Quantum Chemical Calculations on the Interaction between Flavonol and Functional Monomers (Methacrylic Acid and 4-Vinylpyridine) in Molecularly Imprinted Polymers

Abstract
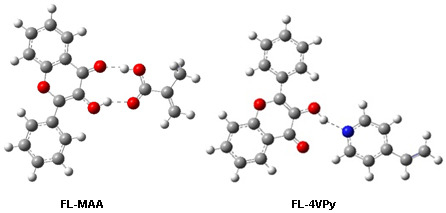
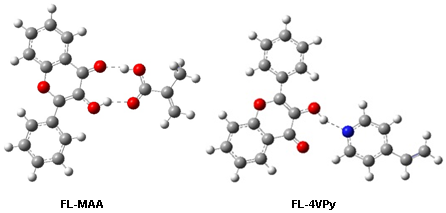
:
1. Introduction
2. Results and Discussion
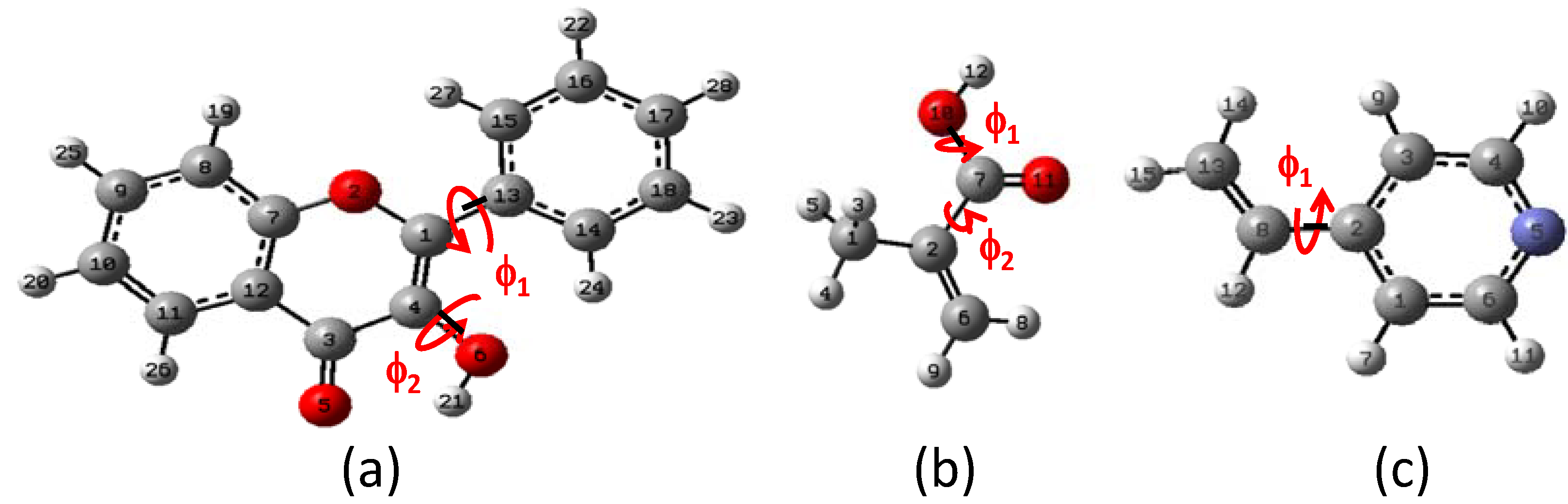
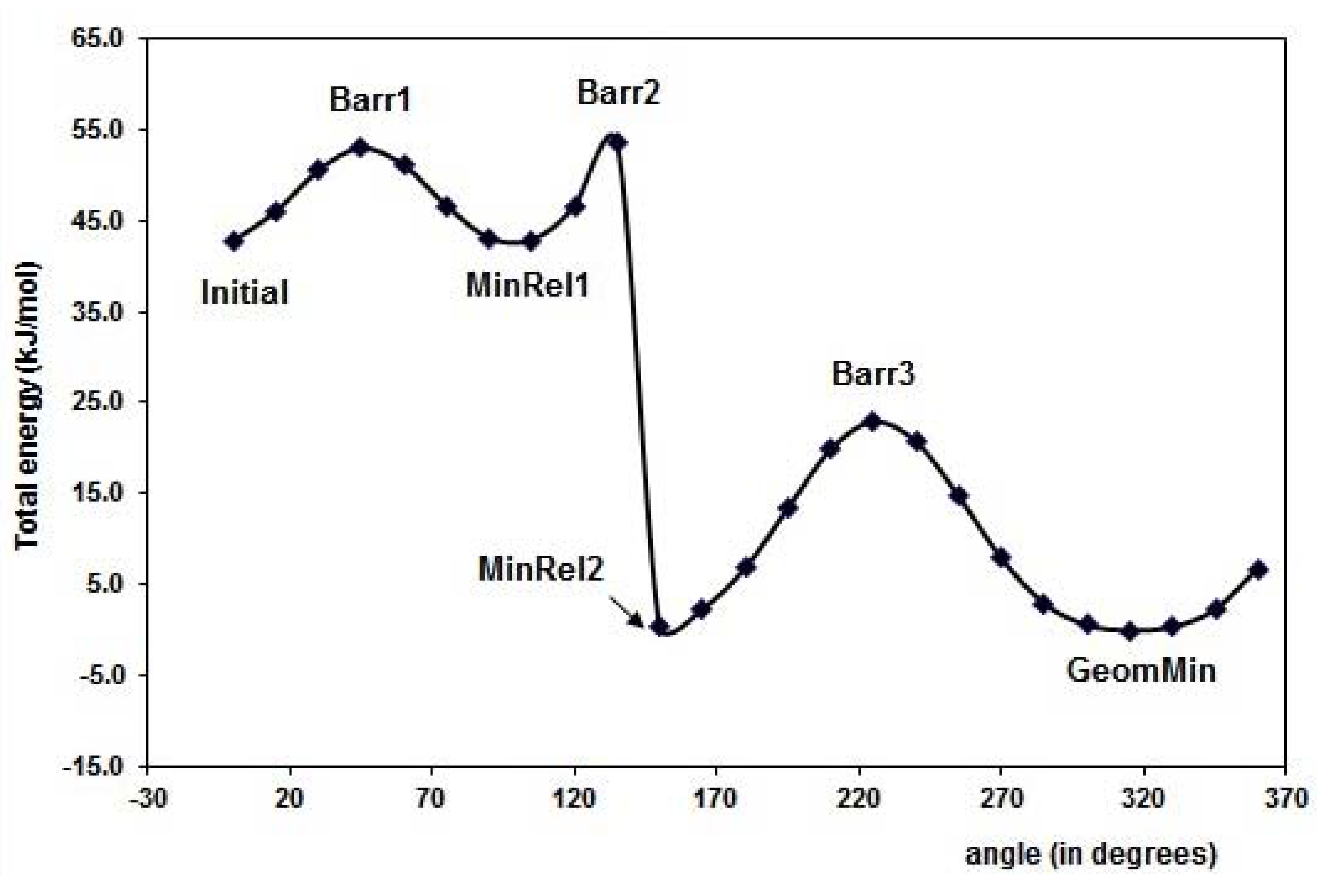
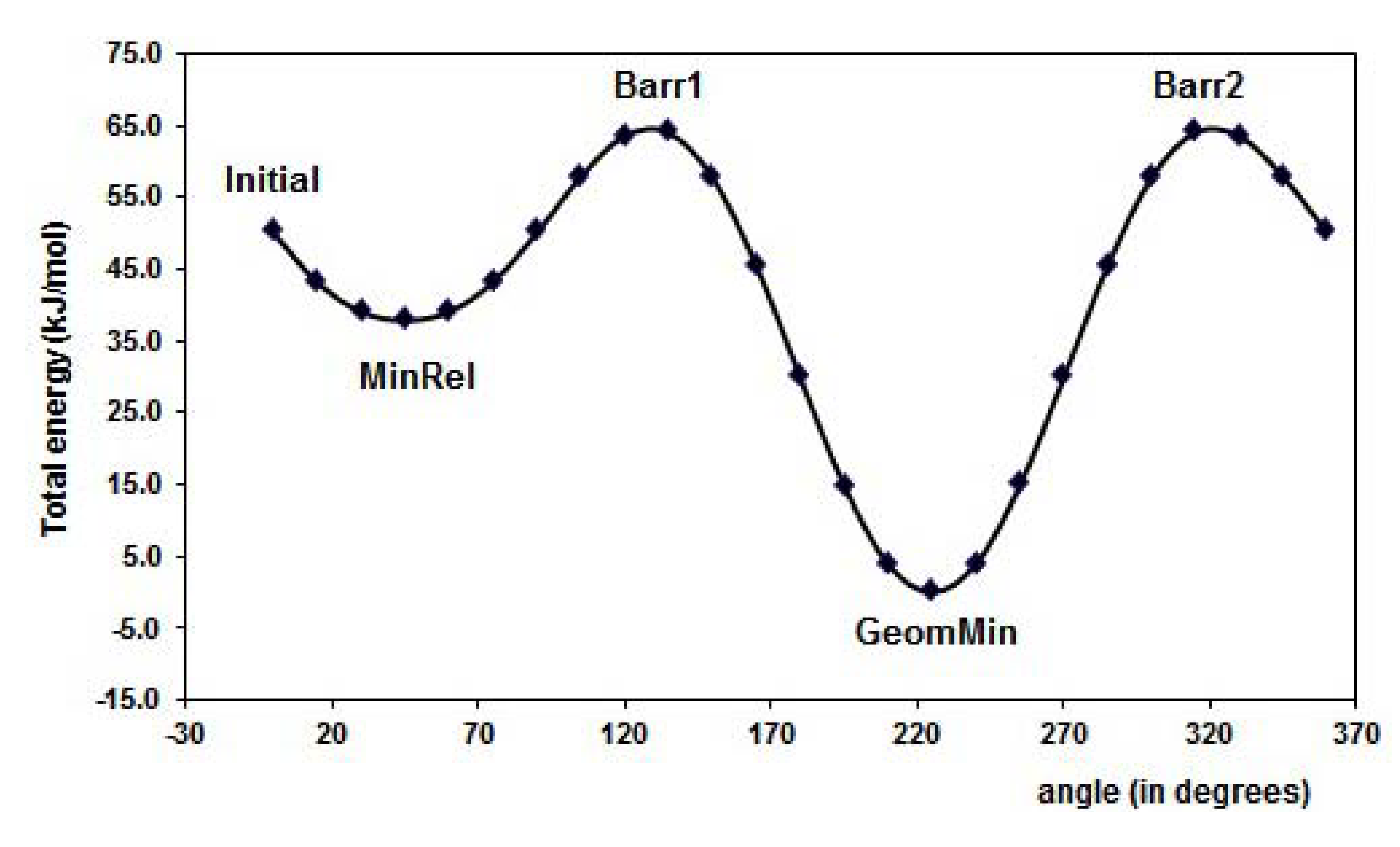
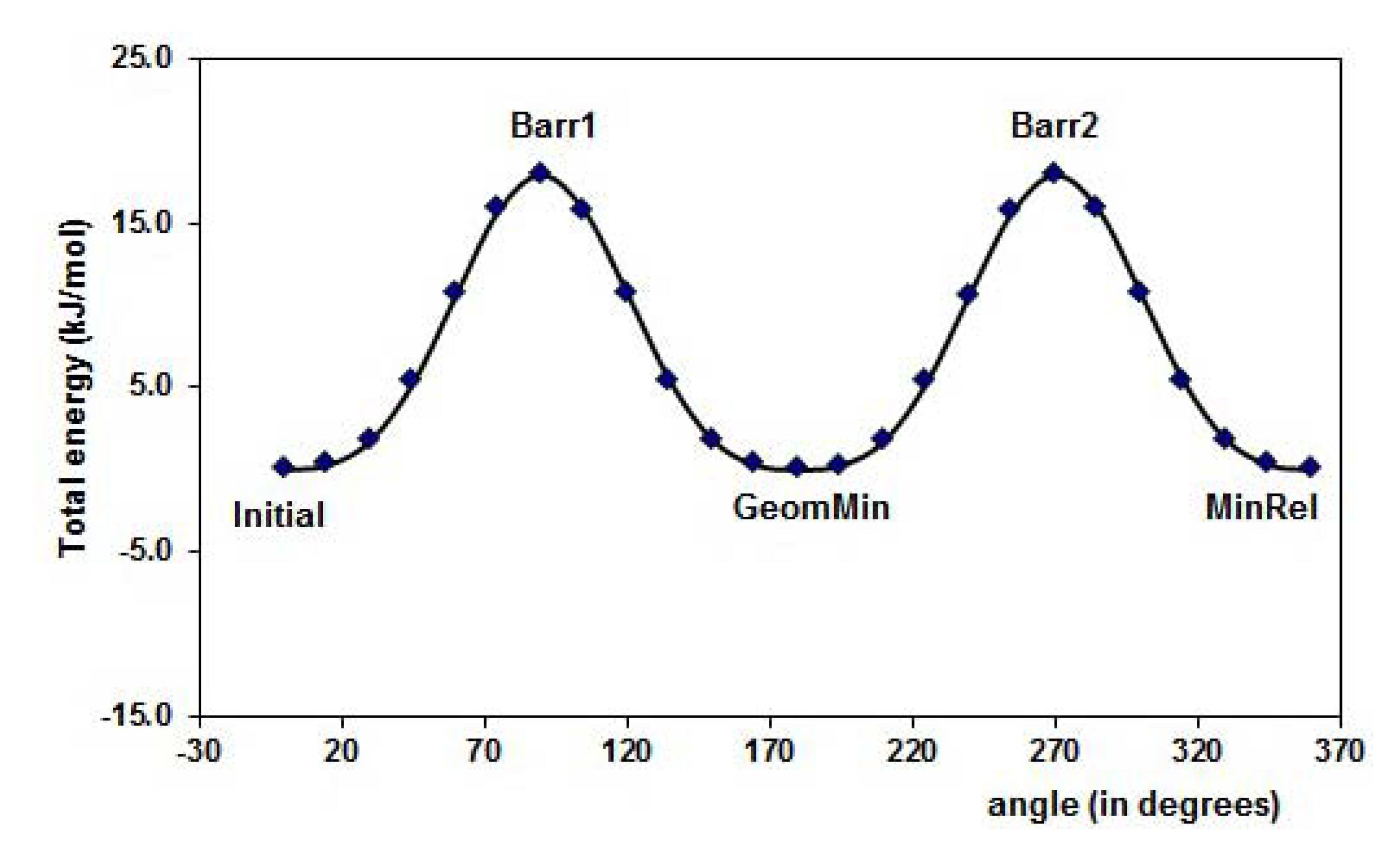
2.1. Conformational analysis
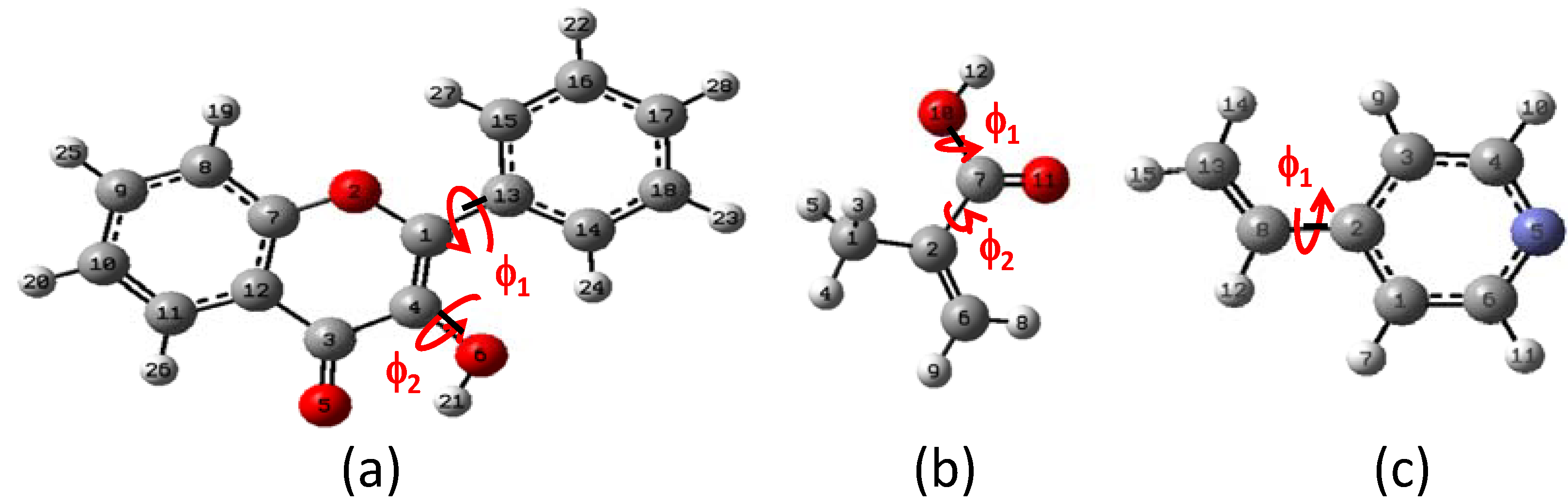


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | FL´s Conformation | MAA´s Conformation | 4VPy Conformation |
|---|---|---|---|
| Initial | 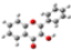 |  | 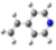 |
| Barr1 | 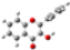 |  |  |
| MinRel1 | 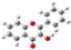 |  | 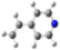 |
| Barr2 | 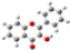 | 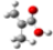 | 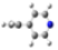 |
| MinRel2 |  | ------- | ------- |
| Barr3 |  | -------- | ------- |
| GeomMin | 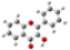 | 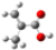 | 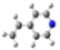 |
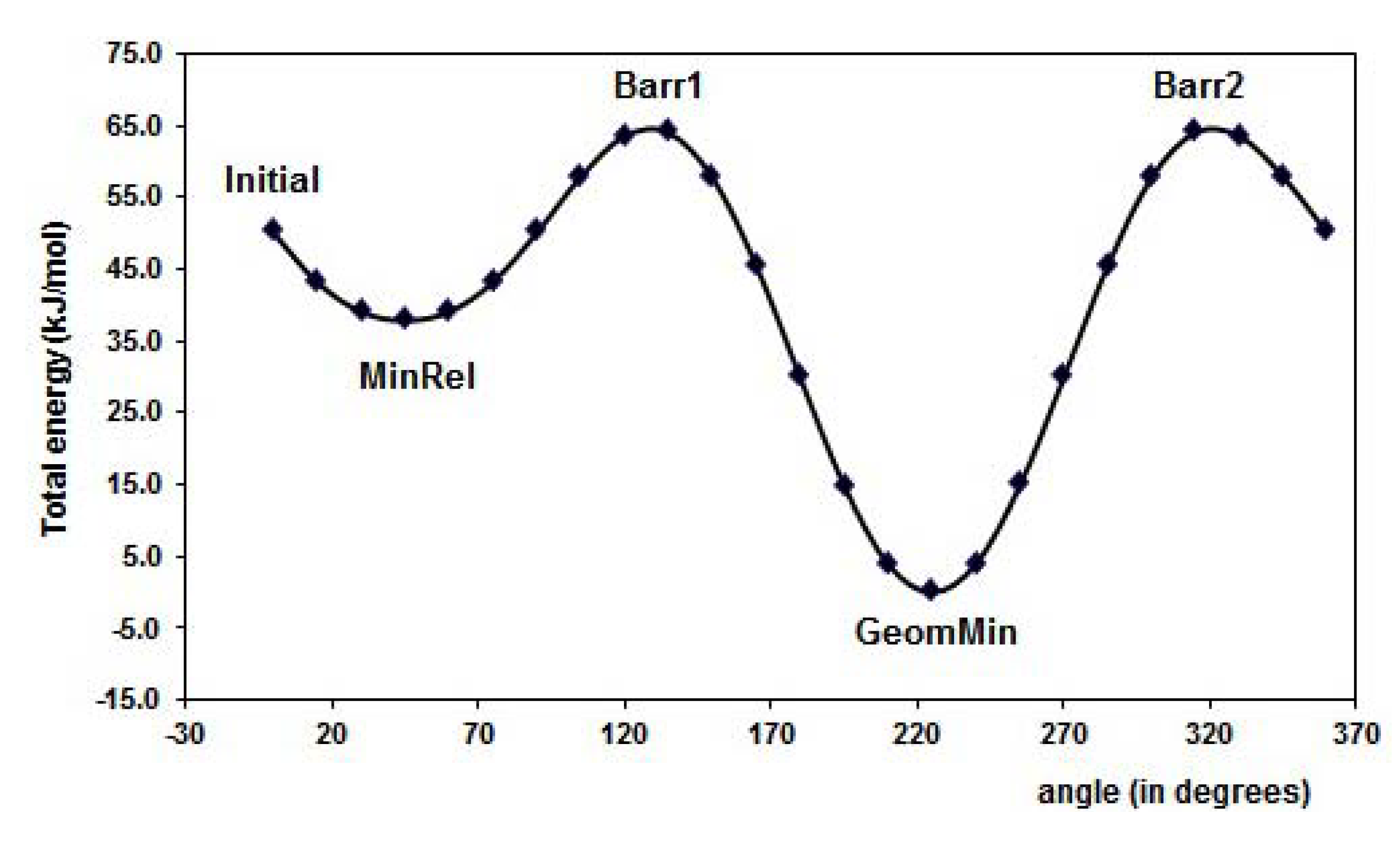
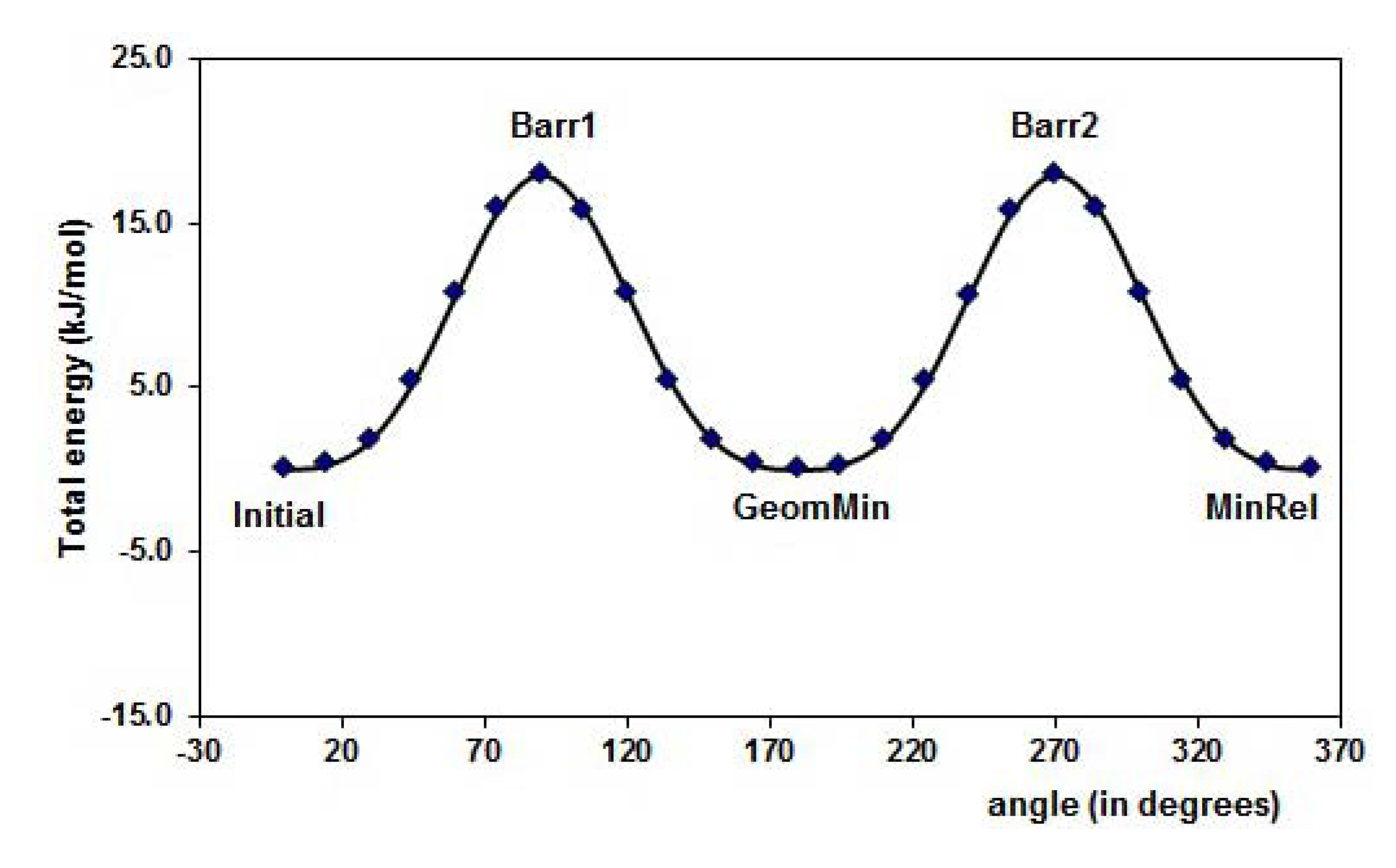
| FL | MAA | 4VPy | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Atom | ESP | Mulliken | Atom | ESP | Mulliken | Atom | ESP | Mulliken | |||
| C1 | 0.029 | 0.548 | C1 | -0.278 | -0.574 | C1 | -0.697 | 0.420 | |||
| O2 | -0.342 | -0.375 | C2 | 0.145 | 0.412 | C2 | 0.764 | -0.007 | |||
| C3 | 0.484 | -0.371 | H3 | 0.095 | 0.172 | C3 | -0.624 | 0.380 | |||
| C4 | 0.111 | -0.085 | H4 | 0.091 | 0.145 | C4 | 0.480 | -0.511 | |||
| O5 | -0.557 | -0.593 | H5 | 0.095 | 0.172 | N5 | -0.721 | -0.156 | |||
| O6 | -0.572 | -0.580 | C6 | -0.505 | -0.473 | C6 | 0.568 | -0.378 | |||
| C7 | 0.495 | -0.606 | C7 | 0.663 | 0.417 | H7 | 0.194 | 0.126 | |||
| C8 | -0.358 | -0.085 | H8 | 0.208 | 0.172 | C8 | -0.434 | -0.368 | |||
| C9 | 0.007 | -0.267 | H9 | 0.214 | 0.139 | H9 | 0.198 | 0.118 | |||
| C10 | -0.243 | 0.200 | O10 | -0.598 | -0.459 | H10 | 0.029 | 0.128 | |||
| C11 | 0.006 | -0.261 | O11 | -0.559 | -0.495 | H11 | 0.007 | 0.128 | |||
| C12 | -0.244 | 1.003 | H12 | 0.428 | 0.374 | H12 | 0.204 | 0.125 | |||
| C13 | 0.333 | 1.008 | C13 | -0.289 | -0.280 | ||||||
| C14 | -0.297 | -0.455 | H14 | 0.156 | 0.132 | ||||||
| C15 | -0.288 | 0.153 | H15 | 0.164 | 0.144 | ||||||
| C16 | -0.101 | -0.289 | |||||||||
| C17 | -0.133 | -0.165 | |||||||||
| C18 | -0.096 | -0.448 | |||||||||
| H19 | 0.177 | 0.140 | |||||||||
| H20 | 0.145 | 0.136 | |||||||||
| H21 | 0.433 | 0.411 | |||||||||
| H22 | 0.129 | 0.128 | |||||||||
| H23 | 0.128 | 0.130 | |||||||||
| H24 | 0.214 | 0.158 | |||||||||
| H25 | 0.114 | 0.137 | |||||||||
| H26 | 0.122 | 0.165 | |||||||||
| H27 | 0.176 | 0.135 | |||||||||
| H28 | 0.129 | 0.128 | |||||||||
2.2. Solvent effect
| Environment | Energy (u. a.) | ∆E (kcal mol-1) | ∆E (kJ mol-1) |
|---|---|---|---|
| In vacuum | -803.3429098 | --------------- | ---------------- |
| CHCl3 | -803.3547000 | -7.3983505 | - 30.9546985 |
| THF | -803.3565292 | -8.5461735 | -35.7571899 |
| ACN | -803.3599984 | -10.7230965 | -44.8654358 |
| Environment | Energy (u. a.) | ∆ E (kcal mol-1) | ∆ E (kJ mol-1) |
|---|---|---|---|
| In vacuum | -306.5084406 | --------------- | ---------------- |
| CHCl3 | -306.5193031 | -6.8162188 | -28.5190595 |
| THF | -306.5209396 | -7.8431225 | -32.8156245 |
| ACN | -306.5237232 | -9.5898315 | -40.1238550 |
| Environment | Energy (u. a.) | ∆ E (kcal mol-1) | ∆ E (kJ mol-1) |
|---|---|---|---|
| In vacuum | -325.709397 | --------------- | ---------------- |
| CHCl3 | -325.716818 | -4.6566775 | -19.4835387 |
| THF | -325.718006 | -5.4021475 | -22.6025851 |
| ACN | -325.720076 | -6.7010725 | -28.0372873 |
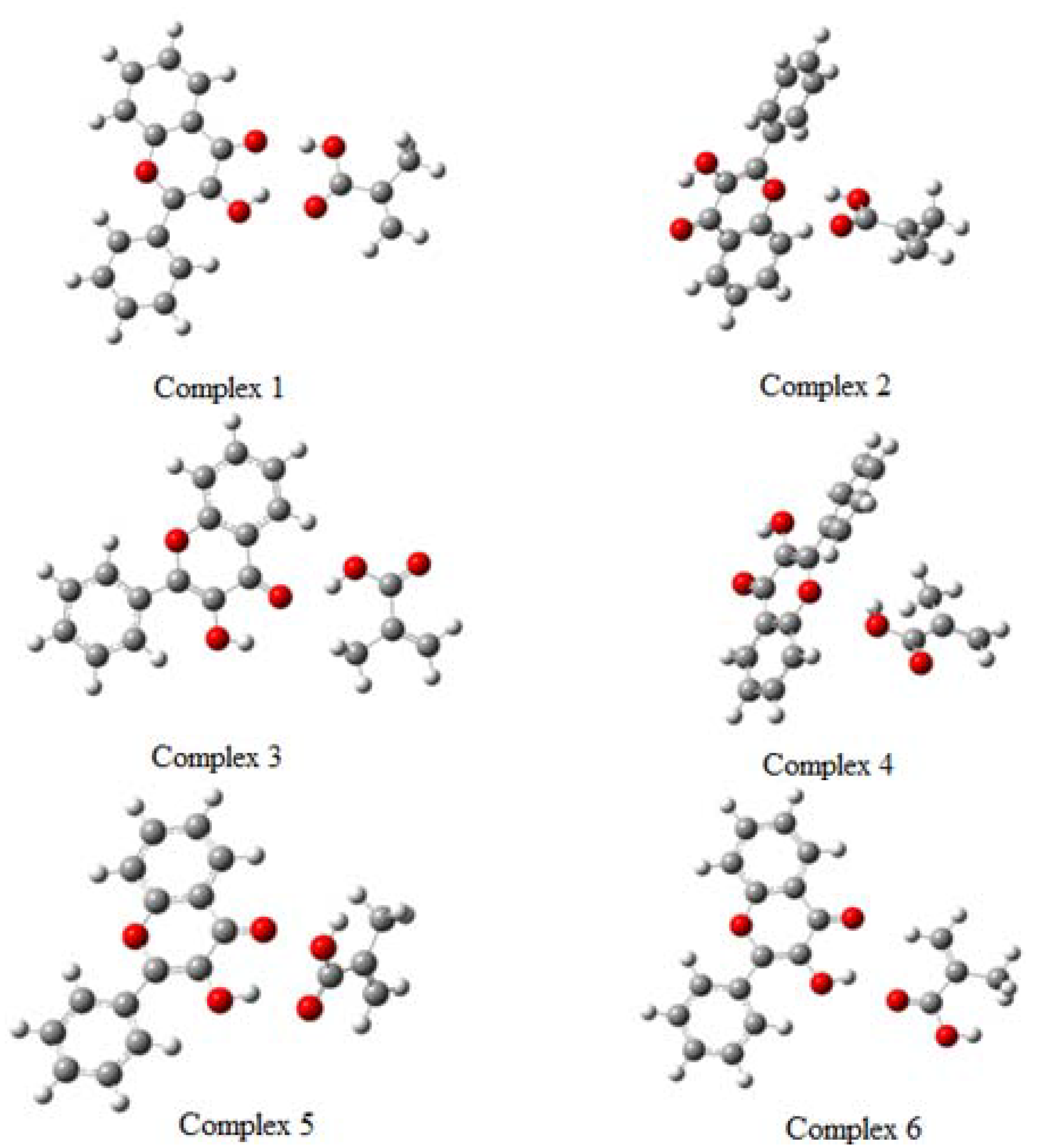
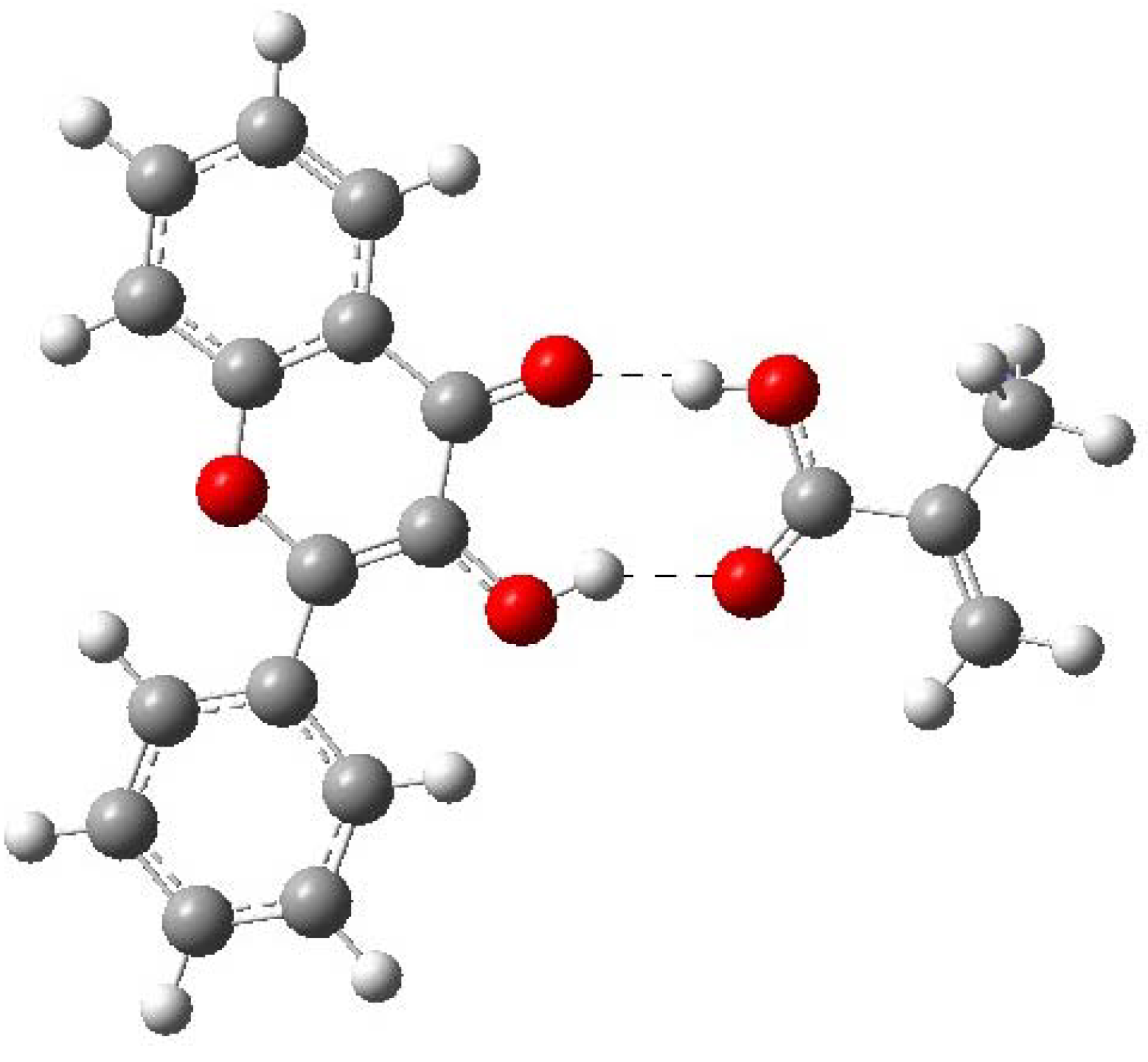
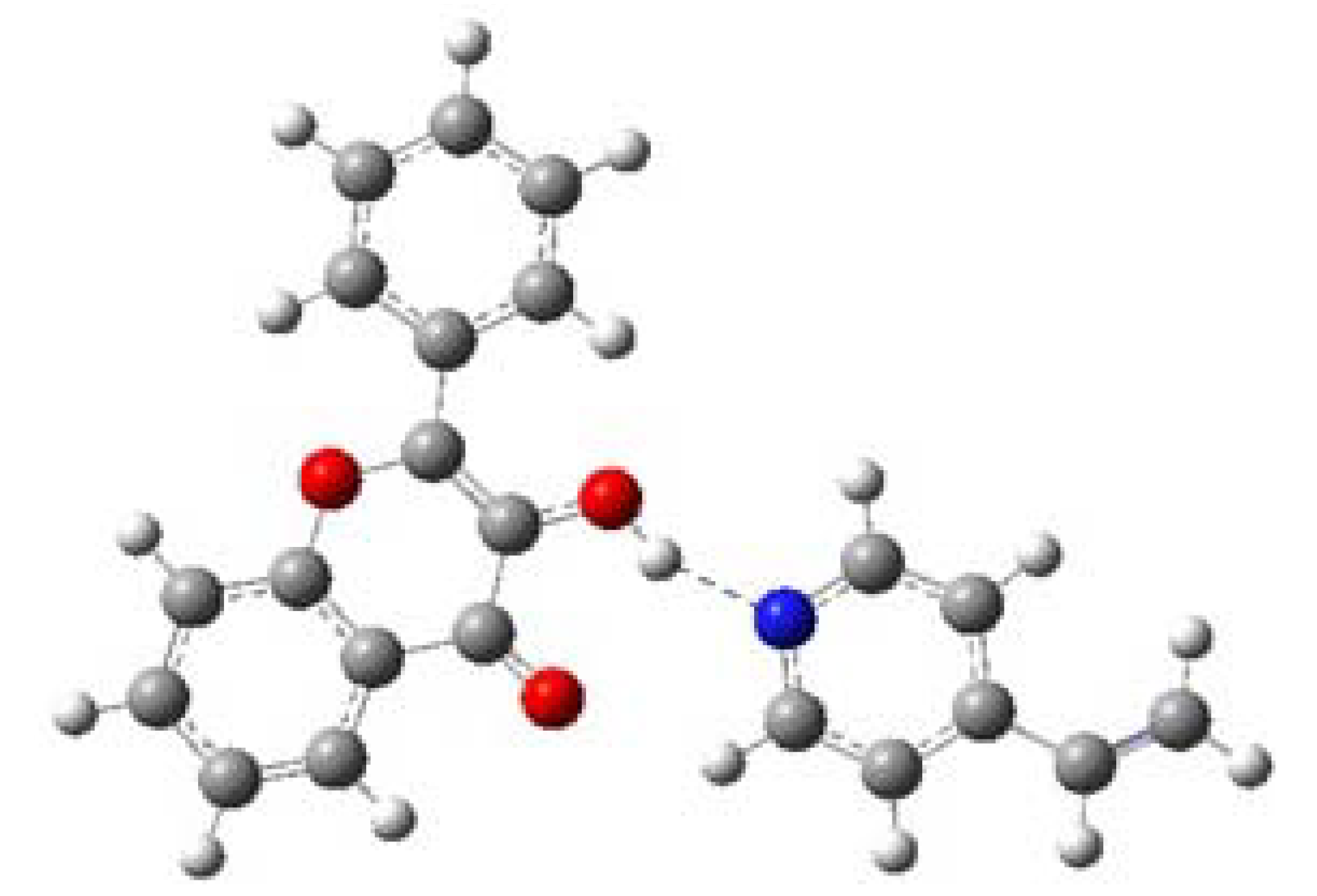
2.3. Pre-polymerization: complex formation stage
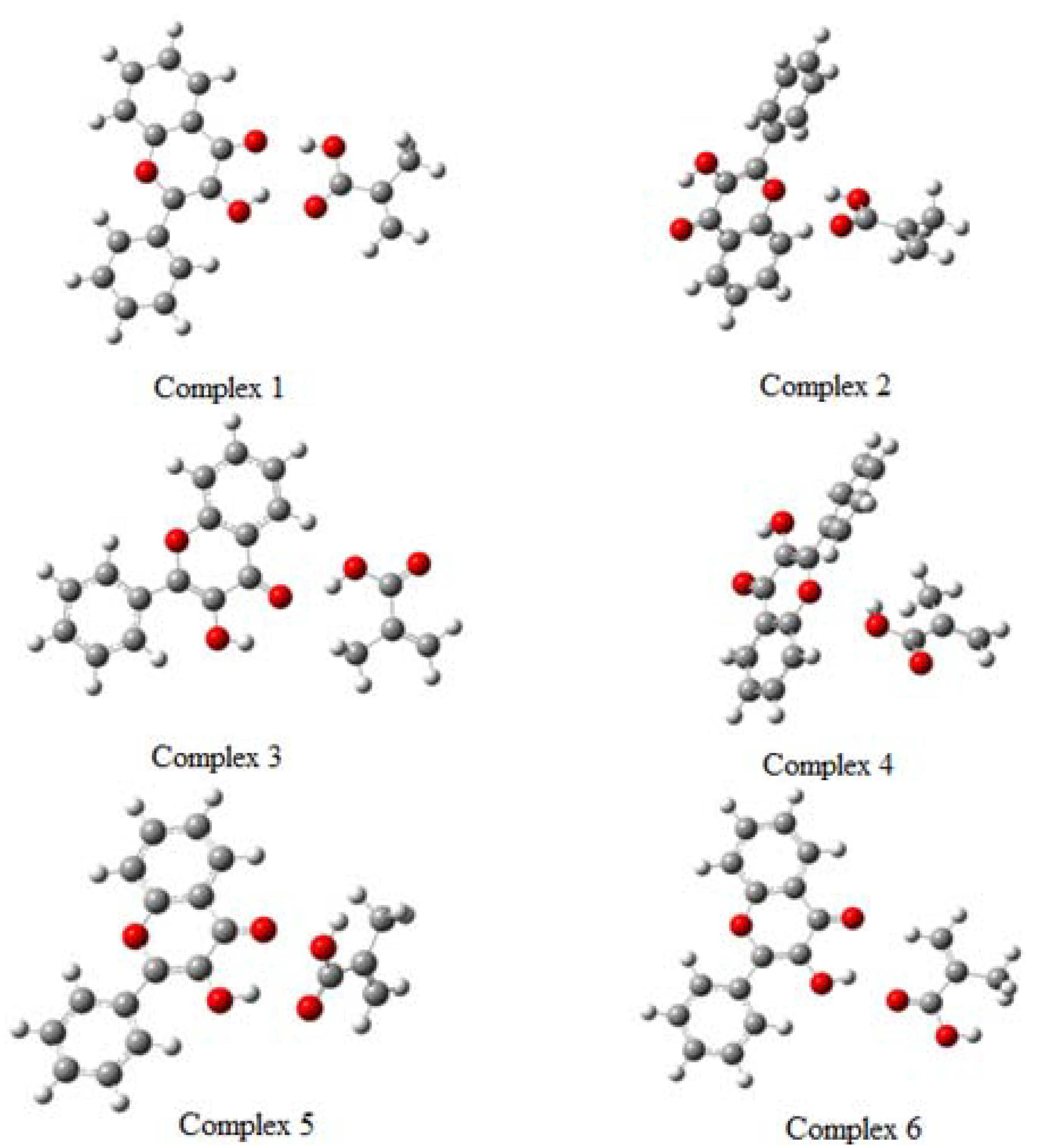
| Species | Energy in Hartrees | ∆E (kcal mol-1) | ∆E (kcal mol-1) |
|---|---|---|---|
| Flavonol | -803.3429098 | ||
| MAA GeomMin | -306.5084406 | ||
| MAA MinRel1 | -306.4977442 | ||
| Complex 1 | -1109.870675 | -12.13 | 0.00 |
| Complex 2 | -1109.854211 | -1.80 | 10.33 |
| Complex 3 | -1109.854015 | -1.67 | 10.45 |
| Complex 4 | -1109.845292 | 3.80 | 15.93 |
| Complex 5 | -1109.846708 | 2.91 | 15.04 |
| Complex 6 | -1109.847501 | 2.42 | 14.54 |
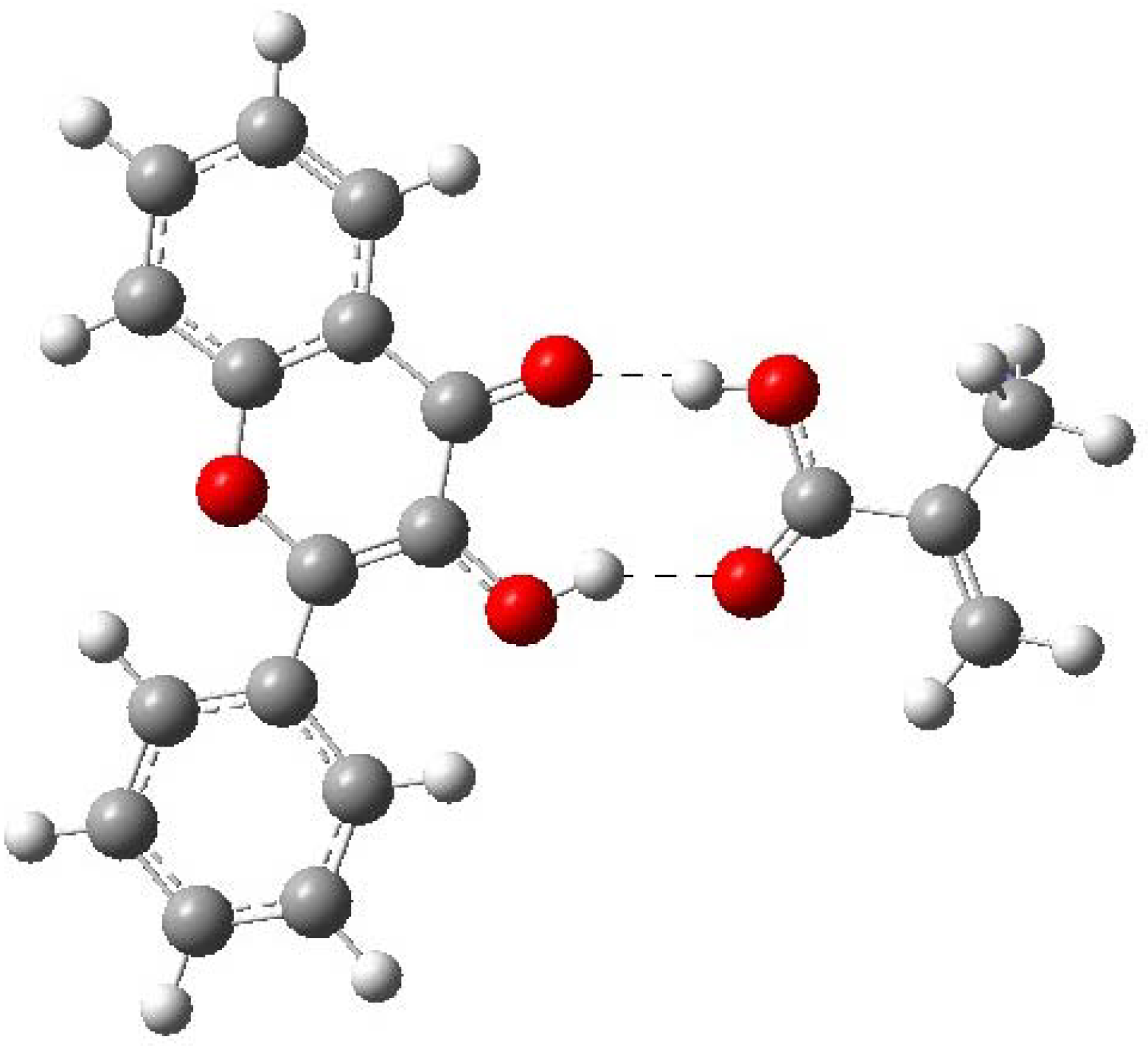
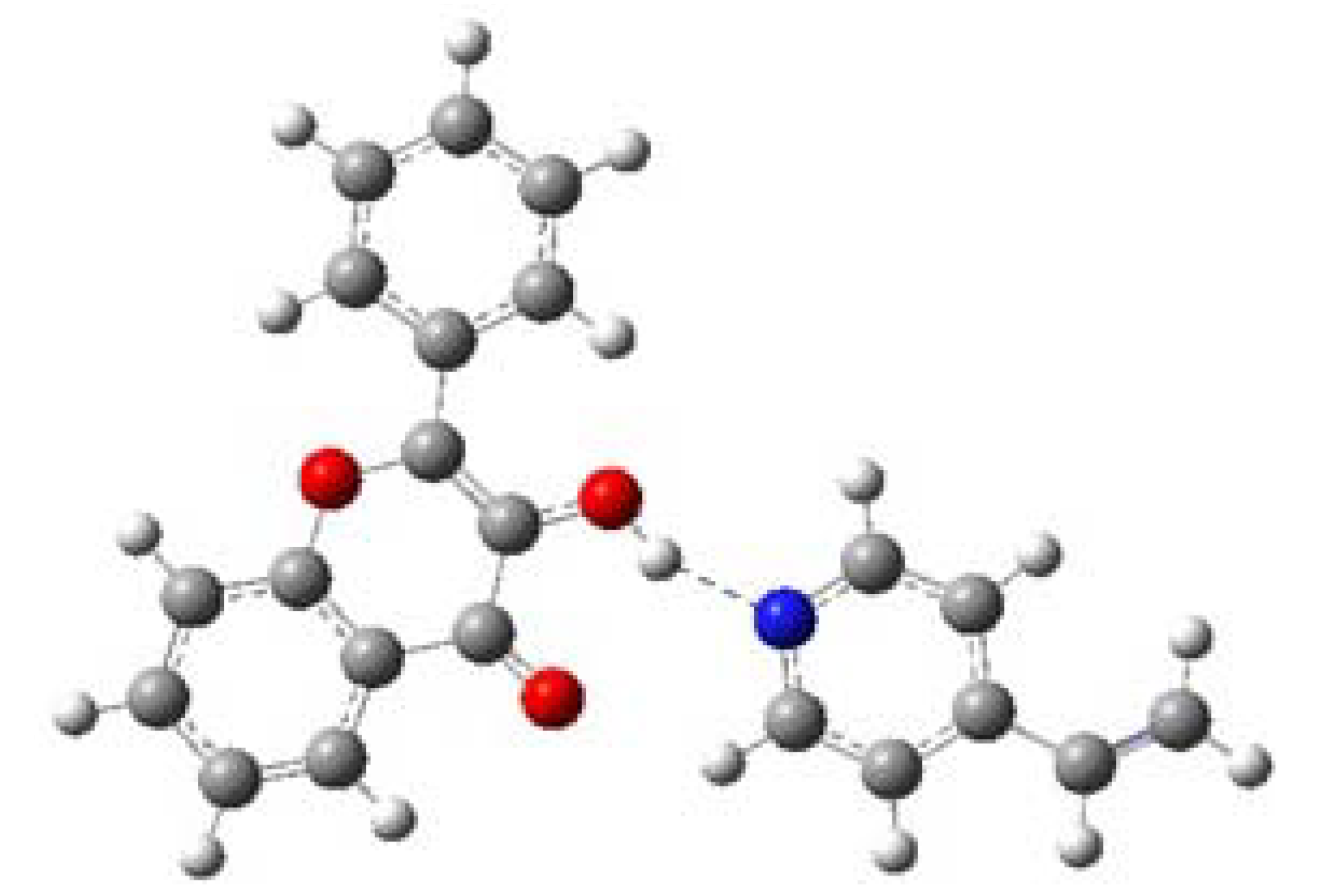
2.4. Evaluation of the binding energy
| Species | Energy (u. a.) | ∆E (kcal mol-1) | ∆E (kJ mol-1) |
|---|---|---|---|
| FL (free) | -803.3429098 | ------------ | ------------ |
| MAA (free) | -306.5084406 | ------------ | ------------ |
| 4VPy (free) | -325.7093970 | ------------ | ------------ |
| Complex 1 (FL-MAA) | -1109.8706749 | -12.125559 | -50.7333389 |
| Complex (FL-4VPy) | - 1129.0627294 | -6.5399305 | -27.3630692 |
| Species | Energy (u. a.) | ∆ E solv (kcal mol-1) | ∆ E solv (kJ mol-1) |
|---|---|---|---|
| In vacuum | |||
| Complex 1 (FL-MAA) | -1109.8706749 | ------------ | ------------ |
| Complex (FL-4VPy) | - 1129.0627294 | ------------ | ------------ |
| With solvent effect (CHCl3) | |||
| Complex 1 (FL-MAA) | -1109.86921 | -0.9062 | -3.79 |
| Complex (FL-4VPy) | - 1129.062447 | -0.0003 | -0.74 |
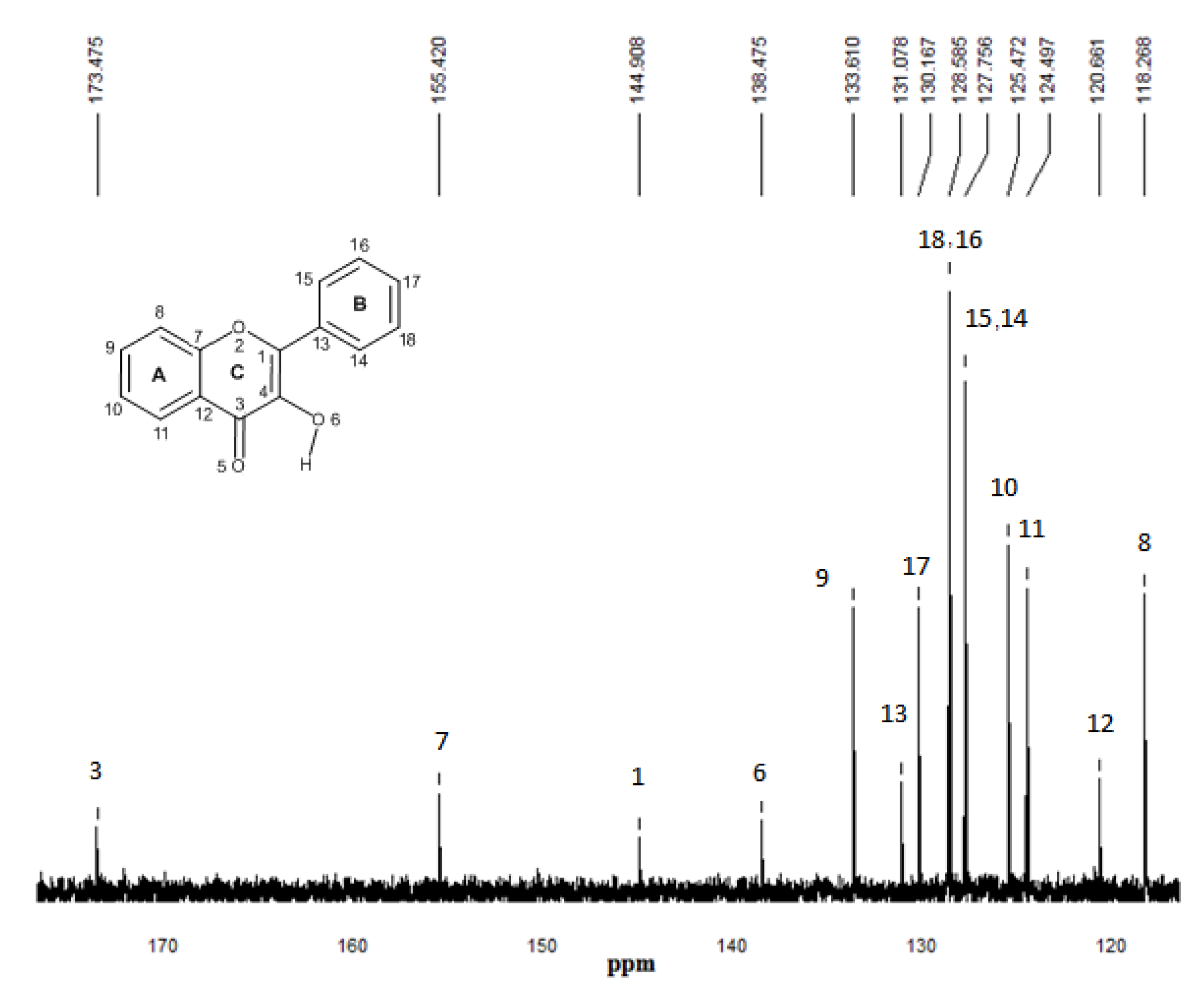
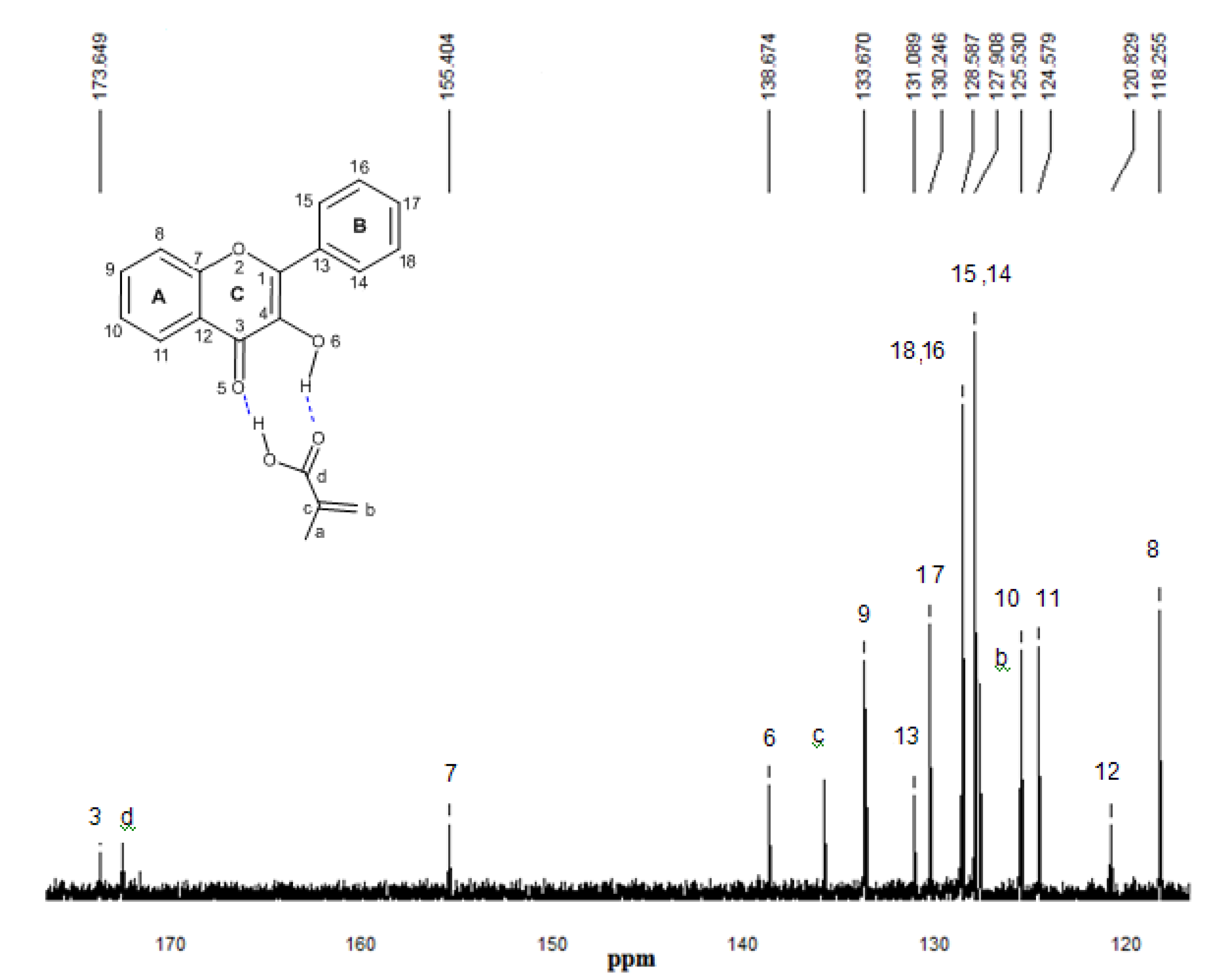
2.5. 13C-NMR spectroscopy
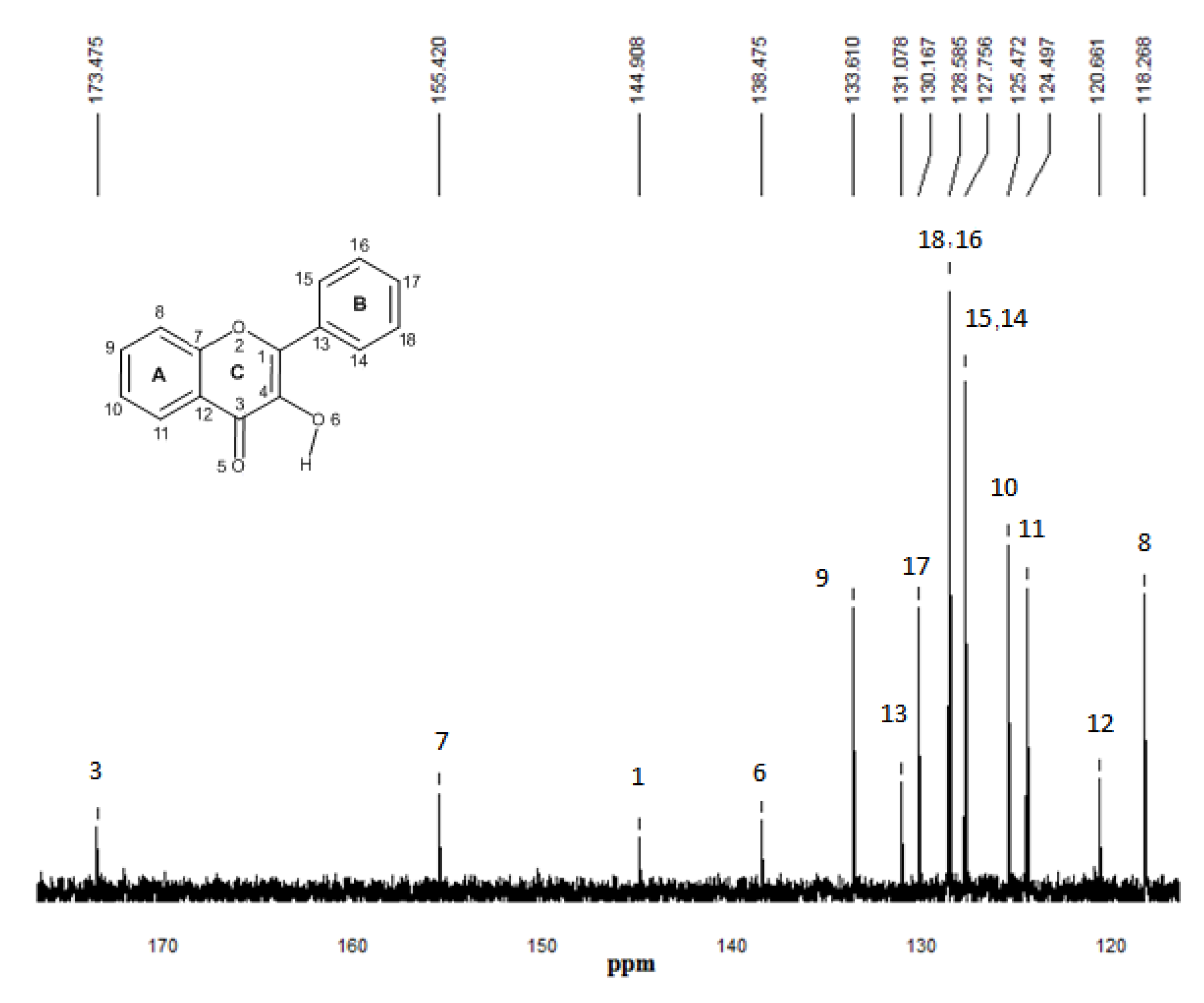
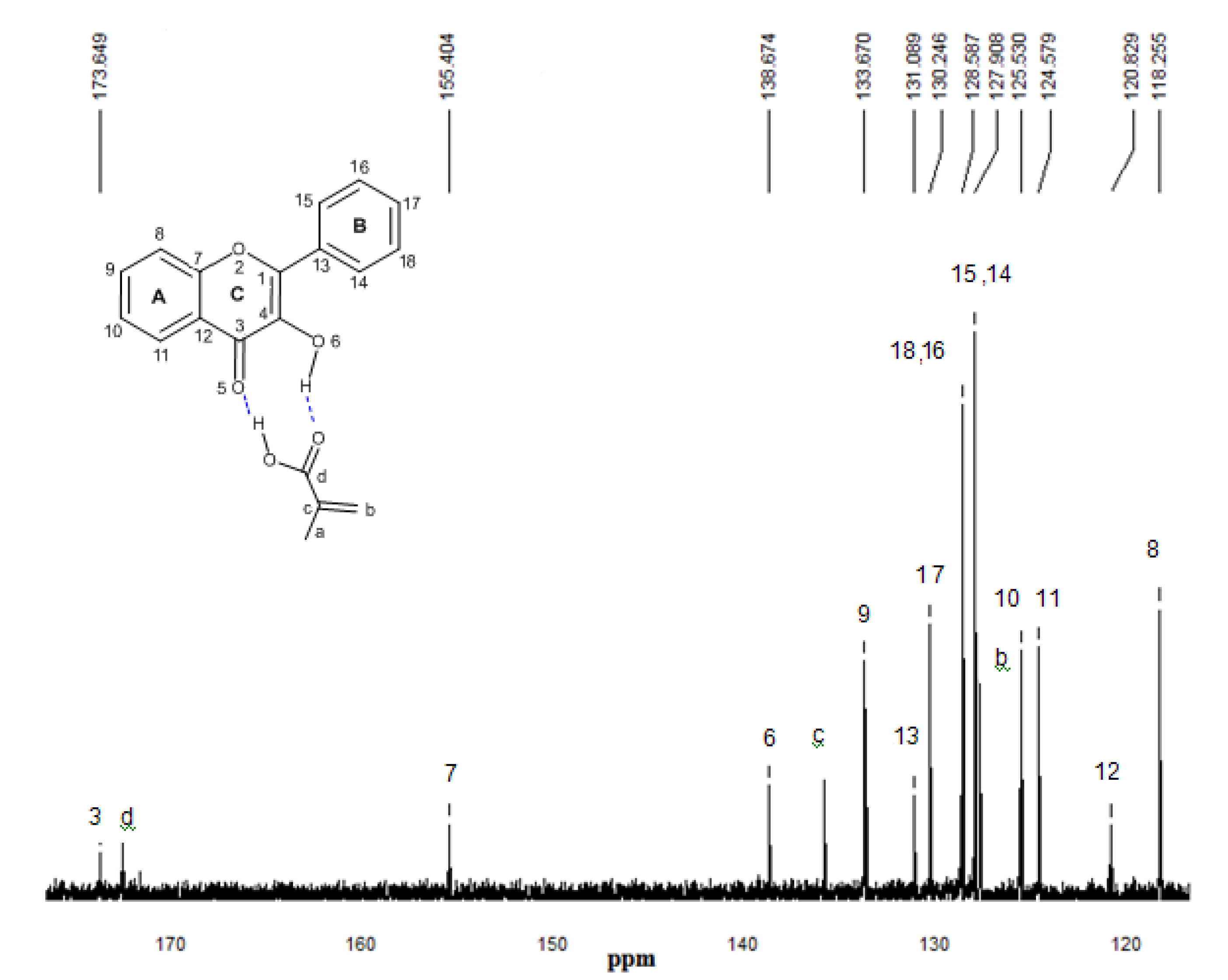
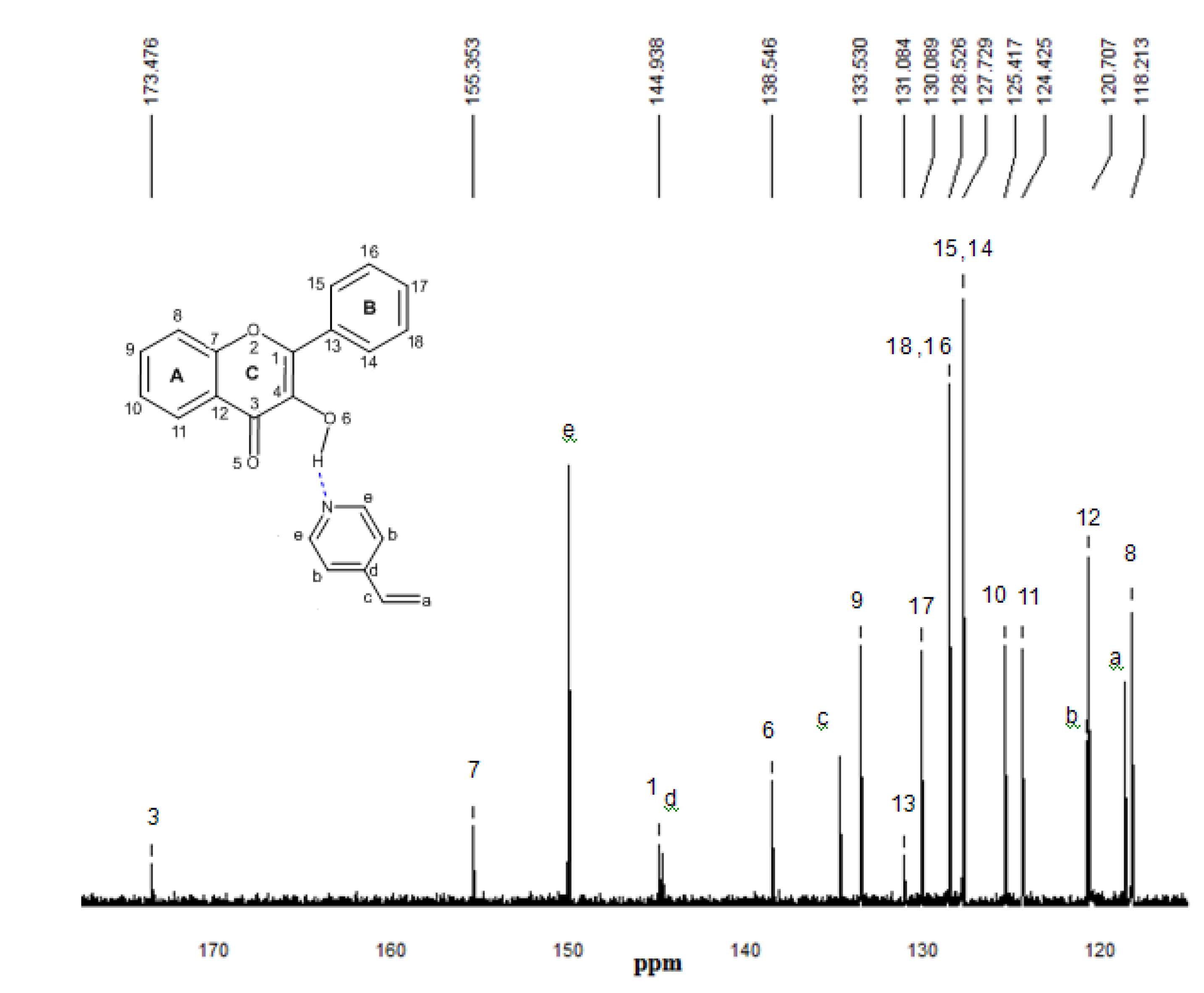
3. Procedures
3.1. Molecular simulation
3.2. 13C-NMR
4. Conclusions
Acknowledgements
- Samples Availability: Contact the authors.
References
- Yu, C.; Mosbach, K. Influence of mobile phase composition and cross-linking density on the enantiomeric recognition properties of molecularly imprinted polymers. J. Chromatogr. A 2000, 888, 63–72. [Google Scholar] [CrossRef]
- Chen, W.Y.; Chen, C.S.; Lin, F.Y. Molecular recognition in imprinted polymers: Thermodynamic investigation of analyte binding using microcalorimetry. J. Chromatogr. A 2001, 923, 1–6. [Google Scholar] [CrossRef]
- Kirsch, N.; Alexander, C.; Lübke, M.; Whitcombe, M.J.; Vulfson, E.N. Enhancement of selectivity of imprinted polymers via post-imprinting modification of recognition sites. Polymer 2000, 41, 5583–5590. [Google Scholar] [CrossRef]
- Andersson, H.S.; Koch-Schmidt, A.C.; Ohlson, S.; Mosbach, K. Study of the nature of recognition in molecularly imprinted polymers. J. Mol. Recognit. 1996, 9, 675–682. [Google Scholar]
- Katz, A.; Davis, M.E. Investigations into the mechanism of molecular recognition with imprinted polymers. Macromolecules 1999, 32, 4113–4121. [Google Scholar] [CrossRef]
- Andersson, H.S.; Karlesson, J.G.; Piletsky, S.A.; Kouch- Schmidt, A.C.; Mosbach, K.; Nicholls, I.A. Study of the nature of recognition in molecularly imprinted polymers: Influence of monomer-template ratio and sample load on retention and selectivity. J. Chromatogr. A 1999, 848, 39–49. [Google Scholar] [CrossRef]
- Nicholls, I.A. Thermodynamic considerations for the design of and ligand recognition by molecularly imprinted polymer. Chem. Lett. 1995, 24, 1035–1036. [Google Scholar] [CrossRef]
- Whitcombe, M.J.; Martin, L.; Vulfson, E.N. Predicting the selectivity of imprinted polymers. Chromatographia 1998, 47, 457–464. [Google Scholar] [CrossRef]
- McNiven, S.; Yokobayashi, Y.; Cheong, S.H.; Karube, I. Enhancing the selectivity of molecularly imprinted polymers. Chem. Lett. 1997, 26, 1297–1298. [Google Scholar]
- Dong, W.; Yan, M.; Zhan, M.; Liu, Z.; Li, Y. A computational and experimental investigation of the interaction between the template molecule and the functional monomer used in the molecularly imprinted polymer. Anal. Chim. Acta 2005, 542, 186–192. [Google Scholar] [CrossRef]
- Chartchalerm, I.; Chanin, N.; Prasit, B.; Theeraphon, P.; Lei, Y.; Leif, B.; Virapong, P. Computational insights on sulfonamide imprinted polymers. Molecules 2008, 13, 3077–3091. [Google Scholar] [CrossRef]
- Diñeiro, Y.; Menéndez, M.I.; Blanco-López, M.C.; Lobo-Castaño, M.J.; Miranda-Ordieres, A.J.; Tuñon-Blanco, P. Computational approach to the rational design of molecularly imprinted polymers for voltammetric sensing of homovanillic acid. Anal. Chem. 2005, 77, 6741–6746. [Google Scholar]
- Dong, W.; Yan, M.; Liu, Z.; Wu, G.; Li, Y. Effects of solvents on the adsorption selectivity of molecularly imprinted polymers: Molecular simulation and experimental validation. Separ. Purif. Technol. 2007, 53, 183–188. [Google Scholar] [CrossRef]
- Bohm, B.A. Introduction to Flavonoids; Harwood Academic Publishers: Singapore, 1998; Volume 2, pp. 121–130. [Google Scholar]
- Middleton, E., Jr.; Kandaswami, C. The Flavonoids; Harborne, J.B., Ed.; Chapman & Hall: London, UK, 1986; Volume Chapter 15, pp. 619–652. [Google Scholar]
- Kaur, C.; Kapoor, H.C. Antioxidants in fruits and vegetables-the millennium’s health. Int. J. Food Sci. Technol. 2001, 36, 703–725. [Google Scholar] [CrossRef]
- Dickert, F.L.; Besenbock, H.; Tortschanoff, M. Molecular imprinting through van der Waals interactions: Fluorescence detection of PAHs in water. Adv. Mater. 1998, 10, 149–151. [Google Scholar] [CrossRef]
- Tóth, J.; Remko, M.; Nagy, M. The ability of molecular modeling methods to reproduce the structure of flavonoids. Acta Facul. Pharm. Univ. Comenianae Tomus LII 2005, 52, 218–225. [Google Scholar]
- Cornard, J.P.; Vrielynck, L.; Merlin, J.C. Structural and vibrational study of 3-hydroxyflavone and 3-methoxyflavone. Spectrochim. Acta 1995, 51A, 913–923. [Google Scholar]
- Cody, V. Crystal and molecular structures of flavonoids. Prog. Clin. Biol. Res. 1988, 28029–28044. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The rol of exact exchange. J. Chem. Phys. 1993, 98, 5648–5653. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Besler, B.H.; Merz, K.M., Jr.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comp. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comp. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.J.A.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Agrawal, P.K.; Thakur, R.S.; Bansal, M.C. Carbon-13 NMR of Flavonoids; Elsevier: Amsterdam, The Netherlands, 1989; Volume Chapter 3, pp. 95–182. [Google Scholar]
- Miertus, S.; Scrocco, G.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Miertus, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Gómez-Pineda, L.E.; Pina-Luis, G.E.; Cortés-Romero, C.M.; Palomar-Pardavé, M.E.; Rosquete-Pina, G.A.; Díaz-García, M.E.; Hernández, M.d.l.A.C. Quantum Chemical Calculations on the Interaction between Flavonol and Functional Monomers (Methacrylic Acid and 4-Vinylpyridine) in Molecularly Imprinted Polymers. Molecules 2010, 15, 4017-4032. https://doi.org/10.3390/molecules15064017
Gómez-Pineda LE, Pina-Luis GE, Cortés-Romero CM, Palomar-Pardavé ME, Rosquete-Pina GA, Díaz-García ME, Hernández MdlAC. Quantum Chemical Calculations on the Interaction between Flavonol and Functional Monomers (Methacrylic Acid and 4-Vinylpyridine) in Molecularly Imprinted Polymers. Molecules. 2010; 15(6):4017-4032. https://doi.org/10.3390/molecules15064017
Chicago/Turabian StyleGómez-Pineda, Luis Enrique, Georgina Esther Pina-Luis, Carlos Martín Cortés-Romero, Manuel Eduardo Palomar-Pardavé, Giselle Alicia Rosquete-Pina, Marta Elena Díaz-García, and María de los Angeles Cuán Hernández. 2010. "Quantum Chemical Calculations on the Interaction between Flavonol and Functional Monomers (Methacrylic Acid and 4-Vinylpyridine) in Molecularly Imprinted Polymers" Molecules 15, no. 6: 4017-4032. https://doi.org/10.3390/molecules15064017
APA StyleGómez-Pineda, L. E., Pina-Luis, G. E., Cortés-Romero, C. M., Palomar-Pardavé, M. E., Rosquete-Pina, G. A., Díaz-García, M. E., & Hernández, M. d. l. A. C. (2010). Quantum Chemical Calculations on the Interaction between Flavonol and Functional Monomers (Methacrylic Acid and 4-Vinylpyridine) in Molecularly Imprinted Polymers. Molecules, 15(6), 4017-4032. https://doi.org/10.3390/molecules15064017