Synthesis of 2-(2-Aminopyrimidine)-2,2-difluoroethanols as Potential Bioisosters of Salicylidene Acylhydrazides

Abstract
:1. Introduction
2. Results and Discussion
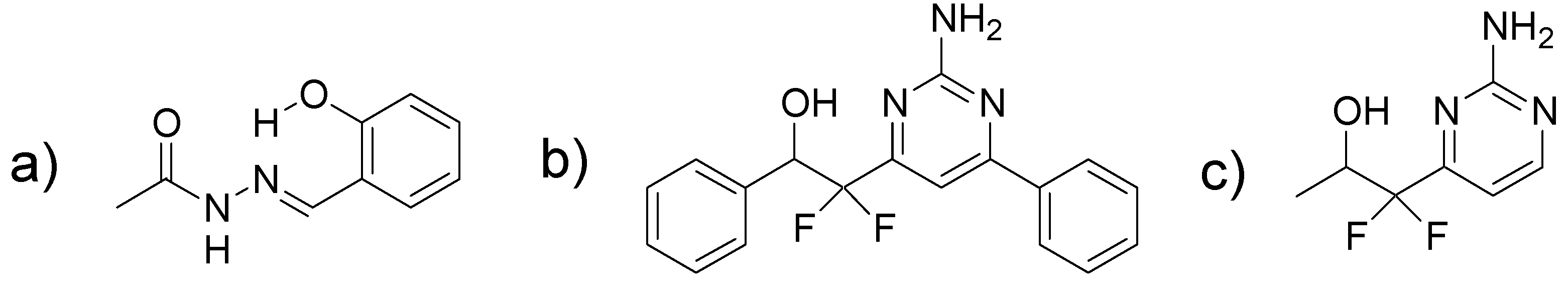
2.1. Scaffold Hopping from a Salicylidene Acylhydrazide
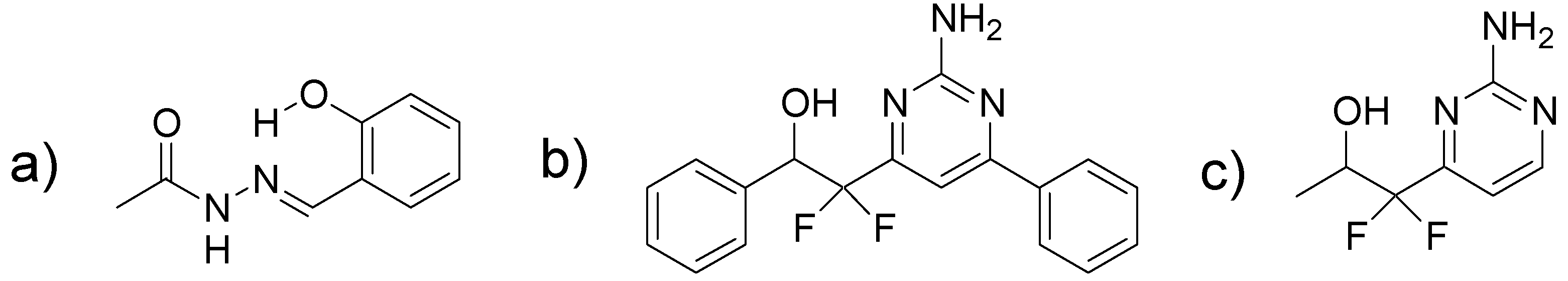
2.2. Synthesis of 2-(2-Amino-pyrimidine)-2,2-difluoro-ethanols
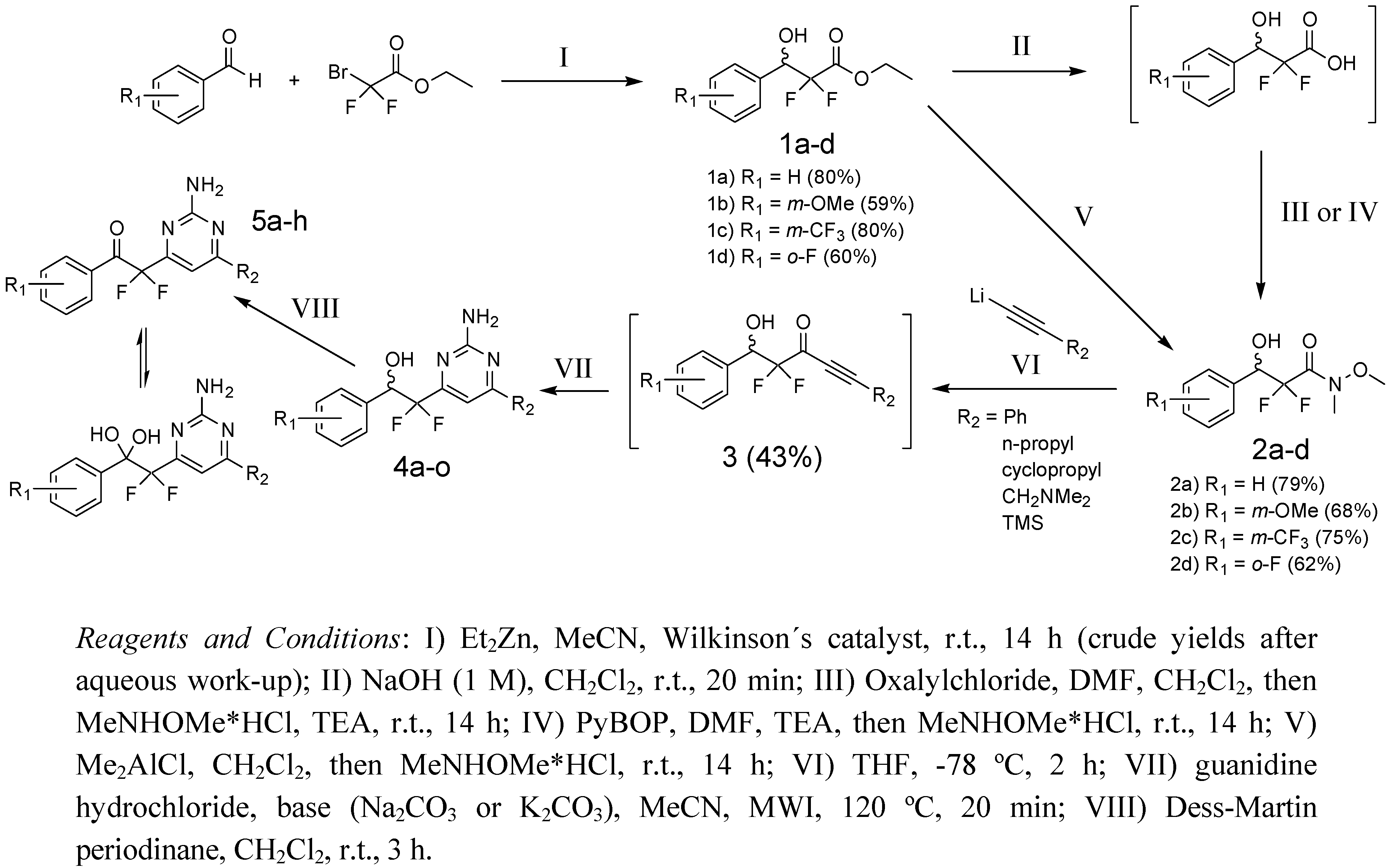

{kind=link}
{kind=link}
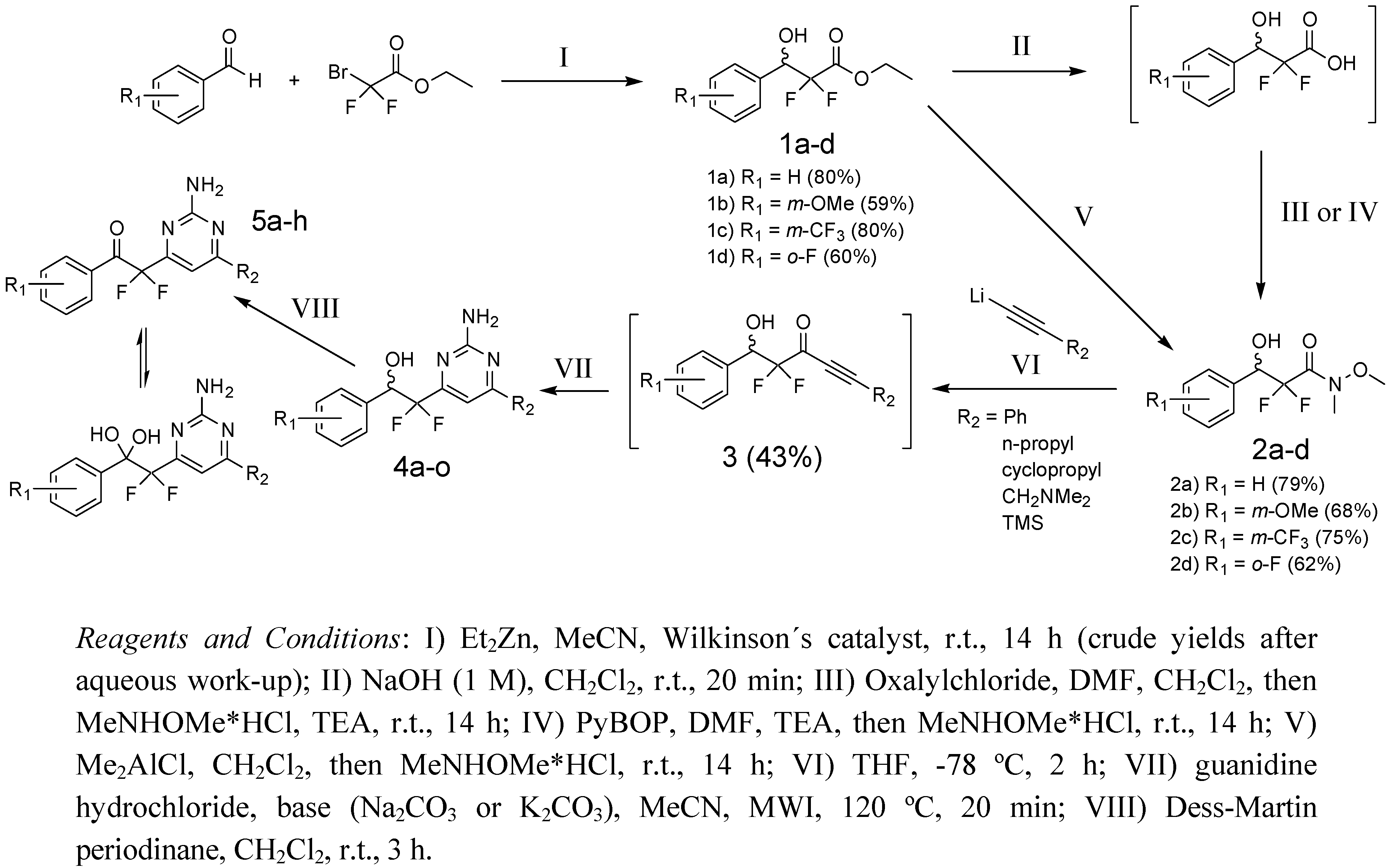{kind=link}
| ID | R1 | R2 | Yield† |
|---|---|---|---|
| 4a | H | Ph | 26% |
| 4b | H | n-Propyl | 19% |
| 4c | H | Cyclopropyl | 11% |
| 4d | H | CH2NME2 | <1% |
| 4e | H | H | 8% |
| 4f | m-OMe | Ph | 25% |
| 4g | m-OMe | n-Propyl | 12% |
| 4h | m-OMe | Cyclopropyl | 11% |
| 4i | m-CF3 | Ph | 42% |
| 4j | m-CF3 | n-Propyl | 19% |
| 4k | m-CF3 | Cyclopropyl | 24% |
| 4l | m-CF3 | H | 28% |
| 4m | o-F | Ph | 9% |
| 4n | o-F | n-Propyl | 3% |
| 4o | o-F | H | 12% |
| ID | R1 | R2 | Yield† |
|---|---|---|---|
| 5a | H | Ph | 98% |
| 5b | H | n-Propyl | 73% |
| 5c | H | Cyclopropyl | 68% |
| 5d | H | H | 27% |
| 5e | m-CF3 | Ph | 54% |
| 5f | m-CF3 | n-Propyl | 55% |
| 5g | m-CF3 | Cyclopropyl | 38% |
| 5h | m-CF3 | H | 66% |
3. Experimental
3.1. General
3.2. Scaffold Hopping
3.3. Synthesis
3.3.1. General procedure for the synthesis of Reformatsky adducts 1a-d
3.3.2. Synthesis of Weinreb amide 2a via acid chloride formation in situ
3.3.3. Typical procedure for the synthesis of Weinreb amides 2a and 2c, using PyBOP, exemplified by the synthesis of 2c
3.3.4. Typical procedure for the synthesis of Weinreb amides 2a, 2b, and 2d, using dimethyl aluminium chloride, exemplified with the synthesis of 2d
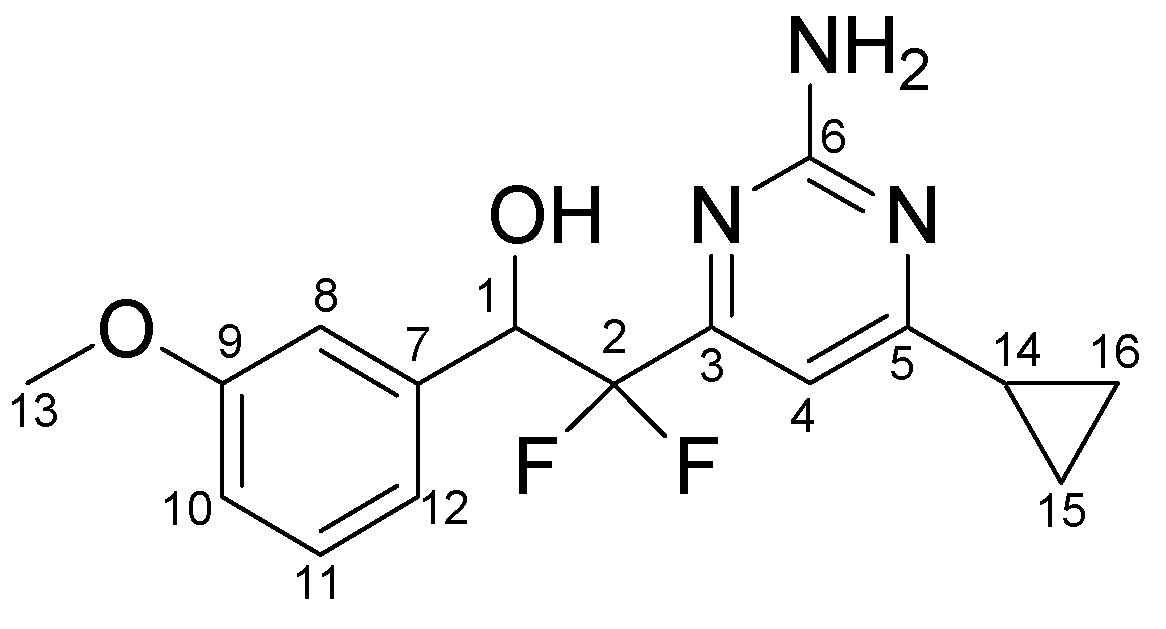
3.3.5. Typical procedure for the synthesis of pyrimidines 4a-o, exemplified by the synthesis of 4j
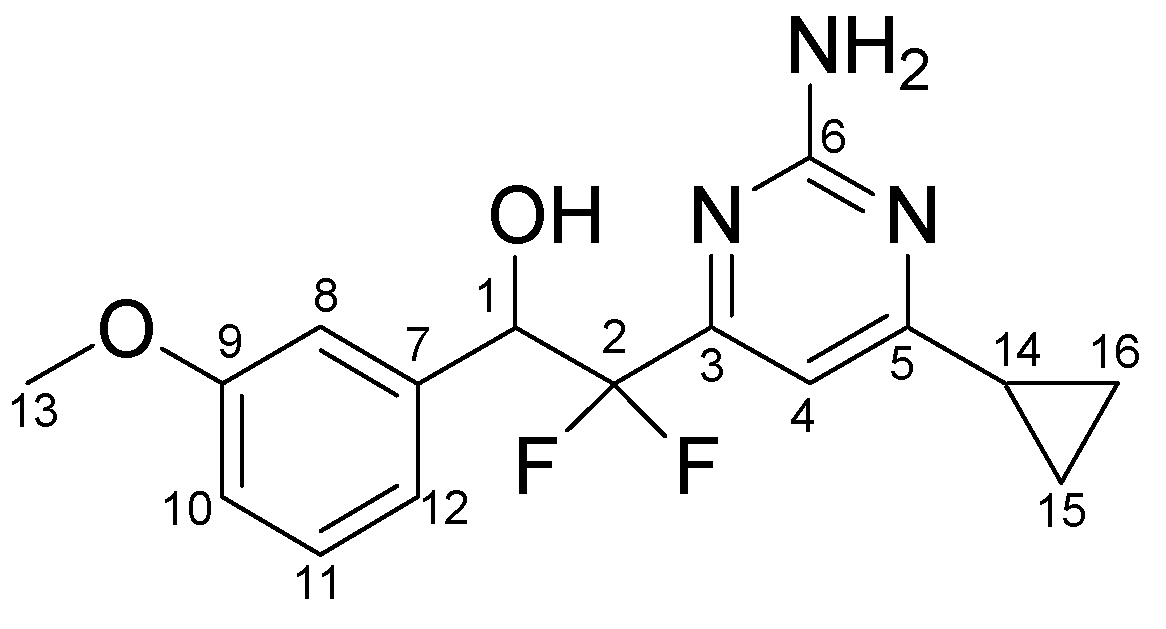
3.3.6. Typical procedure for the synthesis of ketones 5a-h, exemplified by the synthesis of 5f
4. Conclusions
Acknowledgements
References
- Hueck, C.J. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 1998, 62, 379–433. [Google Scholar]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Baron, C. Antivirulence drugs to target bacterial secretion systems. Curr. Opin. Microbiol. 2010, 13, 100–105. [Google Scholar] [CrossRef]
- Kauppi, A.M.; Nordfelth, R.; Uvell, H.; Wolf-Watz, H.; Elofsson, M. Targeting bacterial virulence: Inhibitors of type III secretion in Yersinia. Chem. Biol. 2003, 10, 241–249. [Google Scholar] [CrossRef]
- Nordfelth, R.; Kauppi, A.M.; Norberg, H.A.; Wolf-Watz, H.; Elofsson, M. Small-molecule inhibitors specifically targeting type III secretion. Infect. Immun. 2005, 73, 3104–3114. [Google Scholar] [CrossRef]
- Muschiol, S.; Bailey, L.; Gylfe, A.; Sundin, C.; Hultenby, K.; Bergstrom, S.; Elofsson, M.; Wolf-Watz, H.; Normark, S.; Henriques-Normark, B. A small-molecule inhibitor of type III secretion inhibits different stages of the infectious cycle of Chlamydia trachomatis. Proc. Natl. Acad. Sci. USA 2006, 103, 14566–14571. [Google Scholar]
- Wolf, K.; Betts, H.J.; Chellas-Gery, B.; Hower, S.; Linton, C.N.; Fields, K.A. Treatment of Chlamydia trachomatis with a small molecule inhibitor of the Yersinia type III secretion system disrupts progression of the chlamydial developmental cycle. Mol. Microbiol. 2006, 61, 1543–1555. [Google Scholar] [CrossRef]
- Bailey, L.; Gylfe, A.; Sundin, C.; Muschiol, S.; Elofsson, M.; Nordstrom, P.; Henriques-Normark, B.; Lugert, R.; Waldenstrom, A.; Wolf-Watz, H.; Bergstrom, S. Small molecule inhibitors of type III secretion in Yersinia block the Chlamydia pneumoniae infection cycle. FEBS Lett. 2007, 581, 587–595. [Google Scholar]
- Slepenkin, A.; Enquist, P.A.; Hagglund, U.; de la Maza, L.M.; Elofsson, M.; Peterson, E.M. Reversal of the antichlaraydial activity of putative type III secretion inhibitors by iron. Infect. Immun. 2007, 75, 3478–3489. [Google Scholar] [CrossRef]
- Negrea, A.; Bjur, E.; Ygberg, S.E.; Elofsson, M.; Wolf-Watz, H.; Rhen, M. Salicylidene acylhydrazides that affect type III protein secretion in Salmonella enterica serovar Typhimurium. Antimicrob. Agents Chemother. 2007, 51, 2867–2876. [Google Scholar] [CrossRef]
- Hudson, D.L.; Layton, A.N.; Field, T.R.; Bowen, A.J.; Wolf-Watz, H.; Elofsson, M.; Stevens, M.P.; Galyov, E.E. Inhibition of type III secretion in Salmonella enterica serovar typhimurium by small-molecule inhibitors. Antimicrob. Agents. Chemother. 2007, 51, 2631–2635. [Google Scholar]
- Veenendaal, A.K.J.; Sundin, C.; Blocker, A.J. Small-molecule type III secretion system inhibitors block assembly of the shigella type III secreton. J. Bacteriol. 2009, 191, 563–570. [Google Scholar] [CrossRef]
- Tree, J.J.; Wang, D.; McInally, C.; Mahajan, A.; Layton, A.; Houghton, I.; Elofsson, M.; Stevens, M.P.; Gally, D.L.; Roe, A.J. Characterization of the effects of salicylidene acylhydrazide compounds on type III secretion in Escherichia coli O157:H7. Infect. Immun. 2009, 77, 4209–4220. [Google Scholar] [CrossRef]
- Dahlgren, M.K.; Zetterström, C.E.; Gylfe, A.; Linusson, A.; Elofsson, M. Statistical molecular design of a focused salicylidene acylhydrazide library and multivariate QSAR of inhibition of type III secretion in the Gram-negative bacterium Yersinia. Bioorg. Med. Chem. 2010, 18, 2686–2703. [Google Scholar]
- Manvar, A.; Malde, A.; Verma, J.; Virsodia, V.; Mishra, A.; Upadhyay, K.; Acharya, H.; Coutinho, E.; Shah, A. Synthesis, anti-tubercular activity and 3D-QSAR study of coumarin-4-acetic acid benzylidene hydrazides. Eur. J. Med. Chem. 2008, 43, 2395–2403. [Google Scholar] [CrossRef]
- Yang, H.Y.; Shen, Y.; Chen, J.H.; Jiang, Q.F.; Leng, Y.; Shen, J.H. Structure-based virtual screening for identification of novel 11 beta-HSD1 inhibitors. Eur. J. Med. Chem. 2009, 44, 1167–1171. [Google Scholar] [CrossRef]
- Liu, W.Y.; Li, H.Y.; Zhao, B.X.; Shin, D.S.; Lian, S.; Miao, J.Y. Synthesis of novel ribavirin hydrazone derivatives and anti-proliferative activity against A549 lung cancer cells. Carbohydr. Res. 2009, 344, 1270–1275. [Google Scholar] [CrossRef]
- Vicini, P.; Incerti, M.; La Colla, P.; Loddo, R. Anti-HIV evaluation of benzo[d]isothiazole hydrazones. Eur. J. Med. Chem. 2009, 44, 1801–1807. [Google Scholar] [CrossRef]
- Mircus, G.; Hagag, S.; Levdansky, E.; Sharon, H.; Shadkchan, Y.; Shalit, I.; Osherov, N. Identification of novel cell wall destabilizing antifungal compounds using a conditional Aspergillus nidulans protein kinase C mutant. J. Antimicrob. Chemother. 2009, 64, 755–763. [Google Scholar] [CrossRef]
- Romeiro, N.C.; Aguirre, G.; Hernandez, P.; Gonzalez, M.; Cerecetto, H.; Aldana, I.; Perez-Silanes, S.; Monge, A.; Barreiro, E.J.; Lima, L.M. Synthesis, trypanocidal activity and docking studies of novel quinoxaline-N-acylhydrazones, designed as cruzain inhibitors candidates. Bioorg. Med. Chem. 2009, 17, 641–652. [Google Scholar]
- Cosconati, S.; Marinelli, L.; Trotta, R.; Virno, A.; Mayol, L.; Novellino, E.; Olson, A.J.; Randazzo, A. Tandem application of virtual screening and NMR experiments in the discovery of brand new DNA quadruplex groove binders. J. Am. Chem. Soc. 2009, 131, 16336–16337. [Google Scholar]
- He, L.Y.; Zhang, L.; Liu, X.F.; Li, X.H.; Zheng, M.Y.; Li, H.L.; Yu, K.Q.; Chen, K.X.; Shen, X.; Jiang, H.L.; Liu, H. Discovering potent inhibitors against the beta-hydroxyacyl-acyl carrier protein dehydratase (FabZ) of Helicobacter pylori: Structure-based design, synthesis, bioassay, and crystal structure determination. J. Med. Chem. 2009, 52, 2465–2481. [Google Scholar]
- Patkar, C.G.; Larsen, M.; Owston, M.; Smith, J.L.; Kuhn, R.J. Identification of inhibitors of Yellow fever virus replication using a replicon-based high-throughput assay. Antimicrob. Agents Chemother. 2009, 53, 4103–4114. [Google Scholar]
- Marlo, J.E.; Niswender, C.M.; Days, E.L.; Bridges, T.M.; Xiang, Y.; Rodriguez, A.L.; Shirey, J.K.; Brady, A.E.; Nalywajko, T.; Luo, Q.; Austin, C.A.; Williams, M.B.; Kim, K.; Williams, R.; Orton, D.; Brown, H.A.; Lindsley, C.W.; Weaver, C.D.; Conn, P.J. Discovery and characterization of novel allosteric potentiators of M-1 muscarinic receptors reveals multiple modes of activity. Mol. Pharmacol. 2009, 75, 577–588. [Google Scholar] [CrossRef]
- Patel, V.; Mazitschek, R.; Coleman, B.; Nguyen, C.; Urgaonkar, S.; Cortese, J.; Barker, R.H.; Greenberg, E.; Tang, W.P.; Bradner, J.E.; Schreiber, S.L.; Duraisingh, M.T.; Wirth, D.F.; Clardy, J. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. J. Med. Chem. 2009, 52, 2185–2187. [Google Scholar]
- Peterson, Q.P.; Hsu, D.C.; Goode, D.R.; Novotny, C.J.; Totten, R.K.; Hergenrother, P.J. Procaspase-3 activation as an anti-cancer strategy: Structure-activity relationship of procaspase-activating ompound 1 (PAC-1) and its cellular co-localization with caspase-3. J. Med. Chem. 2009, 52, 5721–5731. [Google Scholar] [CrossRef]
- MacroModel, version 9.5, Schrödinger LLC: 120 West 45th Street, Tower 45, New York, NY, USA; 10036–4041.
- SHOP, version 1.0. Molecular Discovery Ltd.: 215 Marsh Road, HA5 5NE Pinner, Middlesex, U.K.
- Bagley, M.C.; Hughes, D.D.; Lubinu, M.C.; Merritt, E.A.; Taylor, P.H.; Tomkinson, N.C.O. Microwave-assisted synthesis of pyrimidine libraries. Qsar Comb. Sci. 2004, 23, 859–867. [Google Scholar] [CrossRef]
- Iseki, K.; Asada, D.; Kuroki, Y. Preparation of optically active alpha, alpha-difluoro-beta-hydroxyketones. J. Fluorine Chem. 1999, 97, 85–89. [Google Scholar] [CrossRef]
- Sato, K.; Tarui, A.; Kita, T.; Ishida, Y.; Tamura, H.; Omote, M.; Ando, A.; Kumadaki, I. Rhodium-catalyzed Reformatsky-type reaction of ethyl bromodifluoroacetate. Tet. Lett. 2004, 45, 5735–5737. [Google Scholar] [CrossRef]
- Bergmann, R.; Liljefors, T.; Sorensen, M.D.; Zamora, I. SHOP: Receptor-Based Scaffold HOPping by GRID-Based Similarity Searches. J. Chem. Info. Model. 2009, 49, 658–669. [Google Scholar] [CrossRef]
- Molecular Operating Environment, (MOE), version 2008.10, Chemical Computing Group Inc., Suite 910 - 1010 Sherbrooke St. W: Montreal, Quebec, Canada, H3A 2R7.
- Sample Availability: Please contact the corresponding author.
© 2010 by the authors;
Share and Cite
Dahlgren, M.K.; Öberg, C.T.; Wallin, E.A.; Janson, P.G.; Elofsson, M. Synthesis of 2-(2-Aminopyrimidine)-2,2-difluoroethanols as Potential Bioisosters of Salicylidene Acylhydrazides. Molecules 2010, 15, 4423-4438. https://doi.org/10.3390/molecules15064423
Dahlgren MK, Öberg CT, Wallin EA, Janson PG, Elofsson M. Synthesis of 2-(2-Aminopyrimidine)-2,2-difluoroethanols as Potential Bioisosters of Salicylidene Acylhydrazides. Molecules. 2010; 15(6):4423-4438. https://doi.org/10.3390/molecules15064423
Chicago/Turabian StyleDahlgren, Markus K., Christopher T. Öberg, Erika A. Wallin, Pär G. Janson, and Mikael Elofsson. 2010. "Synthesis of 2-(2-Aminopyrimidine)-2,2-difluoroethanols as Potential Bioisosters of Salicylidene Acylhydrazides" Molecules 15, no. 6: 4423-4438. https://doi.org/10.3390/molecules15064423
APA StyleDahlgren, M. K., Öberg, C. T., Wallin, E. A., Janson, P. G., & Elofsson, M. (2010). Synthesis of 2-(2-Aminopyrimidine)-2,2-difluoroethanols as Potential Bioisosters of Salicylidene Acylhydrazides. Molecules, 15(6), 4423-4438. https://doi.org/10.3390/molecules15064423