An Improved in Vivo Deuterium Labeling Method for Measuring the Biosynthetic Rate of Cytokinins



 and
and
Abstract
:1. Introduction

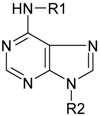
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | R2 | Common name | Abbreviation |
|---|---|---|---|
 | H | isopentenyladenine | iP |
| R | isopentenyladenosine | iPR | |
| RMP | isopentenyladenosine-5´-monophosphate | iPMP | |
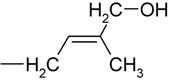 | H | trans-zeatin | tZ |
| R | trans-zeatin riboside | tZR | |
| RMP | trans-zeatin riboside-5´-monophosphate | tZMP | |
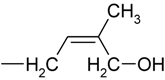 | H | cis-zeatin | cZ |
| R | cis-zeatin riboside | cZR | |
| RMP | cis-zeatin riboside-5´-monophosphate | cZMP | |
 | H | dihydrozeatin | DHZ |
| R | dihydrozeatin riboside | DHZR |
2. Results and Discussion
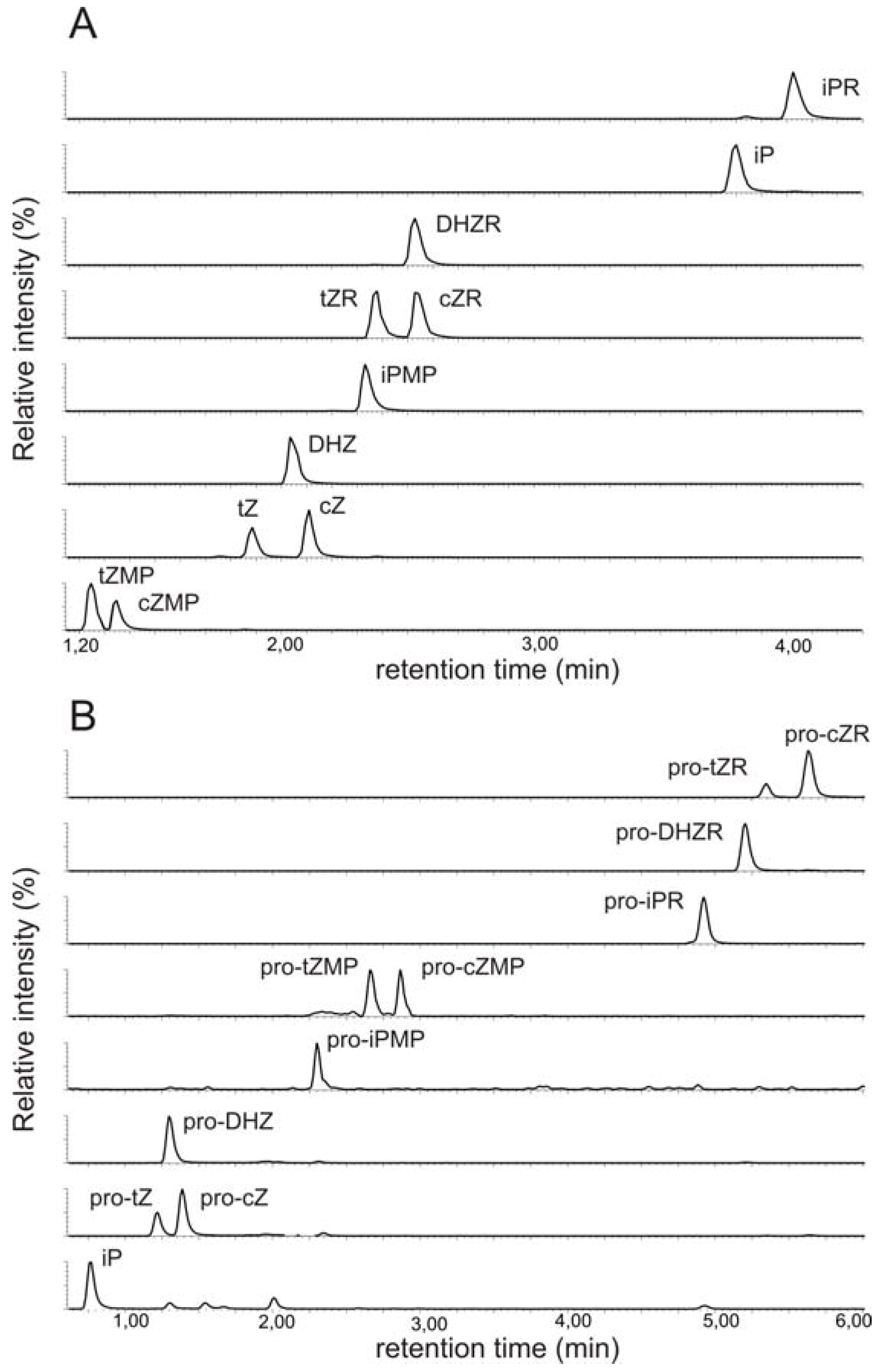
2.1. Implementation of ultra performance liquid chromatography expedites cytokinin analysis
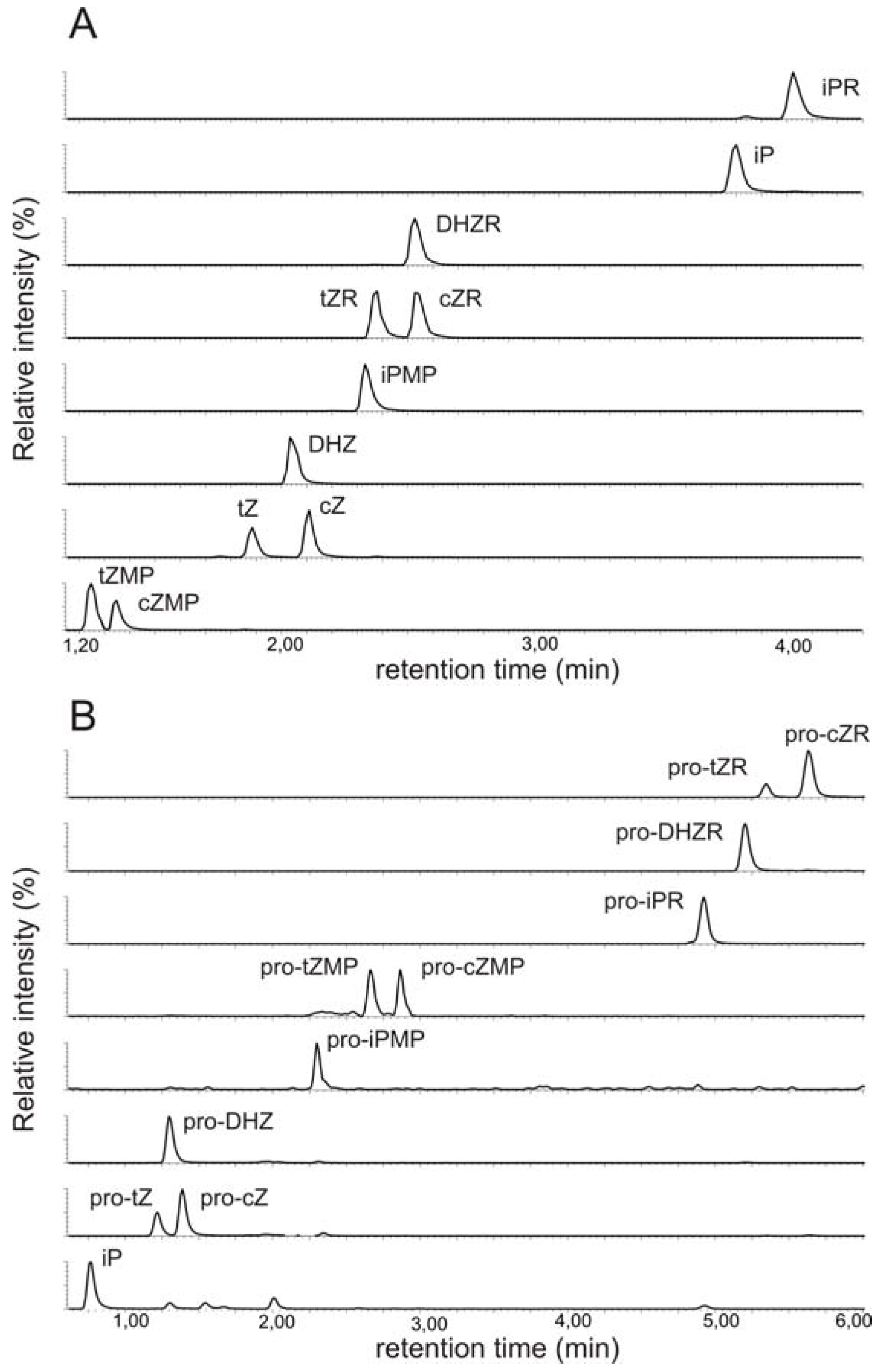
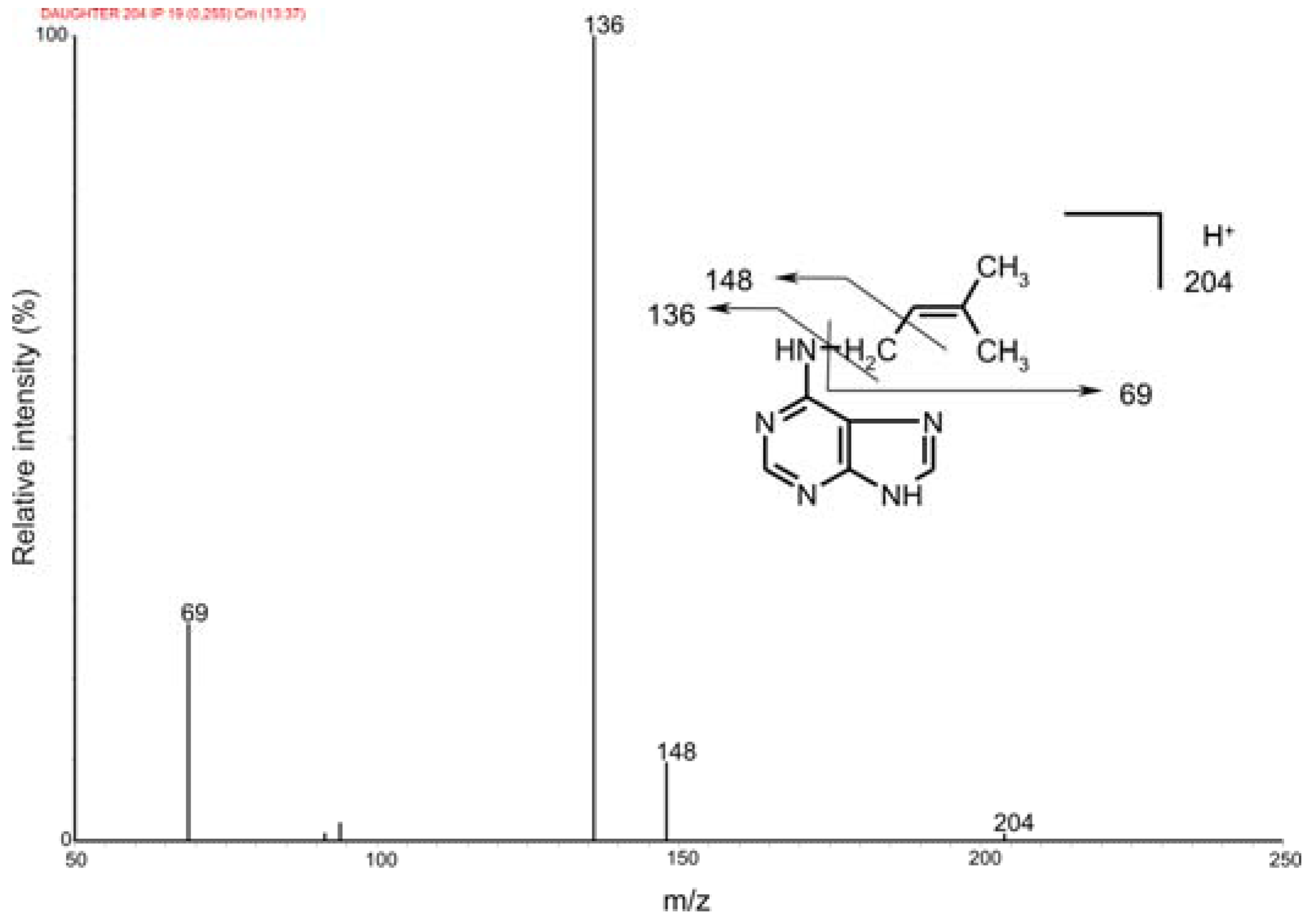
2.2. Mass spectrometric detection
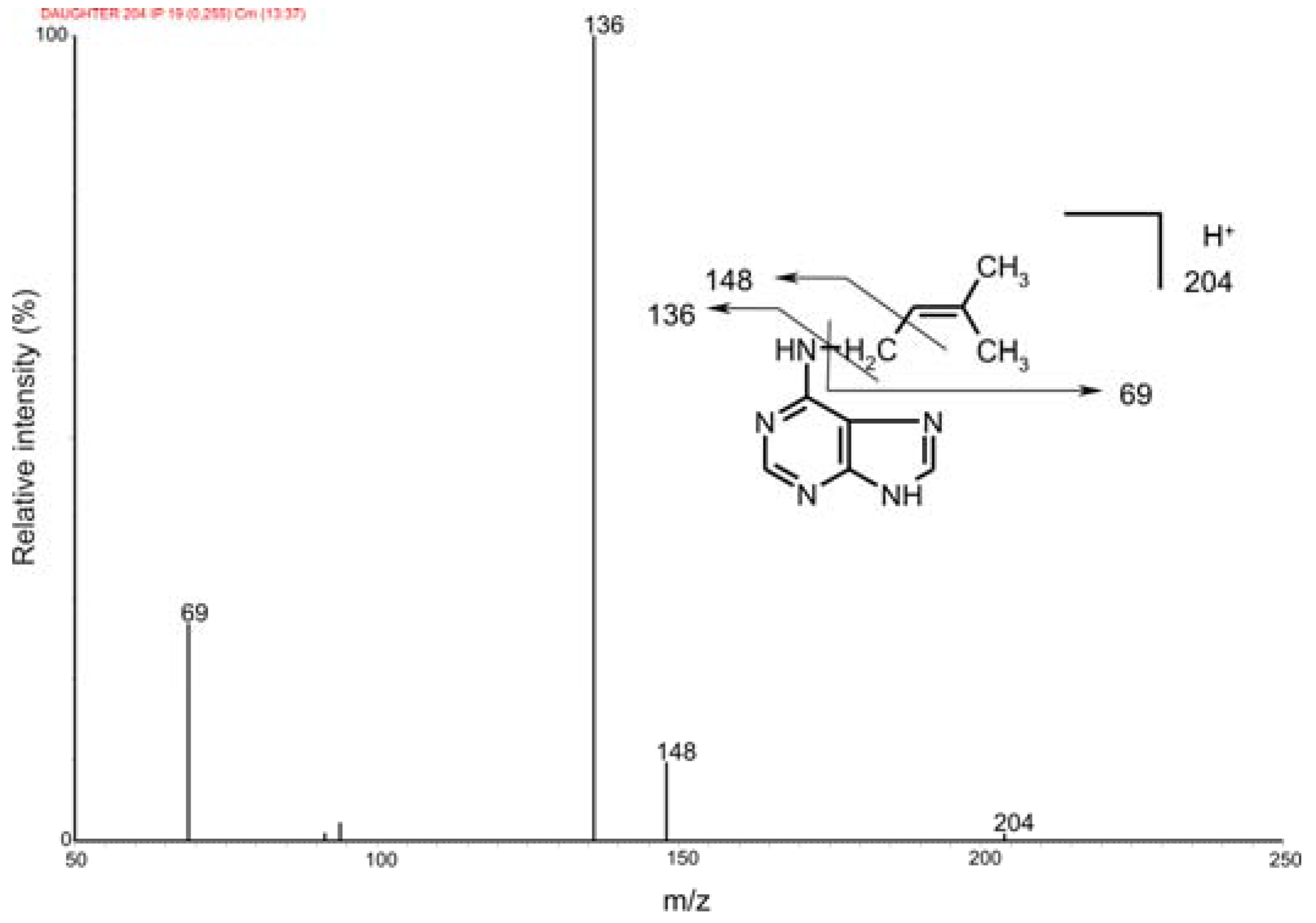
| Features | iP | D6-iP |
|---|---|---|
| Selected fragment formula | C5H9+ | C5H3D6+ |
| Experimental m/z | 69.0703 | 75.1081 |
| Calculated m/z | 69.0704 | 75.1081 |
| Difference (ppm) | -1.4 | 0.0 |
| Derivatized cytokinins | Underivatized cytokinins | ||||
|---|---|---|---|---|---|
| Cytokinin | Isotopomer | Diagnostic transition | Cytokinin | Isotopomer | Diagnostic transition |
| pro-iPR | I0 | 504-204 | iPR | I0 | 336-204 |
| I1 | 505-205 | I1 | 337-205 | ||
| I2 | 506-206 | I2 | 338-206 | ||
| I3 | 507-207 | I3 | 339-207 | ||
| pro-ZR | I0 | 576-276 | ZR | I0 | 352-220 |
| I1 | 577-277 | I1 | 353-221 | ||
| I2 | 578-278 | I2 | 354-222 | ||
| I3 | 579-279 | I3 | 355-223 | ||
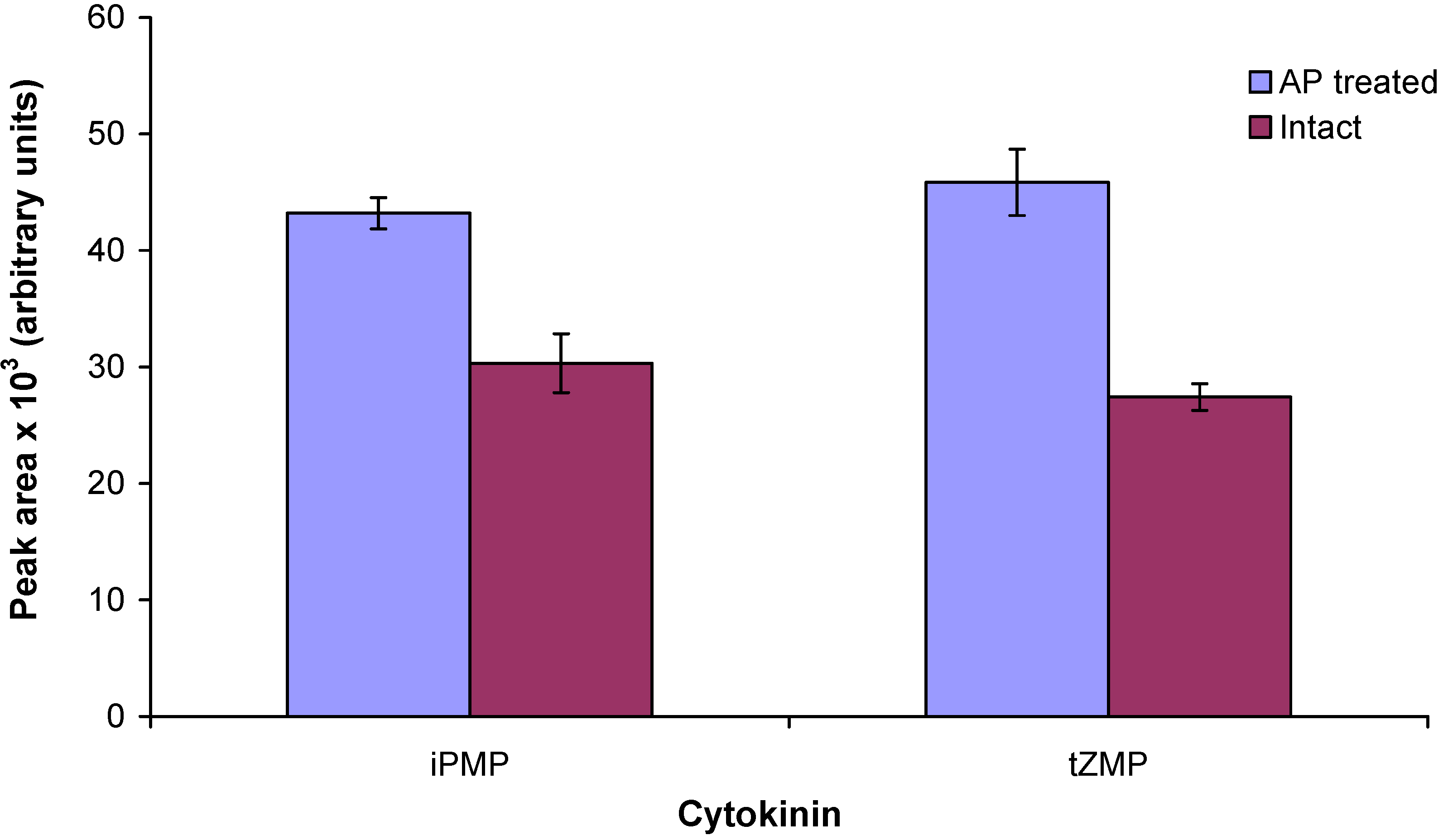
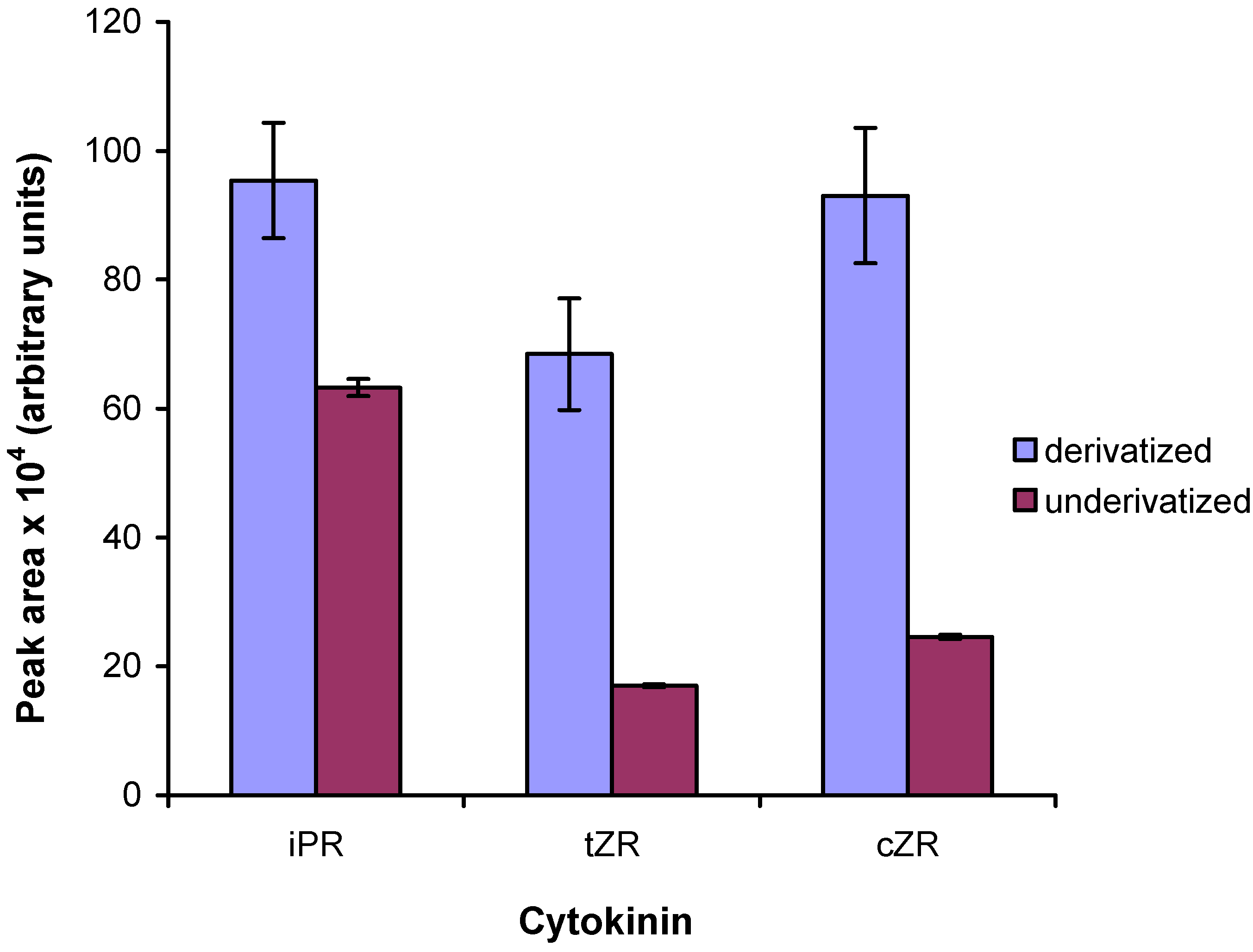
2.3. Enzymatic treatment of cytokinin nucleotides improves the sensitivity of mass spectrometric detection
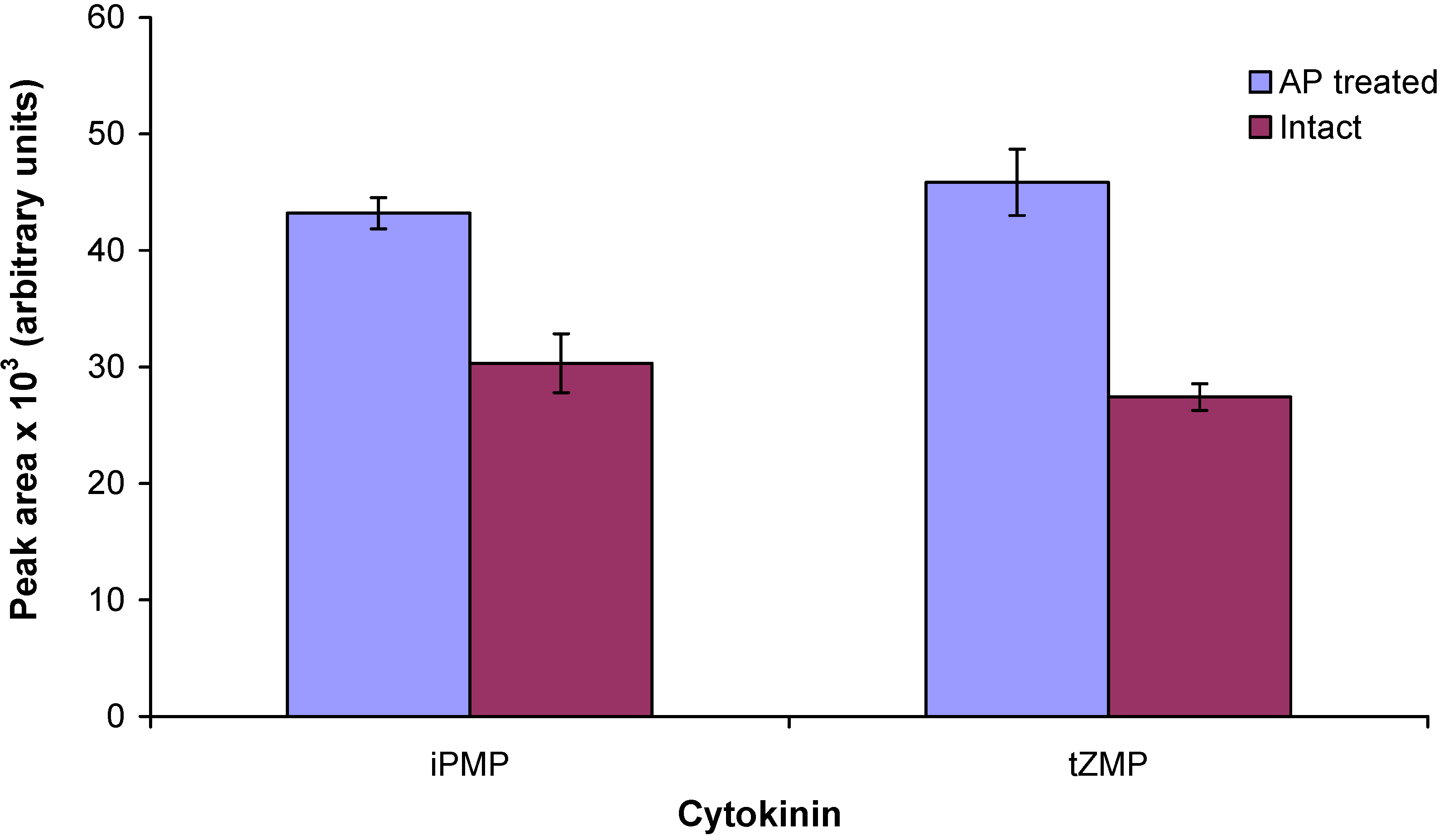
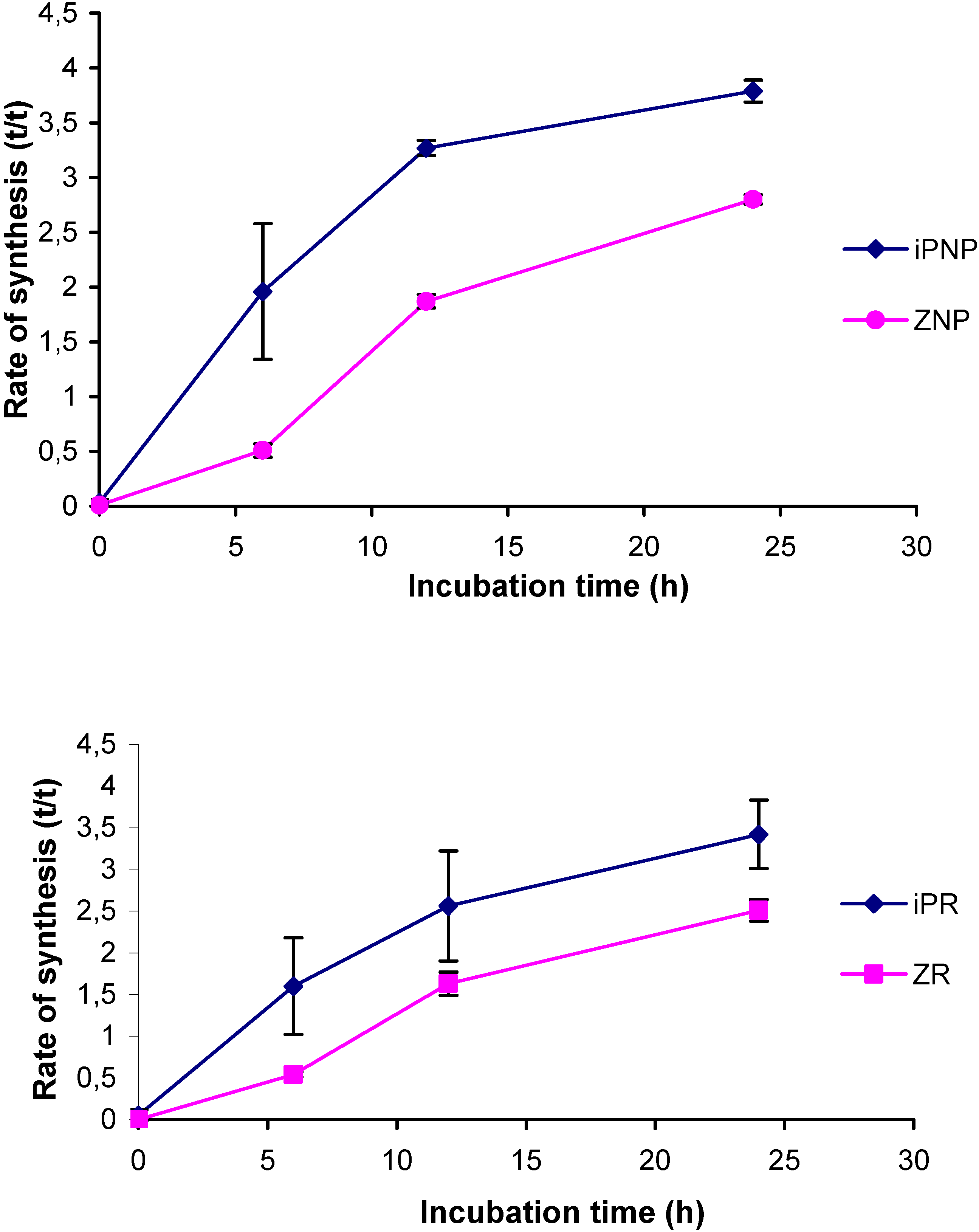
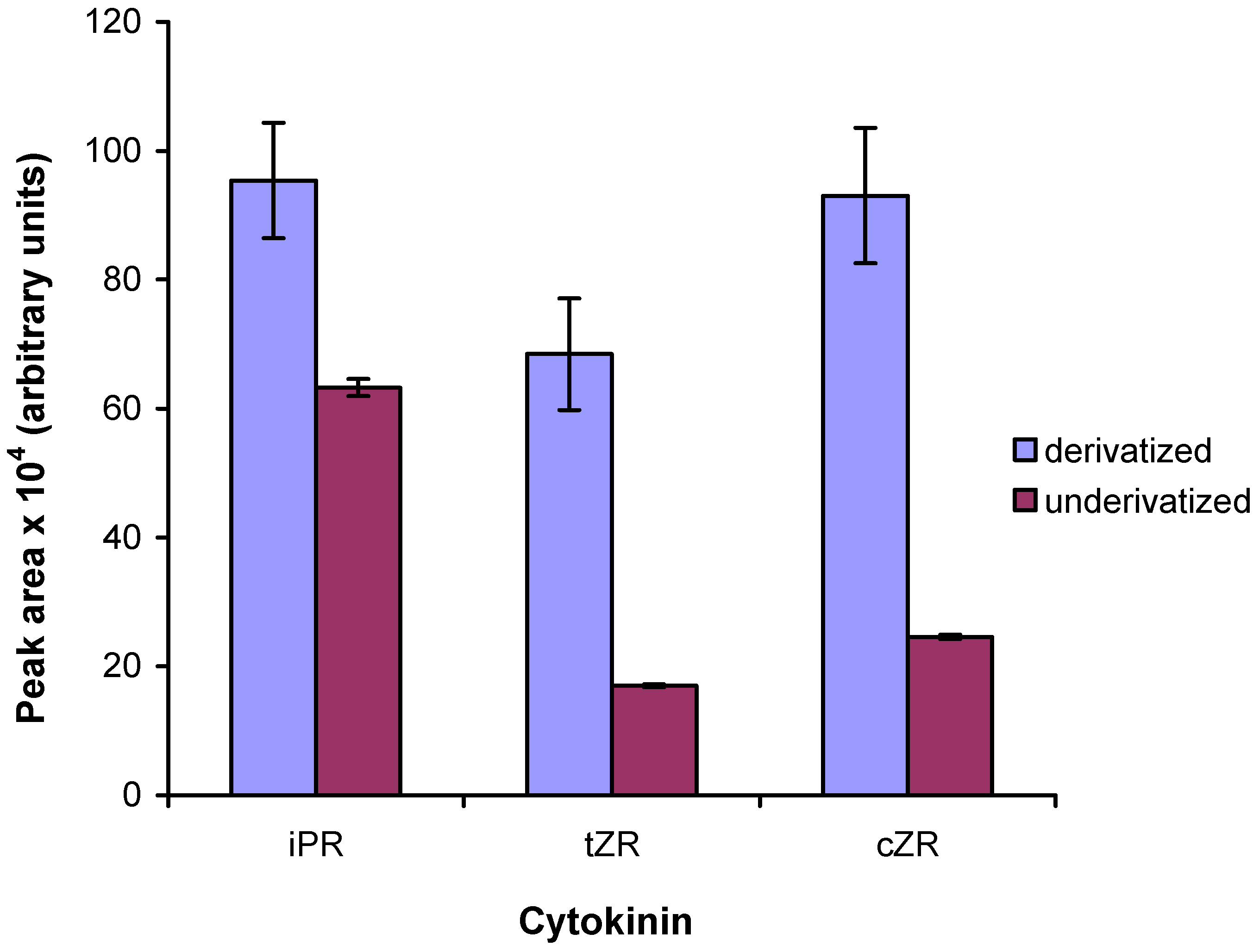
2.4. Application of the new method

2.5. Comparison to existing methods
| Cytokinin | Derivatized | Underivatized | ||
|---|---|---|---|---|
| Rate of synthesis (t/t) | SD | Rate of synthesis (t/t) | SD | |
| iPNP | 1.6843 | 0.2099 | 1.7961 | 0.1750 |
| iPR | 1.2965 | 0.1334 | 1.4538 | 0.1525 |
3. Experimental
3.1. Chemicals
3.2. Biological material
3.3. Sample preparation
3.4. HPLC-Q-TOF MS identification
3.5. UPLC-MS/MS
4. Conclusions
Acknowledgements
References and Notes
- Mok, M.C. Cytokinins and plant development-an overview. In Cytokinins: Chemistry and Function; Mok, D.W.S., Mok, M.C., Eds.; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Sakakibara, H. Cytokinins: activity, biosynthesis, and translocation. Annu. Rev. Plant Biol. 2006, 57, 431–449. [Google Scholar] [CrossRef]
- Spíchal, L.; Rakova, N.Y.; Reifler, M.; Mizuno, T.; Romanov, G.A.; Strnad, M.; Schmulling, T. Two cytokinin receptors of Arabidopsis thaliana, CRE1/AHK4 and AHK3, differ in their ligand specificity in a bacterial assay. Plant Cell Physiol. 2004, 45, 1299–1305. [Google Scholar] [CrossRef]
- Kakimoto, T. Identification of plant cytokinin biosynthetic enzymes as dimethylallyl diphosphate: ATP/ADP isopentenyltransferases. Plant Cell Physiol. 2001, 42, 677–685. [Google Scholar] [CrossRef]
- Takei, K.; Sakakibara, H.; Sugiyama, T. Identification of genes encoding adenylate isopentenyltransferase, a cytokinin biosynthesis enzyme, in Arabidopsis thaliana. J. Biol. Chem. 2001, 276, 26405–26410. [Google Scholar] [CrossRef]
- Takei, K.; Yamaya, T.; Sakakibara, H. Arabidopsis CYP735A1 and CYP735A2 encode cytokinin hydroxylases that catalyze the biosynthesis of trans-zeatin. J. Biol. Chem. 2004, 279, 41866–41872. [Google Scholar]
- Miyawaki, K.; Tarkowski, P.; Matsumoto-Kitano, M.; Kato, T.; Tarkowska, D.; Tabata, S.; Sandberg, G.; Kakimoto, T. In planta roles of Arabidopsis ATP/ADP isopentenyltransferases and tRNA isopentenyltransferases in cytokinin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 16598–16603. [Google Scholar]
- Tarkowski, P.; Ge, L.Y.; Young, J.W.H.; Tan, S.N. Analytical methods for cytokinins. TrAC. Trends Anal. Chem. 2009, 28, 323–335. [Google Scholar] [CrossRef]
- Hocart, C.H.; Letham, D.S. Biosynthesis of cytokinin in germinating seeds of Zea mays. J. Exp. Bot. 1990, 41, 1525–1528. [Google Scholar] [CrossRef]
- Sakakibara, H.; Kasahara, H.; Ueda, N.; Kojima, M.; Takei, K.; Hishiyama, S.; Asami, T.; Okada, K.; Kamiya, Y.; Yamaya, T.; Yamaguchi, S. Agrobacterium tumefaciens increases cytokinin production in plastids by modifying the biosynthetic pathway in the host plant. Proc. Natl. Acad. Sci. USA 2005, 102, 9972–9977. [Google Scholar]
- Åstot, C.; Dolezal, K; Moritz, T.; Sandberg, G. Deuterium in vivo labelling of cytokinins in Arabisopsis thaliana analysed by capillary liquid chromatography/frit-fast atom bombardment mass spectrometry. J. Mass Spectrometry 2000, 35, 13–22. [Google Scholar] [CrossRef]
- Åstot, C.; Dolezal, K; Nordström, A.; Wang, Q.; Kunkel, T.; Moritz, T.; Chua, N.H.; Sandberg, G. An alternative cytokinin biosynthesis pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 14788–14783. [Google Scholar]
- Åstot, C.; Dolezal, K; Moritz, T.; Sandberg, G. Precolumn derivatization and capillary liquid chromatographic/frit-fast atom bombardment mass spectrometric analysis of cytokinins in Arabidopsis thaliana. J. Mass Spectrometry 1998, 33, 892–902. [Google Scholar] [CrossRef]
- Nordström, A.; Tarkowski, P.; Tarkowska, D.; Norbaek, R.; Åstot, C.; Dolezal, K.; Sandberg, G. Auxin regulation of cytokinin biosynthesis in Arabidopsis thaliana: factor of potential importance for auxin-cytokinin-regulated development. Proc. Natl. Acad. Sci. USA 2004, 101, 8039–8044. [Google Scholar]
- Nováková, L.; Vlčková, H. A review of current trends and advantages in modern bio-analytical methods: Chromatography and sample preparation. Anal. Chim. Acta 2009, 656, 8–35. [Google Scholar] [CrossRef]
- Nordström, A.; Tarkowski, P.; Tarkowská, D.; Dolezal, K.; Åstot, C.; Sandberg, G.; Moritz, T. Derivatization for LC-electrospray ionization-MS: a tool for improving reversed-phase separation and ESI response of bases, ribosides and intact nucleotides. Anal. Chem. 2004, 76, 2869–2877. [Google Scholar]
- Novák, O.; Hauserová, E.; Amakorová, P.; Doležal, K.; Strnad, M. Cytokinin profiling in plant tissues using ultra-performance liquid chromatography-electrospray tandem mass spectrometry. Phytochemistry 2008, 69, 2214–2224. [Google Scholar] [CrossRef]
- Novák, O.; Tarkowski, P.; Tarkowská, D.; Doležal, K.; Lenobel, R.; Strnad, M. Quantitative analysis of cytokinins in plants by liquid chromatography-single-quadrupole mass spectrometry. Anal. Chim. Acta 2003, 480, 207–218. [Google Scholar] [CrossRef]
- Cech, N.B.; Enke, C.G. Effect of affinity for droplet surface on the fraction of analyte molecules charged during electrospray droplet fission. Anal. Chem. 2001, 73, 4632–4639. [Google Scholar] [CrossRef]
- Béres, T.; Zatloukal, M.; Voller, J.; Niemann, P.; Gahsche, M.C.; Tarkowski, P.; Novák, O.; Hanuš, J.; Strnad, M.; Doležal, K. Synthesis and LC-MS identification and quantification of cytokinin nucleotides in K-562 human leukemia cells. Anal. Bioanal. Chem. 2010, 398, 2071–2080. [Google Scholar] [CrossRef]
- Sun, J.Q.; Niu, Q.W; Tarkowski, P.; Zheng, B.L.; Tarkowska, D.; Sandberg, G.; Chua, N.H.; Zuo, J. The Arabidopsis AtIPT8/PGA22 gene encodes an isopentenyl transferase that is involved in de novo cytokinin biosynthesis. Plant Physiol. 2003, 131, 167–176. [Google Scholar] [CrossRef]
- Dobrev, P.I.; Novák, O.; Doležal, K.; Trčková, M.; Kamínek, M. Determination of phytohormone biosynthesis in wheat grains by LC/MS. In Proceedings of 34th Symposium on High-Performance Liquid Phase Separations and Related Techniques, Dresden, Germany, June 28–July 2, 2009; Dizdarevic, A., Huber, C., Eds.; GDC: Dresden, Germany, 2009; p. 723. [Google Scholar]
- Stirk, W.A.; Novák, O.; Václavíková, K.; Tarkowski, P.; Strnad, M.; van Staden, J. Spatial and temporal changes in endogenous cytokinin in developing pea roots. Planta 2008, 227, 1279–1289. [Google Scholar] [CrossRef]
- Petersson, S.V.; Johansson, A.I.; Kowalczyk, M.; Makoveychuk, A.; Wang, J.Y.; Moritz, T.; Grebe, M.; Benfey, P.N.; Sandberg, G.; Ljung, K. An auxin gradient and maximum in the Arabidopsis root apex shown by high-resolution cell-specific analysis of IAA distribution and synthesis. Plant Cell 2009, 21, 1659–1668. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds of interest are available from the authors.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tarkowski, P.; Floková, K.; Václavíková, K.; Jaworek, P.; Raus, M.; Nordström, A.; Novák, O.; Doležal, K.; Šebela, M.; Frébortová, J. An Improved in Vivo Deuterium Labeling Method for Measuring the Biosynthetic Rate of Cytokinins. Molecules 2010, 15, 9214-9229. https://doi.org/10.3390/molecules15129214
Tarkowski P, Floková K, Václavíková K, Jaworek P, Raus M, Nordström A, Novák O, Doležal K, Šebela M, Frébortová J. An Improved in Vivo Deuterium Labeling Method for Measuring the Biosynthetic Rate of Cytokinins. Molecules. 2010; 15(12):9214-9229. https://doi.org/10.3390/molecules15129214
Chicago/Turabian StyleTarkowski, Petr, Kristýna Floková, Kateřina Václavíková, Pavel Jaworek, Martin Raus, Anders Nordström, Ondřej Novák, Karel Doležal, Marek Šebela, and Jitka Frébortová. 2010. "An Improved in Vivo Deuterium Labeling Method for Measuring the Biosynthetic Rate of Cytokinins" Molecules 15, no. 12: 9214-9229. https://doi.org/10.3390/molecules15129214
APA StyleTarkowski, P., Floková, K., Václavíková, K., Jaworek, P., Raus, M., Nordström, A., Novák, O., Doležal, K., Šebela, M., & Frébortová, J. (2010). An Improved in Vivo Deuterium Labeling Method for Measuring the Biosynthetic Rate of Cytokinins. Molecules, 15(12), 9214-9229. https://doi.org/10.3390/molecules15129214