Ring Cleavage Reactions of Methyl α-D-Allopyranoside Derivatives with Phenylboron Dichloride and Triethylsilane

Abstract
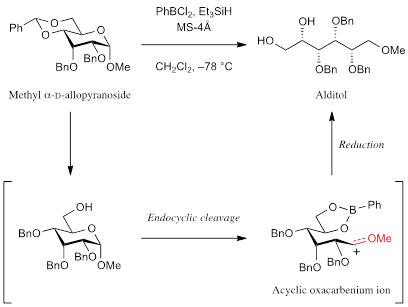
: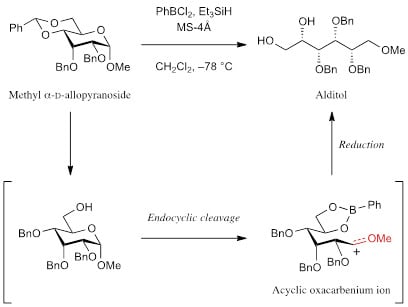
1. Introduction
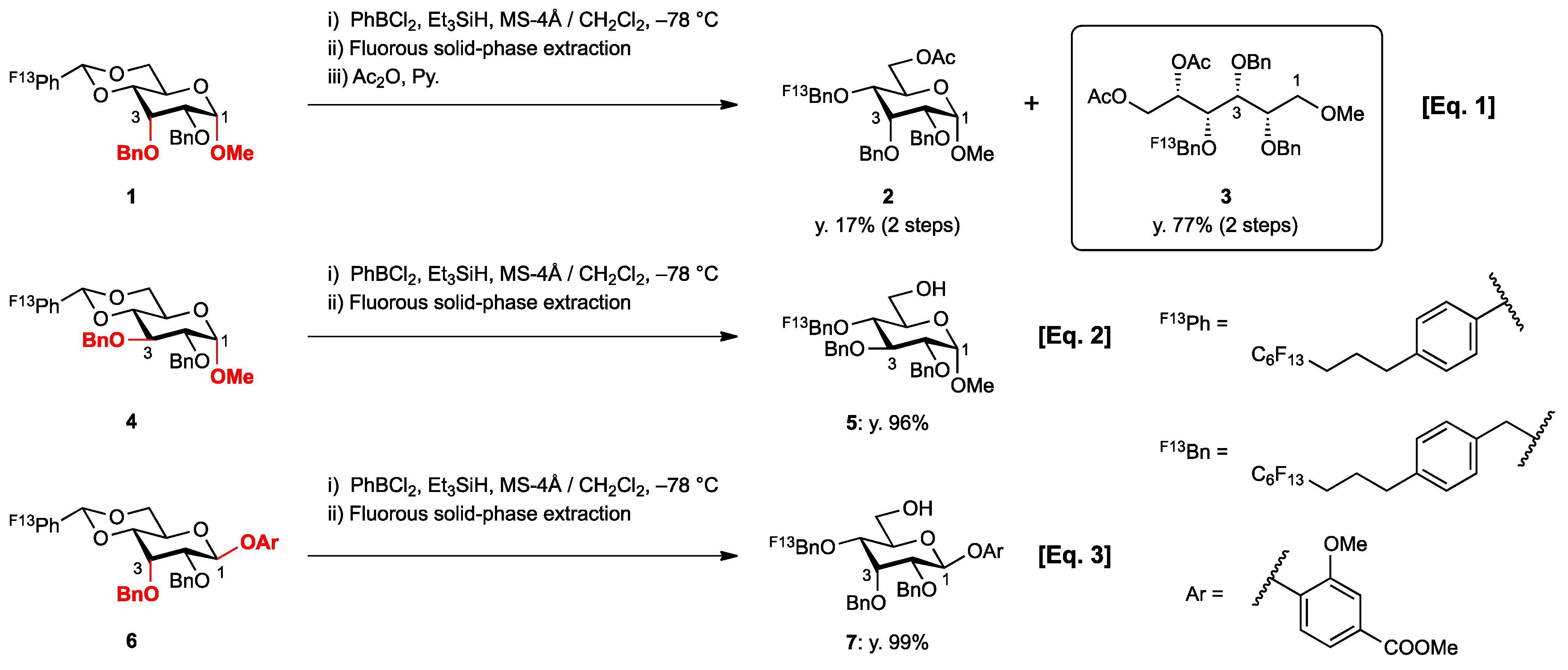
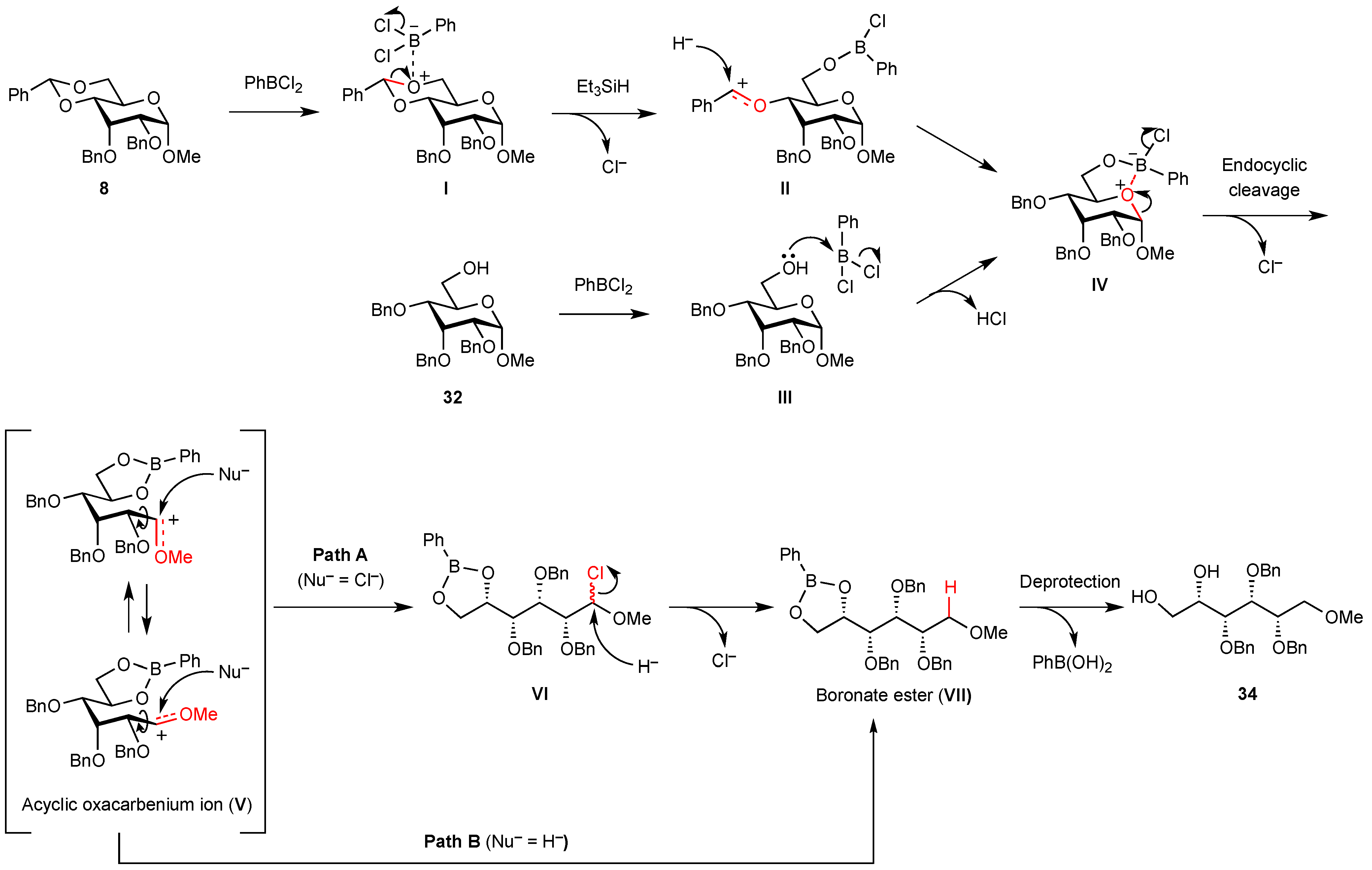
2. Results and Discussion

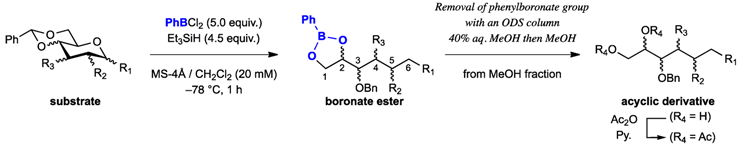
{kind=link}
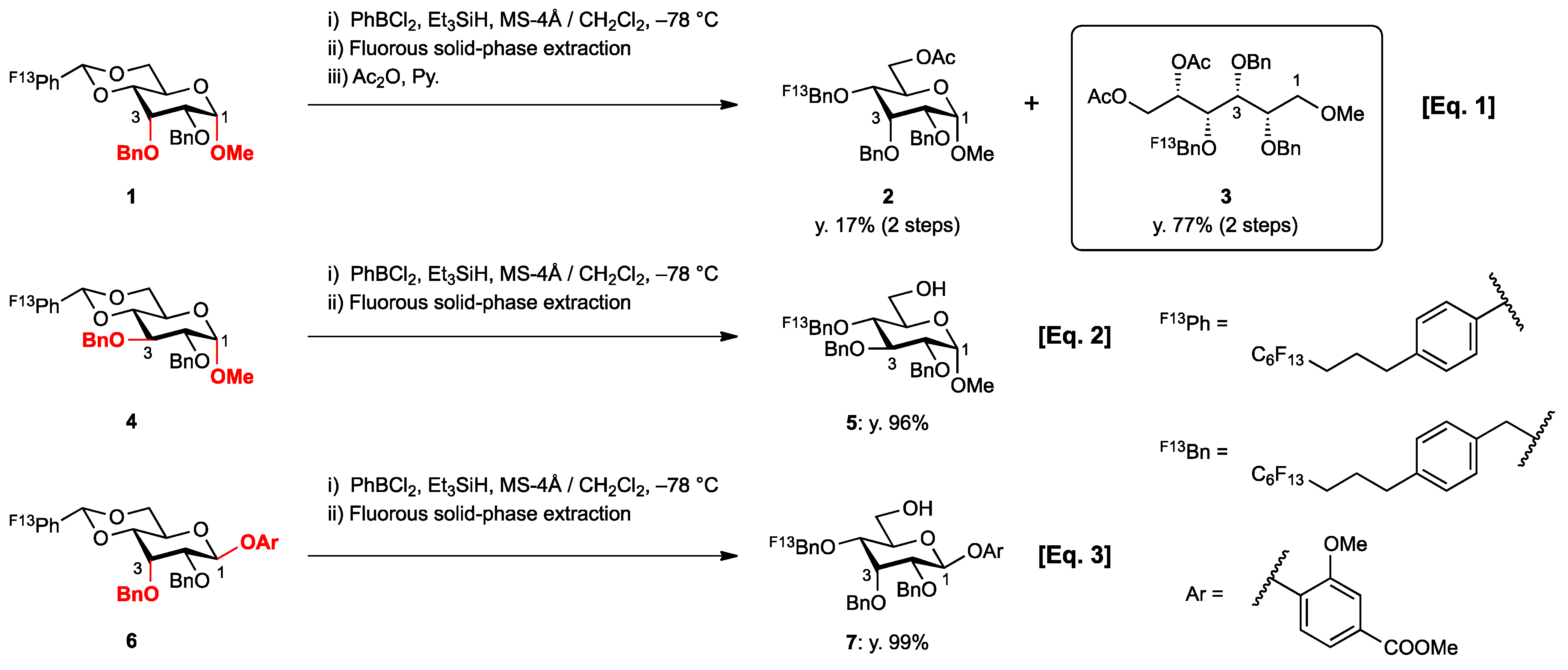{kind=link}
{kind=link}
| Entry | Substrate | Product (isolated yield) | |
|---|---|---|---|
| Acyclic derivative | 4- O-benzylated derivative | ||
 | 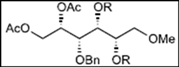 |  | |
| 1 | 8: R = Bn | 19 (R = Bn): y. 78% a | 20 (R = Bn): y. 7% a |
| 2 | 9: R = MOM | 21 (R = MOM): y. - b | |
| 3 | 10: R = Bz | 22 (R = Bz): y. - b | |
| 4 |  11 | 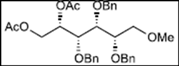 19: y. 17% | 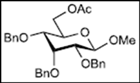 23: y. 46% |
| 5 |  12 | - | 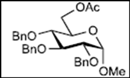 24: y. 78% |
| 6 | 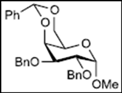 13 | - | 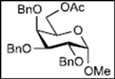 25: y. 80% |
| 7 |  14 | 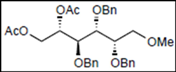 26: y. 71% | - |
| 8 | 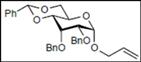 15 | 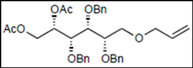 27: y. 77% | - |
| 9 | 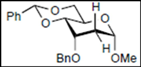 16 | 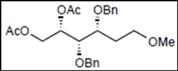 28: y. 86% | - |
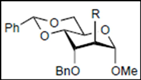 | 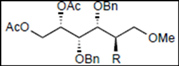 | 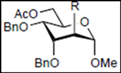 | |
| 10 | 17: R = OBn | 29 (R = OBn): y. 27% c | |
| 11 | 18: R = N3 | (R = N3): - | 30 (R = N3): y. 83% |
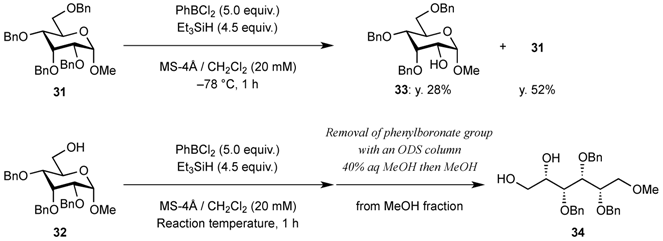
| Entry | Reaction temperature (°C) | Yield of acyclic compound 34 (%) | Yield of recovered starting material 32 (%) |
|---|---|---|---|
| 1 | −78 | 10 | 80 |
| 2 | −60 | 53 | 41 |
| 3 | −50 | 70 | 24 |
| 4 | −40 | 80 | 18 |
| 5 | −19 | 92 | - |
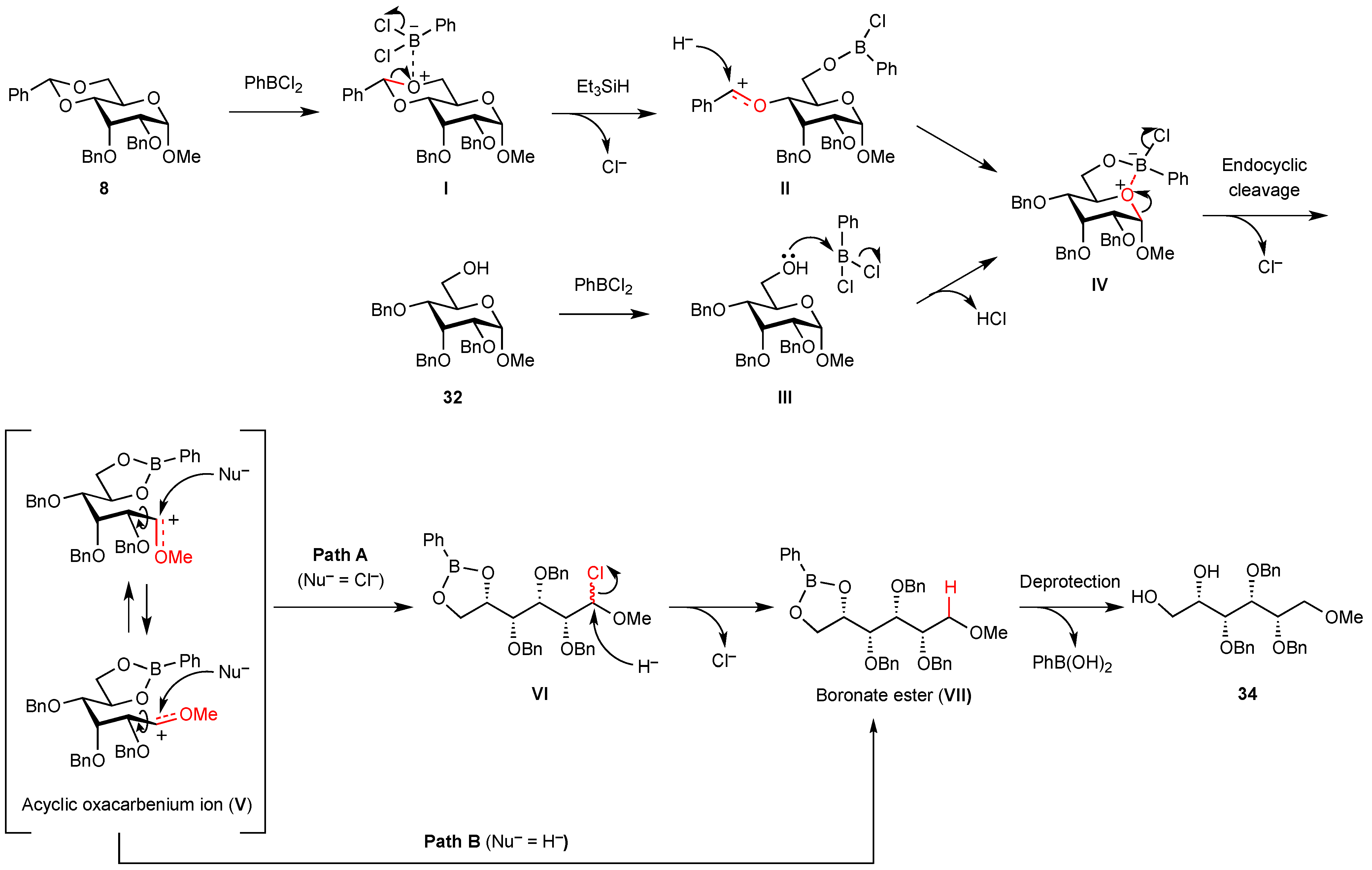
3. Experimental
3.1. General
3.2. General Procedure for Endocyclic Cleavage with PhBCl2 and Et3SiH
4. Conclusions
Acknowledgements
References and Notes
- DeNinno, M.P.; Etienne, J.B.; Duplantier, K.C. A method for the selective reduction of carbohydrate 4,6-O-benzylidene acetals. Tetrahedron Lett. 1995, 36, 669–672. [Google Scholar] [CrossRef]
- Sakagami, M.; Hamana, H. A selective ring opening reaction of 4,6-O-benzylidene acetals in carbohydrates using trialkylsilane derivatives. Tetrahedron Lett. 2000, 41, 5547–5551. [Google Scholar] [CrossRef]
- Shie, C.-R.; Toone, Z.-H.; Kulkarni, S.S.; Uang, B.-J.; Hsu, C.-Y.; Hung, S.-C. Cu(OTf)2 as an efficient and dual-purpose catalyst in the regioselective reductive ring opening of benzylidene acetals. Angew. Chem. Int. Ed. 2005, 44, 1665–1668. [Google Scholar]
- Hernández-Torres, J.M.; Achkar, J.; Wei, A. Temperature-controlled regioselectivity in the reductive cleavage of p-methoxybenzylidene acetals. J. Org. Chem. 2004, 69, 7206–7211. [Google Scholar] [CrossRef]
- Sherman, A.A.; Mironov, Y.V.; Yudina, O.N.; Nifantiev, N.E. The presence of water improves reductive openings of benzylidene acetals with trimethylaminoborane and aluminium chloride. Carbohydr. Res. 2003, 338, 697–703. [Google Scholar]
- Wang, C.-C.; Luo, S.-Y.; Shie, C.-R.; Hung, S.-C. Metal trifluoromethanesulfonate-catalyzed regioselective borane-reductive ring opening of benzylidene acetals: A concise synthesis of 1,4-dideoxy-1,4-imino-L-xylitol. Org. Lett. 2002, 4, 847–849. [Google Scholar]
- Debenham, A.D.; Toone, E.J. Regioselective reduction of 4,6-O-benzylidenes using triethylsilane and BF3·Et2O. Tetrahedron:Asymmetry 2000, 11, 385–387. [Google Scholar] [CrossRef]
- Rao, K.V.; Patil, P.R.; Atmakuri, S.; Kartha, K.P.R. Iodide-sodium cyanoborohydride-mediated reductive ring opening of 4,6-benzylidene acetals of hexopyranosides. Carbohydr. Res. 2010, 345, 2709–2713. [Google Scholar] [CrossRef]
- Panchadhayee, R.; Misra, A.K. Regioselective reductive ring opening of benzylidene acetals using triethylsilane and iodide. Synlett 2010, 8, 1193–1196. [Google Scholar]
- Daragics, K.; Fügedi, P. Regio- and chemoselective reductive cleavage of 4,6-O-benzylidene-type acetals of hexopyranosides using BH3·THF−TMSOTf. Tetrahedron Lett. 2009, 50, 2914–2916. [Google Scholar] [CrossRef]
- Saito, S.; Kuroda, A.; Tanaka, K.; Kimura, R. A novel reducing system for acetal cleavage: BH3·S(CH3)2−BF3·O(C2H5)2 combination. Synlett 1996, 3, 231–233. [Google Scholar]
- Tani, S.; Sawadi, S.; Kojima, M.; Akai, S.; Sato, K. A novel method for regioselective ring-opening reduction of 4,6-O-benzylidene hexopyranoside derivatives using CoCl2 and BH3·THF. Tetrahedron Lett. 2007, 48, 3103–3104. [Google Scholar]
- Ohlin, M.; Johnsson, R.; Ellervik, U. Regioselective reductive openings of 4,6-benzylidene acetals: synthetic and mechanistic aspects. Carbohydr. Res. 346, 2011, 1358–1370. [Google Scholar]
- Denmark, S.E.; Almstead, N.G. Studies on the mechanism and origin of stereoselective opening of chiral dioxane acetals. J. Am. Chem. Soc. 1991, 113, 8089–8110. [Google Scholar] [CrossRef]
- Kojima, M.; Nakamura, Y.; Takeuchi, S. A practical fluorous benzylidene acetal protecting group for a quick synthesis of disaccharides. Tetrahedron Lett. 2007, 48, 4431–4436. [Google Scholar]
- Kojima, M.; Nakamura, Y.; Ito, S.; Takeuchi, S. Total synthesis of macrocyclic glycosides, clemochinenosides A and B, and berchemolide, by fluorous mixture synthesis. Tetrahedron Lett. 2009, 50, 6143–6149. [Google Scholar] [CrossRef]
- Lidberg, B. Action of strong acids on acetylated glucosides. Acta Chem. Scand. 1949, 3, 1153–1169. [Google Scholar] [CrossRef]
- Morishima, N.; Koto, S.; Zen, S. A rapid anomerization of alkyl per-O-benzyl-β-D-glucopyranosides by titanium tetrachloride. Chem. Lett. 1979, 8, 749–750. [Google Scholar] [CrossRef]
- O’Brien, C.; Poláková, M.; Pitt, N.; Tosin, M.; Murphy, P.V. Glycosidation-anomerization reactions of 6,1-anhydroglucopyranuronic acid and anomerization of β-D-glucopyranosiduronic acids promoted by SnCl4. Chem. Eur. J. 2007, 13, 902–909. [Google Scholar] [CrossRef]
- Manabe, S.; Ishii, K.; Hashizume, D.; Koshino, H.; Ito, Y. Evidence for endocyclic cleavage of conformationally restricted glycopyranosides. Chem. Eur. J. 2009, 15, 6894–6901. [Google Scholar] [CrossRef]
- Satoh, H.; Hutter, J.; Lüthi, H.P.; Manabe, S.; Ishii, K.; Ito, Y. Low-barrier pathway for endo-cleavage induced anomerization of pyranosides with N-benzyl-2,3-trans-oxazolidinone groups. Eur. J. Org. Chem. 2009, 1127–1131. [Google Scholar]
- Manabe, S.; Ito, Y. Significant solvent effect in anomerization reaction of pyranosides with 2,3-trans carbamate and carbonate. Tetrahedron Lett. 2009, 50, 4827–4829. [Google Scholar] [CrossRef]
- Pilgrim, W.; Murphy, P. SnCl4- and TiCl4-catalyzed anomerization of acylated O- and S-glycosides: Analysis of factors that lead to higher α:β anomer ratios and reaction rates. J. Org. Chem. 2010, 75, 6747–6755. [Google Scholar] [CrossRef]
- Satoh, H.; Manabe, S.; Ito, Y.; Lüthi, H.P.; Laino, T.; Hutter, J. Endocyclic cleavage in glycosides with 2,3-trans cyclic protecting groups. J. Am. Chem. Soc. 2011, 133, 5610–5619. [Google Scholar]
- Köster, R.; Penadés-Ullate, S.; Dahlhoff, W.V. Catalyzed acetal reduction with >BH boranes−1-O-alkyl(aryl)alditols, anhydroalditols, and 1-O-alditylalditols from O-glycopyranoside. Angew. Chem. Int. Ed. 1985, 24, 519–521. [Google Scholar]
- Guindon, Y.; Anderson, P.C. Stereoelectronic effects in the ring cleavage of methyl glycopyranosides using dimethylboron bromide. Tetrahedron Lett. 1987, 28, 2485–2488. [Google Scholar] [CrossRef]
- Inghardt, T.; Frejd, T. Organoaluminum-induced opening of the pyranosidic ring of benzyl 2-deoxy-2-C-methylpentopyranosides. J. Org. Chem. 1989, 54, 5539–5543. [Google Scholar] [CrossRef]
- Hashimoto, H.; Hayakawa, M. Synthesis of a new fucosidase inhibitor, 1,5-dideoxy-1,5-imino-L-talitol, via cyanotrimethylsilanolysis of a β-D-ribofuranoside and its inhibitory activities. Chem. Lett. 1989, 18, 1881–1884. [Google Scholar] [CrossRef]
- Hashimoto, H.; Kawanishi, M.; Yuasa, H. New and facile synthetic routes to 5-thioaldohexopyranosides via aldose monothioacetal derivatives. Tetrahedron Lett. 1991, 32, 7087–7090. [Google Scholar]
- Martin, O.R.; Rao, S.P.; Yang, T.-F.; Fotia, F. Chelation-controlled regio- and stereoselective C-allylation of glycosides and related cyclic hemiacetals. Synlett 1991, 702–704. [Google Scholar]
- Olsson, R.; Rundström, P.; Frejd, T. Chelation-controlled regioselective endo cleavage and stereoselective C-1 alkylation of pentofuranosides. J. Chem. Soc. Perkin Trans. 1 1998, 785–790. [Google Scholar]
- Olsson, R.; Berg, U.; Frejd, T. Endocyclic cleavage of glycosides. VI. Substituent effects of the alkylative endocyclic cleavage of glycosides. Tetrahedron 1998, 54, 3935–3954. [Google Scholar] [CrossRef]
- 1,2-Boronate ester: Colorless syrup; Rf = 0.50 (hexane/EtOAc = 1:1 v/v); IR (NaCl, neat): 1602, 1441, 1397, 1327cm−1; 1H-NMR (250 MHz, CDCl3): δ 7.81−7.77 (2H, m, BC6H5), 7.50−7.18 (18H, m, ArH), 4.81 (ddd, J2,3 = 3.7 Hz, J2,1 = 7.3 Hz, J2,1′ = 8.3 Hz, H-2), 4.72, 4.57 (2H, each d, J = 11.7 Hz, PhCH2), 4.69, 4.60 (2H, each d, J = 11.5 Hz, PhCH2), 4.68, 4.63 (2H, each d, J = 11.4 Hz, PhCH2), 4.51 (1H, dd, J1,2 = 7.3 Hz, J1,1′ = 9.1 Hz, H-1), 4.18 (1H, dd, J1′,2 = 8.4 Hz, J1′,1 = 9.0 Hz, H-1′), 4.08 (1H, dd t-like, J3,4 = 3.6 Hz, J3,2 = 3.7 Hz, H-3), 3.88 (1H, dd, J4,3 = 3.6 Hz, J4,5 = 6.2 Hz, H-4), 3.81 (1H, ddd, J5,4 = 6.2 Hz, J5,6 = 3.3 Hz, J5,6′ = 4.5 Hz, H-5), 3.66 (1H, dd, J6,5 = 3.3 Hz, J6,6′ = 10.4 Hz, H-1), 3.59 (1H, dd, J6′,5 = 4.5 Hz, J6’,6 = 10.4 Hz, H-1′), 3.35 (3H, s, OCH3); 13C-NMR (63 MHz, CDCl3): δ 138.2, 138.1, 138.0, 134.9(BC6H5), 131.3(BC6H5), 128.4, 128.3, 128.24, 128.17, 128.0, 127.9, 127.8, 127.7, 127.61, 127.57, 80.2, 78.2, 78.0, 77.3, 74.0, 73.5, 72.4, 71.8, 67.5, 59.1. The structure of boronate ester was established by comparing 1H- and 13C-NMR chemical shifts with those of similar compounds reported in the following literatures [34,35,36,37]
- Crinch, D.; de la Mora, M.; Vinod, A.U. Influence of the 4,6-O-benzylidene, 4,6-O-phenylboronate, and 4,6-O-polystyrylboronate protecting groups on the stereochemical outcome of thioglycoside-based glycosylations mediated by 1-benzenesulfinyl piperidine/triflic anhydride and N-iodosuccinimide/trimethylsilyl triflate. J. Org. Chem. 2003, 68, 8142–8148. [Google Scholar] [CrossRef]
- Smith, J.M.; Borsenberger, V.; Raftery, J.; Sutherland, J.D. Exploratory studies to investigate a linked prebiotic origin of RNA and coded peptides. 2nd communication. Chem. Biodiv. 2004, 1, 1418–1451. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Martino, E.D.; Marcantoni, E.; Sambri, L. Highly stereoselective and efficient addition of organocerium reagents to syn-β-alkyl-β-hydroxy-α-methyl ketones by way of their titanium alkoxides-Synthesis of complex 1,3-diol units with three streodefined centres. Eur. J. Org. Chem. 2001, 15, 2901–2909. [Google Scholar]
- Meiland, M.; Heinze, T.; Guenther, W.; Liebert, T. Seven-membered ring boronates at trans-diol moieties of carbohydrates. Tetrahedron Lett. 2009, 50, 469–472. [Google Scholar]
- Sample Availability: Samples of the compounds 8, 11-20 and 23-34 are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kojima, M.; Nakamura, Y.; Ito, Y.; Takeuchi, S. Ring Cleavage Reactions of Methyl α-D-Allopyranoside Derivatives with Phenylboron Dichloride and Triethylsilane. Molecules 2011, 16, 10303-10313. https://doi.org/10.3390/molecules161210303
Kojima M, Nakamura Y, Ito Y, Takeuchi S. Ring Cleavage Reactions of Methyl α-D-Allopyranoside Derivatives with Phenylboron Dichloride and Triethylsilane. Molecules. 2011; 16(12):10303-10313. https://doi.org/10.3390/molecules161210303
Chicago/Turabian StyleKojima, Masaru, Yutaka Nakamura, Yuusuke Ito, and Seiji Takeuchi. 2011. "Ring Cleavage Reactions of Methyl α-D-Allopyranoside Derivatives with Phenylboron Dichloride and Triethylsilane" Molecules 16, no. 12: 10303-10313. https://doi.org/10.3390/molecules161210303
APA StyleKojima, M., Nakamura, Y., Ito, Y., & Takeuchi, S. (2011). Ring Cleavage Reactions of Methyl α-D-Allopyranoside Derivatives with Phenylboron Dichloride and Triethylsilane. Molecules, 16(12), 10303-10313. https://doi.org/10.3390/molecules161210303