In Vitro and Ex Vivo Selection Procedures for Identifying Potentially Therapeutic DNA and RNA Molecules




Abstract
:
1. Introduction
2. General Principles of in Vitro Selection Methods
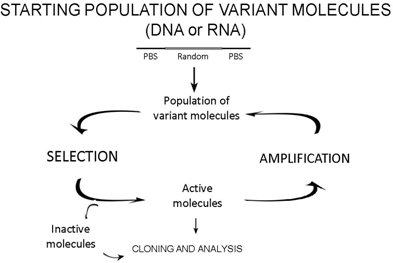
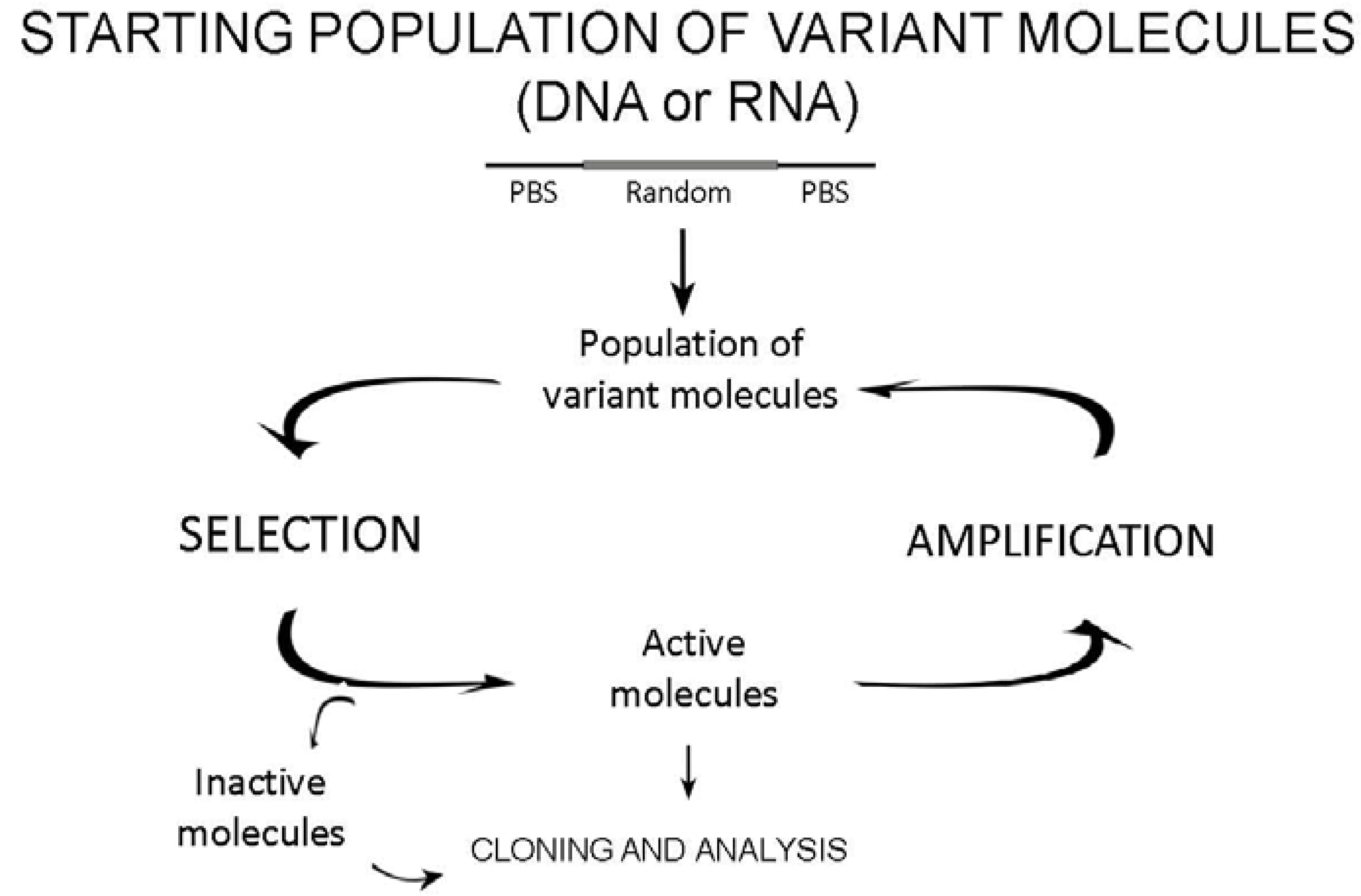
2.1. The design of starting variant populations
2.2. Selection
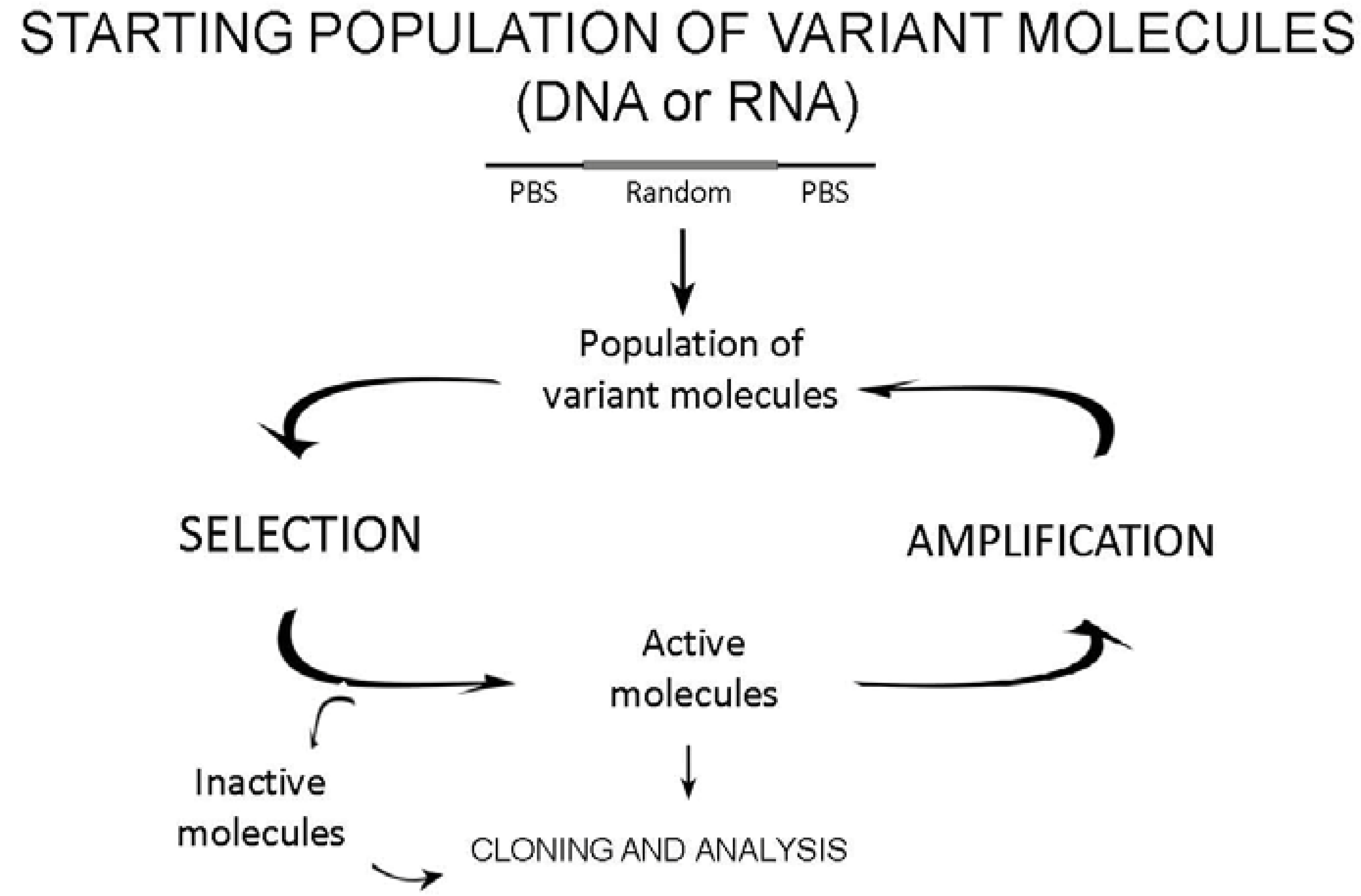
2.3. Amplification of active molecules
3. Ex Vivo Selection
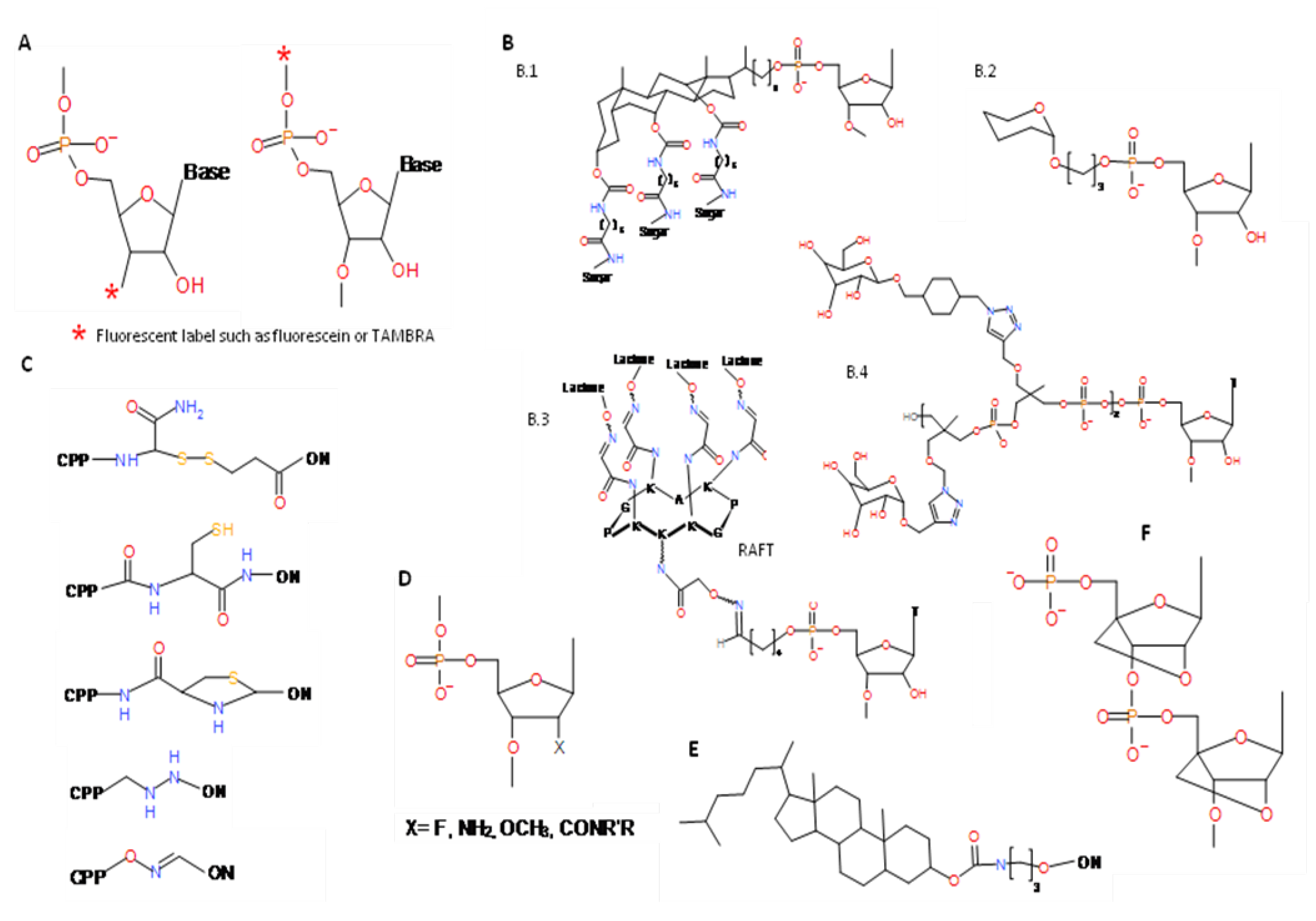
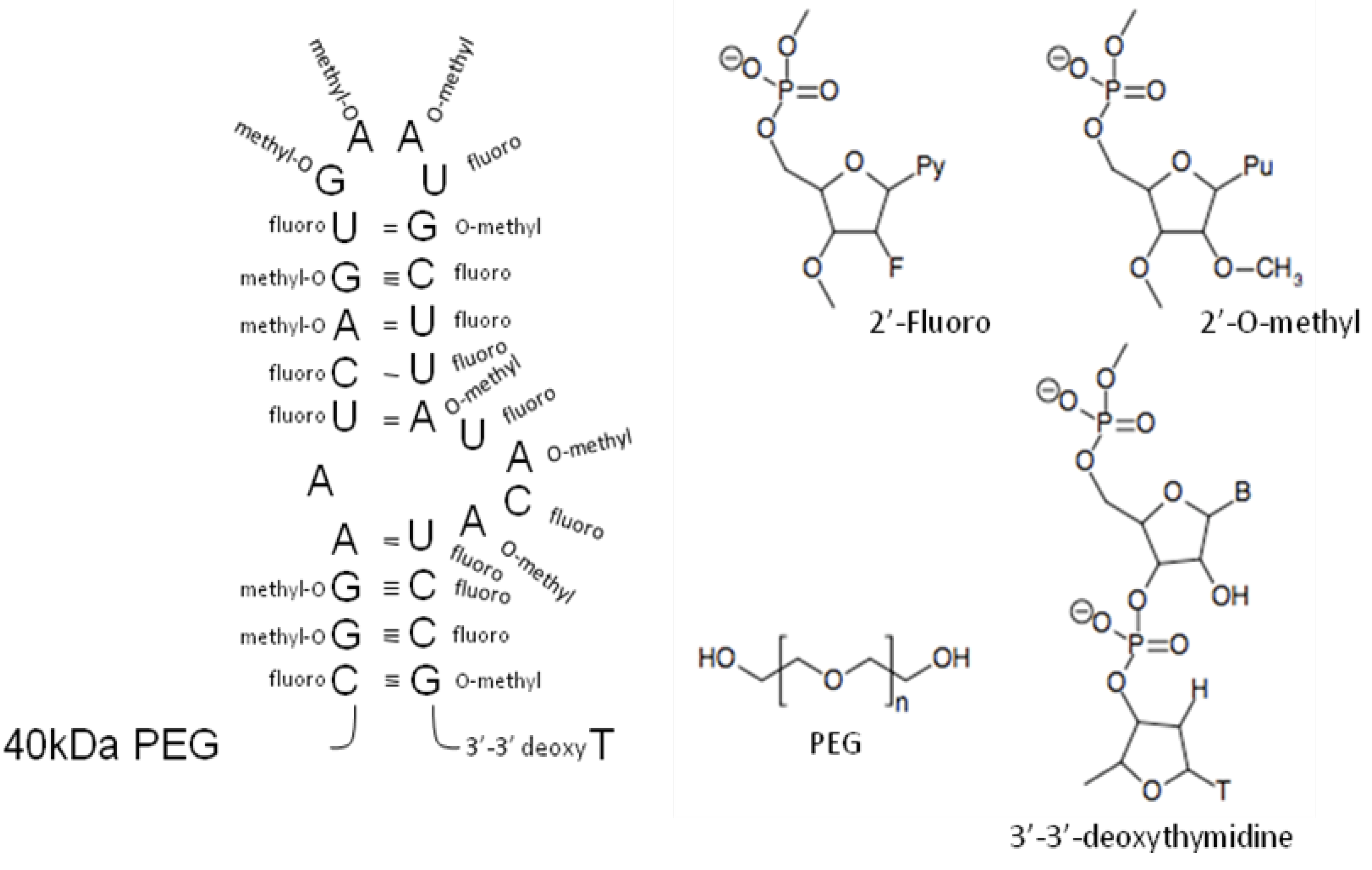
4. Post-selection Modifications
5. Therapeutic Applications of Nucleic Acid Selection Procedures
6. Aptamers as Therapeutic Agents

{kind=link}
{kind=link}
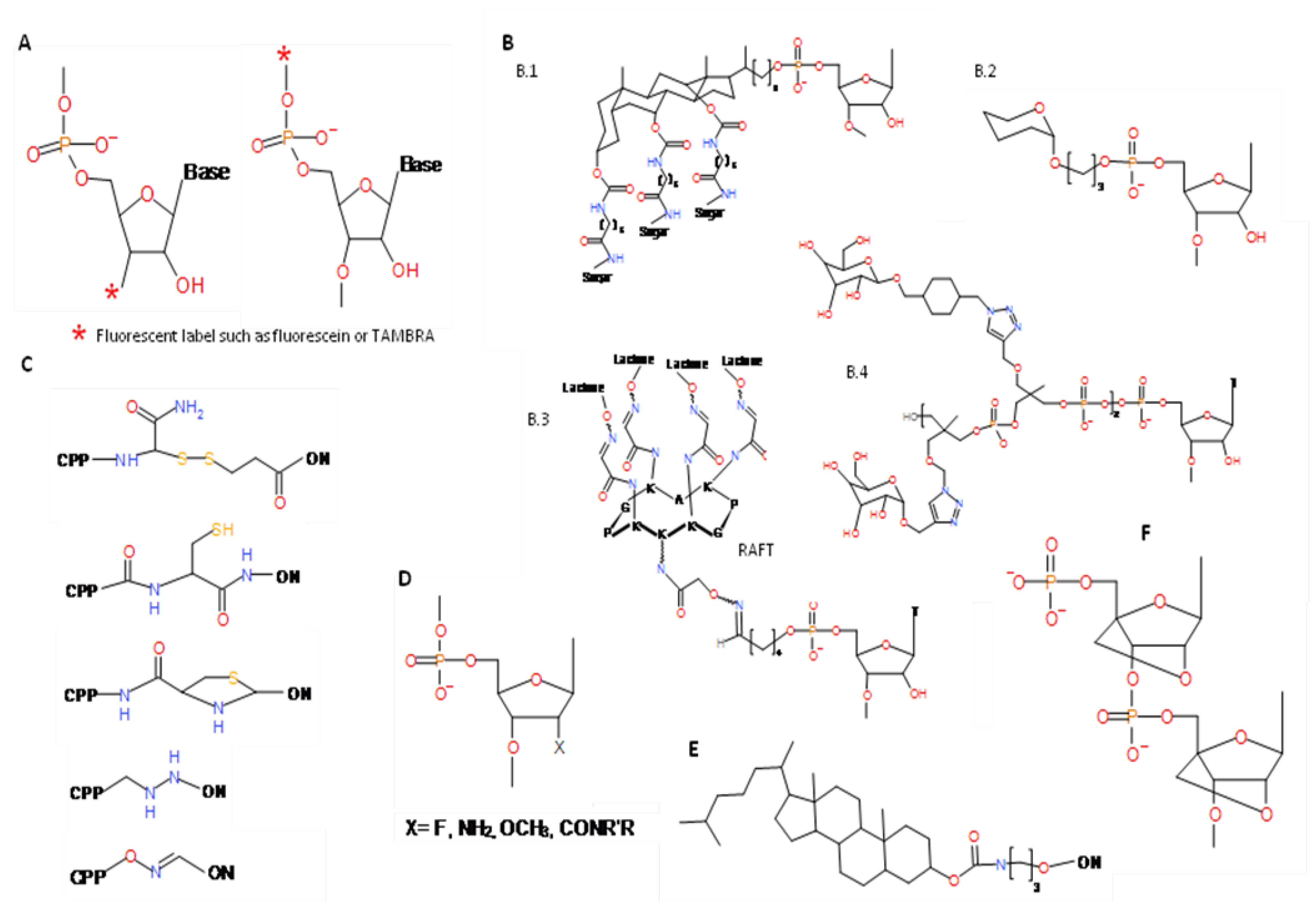{kind=link}
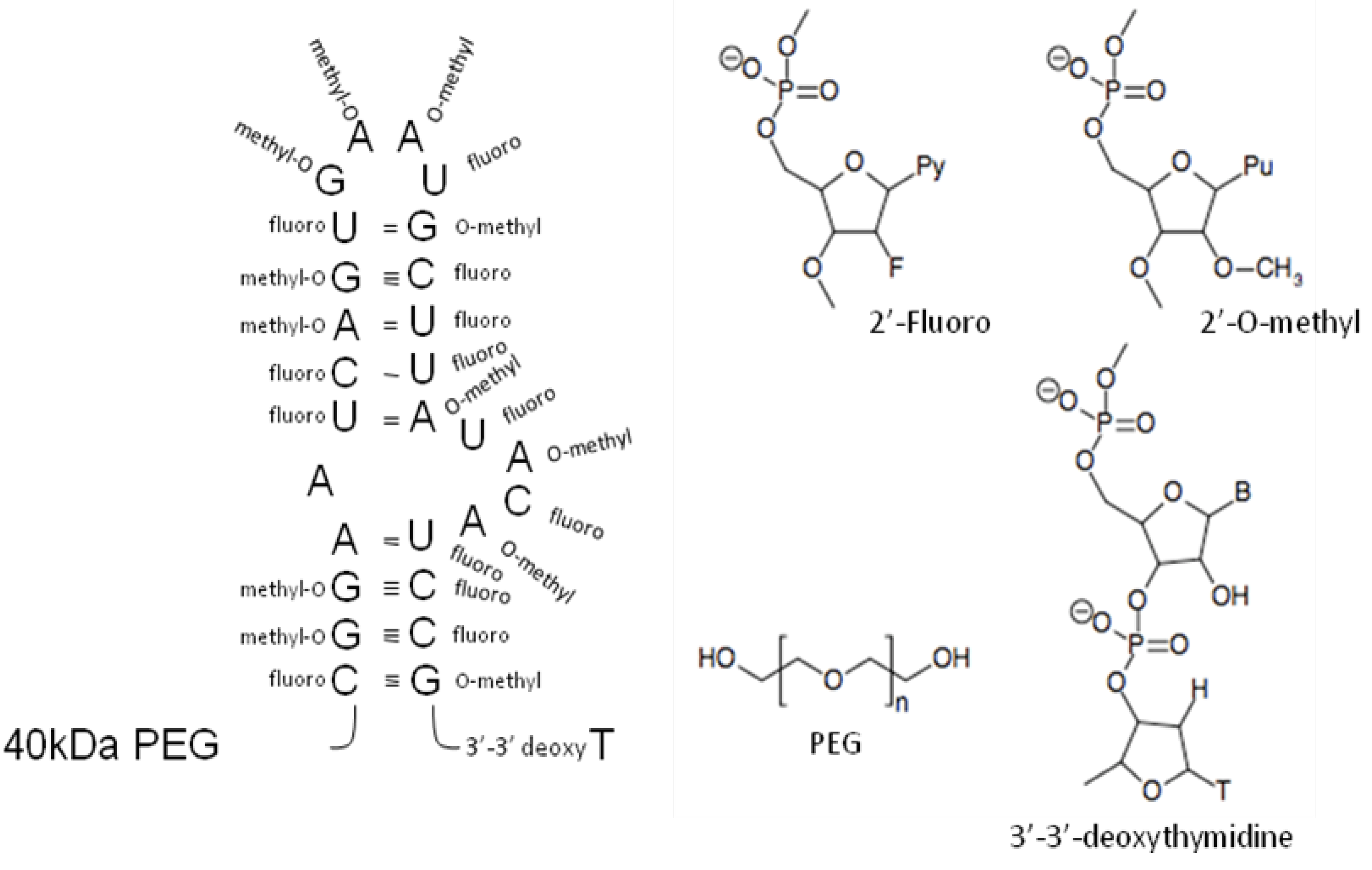{kind=link}
{kind=link}
| Therapeutic target | Aptamers | Type | Disease indication | Clinical status | Reference |
|---|---|---|---|---|---|
| VEGF | MacugenTM (Pegaptanib Sodium) | 2’-Fluoro- 2’-O-methyl RNA+PEG | Macular degeneration | Market | [57,58] |
| Von Willebrand factor | ARC1779 | DNA/RNA+PEG | Thrombotic microangiopathy Adjunct to carotid endarterectomy | Phase 2 Phase 2 | [59,60] |
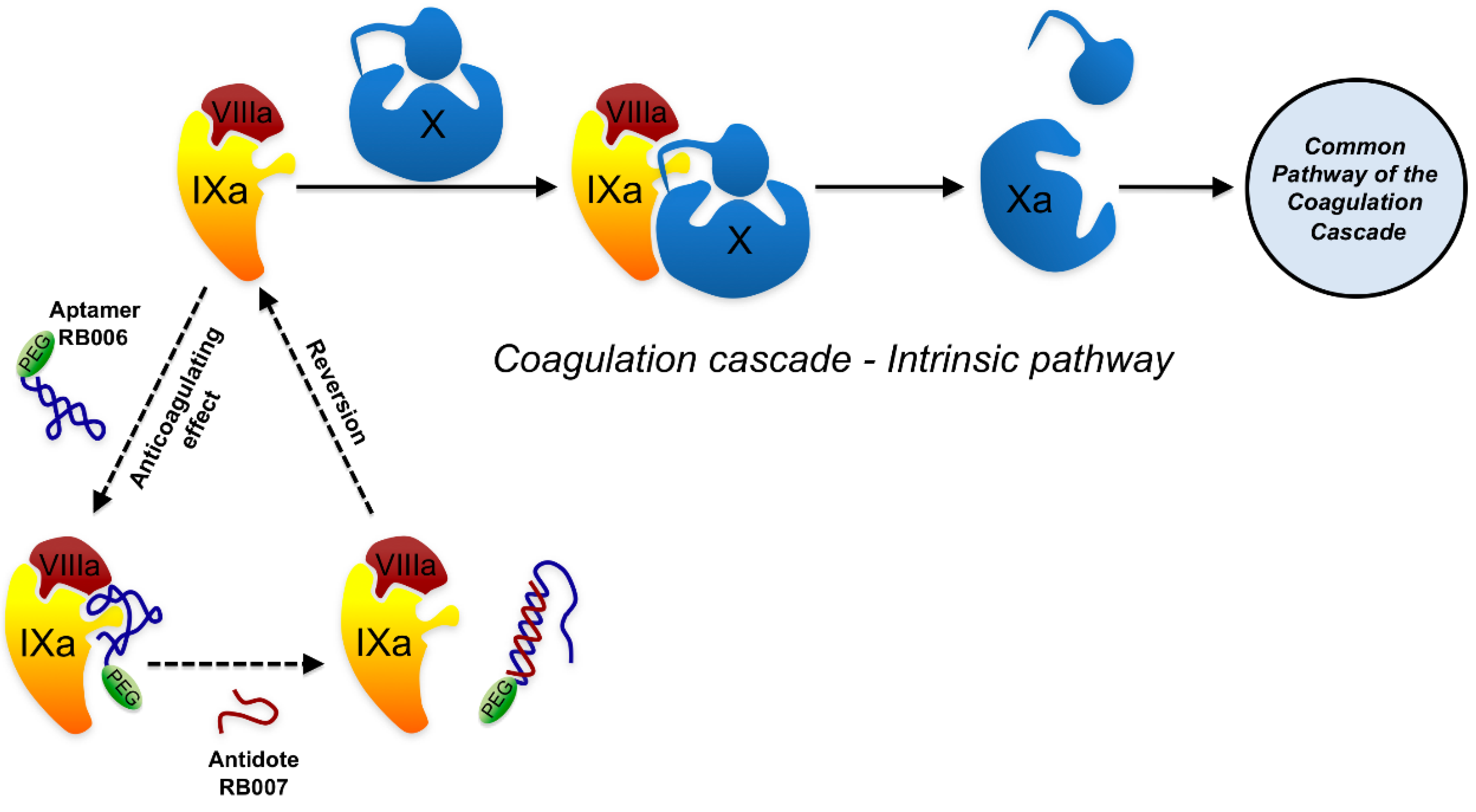
| Factor IXa | REG-1 (RB006 aptamer + RB007 antidote) | RB006 2’-Fluoro RNA+PEG and RB007 2’-O-methyl RNA | Coronary artery bypass Percutaneous coronary intervention | Phase 2 Phase 2 | [61,62,63] |
| Nucleolin | AS1411 | DNA | Acute myeologenous leukemiaRenal cell carcinoma | Phase 2 Phase 2 | [64,65,66] |
| PDGF-b | E10030 | DNA | Macular degeneration | Phase 1 | [67,68] |
| Complement factor 5 | ARC1905 | 2’-Fluoro RNA | Macular degeneration | Phase 1 | [69] |
| Thrombin | NU172 | DNA | Coronary artery bypass | Phase 1 | [70,71,72] |
6.1. Aptamer-based anti-degenerative disease agents
6.2. Anti-inflammatory aptamers
6.3. Anti-immunoglobulin E aptamers
6.4. Aptamer-based therapy against cancer
6.5. Anti-vascular diseases aptamers
6.6. Anti-pathogen applications of selection procedures.
6.7. Aptamers working as antidotes.
6.8. Aptamers as delivery tools
7. Conclusions
Acknowledgements
References
- Mills, D.R.; Peterson, R.L.; Spiegelman, S. An extracellular Darwinian experiment with a self-duplicating nucleic acid molecule. Proc. Natl. Acad. Sci. USA 1967, 58, 217–224. [Google Scholar] [CrossRef]
- Green, R.; Ellington, A.D.; Szostak, J.W. In vitro genetic analysis of the Tetrahymena self-splicing intron. Nature 1990, 347, 406–408. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Burke, J.M.; Berzal-Herranz, A. In vitro selection and evolution of RNA: applications for catalytic RNA, molecular recognition, and drug discovery. FASEB J. 1993, 7, 106–112. [Google Scholar]
- Berzal-Herranz, A. In vitro selection of hairpin ribozymes. J. Hepatol. 1996, 25, 1002–1003. [Google Scholar] [CrossRef]
- Breaker, R.R. In Vitro Selection of Catalytic Polynucleotides. Chem. Rev. 1997, 97, 371–390. [Google Scholar]
- Wilson, D.S.; Szostak, J.W. In vitro selection of functional nucleic acids. Annu. Rev. Biochem. 1999, 68, 611–647. [Google Scholar] [CrossRef]
- Nieuwlandt, D. In vitro selection of functional nucleic acid sequences. Curr. Issues Mol. Biol. 2000, 2, 9–16. [Google Scholar]
- Dausse, E.; Cazenave, C.; Rayner, B.; Toulme, J.J. In vitro selection procedures for identifying DNA and RNA aptamers targeted to nucleic acids and proteins. Methods Mol. Biol. 2005, 288, 391–410. [Google Scholar]
- Romero-López, C.; Díaz-González, R.; Berzal-Herranz, A. RNA selection and evolution in vitro: powerful techniques for the analysis and identification of new molecular tools. Biotechnol. Biotechnol. Equip. 2007, 21, 272–282. [Google Scholar]
- Dausse, E.; Da Rocha Gomes, S.; Toulme, J.J. Aptamers: a new class of oligonucleotides in the drug discovery pipeline? Curr. Opin. Pharmacol. 2009, 9, 602–607. [Google Scholar] [CrossRef]
- Gevertz, J.; Gan, H.H.; Schlick, T. In vitro RNA random pools are not structurally diverse: a computational analysis. RNA 2005, 11, 853–863. [Google Scholar] [CrossRef]
- Jarosch, F.; Buchner, K.; Klussmann, S. In vitro selection using a dual RNA library that allows primerless selection. Nucleic Acids Res. 2006, 34, e86. [Google Scholar] [CrossRef]
- Vater, A.; Jarosch, F.; Buchner, K.; Klussmann, S. Short bioactive Spiegelmers to migraine-associated calcitonin gene-related peptide rapidly identified by a novel approach: tailored-SELEX. Nucleic Acids Res. 2003, 31, e130. [Google Scholar] [CrossRef]
- Pan, W.; Xin, P.; Clawson, G.A. Minimal primer and primer-free SELEX protocols for selection of aptamers from random DNA libraries. Biotechniques 2008, 44, 351–360. [Google Scholar]
- Berzal-Herranz, A.; Joseph, S.; Burke, J.M. In vitro selection of active hairpin ribozymes by sequential RNA-catalyzed cleavage and ligation reactions. Genes Dev. 1992, 6, 129–134. [Google Scholar]
- Chen, X.; Li, N.; Ellington, A.D. Ribozyme catalysis of metabolism in the RNA world. Chem. Biodivers. 2007, 4, 633–655. [Google Scholar] [CrossRef]
- Fitzwater, T.; Polisky, B. A SELEX primer. Methods Enzymol. 1996, 267, 275–301. [Google Scholar]
- Naimuddin, M.; Kitamura, K.; Kinoshita, Y.; Honda-Takahashi, Y.; Murakami, M.; Ito, M.; Yamamoto, K.; Hanada, K.; Husimi, Y.; Nishigaki, K. Selection-by-function: efficient enrichment of cathepsin E inhibitors from a DNA library. J. Mol. Recognit. 2007, 20, 58–68. [Google Scholar] [CrossRef]
- Wu, L.; Curran, J.F. An allosteric synthetic DNA. Nucleic Acids Res. 1999, 27, 1512–1516. [Google Scholar] [CrossRef]
- Buskirk, A.R.; Landrigan, A.; Liu, D.R. Engineering a ligand-dependent RNA transcriptional activator. Chem. Biol. 2004, 11, 1157–1163. [Google Scholar] [CrossRef]
- Buskirk, A.R.; Kehayova, P.D.; Landrigan, A.; Liu, D.R. In vivo evolution of an RNA-based transcriptional activator. Chem. Biol. 2003, 10, 533–540. [Google Scholar] [CrossRef]
- Wieland, M.; Berschneider, B.; Erlacher, M.D.; Hartig, J.S. Aptazyme-mediated regulation of 16S ribosomal RNA. Chem. Biol. 2010, 17, 236–242. [Google Scholar] [CrossRef]
- Nomura, Y.; Yokobayashi, Y. Reengineering a natural riboswitch by dual genetic selection. J. Am. Chem. Soc. 2007, 129, 13814–13815. [Google Scholar] [CrossRef]
- Chen, X.; Denison, L.; Levy, M.; Ellington, A.D. Direct selection for ribozyme cleavage activity in cells. RNA 2009, 15, 2035–2045. [Google Scholar] [CrossRef]
- Behlke, M.A. Chemical modification of siRNAs for in vivo use. Oligonucleotides 2008, 18, 305–319. [Google Scholar] [CrossRef]
- Thiel, K.W.; Giangrande, P.H. Therapeutic applications of DNA and RNA aptamers. Oligonucleotides 2009, 19, 209–222. [Google Scholar] [CrossRef]
- Singh, Y.; Murat, P.; Defrancq, E. Recent developments in oligonucleotide conjugation. Chem. Soc. Rev. 2010.
- Said Hassane, F.; Saleh, A.F.; Abes, R.; Gait, M.J.; Lebleu, B. Cell penetrating peptides: overview and applications to the delivery of oligonucleotides. Cell Mol. Life Sci. 2010, 67, 715–726. [Google Scholar] [CrossRef]
- Degols, G.; Leonetti, J.P.; Lebleu, B. Sequence-specific activity of antisense oligonucleotides conjugated to poly (L-lysine) carriers. Ann. N.Y. Acad. Sci. 1992, 660, 331–333. [Google Scholar] [CrossRef]
- Lundquist, J.J.; Toone, E.J. The cluster glycoside effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar]
- Gissot, A.; Camplo, M.; Grinstaff, M.W.; Barthelemy, P. Nucleoside, nucleotide and oligonucleotide based amphiphiles: a successful marriage of nucleic acids with lipids. Org. Biomol. Chem. 2008, 6, 1324–1333. [Google Scholar] [CrossRef]
- Proske, D.; Gilch, S.; Wopfner, F.; Schatzl, H.M.; Winnacker, E.L.; Famulok, M. Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem 2002, 3, 717–725. [Google Scholar] [CrossRef]
- White, R.R.; Roy, J.A.; Viles, K.D.; Sullenger, B.A.; Kontos, C.D. A nuclease-resistant RNA aptamer specifically inhibits angiopoietin-1-mediated Tie2 activation and function. Angiogenesis 2008, 11, 395–401. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, S.K.; Koshkin, A.A.; Rajwanshi, V.K.; Meldgaard, M.; Wengel, J. The first analogues of LNA (locked nucleic acids): phosphorothioate-LNA and 2'-thio-LNA. Bioorg. Med. Chem. Lett. 1998, 8, 2219–2222. [Google Scholar] [CrossRef]
- Bondensgaard, K.; Petersen, M.; Singh, S.K.; Rajwanshi, V.K.; Kumar, R.; Wengel, J.; Jacobsen, J.P. Structural studies of LNA:RNA duplexes by NMR: conformations and implications for RNase H activity. Chemistry 2000, 6, 2687–2695. [Google Scholar] [CrossRef]
- Petersen, M.; Nielsen, C.B.; Nielsen, K.E.; Jensen, G.A.; Bondensgaard, K.; Singh, S.K.; Rajwanshi, V.K.; Koshkin, A.A.; Dahl, B.M.; Wengel, J.; Jacobsen, J.P. The conformations of locked nucleic acids (LNA). J. Mol. Recognit. 2000, 13, 44–53. [Google Scholar] [CrossRef]
- Veedu, R.N.; Wengel, J. Locked nucleic acids: promising nucleic acid analogs for therapeutic applications. Chem. Biodivers. 2010, 7, 536–542. [Google Scholar] [CrossRef]
- Veedu, R.N.; Vester, B.; Wengel, J. In vitro incorporation of LNA nucleotides. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1207–1210. [Google Scholar] [CrossRef]
- Veedu, R.N.; Vester, B.; Wengel, J. Polymerase chain reaction and transcription using locked nucleic acid nucleotide triphosphates. J. Am. Chem. Soc. 2008, 130, 8124–8125. [Google Scholar] [CrossRef]
- Veedu, R.N.; Vester, B.; Wengel, J. Efficient enzymatic synthesis of LNA-modified DNA duplexes using KOD DNA polymerase. Org. Biomol. Chem. 2009, 7, 1404–1409. [Google Scholar] [CrossRef]
- Klussmann, S.; Nolte, A.; Bald, R.; Erdmann, V.A.; Furste, J.P. Mirror-image RNA that binds D-adenosine. Nat. Biotechnol. 1996, 14, 1112–1115. [Google Scholar] [CrossRef]
- Nolte, A.; Klussmann, S.; Bald, R.; Erdmann, V.A.; Furste, J.P. Mirror-design of L-oligonucleotide ligands binding to L-arginine. Nat. Biotechnol. 1996, 14, 1116–1119. [Google Scholar] [CrossRef]
- Wlotzka, B.; Leva, S.; Eschgfaller, B.; Burmeister, J.; Kleinjung, F.; Kaduk, C.; Muhn, P.; Hess-Stumpp, H.; Klussmann, S. In vivo properties of an anti-GnRH Spiegelmer: an example of an oligonucleotide-based therapeutic substance class. Proc. Natl. Acad. Sci. USA 2002, 99, 8898–8902. [Google Scholar]
- Helmling, S.; Maasch, C.; Eulberg, D.; Buchner, K.; Schroder, W.; Lange, C.; Vonhoff, S.; Wlotzka, B.; Tschop, M.H.; Rosewicz, S.; Klussmann, S. Inhibition of ghrelin action in vitro and in vivo by an RNA-Spiegelmer. Proc. Natl. Acad. Sci. USA 2004, 101, 13174–13179. [Google Scholar]
- Santoro, S.W.; Joyce, G.F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 4262–4266. [Google Scholar] [CrossRef]
- Doherty, E.A.; Doudna, J.A. Ribozyme structures and mechanisms. Annu. Rev. Biochem. 2000, 69, 597–615. [Google Scholar] [CrossRef]
- Schubert, S.; Furste, J.P.; Werk, D.; Grunert, H.P.; Zeichhardt, H.; Erdmann, V.A.; Kurreck, J. Gaining target access for deoxyribozymes. J. Mol. Biol. 2004, 339, 355–363. [Google Scholar] [CrossRef]
- Isaka, Y. DNAzymes as potential therapeutic molecules. Curr. Opin. Mol. Ther. 2007, 9, 132–136. [Google Scholar]
- Reyes-Darias, J.A.; Sanchez-Luque, F.J.; Berzal-Herranz, A. Inhibition of HIV-1 replication by RNA-based strategies. Curr. HIV Res. 2008, 6, 500–514. [Google Scholar] [CrossRef]
- Cullen, B.R.; Greene, W.C. Regulatory pathways governing HIV-1 replication. Cell 1989, 58, 423–426. [Google Scholar] [CrossRef]
- Marciniak, R.A.; Garcia-Blanco, M.A.; Sharp, P.A. Identification and characterization of a HeLa nuclear protein that specifically binds to the trans-activation-response (TAR) element of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1990, 87, 3624–3628. [Google Scholar] [CrossRef]
- Sullenger, B.A.; Gallardo, H.F.; Ungers, G.E.; Gilboa, E. Overexpression of TAR sequences renders cells resistant to human immunodeficiency virus replication. Cell 1990, 63, 601–608. [Google Scholar] [CrossRef]
- White, R.; Rusconi, C.; Scardino, E.; Wolberg, A.; Lawson, J.; Hoffman, M.; Sullenger, B. Generation of species cross-reactive aptamers using "toggle" SELEX. Mol. Ther. 2001, 4, 567–573. [Google Scholar] [CrossRef]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T., Jr.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar]
- Ciulla, T.A.; Rosenfeld, P.J. Antivascular endothelial growth factor therapy for neovascular age-related macular degeneration. Curr. Opin. Ophthalmol. 2009, 20, 158–165. [Google Scholar] [CrossRef]
- Diener, J.L.; Daniel Lagasse, H.A.; Duerschmied, D.; Merhi, Y.; Tanguay, J.F.; Hutabarat, R.; Gilbert, J.; Wagner, D.D.; Schaub, R. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J. Thromb. Haemost. 2009, 7, 1155–1162. [Google Scholar]
- Gilbert, J.C.; DeFeo-Fraulini, T.; Hutabarat, R.M.; Horvath, C.J.; Merlino, P.G.; Marsh, H.N.; Healy, J.M.; Boufakhreddine, S.; Holohan, T.V.; Schaub, R.G. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation 2007, 116, 2678–2686. [Google Scholar] [CrossRef]
- Dyke, C.K.; Steinhubl, S.R.; Kleiman, N.S.; Cannon, R.O.; Aberle, L.G.; Lin, M.; Myles, S.K.; Melloni, C.; Harrington, R.A.; Alexander, J.H.; Becker, R.C.; Rusconi, C.P. First-in-human experience of an antidote-controlled anticoagulant using RNA aptamer technology: a phase 1a pharmacodynamic evaluation of a drug-antidote pair for the controlled regulation of factor IXa activity. Circulation 2006, 114, 2490–2497. [Google Scholar] [CrossRef]
- Chan, M.Y.; Rusconi, C.P.; Alexander, J.H.; Tonkens, R.M.; Harrington, R.A.; Becker, R.C. A randomized, repeat-dose, pharmacodynamic and safety study of an antidote-controlled factor IXa inhibitor. J. Thromb. Haemost. 2008, 6, 789–796. [Google Scholar] [CrossRef]
- Chan, M.Y.; Cohen, M.G.; Dyke, C.K.; Myles, S.K.; Aberle, L.G.; Lin, M.; Walder, J.; Steinhubl, S.R.; Gilchrist, I.C.; Kleiman, N.S.; Vorchheimer, D.A.; Chronos, N.; Melloni, C.; Alexander, J.H.; Harrington, R.A.; Tonkens, R.M.; Becker, R.C.; Rusconi, C.P. Phase 1b randomized study of antidote-controlled modulation of factor IXa activity in patients with stable coronary artery disease. Circulation 2008, 117, 2865–2874. [Google Scholar]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef]
- Soundararajan, S.; Chen, W.; Spicer, E.K.; Courtenay-Luck, N.; Fernandes, D.J. The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res. 2008, 68, 2358–2365. [Google Scholar] [CrossRef]
- Ireson, C.R.; Kelland, L.R. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 2006, 5, 2957–2962. [Google Scholar] [CrossRef]
- Jo, N.; Mailhos, C.; Ju, M.; Cheung, E.; Bradley, J.; Nishijima, K.; Robinson, G.S.; Adamis, A.P.; Shima, D.T. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am. J. Pathol. 2006, 168, 2036–2053. [Google Scholar] [CrossRef]
- Sennino, B.; Falcon, B.L.; McCauley, D.; Le, T.; McCauley, T.; Kurz, J.C.; Haskell, A.; Epstein, D.M.; McDonald, D.M. Sequential loss of tumor vessel pericytes and endothelial cells after inhibition of platelet-derived growth factor B by selective aptamer AX102. Cancer Res. 2007, 67, 7358–7367. [Google Scholar]
- Biesecker, G.; Dihel, L.; Enney, K.; Bendele, R.A. Derivation of RNA aptamer inhibitors of human complement C5. Immunopharmacology 1999, 42, 219–230. [Google Scholar] [CrossRef]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar]
- Griffin, L.C.; Tidmarsh, G.F.; Bock, L.C.; Toole, J.J.; Leung, L.L. In vivo anticoagulant properties of a novel nucleotide-based thrombin inhibitor and demonstration of regional anticoagulation in extracorporeal circuits. Blood 1993, 81, 3271–3276. [Google Scholar]
- DeAnda, A., Jr.; Coutre, S.E.; Moon, M.R.; Vial, C.M.; Griffin, L.C.; Law, V.S.; Komeda, M.; Leung, L.L.; Miller, D.C. Pilot study of the efficacy of a thrombin inhibitor for use during cardiopulmonary bypass. Ann. Thorac. Surg. 1994, 58, 344–350. [Google Scholar] [CrossRef]
- Shukla, D.; Namperumalsamy, P.; Goldbaum, M.; Cunningham, E.T., Jr. Pegaptanib sodium for ocular vascular disease. Indian J. Ophthalmol. 2007, 55, 427–430. [Google Scholar] [CrossRef]
- Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjic, N. 2'-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998, 273, 20556–20567. [Google Scholar]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Cunningham, E.T., Jr.; Adamis, A.P.; Altaweel, M.; Aiello, L.P.; Bressler, N.M.; D'Amico, D.J.; Goldbaum, M.; Guyer, D.R.; Katz, B.; Patel, M.; Schwartz, S.D. A phase II randomized double-masked trial of pegaptanib, an anti-vascular endothelial growth factor aptamer, for diabetic macular edema. Ophthalmology 2005, 112, 1747–1757. [Google Scholar] [CrossRef]
- Lee, K.S.; Park, S.J.; Kim, S.R.; Min, K.H.; Lee, K.Y.; Choe, Y.H.; Hong, S.H.; Lee, Y.R.; Kim, J.S.; Hong, S.J.; Lee, Y.C. Inhibition of VEGF blocks TGF-beta1 production through a PI3K/Akt signalling pathway. Eur. Respir. J. 2008, 31, 523–531. [Google Scholar] [CrossRef]
- Huang, Z.; Pei, W.; Jayaseelan, S.; Shi, H.; Niu, L. RNA aptamers selected against the GluR2 glutamate receptor channel. Biochemistry 2007, 46, 12648–12655. [Google Scholar] [CrossRef]
- Huang, Z.; Pei, W.; Han, Y.; Jayaseelan, S.; Shekhtman, A.; Shi, H.; Niu, L. One RNA aptamer sequence, two structures: a collaborating pair that inhibits AMPA receptors. Nucleic Acids Res. 2009, 37, 4022–4032. [Google Scholar] [CrossRef]
- Wang, Y.; Khaing, Z.Z.; Li, N.; Hall, B.; Schmidt, C.E.; Ellington, A.D. Aptamer antagonists of myelin-derived inhibitors promote axon growth. PLoS One 2010, 5, e9726. [Google Scholar]
- Rhie, A.; Kirby, L.; Sayer, N.; Wellesley, R.; Disterer, P.; Sylvester, I.; Gill, A.; Hope, J.; James, W.; Tahiri-Alaoui, A. Characterization of 2'-fluoro-RNA aptamers that bind preferentially to disease-associated conformations of prion protein and inhibit conversion. J. Biol. Chem. 2003, 278, 39697–39705. [Google Scholar]
- Doring, G. The role of neutrophil elastase in chronic inflammation. Am. J. Respir. Crit. Care Med. 1994, 150, S114–117. [Google Scholar]
- Smith, D.; Kirschenheuter, G.P.; Charlton, J.; Guidot, D.M.; Repine, J.E. In vitro selection of RNA-based irreversible inhibitors of human neutrophil elastase. Chem. Biol. 1995, 2, 741–750. [Google Scholar] [CrossRef]
- Charlton, J.; Kirschenheuter, G.P.; Smith, D. Highly potent irreversible inhibitors of neutrophil elastase generated by selection from a randomized DNA-valine phosphonate library. Biochemistry 1997, 36, 3018–3026. [Google Scholar] [CrossRef]
- Bless, N.M.; Smith, D.; Charlton, J.; Czermak, B.J.; Schmal, H.; Friedl, H.P.; Ward, P.A. Protective effects of an aptamer inhibitor of neutrophil elastase in lung inflammatory injury. Curr. Biol. 1997, 7, 877–880. [Google Scholar] [CrossRef]
- Gounni, A.S.; Lamkhioued, B.; Ochiai, K.; Tanaka, Y.; Delaporte, E.; Capron, A.; Kinet, J.P.; Capron, M. High-affinity IgE receptor on eosinophils is involved in defence against parasites. Nature 1994, 367, 183–186. [Google Scholar]
- Sutton, B.J.; Gould, H.J. The human IgE network. Nature 1993, 366, 421–428. [Google Scholar] [CrossRef]
- Wiegand, T.W.; Williams, P.B.; Dreskin, S.C.; Jouvin, M.H.; Kinet, J.P.; Tasset, D. High-affinity oligonucleotide ligands to human IgE inhibit binding to Fc epsilon receptor I. J. Immunol. 1996, 157, 221–230. [Google Scholar]
- Cerchia, L.; Duconge, F.; Pestourie, C.; Boulay, J.; Aissouni, Y.; Gombert, K.; Tavitian, B.; de Franciscis, V.; Libri, D. Neutralizing aptamers from whole-cell SELEX inhibit the RET receptor tyrosine kinase. PLoS Biol. 2005, 3, e123. [Google Scholar] [CrossRef]
- Vento, M.T.; Iuorio, M.; Netti, P.A.; Duconge, F.; Tavitian, B.; Franciscis, V.; Cerchia, L. Distribution and bioactivity of the Ret-specific D4 aptamer in three-dimensional collagen gel cultures. Mol. Cancer Ther. 2008, 7, 3381–3388. [Google Scholar] [CrossRef]
- Chen, C.H.; Chernis, G.A.; Hoang, V.Q.; Landgraf, R. Inhibition of heregulin signaling by an aptamer that preferentially binds to the oligomeric form of human epidermal growth factor receptor-3. Proc. Natl. Acad. Sci. USA 2003, 100, 9226–9231. [Google Scholar] [CrossRef]
- Blake, C.M.; Sullenger, B.A.; Lawrence, D.A.; Fortenberry, Y.M. Antimetastatic potential of PAI-1-specific RNA aptamers. Oligonucleotides 2009, 19, 117–128. [Google Scholar] [CrossRef]
- Madsen, J.B.; Dupont, D.M.; Andersen, T.B.; Nielsen, A.F.; Sang, L.; Brix, D.M.; Jensen, J.K.; Broos, T.; Hendrickx, M.L.; Christensen, A.N.; Kjems, J.; Andreasen, P.A. RNA Aptamers as Conformational Probes and Regulatory Agents for Plasminogen Activator Inhibitor-1. Biochemistry 2010, 49, 4103–4115. [Google Scholar]
- Lebruska, L.L.; Maher, L.J., 3rd. Selection and characterization of an RNA decoy for transcription factor NF-kappa B. Biochemistry 1999, 38, 3168–3174. [Google Scholar] [CrossRef]
- Wurster, S.E.; Maher, L.J., 3rd. Selection and characterization of anti-NF-kappaB p65 RNA aptamers. RNA 2008, 14, 1037–1047. [Google Scholar] [CrossRef]
- Cassiday, L.A.; Maher, L.J., 3rd. Yeast genetic selections to optimize RNA decoys for transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 2003, 100, 3930–3935. [Google Scholar] [CrossRef]
- Wurster, S.E.; Bida, J.P.; Her, Y.F.; Maher, L.J., 3rd. Characterization of anti-NF-kappaB RNA aptamer-binding specificity in vitro and in the yeast three-hybrid system. Nucleic Acids Res. 2009, 37, 6214–6224. [Google Scholar] [CrossRef]
- Mi, J.; Zhang, X.; Rabbani, Z.N.; Liu, Y.; Su, Z.; Vujaskovic, Z.; Kontos, C.D.; Sullenger, B.A.; Clary, B.M. H1 RNA polymerase III promoter-driven expression of an RNA aptamer leads to high-level inhibition of intracellular protein activity. Nucleic Acids Res. 2006, 34, 3577–3584. [Google Scholar] [CrossRef]
- Mi, J.; Zhang, X.; Rabbani, Z.N.; Liu, Y.; Reddy, S.K.; Su, Z.; Salahuddin, F.K.; Viles, K.; Giangrande, P.H.; Dewhirst, M.W.; Sullenger, B.A.; Kontos, C.D.; Clary, B.M. RNA aptamer-targeted inhibition of NF-kappa B suppresses non-small cell lung cancer resistance to doxorubicin. Mol. Ther. 2008, 16, 66–73. [Google Scholar] [CrossRef]
- Jian, Y.; Gao, Z.; Sun, J.; Shen, Q.; Feng, F.; Jing, Y.; Yang, C. RNA aptamers interfering with nucleophosmin oligomerization induce apoptosis of cancer cells. Oncogene 2009, 28, 4201–4211. [Google Scholar] [CrossRef]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar]
- McNamara, J.O.; Kolonias, D.; Pastor, F.; Mittler, R.S.; Chen, L.; Giangrande, P.H.; Sullenger, B.; Gilboa, E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Invest. 2008, 118, 376–386. [Google Scholar] [CrossRef]
- Dollins, C.M.; Nair, S.; Boczkowski, D.; Lee, J.; Layzer, J.M.; Gilboa, E.; Sullenger, B.A. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 2008, 15, 675–682. [Google Scholar] [CrossRef]
- White, R.R.; Shan, S.; Rusconi, C.P.; Shetty, G.; Dewhirst, M.W.; Kontos, C.D.; Sullenger, B.A. Inhibition of rat corneal angiogenesis by a nuclease-resistant RNA aptamer specific for angiopoietin-2. Proc. Natl. Acad. Sci. USA 2003, 100, 5028–5033. [Google Scholar]
- Sarraf-Yazdi, S.; Mi, J.; Moeller, B.J.; Niu, X.; White, R.R.; Kontos, C.D.; Sullenger, B.A.; Dewhirst, M.W.; Clary, B.M. Inhibition of in vivo tumor angiogenesis and growth via systemic delivery of an angiopoietin 2-specific RNA aptamer. J. Surg. Res. 2008, 146, 16–23. [Google Scholar] [CrossRef]
- Nimjee, S.M.; Rusconi, C.P.; Sullenger, B.A. Aptamers: an emerging class of therapeutics. Annu. Rev. Med. 2005, 56, 555–583. [Google Scholar] [CrossRef]
- Bates, P.J.; Kahlon, J.B.; Thomas, S.D.; Trent, J.O.; Miller, D.M. Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J. Biol. Chem. 1999, 274, 26369–26377. [Google Scholar]
- Girvan, A.C.; Teng, Y.; Casson, L.K.; Thomas, S.D.; Juliger, S.; Ball, M.W.; Klein, J.B.; Pierce, W.M., Jr.; Barve, S.S.; Bates, P.J. AGRO100 inhibits activation of nuclear factor-kappaB (NF-kappaB) by forming a complex with NF-kappaB essential modulator (NEMO) and nucleolin. Mol. Cancer Ther. 2006, 5, 1790–1799. [Google Scholar] [CrossRef]
- Floege, J.; Ostendorf, T.; Janssen, U.; Burg, M.; Radeke, H.H.; Vargeese, C.; Gill, S.C.; Green, L.S.; Janjic, N. Novel approach to specific growth factor inhibition in vivo: antagonism of platelet-derived growth factor in glomerulonephritis by aptamers. Am. J. Pathol. 1999, 154, 169–179. [Google Scholar] [CrossRef]
- Hicke, B.J.; Marion, C.; Chang, Y.F.; Gould, T.; Lynott, C.K.; Parma, D.; Schmidt, P.G.; Warren, S. Tenascin-C aptamers are generated using tumor cells and purified protein. J. Biol. Chem. 2001, 276, 48644–48654. [Google Scholar]
- Lindner, V.; Giachelli, C.M.; Schwartz, S.M.; Reidy, M.A. A subpopulation of smooth muscle cells in injured rat arteries expresses platelet-derived growth factor-B chain mRNA. Circ. Res. 1995, 76, 951–957. [Google Scholar] [CrossRef]
- Iida, H.; Seifert, R.; Alpers, C.E.; Gronwald, R.G.; Phillips, P.E.; Pritzl, P.; Gordon, K.; Gown, A.M.; Ross, R.; Bowen-Pope, D.F.; Johnson, R.J. Platelet-derived growth factor (PDGF) and PDGF receptor are induced in mesangial proliferative nephritis in the rat. Proc. Natl. Acad. Sci. USA 1991, 88, 6560–6564. [Google Scholar]
- Heldin, C.H. Structural and functional studies on platelet-derived growth factor. EMBO J. 1992, 11, 4251–4259. [Google Scholar]
- Green, L.S.; Jellinek, D.; Jenison, R.; Ostman, A.; Heldin, C.H.; Janjic, N. Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry 1996, 35, 14413–14424. [Google Scholar]
- Morishita, R.; Sugimoto, T.; Aoki, M.; Kida, I.; Tomita, N.; Moriguchi, A.; Maeda, K.; Sawa, Y.; Kaneda, Y.; Higaki, J.; Ogihara, T. In vivo transfection of cis element "decoy" against nuclear factor-kappaB binding site prevents myocardial infarction. Nat. Med. 1997, 3, 894–899. [Google Scholar] [CrossRef]
- Ostendorf, T.; Kunter, U.; Grone, H.J.; Bahlmann, F.; Kawachi, H.; Shimizu, F.; Koch, K.M.; Janjic, N.; Floege, J. Specific antagonism of PDGF prevents renal scarring in experimental glomerulonephritis. J. Am. Soc. Nephrol. 2001, 12, 909–918. [Google Scholar]
- Ostendorf, T.; Kunter, U.; van Roeyen, C.; Dooley, S.; Janjic, N.; Ruckman, J.; Eitner, F.; Floege, J. The effects of platelet-derived growth factor antagonism in experimental glomerulonephritis are independent of the transforming growth factor-beta system. J. Am. Soc. Nephrol. 2002, 13, 658–667. [Google Scholar]
- Jain, R.K. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987, 47, 3039–3051. [Google Scholar]
- Jain, R.K. Delivery of molecular medicine to solid tumors. Science 1996, 271, 1079–1080. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A.; Sjoquist, M.; Buchdunger, E.; Reed, R.K.; Heldin, C.H.; Rubin, K. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res. 2001, 61, 2929–2934. [Google Scholar]
- Ni, Z.; Hui, P. Emerging pharmacologic therapies for wet age-related macular degeneration. Ophthalmologica 2009, 223, 401–410. [Google Scholar] [CrossRef]
- Hunter, T. Braking the cycle. Cell 1993, 75, 839–841. [Google Scholar] [CrossRef]
- Nevins, J.R. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 1992, 258, 424–429. [Google Scholar]
- Giangrande, P.H.; Zhang, J.; Tanner, A.; Eckhart, A.D.; Rempel, R.E.; Andrechek, E.R.; Layzer, J.M.; Keys, J.R.; Hagen, P.O.; Nevins, J.R.; Koch, W.J.; Sullenger, B.A. Distinct roles of E2F proteins in vascular smooth muscle cell proliferation and intimal hyperplasia. Proc. Natl. Acad. Sci. USA 2007, 104, 12988–12993. [Google Scholar]
- Ishizaki, J.; Nevins, J.R.; Sullenger, B.A. Inhibition of cell proliferation by an RNA ligand that selectively blocks E2F function. Nat. Med. 1996, 2, 1386–1389. [Google Scholar] [CrossRef]
- Rusconi, C.P.; Yeh, A.; Lyerly, H.K.; Lawson, J.H.; Sullenger, B.A. Blocking the initiation of coagulation by RNA aptamers to factor VIIa. Thromb. Haemost. 2000, 84, 841–848. [Google Scholar]
- Rusconi, C.P.; Scardino, E.; Layzer, J.; Pitoc, G.A.; Ortel, T.L.; Monroe, D.; Sullenger, B.A. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 2002, 419, 90–94. [Google Scholar]
- Rusconi, C.P.; Roberts, J.D.; Pitoc, G.A.; Nimjee, S.M.; White, R.R.; Quick, G., Jr.; Scardino, E.; Fay, W.P.; Sullenger, B.A. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat. Biotechnol. 2004, 22, 1423–1428. [Google Scholar] [CrossRef]
- Becker, R.C.; Povsic, T.; Cohen, M.G.; Rusconi, C.P.; Sullenger, B. Nucleic acid aptamers as antithrombotic agents: Opportunities in extracellular therapeutics. Thromb. Haemost. 2010, 103, 586–595. [Google Scholar] [CrossRef]
- Blann, A. von Willebrand factor and the endothelium in vascular disease. Br. J. Biomed. Sci. 1993, 50, 125–134. [Google Scholar]
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef]
- Sawa, Y.; Morishita, R.; Suzuki, K.; Kagisaki, K.; Kaneda, Y.; Maeda, K.; Kadoba, K.; Matsuda, H. A novel strategy for myocardial protection using in vivo transfection of cis element 'decoy' against NFkappaB binding site: evidence for a role of NFkappaB in ischemia-reperfusion injury. Circulation 1997, 96, II-280-284, discussion II-285. [Google Scholar]
- Tomita, T.; Takano, H.; Tomita, N.; Morishita, R.; Kaneko, M.; Shi, K.; Takahi, K.; Nakase, T.; Kaneda, Y.; Yoshikawa, H.; Ochi, T. Transcription factor decoy for NFkappaB inhibits cytokine and adhesion molecule expressions in synovial cells derived from rheumatoid arthritis. Rheumatology (Oxford) 2000, 39, 749–757. [Google Scholar] [CrossRef]
- Ueno, T.; Sawa, Y.; Kitagawa-Sakakida, S.; Nishimura, M.; Morishita, R.; Kaneda, Y.; Kohmura, E.; Yoshimine, T.; Matsuda, H. Nuclear factor-kappa B decoy attenuates neuronal damage after global brain ischemia: a future strategy for brain protection during circulatory arrest. J. Thorac. Cardiovasc. Surg. 2001, 122, 720–727. [Google Scholar] [CrossRef]
- Barfod, A.; Persson, T.; Lindh, J. In vitro selection of RNA aptamers against a conserved region of the Plasmodium falciparum erythrocyte membrane protein 1. Parasitol. Res. 2009, 105, 1557–1566. [Google Scholar] [CrossRef]
- Unwalla, H.J.; Li, H.; Li, S.Y.; Abad, D.; Rossi, J.J. Use of a U16 snoRNA-containing ribozyme library to identify ribozyme targets in HIV-1. Mol. Ther. 2008, 16, 1113–1119. [Google Scholar] [CrossRef]
- Sriram, B.; Banerjea, A.C. In vitro-selected RNA cleaving DNA enzymes from a combinatorial library are potent inhibitors of HIV-1 gene expression. Biochem. J. 2000, 352 Pt 3, 667–673. [Google Scholar]
- Tuerk, C.; MacDougal, S.; Gold, L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 1992, 89, 6988–6992. [Google Scholar] [CrossRef]
- Burke, D.H.; Scates, L.; Andrews, K.; Gold, L. Bent pseudoknots and novel RNA inhibitors of type 1 human immunodeficiency virus (HIV-1) reverse transcriptase. J. Mol. Biol. 1996, 264, 650–666. [Google Scholar] [CrossRef]
- Khati, M.; Schuman, M.; Ibrahim, J.; Sattentau, Q.; Gordon, S.; James, W. Neutralization of infectivity of diverse R5 clinical isolates of human immunodeficiency virus type 1 by gp120-binding 2'F-RNA aptamers. J. Virol. 2003, 77, 12692–12698. [Google Scholar] [CrossRef]
- Kanamori, H.; Yuhashi, K.; Uchiyama, Y.; Kodama, T.; Ohnishi, S. In vitro selection of RNA aptamers that bind the RNA-dependent RNA polymerase of hepatitis C virus: a possible role of GC-rich RNA motifs in NS5B binding. Virology 2009, 388, 91–102. [Google Scholar] [CrossRef]
- Jang, K.J.; Lee, N.R.; Yeo, W.S.; Jeong, Y.J.; Kim, D.E. Isolation of inhibitory RNA aptamers against severe acute respiratory syndrome (SARS) coronavirus NTPase/Helicase. Biochem. Biophys. Res. Commun. 2008, 366, 738–744. [Google Scholar] [CrossRef]
- Gopinath, S.C.; Sakamaki, Y.; Kawasaki, K.; Kumar, P.K. An efficient RNA aptamer against human influenza B virus hemagglutinin. J. Biochem. 2006, 139, 837–846. [Google Scholar] [CrossRef]
- Jing, N.; Marchand, C.; Liu, J.; Mitra, R.; Hogan, M.E.; Pommier, Y. Mechanism of inhibition of HIV-1 integrase by G-tetrad-forming oligonucleotides in vitro. J. Biol. Chem. 2000, 275, 21460–21467. [Google Scholar]
- Jing, N.; Hogan, M.E. Structure-activity of tetrad-forming oligonucleotides as a potent anti-HIV therapeutic drug. J. Biol. Chem. 1998, 273, 34992–34999. [Google Scholar] [CrossRef]
- Bellecave, P.; Cazenave, C.; Rumi, J.; Staedel, C.; Cosnefroy, O.; Andreola, M.L.; Ventura, M.; Tarrago-Litvak, L.; Astier-Gin, T. Inhibition of hepatitis C virus (HCV) RNA polymerase by DNA aptamers: mechanism of inhibition of in vitro RNA synthesis and effect on HCV-infected cells. Antimicrob. Agents Chemother. 2008, 52, 2097–2110. [Google Scholar] [CrossRef]
- Duconge, F.; Toulme, J.J. In vitro selection identifies key determinants for loop-loop interactions: RNA aptamers selective for the TAR RNA element of HIV-1. RNA 1999, 5, 1605–1614. [Google Scholar] [CrossRef]
- Kolb, G.; Reigadas, S.; Castanotto, D.; Faure, A.; Ventura, M.; Rossi, J.J.; Toulme, J.J. Endogenous expression of an anti-TAR aptamer reduces HIV-1 replication. RNA Biol. 2006, 3, 150–156. [Google Scholar] [CrossRef]
- Boucard, D.; Toulme, J.J.; Di Primo, C. Bimodal loop-loop interactions increase the affinity of RNA aptamers for HIV-1 RNA structures. Biochemistry 2006, 45, 1518–1524. [Google Scholar] [CrossRef]
- Aldaz-Carroll, L.; Tallet, B.; Dausse, E.; Yurchenko, L.; Toulme, J.J. Apical loop-internal loop interactions: a new RNA-RNA recognition motif identified through in vitro selection against RNA hairpins of the hepatitis C virus mRNA. Biochemistry 2002, 41, 5883–5893. [Google Scholar] [CrossRef]
- Kikuchi, K.; Umehara, T.; Fukuda, K.; Hwang, J.; Kuno, A.; Hasegawa, T.; Nishikawa, S. RNA aptamers targeted to domain II of hepatitis C virus IRES that bind to its apical loop region. J. Biochem. 2003, 133, 263–270. [Google Scholar] [CrossRef]
- Kikuchi, K.; Umehara, T.; Fukuda, K.; Kuno, A.; Hasegawa, T.; Nishikawa, S. A hepatitis C virus (HCV) internal ribosome entry site (IRES) domain III-IV-targeted aptamer inhibits translation by binding to an apical loop of domain IIId. Nucleic Acids Res. 2005, 33, 683–692. [Google Scholar] [CrossRef]
- Kikuchi, K.; Umehara, T.; Nishikawa, F.; Fukuda, K.; Hasegawa, T.; Nishikawa, S. Increased inhibitory ability of conjugated RNA aptamers against the HCV IRES. Biochem. Biophys. Res. Commun. 2009, 386, 118–123. [Google Scholar] [CrossRef]
- Konno, K.; Fujita, S.; Iizuka, M.; Nishikawa, S.; Hasegawa, T.; Fukuda, K. Isolation and characterization of RNA aptamers specific for the HCV minus-IRES domain I. Nucleic Acids Symp Ser (Oxf) 2008, 52, 493–494. [Google Scholar] [CrossRef]
- Romero-Lopez, C.; Barroso-delJesus, A.; Puerta-Fernandez, E.; Berzal-Herranz, A. Interfering with hepatitis C virus IRES activity using RNA molecules identified by a novel in vitro selection method. Biol. Chem. 2005, 386, 183–190. [Google Scholar]
- Romero-Lopez, C.; Diaz-Gonzalez, R.; Berzal-Herranz, A. Inhibition of hepatitis C virus internal ribosome entry site-mediated translation by an RNA targeting the conserved IIIf domain. Cell Mol. Life Sci. 2007, 64, 2994–3006. [Google Scholar] [CrossRef]
- Romero-Lopez, C.; Diaz-Gonzalez, R.; Barroso-delJesus, A.; Berzal-Herranz, A. Inhibition of hepatitis C virus replication and internal ribosome entry site-dependent translation by an RNA molecule. J. Gen. Virol. 2009, 90, 1659–1669. [Google Scholar] [CrossRef]
- Fan, S.; Wu, F.; Martiniuk, F.; Hale, M.L.; Ellington, A.D.; Tchou-Wong, K.M. Protective effects of anti-ricin A-chain RNA aptamer against ricin toxicity. World J. Gastroenterol. 2008, 14, 6360–6365. [Google Scholar] [CrossRef]
- Ulrich, H.; Ippolito, J.E.; Pagan, O.R.; Eterovic, V.A.; Hann, R.M.; Shi, H.; Lis, J.T.; Eldefrawi, M.E.; Hess, G.P. In vitro selection of RNA molecules that displace cocaine from the membrane-bound nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 1998, 95, 14051–14056. [Google Scholar]
- Hess, G.P.; Ulrich, H.; Breitinger, H.G.; Niu, L.; Gameiro, A.M.; Grewer, C.; Srivastava, S.; Ippolito, J.E.; Lee, S.M.; Jayaraman, V.; Coombs, S.E. Mechanism-based discovery of ligands that counteract inhibition of the nicotinic acetylcholine receptor by cocaine and MK-801. Proc. Natl. Acad. Sci. USA 2000, 97, 13895–13900. [Google Scholar]
- Sivaprakasam, K.; Pagan, O.R.; Hess, G.P. Minimal RNA aptamer sequences that can inhibit or alleviate noncompetitive inhibition of the muscle-type nicotinic acetylcholine receptor. J. Membr. Biol. 2010, 233, 1–12. [Google Scholar] [CrossRef]
- Daniels, D.A.; Chen, H.; Hicke, B.J.; Swiderek, K.M.; Gold, L. A tenascin-C aptamer identified by tumor cell SELEX: systematic evolution of ligands by exponential enrichment. Proc. Natl. Acad Sci. USA 2003, 100, 15416–15421. [Google Scholar]
- Hicke, B.J.; Stephens, A.W.; Gould, T.; Chang, Y.F.; Lynott, C.K.; Heil, J.; Borkowski, S.; Hilger, C.S.; Cook, G.; Warren, S.; Schmidt, P.G. Tumor targeting by an aptamer. J. Nucl. Med. 2006, 47, 668–678. [Google Scholar]
- Lupold, S.E.; Hicke, B.J.; Lin, Y.; Coffey, D.S. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002, 62, 4029–4033. [Google Scholar]
- McNamara, J.O., 2nd; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef]
- Wullner, U.; Neef, I.; Eller, A.; Kleines, M.; Tur, M.K.; Barth, S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr. Cancer Drug Targets 2008, 8, 554–565. [Google Scholar] [CrossRef]
- Bagalkot, V.; Farokhzad, O.C.; Langer, R.; Jon, S. An aptamer-doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew. Chem. Int. Ed. Engl. 2006, 45, 8149–8152. [Google Scholar] [CrossRef]
- Chu, T.C.; Marks, J.W., 3rd; Lavery, L.A.; Faulkner, S.; Rosenblum, M.G.; Ellington, A.D.; Levy, M. Aptamer:toxin conjugates that specifically target prostate tumor cells. Cancer Res. 2006, 66, 5989–5992. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhzad, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 17356–17361. [Google Scholar]
- Zhou, J.; Li, H.; Li, S.; Zaia, J.; Rossi, J.J. Novel dual inhibitory function aptamer-siRNA delivery system for HIV-1 therapy. Mol. Ther. 2008, 16, 1481–1489. [Google Scholar] [CrossRef]
- Zhou, J.; Swiderski, P.; Li, H.; Zhang, J.; Neff, C.P.; Akkina, R.; Rossi, J.J. Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res. 2009, 37, 3094–3109. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Marton, S.; Reyes-Darias, J.A.; Sánchez-Luque, F.J.; Romero-López, C.; Berzal-Herranz, A. In Vitro and Ex Vivo Selection Procedures for Identifying Potentially Therapeutic DNA and RNA Molecules. Molecules 2010, 15, 4610-4638. https://doi.org/10.3390/molecules15074610
Marton S, Reyes-Darias JA, Sánchez-Luque FJ, Romero-López C, Berzal-Herranz A. In Vitro and Ex Vivo Selection Procedures for Identifying Potentially Therapeutic DNA and RNA Molecules. Molecules. 2010; 15(7):4610-4638. https://doi.org/10.3390/molecules15074610
Chicago/Turabian StyleMarton, Soledad, José A. Reyes-Darias, Francisco J. Sánchez-Luque, Cristina Romero-López, and Alfredo Berzal-Herranz. 2010. "In Vitro and Ex Vivo Selection Procedures for Identifying Potentially Therapeutic DNA and RNA Molecules" Molecules 15, no. 7: 4610-4638. https://doi.org/10.3390/molecules15074610
APA StyleMarton, S., Reyes-Darias, J. A., Sánchez-Luque, F. J., Romero-López, C., & Berzal-Herranz, A. (2010). In Vitro and Ex Vivo Selection Procedures for Identifying Potentially Therapeutic DNA and RNA Molecules. Molecules, 15(7), 4610-4638. https://doi.org/10.3390/molecules15074610