Synthesis and Use of Stable Isotope Enriched Retinals in the Field of Vitamin A

{kind=link}
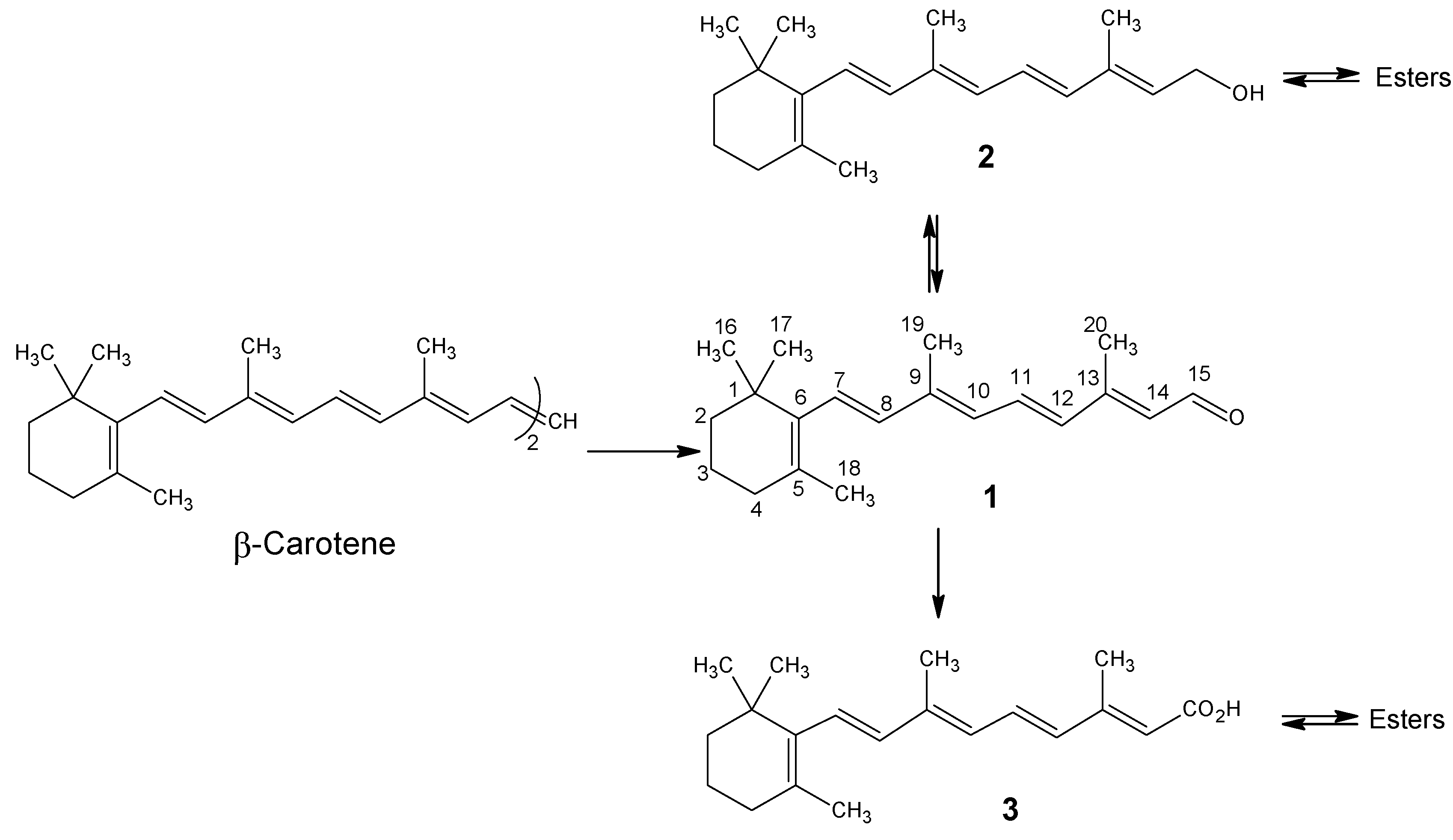{kind=link}
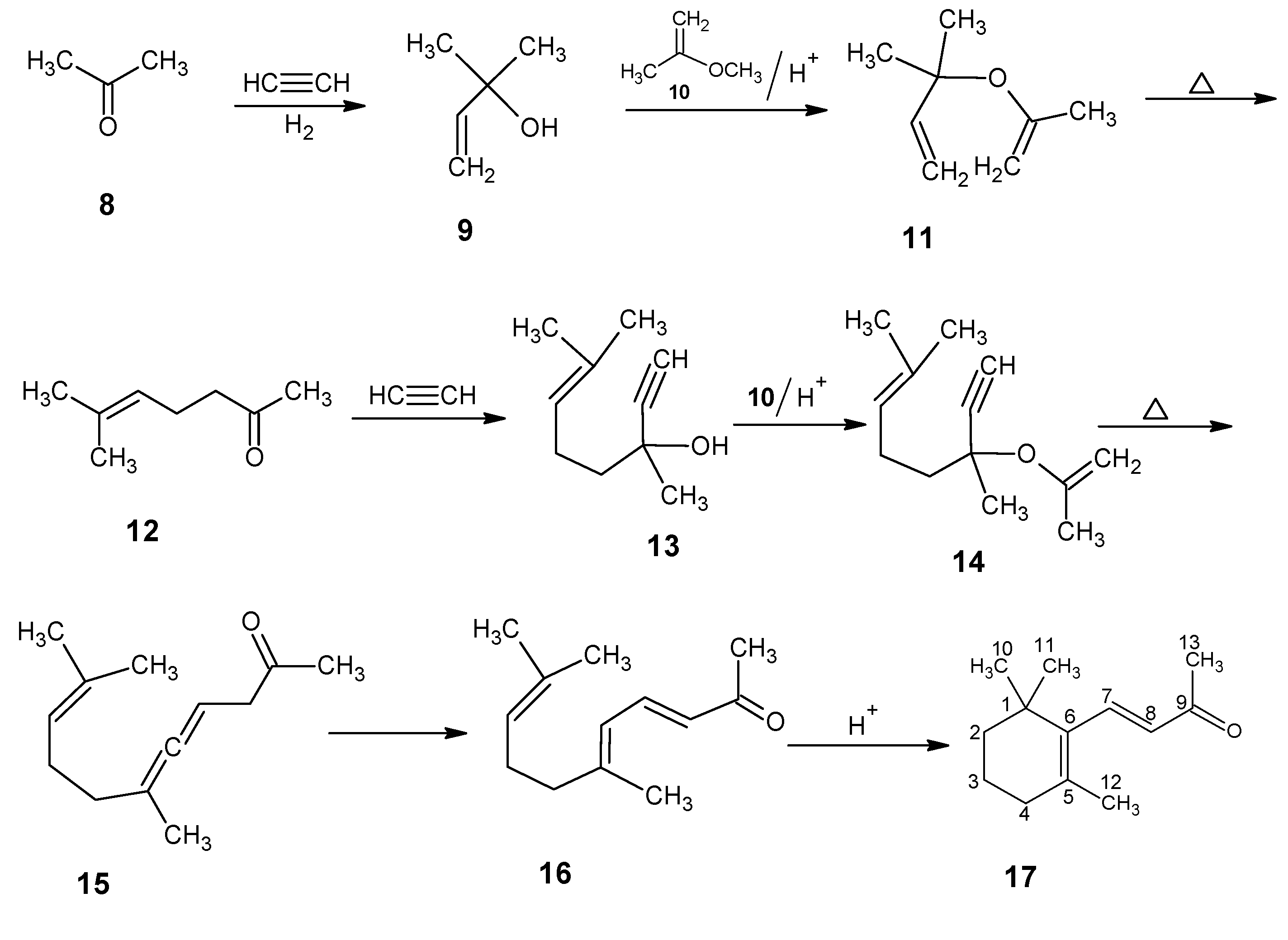{kind=link}
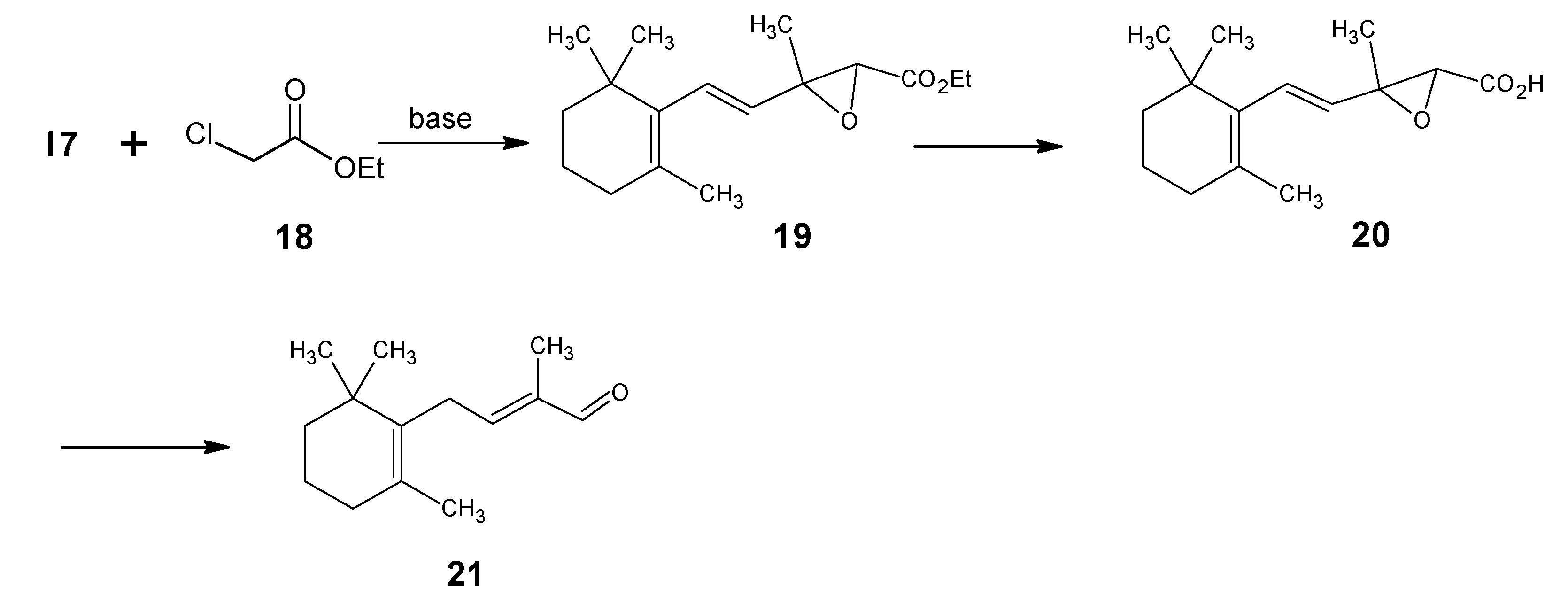{kind=link}
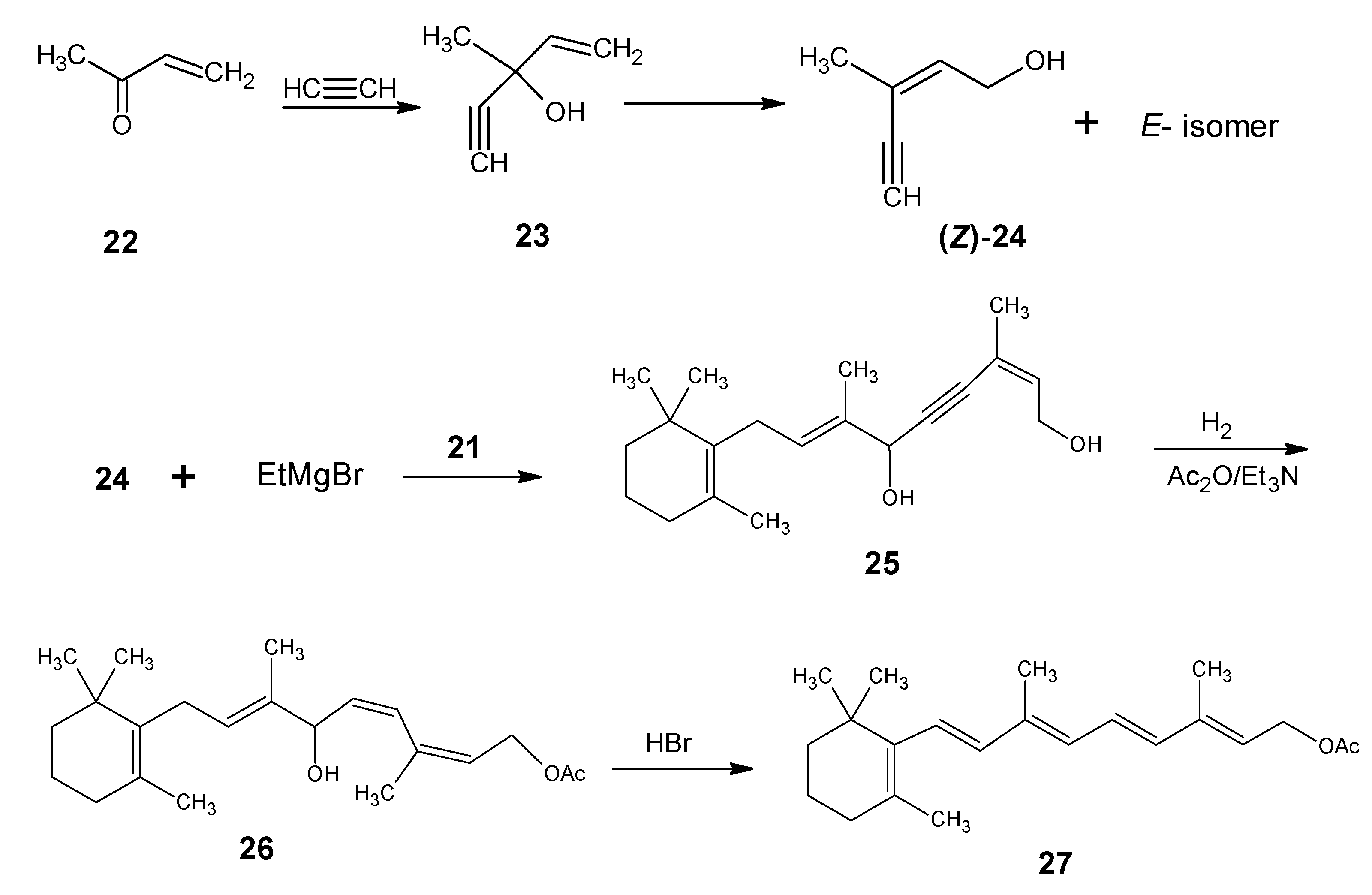{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
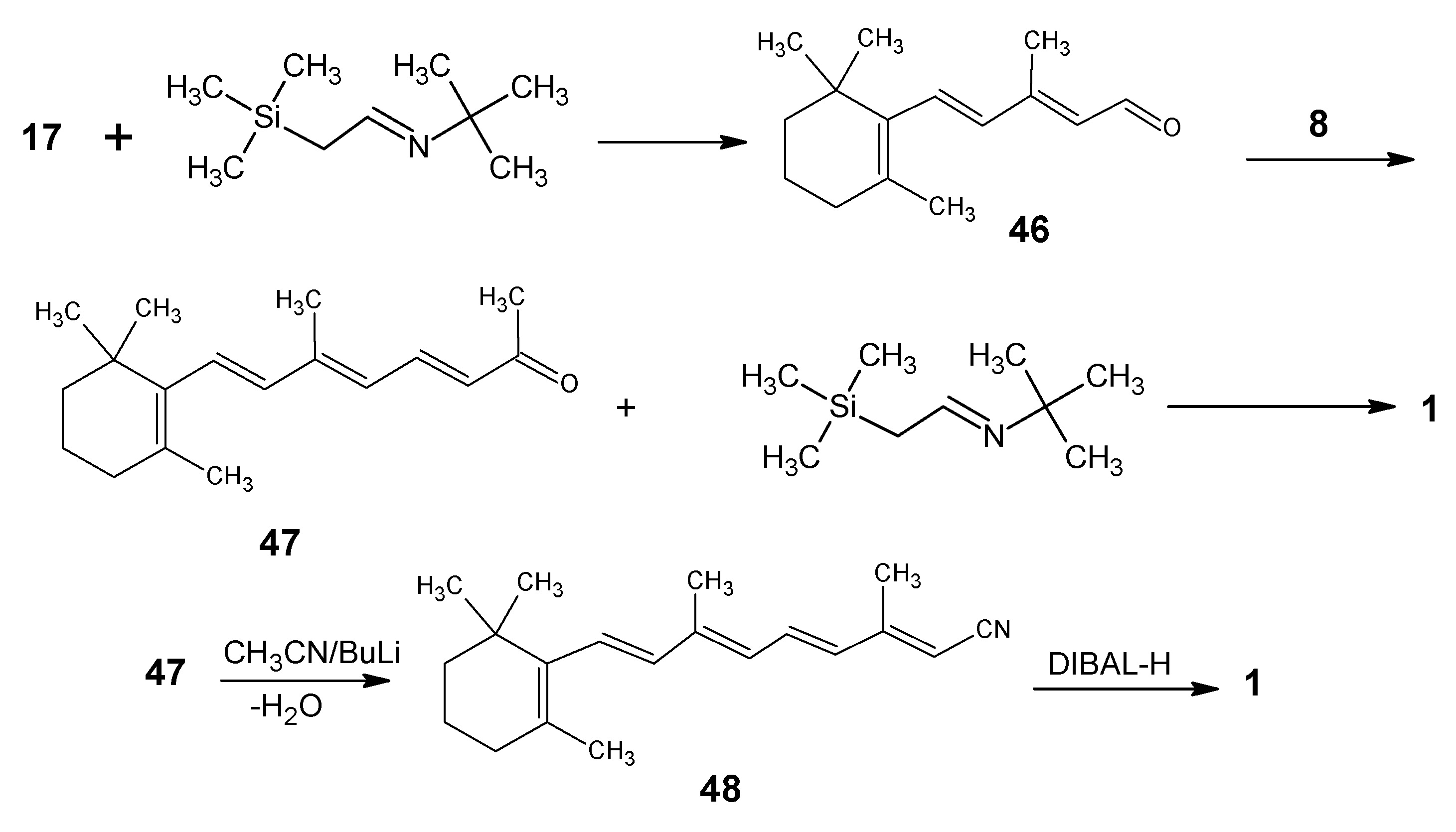{kind=link}
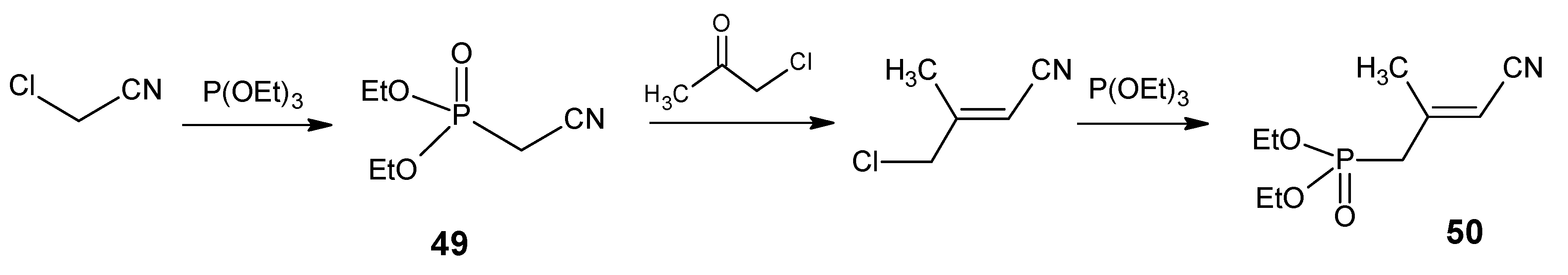{kind=link}
{kind=link}
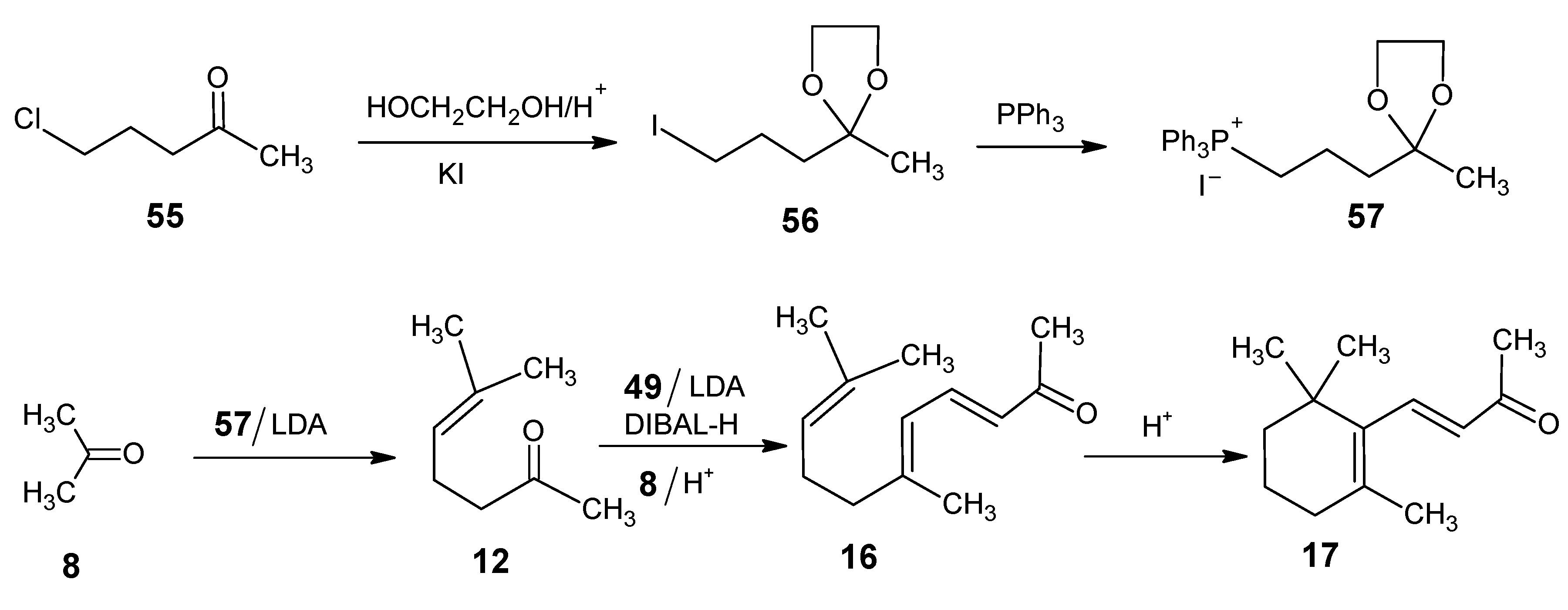{kind=link}
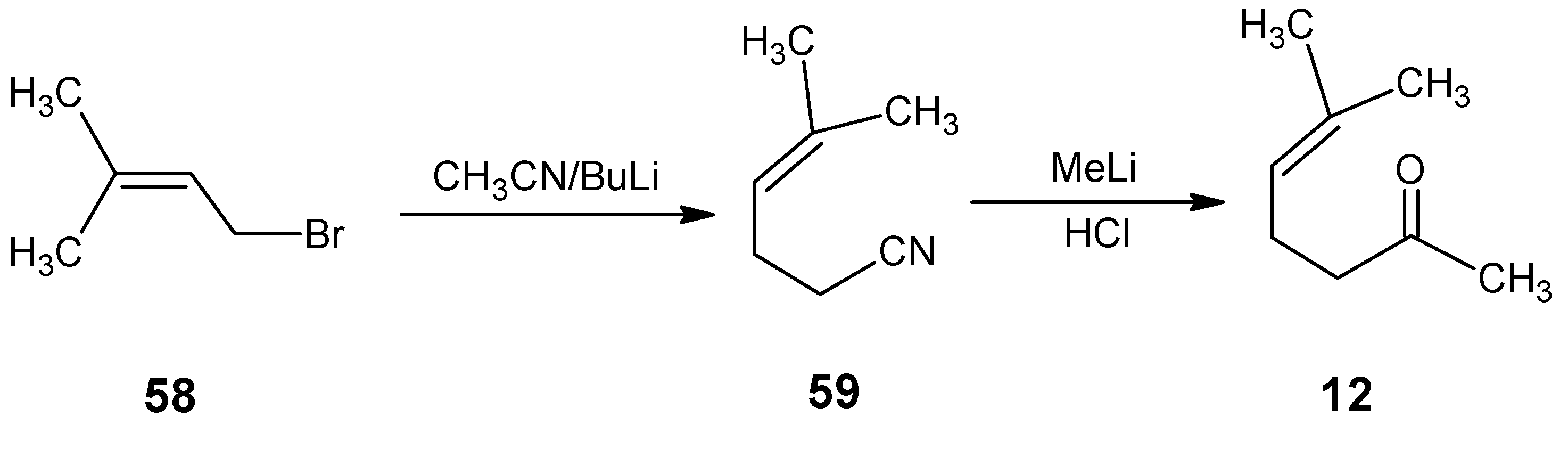{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Contents
Part A. Introduction
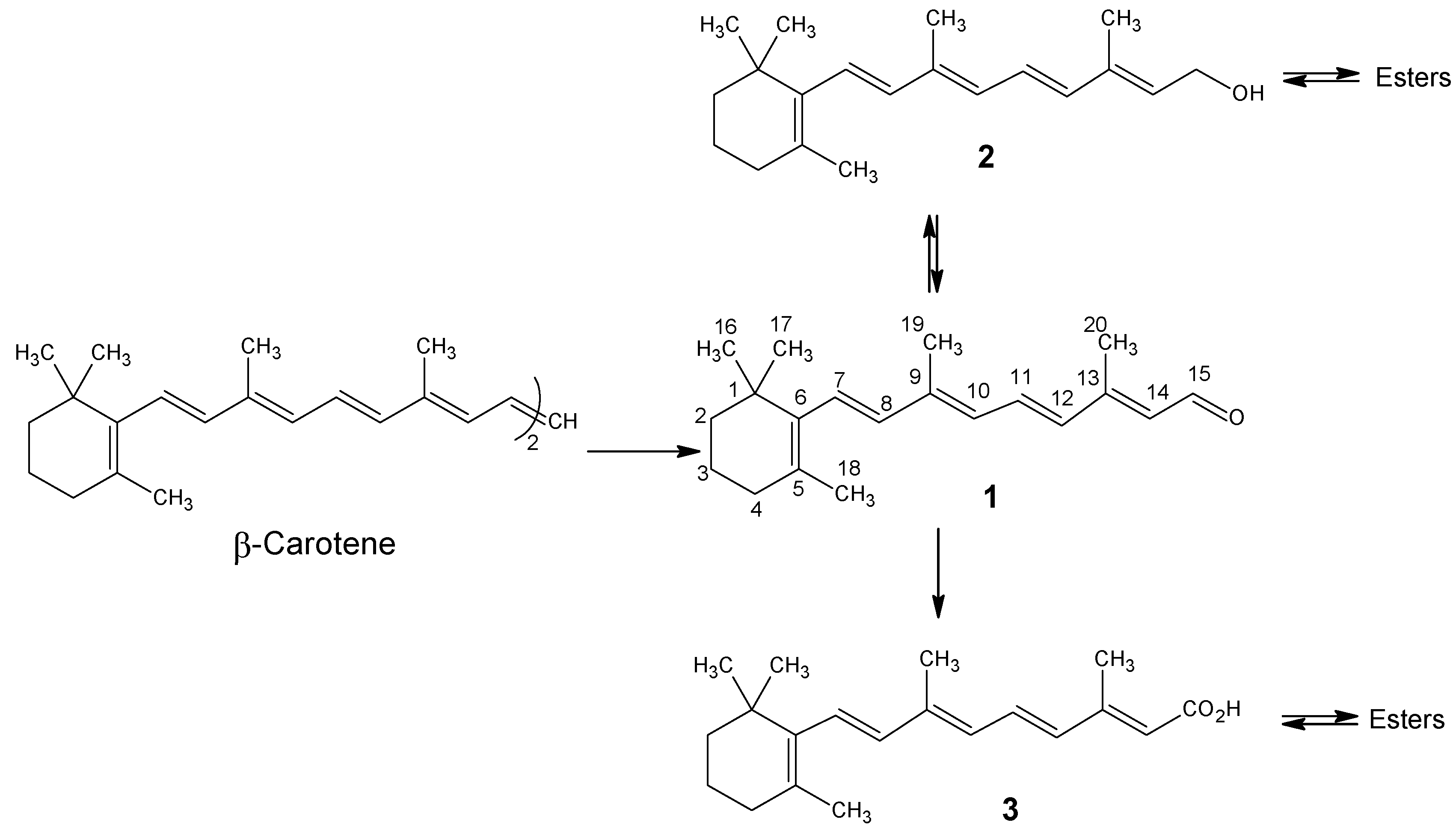
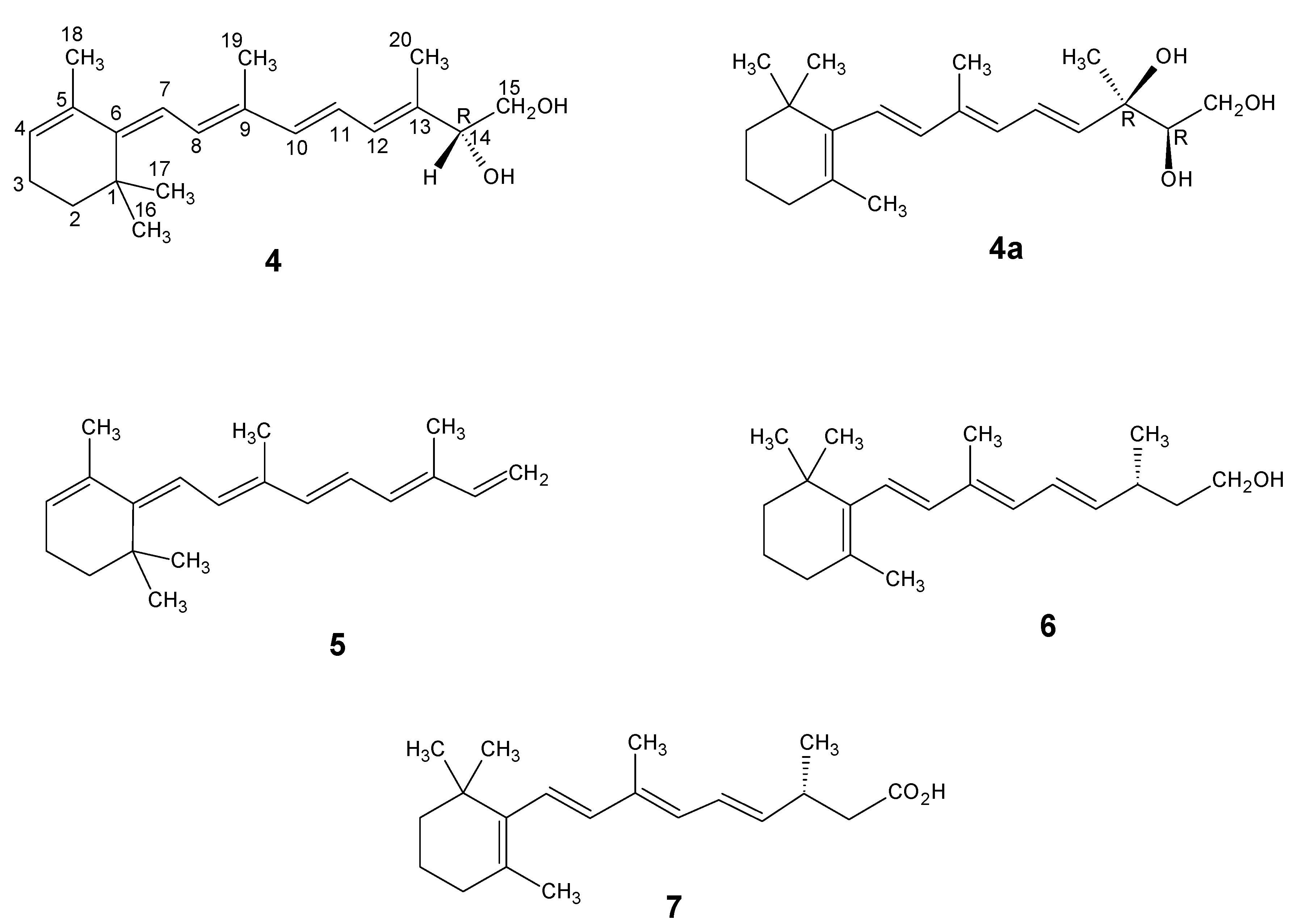
Part B. Technical Syntheses
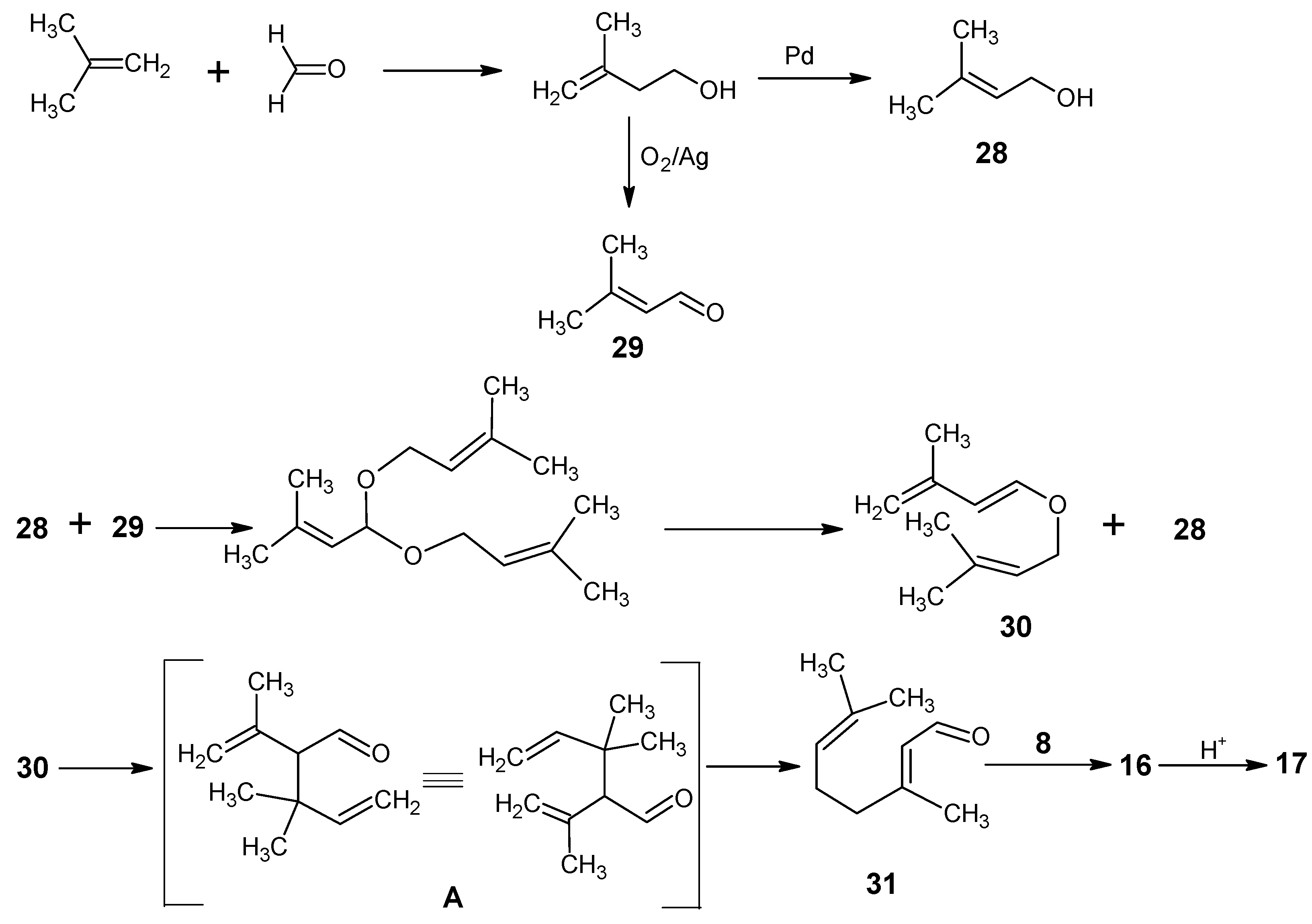
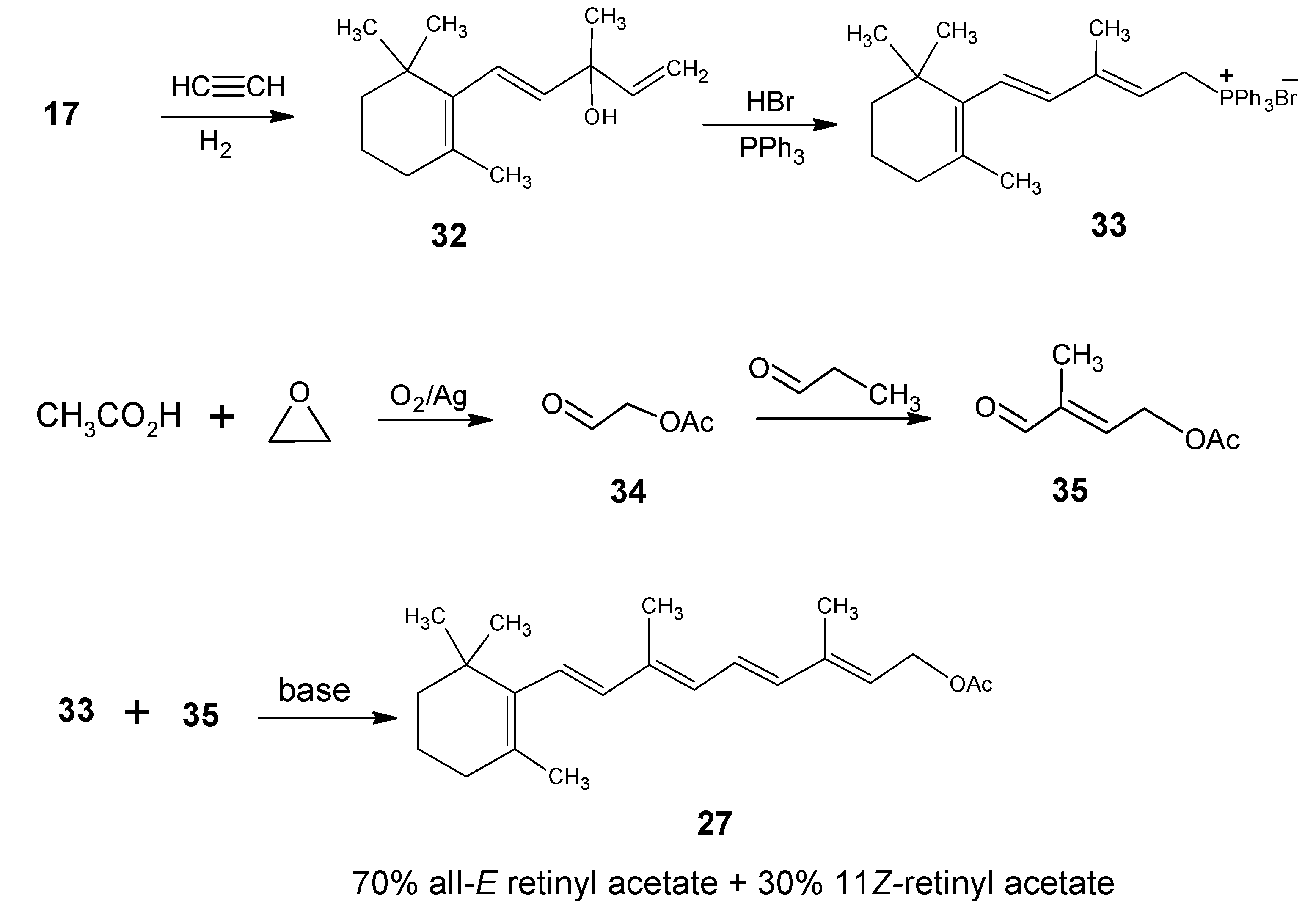
1. Hoffmann-La Roche, DSM
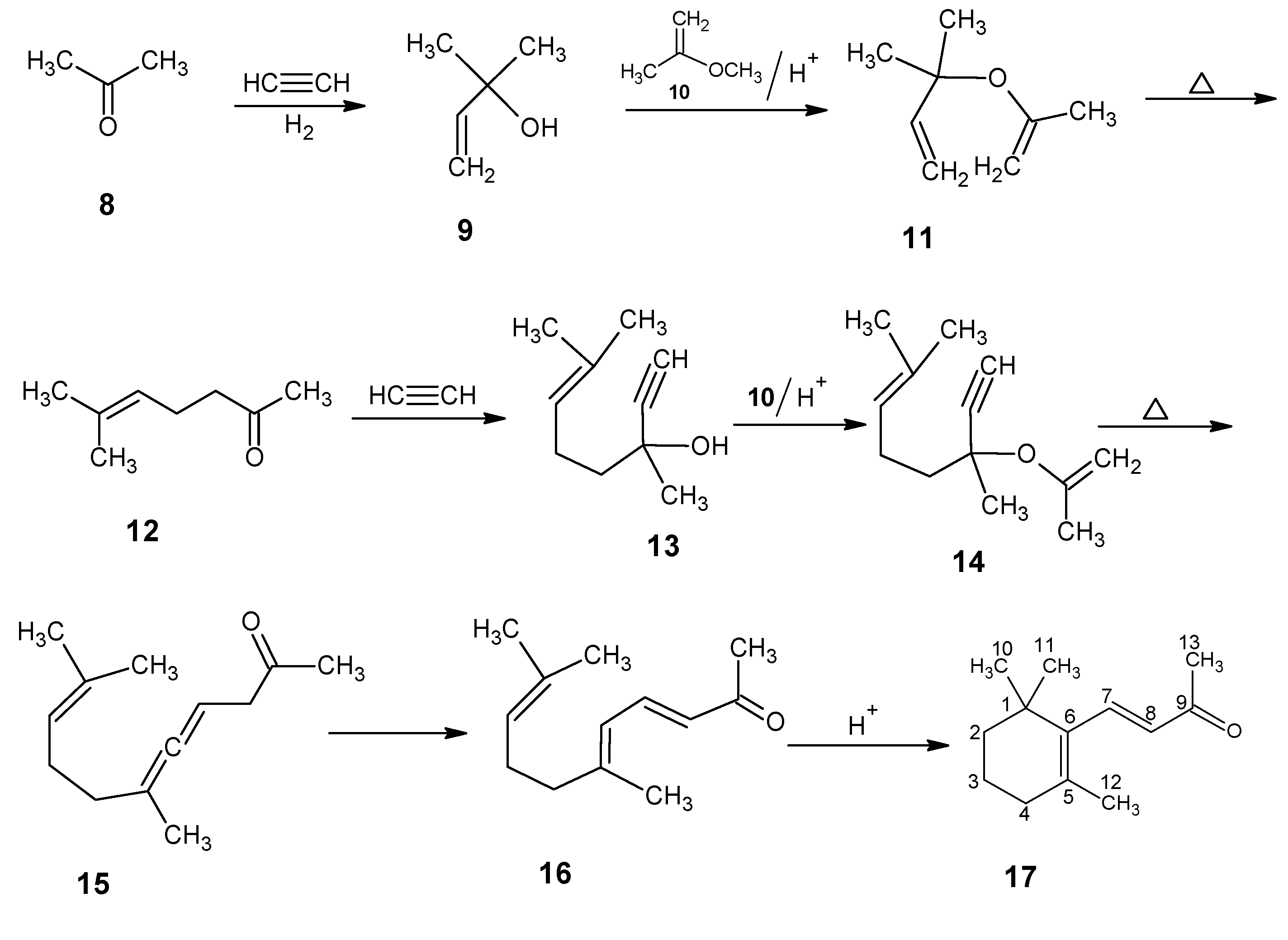
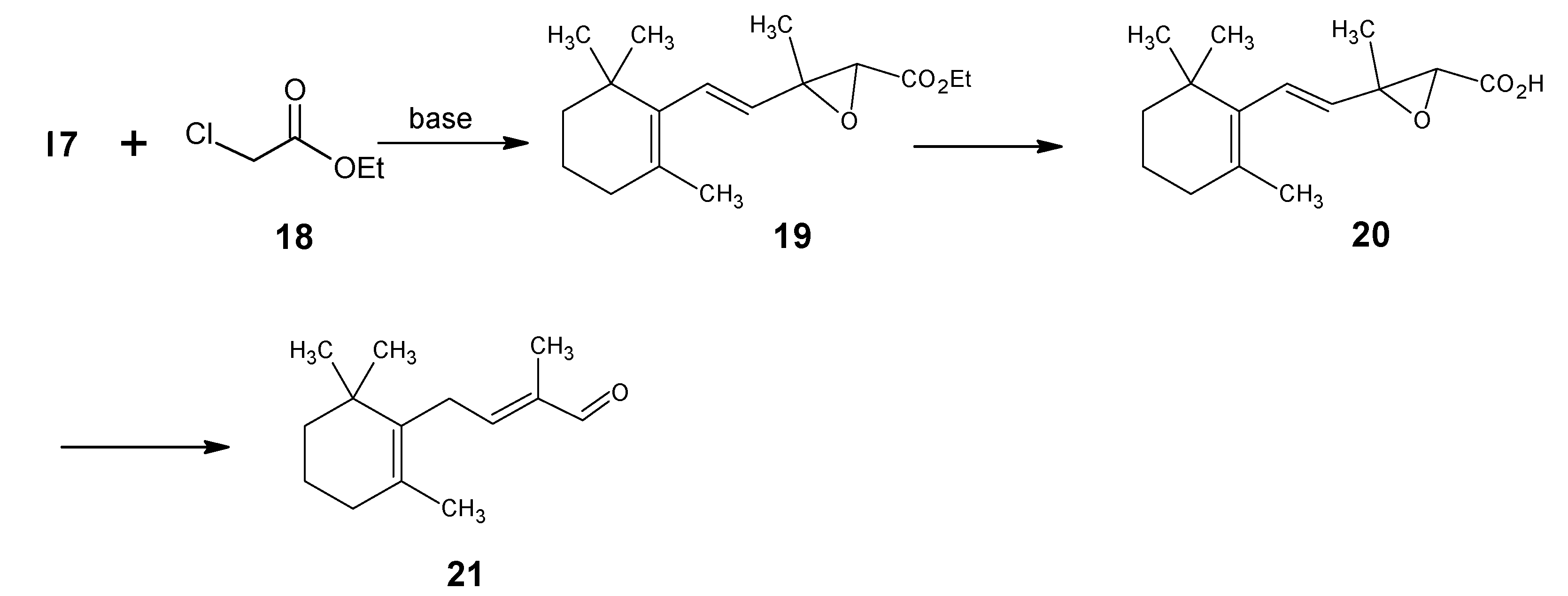
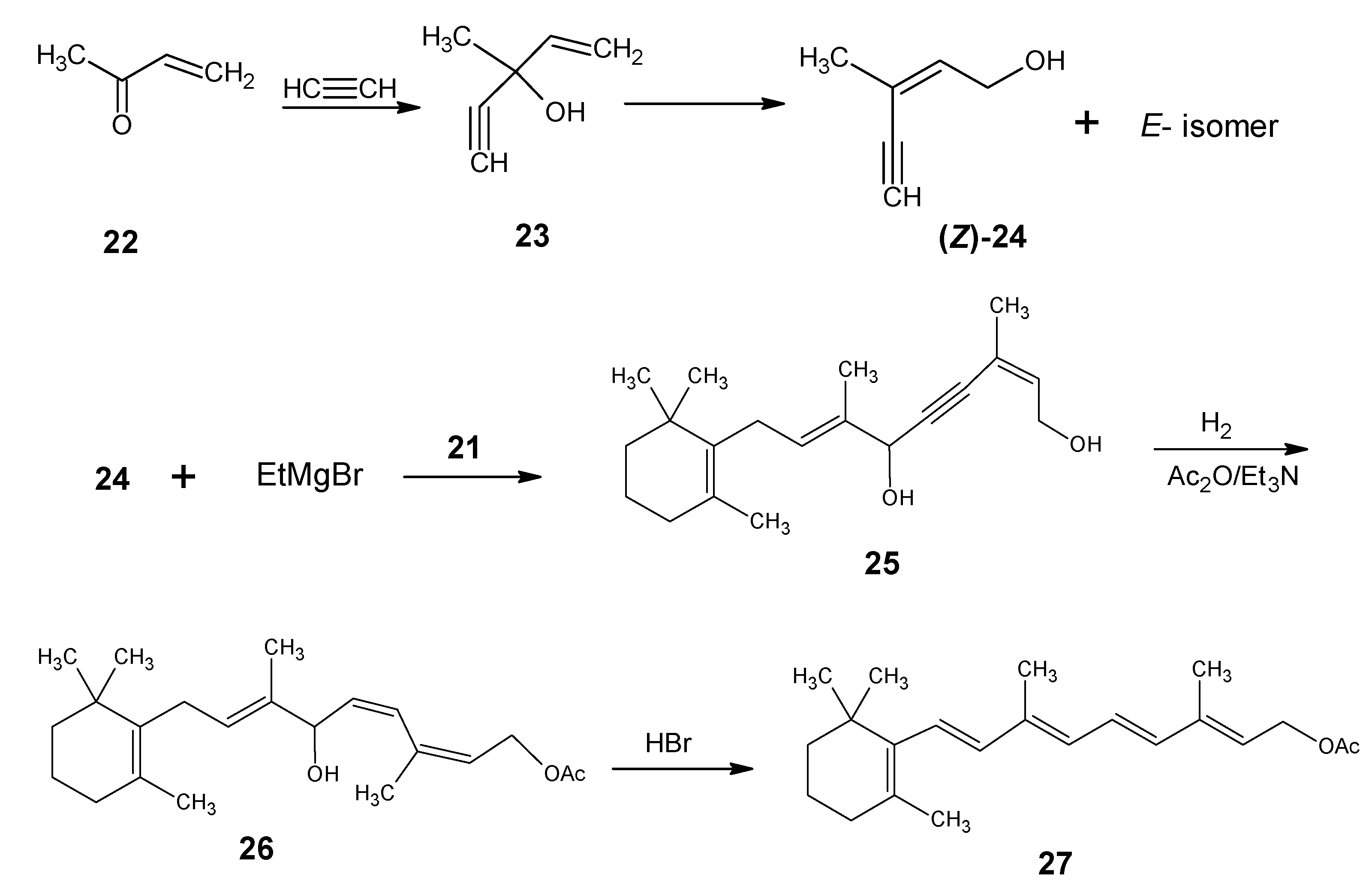
2. BASF
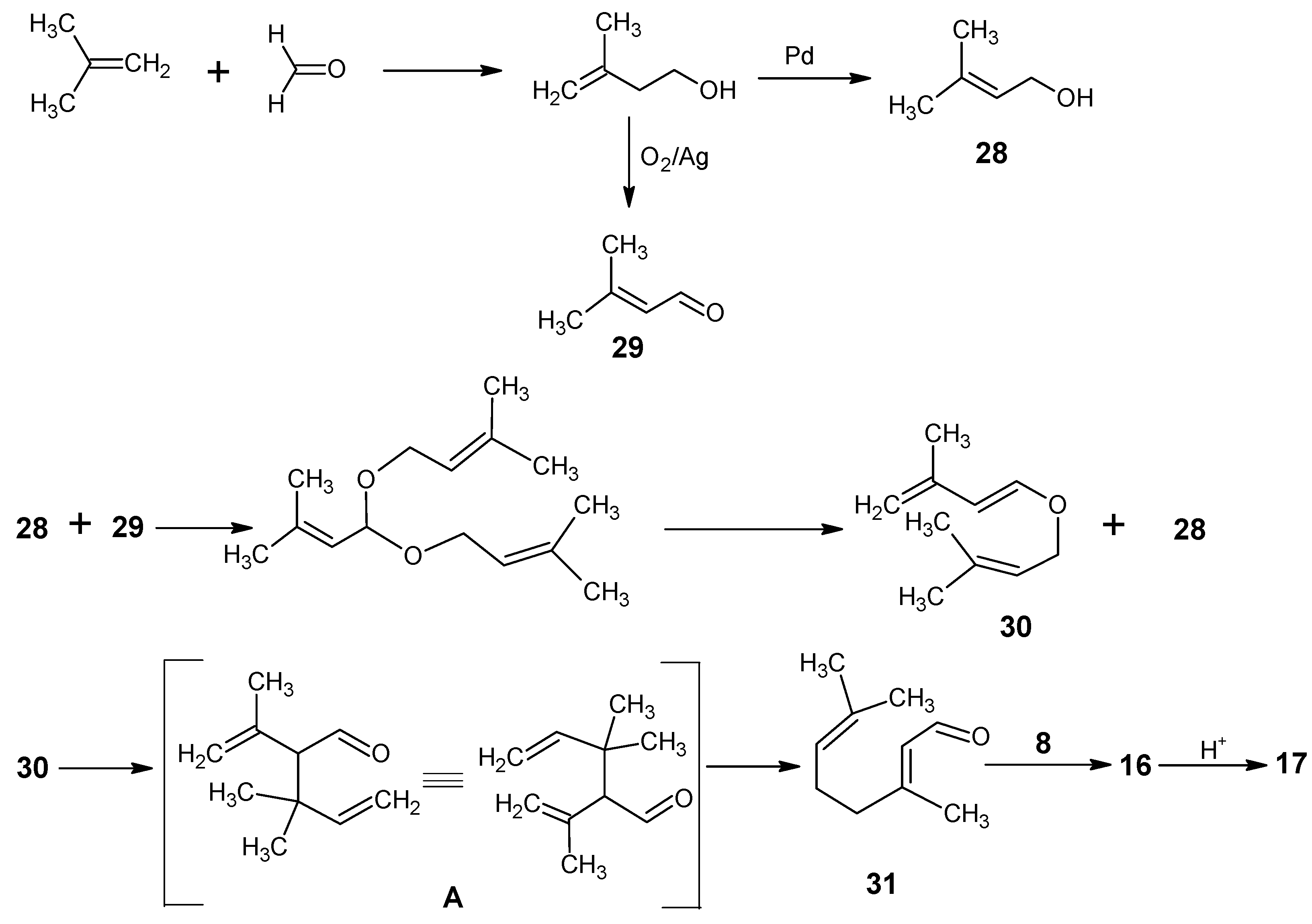
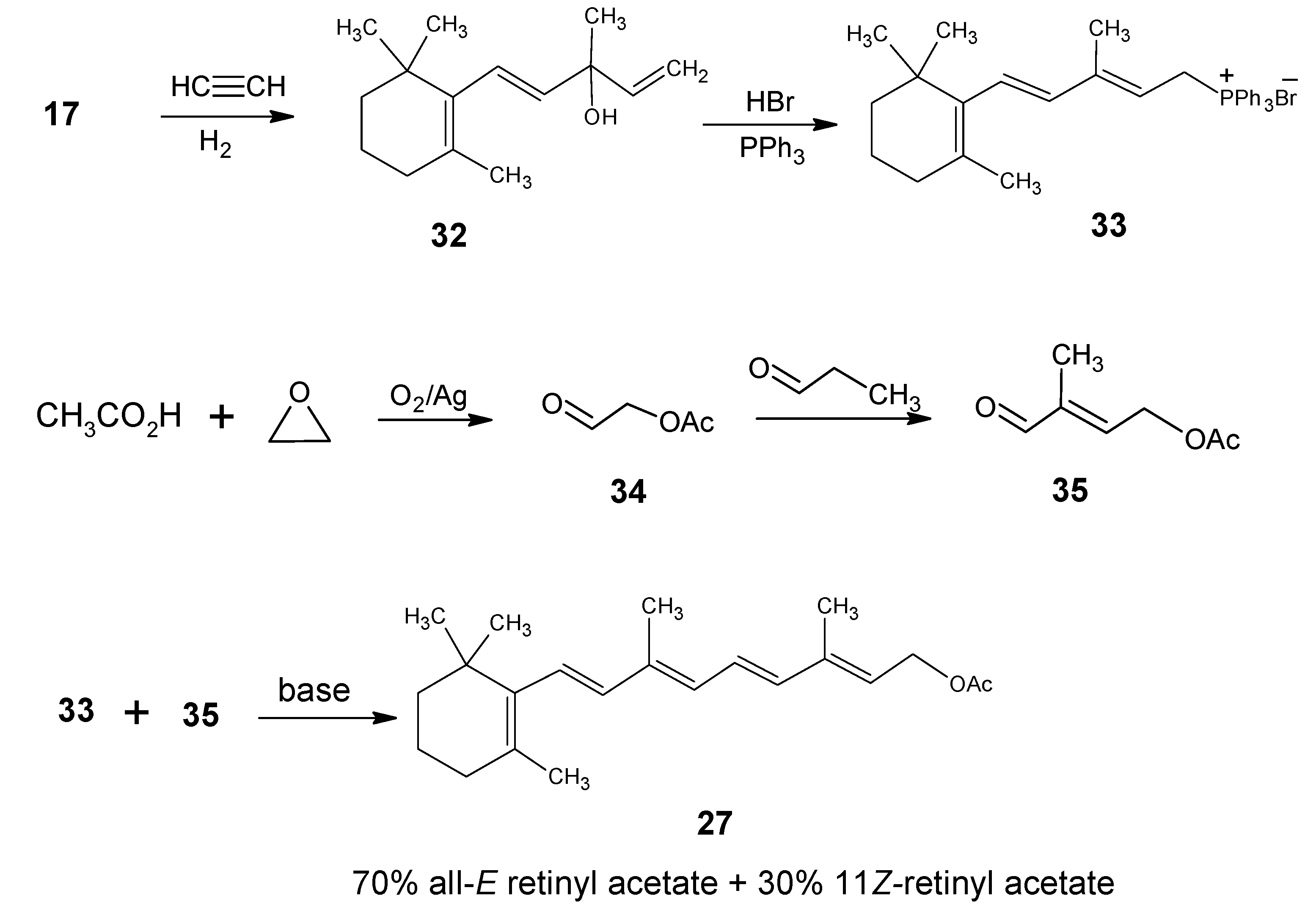
Part C. Site-Directed Highly Stable Isotope Enriched Retinals
1. Deuterium Labeled Retinals
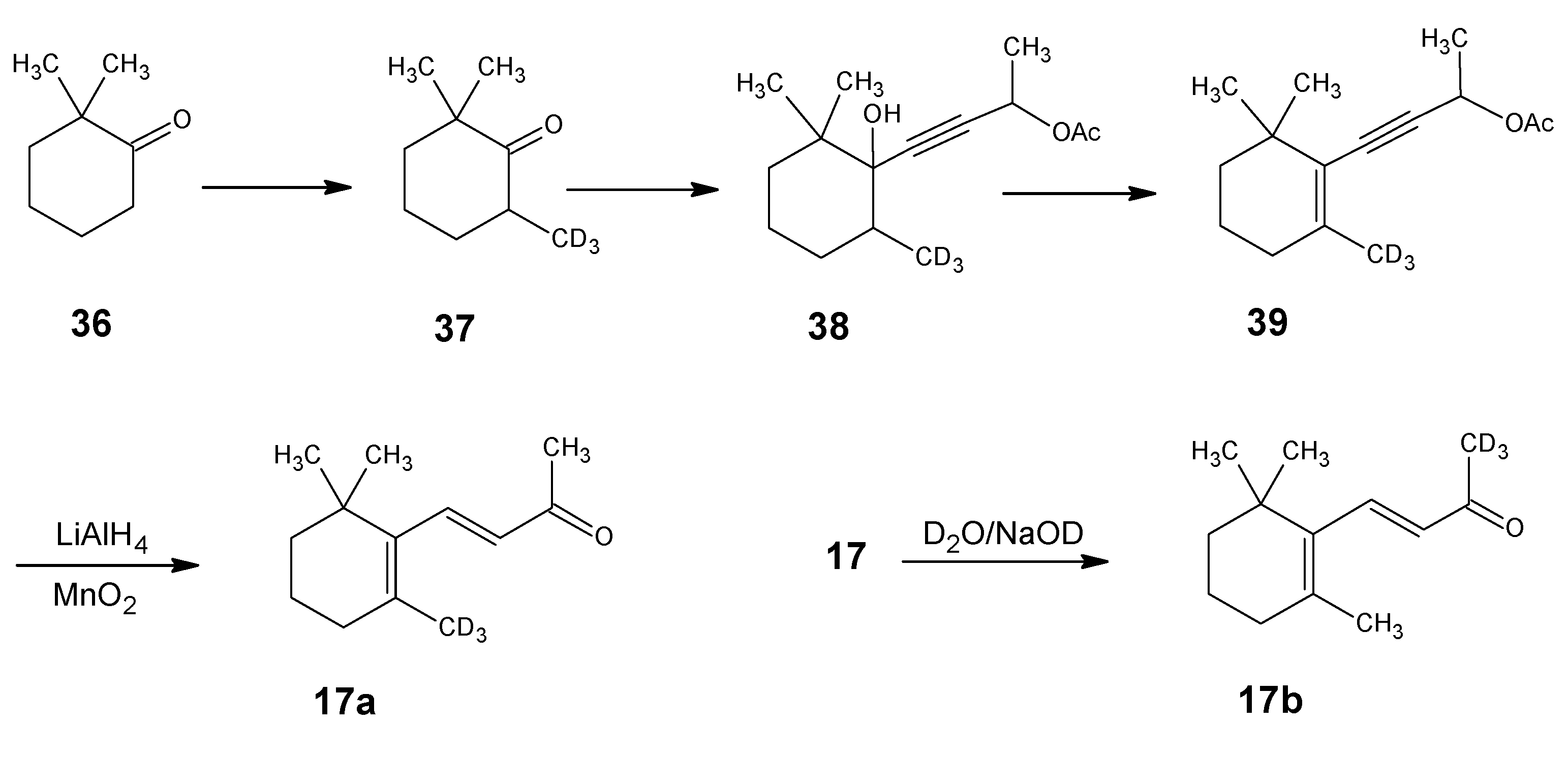
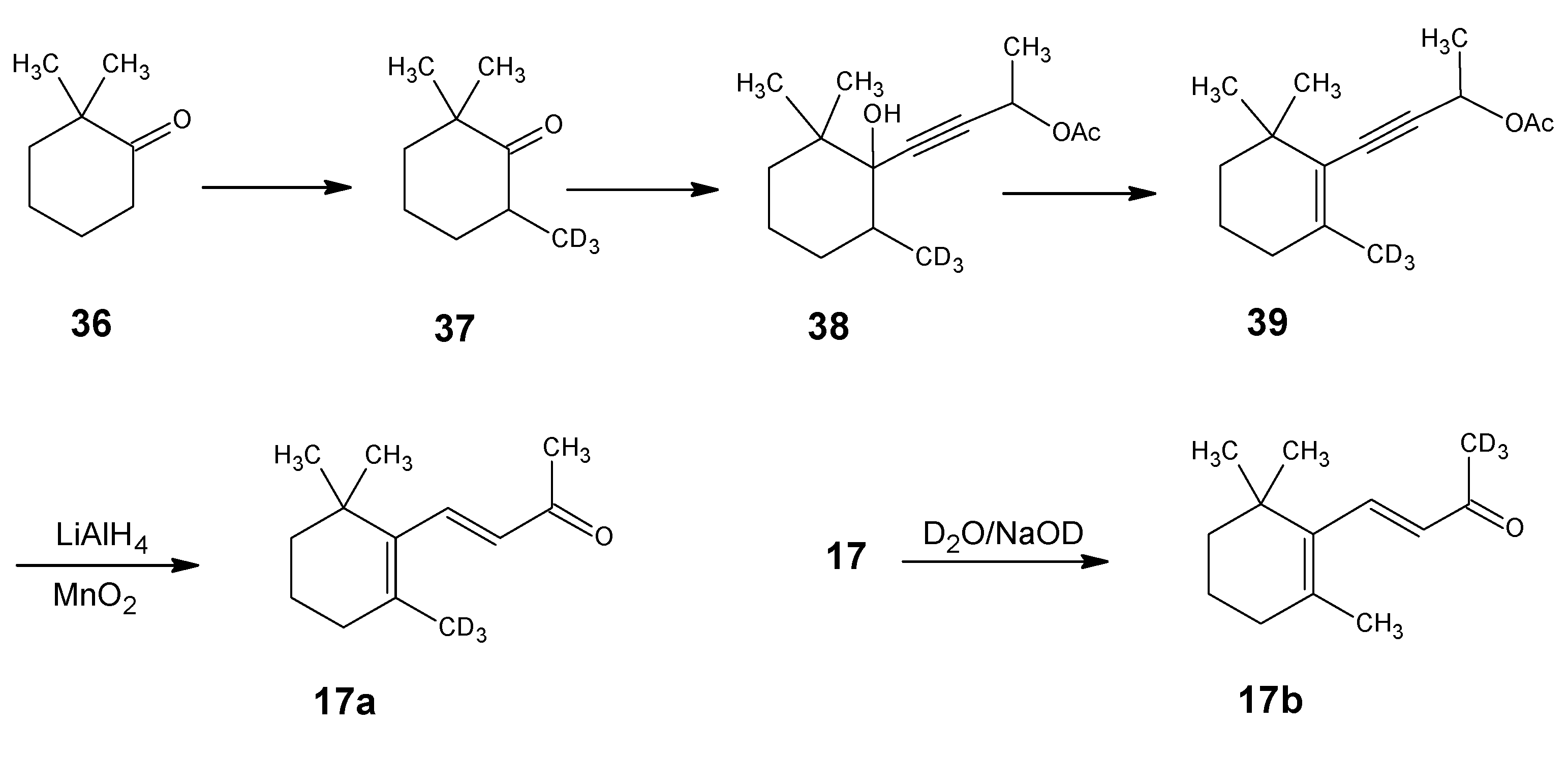
1.1. Incorporation of deuterium at positions 18 and 19 of retinal 1: Preparation of 11Z- [18-D3]-retinal and 11Z-[19-D3]-retinal via [12-D3]-β-ionone 17a and [13-D3]-β-ionone 17b, respectively

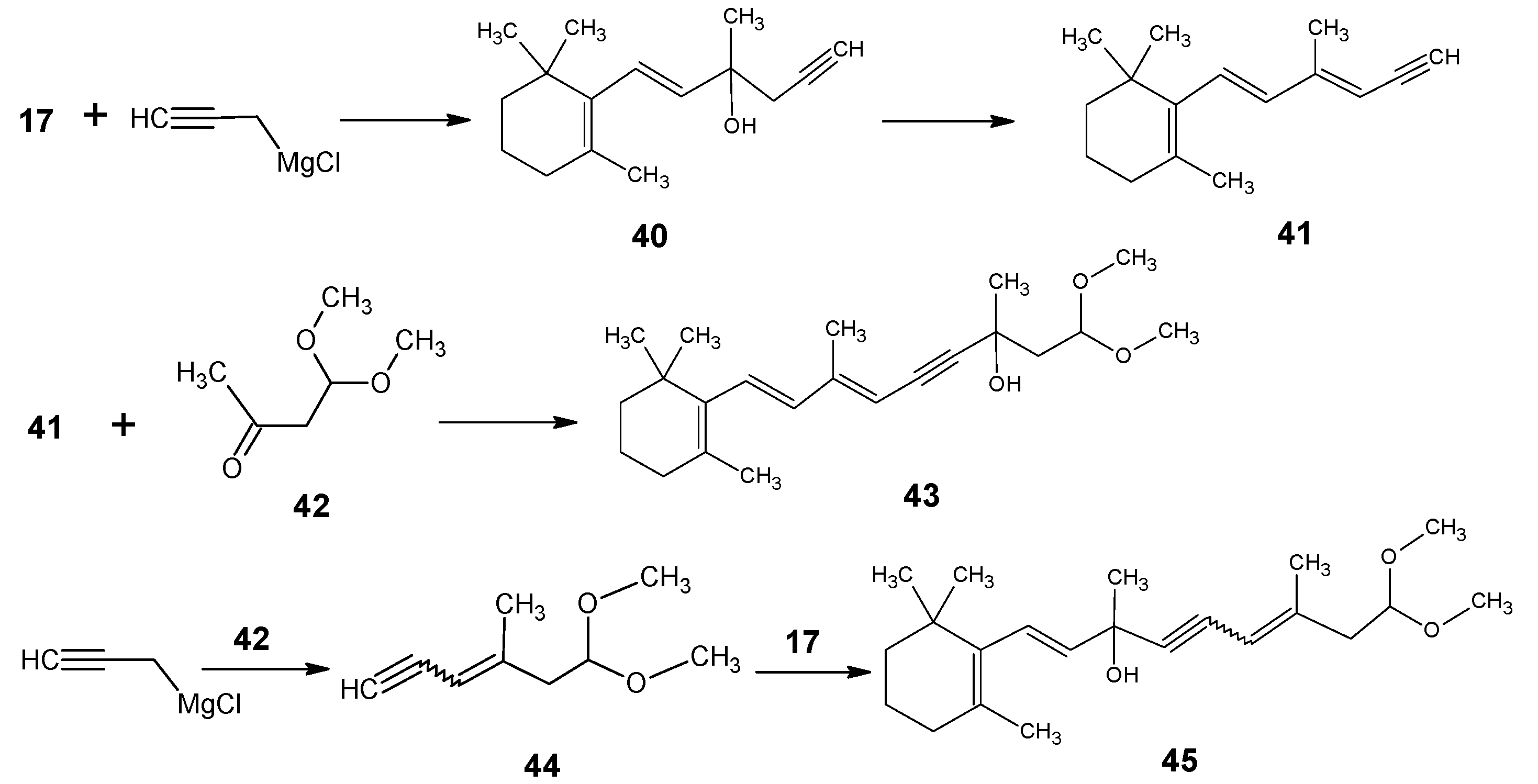
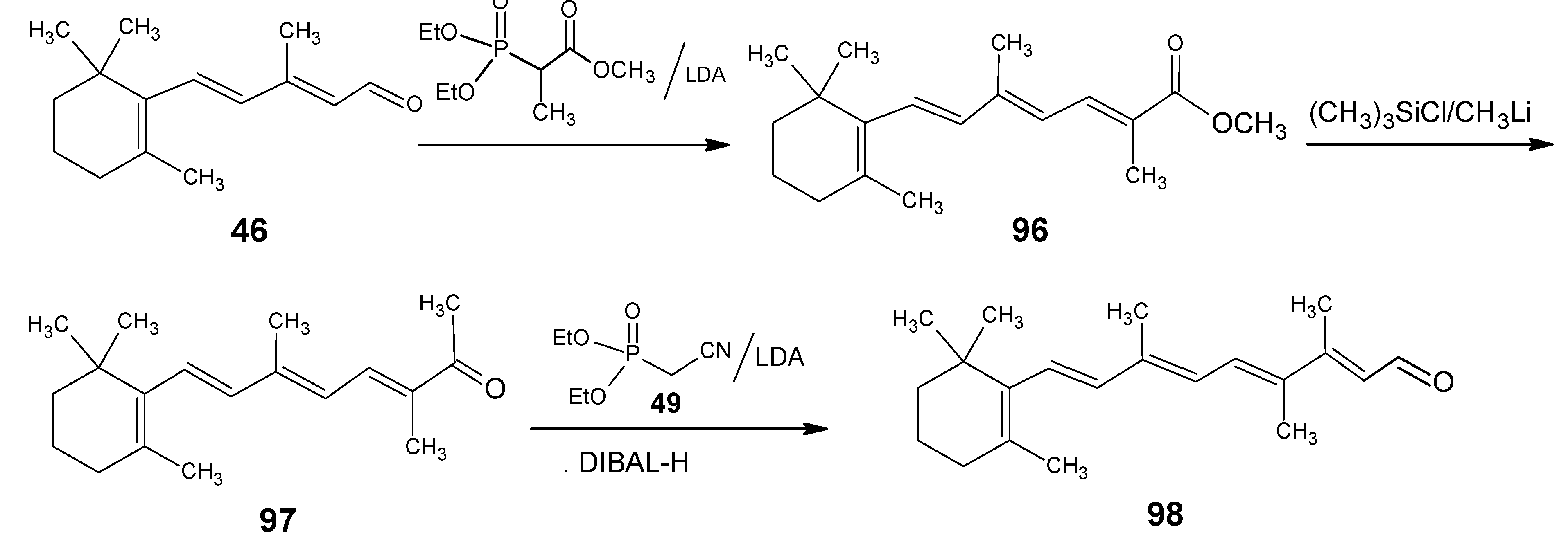
1.2. Incorporation of deuterium at positions 10, 11, 12, 14 and 20 of retinal 1: Preparation of all-E [10-D]-retinal, all-E [11-D]-retinal, all-E [12-D]-retinal, all-E [11,12-D2]-retinal, all-E [10,11-D2]-retinal, all-E [14,20,20,20-D4]-retinal
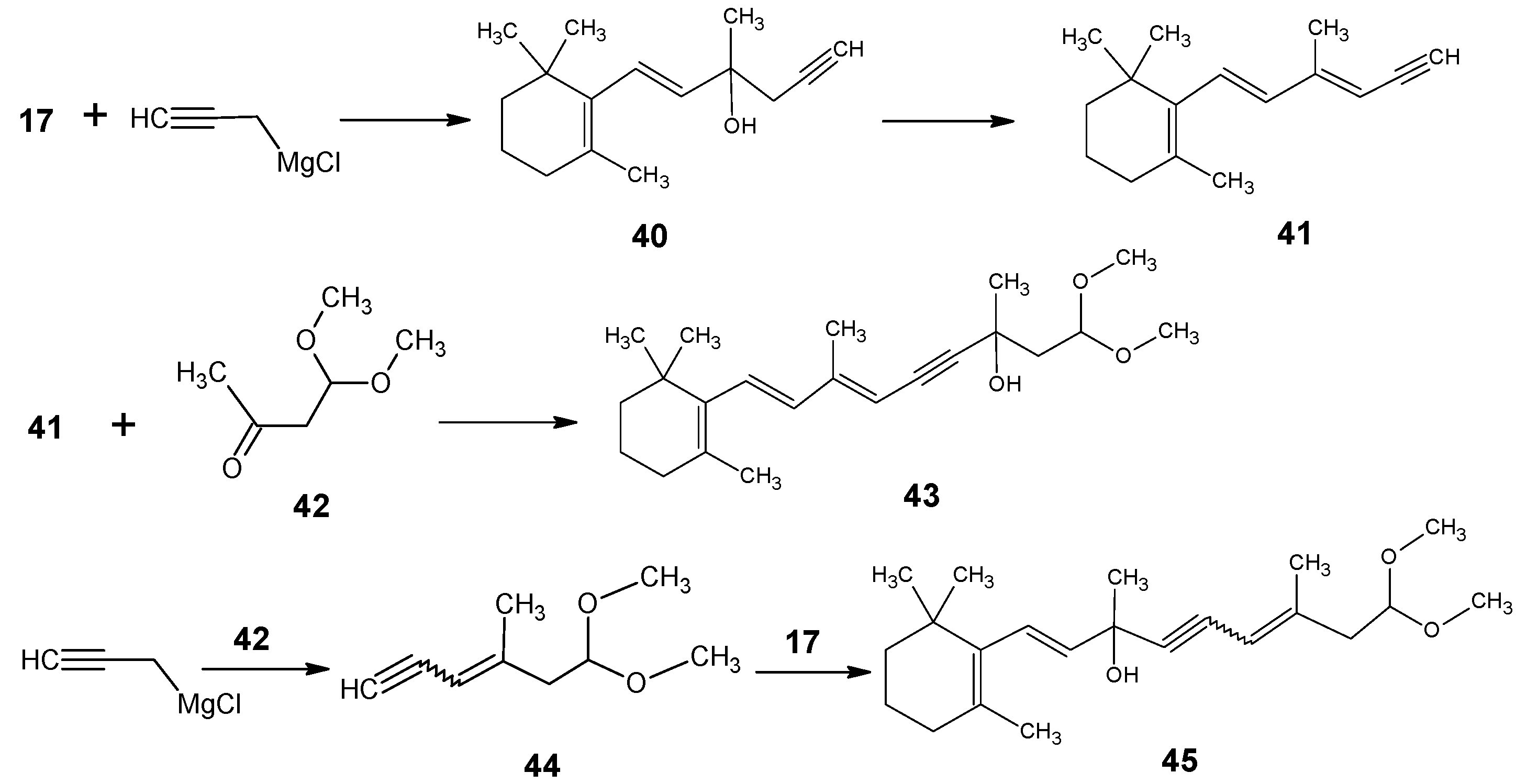
1.3. Incorporation of deuterium at positions 14,15, and 20 of retinal 1: Preparation of all-E [14-D]-retinal, all-E [15-D]-retinal, all-E [14,15-D2]-retinal and all-E [20,20,20-D3]-retinal.
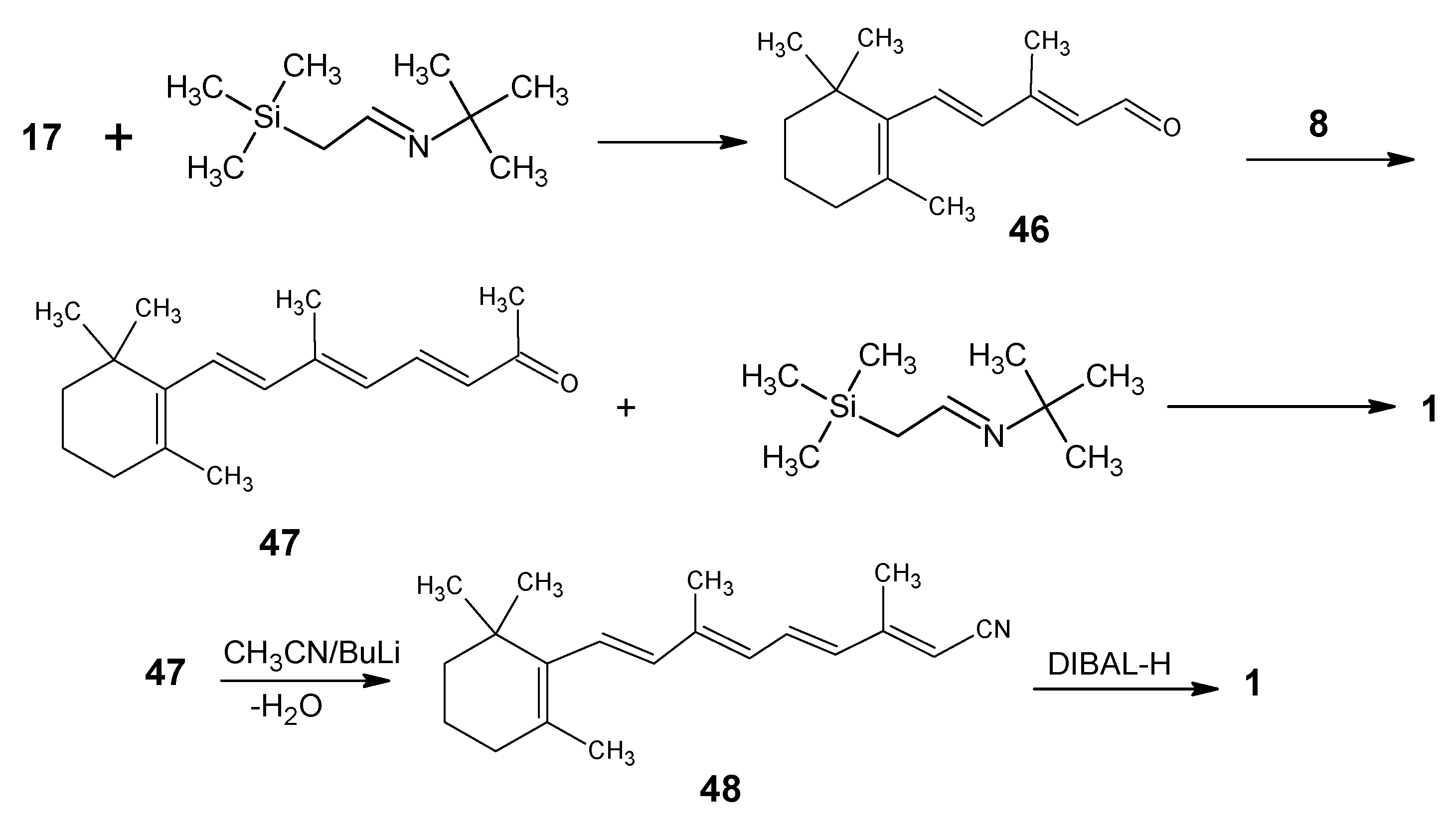
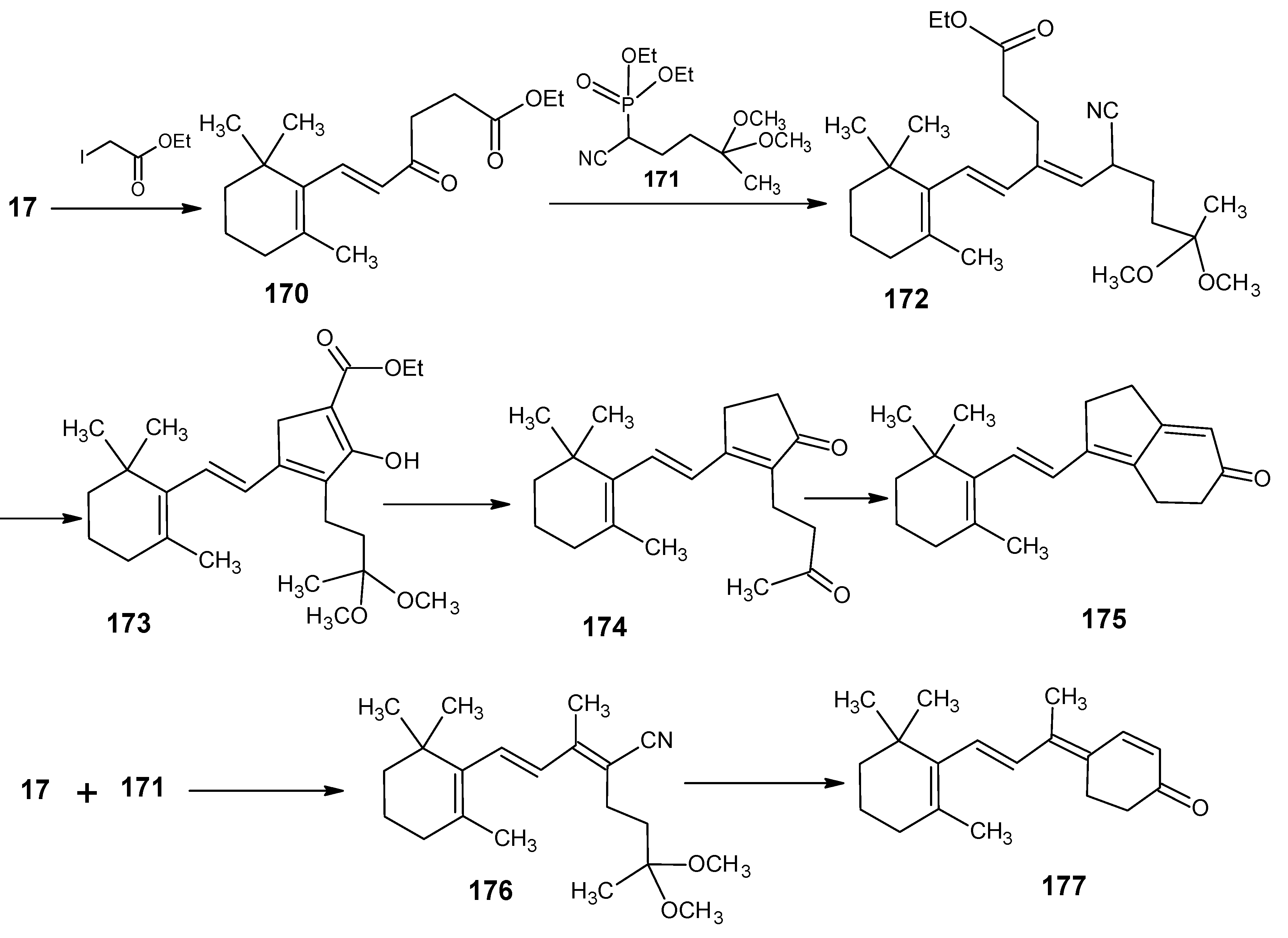
2. 13C-Labeled Retinals
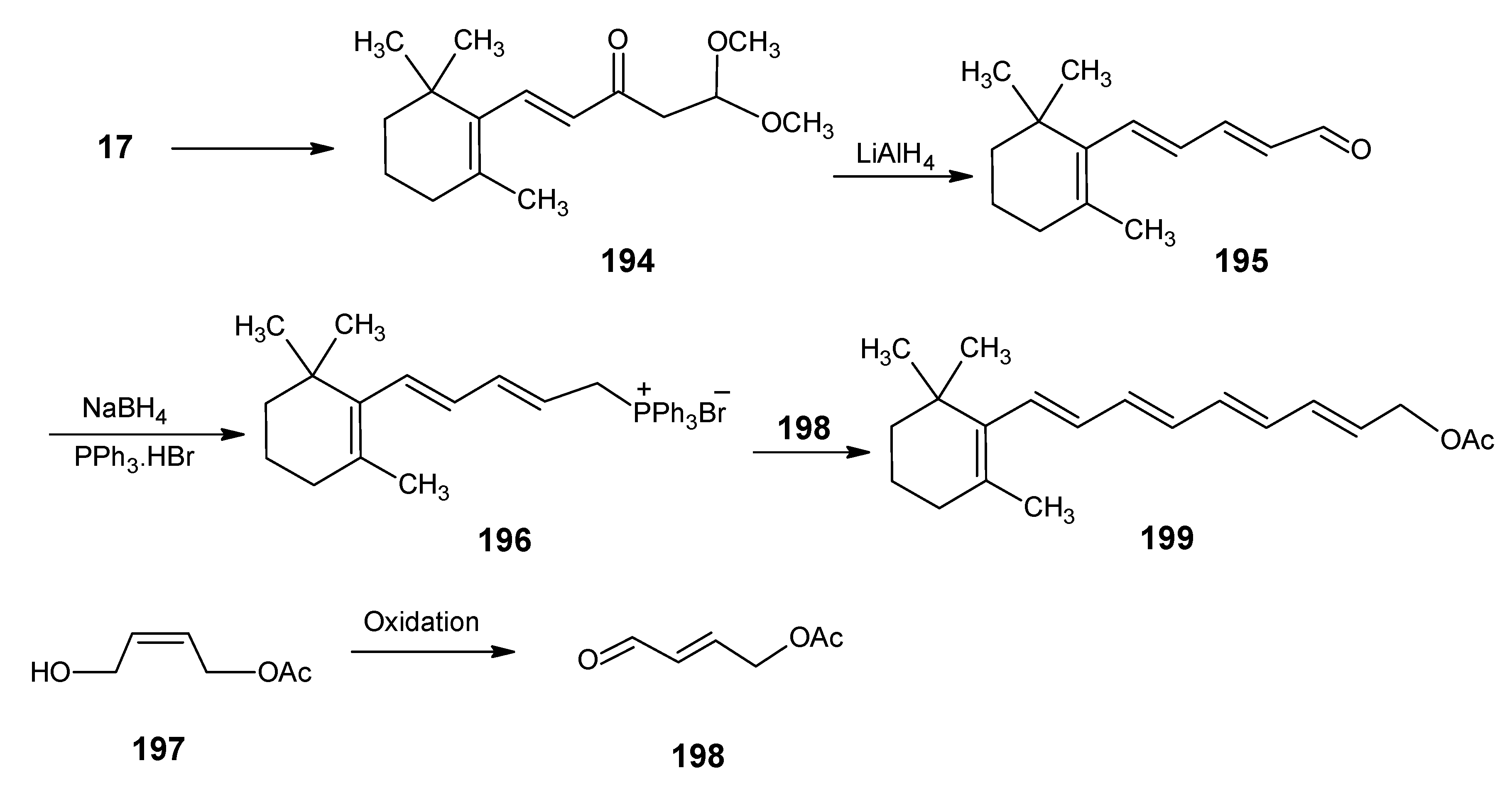
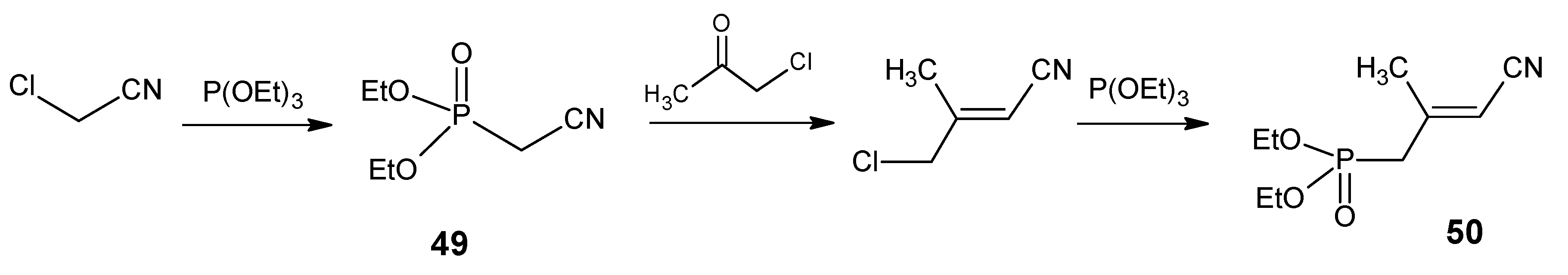
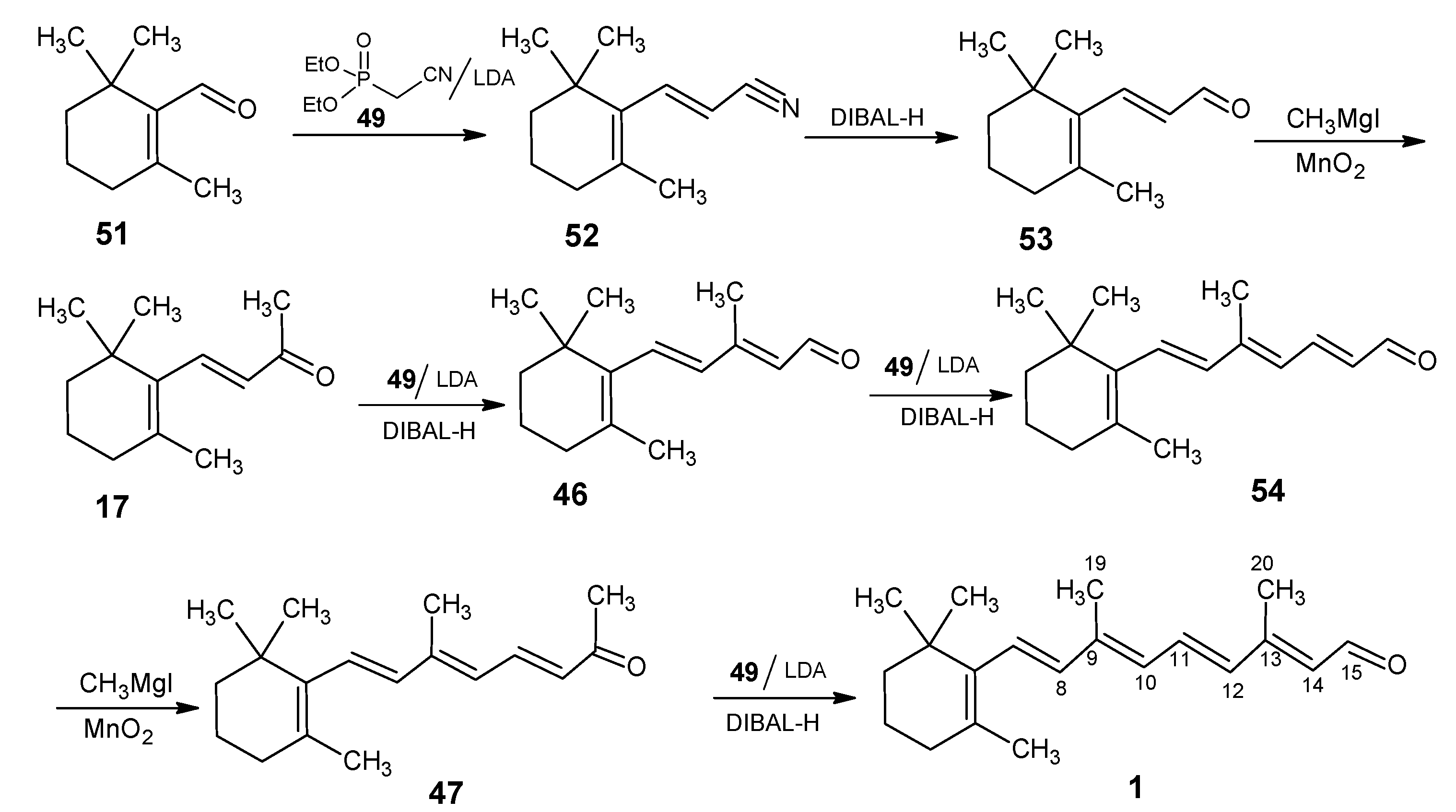
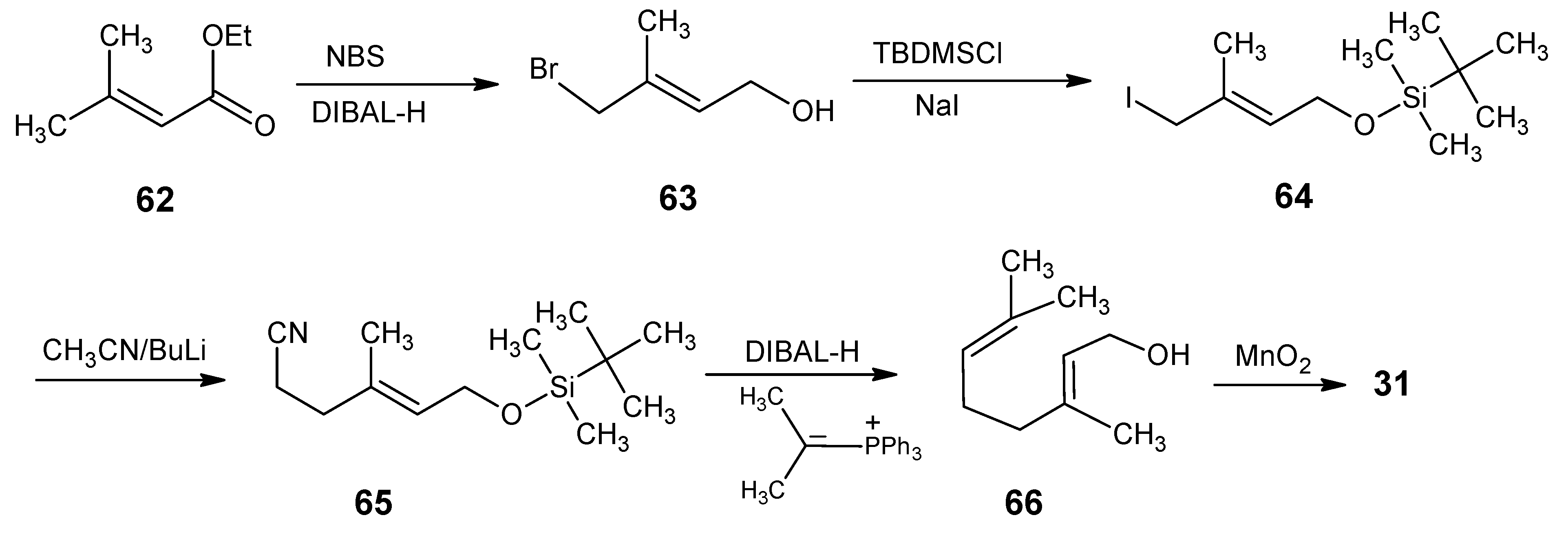
2.1. Incorporation of 13C at positions 14 and 15 of retinal 1: Preparation of [14-13C]-retinal, [15-13C]-retinal and [14,15-13C2]-retinal
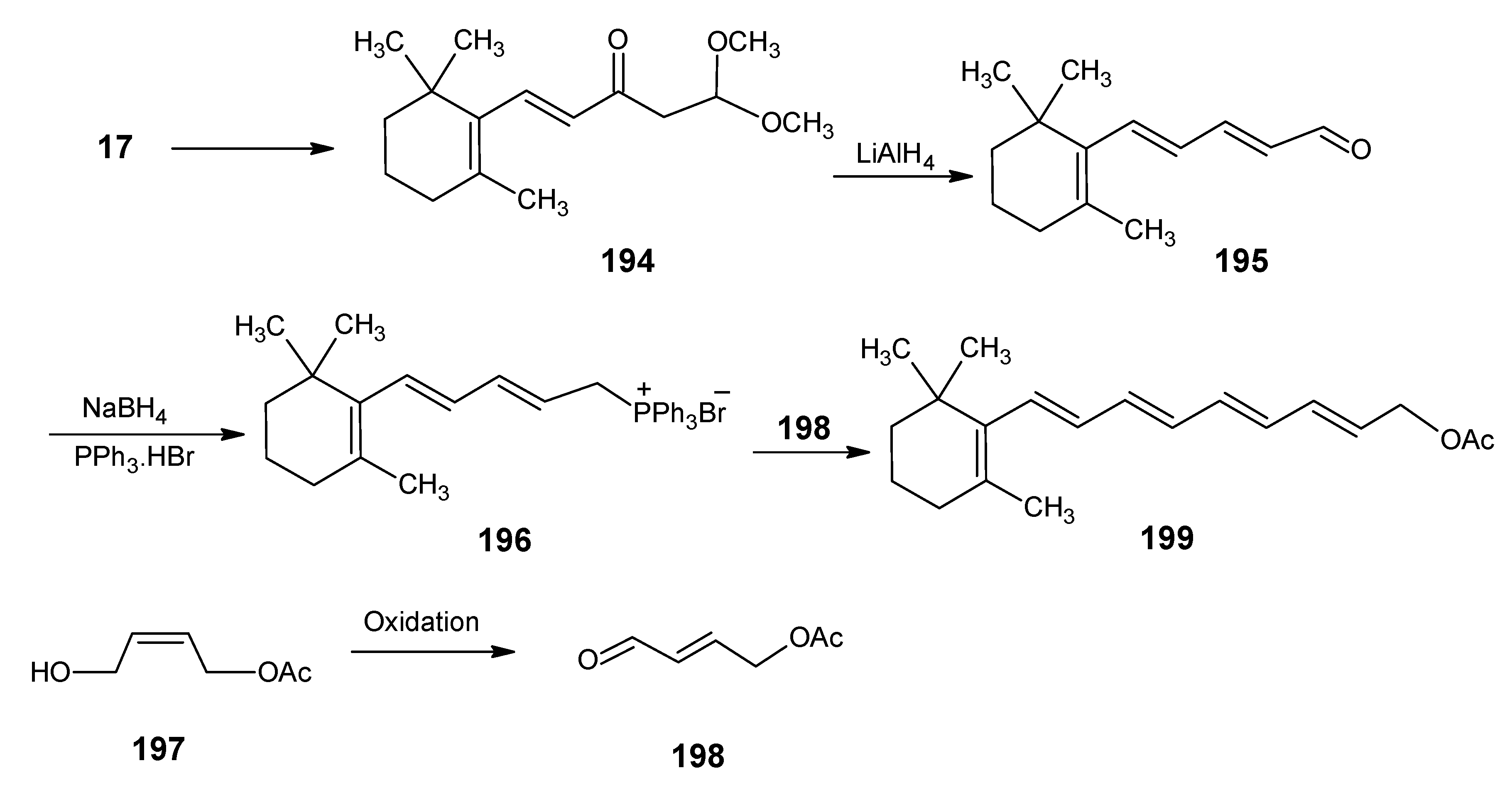
2.2. Incorporation of 13C in the six membered ring of retinal 1: Preparation of [1-13C]-, [2-13C]-, [1,3-13C2]- and [1,2, 3-13C3]-, [3-13C]-, [4-13C]-, [5-13C]>-, [4,5-13C2]-, [6-13C]-, [7-13C]-, and [18-13C]- retinal

2.3. Incorporation of 13C at all positions of retinal 1: Preparation of [U-13C]-retinal
3. Isotope Enriched Chemically Modified Retinals
3.1. Preparation of (11Z)-3,4-didehydroretinal, (3R)-(11Z)-3-hydroxyretinal and (4R)-(11Z)-4-hydroxyretinal
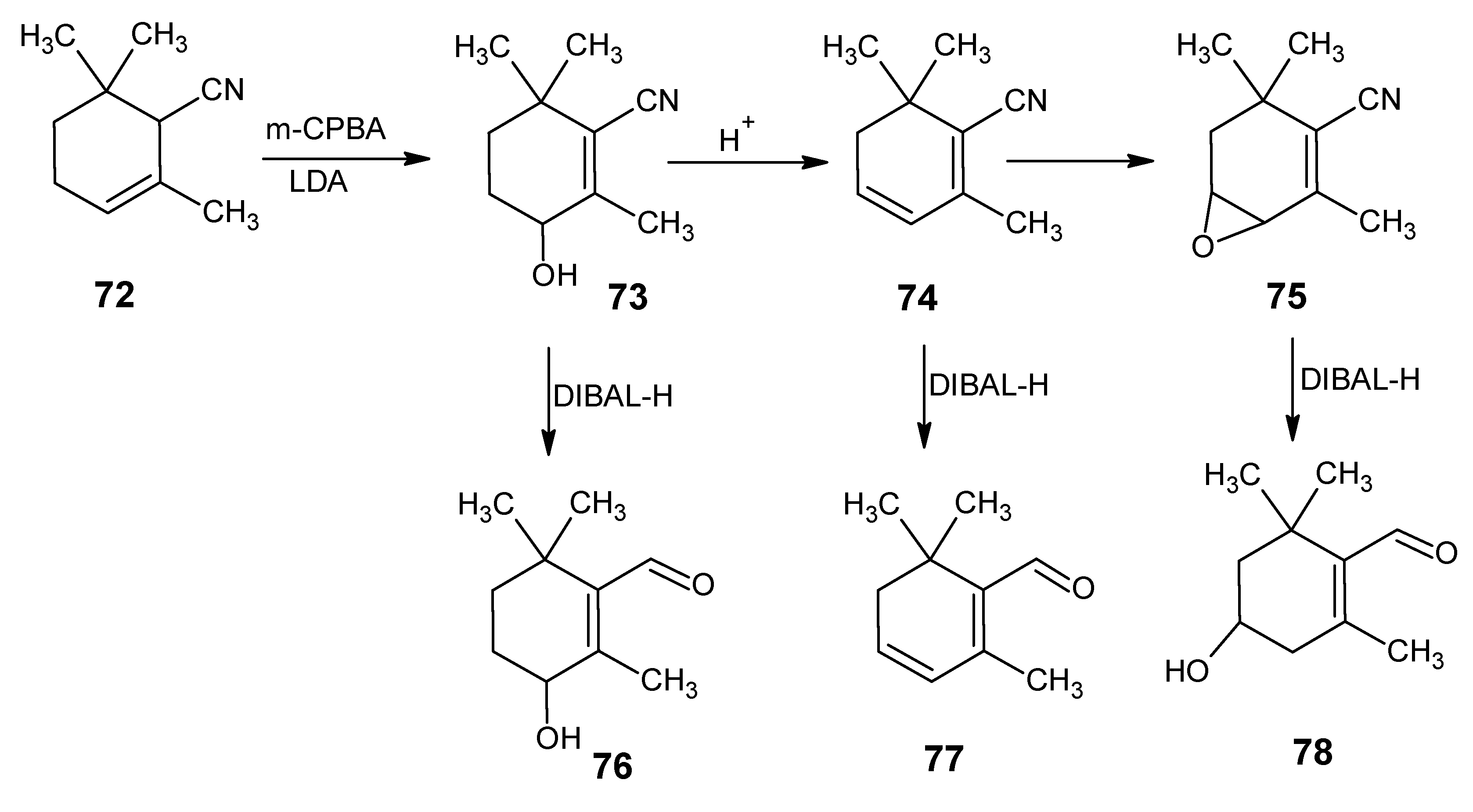
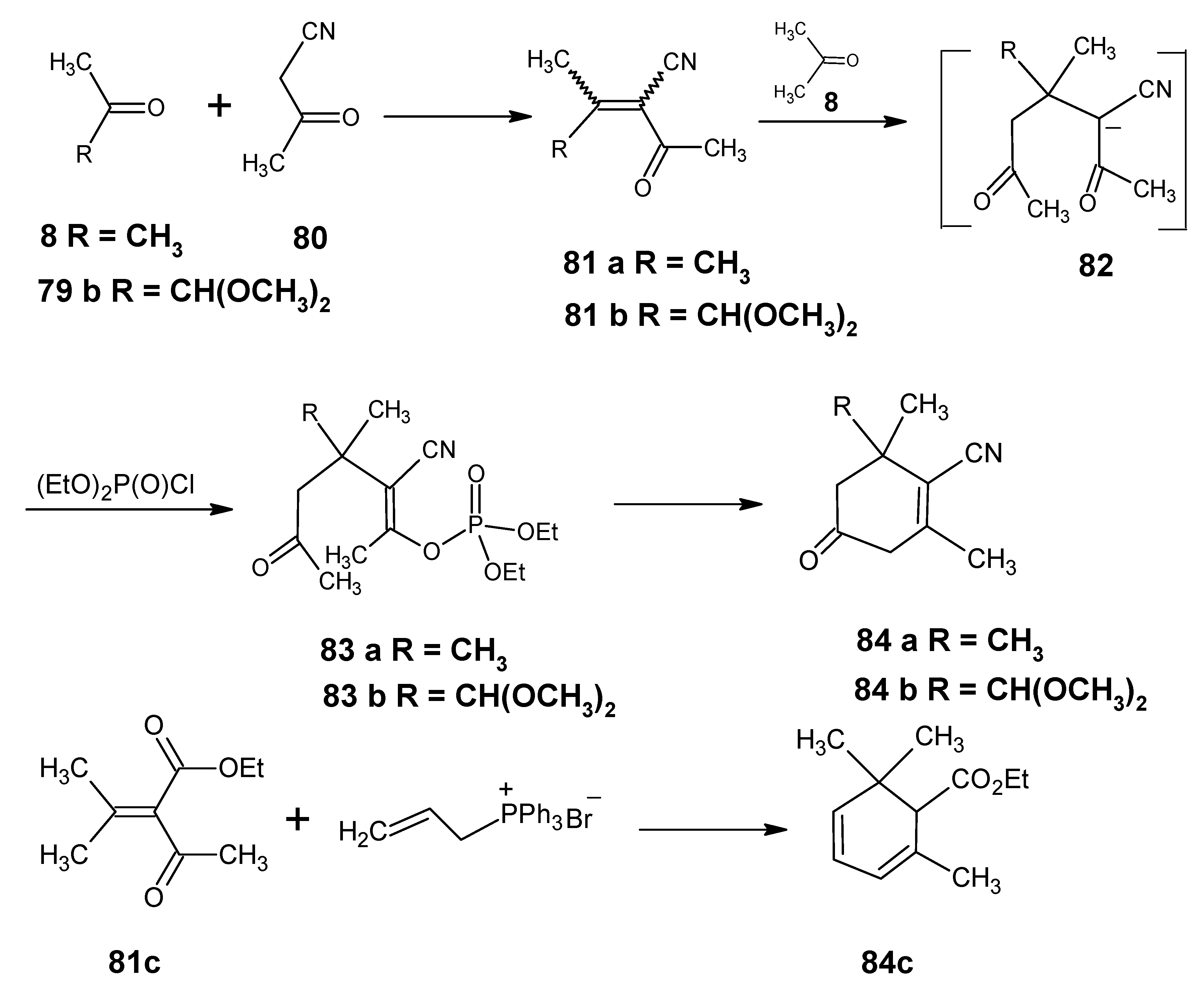
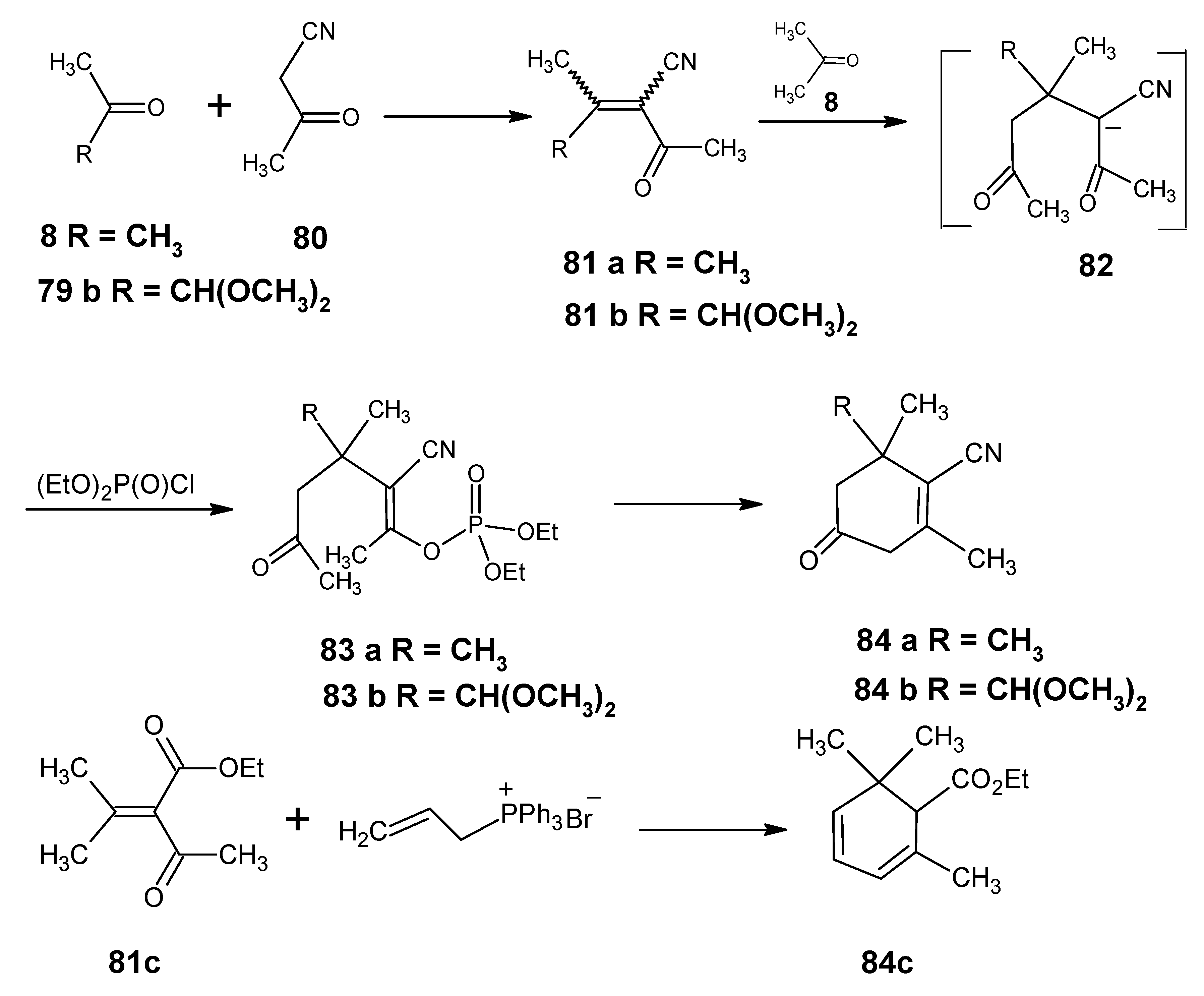
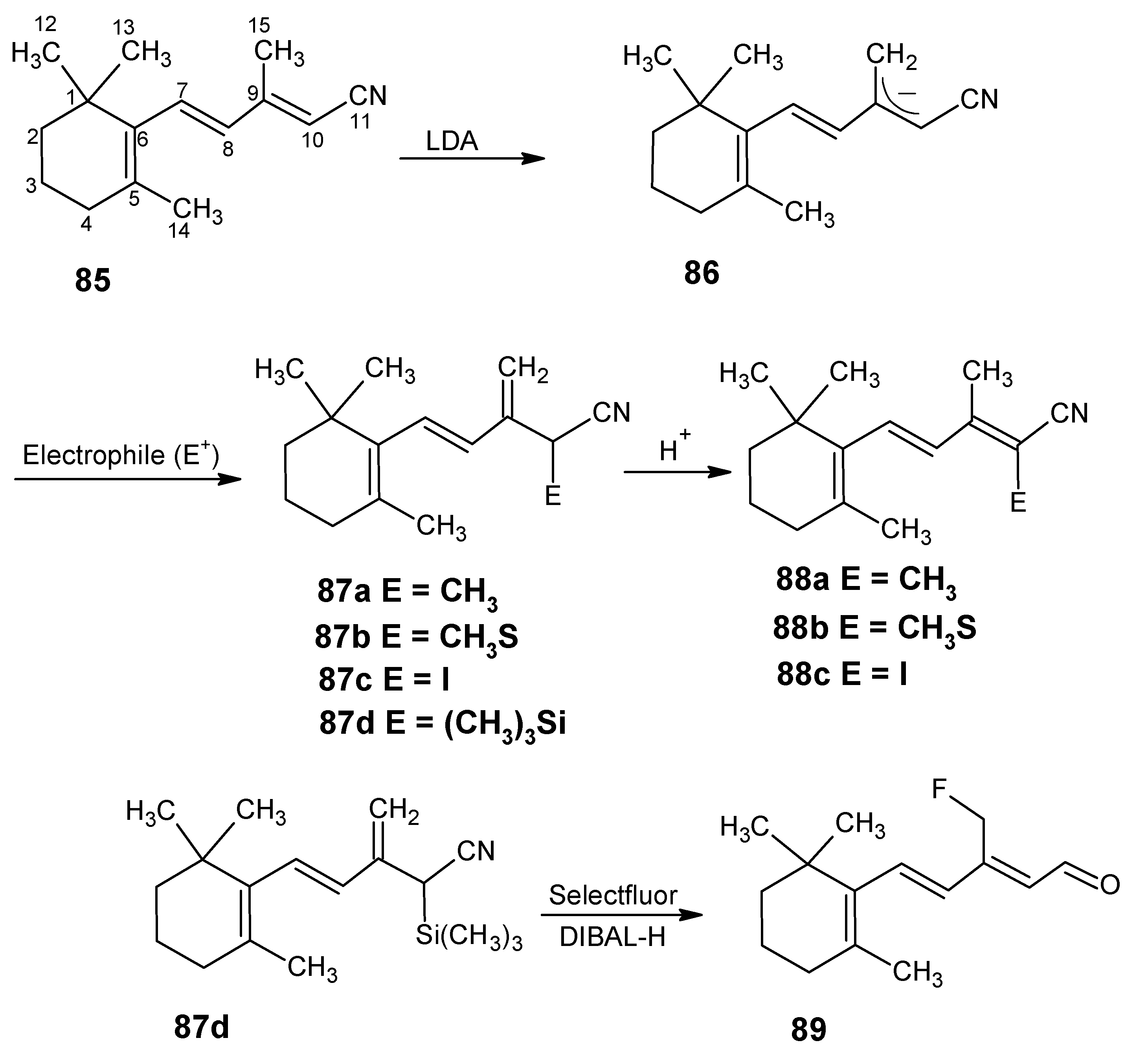
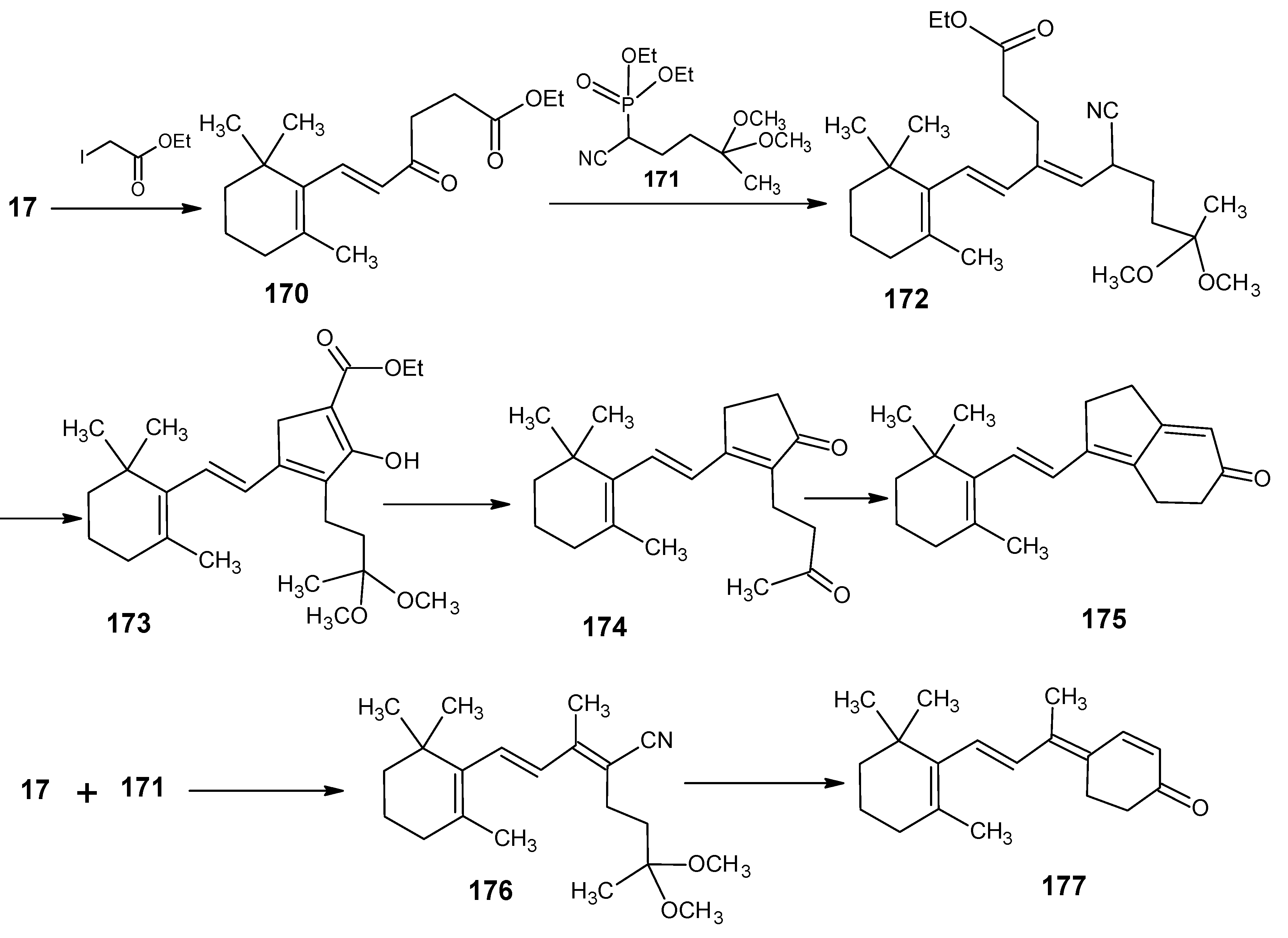
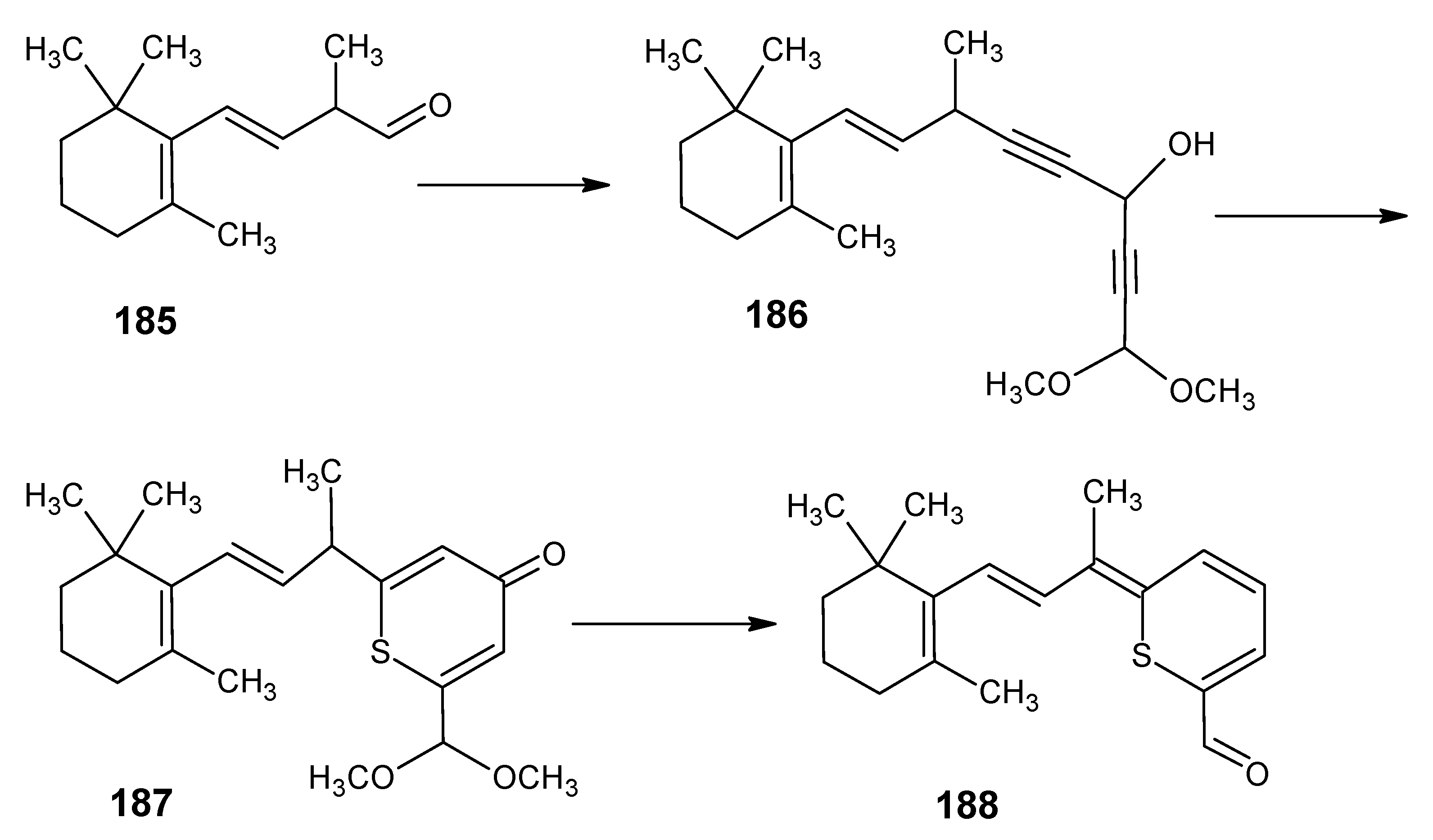
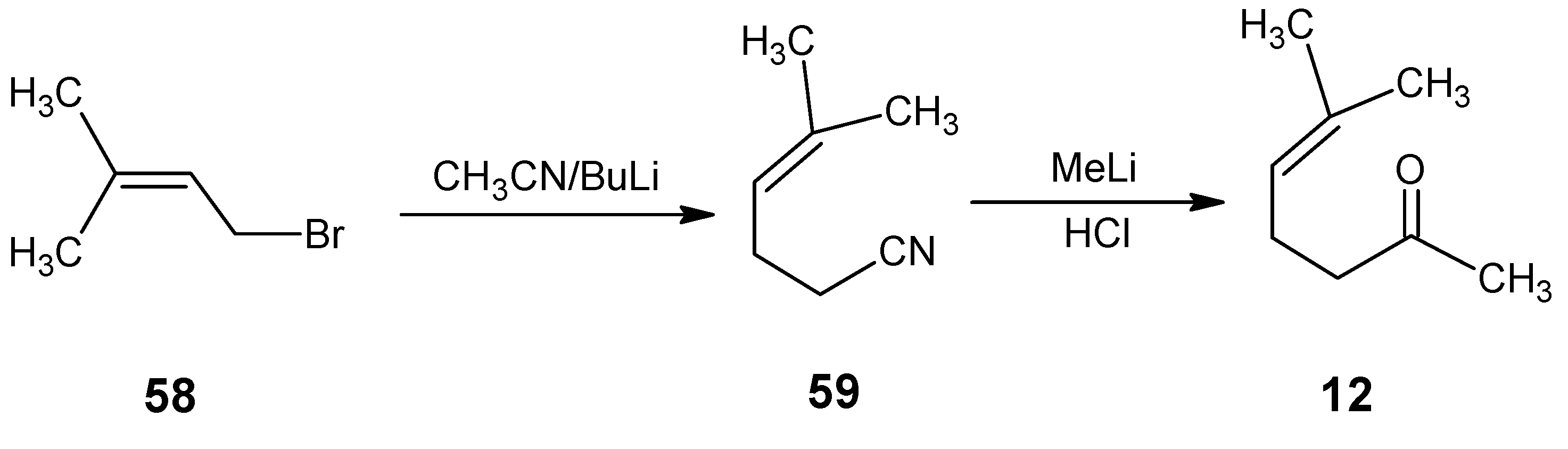
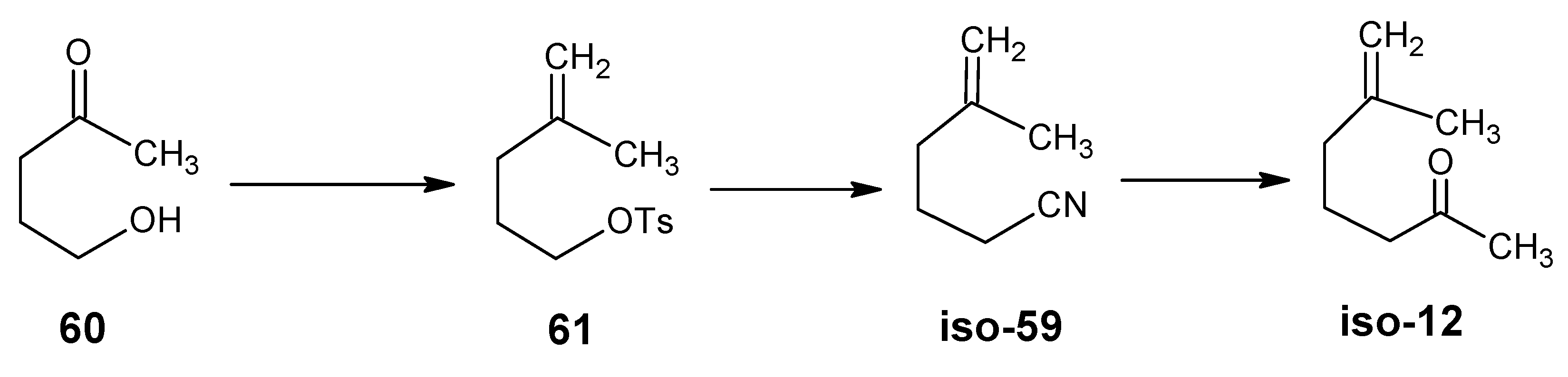
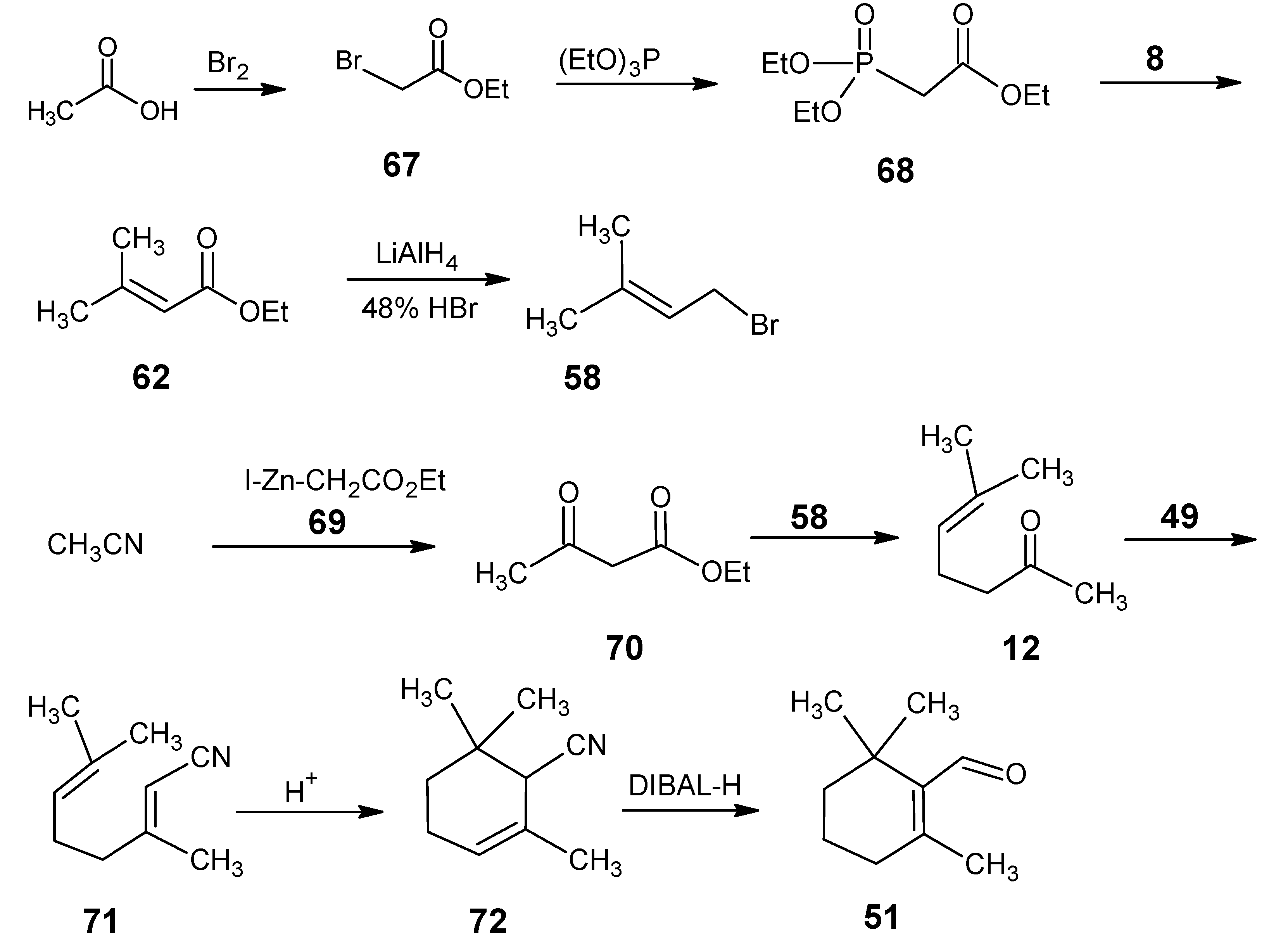
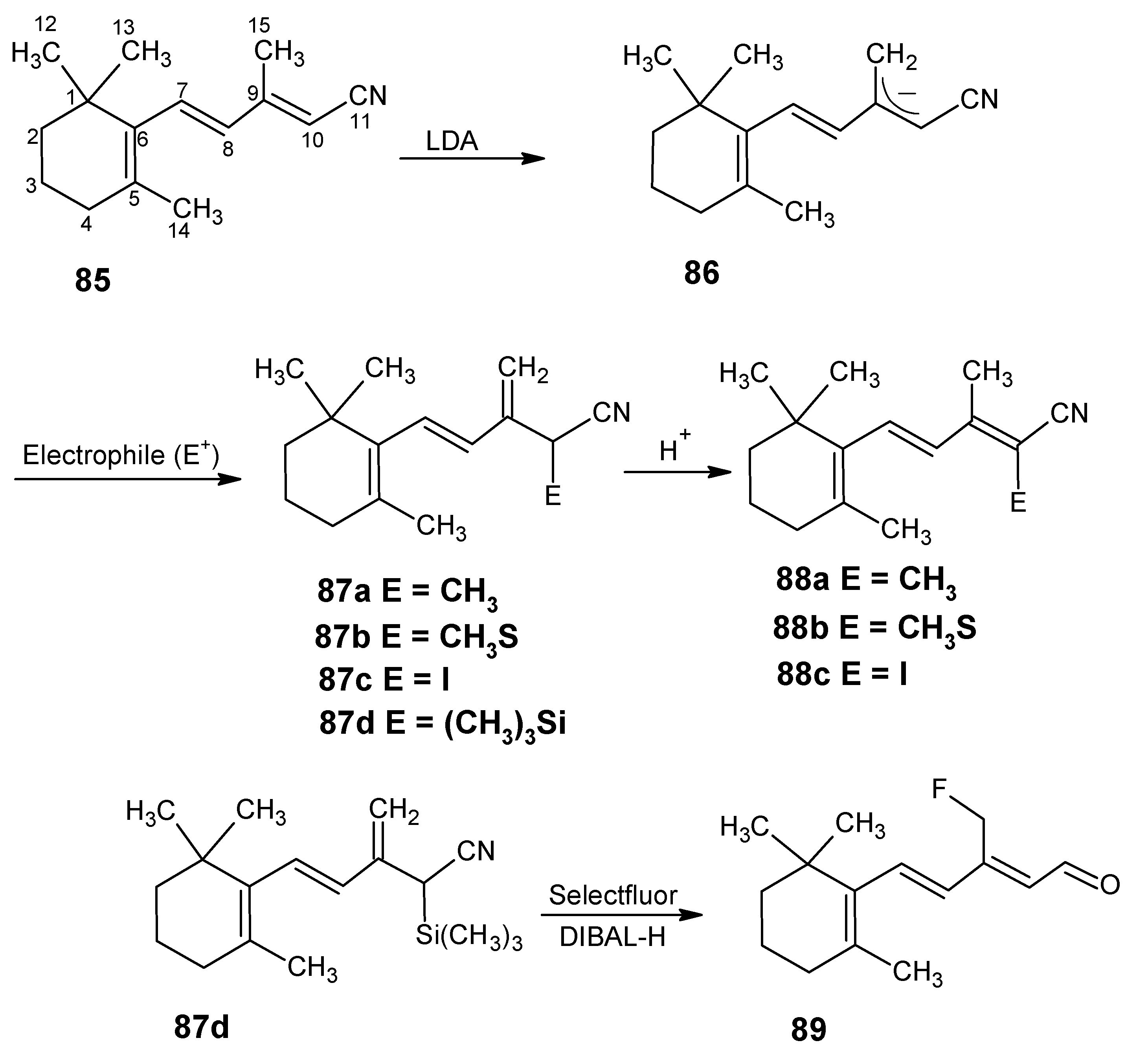
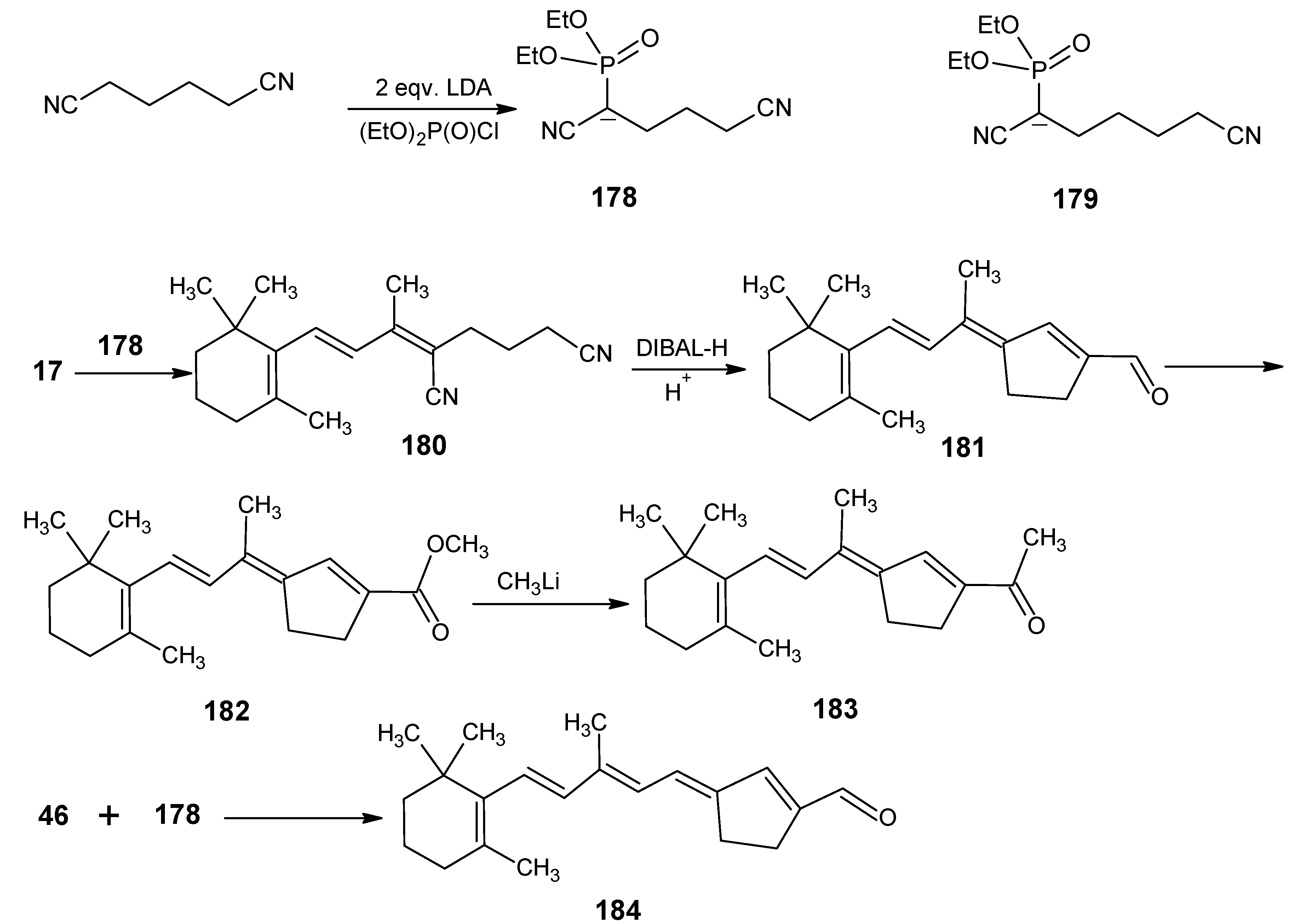
3.2. Preparation of 10-methylretinal, 10-methylthioretinal, 10-iodoretinal and 19-fluororetinal via β-ionylidene acetonitrile 85
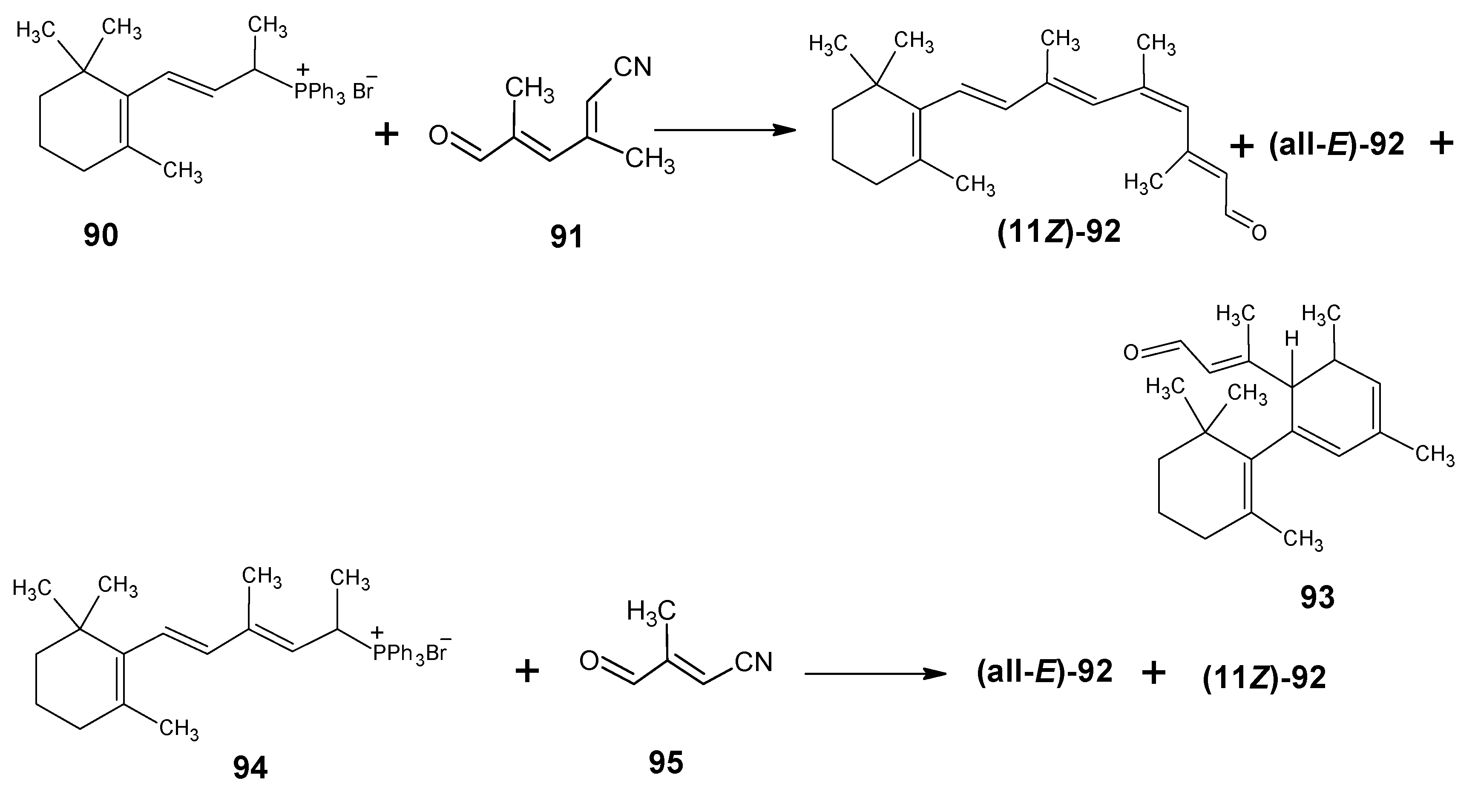
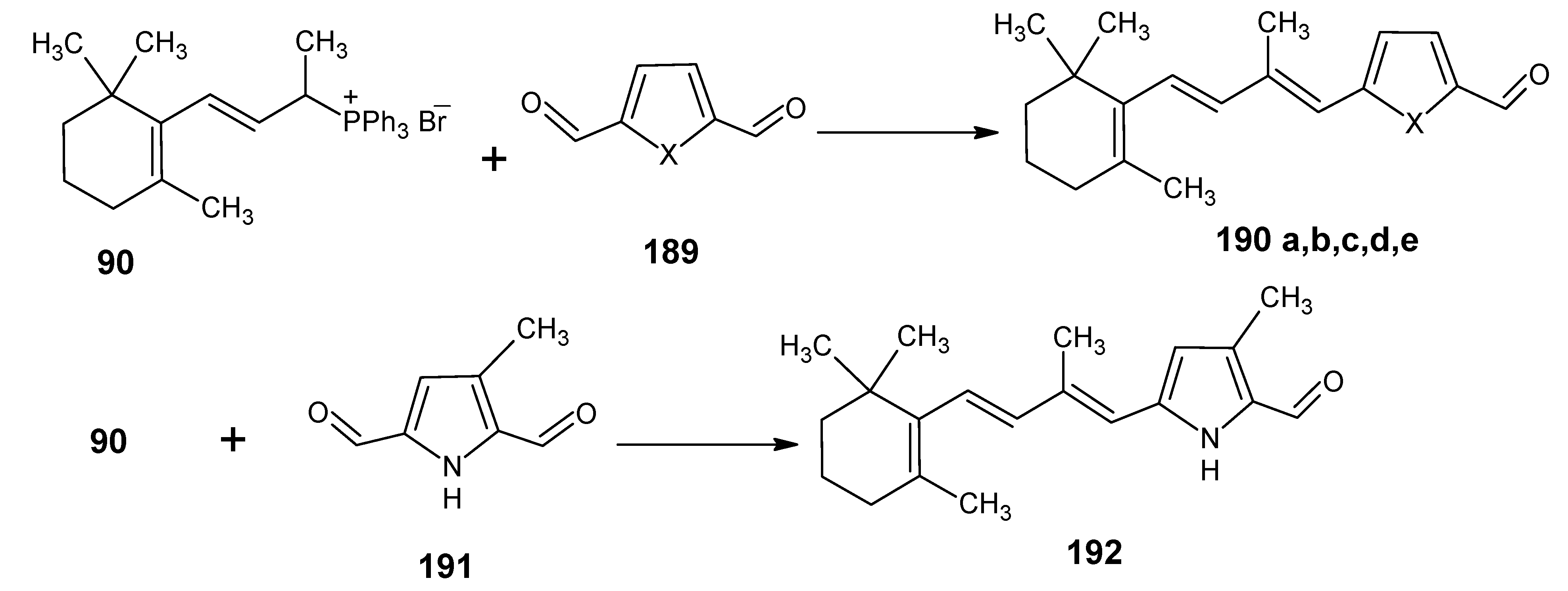
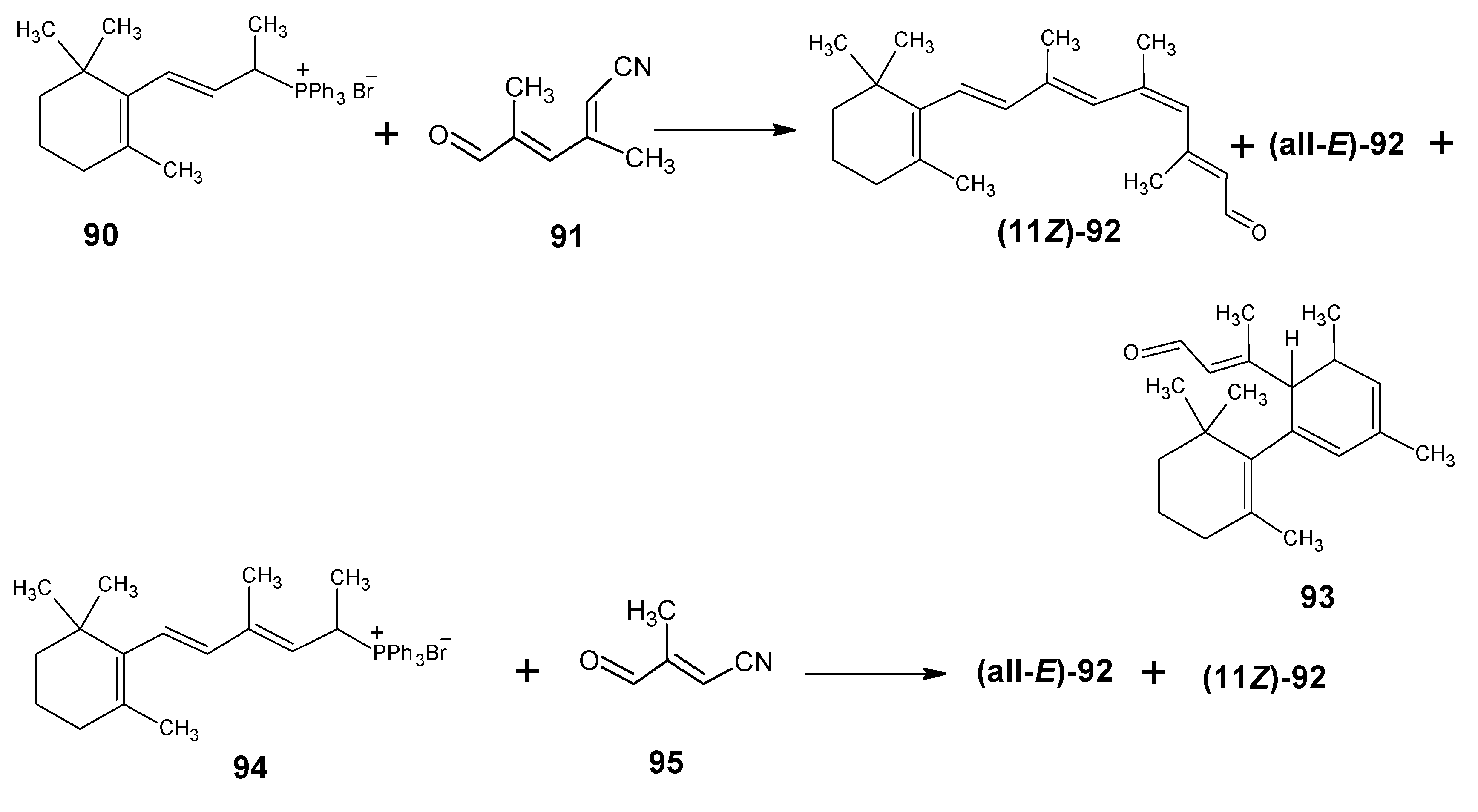
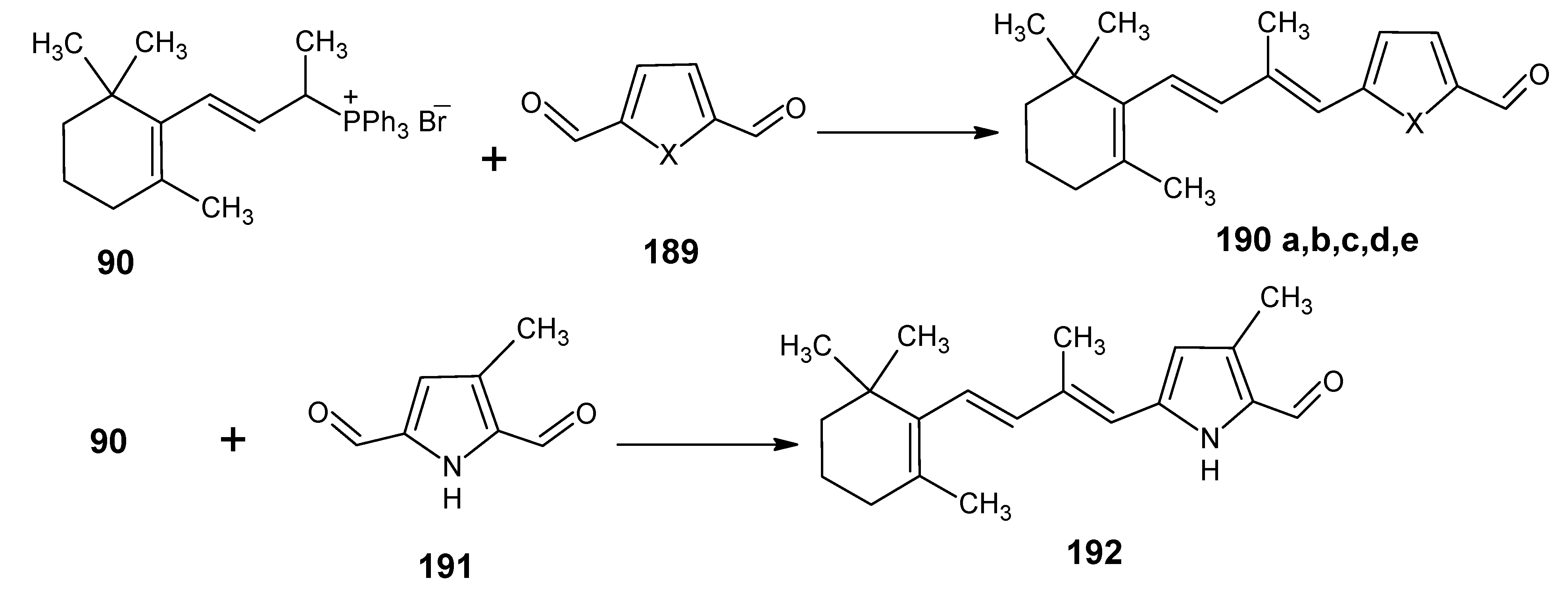
3.3.Preparation of 11-methylretinal and 12-methylretinal via β-ionyl triphenylphosphonium bromide 90
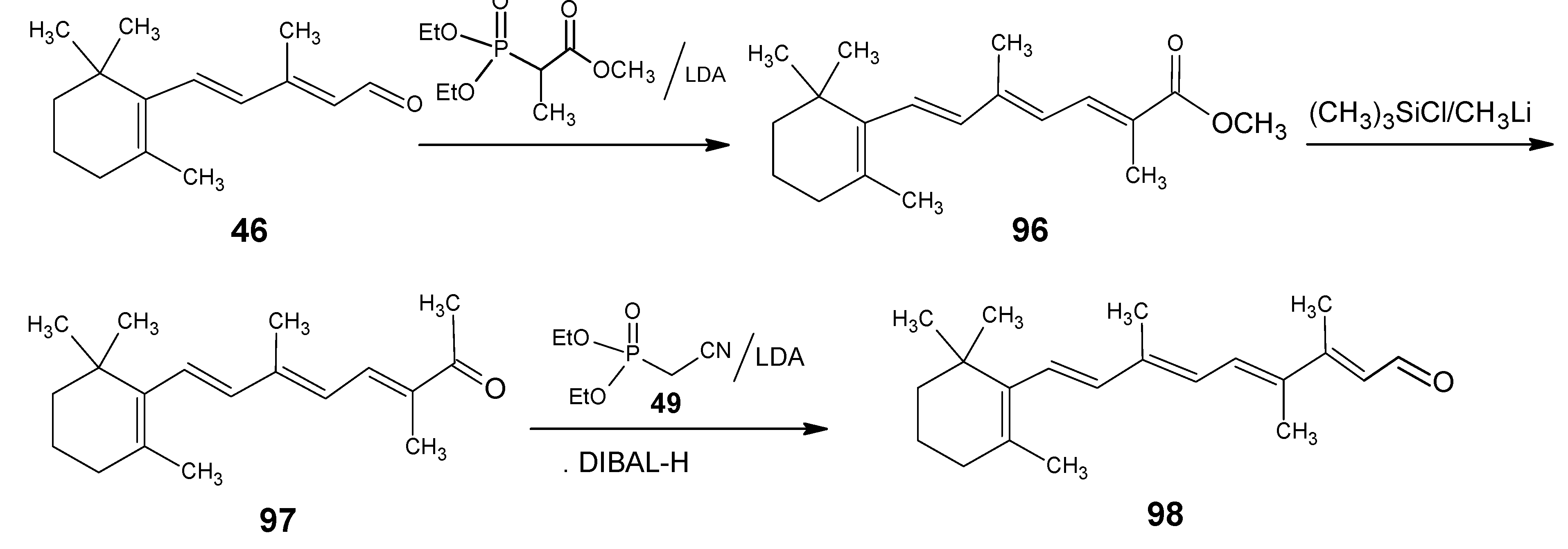
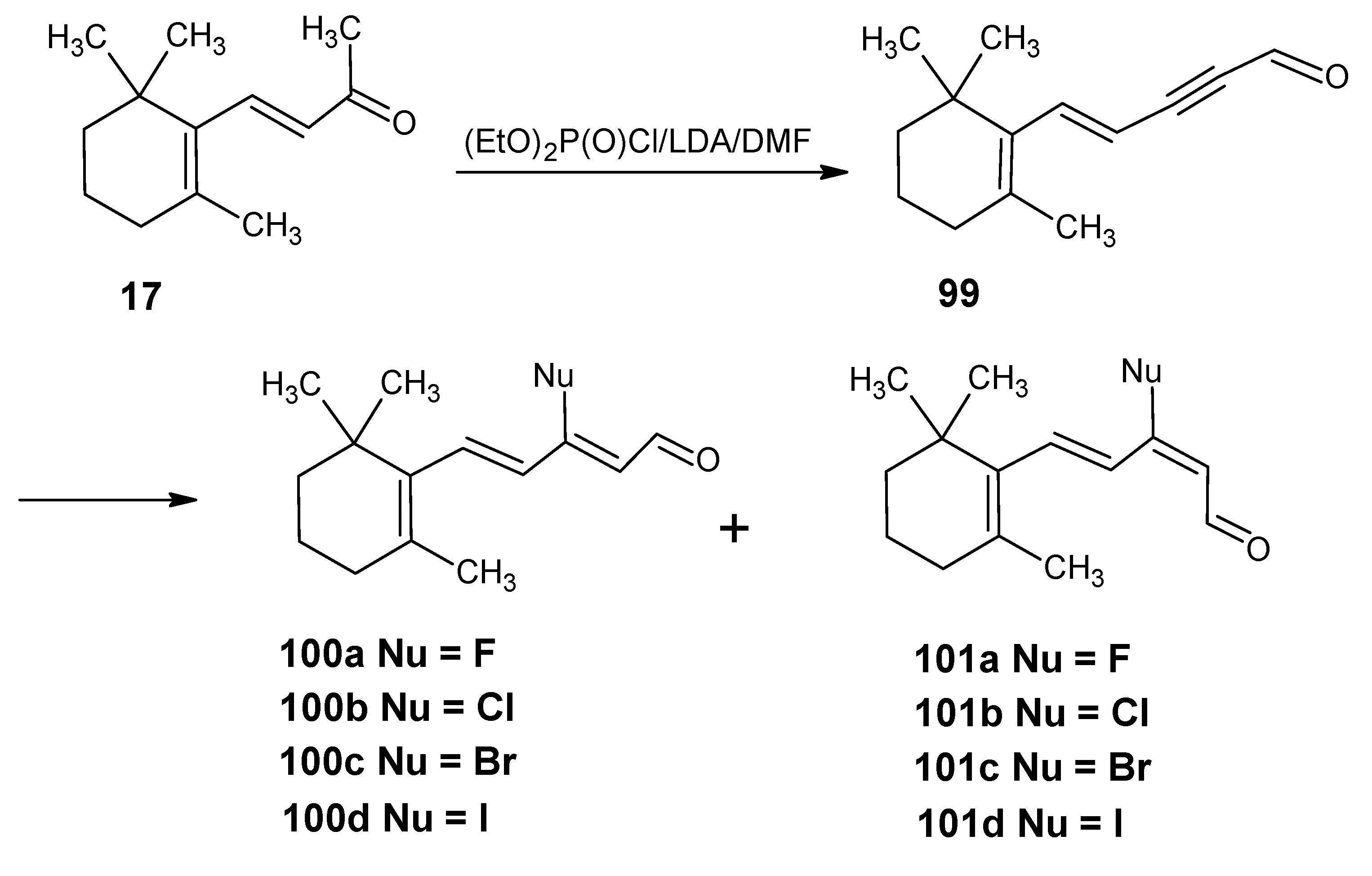
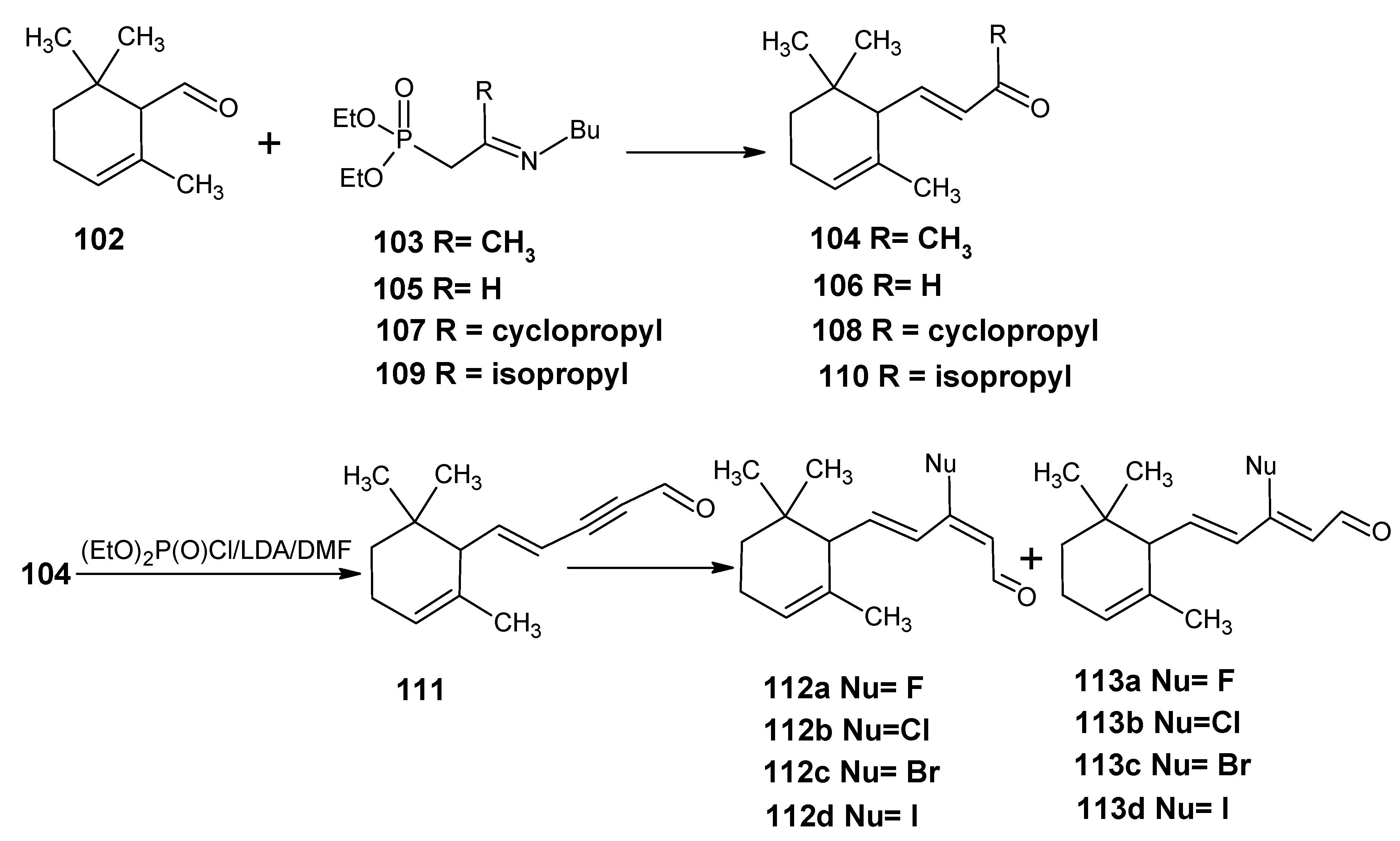
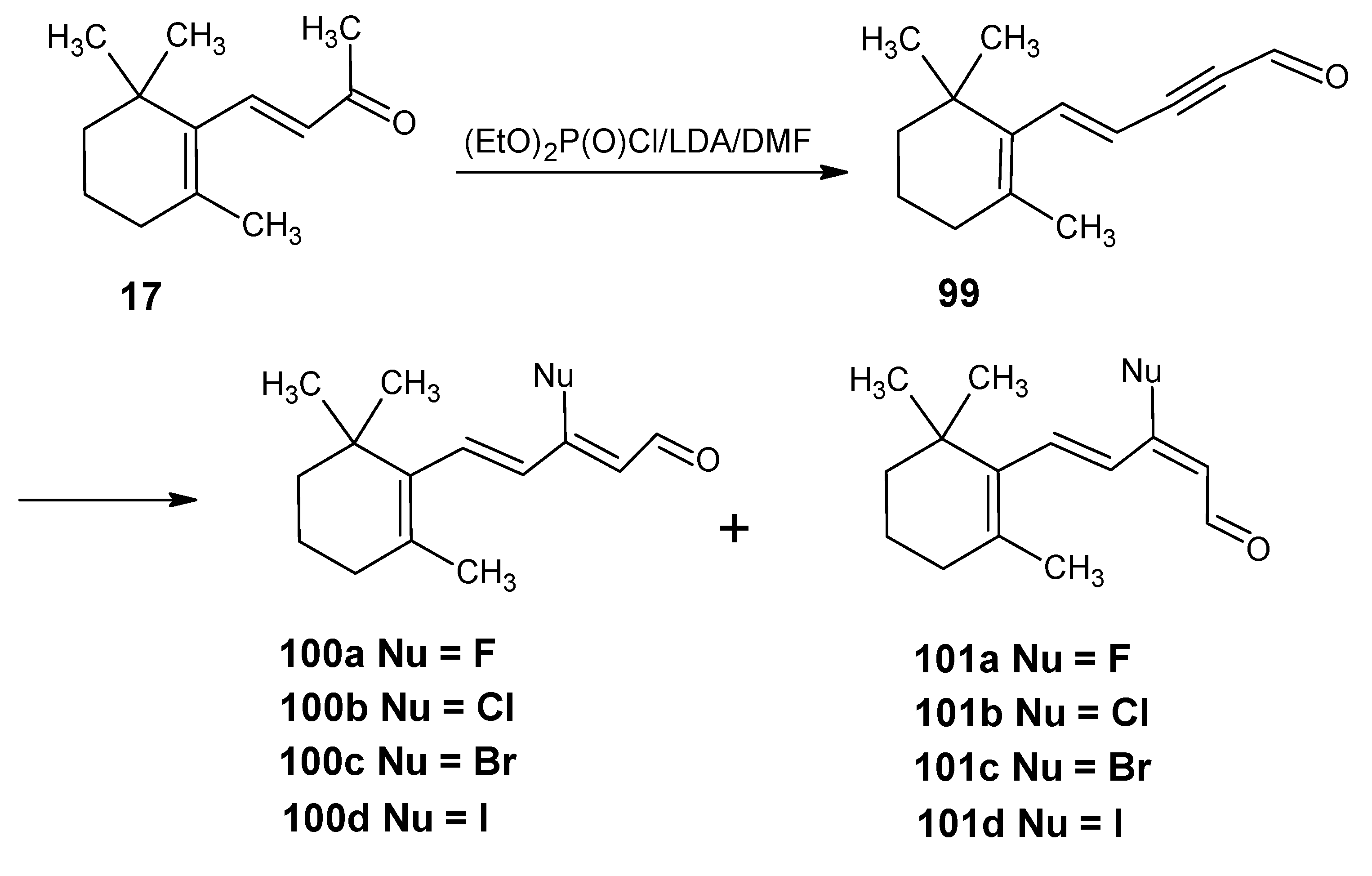
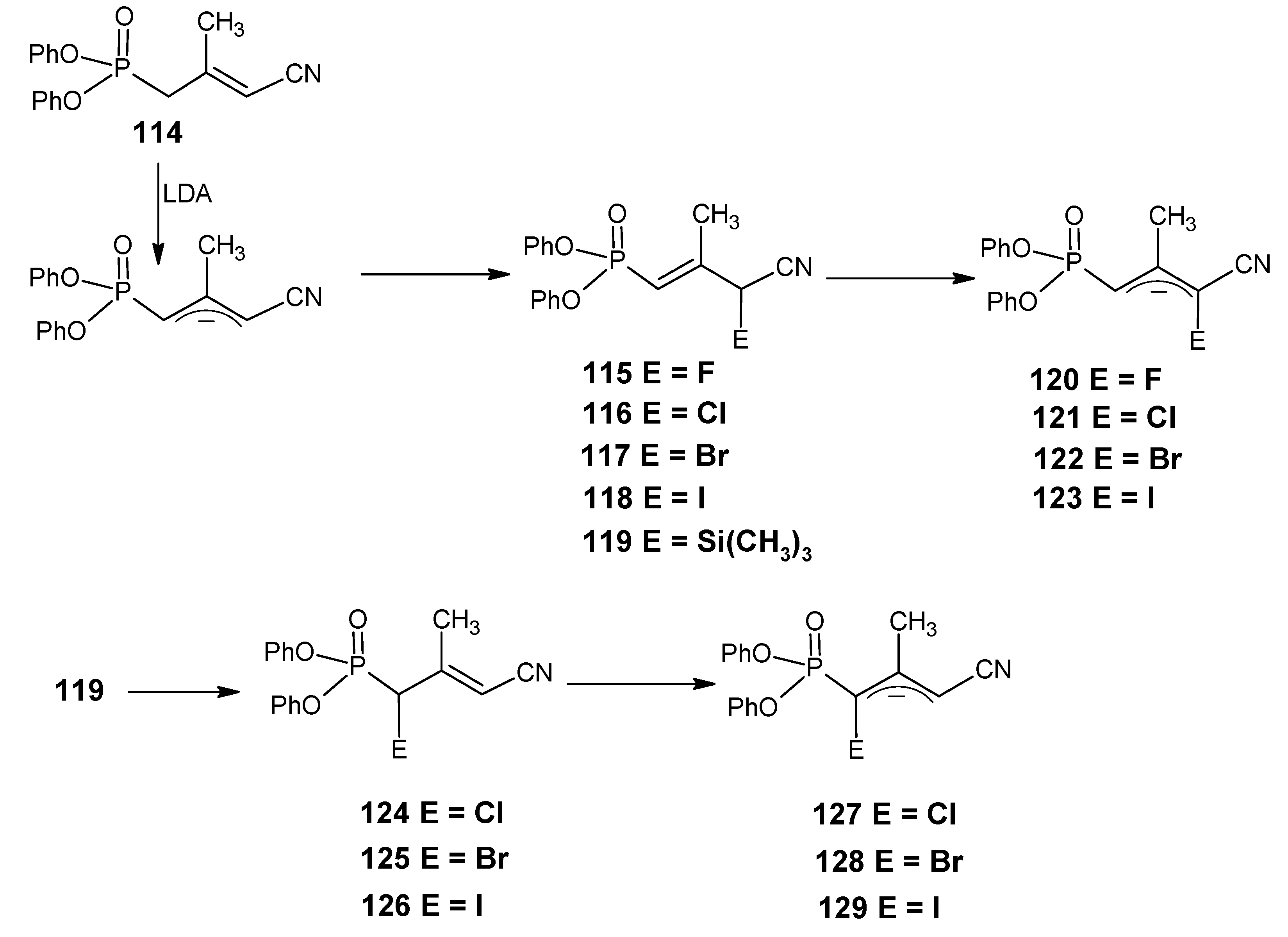
3.4. Preparation of 9-demethyl-9-haloretinals and 13-demethyl-13-haloretinals
4. α-Retinals via α-Ionone
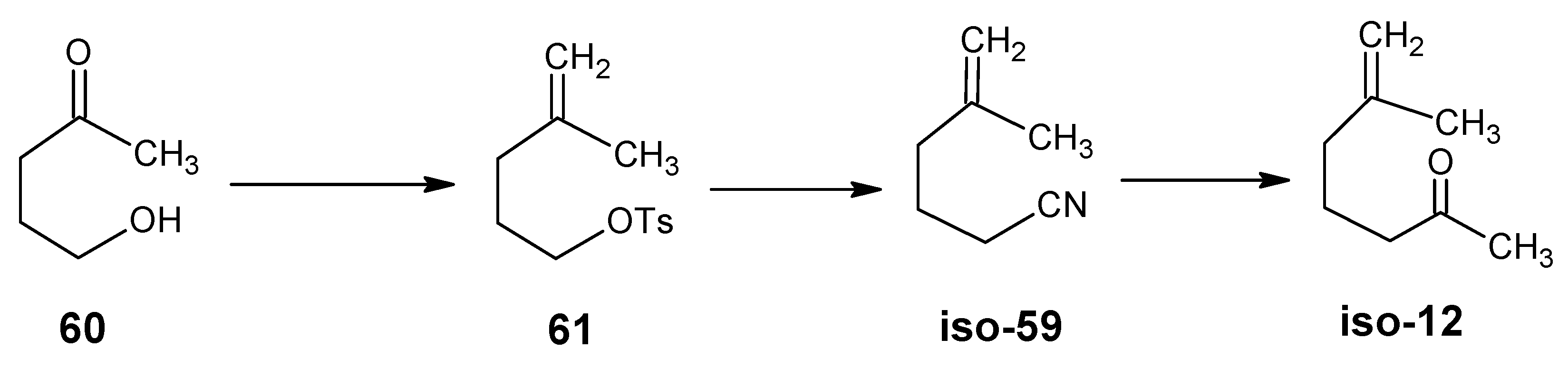
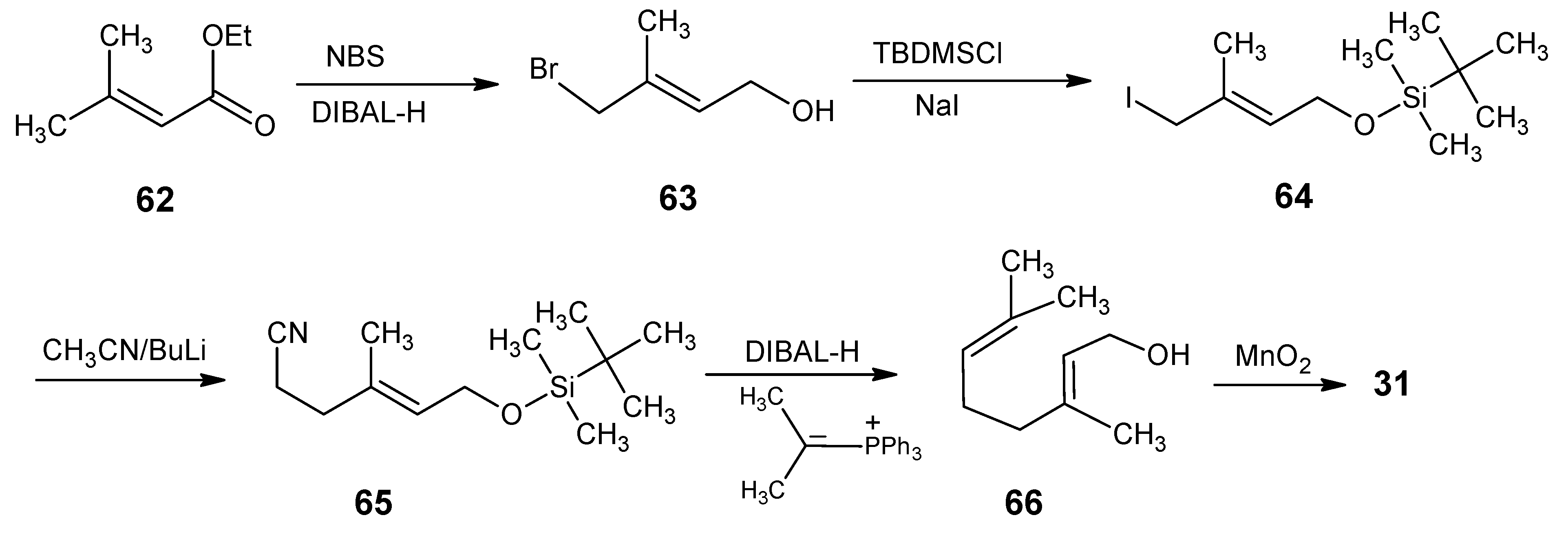
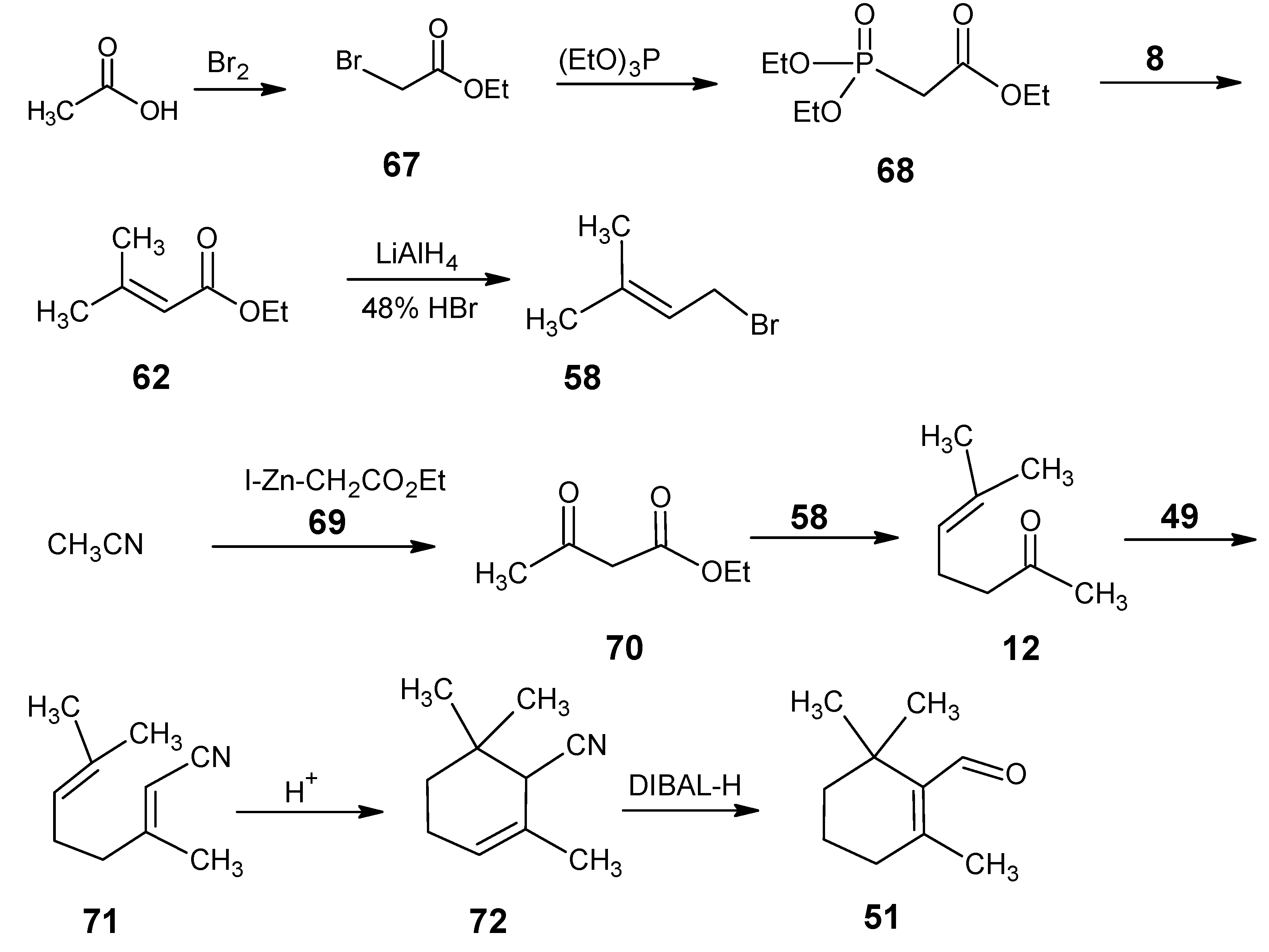
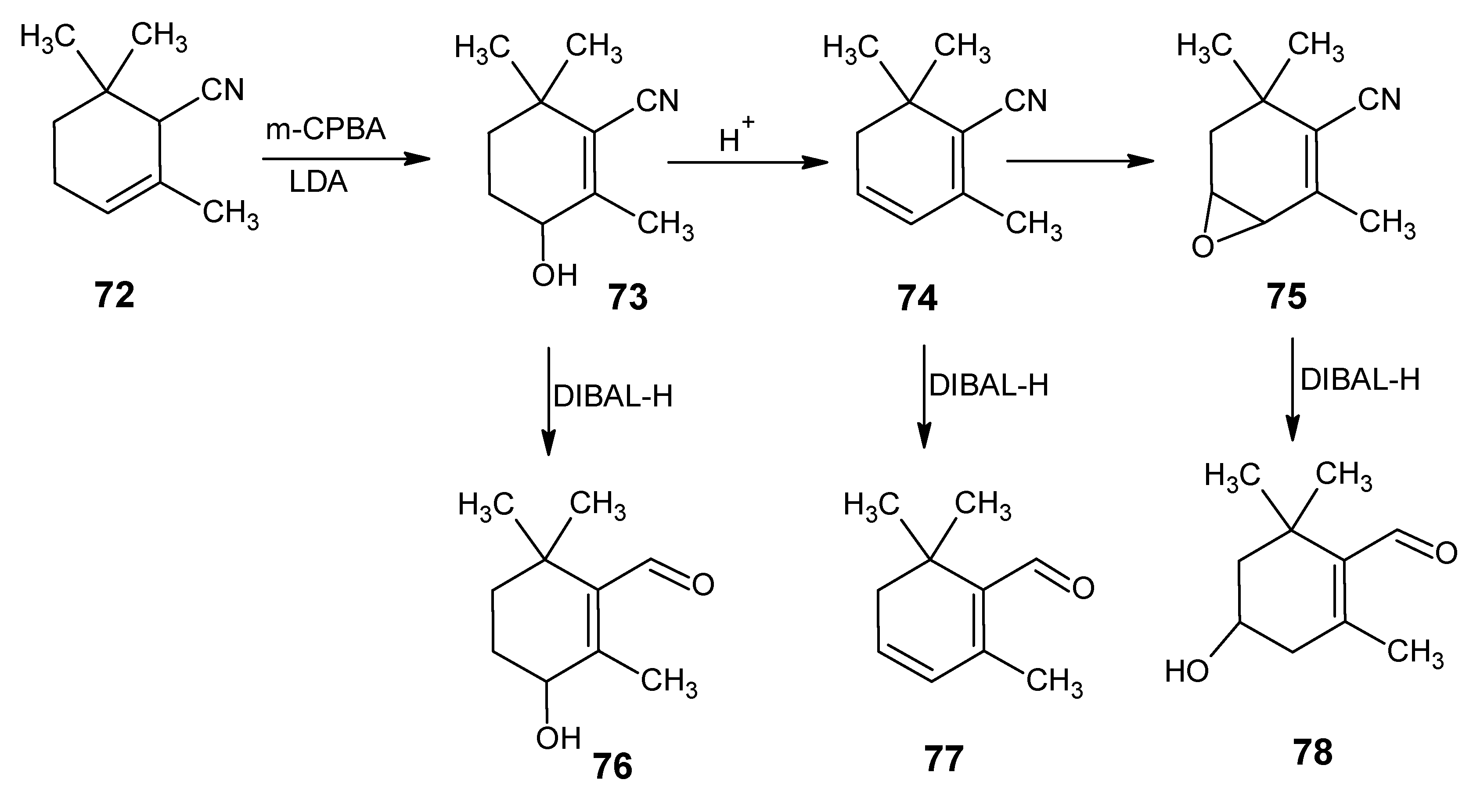
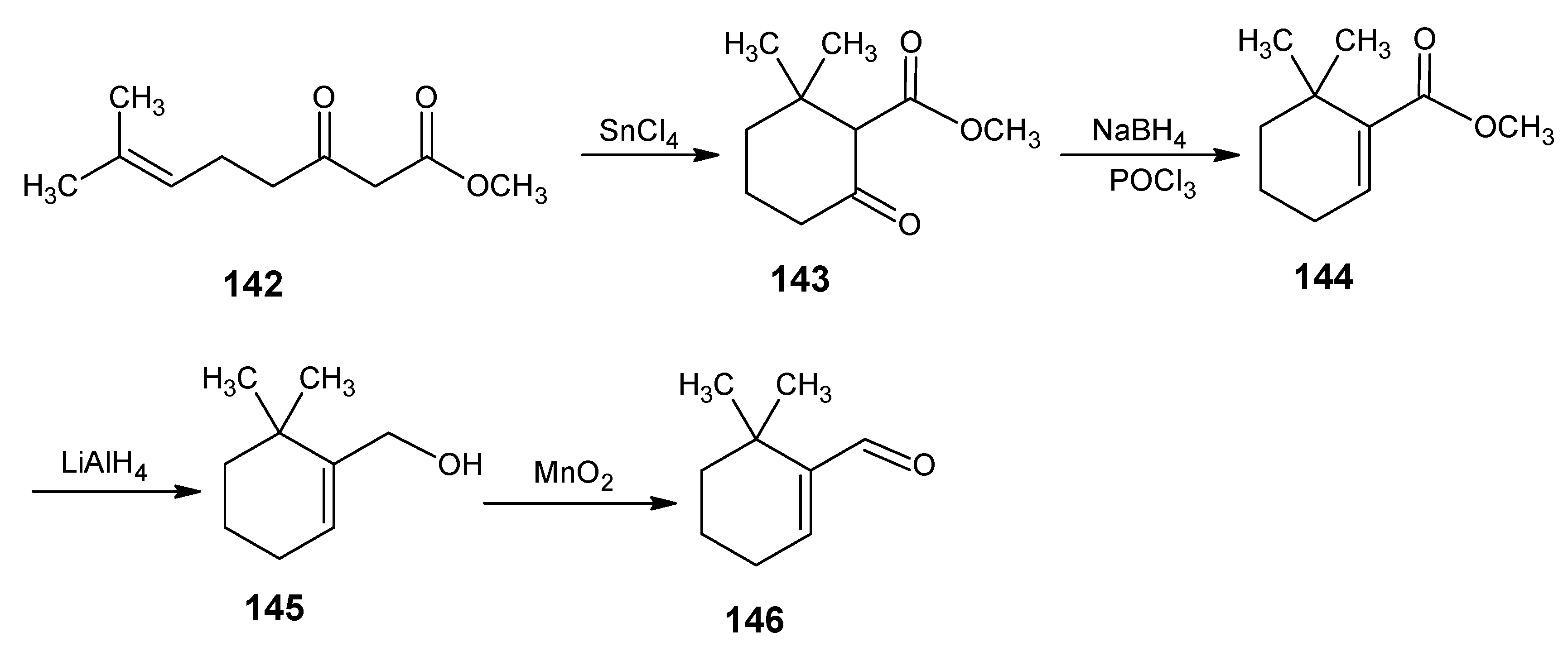
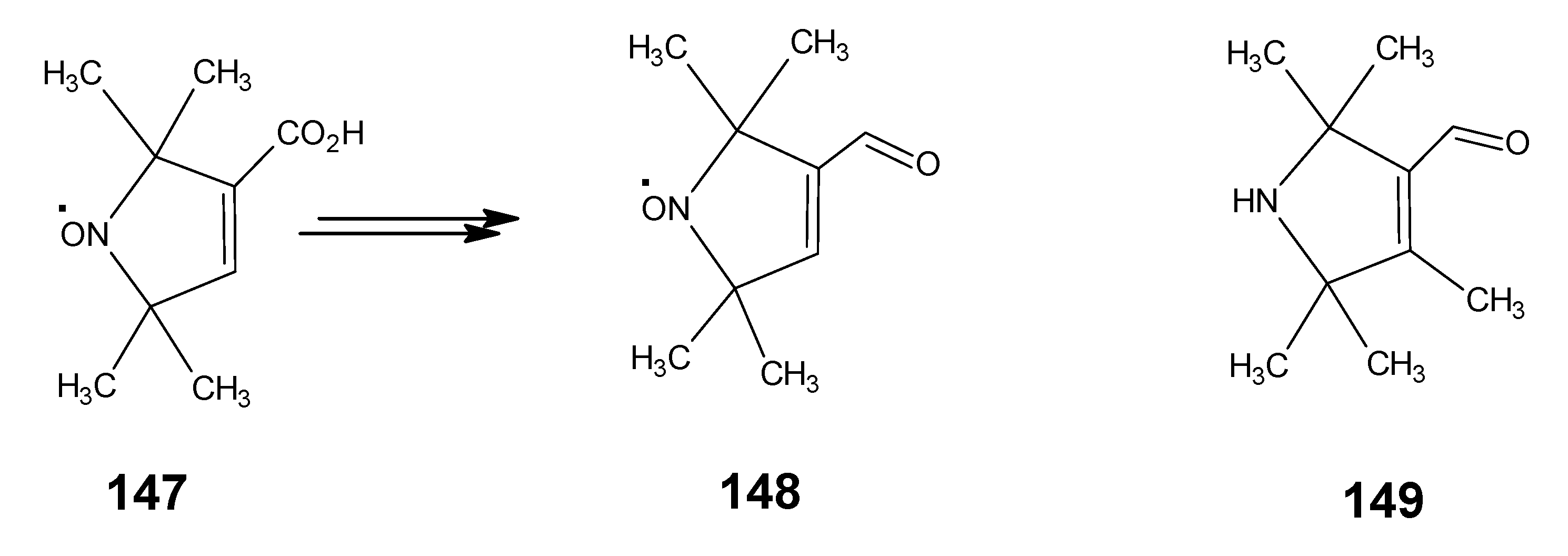
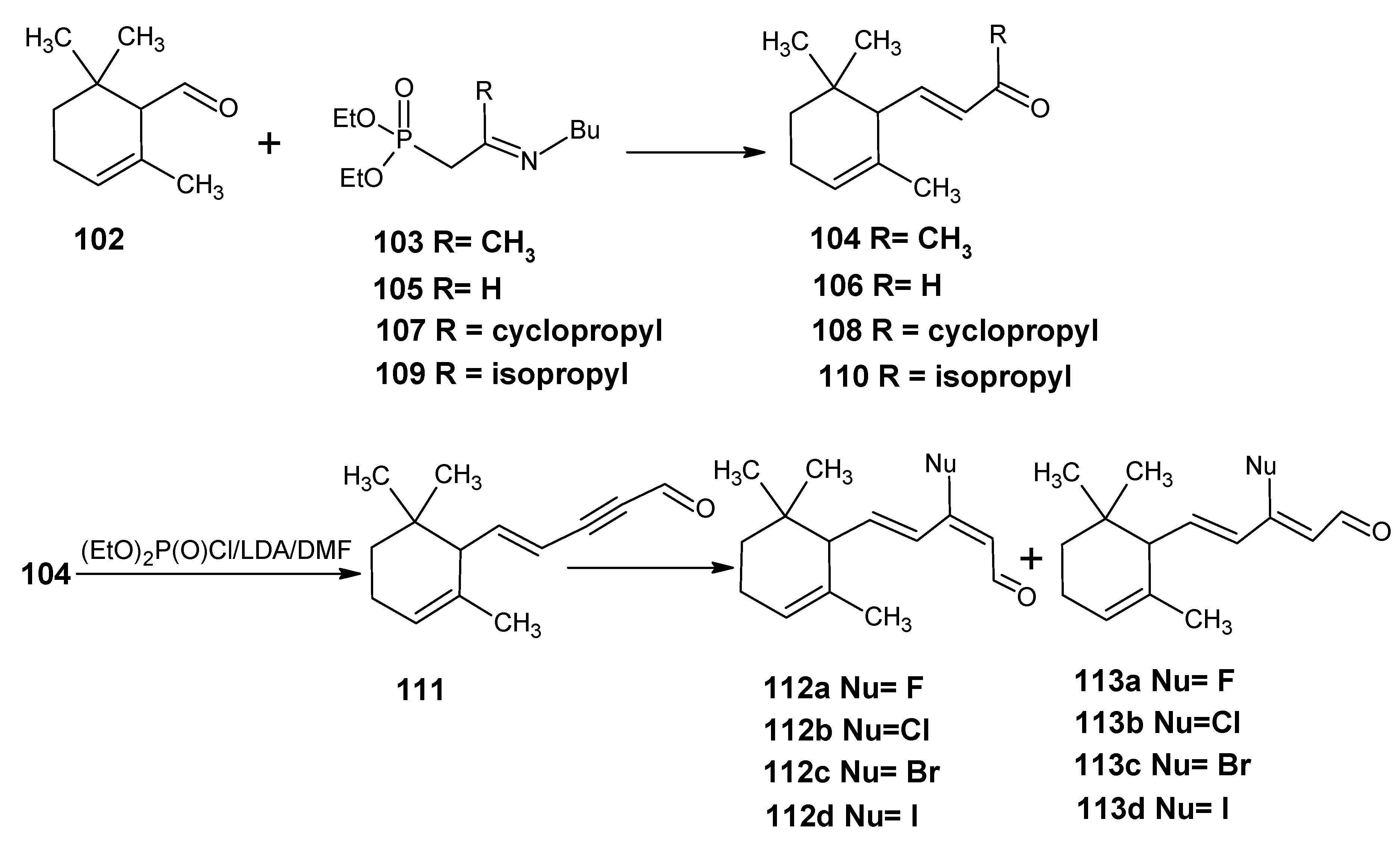
4.1. 9-Demethyl-9-halo-α-retinals, 9-substituted α-retinals, 9-demethyl α-retinal, 19,19-ethano-α-retinal, 19,19-dimethyl α-retinal and 12- and 14-halo substituted α-retinals
5. Nor-Retinals
5.1. 16,17,18-Trinor-retinal, 16,17-dinor-retinal and 16-nor-retinal
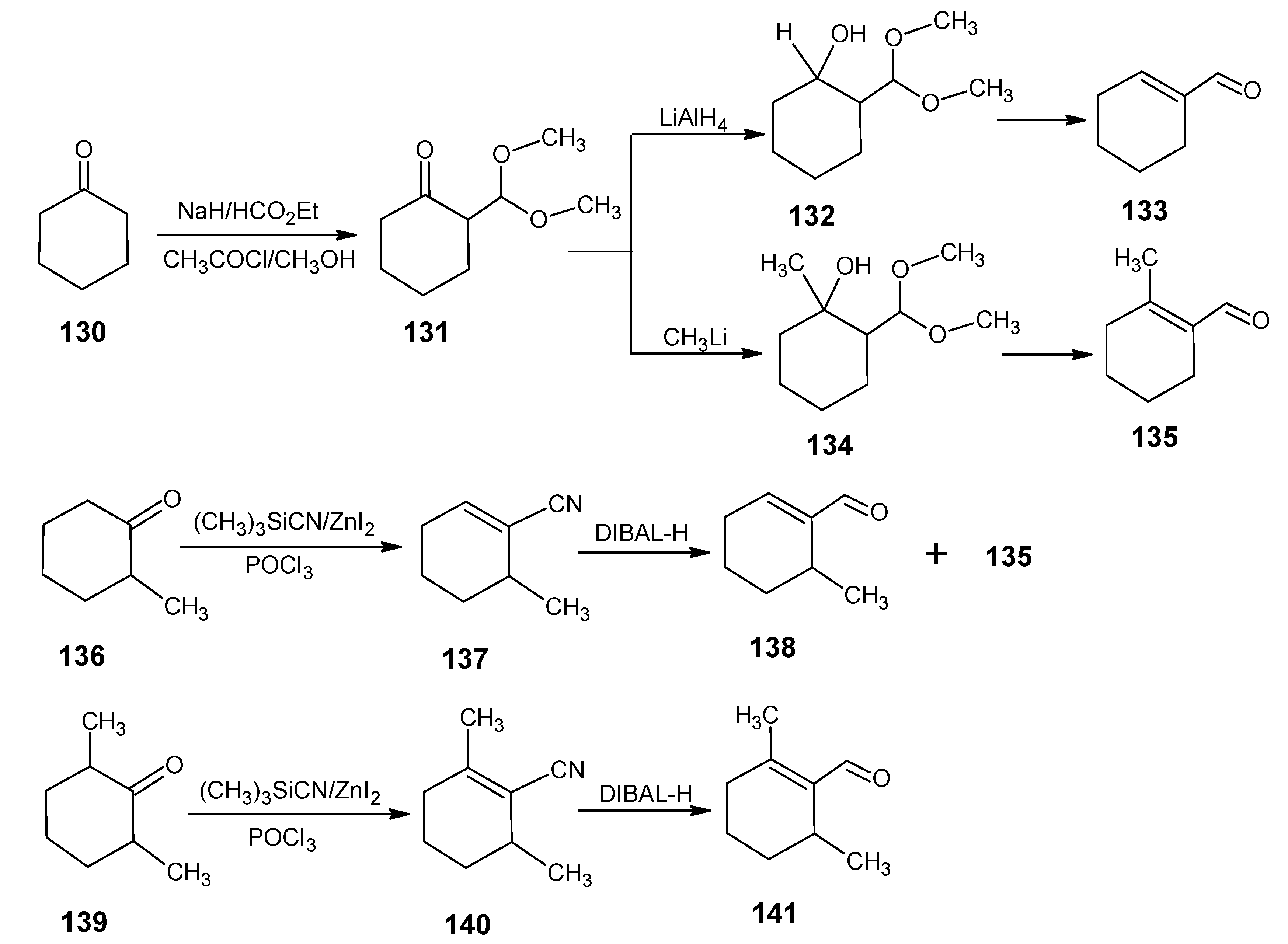
6. Bridged and Demethyl Retinals
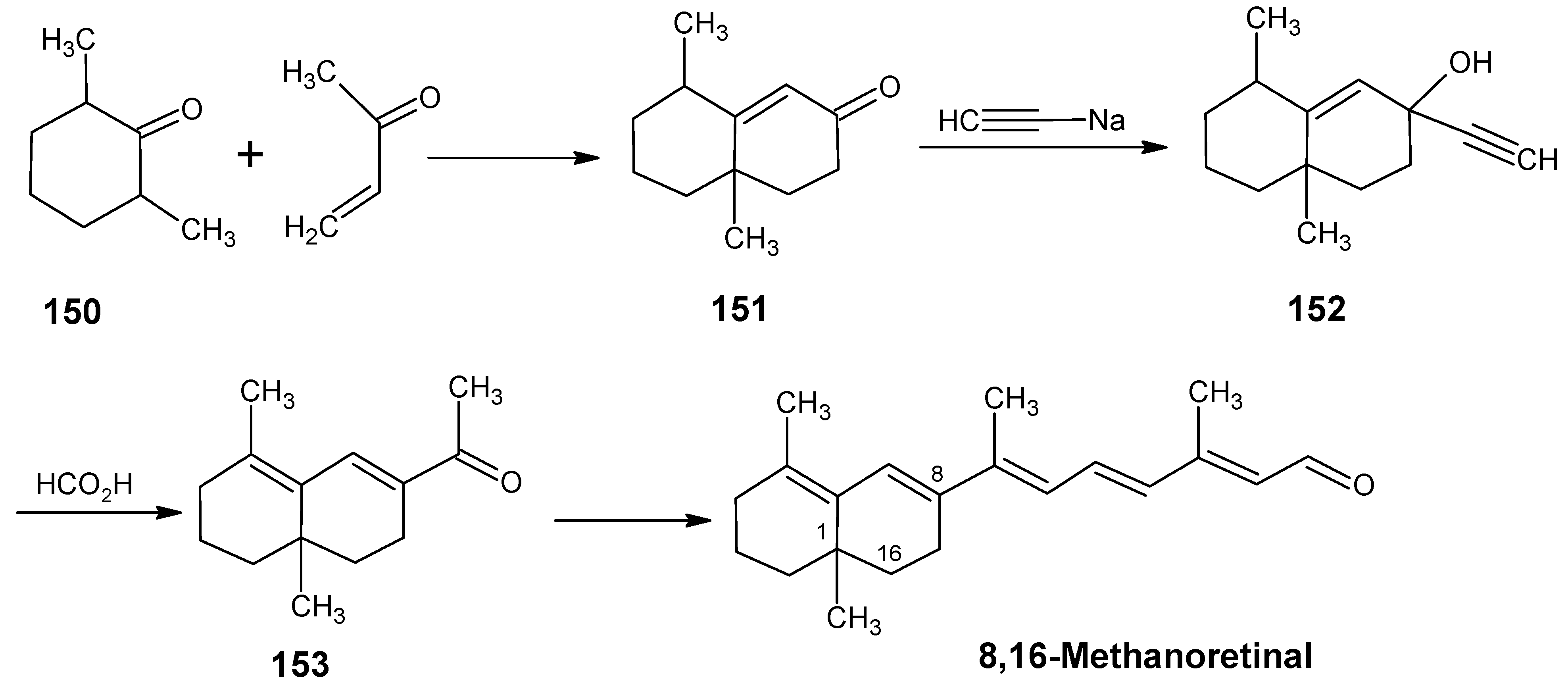
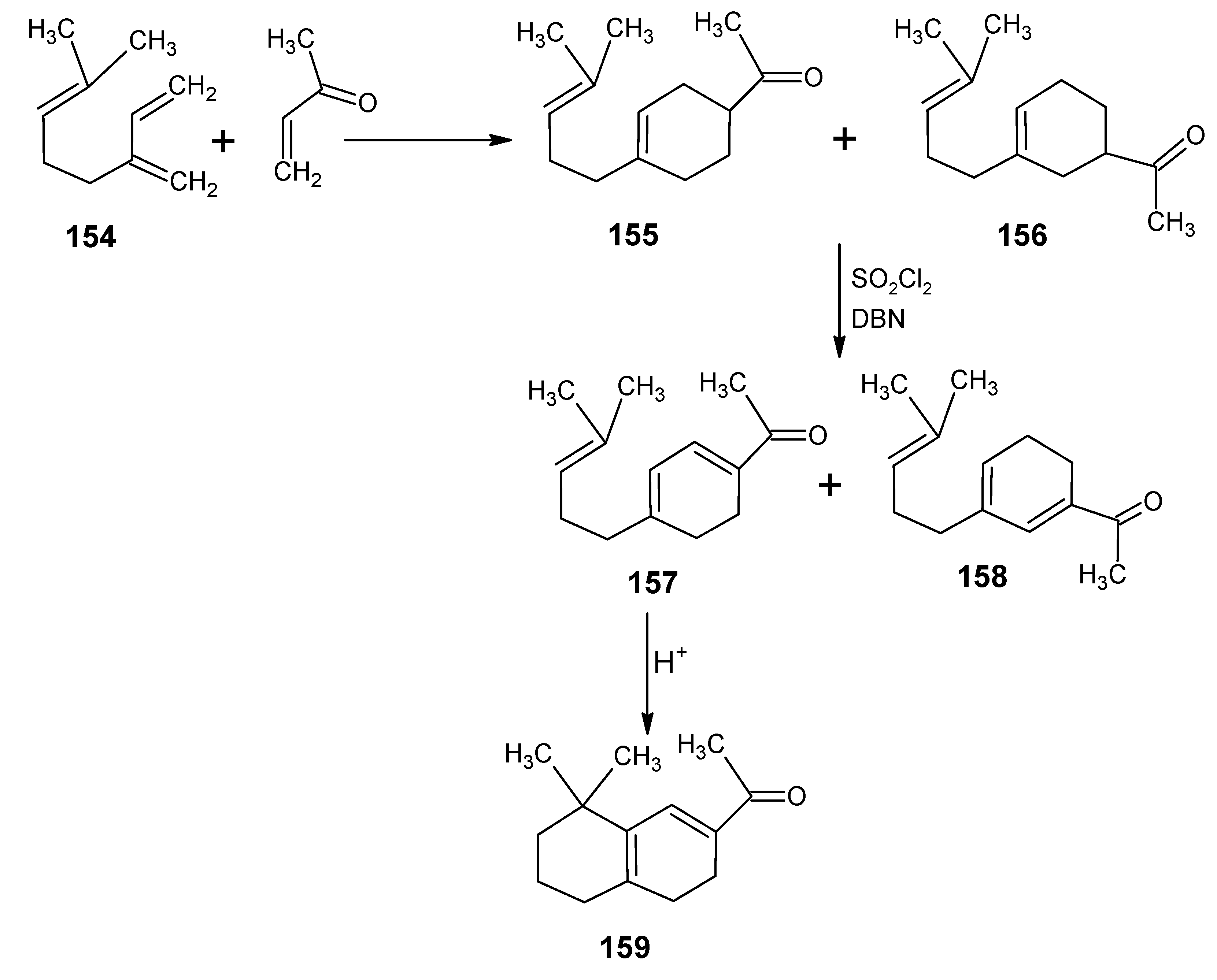
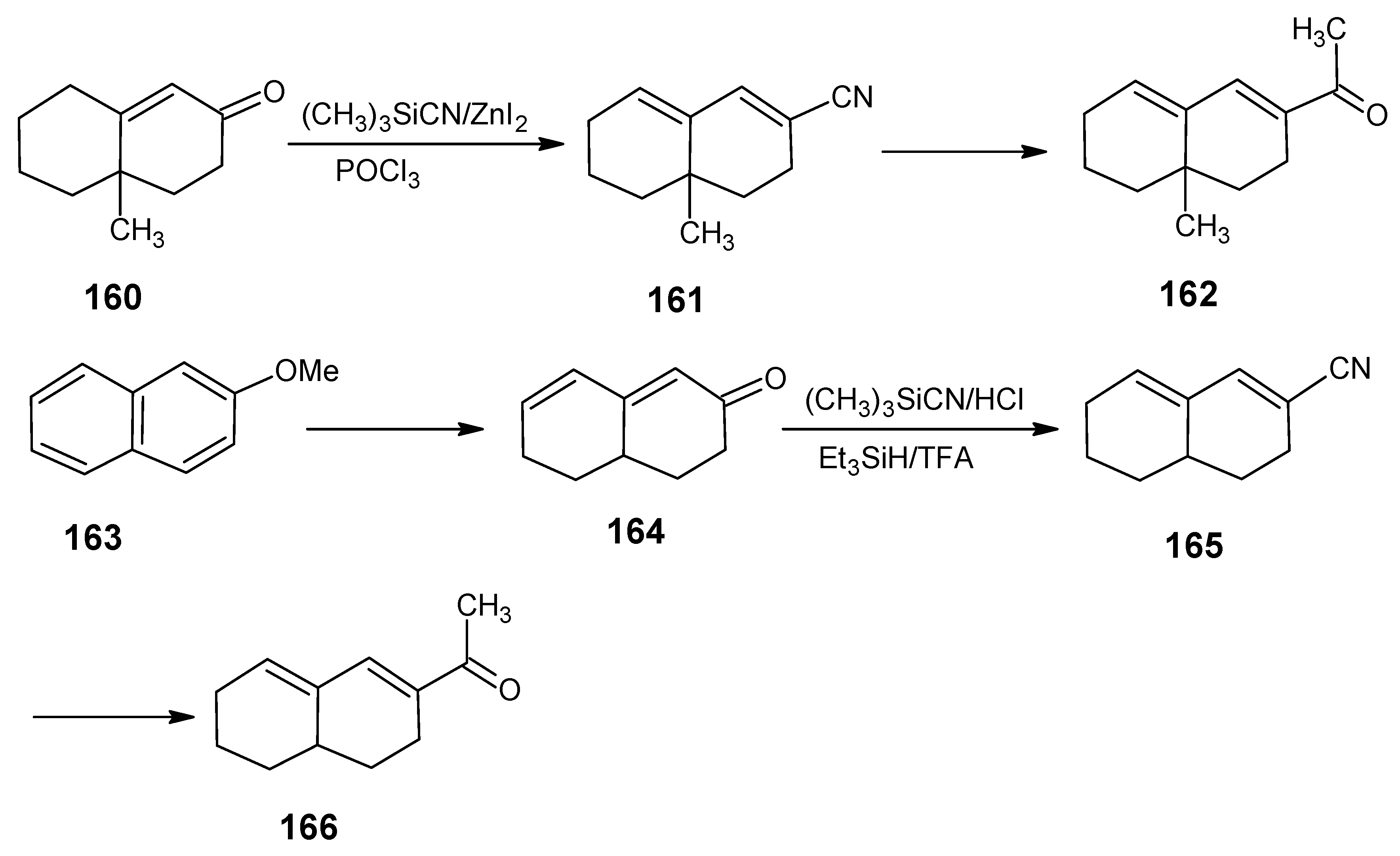
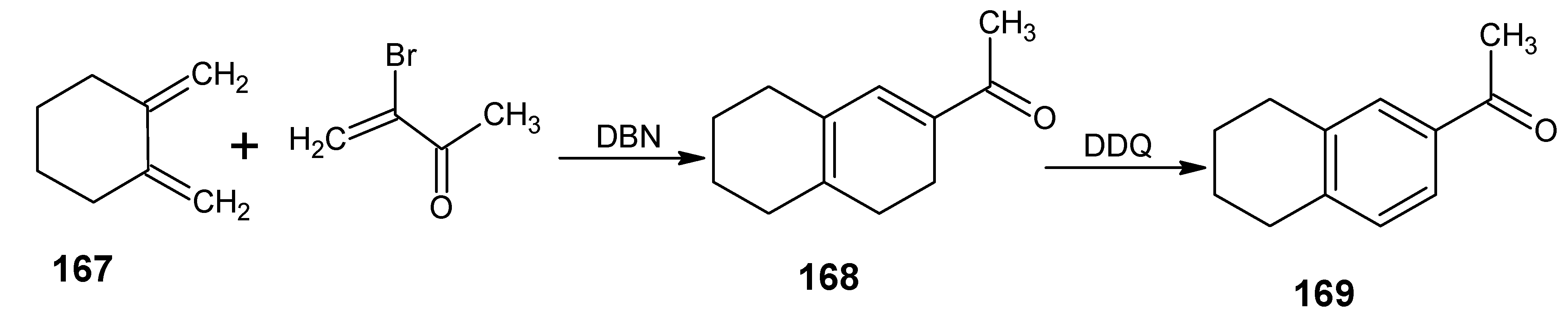
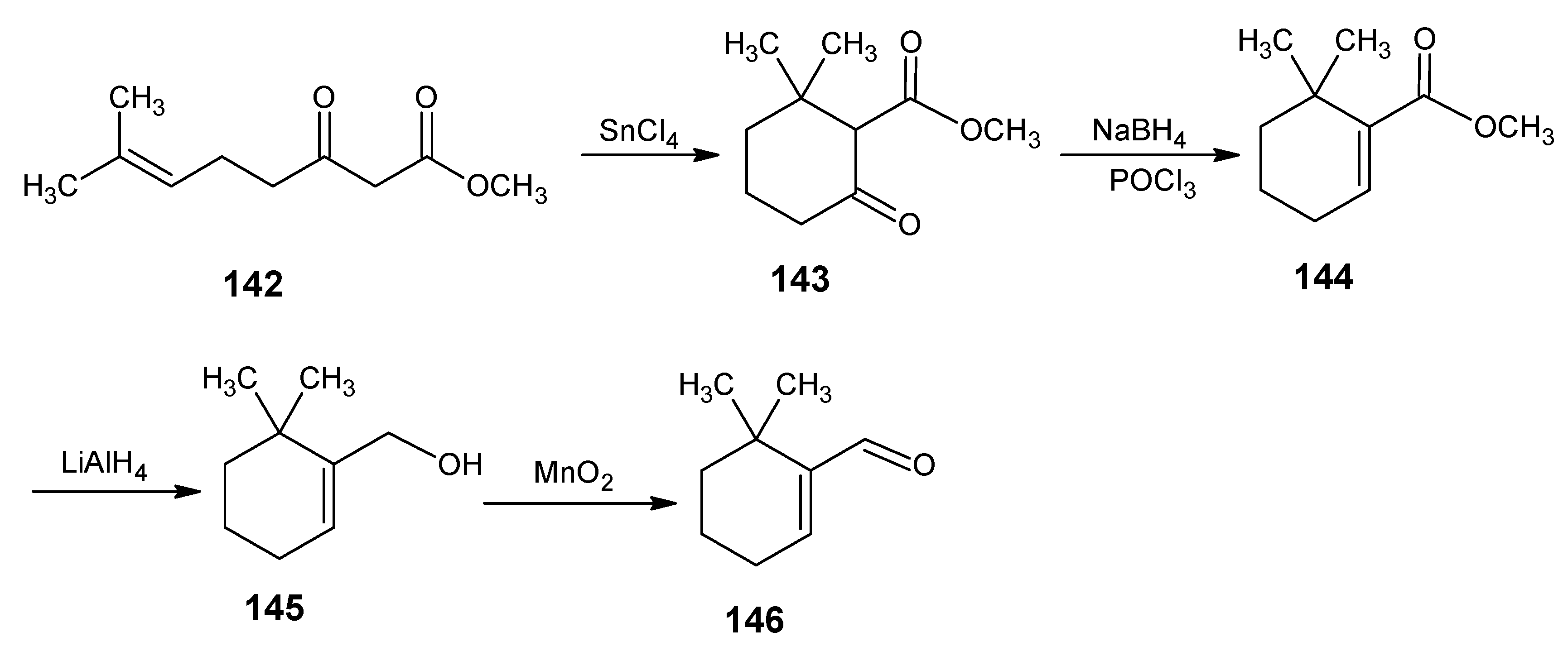
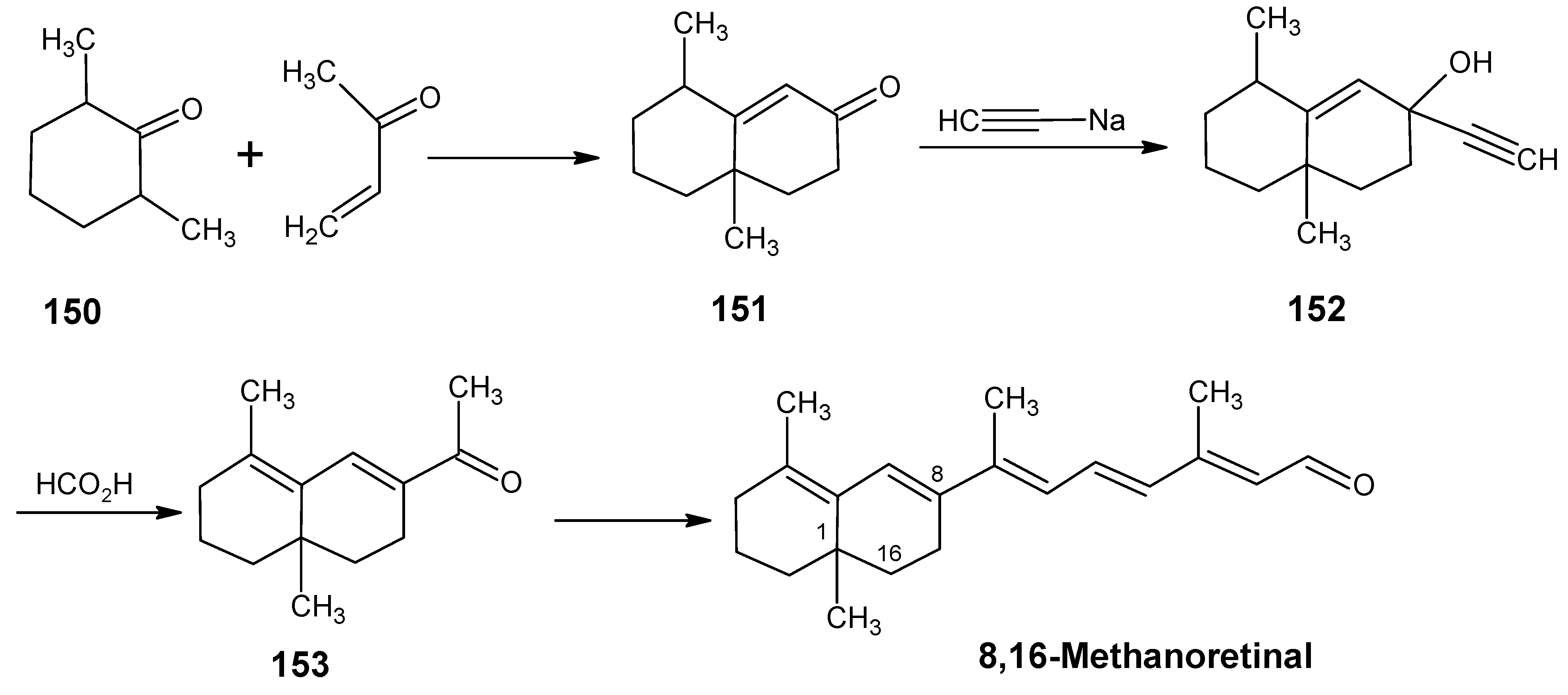
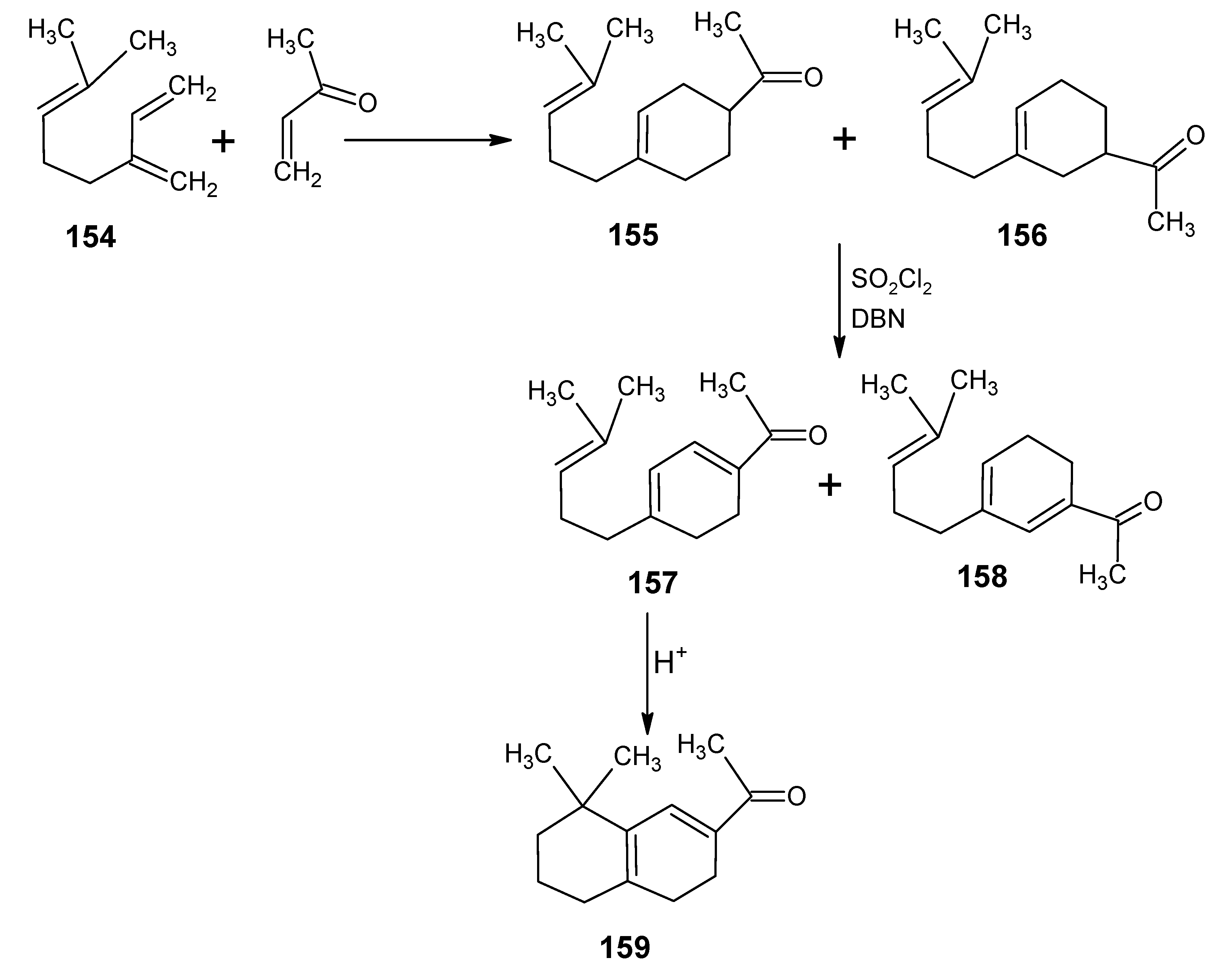
6.1. DL-8,16-Methanoretinal, 8,18-methanoretinal, (R)-5-demethyl-8,16-methanoretinal, 1,5-didemethyl-8,16-methanoretinal, 1,1-didemethyl-8,18-methanoretinal, 1,1-didemethyl-18-didehydro-8,18-methanoretinal

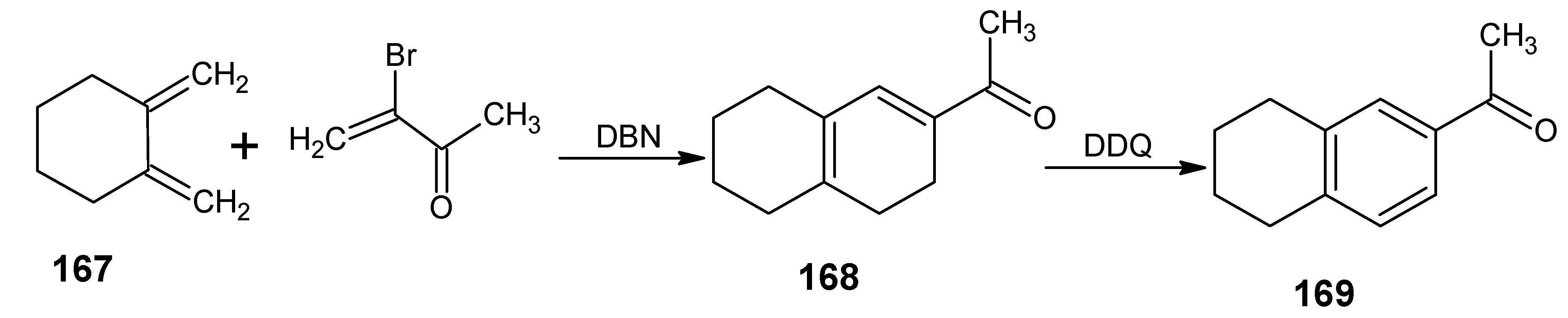
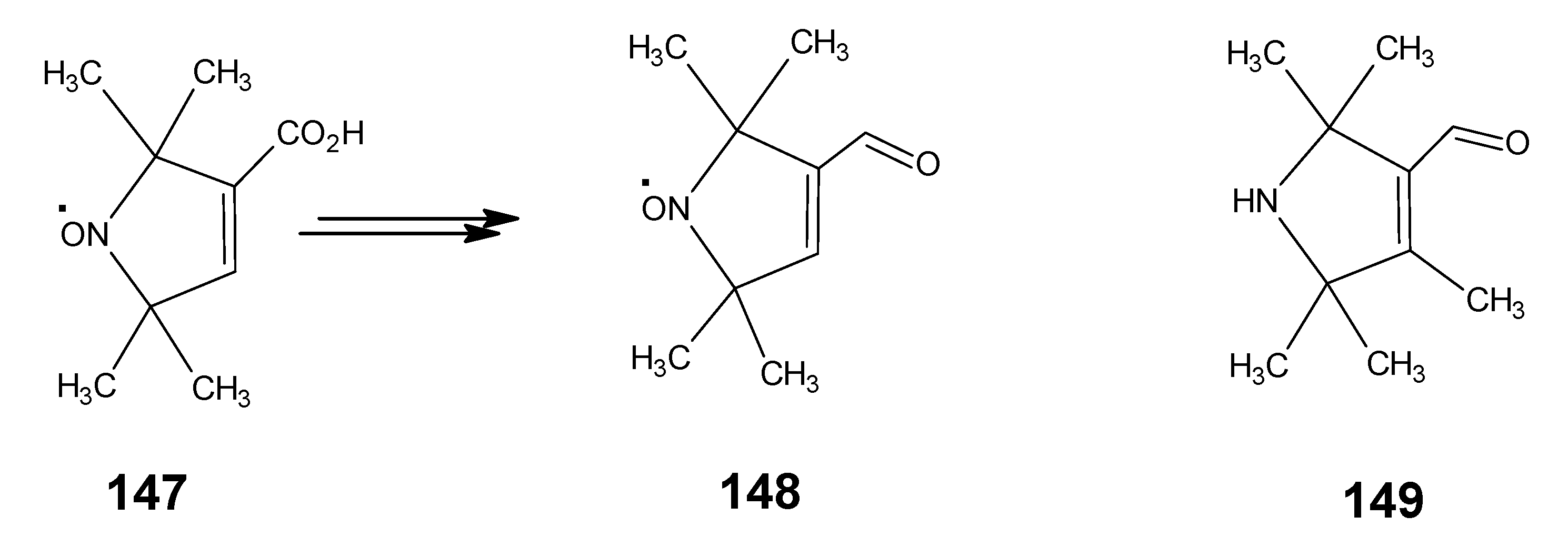
6.2. 11,19-10,20-Dimethanoretinal, 10,20-methanoretinal, 13-demethyl-10,12-ethanoretinal, 13-demethyl-12,14-ethanoretinal, 13-demethyl-10,12-propanoretinal and 13-demethyl-12,14-propanoretinal
6.3. 13-Demethyl-10,14-thiaretinal and 11,14-bridged 13-demethyl retinals
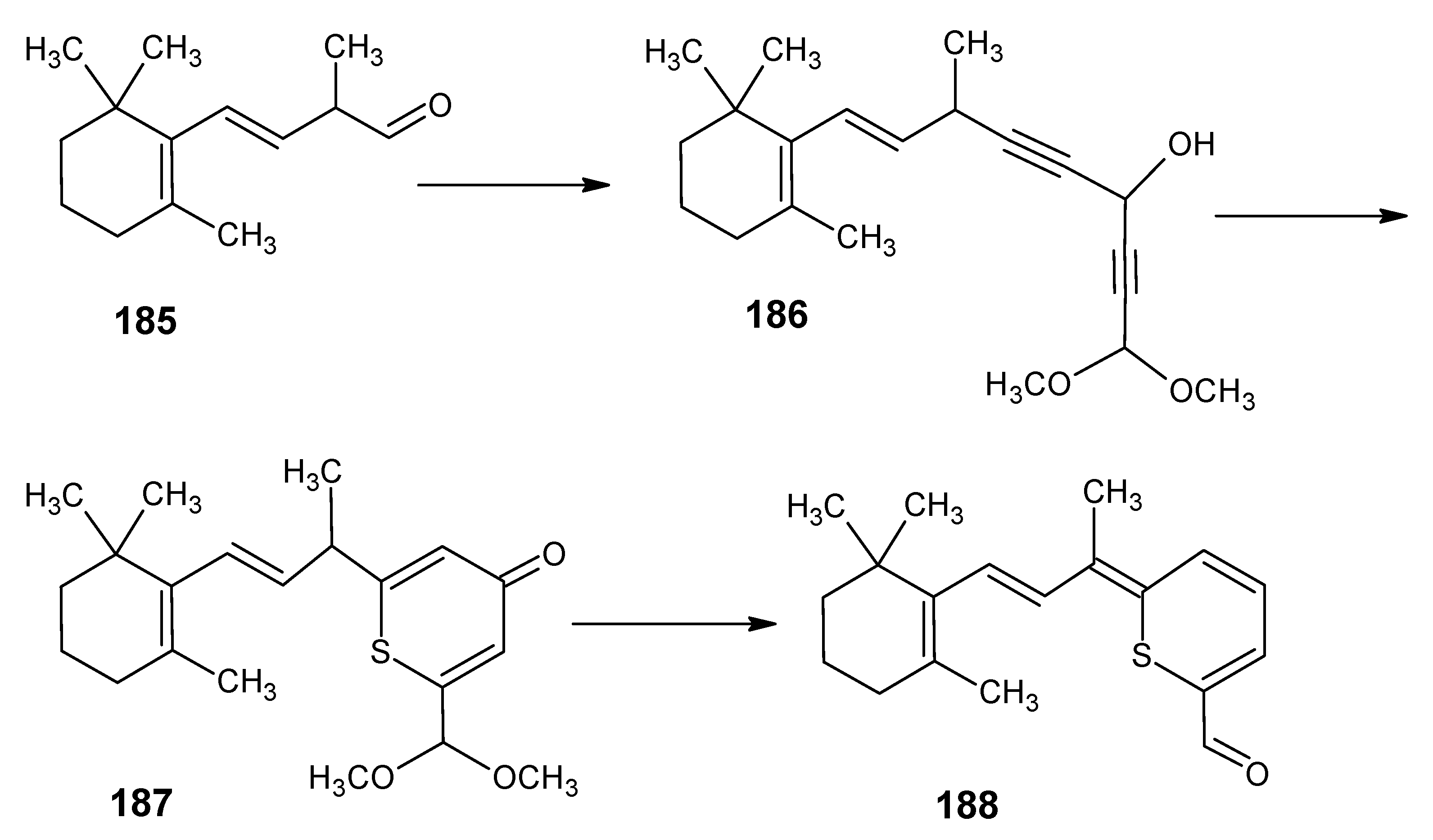
6.4. 9-Demethyl retinal, 13-demethyl retinal and 9,13-didemethyl retinal
Conclusions
Acknowledgments
- Samples Availability: Samples of the compounds are available from authors.
References
- Stepp, W. Versuche über Fütterung mit lipoidfreier Nahrung. Biochem. Zeitschrift 1909, 22, 452–460. [Google Scholar]
- Osborne, T.B.; Mendel, L.B. The influence of butter fat on growth. J. Biol. Chem. 1913, 16, 423–437. [Google Scholar]
- Steenbock, H. White corn vs. yellow corn and a probable relation between the fat-soluble vitamine and yellow plant pigments. Science 1919, 50, 352–353. [Google Scholar]
- Khachik, F. Analysis of carotenoids in nutritional studies. In Carotenoids Volume 5: Nutrition and Health; Britton, G., Liaaen-Jensen, S., Pfander, H., Eds.; Birkhauser Verlag: Basel, Switzerland, 2009; pp. 7–43. [Google Scholar]
- Tang, G.; Russell, R.M. Carotenoids as provitamin A. In Carotenoids Volume 5: Nutrition and Health; Britton, G., Liaaen-Jensen, S., Pfander, H., Eds.; Birkhauser Verlag: Basel, Switzerland, 2009; pp. 149–172. [Google Scholar]
- Thibodeux, C.J.; Liu, H.W. Unraveling the mechanisms of isoprenoid biosynthetic enzymes: Mechanistic studies of the early stage enzymes. Chimia 2009, 63, 334–339. [Google Scholar] [CrossRef]
- Karrer, P.; Morf, R. Pflanzenfarbstoffe XXXV. Zur Konstitution des β-Carotins und β-Dihydro-carotins. Helv. Chim. Acta 1931, 14, 1033–1036. [Google Scholar]
- Karrer, P.; Morf, R.; Schoepp, K. Zur Kenntnis des Vitamins A aus Fischtranen. Helv. Chim. Acta 1931, 14, 1431–1436. [Google Scholar] [CrossRef]
- Moore, T. The Relation of carotin to vitamin A. Lancet 1929, 2, 380–381. [Google Scholar]
- Morton, R.A. Chemical aspects of the visual process. Nature 1944, 153, 69–71. [Google Scholar] [CrossRef]
- Oroshnik, W. The synthesis and configuration of neo-b vitamin A and neoretinene b. J. Am. Chem. Soc. 1956, 78, 2651–2652. [Google Scholar] [CrossRef]
- DeGrip, W.J.; Rothschild, K.J. Structure and mechanism of vertebrate visual pigments. In Molecular Mechanisms in Visual Transduction; Stavenga, D.G., DeGrip, W.J., Pugh, E.N., Jr., Eds.; Elsevier: Amsterdam, The Netherland, 2000; Volume 3, pp. 1–54. [Google Scholar]
- Van Wijk, A.A.C.; van de Weerd, M.B.; Lugtenburg, J. Synthetic scheme for the preparation of 13C-labeled 3,4-didehydroretinal, 3-hydroxyretinal and 4-hydroxyretinal up to uniform 13C-enrichment. Eur. J. Org. Chem. 2003, 863–868. [Google Scholar]
- Gaertner, W. Invertebrate visual pigments, vision (I). In Molecular Mechanisms in Visual Transduction; Stavenga, D.G., DeGrip, W.J., Pugh, E.N., Jr., Eds.; Elsevier: Amsterdam, The Netherland, 2000; pp. 297–388. [Google Scholar]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar]
- Kono, M.; Goletz, P.W.; Crouch, R.K. 11-Cis- and all-trans-retinols can activate rod opsin: Rational design of the visual cycle. Biochemistry 2008, 47, 7567–7571. [Google Scholar]
- Mangeldorf, D.J.; Umesono, K.; Evans, R.M. The retinoid receptors. In TheRetinoids: Biology, Chemistry, and Medicine; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 319–349. [Google Scholar]
- Buck, J.; Derguini, F.; Levi, E.; Nakanishi, K.; Hämmerling, U. Intracellular signaling by 14-hydroxy-4,14-retro-retinol. Science 1991, 254, 1654–1656. [Google Scholar]
- Buck, J.; Grün, F.; Derguini, F.; Chen, Y.; Kimura, S.; Noy, N.; Hämmerling, U. Anhydroretinol: A naturally occurring inhibitor of lymphocyte physiology. J. Exp. Med. 1993, 178, 675–680. [Google Scholar] [CrossRef]
- Derguini, F.; Nakanishi, K.; Hämmerling, U.; Chua, R.; Eppinger, T.; Levi, E.; Buck, J. 13,14-Dihydroxyretinol, a new bioactive retinol metabolite. J. Bio. Chem. 1995, 270, 18875–18880. [Google Scholar]
- Moise, A.R.; Kuska, V.; Imanishi, Y.; Palczewshi, K. Identification of all-trans-retinol: All-trans-13,14-dihydroretinol saturase. J. Biol. Chem. 2004, 279, 50230–50242. [Google Scholar]
- Moise, A.R.; Domínguez, M.; Alvarez, S.; Alvarez, R.; Schupp, M.; Cristancho, A.G.; Kiser, P.D.; De Lera, A.R.; Lazar, M.A.; Palczewski, K. Stereospecificity of retinol saturase: Absolute configuration, synthesis, and biological evaluation of dihydroretinoids. J. Am. Chem. Soc. 2008, 130, 1154–1155. [Google Scholar]
- Moise, A.R.; Kuska, V.; Blaner, W.S.; Baehr, W.; Palczewski, K. Metabolism and transactivation activity of 13,14-dihydroretinoic acid. J. Biol. Chem. 2005, 280, 27815–27825. [Google Scholar]
- Olson, J.A. Recommended dietary intakes (RDI) of vitamin A in humans. Am. J. Clin. Nutr. 1987, 45, 704–716. [Google Scholar]
- Ubels, J.L.; Macrae, S.M. Vitamin A is present as retinol in the tears of humans and rabbits. Curr. Eye Res. 1984, 3, 815–822. [Google Scholar] [CrossRef]
- Gasymov, O.K.; Abduragimov, A.R.; Yusifov, T.N.; Glasgow, B.J. Relaxation of ß-structure in tear lipocalin and enhancement of retinoid binding. Invest. Ophthalmol. Vis. Sci. 2002, 43, 3165–3173. [Google Scholar]
- Underwood, B.A. Vitamin A in human nutrition: Public health considerations. In The Retinoids: Biology, Chemistry, and Medicine; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 211–227. [Google Scholar]
- Billson, F.; Pararajasegaram, R. Prevention of childhood blindness. In Based on the WHO Meeting on the Prevention of Childhood Blindness, London, UK, 29 May–1 June 1990; World Health Organization: Geneva, Switzerland, 1992. [Google Scholar]
- Underwood, B.A.; Arthur, P. The contribution of vitamin A to public health. Faseb J. 1996, 10, 1040–1048. [Google Scholar]
- Isler, O. History and industrial application of carotenoids and vitamin A. Pure Appl. Chem. 1979, 51, 447–462. [Google Scholar] [CrossRef]
- Reif, W.; Grassner, H. Die technische vitamin-A-synthese der BASF. Chem. Ing. Tech. 1973, 45, 646–652. [Google Scholar] [CrossRef]
- Paust, J. Recent progress in commercial retinoids and carotenoids. Pure Appl. Chem. 1991, 63, 45–58. [Google Scholar] [CrossRef]
- Isler, O.; Kienzle, F. Vitamin A. In Kirk-Othmer Encyclopedia of Chemical Technology, 3rd ed.; John Wiley & Sons: New York, NY, USA; Volume 24, pp. 140–158.
- Schoenheimer, R. The Dynamic State of Body Constituents; Harvard University Press: Cambridge, USA, 1942. [Google Scholar]
- Kaegi, H.H. Synthesis of retinoids labeled with radioisotopes. In The Retinoids; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Academic Press Inc.: San Diego, USA, 1984; Volume 1, pp. 1147–1181. [Google Scholar]
- Olsen, J.A. Isotope-dilution techniques: A wave of the future in human nutrition. Am. J. Clin. Nutr. 1997, 66, 186–187. [Google Scholar]
- Dueker, S.R.; Jones, A.D.; Smith, G.M.; Clifford, A.J. Stable isotope methods for the study of β-carotene-d8 metabolism in humans utilizing tandem mass spectrometry and high-performance liquid chromatography. Anal. Chem. 1994, 66, 4177–4185. [Google Scholar]
- Tanumihardjo, S.A. Vitamin A status assessment in rats with 13C4-retinyl acetate and gas chromatography/combustion/isotope ratio mass spectrometry. J. Nutr. 2000, 130, 2844–2849. [Google Scholar]
- Wang, J.; Wang, Y.; Wang, Z.; Li, L.; Qin, J.; Lai, W.; Fu, Y.; Suter, P.M.; Russell, R.M.; Grusak, M. A.; Tang, G.; Yin, S. Vitamin A equivalence of spirulina β-carotene in Chinese adults as assessed by using a stable-isotope reference method. Am. J. Clin. Nutr. 2008, 87, 1730–1737. [Google Scholar]
- Tang, G.; Qin, J.; Dolnikowski, G.G.; Russell, R.M.; Grusak, M.A. Golden rice is an effective source of vitamin A. Am. J. Clin. Nutr. 2009, 89, 1776–1783. [Google Scholar] [CrossRef]
- Mathies, R.A.; Lugtenburg, J. The primary photoreaction of rhodopsin. In Molecular Mechanisms in Visual Transduction; Stavenga, D.G., DeGrip, W.J., Pugh, E.N., Jr., Eds.; Elsevier: Amsterdam, The Netherland, 2000; Volume 3, pp. 355–390. [Google Scholar]
- Siebert, F.; Hildebrandt, P. Vibrational Spectroscopy in Life Science; WILEY-VCH Verlag gmbh & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Sudo, Y.; Furutani, Y.; Wada, A.; Ito, M.; Kamo, N.; Kandori, H. Steric constraint in the primary photoproduct of an archaeal rhodopsin from regiospecific perturbation of C-D stretching vibration of the retinyl chromophore. J. Am. Chem. Soc. 2005, 127, 16036–16037. [Google Scholar]
- Verhoeven, M.A.; Creemers, A.F.L.; Bovee-Geurts, P.H.M.; DeGrip, W.J.; Lugtenburg, J.; De Groot, H.J.M. Ultra-high-field MAS NMR assay of a multispin labeled ligand bound to its G-protein receptor target in the natural membrane environment: Electronic structure of the retinylidene chromophore in rhodopsin. Biochemistry 2001, 40, 3282–3288. [Google Scholar]
- Concistré, M.; Gansmüller, A.; McLean, N.; Johannessen, O.G.; Montesinos, I.M.; Bovee-Geurts, P.H.M.; Verdegem, P.; Lugtenburg, J.; Brown, R.C.D.; De Grip, W.J.; Levitt, M.H. Double-quantum 13C nuclear magnetic resonance of bathorhodopsin, the first photointermediate in mammalian vision. J. Am. Chem. Soc. 2008, 130, 10490–10491. [Google Scholar]
- Lansing, J.C.; Hohwy, M.; Jaroniec, C.P.; Creemers, A.F.L.; Lugtenburg, J.; Herzfeld, J.; Griffin, R.G. Determination of Torsion Angles in Membrane Proteins in Perspectives in Solid State NMR in Biology; Kiihne, S.R., De Groot, H.J.M., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; pp. 185–190. [Google Scholar]
- Carravetta, M.; Edén, M.; Johannessen, O.G.; Luthman, H.; Verdegem, P.J.E.; Lugtenburg, J.; Sebald, A; Levitt, M.H. Estimation of carbon-carbon bond lengths and medium-range internuclear distances by solid-state nuclear magnetic resonance. J. Am. Chem. Soc. 2001, 123, 10628–10638. [Google Scholar]
- Kiihne, S.R.; Creemers, A.F.L.; DeGrip, W.J.; Bovee-Geurts, P.H.M.; Lugtenburg, J.; De Groot, H.J.M. Selective Interface Detection: Mapping binding site contacts in membrane proteins by NMR Spectroscopy. J. Am. Chem. Soc. 2005, 127, 5734–5735. [Google Scholar]
- Venter, J.C.; Adams, M.D.; Myers, E. W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A. et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar]
- Murhammer, D.W. Review and patents and literature. The use of insect cell cultures for recombinant protein synthesis: Engineering aspects. Appl. Biochem. Biotechnol. 1991, 31, 283–310. [Google Scholar]
- Vissers, P.M.A.M.; DeGrip, W.J. Functional expression of human cone pigments using recombinant baculovirus: Compatibility with histidine tagging and evidence for N-glycosylation. FEBS Lett. 1996, 396, 26–30. [Google Scholar] [CrossRef]
- Creemers, A.F.L.; Lugtenburg, J. The preparation of all-trans uniformly 13C-labeled retinal via a modular total organic synthetic strategy. Emerging central contribution of organic synthesis toward the structure and function study with atomic resolution in protein research. J. Am. Chem. Soc. 2002, 124, 6324–6334. [Google Scholar]
- Lockhart, I.M. Isotopes: Essential Chemistry and Applications; Elvidge, J.A., Jones, J.R., Eds.; The Chemical Society: London, UK, 1979. [Google Scholar]
- Torres, J.; Adams, P.D.; Arkin, I.T. Use of a new label, 13C=18O, in the determination of a structural model of phospholamban in a lipid bilayer. Spatial restraints resolve the ambiguity arising from interpretations of mutagenesis data. J. Mol. Biol. 2000, 300, 677–685. [Google Scholar] [CrossRef]
- Eyring, G.; Curry, B.; Broek, A.; Lugtenburg, J.; Mathies, R.A. Assignment and interpretation of hydrogen out-of-plane vibrations in the resonance Raman spectra of rhodopsin and bathorhodopsin. Biochemistry 1982, 21, 384–393. [Google Scholar]
- Verdegem, P.J.E.; Bovee-Geurts, P.H.M.; DeGrip, W.J.; Lugtenburg, J.; De Groot, H.J.M. Retinylidene ligand structure in bovine rhodopsin, metarhodopsin-I, and 10-methylrhodopsin from internuclear distance measurements using 13C-labeling and 1-D rotational resonance MAS NMR. Biochemistry 1999, 38, 11316–11324. [Google Scholar]
- DeLange, F.; Bovee-Geurts, P.H.M.; VanOostrum, J.; Portier, M.D.; Verdegem, P.J.E.; Lugtenburg, J.; DeGrip, W.J. An additional methyl group at the 10-position of retinal dramatically slows down the kinetics of the rhodopsin photocascade. Biochemistry 1998, 37, 1411–1420. [Google Scholar]
- Winkler, F.J.; Kühnl, K.; Medina, R.; Schwarz Kaske, R.; Schmidt, H.L. Principles and Results of stable isotope labelling of L-α-aminoacids by combined chemical and enzymatic methods. Isot. Environ. Health Stud. 1995, 31, 161–190. [Google Scholar] [CrossRef]
- Wald, G. The molecular basis of visual excitation. Nature 1968, 219, 800–807. [Google Scholar] [CrossRef]
- Durston, A.J.; Van der Wees, J.; Pijnappel, W.W.M.; Schilthuis, J.G.; Godsave, S.F. Retinoid signalling and axial patterning during early vertebrate embryogenesis. Cell. Mol. Life Sci. 1997, 53, 339–349. [Google Scholar] [CrossRef]
- Pijnappel, W.W.M.; Folkers, G.E.; De Jonge, W.J.; Verdegem, P.J.M.; De Laat, S.W.; Lugtenburg, J.; Hendriks, H.F.J.; Van der Saag, P.T.; Durston, A.J. Metabolism to a response pathway selective retinoid ligand during axial pattern formation. Proc. Natl. Acad. Sci. USA 1998, 95, 15424–15429. [Google Scholar]
- Smith, S.O.; Palings, I.; Miley, M.E.; Courtin, J.; De Groot, H.J.M.; Lugtenburg, J.; Mathies, R.A.; Griffin, R.G. Solid-state NMR studies on the mechanism of the opsin shift in the visual pigment rhodopsin. Biochemistry 1990, 29, 8158–8164. [Google Scholar]
- Creemers, A.F.L.; Klaassen, C.H.W.; Bovee-Geurts, P.H.M.; Kelle, R.; Kragl, U.; Raap, J.; DeGrip, W.J.; Lugtenburg, J.; De Groot, H.J.M. Solid state 15N NMR evidence for a complex schiff base counterion in the visual G-protein-coupled receptor rhodopsin. Biochemistry 1999, 38, 7195–7199. [Google Scholar]
- Baldwin, J.M.; Schertler, G.F.X.; Unger, V.M. An alpha-carbon template for the transmembrane helices in the rhodopsin family of G-protein-coupled receptors. J. Mol. Biol. 1997, 272, 144–164. [Google Scholar] [CrossRef]
- Ji, T.H.; Grossmann, M.; Ji, I. G-Protein-coupled receptors I. Diversity of receptor-ligand interactions. J. Biol. Chem. 1998, 273, 17299–17302. [Google Scholar]
- Hubbart, R.; Kropf, A. The action of light on rhodopsin. Proc. Natl. Acad. Sci. 1958, 44, 130–139. [Google Scholar]
- Palings, I.; Pardoen, J.A.; Van den Berg, E.M.M; Winkel, C.; Lugtenburg, J.; Mathies, R.A. Assignment of fingerprint vibrations in the resonance Raman spectra of rhodopsin, isorhodopsin, and bathorhodopsin: Implications for chromophore structure and environment. Biochemistry 1987, 26, 2544–2556. [Google Scholar]
- Feng, X.; Verdegem, P.J.E.; Lee, Y.K.; Sandström, D.; Edén, M.; Bovee-Geurts, P.; DeGrip, W.J.; Lugtenburg, J.; De Groot, H.J.M.; Levitt, M.H. Direct determination of a molecular torsional angle in the membrane protein rhodopsin by solid-state NMR. J. Am. Chem. Soc. 1997, 119, 6853–6857. [Google Scholar]
- Creemers, A.F.L.; Kiihne, S.; Bovee-Geurts, P.H.M.; DeGrip, W.J.; Lugtenburg, J.; De Groot, H.J.M. 1H and 13C MAS NMR evidence for pronounced ligand-protein interactions involving the ionone ring of the retinylidene chromophore in rhodopsin. Proc. Natl. Acad. Sci.USA 2002, 99, 9101–9106. [Google Scholar]
- Vissers, P.M.A.M.; Bovee-Geurts, P.H.M.; Portier, M.D.; Klaassen, C.H.W.; DeGrip, W.J. Large-scale production and purification of the human green cone pigment: Characterization of late photo-intermediates. Biochem. J. 1998, 330, 1201–1208. [Google Scholar]
- Dawson, M.I.; Okamura, W.H. Chemistry and Biology of Synthetic Retinoids; CRC Press Inc.: Boca Raton, FL, USA, 1990. [Google Scholar]
- Sporn, M.B.; Roberts, A.B.; Goodman, D.S. Chemistry and physical properties of retinoids. In The Retinoids; Academic press, Inc.: Orlando, FL, USA, 1984; Volume 1, pp. 7–146. [Google Scholar]
- Barnard, J.H.; Collings, J.C.; Whiting, A.; Przyborski, A.A.; Marder, T.B. Synthetic retinoids: Structure-activity relationships. Chem. Eur. J. 2009, 15, 11430–11442. [Google Scholar]
- Sporn, M.B.; Roberts, A.B. ; Goodman D.S. The Retinoids: Biology, Chemistry, and Medicine; Raven Press: New York, NY, USA, 1994. [Google Scholar]
- Isler, O.; Huber, W.; Ronco, A.; Kofler, M. Synthese des vitamin A. Helv. Chim. Acta 1947, 30, 1911–1927. [Google Scholar]
- Schwieter, U.; Planta, C.V.; Rüegg, R.; Isler, O. Synthesen in der Vitamen-A2-Reihe. Die darstellung von sterisch ungehinderten vitamin-A2-isomeren. Helv. Chim. Acta. 1962, 45, 528–541. [Google Scholar]
- Saucy, G.; Marbet, R. ber die reaktion von tertiären vinylcarbinolen mit isopropenyläther eine neue methode zur herstellung von γ,δ,-ungesättigten ketonen. Helv. Chim. Acta 1967, 50, 2091–2095. [Google Scholar] [CrossRef]
- Saucy, G.; Marbet, R. Über eine neuartige synthese von β-ketoallenen durch reaktion von tertiären acetylencarbinolen mit vinyläthern eine ergiebige methode zur herstellung des pseudojonons und verwandter verbindungen. Helv. Chim. Acta. 1967, 50, 1158–1167. [Google Scholar] [CrossRef]
- Kildahl-Andersen, G.; Konovalova, T.A.; Focsan, A.L.; Kispert, L.D.; Anthonsen, T.; Liaaen-Jensen, S. Comparative studies on radical cation formation from carotenoids and retinoids. Tetrahedron Lett. 2007, 48, 8196–8199. [Google Scholar]
- Pommer, H. Synthesen in der vitamin-A-reihe. Angew. Chem. 1960, 72, 811–819. [Google Scholar] [CrossRef]
- Reif, W.; Grassner, H. Die technische vitamin A synthese der BASF. Chem. Ing. Tech. 1975, 45, 646–652. [Google Scholar]
- Paust, J. Recent progress in commercial retinoids and carotenoids. Pure Appl. Chem. 1991, 63, 45–58. [Google Scholar] [CrossRef]
- Ho, T.L. Hard and Soft Acid and Base Principle and Organic Chemistry; Academic Press: New York, NY, USA, 1977. [Google Scholar]
- Rigassi, N.; Schweitzer, U. Process for the manufacture of polyene compounds. Patent CA 914201 (A), 07 November 1972. [Google Scholar]
- Fransen, M.R.; Palings, I.; Lugtenburg, J.; Jansen, P.A.A.; Gronendijk, G.W.T. Preparation and photolysis of deuterium labeled rhodopsin. Recl. Trav. Chim. Pays-Bas. 1980, 99, 384–391. [Google Scholar]
- Broek, A.D.; Lugtenburg, J. 10-Mono-, 11-mono-, 22-mono- and 11,12-dideutero retinal. Recl. Trav. Chim. Pays-Bas. 1981, 99, 363–366. [Google Scholar]
- Broek, A.D.; Lugtenburg, J. Preparation of deuterium labeled retinals having high deuterium content on specific positions 10-mono-, 11-mono-, 10, 11-di- and 14,20,20,20-tetradeutero retinal. Recl. Trav. Chim. Pays-Bas. 1982, 101, 103–105. [Google Scholar]
- Pardoen, J.A.; Winkel, C.; Mulder, P.P.J.; Lugtenburg, J. Synthesis of retinals labelled at positions 14 and 15 with 13C and/or 2H. Recl. Trav. Chim. Pays-Bas. 1984, 103, 135–141. [Google Scholar]
- Pardoen, J.A.; Neijenesch, H.N.; Mulder, P.P.J.; Lugtenburg, J. Synthesis of 10-, 11-, 14- and 20-mono 13C-retinal. Recl. Trav. Chim. Pays-Bas. 1983, 102, 341–347. [Google Scholar]
- Pardoen, J.A.; Van den Berg, E.M.M.; Mulder, P.P.J.; Lugtenburg, J. Synthesis of 8-, 9-, 12- and 13-mono 13C-retinal. Can. J. Chem. 1985, 63, 1431–1435. [Google Scholar]
- Gebhard, R.; Courtin, J.M.L.; Shadid, L.B.; Van Haveren, C.J.; Van Haeringen, C.J.; Lugtenburg, J. Synthesis of retinals labelled with 13C in the cyclohexene ring. Recl. Trav. Chim. Pays-Bas. 1989, 108, 207–214. [Google Scholar]
- Courtin, J.M.L.; ‘t Lam, A.J.M.; Peters, A.J.M.; Lugtenburg, J. Synthesis of 5-,6-,7- and 18-mono 13C labelled retinals. Recl. Trav. Chim. Pays-Bas. 1985, 104, 281–288. [Google Scholar]
- Wang, Y.; Woo, W.S.; Van der Hoef, I.; Lugtenburg, J. 9-Demethyl-9-haloretinals by Wadsworth-Emmons Coupling- Easy preparation of pure (all-E), (9Z) and (11Z) isomers. Eur. J. Org. Chem. 2004, 2166–2175. [Google Scholar]
- Corey, E.J.; Erickson, B.W. γ-Condensation of an allylic phosphonium ylide. J. Org. Chem. 1974, 39, 821–825. [Google Scholar] [CrossRef]
- Still, W.C.; Gennari, C. Direct synthesis of Z-unsaturated esters. A useful modification of the Horner-Emmons olefination. Tetrahedron Lett. 1983, 24, 4405–4408. [Google Scholar]
- Ando, K. Z-Selective Horner-Wadsworth-Emmons reaction of α-substituted ethyl (diarylphosphono)acetates with aldehydes. J. Org. Chem. 1998, 63, 8411–8416. [Google Scholar] [CrossRef]
- Ando, K.A. Mechanistic study of the Horner-Wadsworth-Emmons reaction: Computational investigation on the reaction pass and the stereochemistry in the reaction of lithium enolate derived from trimethyl phosphonoacetate with acetaldehyde. J. Org. Chem. 1999, 64, 6815–6821. [Google Scholar]
- Mead, D.; Asato, A.E.; Denny, M.; Liu, R.S.H.; Hanzawa, Y.; Taguchi, T.; Yamada, A.; Kobayashi, N.; Hosoda, A.; Kobayashi, Y. 9-Cis 11-cis isomers of 18,18,18-, 19,19,19- and 20,20,20-trifluororetinal. Tetrahedron Lett. 1987, 28, 259–262. [Google Scholar]
- Trehan, A.; Liu, R.S.H. All-cis-retinal and 7-cis,9-cis,11-cis-retinal. Tetrahedron Lett. 1988, 29, 419–422. [Google Scholar] [CrossRef]
- Wada, A.; Wang, F.; Ito, M. A convenient and stereoselective synthesis of 11Z-3,4-didehydroretinal by Horner-Emmons reaction using diphenyl phosphonate. Chem. Pharm. Bull. 2008, 56, 112–114. [Google Scholar]
- DeGrip, W.J.; Bovee-Geurts, P.H.M.; Van der Hoef, I.; Lugtenburg, J. 7,8 -Dihydro retinals outperform the native retinals in conferring photosensitivity to visual opsin. J. Am. Chem. Soc. 2007, 129, 13265–13269. [Google Scholar]
- Dawadi, P.B.S.; Lugtenburg, J. Efficient preparation of [2-13C]- and [3-13C]-3-cyano-4-methyl-3-pyrrolin-2-one. Eur. J. Org. Chem. 2007, 1294–1300. [Google Scholar]
- Büchi, G.; Wuest, H. Synthetic studies on damascenones. Helv. Chim. Acta. 1971, 54, 1767–1776. [Google Scholar] [CrossRef]
- Kok, J.G.J.; Van Moorselaar, R. Technische synthese von vitamin A. Chem. Weekbl. 1973, 69, V8–V9. [Google Scholar]
- Verdegem, P.J.E.; Monnee, M.C.F.; Lugtenburg, J. Simple and efficient preparation of [10,20-13C2]- and [10-CH3, 13-13C2]-10-methylretinal: Introduction of substituents at the 2-position of 2,3-unsaturated nitriles. J. Org. Chem. 2001, 66, 1269–1282. [Google Scholar] [CrossRef]
- Wang, Y.; Lugtenburg, J. Preparation of (all-E)- and (11Z)-12-haloretinals and (11Z,13Z)- and (13Z)-14-haloretinals by the C15 + C5 route - exploring the possibility of preparing any retinoid rationally chemically modified at any position in the conjugated tail. Eur. J. Org. Chem. 2004, 5100–5110. [Google Scholar]
- Verhoeven, M.A.; Bovee-Geurts, P.H.M.; De Groot, H.J.M.; Lugtenburg, J.; DeGrip, W.J. Methyl substituents at the 11 or 12 position of retinal profoundly and differentially affect photochemistry and signalling activity of rhodopsin. J. Mol. Biol. 2006, 363, 98–113. [Google Scholar] [CrossRef]
- Freeman, J.P. Conjugate addition of the Wittig reagent. J. Org. Chem. 1966, 31, 538–541. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C. Eine neue synthese von Cyclohexadien-Derivaten. Chem. Ber. 1973, 106, 3779–3787. [Google Scholar] [CrossRef]
- Dauben, W.G.; Hart, D.J.; Ipaktschi, J.; Kozikowski, A.P. A facile synthesis of substituted conjugated cyclohexadienes. Tetrahedron Lett. 1973, 4425–4428. [Google Scholar]
- Dauben, W.G.; Ipaktschi, J. Facile synthesis of strained bridgehead olefins via the intramolecular Wittig reaction. J. Am. Chem. Soc. 1973, 5088–5089. [Google Scholar]
- Padwa, A.; Brodsky, L. Utilization of the 1,4-conjugated Wittig reaction for the synthesis of substituted 1,3-cyclohexadines. J. Org. Chem. 1974, 39, 1318–1320. [Google Scholar] [CrossRef]
- Wang, Y.; Lugtenburg, J. 4,5-Didehydro-9-demethyl-9-halo-5,6-dihydroretinals and their 9-cyclopropyl and 9-isopropyl derivatives- Simple preparation of α-ionone derivatives and pure (all-E)-, (9Z)- and (11Z)- α-retinals. Eur. J. Org. Chem. 2004, 3497–3510. [Google Scholar]
- Sharma, A.S. On the regiochemistry of the reaction of lithioketeneimines with chlorotrimethylsilane. Ind. J. Chem. 1979, 18B, 84–86. [Google Scholar]
- Al-Badri, H.; About-Jaudet, E.; Collignon, N. New and efficient synthesis of (E)-4-diethoxyphosphonyl-2-methyl-2-butenal and of ethyl (E)-4-diethoxyphosphonyl-2-methyl-2-butenoate, important building blocks in retinoid chemistry. Tetrahedron Lett. 1995, 36, 393–396. [Google Scholar] [CrossRef]
- Courtin, J.M.L.; Verhagen, L.; Biesheuvel, P.L.; Lugtenburg, J.; Van der Bend, R.L.; Van Dam, K. Bacteriorhodopsin, the influence of cyclohexene-ring methyls. Recl. Trav. Chim. Pays-Bas. 1987, 106, 112–119. [Google Scholar]
- Spijker-Assink, M.B.; Winkel, C.; Baldwin, G.S.; Lugtenburg, J. 5-Demethylretinal and its 5-2H-, 7-2H-, 5,7-2H2-isotopomers, synthesis, photochemistry and spectroscopy. Recl. Trav. Chim. Pays-Bas. 1988, 107, 125–131. [Google Scholar]
- Groesbeek, M.; Lugtenburg, J. Synthesis of nitroxide containing polyenes: Two commercially modified retinals and their interaction with bacetriorhodopsin. Recl. Trav. Chim. Pays-Bas. 1995, 114, 403–409. [Google Scholar]
- Van der Steen, R.; Biesheuvel, P.L.; Erkelens, C.; Mathies, R.A.; Lugtenburg, J. 8,16- and 8,18-Methanobacteriorhodopsin synthesis and spectroscopy of 8,16- and 8,18-methanoretinals and their interaction with bacteriopsin. Recl. Trav. Chim. Pays-Bas. 1989, 108, 83–93. [Google Scholar]
- Spijker-Assink, M.B.; Robijn, G.W.; Ippel, J.H.; Lugtenburg, J.; Groen, B.H.; Van Dam, K. (1R)- and (1S)-5-Dethyl-8,16-methanobacteriorhodopsin and its properties. The synthesis and spectroscopy of 5-demethyl-8,16-methanoretinal in optically active and isotopic forms. Recl. Trav. Chim. Pays-Bas. 1992, 111, 29–40. [Google Scholar]
- Groesbeek, M.; Van Galen, A.J.J.; Ippel, J.H.; Berden, J.A.; Lugtenburg, J. Three bacteriorhodopsin with ring-didemethylated 6-s locked chromophores and their properties. Recl. Trav. Chim. Pays-Bas. 1993, 112, 237–246. [Google Scholar]
- Groesbeek, M.; Robijn, G.W.; Lugtenburg, J. Synthesis and spectroscopic characterization of the double locked 9E, 11Z retinal model systems 7E,13E-11.19-10.20-dimethanoretinal and its 13Z isomer. Recl. Trav. Chim. Pays-Bas. 1992, 111, 92–98. [Google Scholar]
- Groesbeek, M.; Kirillova, Y.J.; Boeff, R.; Lugtenburg, J. Syntheis of six novel retinals and their interaction with bacteriorhodopsin. Recl. Trav. Chim. Pays-Bas. 1994, 113, 45–52. [Google Scholar]
- Van Amsterdam, L.J.P.; Lugtenburg, J. Total synthesis of a 10,14-sulphur-bridged 11-cis retinal analogue: 6-formyl-2-[4-(2,6,6-trimethylcyclohex-1-en-1yl)but-3-en-2-ylidene]-2H-thiopyran. J. Chem. Soc. Chem. Commun. 1982, 946–957. [Google Scholar] [CrossRef]
- Muradin-Szweykowska, M.; Peters, A.J.M.; Lugtenburg, J. The interaction of bacteriorhodopsin with 11,14-bridged retinals. The synthesis of 13-demethyl-11,14-imino-, 13-demethyl-N-methyl-11,14-imino-, 13-demethyl-11,14-thio-, 13-demethyl-11,14-etheno and their binding with the baceteriopsin. Recl. Trav. Chim. Pays-Bas. 1984, 103, 105–109. [Google Scholar]
- Broek, A.D.; Muradin-Szweykowska, M.; Courtin, J.M.L.; Lugtenburg, J. Preparation of 11,14 epoxy-bridged and isomeric chain demethylated retinals. 9-demethyl-, 13-demethyl-, and 9,13-bisdemethyl retinals. Recl. Trav. Chim. Pays-Bas. 1983, 102, 46–51. [Google Scholar]
- Van den Berg, E.M.M.; Van der Bent, A.; Lugtenburg, J. Synthesis of specifically deuterated 9- and 13-demethyl retinals. Recl. Trav. Chim. Pays-Bas. 1990, 109, 160–167. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dawadi, P.B.S.; Lugtenburg, J. Synthesis and Use of Stable Isotope Enriched Retinals in the Field of Vitamin A. Molecules 2010, 15, 1825-1872. https://doi.org/10.3390/molecules15031825
Dawadi PBS, Lugtenburg J. Synthesis and Use of Stable Isotope Enriched Retinals in the Field of Vitamin A. Molecules. 2010; 15(3):1825-1872. https://doi.org/10.3390/molecules15031825
Chicago/Turabian StyleDawadi, Prativa B.S., and Johan Lugtenburg. 2010. "Synthesis and Use of Stable Isotope Enriched Retinals in the Field of Vitamin A" Molecules 15, no. 3: 1825-1872. https://doi.org/10.3390/molecules15031825
APA StyleDawadi, P. B. S., & Lugtenburg, J. (2010). Synthesis and Use of Stable Isotope Enriched Retinals in the Field of Vitamin A. Molecules, 15(3), 1825-1872. https://doi.org/10.3390/molecules15031825