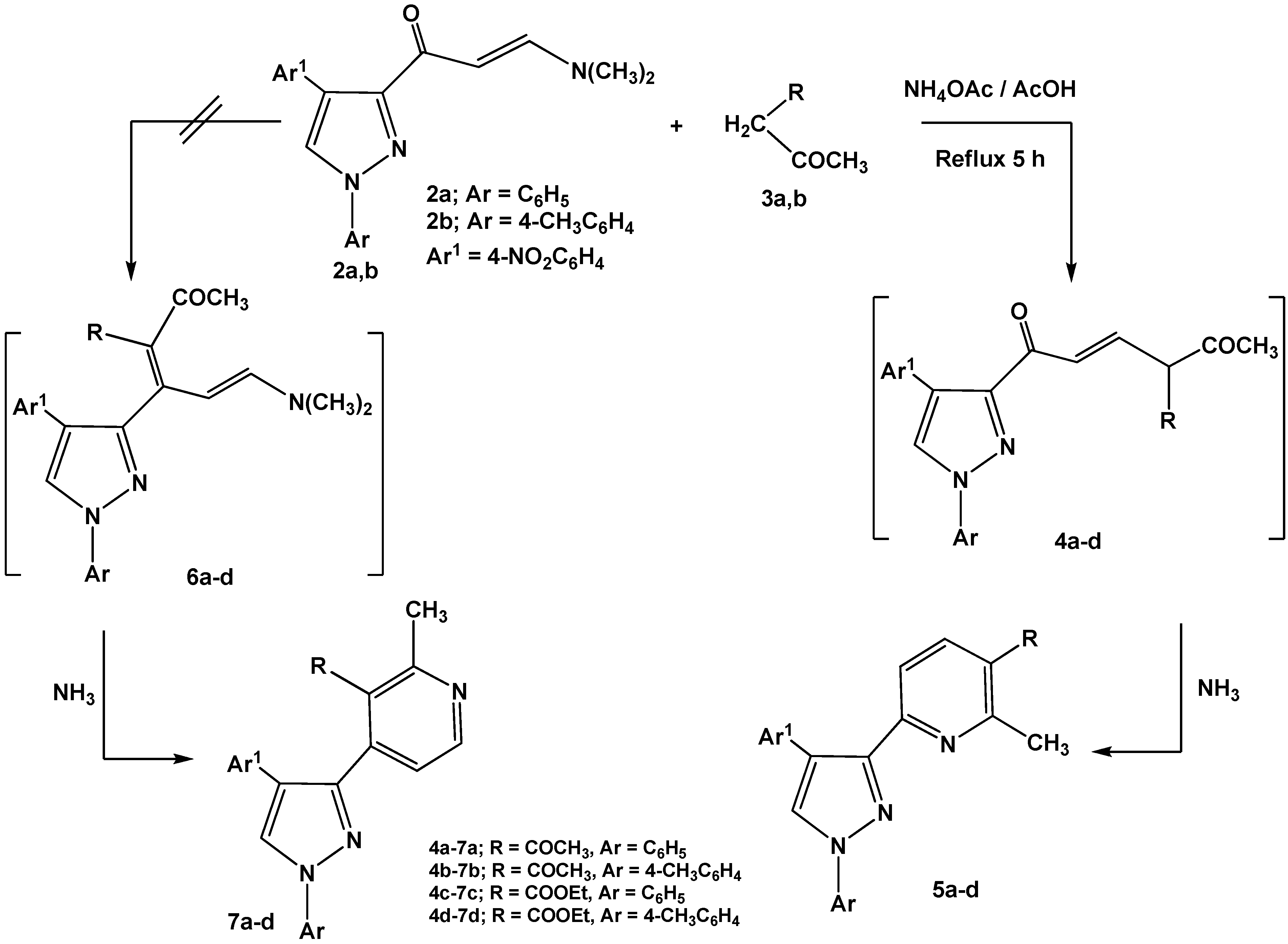
Reactions of enaminones
2a,b with
C-nucleophiles such as 2,4-pentanedione and ethyl 3-oxo-butanoate were carried out in glacial acetic acid in the presence of ammonium acetate and led to formation of 6-(pyrazol-3-yl)-pyridine derivatives
5a-d via nucleophilic displacement of active methylene to the dimethylamino group followed by concurrent elimination of water molecule from non-isolable intermediates
4a-d (
Scheme 2). The other possible isomeric structures 4-(pyrazol-3-yl)-pyridines
7a-d were discarded based on
1H-NMR data that revealed pyridyl hydrogens at C-4, C-5 as a pair of doublets at
δ 7.5, 7.7 ppm, respectively, with
J = 8 Hz assignable to 6-substituted-pyridines
5a-d. The isomeric structures
7a-d should display pair of doublets corresponding to C-5, C-6 with a lower coupling constant (
J = 2–3 Hz) [
28].
Scheme 2.
Reactions of enaminones 2a,b with active methylene compounds.
Scheme 2.
Reactions of enaminones 2a,b with active methylene compounds.
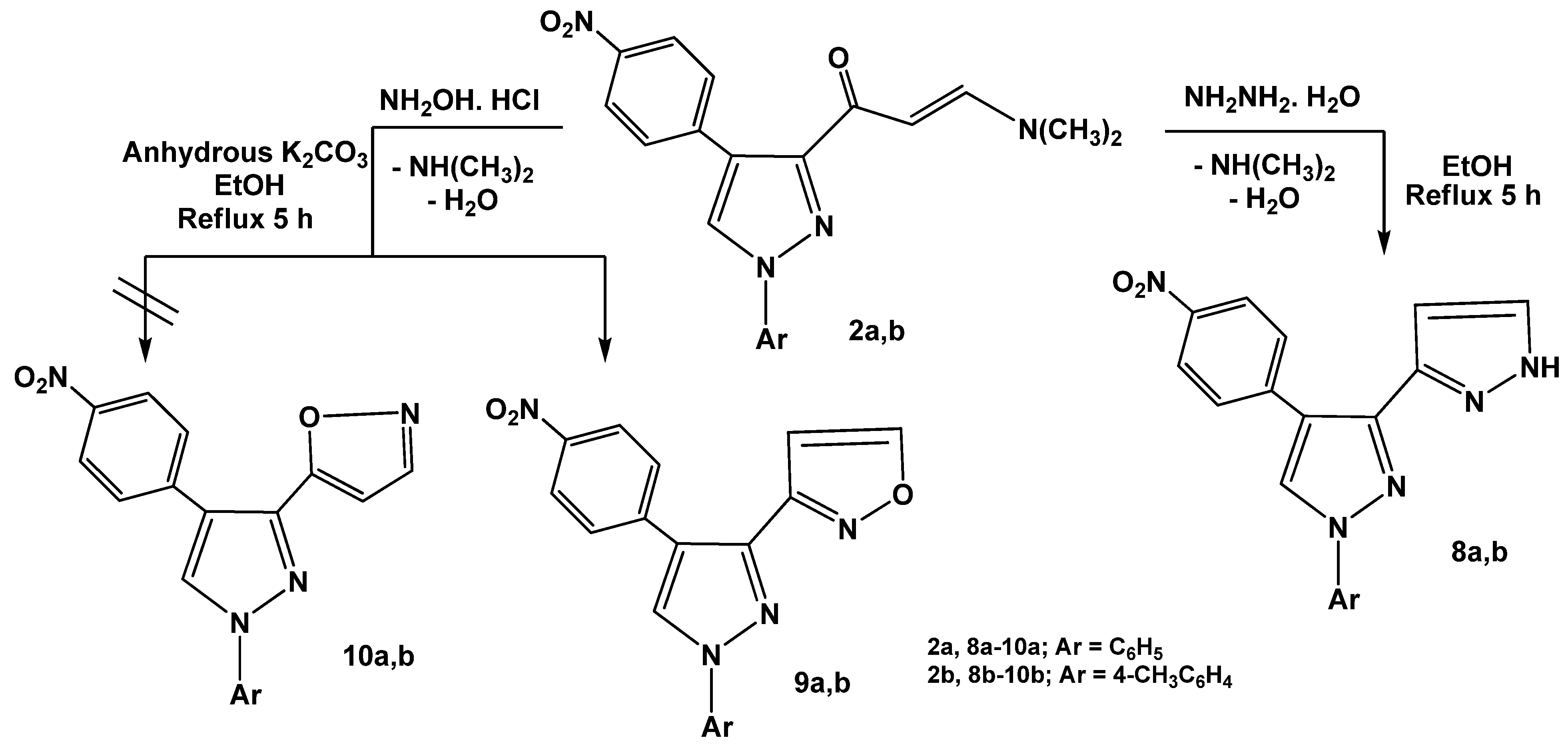
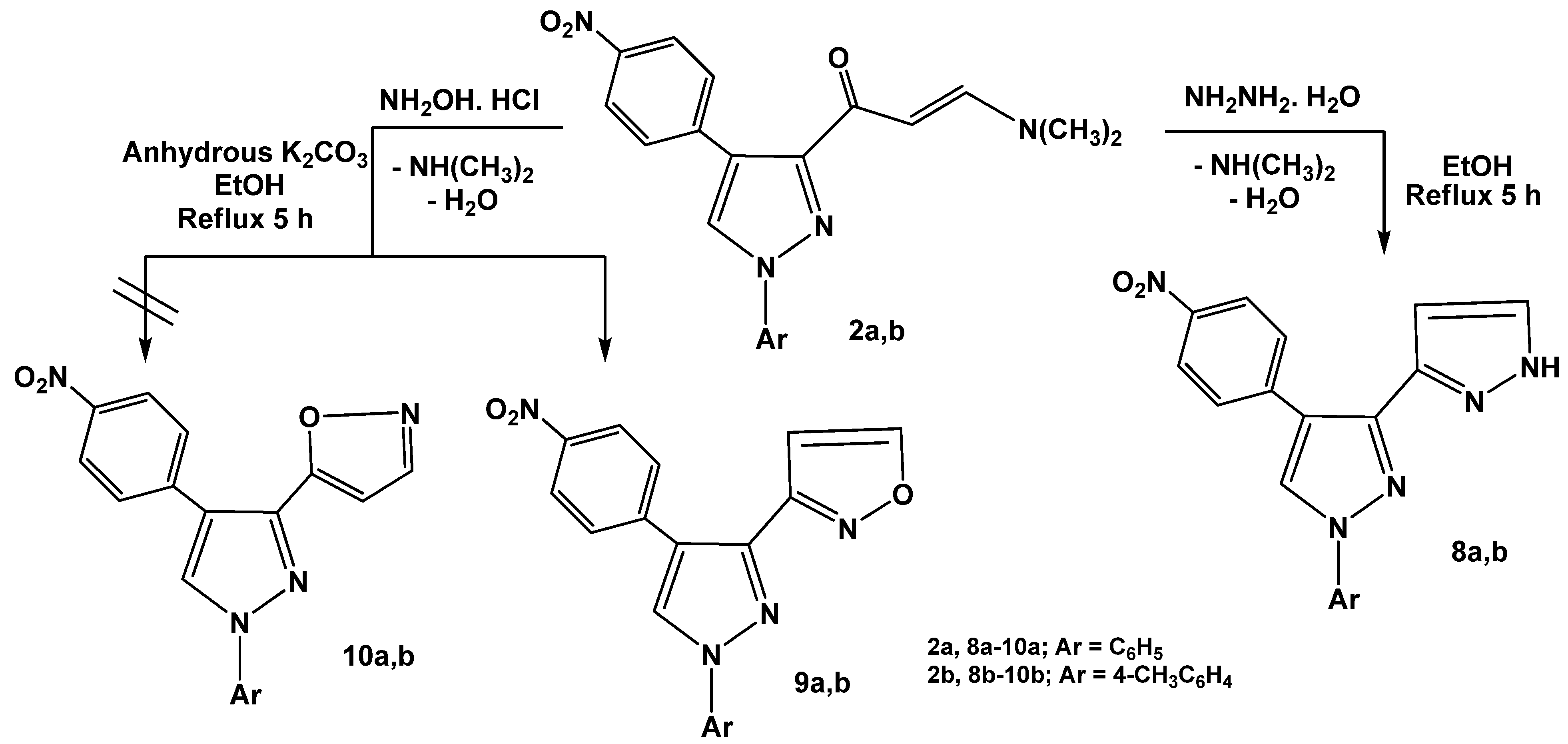
Treatment of enaminones
2a,b with a
N-nucleophile such as hydrazine hydrate in absolute ethanol under reflux afforded 1
H,1'
H-3,3'-bipyrazoles
8a,b. The structures of the products were substantiated by the
1H-NMR spectra which displayed new pair of doublets at
δ 7.53 and 7.58 ppm with (
J = 7.5 Hz) corresponding to pyrazole protons at positions 4 and 5, respectively and another D
2O exchangeable proton at
δ 13 ppm assignable to the NH group. The products were formed
via initial addition of the amino group in hydrazine to the enaminone double bond, followed by elimination of dimethylamine and water molecules to give the final isolable products
8a,b as previously mentioned [
29] (
Scheme 3). Similarly, enaminones
2a,b reacted with hydroxylamine hydrochloride in refluxing absolute ethanol in the presence of anhydrous potassium carbonate to yield products that may be formulated as pyrazolylisoxazoles
9a,b or its isomeric forms
10a,b. Structure
9 was assigned for the reaction products on the basis of the
1H-NMR spectral data in which a resonance for H-4 and H-5 of isoxazole appeared typically at
δ 6.78 and 8.50 ppm, respectively (see Experimental). The other isomeric structures
10a,b were ruled out as the isoxazole H-3 would be expected to resonate at a higher field of
δ 8.0 ppm [
30]. It is thus assumed that, the products
9a,b were formed
via initial condensation of amino group of hydroxylamine with carbonyl group of enaminones
2a,b followed by elimination of dimethylamine (cf.
Scheme 3).
Scheme 3.
Reactions of enaminones 2a,b with hydrazine hydrate and hydroxylamine.
Scheme 3.
Reactions of enaminones 2a,b with hydrazine hydrate and hydroxylamine.
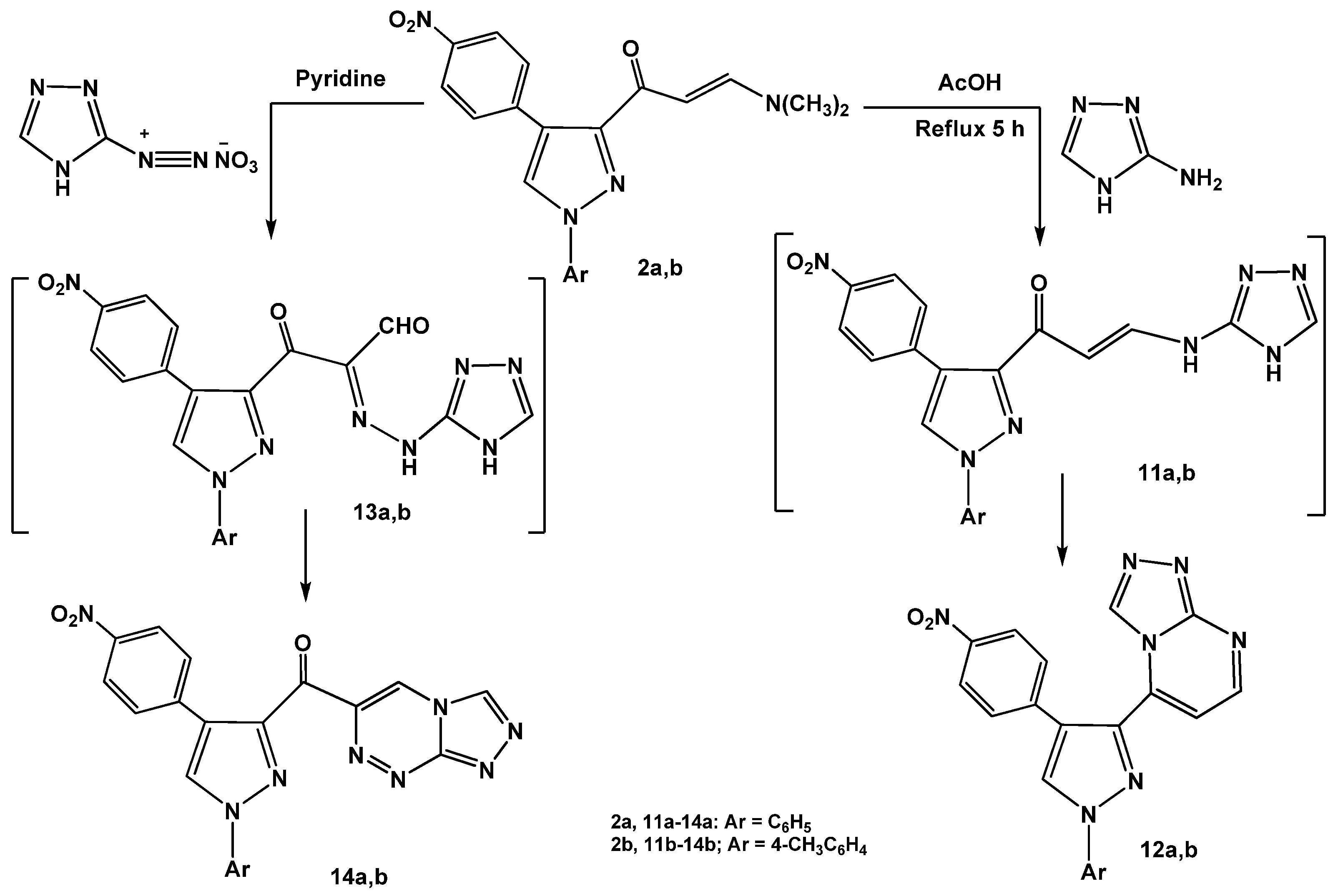
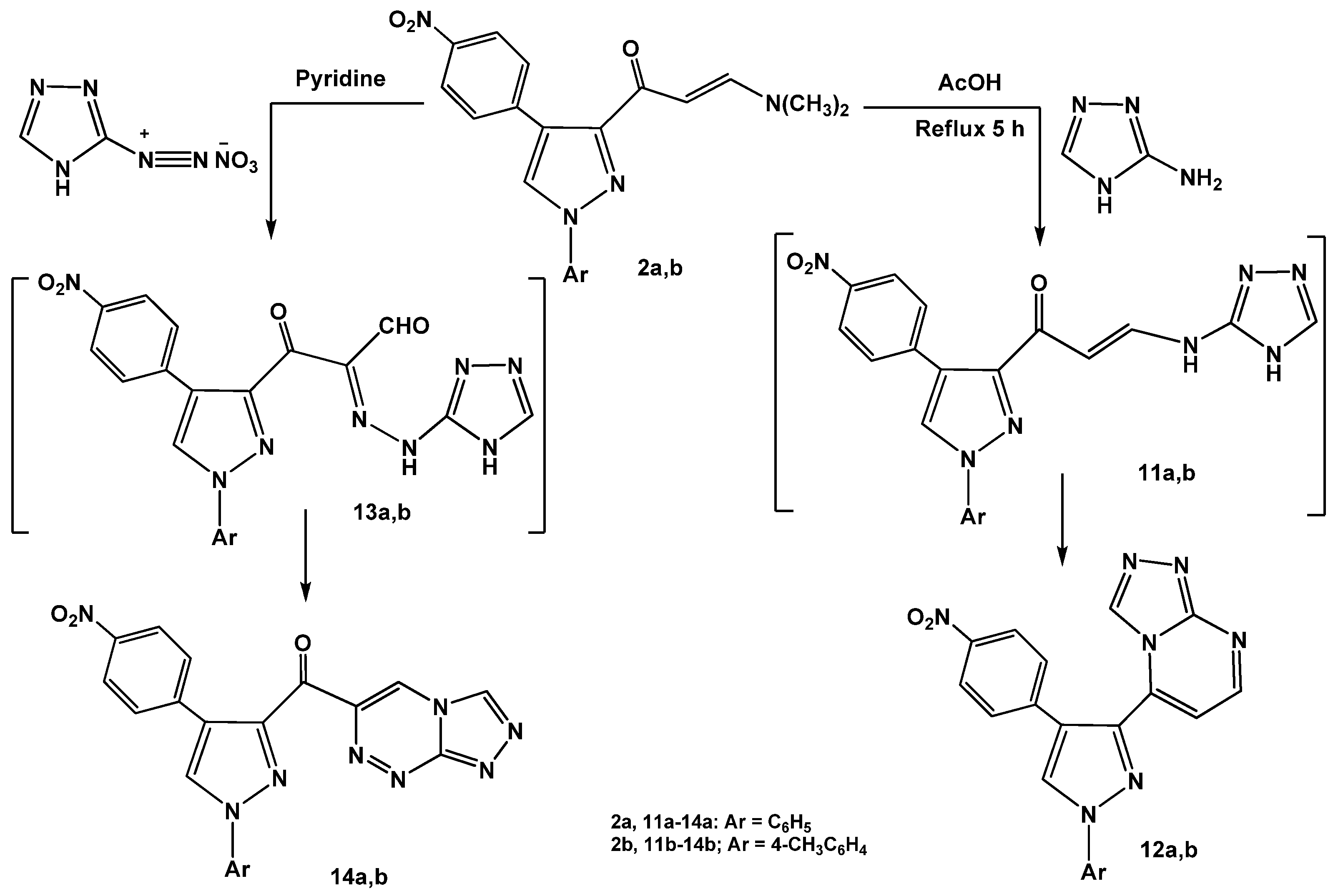
Next, the reactions of enaminones
2a,b with heterocyclic amines were investigated. Refluxing of enaminones
2a,b with 3-amino-1
H-[1,2,4]triazole in glacial acetic acid gave the corresponding [1,2,4]triazolo[4,3-
a]pyrimidines
12a,b via non-isolable intermediates
11a,b (
Scheme 4).
The structures of the products were confirmed by spectral (IR, MS and
1H-NMR) and elemental analyses (see Experimental). On the other hand, coupling of enaminones
2a,b with diazotized 3-amino-1
H-[1,2,4]triazole in pyridine at low temperature afforded the respective pyrazolylcarbonyl- [1,2,4]triazolo[3,4-
c][1,2,4]triazines
14a,b. The reactions proceeded by initial formation of non-isolable hydrazonals [
31,
32,
33]
13a,b followed by elimination of water molecules to give the desired products
14a,b.
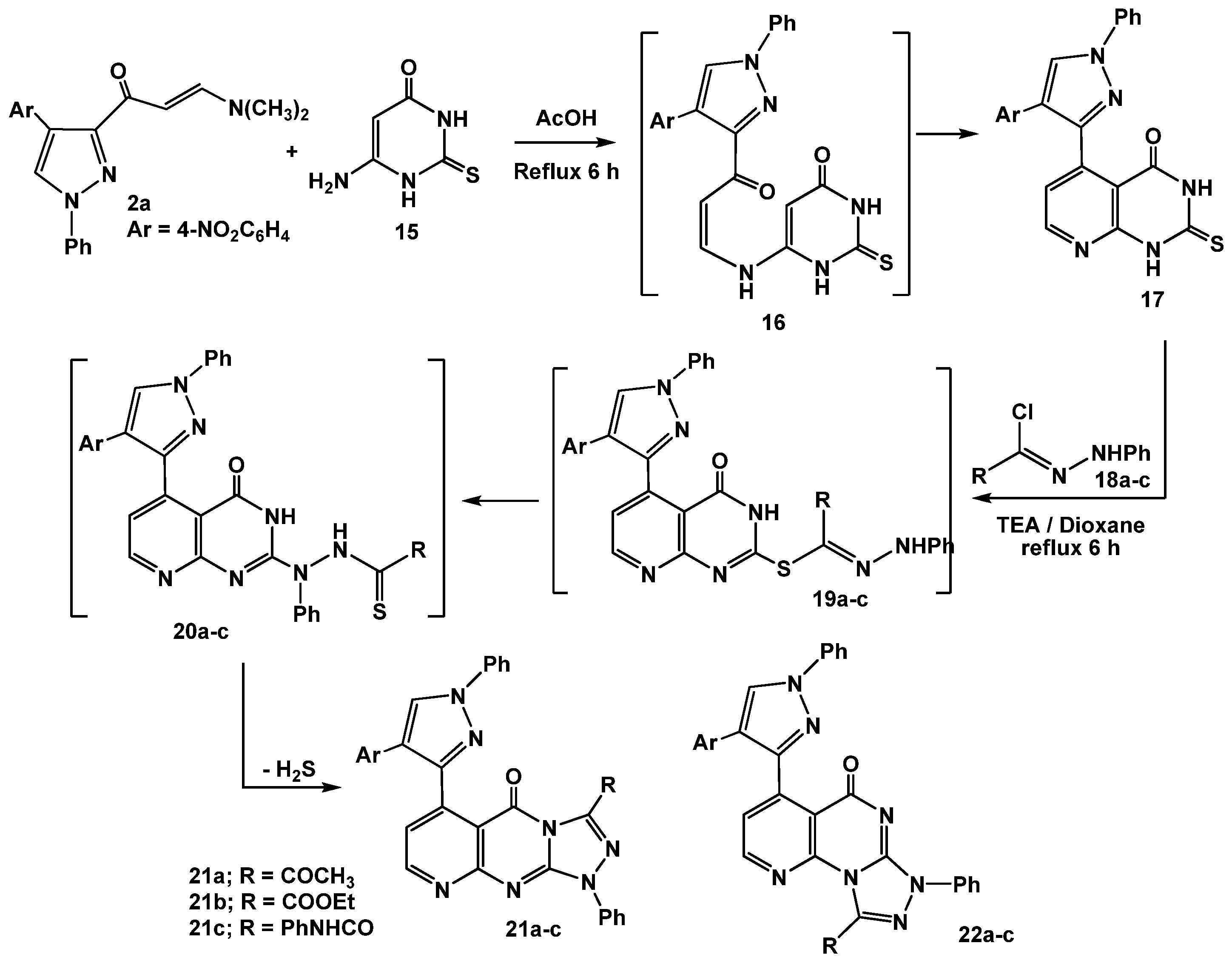
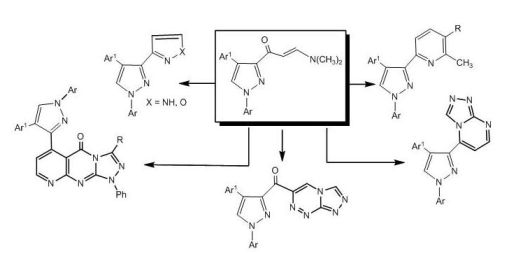
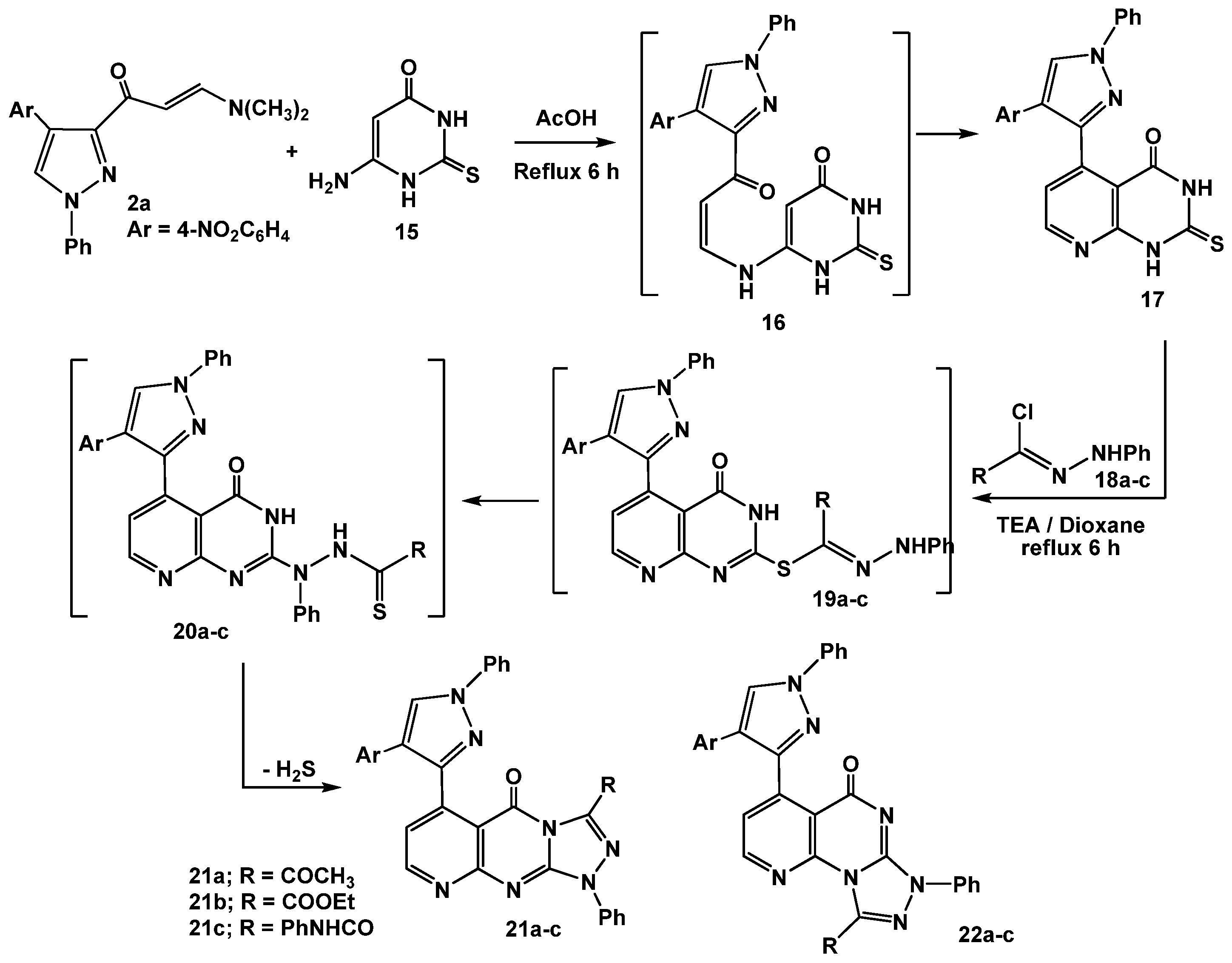
The utility of enaminone 2a in the synthesis of annelated heterocycles was further explored via its reaction with 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (15) in glacial acetic acid under reflux for 6 hours. This reaction afforded the 2-thioxo-2,3-dihydro-1H-pyrido[2,3-d]pyrimidin-4-one 17 via intermediate 16. Spectral (IR, MS, 1H-NMR) data and elemental analysis were in consistent with the isolated product 17.
Scheme 4.
Reactions of enaminones 2a,b with heterocyclic amines.
Scheme 4.
Reactions of enaminones 2a,b with heterocyclic amines.
For example, IR revealed three absorption bands at 3261, 3245, 1677 cm
−1 assignable for 2 NH, and a C=O, respectively. The
1H-NMR spectrum also displayed a characteristic pair of doublet signals at
δ 8.29, 8.48 ppm assigned to the pyridine H-2, H-3 protons, respectively [
34]. Treatment of 2-thioxo-2,3-dihydro-1
H-pyrido[2,3-
d]pyrimidin-4-one (
17) with the appropriate hydrazonoyl chlorides
18a-c in dioxane in the presence of triethylamine under reflux conditions furnished the corresponding pyrido[2,3-
d][1,2,4]triazolo[4,3-
a]pyrimidinones
21a-c as the end products (
Scheme 5).
Scheme 5.
Reactions of enaminone 2a with pyrimidinethione.
Scheme 5.
Reactions of enaminone 2a with pyrimidinethione.
The reactions proceeded through
S-alkylation [
35] to give
S-alkylated products
19a-c followed by Smiles rearrangement [
36], affording intermediates
20a-c which cyclized
in situ under the employed reaction conditions
via elimination of hydrogen sulfide gas to give the desired products
21a-c (cf.
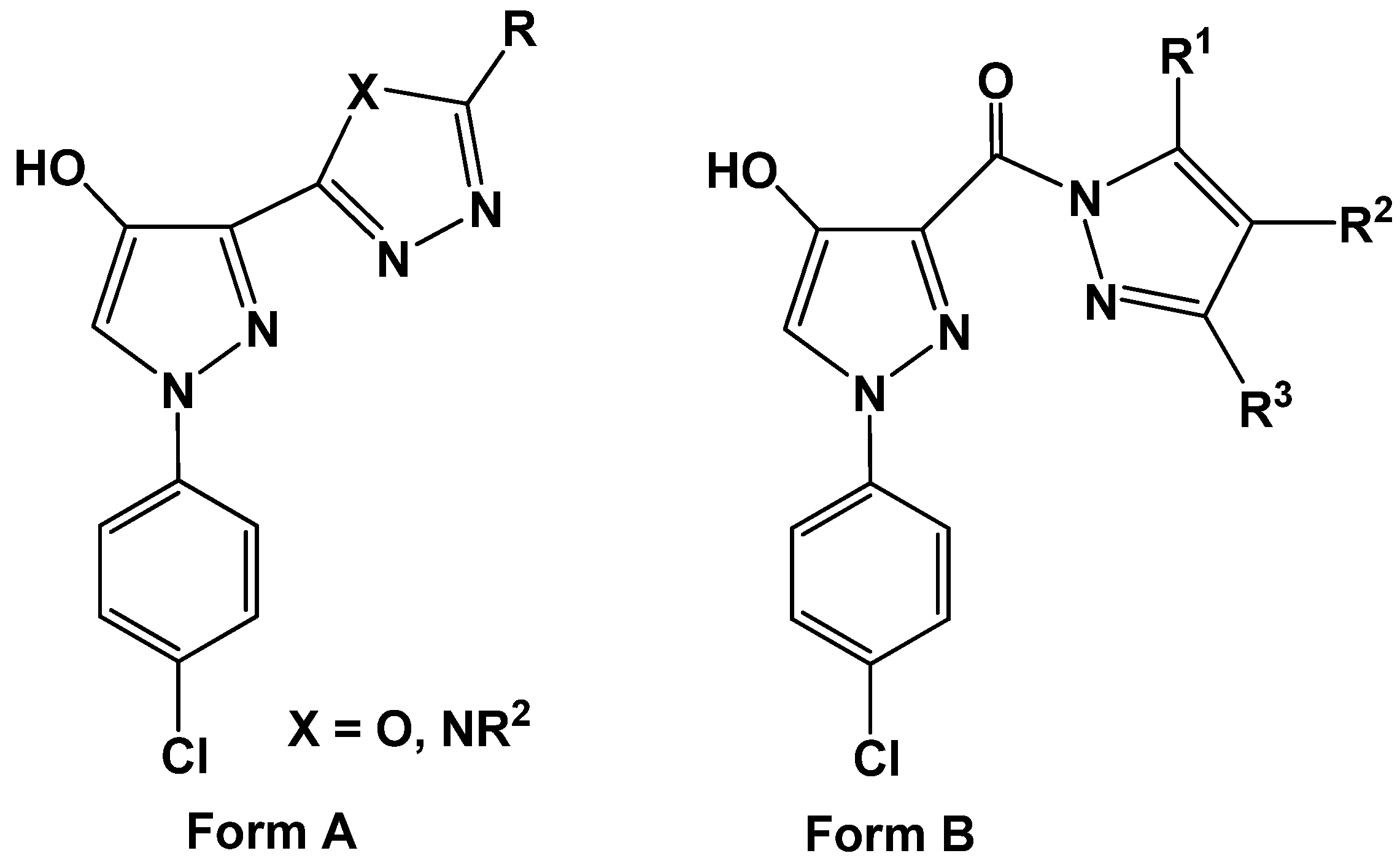
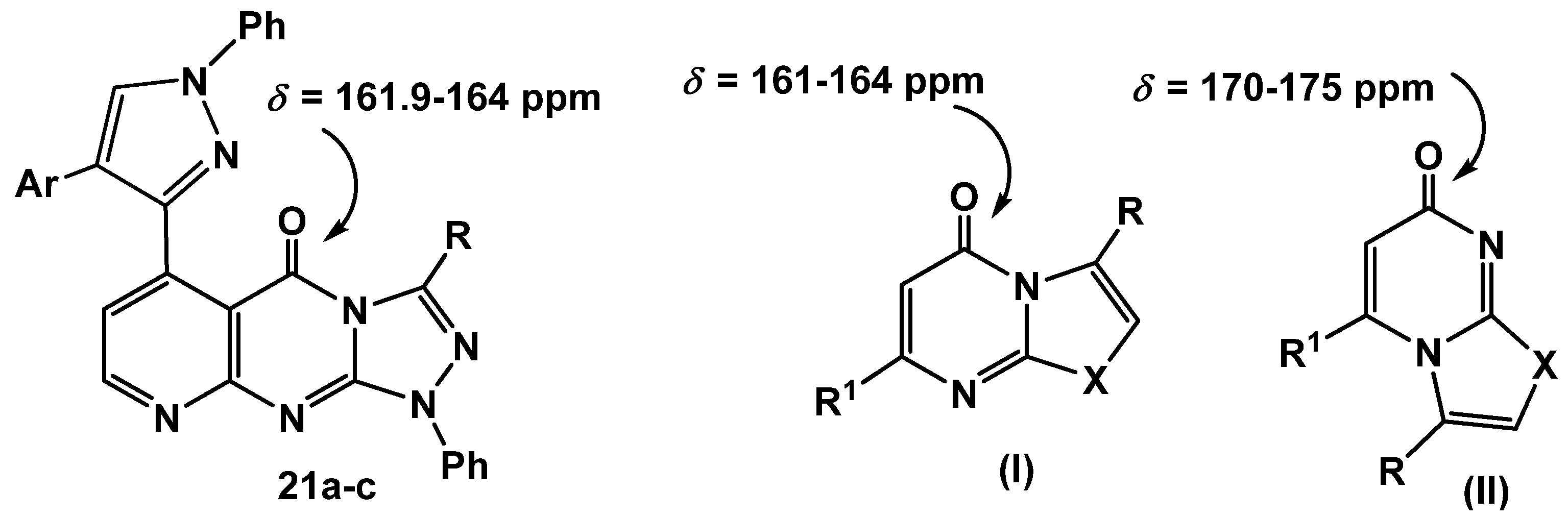
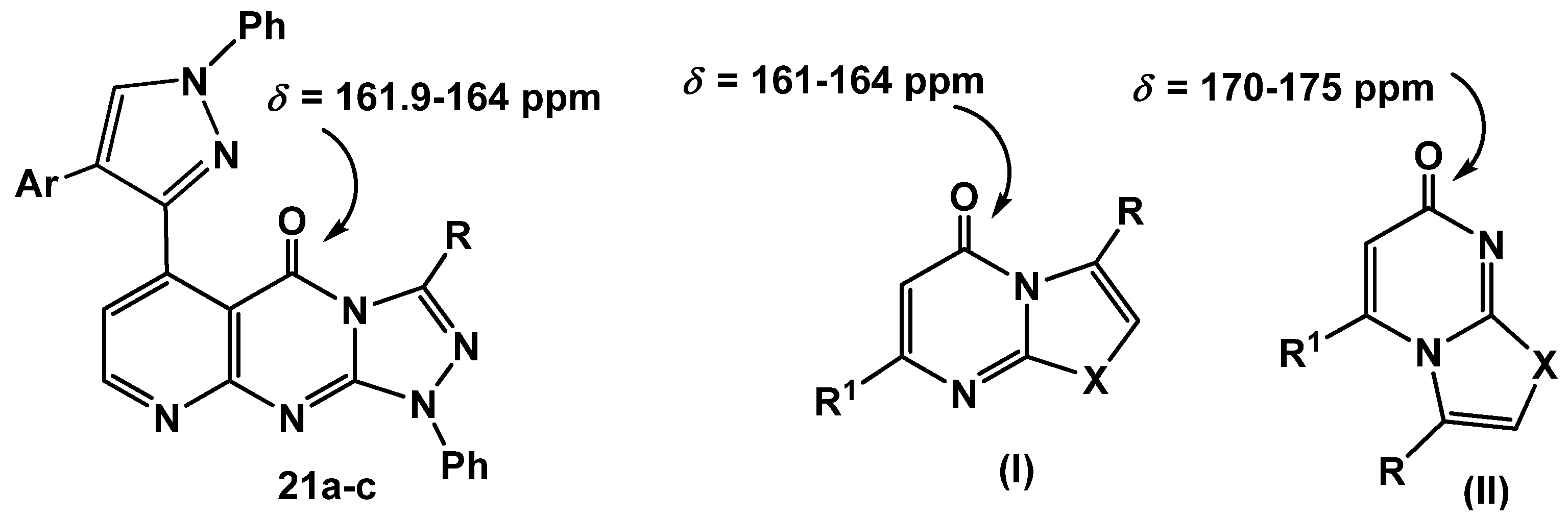
Scheme 5). The other isomeric structures, pyrido[2,3-
d][1,2,4]triazolo[3,4-
a]pyrimidinones
22a-c, were ruled out based on the
13C-NMR which revealed a signal for a carbonyl group at
δ = 161.9–164 ppm which is similar to that of
I (
δ = 161–164 ppm) and different from its isomeric structure
II (
δ = 170–175) [
37] (
Figure 2).
Figure 2.
13C NMR for azolopyrimidinones.
Figure 2.
13C NMR for azolopyrimidinones.
2.1. Antitumor Screening Test
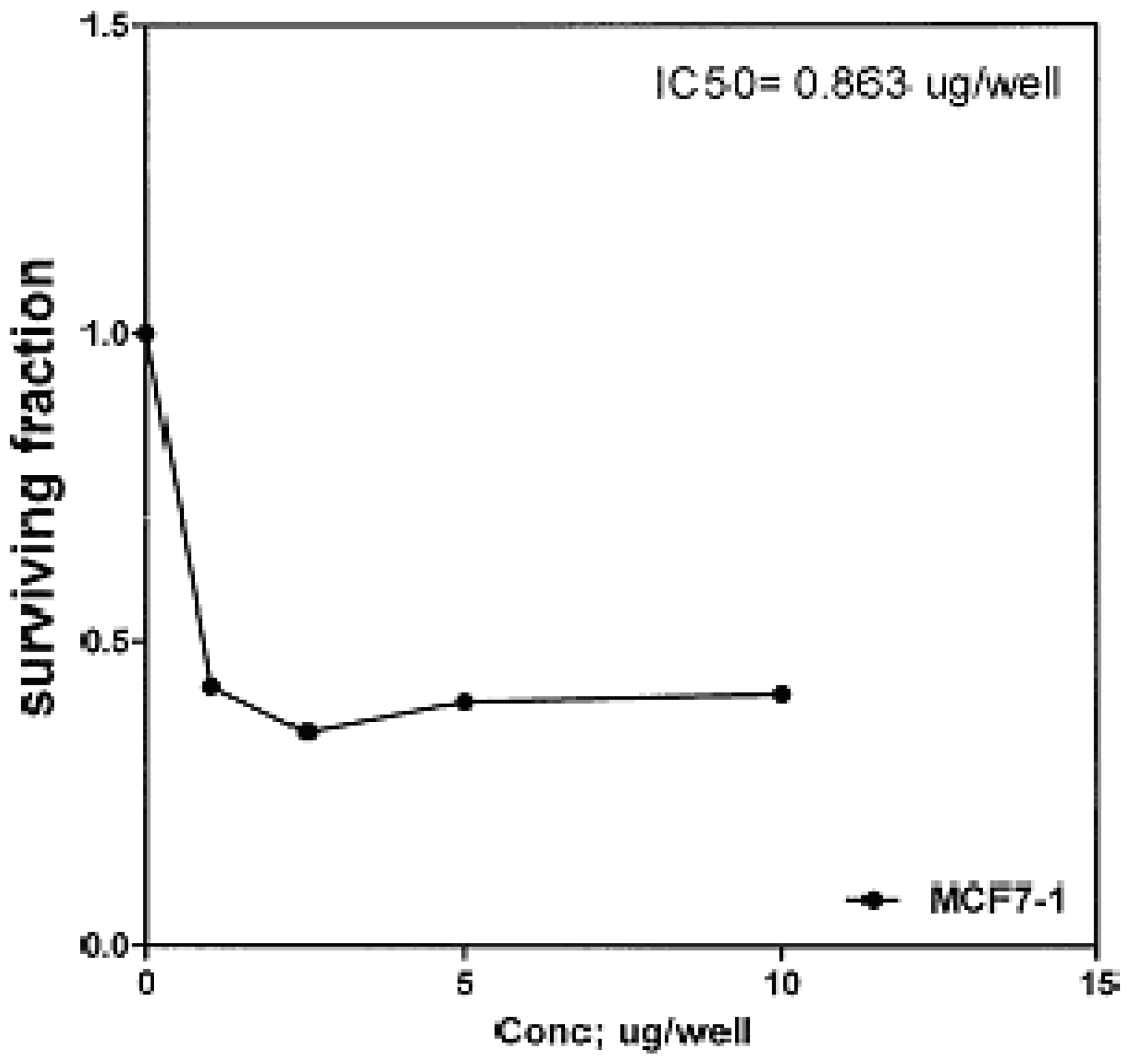
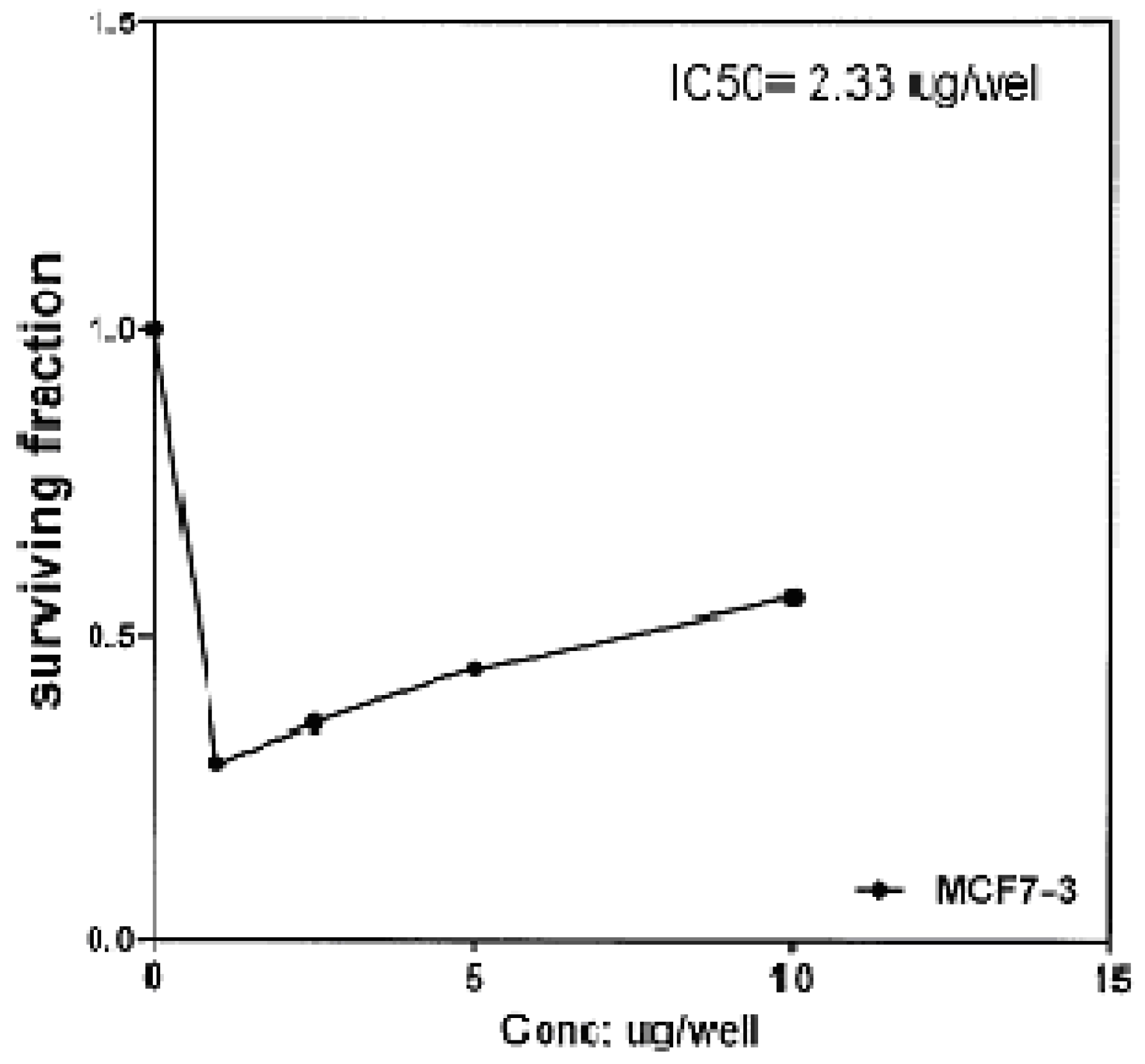
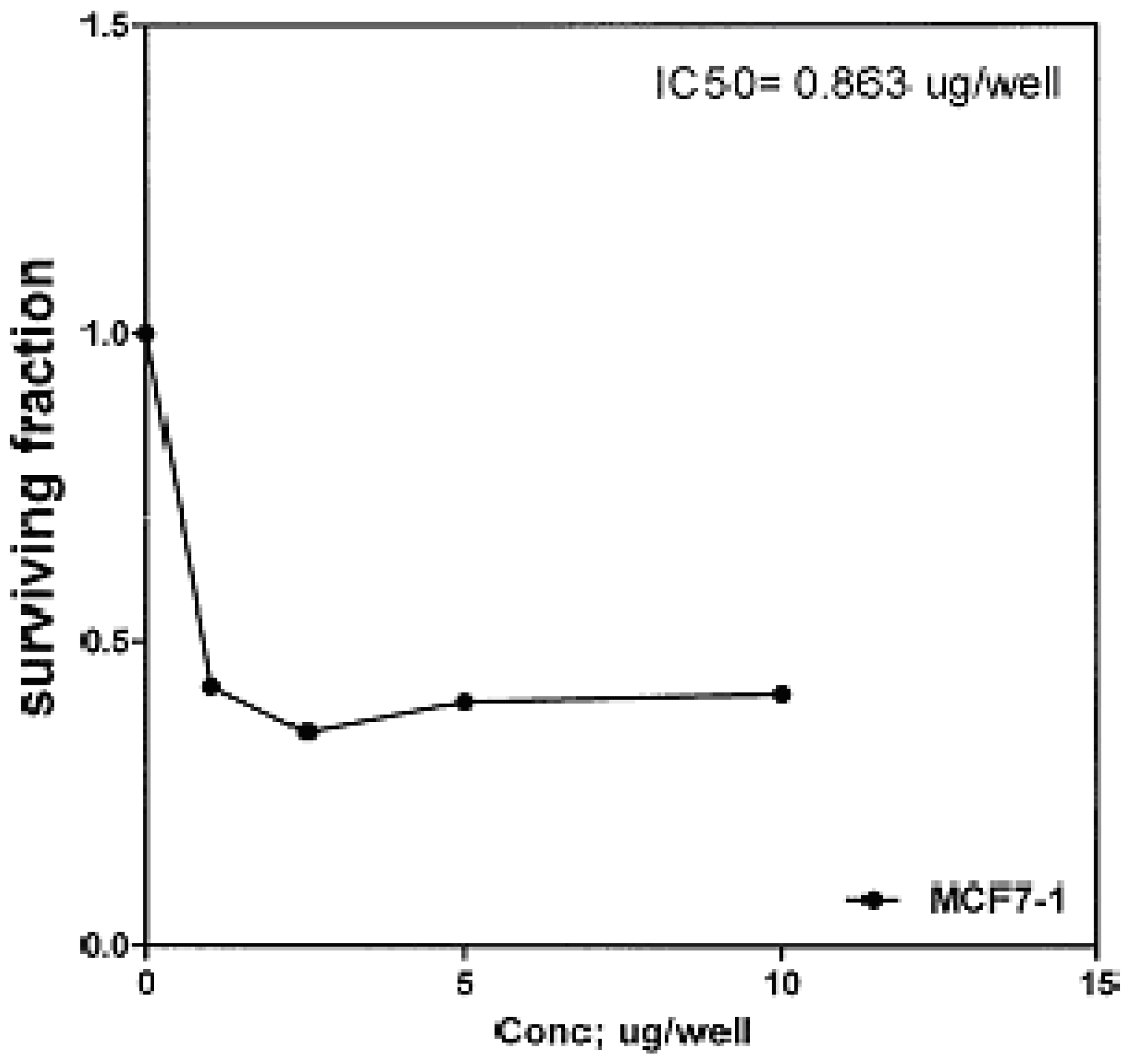
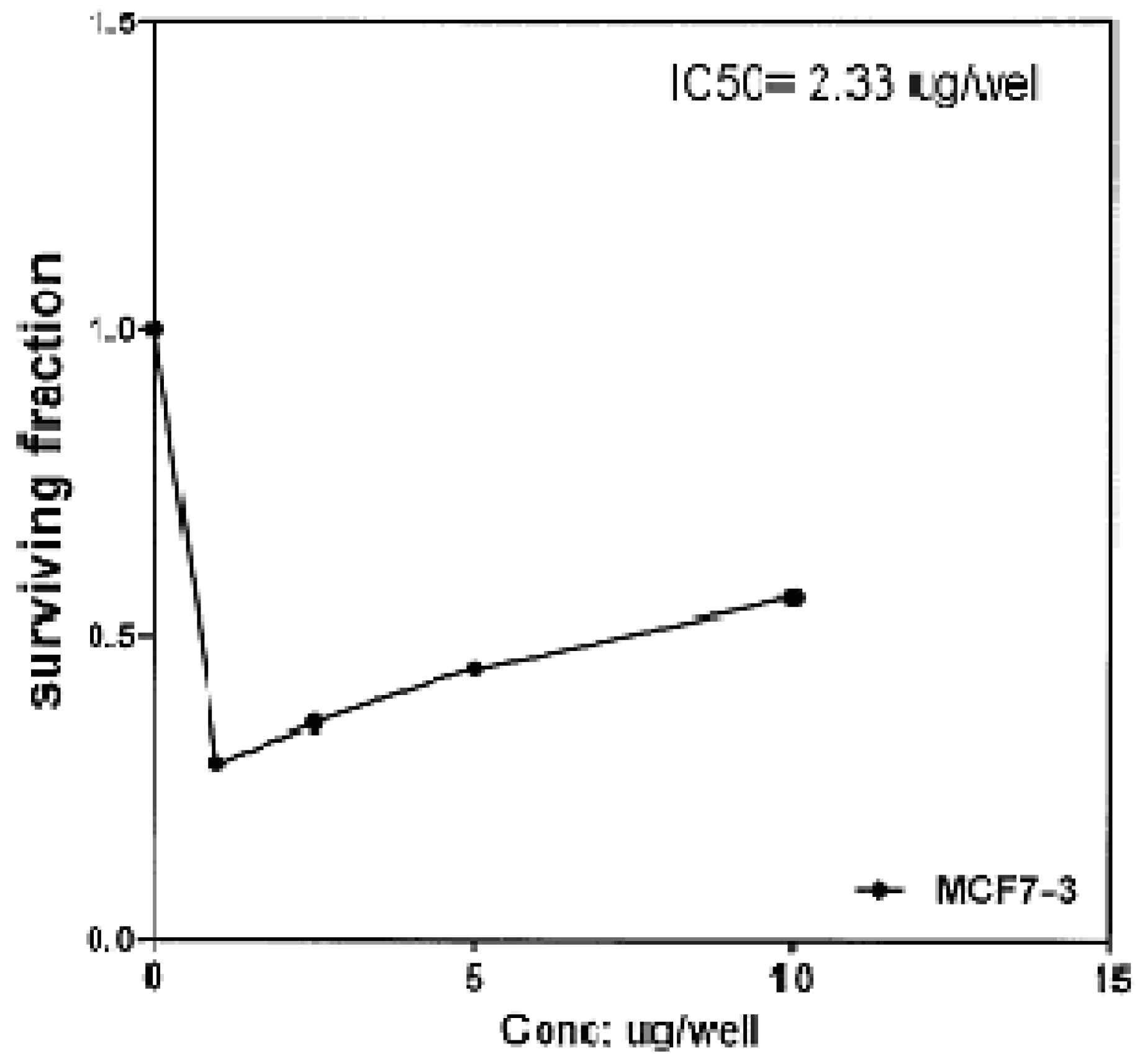
The cytotoxic effects of compounds
2b,
14a and
17 against human breast cell line (MCF-7) and liver carcinoma cell line (HEPG2) were evaluated using 5-fluorouracil as a standard sample in both lines. These compounds were selected by the National Cancer Institute (NCI), Cairo, Egypt. The analysis of the data obtained indicated that the values of IC
50 for such compounds against human breast cell MCF-7 line are 0.863 μg/well (
Figure 3), 2.33 μg/well (
Figure 4), and 2.33 μg/well (
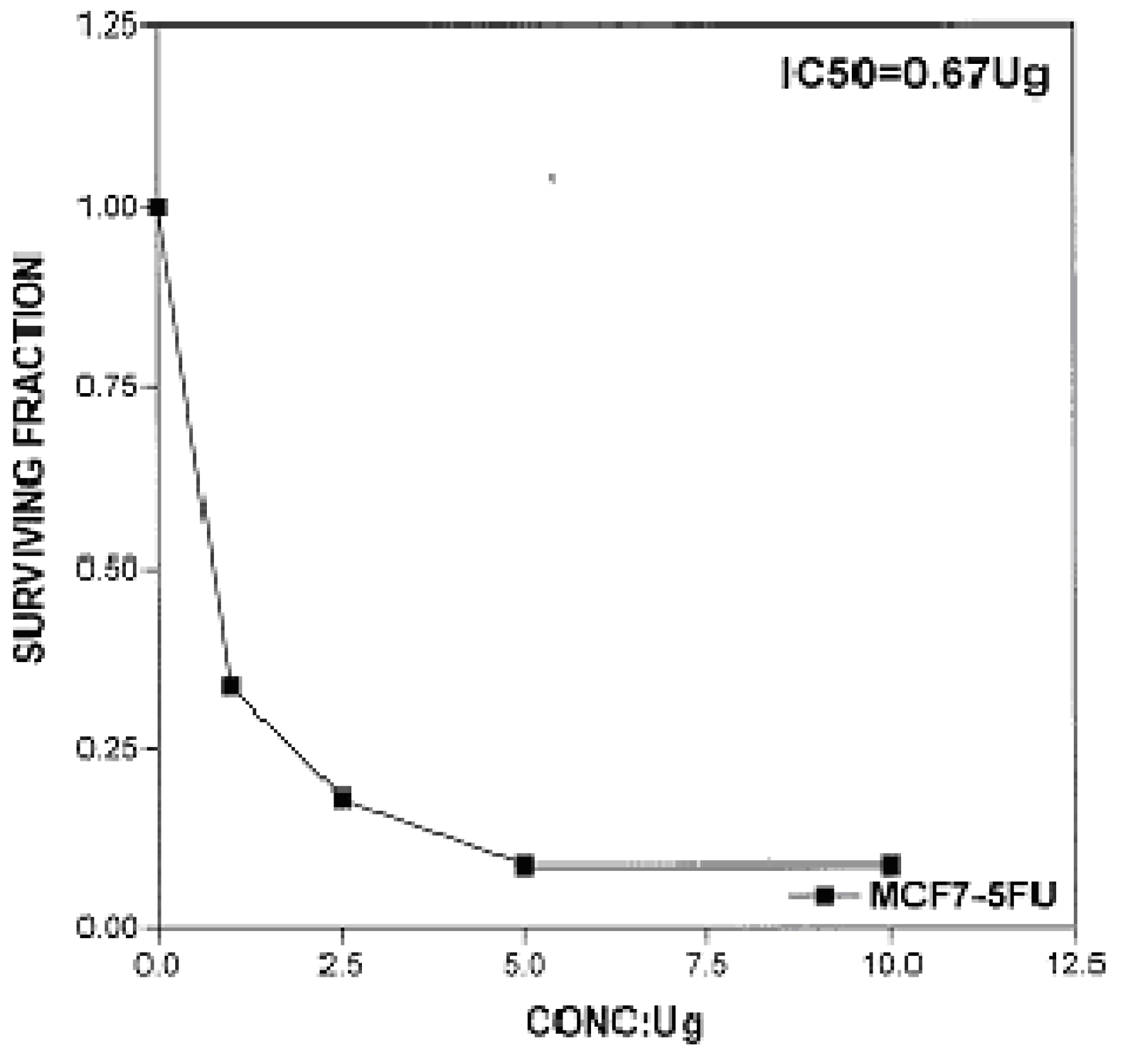
Figure 5), respectively [IC
50 of 5-fluorouracil as a standard sample = 0.67 μg] (
Figure 6). The results indicated that biologically active compound
2b has almost the same activity as the reference drug (5-fluorouracil).
Figure 3.
Effect of conc. of 2b on MCF-7 line.
Figure 3.
Effect of conc. of 2b on MCF-7 line.
Figure 4.
Effect of conc. of 14a on MCF-7 line.
Figure 4.
Effect of conc. of 14a on MCF-7 line.
Figure 5.
Effect of conc. of 17 on MCF-7 line.
Figure 5.
Effect of conc. of 17 on MCF-7 line.
Figure 6.
Effect of conc. of 5-fluorouracil (standard) on MCF-7 line.
Figure 6.
Effect of conc. of 5-fluorouracil (standard) on MCF-7 line.
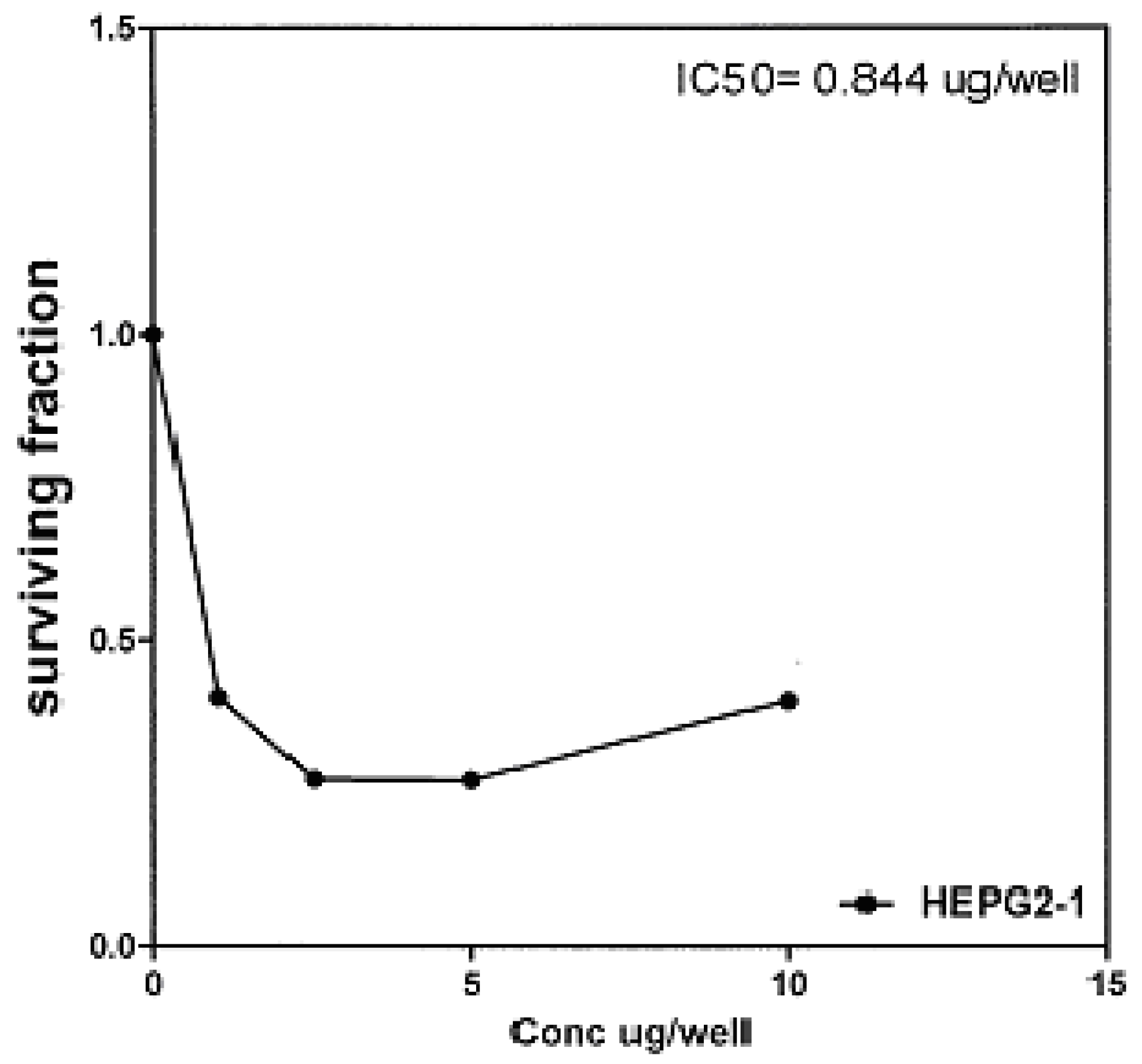
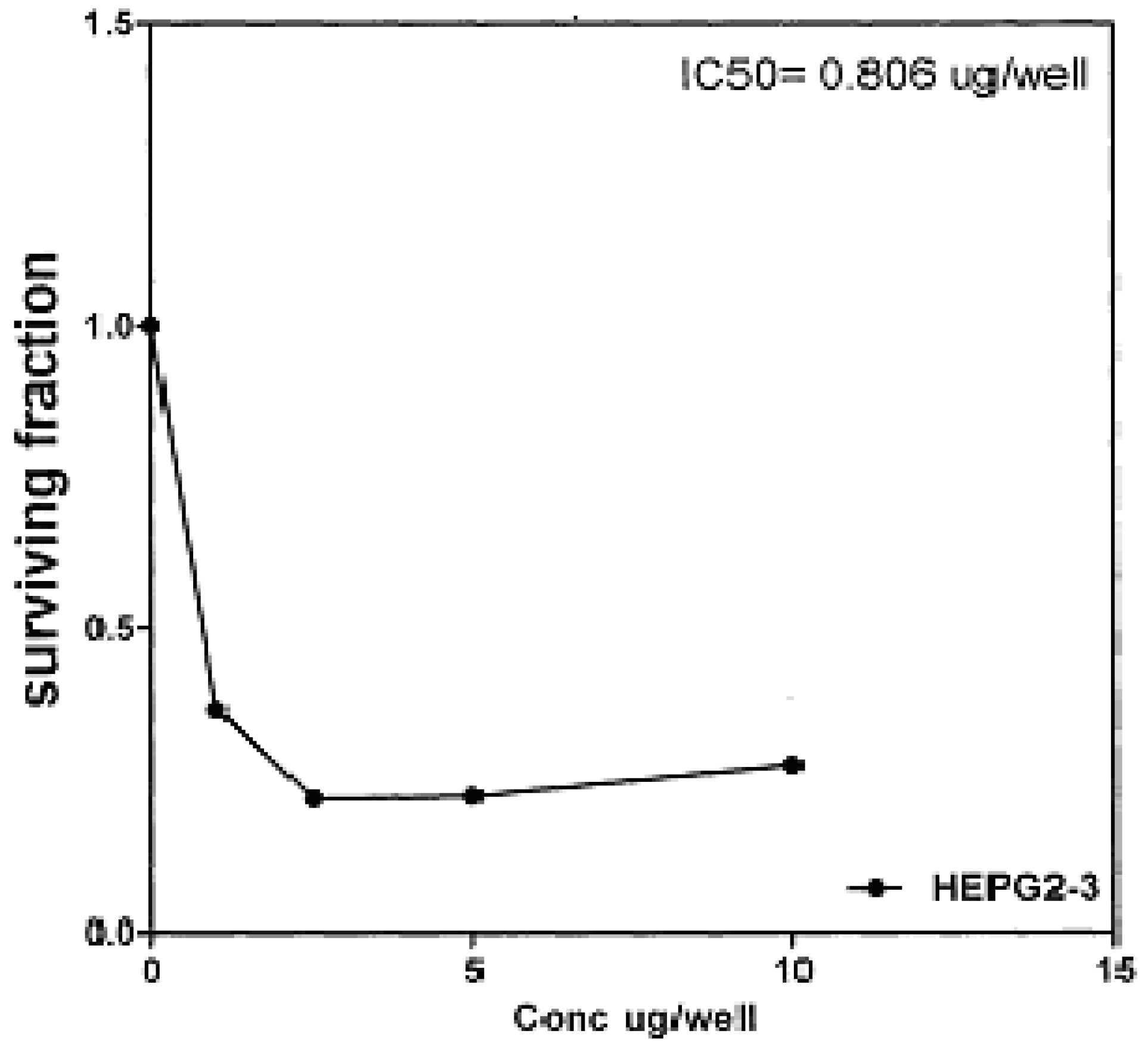
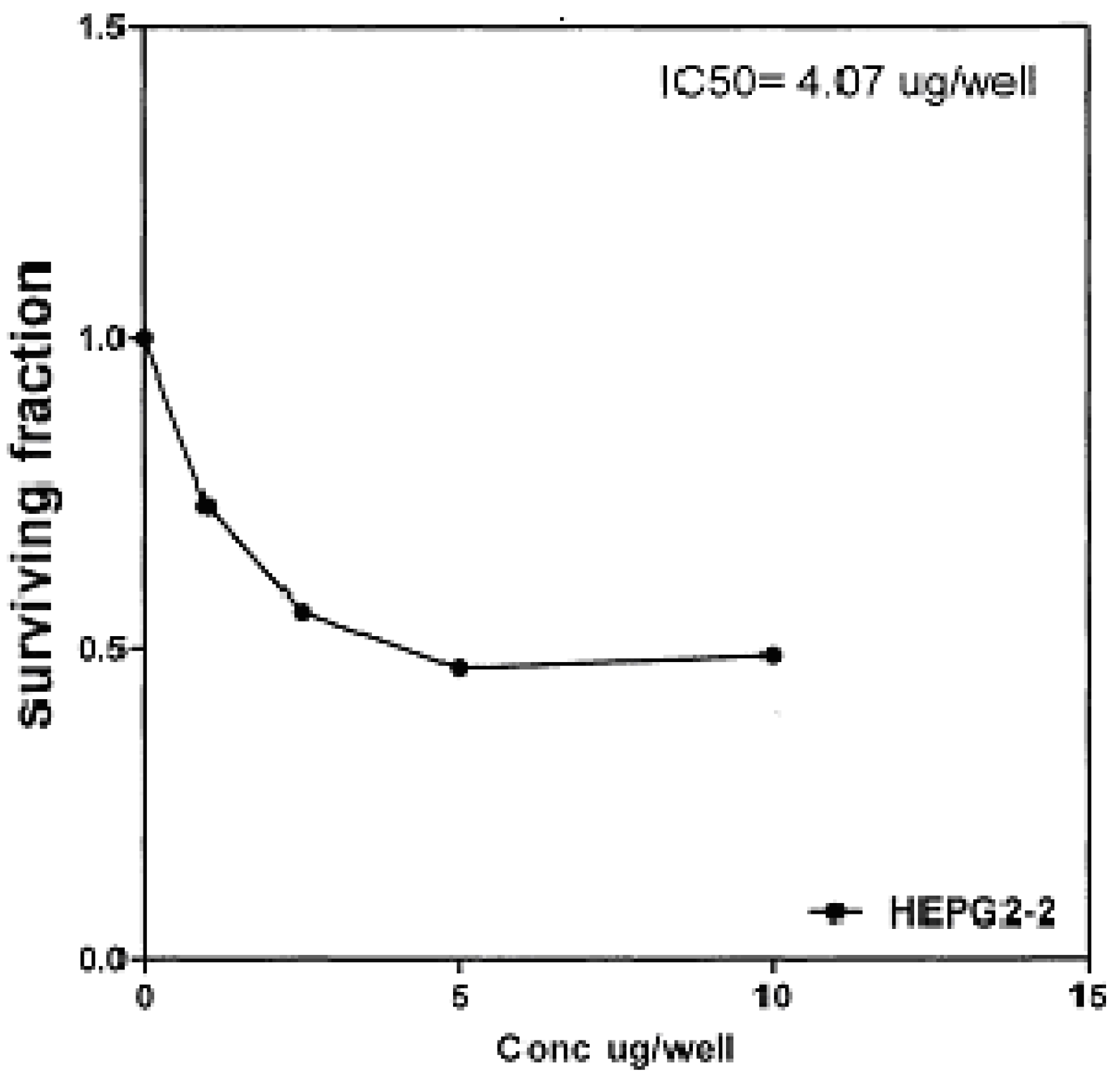
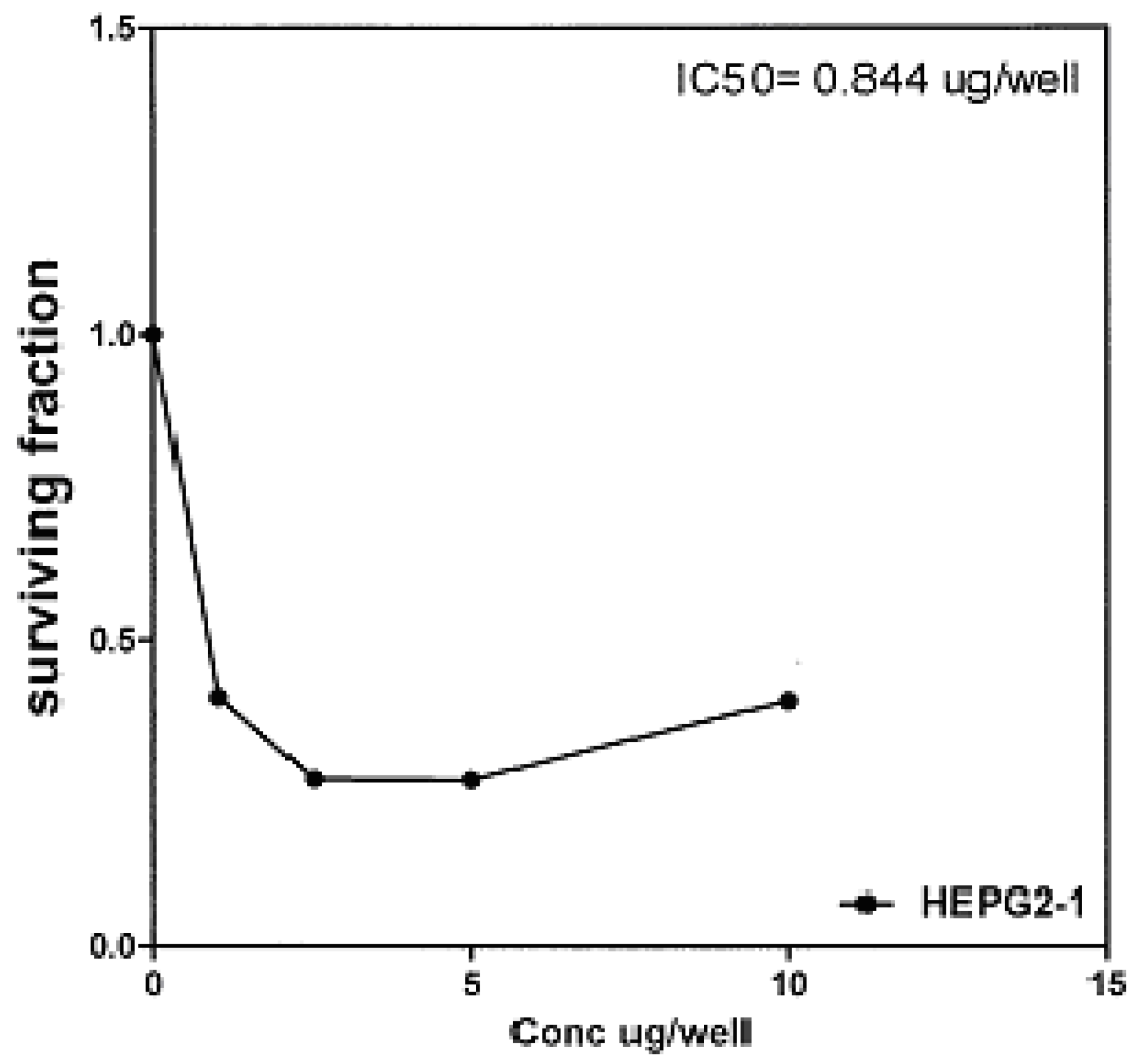
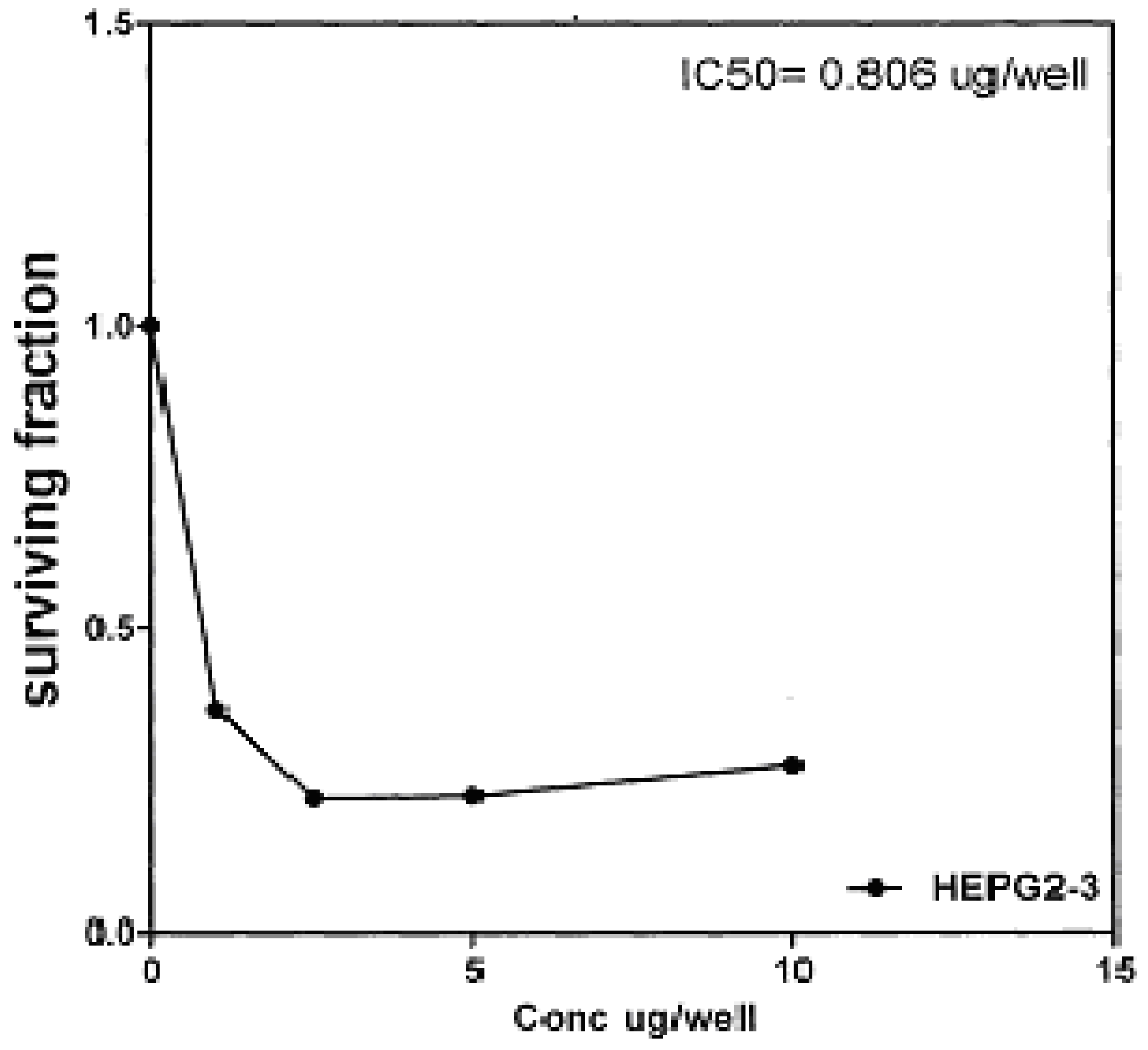
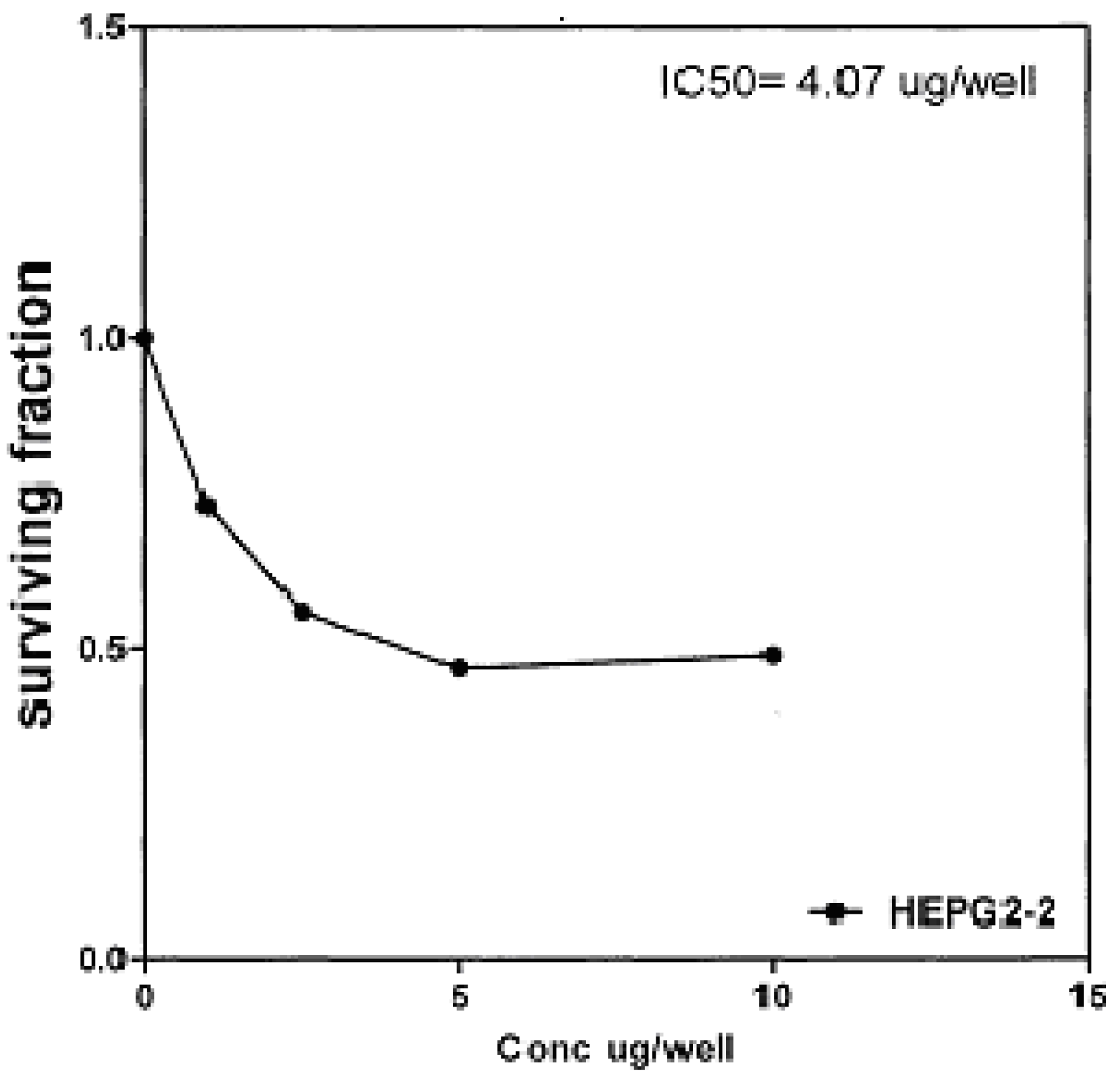
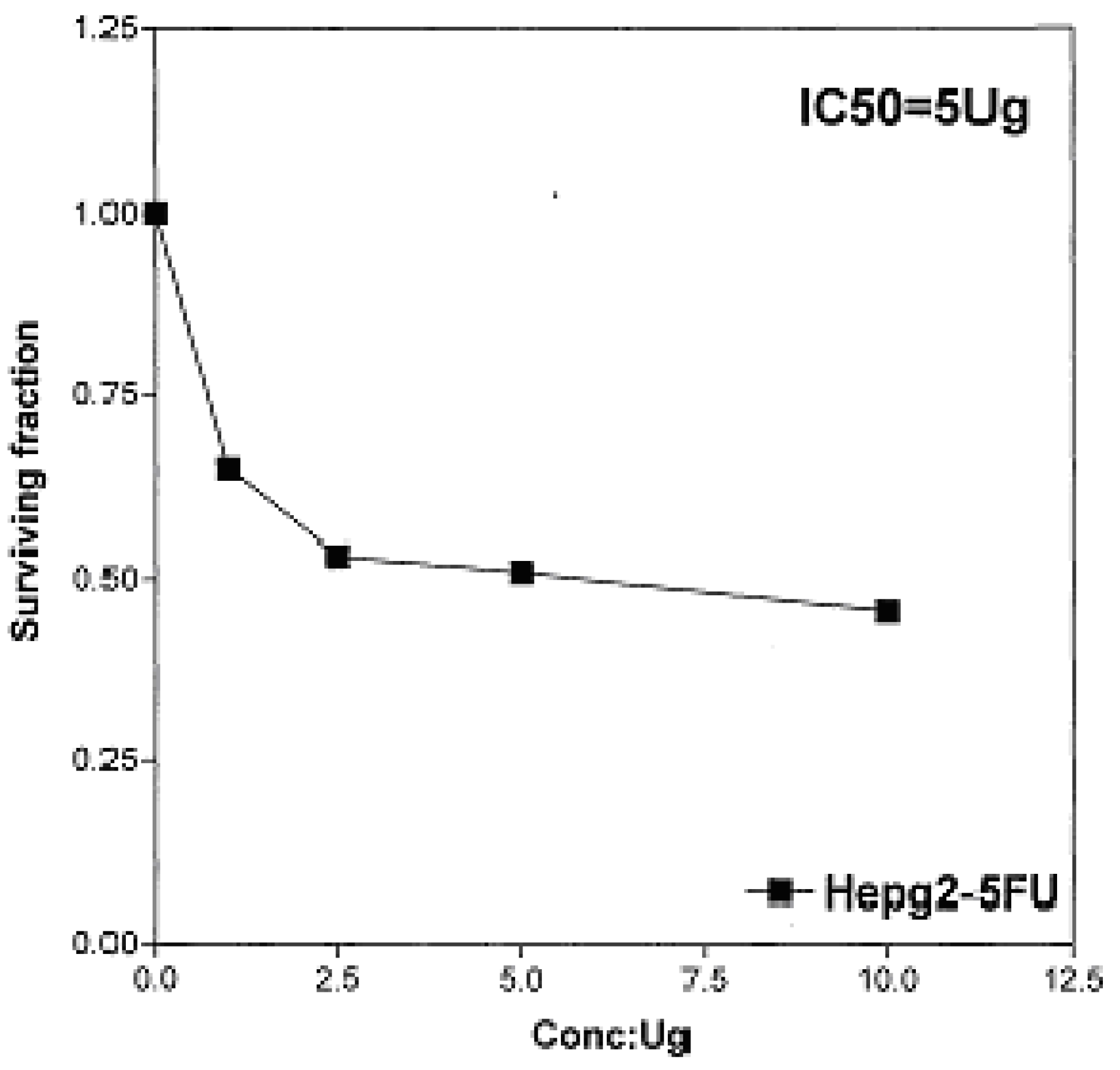
On the other hand, IC
50 of compounds
2b, 14a and
17 against liver carcinoma cell line (HEPG2) are 0.884 μg/well (
Figure 7), 0.806 μg/well (
Figure 8), and 4.07 μg/well (
Figure 9), respectively. [IC
50 of 5-fluorouracil as standard sample = 5 μg] (
Figure 10). The values of IC
50 indicated that the tested compounds
2b, 14a and
17 have higher cytotoxic activities against liver carcinoma cell line (HEPG2) than standard drug (5-fluorouracil). The cytotoxic activity was measured by the Skehan
et al. method (see Experimental).
Figure 7.
Effect of conc. of 2b on HEPG2 line.
Figure 7.
Effect of conc. of 2b on HEPG2 line.
Figure 8.
Effect of conc. of 14a on HEPG2 line.
Figure 8.
Effect of conc. of 14a on HEPG2 line.
Figure 9.
Effect of conc. of 17 on HEPG2 line.
Figure 9.
Effect of conc. of 17 on HEPG2 line.
Figure 10.
Effect of conc. of 5-fluorouracil (standard) on HEPG2 line.
Figure 10.
Effect of conc. of 5-fluorouracil (standard) on HEPG2 line.
The results of biological screening allow the following assumptions about the structure activity relationships (SAR) of these compounds:
The presence of nitrogenous fused heterocycles at position 3 of the main pyrazole moiety, linked directly or through carbonyl group, with multicenters for hydrogen accepting properties are essential for activity where it can intercalate within the DNA strands.
The 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles 2a,b are essential for antitumor activity.
2.2. Antimicrobial Activity
The newly synthesized products
2a,
2b,
5b,
5c,
8b,
9b,
12b,
14a,
21a and
21b were tested for their antimicrobial activities using four species of fungi, namely
Aspergillus fumigatus AF,
Penicillium italicum PI,
Syncephalastrum racemosum SR and
Candida albicans CA, in addition to four bacterial species, namely
Staphylococcus aureus SA,
Pesudomonas aeruginosa PA,
Bacillus subtilis BS and
Escherichia coli EC. The organisms were tested against the activity of solutions of three different concentrations [5 mg/mL, 2.5 mg/mL, 1.25 mg/mL] of each compound and using inhibition zone diameter (IZD) in mm as criterion for the antimicrobial activity. The fungicide
terbinafine and the bactericide
chloramphenicol were used as references to evaluate the potency of the tested compounds under the same conditions. The results, depicted in
Table 1,
Table 2,
Table 3, revealed that compounds
2a and
5c exhibited high degree of inhibition against
SA, and
BS. Compounds
9b,
12b,
14a,
21a and
21b have high inhibition effects against
AF,
PI,
SR and
SA. These compounds also exhibited moderate inhibition effect against
CA and
BS. All the tested compounds were reflecting no inhibition of growth against
PA and
EC.
Table 1.
Antimicrobial activity of products 2a, 2b, 5b, and 5c.
Table 1.
Antimicrobial activity of products 2a, 2b, 5b, and 5c.
| (Sample) | 2a (mg/mL) | 2b (mg/mL) | 5b (mg/mL) | 5c (mg/mL) | Standard* |
|---|
| Tested Microorganism | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 |
| Aspergillus fumigatus (AF) | 9 | 5 | 0 | 8 | 0 | 0 | 7 | 0 | 0 | 6 | 4 | 0 | 24 | 18 | 11 |
| Penicillium italicum (PI) | 7 | 3 | 0 | 6 | 0 | 0 | 5 | 0 | 0 | 5 | 3 | 0 | 19 | 9 | 4 |
| Syncephalastrum racemosum (SR) | 12 | 7 | 3 | 12 | 9 | 5 | 14 | 9 | 0 | 11 | 9 | 0 | 21 | 13 | 9 |
| Candida albicans (CA) | 9 | 6 | 3 | 7 | 4 | 0 | 9 | 4 | 0 | 12 | 9 | 5 | 19 | 10 | 6 |
| Staphylococcus aureus (SA) | 11 | 7 | 4 | 11 | 8 | 5 | 14 | 8 | 5 | 11 | 8 | 5 | 15 | 6 | 4 |
| Pesudomonas aeruginosa (PA) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 11 | 5 | 0 |
| Bacillus subtilis (BS) | 15 | 8 | 6 | 12 | 7 | 4 | 14 | 9 | 4 | 18 | 13 | 9 | 22 | 18 | 11 |
| Escherichia coli (EC) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 27 | 20 | 13 |
Table 2.
Antimicrobial activity of products 8b, 9b, 12b, and 14a.
Table 2.
Antimicrobial activity of products 8b, 9b, 12b, and 14a.
| (Sample) | 8b (mg/mL) | 9b (mg/mL) | 12b (mg/mL) | 14a (mg/mL) | Standard* |
|---|
| Tested Microorganism | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 |
| Aspergillus fumigatus (AF) | 9 | 7 | 3 | 22 | 14 | 9 | 18 | 11 | 6 | 16 | 7 | 3 | 24 | 18 | 11 |
| Penicillium italicum (PI) | 10 | 6 | 3 | 14 | 6 | 3 | 13 | 6 | 4 | 0 | 0 | 0 | 19 | 9 | 4 |
| Syncephalastrum racemosum (SR) | 9 | 7 | 4 | 19 | 12 | 8 | 16 | 9 | 6 | 18 | 12 | 7 | 21 | 13 | 9 |
| Candida albicans (CA) | 10 | 8 | 4 | 9 | 6 | 2 | 10 | 7 | 3 | 9 | 6 | 2 | 19 | 10 | 6 |
| Staphylococcus aureus (SA) | 12 | 7 | 4 | 12 | 8 | 5 | 13 | 8 | 5 | 11 | 8 | 5 | 15 | 6 | 4 |
| Pesudomonas aeruginosa (PA) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 11 | 5 | 0 |
| Bacillus subtilis (BS) | 16 | 9 | 5 | 15 | 8 | 5 | 14 | 10 | 7 | 10 | 8 | 4 | 22 | 18 | 11 |
| Escherichia coli (EC) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 27 | 20 | 13 |
Table 3.
Antimicrobial activity of products 21a and 21b.
Table 3.
Antimicrobial activity of products 21a and 21b.
| (Sample) | 21a (mg/mL) | 21b (mg/mL) | Standard* |
|---|
| Tested Microorganism | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 | 5 | 2.5 | 1.25 |
| Aspergillus fumigatus (AF) | 20 | 11 | 6 | 19 | 14 | 9 | 24 | 18 | 11 |
| Penicillium italicum (PI) | 17 | 6 | 4 | 13 | 6 | 3 | 19 | 9 | 4 |
| Syncephalastrum racemosum (SR) | 15 | 8 | 6 | 17 | 12 | 8 | 21 | 13 | 9 |
| Candida albicans (CA) | 10 | 7 | 3 | 9 | 6 | 2 | 19 | 10 | 6 |
| Staphylococcus aureus (SA) | 13 | 7 | 5 | 10 | 8 | 5 | 15 | 6 | 4 |
| Pesudomonas aeruginosa (PA) | 0 | 0 | 0 | 0 | 0 | 0 | 11 | 5 | 0 |
| Bacillus subtilis (BS) | 12 | 9 | 7 | 13 | 10 | 8 | 22 | 18 | 11 |
| Escherichia coli (EC) | 0 | 0 | 0 | 0 | 0 | 0 | 27 | 20 | 13 |
3. Experimental
3.1. General
All melting points were determined on an electrothermal Gallenkamp apparatus and are uncorrected. Solvents were generally distilled and dried by standard literature procedures prior to use. The IR spectra were measured on a Pye-Unicam SP300 instrument in potassium bromide discs. The
1H NMR spectra were recorded on a Varian Mercury VXR-300 spectrometer (300 MHz) and the chemical shifts were related to that of the solvent DMSO-d
6. The mass spectra were recorded on a GCMS-Q1000-EX Shimadzu and GCMS 5988-A HP spectrometers, the ionizing voltage was 70 eV. Elemental analyses were carried out by the Microanalytical Center of Cairo University, Giza, Egypt. Antitumor activity was evaluated by the National Institute of Cancer, Biology Department, Cairo University, Egypt. Antimicrobial activity was carried out at the Regional Center for Mycology and Biotechnology at Al-Azhar University, Cairo, Egypt. 3-Acetyl-4-(4-nitrophenyl)-1-aryl-1
H-pyrazoles
1a,b [
26] and hydrazonoyl halides [
38,
39,
40,
41,
42,
43]
18a-c were prepared following literature methods.
3.2. Synthesis of 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles 2a,b
A mixture of 3-acetyl-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles (1a or 1b) (0.01 mol) and dimethyl-formamide dimethylacetal (DMF-DMA) (5 mL) was refluxed for 10 hours. After cooling, methanol was added and the solid product was collected by filtration and crystallized from ethanol. The physical constants and the spectral data are shown below.
3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-phenyl-1H-pyrazole (2a). Yellow crystals, (2.89 g, 80%), m.p. 132–134 °C; IR (KBr) υ = 1,642 (CO) cm−1; 1H-NMR (CDCl3) δ = 2.82 (s, 3H, CH3), 3.00 (s, 3H, CH3), 5.88 (d, 1H, J = 13 Hz, CH=), 7.27 (d, 2H, J = 8 Hz, Ar-H), 7.67 (d, 1H, J = 13 Hz, CH=), 7.35–7.68 (m, 5H, Ar-H), 8.05 (d, 2H, J = 8 Hz, Ar-H), 8.90 (s, 1H, pyrazole-H-5) ppm; MS, m/z (%) 362 (M+, 25), 292 (30), 264 (20), 122 (15), 98 (40), 77 (100), 70 (40). Anal. Calcd. for C20H18N4O3 (362.14): C, 66.29; H, 5.01; N, 15.46. Found: C, 66.18; H, 4.93; N, 15.58%.
3-[E-3-(N,N-dimethylamino)acryloyl]-1-(4-methylphenyl)-4-(4-nitrophenyl)-1H-pyrazole (2b). Yellow crystals, (3.31 g, 88%), m.p. 148–150 °C; IR (KBr) υ = 1647 (CO) cm−1; 1H-NMR (DMSO-d6) δ = 2.37 (s, 3H, Ar-CH3), 2.89 (s, 3H, CH3), 3.13 (s, 3H, CH3), 5.88 (d, 1H, J = 13 Hz, CH=), 7.35 (d, 2H, J = 8 Hz, Ar-H), 7.66 (d, 1H, J = 13 Hz, CH=), 7.82 (d, 2H, J = 8 Hz, Ar-H), 7.91 (d, 2H, J = 8 Hz, Ar-H), 8.21 (d, 2H, J = 8 Hz, Ar-H), 8.92 (s, 1H, pyrazole-H-5) ppm; MS, m/z (%) 376 (M+, 25), 306 (40), 278 (20), 98 (40), 92 (85), 77 (100), 70 (40). Anal. Calcd. for C21H20N4O3 (376.15): C, 67.01; H, 5.36; N, 14.88. Found: C, 67.12; H, 5.23; N, 14.71%.
3.3. Reactions of 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles 2a,b with Active Methylene Compounds
To a solution of 2a or 2b (1 mmol) and 2,4-pentanedione (3a) or ethyl 3-oxobutanoate (3b) (1 mmol) in acetic acid (20 mL) was added ammonium acetate (0.156 g, 2 mmol). The reaction mixture was heated under reflux for 5 hours. After cooling, the reaction mixture was poured onto ice and the solid product was collected by filtration and crystallized from an ethanol/dioxane mixture (1:1). The physical constants, together with the spectral data for products 5a-d, are shown below.
3-Acetyl-2-methyl-6-[4-(4-nitrophenyl)-1-phenyl-1H-pyrazol-3-yl]pyridine (5a). Yellow crystals, (0.34 g, 85%), m.p. 318–320 °C; IR (KBr) υ = 1,691 (CO) cm−1; 1H-NMR (DMSO-d6) δ = 2.32 (s, 3H, CH3), 2.42 (s, 3H, COCH3), 7.35 (d, 2H, J = 8 Hz, Ar-H), 7.55 (d, 1H, J = 8 Hz, pyridyl H-4), 7.67 (d, 1H, J = 8 Hz, pyridyl H-5), 7.71–8.20 (m, 5H, Ar-H), 8.26 (d, 2H, J = 8 Hz, Ar-H), 9.01 (s, 1H, pyrazole-H-5) ppm; MS, m/z (%) 398 (M+, 60), 355 (30), 122 (25), 77 (100). Anal. Calcd. for C23H18N4O3 (398.14): C, 69.34; H, 4.55; N, 14.06. Found: C, 69.27; H, 4.68; N, 14.11%.
3-Acetyl-2-methyl-6-[4-(4-nitrophenyl)-1-(4-methylphenyl)-1H-pyrazol-3-yl]pyridine (5b). Yellow crystals, (0.35 g, 85%), m.p. 322–325 °C; IR (KBr) υ = 1,692 (CO) cm−1; 1H-NMR (DMSO-d6) δ = 2.32 (s, 3H, CH3), 2.38 (s, 3H, Ar-CH3), 2.44 (s, 3H, COCH3), 7.37 (d, 2H, J = 8 Hz, Ar-H), 7.53 (d, 1H, J = 8 Hz, pyridyl H-4), 7.61 (d, 1H, J = 8 Hz, pyridyl H-5), 7.76–8.26 (m, 4H, Ar-H), 8.29 (d, 2H, J = 8 Hz, Ar-H), 9.11 (s, 1H, pyrazole H-5) ppm; MS, m/z (%) 412 (M+, 75), 369 (30), 122 (25), 91 (50), 77 (100). Anal. Calcd. for C24H20N4O3 (412.15): C, 69.89; H, 4.89; N, 13.58. Found: C, 69.77; H, 4.78; N, 13.41%.
Ethyl 2-methyl-6-[4-(4-nitrophenyl)-1-phenyl-1H-pyrazol-3-yl]nicotinate (5c). Pale yellow crystals, (0.35 g, 82%), m.p. 180–182 °C; IR (KBr) υ = 1,706 (CO) cm−1; 1H-NMR (DMSO-d6) δ = 1.31 (t, 3H, J = 7 Hz, CH3), 2.38 (s, 3H, CH3), 4.35 (q, 2H, J = 7 Hz, CH2), 7.36 (d, 2H, J = 8 Hz, Ar-H), 7.52 (d, 1H, J = 8 Hz, pyridyl H-4), 7.62 (d, 1H, J = 8 Hz, pyridyl H-5), 7.81-8.20 (m, 5H, Ar-H), 8.28 (d, 2H, J = 8 Hz, Ar-H), 8.97 (s, 1H, pyrazole H-5) ppm; MS, m/z (%) 428 (M+, 60), 355 (30), 122 (25), 77 (100). Anal. Calcd. for C24H20N4O4 (428.15): C, 67.28; H, 4.71; N, 13.08. Found: C, 67.19; H, 4.62; N, 13.16%.
Ethyl 2-methyl-6-[4-(4-nitrophenyl)-1-(4-methylphenyl)-1H-pyrazol-3-yl]nicotinate (5d). Pale yellow crystals, (0.37 g, 85%), m.p. 186–188 °C; IR (KBr) υ = 1,709 (CO) cm−1; 1H-NMR (DMSO-d6) δ = 1.33 (t, 3H, J = 7 Hz, CH3), 2.38 (s, 3H, CH3), 2.41 (s, 3H, Ar-CH3), 4.39 (q, 2H, J = 7 Hz, CH2), 7.38 (d, 2H, J = 8 Hz, Ar-H), 7.51 (d, 1H, J = 8 Hz, pyridyl H-4), 7.59 (d, 1H, J = 8 Hz, pyridyl H-5), 7.72–8.10 (m, 4H, Ar-H), 8.29 (d, 2H, J = 8 Hz, Ar-H), 8.99 (s, 1H, pyrazole H-5) ppm; MS, m/z (%) 442 (M+, 50), 369 (30), 122 (25), 91 (100), 77 (60). Anal. Calcd. for C25H22N4O4 (442.16): C, 67.86; H, 5.01; N, 12.66. Found: C, 67.74; H, 4.92; N, 12.56%.
3.4. Reactions of 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles 2a,b with Hydrazine Hydrate
To a solution of the enaminone (2a or 2b) (1 mmol) in ethanol (10 mL) was added hydrazine hydrate (1 mL) and the mixture was heated under reflux for 5 hours. The reaction mixture was acidified by HCl/ice mixture and the formed product was filtered and crystallized from ethanol.
4-(4-Nitrophenyl)-1-phenyl-1H,1'H-3,3'-bipyrazole (8a). Yellow crystals, (0.30 g, 90%), m.p. 200–202 °C; IR (KBr) υ = 3,246 (NH) cm–1; 1H-NMR (DMSO-d6) δ = 7.25 (d, 2H, J = 8 Hz, Ar-H), 7.35–7.97 (m, 5H, Ar-H), 7.53 (d, 1H, J = 7.5 Hz, pyrazole H-4), 7.58 (d, 1H, J = 7.5 Hz, pyrazole H-5), 8.23 (d, 2H, J = 8 Hz, Ar-H), 9.01 (s, 1H, pyrazole H-5), 13.00 (D2O-exchangeable) (s, 1H, NH) ppm; MS, m/z (%) 331 (M+, 60), 284 (20), 122 (25), 77 (100). Anal. Calcd. for C18H13N5O2 (331.11): C, 65.25; H, 3.95; N, 21.14. Found: C, 65.37; H, 3.88; N, 21.21%.
4-(4-Nitrophenyl)-1-(4-methylphenyl)-1H,1'H-3,3'-bipyrazole (8b). Yellow crystals, (0.31 g, 90%), m.p. 172–174 °C; IR (KBr) υ = 3,226 (NH) cm−1; 1H-NMR (DMSO-d6) δ = 2.36 (s, 3H, Ar-CH3), 7.23 (d, 2H, J = 8 Hz, Ar-H), 7.25–7.91 (m, 4H, Ar-H), 7.51 (d, 1H, J = 7.5 Hz, pyrazole H-4), 7.56 (d, 1H, J = 7.5 Hz, pyrazole H-5), 8.20 (d, 2H, J = 8 Hz, Ar-H), 9.00 (s, 1H, pyrazole H-5), 12.97 (D2O-exchangeable) (s, 1H, NH) ppm; MS, m/z (%) 345 (M+, 70), 299 (20), 122 (25), 91 (100), 77 (80). Anal. Calcd. for C19H15N5O2 (345.12): C, 66.08; H, 4.38; N, 20.28. Found: C, 66.12; H, 4.46; N, 20.18%.
3.5. Reactions of 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles 2a,b with Hydroxylamine Hydrochloride
Hydroxylamine hydrochloride (0.07 g, 1 mmol) was added to a mixture of enaminone 2a or 2b (1 mmol) and anhydrous potassium carbonate (0.5 g) in absolute ethanol (20 mL). The mixture was heated under reflux for 5 hours and poured onto water. The solid product was filtered and crystallized from ethanol.
3-[4-(4-Nitrophenyl)-1-phenyl-1H-pyrazol-3-yl]isoxazole (9a). Yellow crystals, (0.25 g, 75%), m.p. 160–162 °C; IR (KBr) υ = 1,600 (C=N), cm−1; 1H-NMR (DMSO-d6) δ = 6.78 (d, 1H, J = 5 Hz, isoxazole H-4), 7.29 (d, 2H, J = 8 Hz, Ar-H), 7.36–8.21 (m, 5H, Ar-H), 8.48 (d, 2H, J = 8 Hz, Ar-H), 8.72 (d, 1H, J = 5 Hz, isoxazole H-5), 9.05 (s, 1H, pyrazole H-5) ppm; MS, m/z (%) 332 (M+, 60), 286 (20), 122 (25), 77 (100). Anal. Calcd. for C18H12N4O3 (332.09): C, 65.06; H, 3.64; N, 16.86. Found: C, 65.17; H, 3.58; N, 16.71%.
3-[4-(4-Nitrophenyl)-1-(4-methylphenyl)-1H-pyrazol-3-yl]isoxazole (9b). Yellow crystals, (0.26 g, 75%), m.p. 170–172 °C; IR (KBr) υ = 1,601 (C=N), cm−1; 1H-NMR (DMSO-d6) δ = 2.37 (s, 3H, Ar-CH3), 6.78 (d, 1H, J = 5 Hz, isoxazole H-4), 7.35 (d, 2H, J = 8 Hz, Ar-H), 7.39–8.32 (m, 4H, Ar-H), 8.68 (d, 2H, J = 8 Hz, Ar-H), 8.79 (d, 1H, J = 5 Hz, isoxazole H-5), 9.04 (s, 1H, pyrazole H-5) ppm; MS, m/z (%) 346 (M+, 50), 300 (20), 122 (25), 91 (100), 77 (60). Anal. Calcd. for C19H14N4O3 (346.11): C, 65.89; H, 4.07; N, 16.18. Found: C, 65.77; H, 3.98; N, 16.11%.
3.6. Reactions of 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles 2a,b with 3-amino-1H-[1,2,4]triazole
A mixture of enaminone 2a or 2b (1 mmol) and 3-amino-1H-[1,2,4]triazole (0.085 g, 1 mmol), in glacial acetic acid (20 mL), was refluxed for 5 hours. The solid that formed was filtered off, and crystallized from dioxane to afford compounds 12a,b.
5-[4-(4-Nitrophenyl)-1-phenyl-1H-pyrazol-3-yl][1,2,4]triazolo[4,3-a]pyrimidine (12a). Yellow crystals, (0.32 g, 85%), m.p. 290–292 °C; IR (KBr) υ = 1,596 (C=N) cm−1; 1H-NMR (DMSO-d6) δ = 7.45 (d, 2H, J = 8 Hz, Ar-H), 7.59–8.01 (m, 5H, Ar-H), 7.71 (d, 1H, J = 5 Hz, pyrimidine H-5), 8.11 (d, 2H, J = 8 Hz, Ar-H), 8.47 (s, 1H, triazole H-5), 9.01 (s, 1H, pyrazole H-5), 9.34 (d, 1H, J = 5 Hz, pyrimidine H-4) ppm; MS, m/z (%) 383 (M+, 50), 337 (40), 122 (25), 77 (100). Anal. Calcd. for C20H13N7O2 (383.11): C, 62.66; H, 3.42; N, 25.58. Found: C, 62.77; H, 3.58; N, 25.71%.
5-[4-(4-Nitrophenyl)-1-(4-methylphenyl)-1H-pyrazol-3-yl][1,2,4]triazolo[4,3-a]pyrimidine (12b). Yellow crystals, (0.34 g, 85%), m.p. 310–312 °C; IR (KBr) υ = 1,598 (C=N) cm−1; 1H-NMR (DMSO-d6) δ = 2.41 (s, 3H, Ar-CH3), 7.37 (d, 2H, J = 8 Hz, Ar-H), 7.49–8.01 (m, 4H, Ar-H), 7.74 (d, 1H, J = 5 Hz, pyrimidine H-5), 8.16 (d, 2H, J = 8 Hz, Ar-H), 8.49 (s, 1H, triazole H-5), 9.03 (s, 1H, pyrazole H-5), 9.36 (d, 1H, J = 5 Hz, pyrimidine H-4) ppm; MS, m/z (%) 397 (M+, 50), 351 (40), 122 (25), 91 (70), 77 (100). Anal. Calcd. for C21H15N7O2 (397.13): C, 63.47; H, 3.80; N, 24.67. Found: C, 63.58; H, 3.62; N, 24.77%.
3.7. Coupling of 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-aryl-1H-pyrazoles 2a,b with diazonium salt of 3-amino-1H-[1,2,4]triazole
To a cold solution of enaminone 2a or 2b (1 mmol) in pyridine (25 mL) was added the heterocyclic diazonium salt [prepared by diazotizing 3-amino-1H-[1,2,4]triazole (0.085 g, 1 mmol) dissolved in concentrated nitric acid (2 mL) with a solution of sodium nitrite (0.07 g, 1 mmol) in water (2 mL)]. After complete addition of the diazonium salt, the reaction mixture was stirred for a further 30 min in an ice bath, and then poured onto ice/HCl mixture. The solid precipitated was filtered off, washed with water, dried and crystallized from ethanol/dioxane mixture to give the respective products 14a and 14b.
[4-(4-Nitrophenyl)-1-phenyl-1H-3-pyrazolyl]carbonyl[1,2,4]triazolo[3,4-c][1,2,4]triazine (14a). Pale yellow crystals, (0.32 g, 80%), m.p. 290–292 °C; IR (KBr) υ = 1,662 (CO) cm−1; 1H-NMR (DMSO-d6) δ = 7.43 (d, 2H, J = 8 Hz, Ar-H), 7.56–7.96 (m, 5H, Ar-H), 7.77 (s, 1H, triazine H-5), 8.22 (d, 2H, J = 8 Hz, Ar-H), 8.48 (s, 1H, triazole H-5), 9.17 (s, 1H, pyrazole H-5) ppm; MS, m/z (%) 412 (M+, 50), 292 (20), 122 (50), 77 (100). Anal. Calcd. for C20H12N8O3 (412.10): C, 58.25; H, 2.93; N, 27.17. Found: C, 58.37; H, 3.02; N, 27.31%.
[4-(4-Nitrophenyl)-1-(4-methylphenyl)-1H-3-pyrazolyl]{[1,2,4]triazolo[3,4-c][1,2,4]triazin-6-yl}-methanone (14b). Pale yellow crystals, (0.34 g, 80%), m.p. 198–200 °C; IR (KBr) υ = 1,664 (CO) cm−1; 1H-NMR (DMSO-d6) δ = 2.39 (s, 3H, Ar-CH3), 7.46 (d, 2H, J = 8 Hz, Ar-H), 7.51–7.99 (m, 4H, Ar-H), 7.78 (s, 1H, triazine H-5), 8.26 (d, 2H, J = 8 Hz, Ar-H), 8.49 (s, 1H, triazole H-5), 9.13 (s, 1H, pyrazole H-5) ppm; MS, m/z (%) 426 (M+, 50), 306 (20), 148 (60), 122 (50), 77 (100). Anal. Calcd. for C21H14N8O3 (426.12): C, 59.15; H, 3.31; N, 26.28. Found: C, 59.39; H, 3.22; N, 26.38%.
3.8. Synthesis of 5-[4-(4-nitrophenyl)-1-phenyl-1H-pyrazol-3-yl]-2-thioxo-2,3-dihydro-1H-pyrido[2,3-d]pyrimidin-4-one (17)
A mixture of 3-[E-3-(N,N-dimethylamino)acryloyl]-4-(4-nitrophenyl)-1-phenyl-1H-pyrazole (2a) (1.81 g, 5 mmol) and 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (15, 0.715 g, 5 mmol) in acetic acid (20 mL) was refluxed for 6 hours. The reaction mixture was cooled and diluted with methanol and the solid product was collected by filtration and recrystallized from dioxane to give 17. Yellow crystals (0.35 g, 80%), m.p. 310–313 °C; IR (KBr) υ = 3,261, 3,245 (2 NH), 1,677 (CO), cm−1; 1H-NMR (DMSO-d6) δ = 7.42 (d, 2H, J = 8 Hz, Ar-H), 7.49–8.20 (m, 5H, Ar-H), 8.24 (d, 2H, J = 8 Hz, Ar-H), 8.29 (d, 1H, J = 7 Hz, pyridine-H), 8.48 (d, 1H, J = 7 Hz, pyridine-H), 9.05 (s, 1H, pyrazole H-5), 12.62 (s, 1H, NH), 13.14 (s, 1H, NH) ppm; MS, m/z (%) 442 (M+, 40), 396 (20), 122 (40), 77 (100). Anal. Calcd. for C22H14N6O3S (442.08): C, 59.72; H, 3.19; N, 18.99; S, 7.25. Found: C, 59.81; H, 3.14; N, 19.04; S, 7.20%.
3.9. Synthesis of pyrido[2,3-d][1,2,4]triazolo[4,3-a]pyrimidin-5-one derivatives 21a-c
To a mixture of equimolar amounts of 17 and the appropriate hydrazonoyl chlorides 18a-c (1 mmol) in dioxane (15 mL) was added triethylamine (0.14 mL, 1 mmol). The reaction mixture was refluxed until all of the starting materials have disappeared and hydrogen sulfide gas ceased to evolve (6 hours, monitored by TLC). The solvent was evaporated and the residue was triturated with methanol. The solid that formed was filtered and crystallized from methanol/dioxane mixture to give compounds 21a-c.
3-Acetyl-6-[4-(4-nitrophenyl)-1-phenyl-1H-pyrazol-3-yl]-1-phenyl-1,5-dihydropyrido[2,3-d][1,2,4] triazolo[4,3-a]pyrimidin-5-one (21a). Yellow crystals, (0.45 g, 80%), m.p. 280–282 °C; IR (KBr) υ = 1,707, 1,650 (2 CO), cm−1; 1H-NMR (DMSO-d6) δ = 2.84 (s, 3H, COCH3), 7.26 (d, 2H, J = 8 Hz, Ar-H), 7.39–7.85 (m, 10H, Ar-H), 8.03 (d, 1H, J = 7 Hz, pyridine-H), 8.27 (d, 2H, J = 8 Hz, Ar-H), 8.69 (d, 1H, J = 7 Hz, pyridine-H), 9.05 (s, 1H, pyrazole H-5) ppm; 13C-NMR (DMSO-d6) δ = 31.3, 119.8, 121.7, 122.4, 123.3, 124.5, 125.3, 127.4, 128.5, 129.1, 129.8, 131.2, 139.4, 142.5, 143.9, 146.8, 147.8, 148.1, 148.8, 152.1, 153.8, 155.3, 159.5, 164.0, 176 ppm; MS, m/z (%) 568 (M+, 25), 525 (20), 497 (40), 122 (30), 77 (100). Anal. Calcd. for C31H20N8O4 (568.16): C, 65.49; H, 3.55; N, 19.71. Found: C, 65.34; H, 3.42; N, 19.64%.
Ethyl 5-oxo-6-[4-(4-nitrophenyl)-1-phenyl-1H-pyrazol-3-yl]-1-phenyl-1,5-dihydropyrido[2,3-d] [1,2,4]triazolo[4,3-a]pyrimidine-3-carboxylate (21b). Yellow crystals, (0.47 g, 80%), m.p. 250–253 °C; IR (KBr) υ = 1,719, 1,645 (2 CO), cm−1; 1H-NMR (DMSO-d6) δ = 1.45 (t, J = 7 Hz, 3H, CH3,), 4.57 (q, J = 7 Hz, 2H, CH2,), 7.26 (d, 2H, J = 8 Hz, Ar-H), 7.27–7.81 (m, 10H, Ar-H), 8.16 (d, 1H, J = 7 Hz, pyridine-H), 8.21 (d, 2H, J = 8 Hz, Ar-H), 8.62 (d, 1H, J = 7 Hz, pyridine-H), 9.07 (s, 1H, pyrazole H-5) ppm; 13C-NMR (DMSO-d6) δ = 31.6, 35.8, 118.9, 120.7, 122.4, 123.3, 124.6, 125.7, 127.2, 128.5, 129.3, 129.9, 131.2, 139.4, 142.5, 143.7, 146.8, 147.9, 148.2, 148.8, 152.1, 153.8, 155.3, 159.5, 163.4, 177 ppm; MS, m/z (%) 598 (M+, 25), 525 (40), 479 (40), 122 (30), 77 (100). Anal. Calcd. for C32H22N8O5 (598.17): C, 64.21; H, 3.70; N, 18.72. Found: C, 64.34; H, 3.62; N, 18.62%.
N3,1-Diphenyl-5-oxo-6-[4-(4-nitrophenyl)-1-phenyl-1H-pyrazol-3-yl]-1,5-dihydropyrido[2,3-d] [1,2,4]triazolo[4,3-a]pyrimidine-3-carboxamide (21c). Yellow crystals, (0.48 g, 75%), m.p. 325−327 °C; IR (KBr) υ = 3,388 (NH), 1,697, 1,651 (2 CO), cm−1; 1H-NMR (DMSO-d6) δ = 7.23 (d, 2H, J = 8 Hz, Ar-H), 7.39–8.15 (m, 15H, Ar-H), 8.01 (d, 1H, J = 7 Hz, pyridine-H), 8.24 (d, 2H, J = 8 Hz, Ar-H), 8.62 (d, 1H, J = 7 Hz, pyridine-H), 9.01 (s, 1H, pyrazole H-5), 10.92 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6) δ = 111.8, 119.8, 120.7, 121.4, 121.7, 122.4, 123.3, 124.5, 125.3, 125.9, 127.4, 128.5, 129.1, 129.8, 131.2, 139.4, 142.5, 143.9, 146.8, 147.8, 148.1, 148.8, 152.1, 153.8, 155.3, 159.5, 161.9, 168 ppm; MS, m/z (%) 645 (M+, 25), 525 (40), 122 (20), 77 (100). Anal. Calcd. for C36H23N9O4 (645.19): C, 66.97; H, 3.59; N, 19.53. Found: C, 66.84; H, 3.46; N, 19.61%.
3.10. Agar diffusion well method to determine the antimicrobial activity
The microorganism inoculums were uniformly spread using sterile cotton swab on a sterile Petri dish Malt extract agar (for fungi) and nutrient agar (for bacteria). One hundred μL of each sample was added to each well (10 mm diameter holes cut in the agar gel, 20 mm apart from one another). The systems were incubated for 24–48 h at 37 °C (for bacteria) and at 28 °C (for fungi). After incubation, the microorganism's growth was observed. Inhibition of the bacterial and fungal growth were measured in mm. Tests were performed in triplicate [
44].
3.11. Cytotoxic activity
The method applied is similar to that reported by Skehan
et al. [
45] using Sulfo-Rhodamine-B stain (SRB). Cells were plated in 96-multiwill plate (10
4 cells/well) for 24 h before treatment with the tested compounds to allow attachment of cell to the wall of the plate. Different concentrations of the compound under test (0, 2.5, 5, and 10 µg/mL) were added to the cell monolayer in triplicate wells individual dose, monolayer cells were incubated with the compounds for 48 h at 37 °C and in atmosphere of 5% CO
2. After 48 h, cells were fixed, washed and stained with SRB stain, excess stain was washed with acetic acid and attached stain was recovered with
tris-EDTA buffer, color intensity was measured in an ELISA reader. The relation between surviving fraction and drug concentration is plotted to get the survival curve of each tumor cell line after the specified compound. The response parameter calculated was the IC
50 value, which corresponds to the compound concentration causing 50% mortality in net cells (Figures 3-10).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}