Synthesis of Non-Cytotoxic Poly(Ester-Amine) Dendrimers as Potential Solubility Enhancers for Drugs: Methotrexate as a Case Study

Abstract
:
1. Introduction
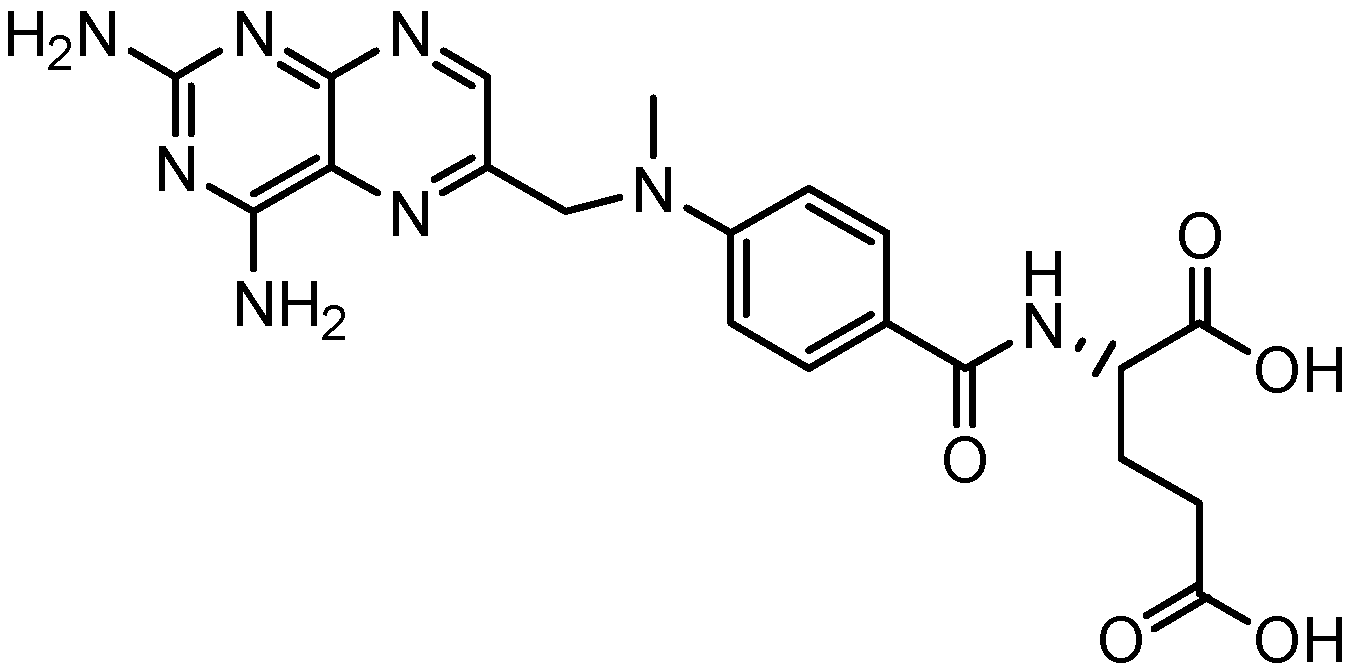
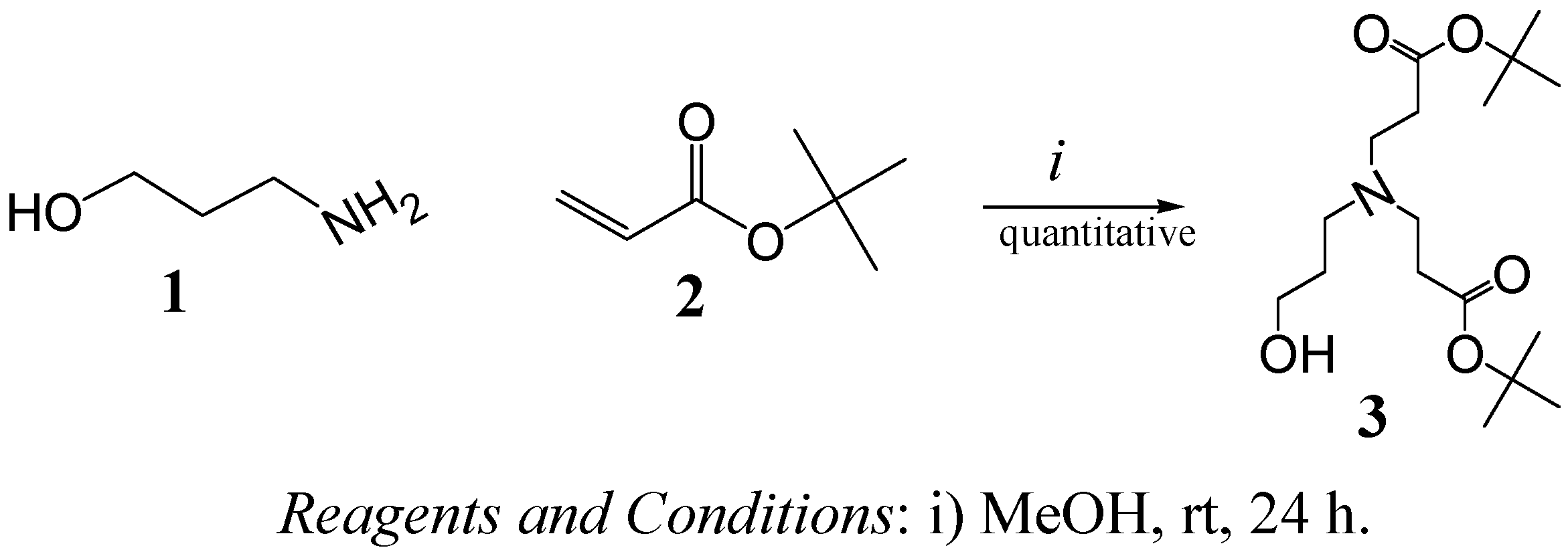
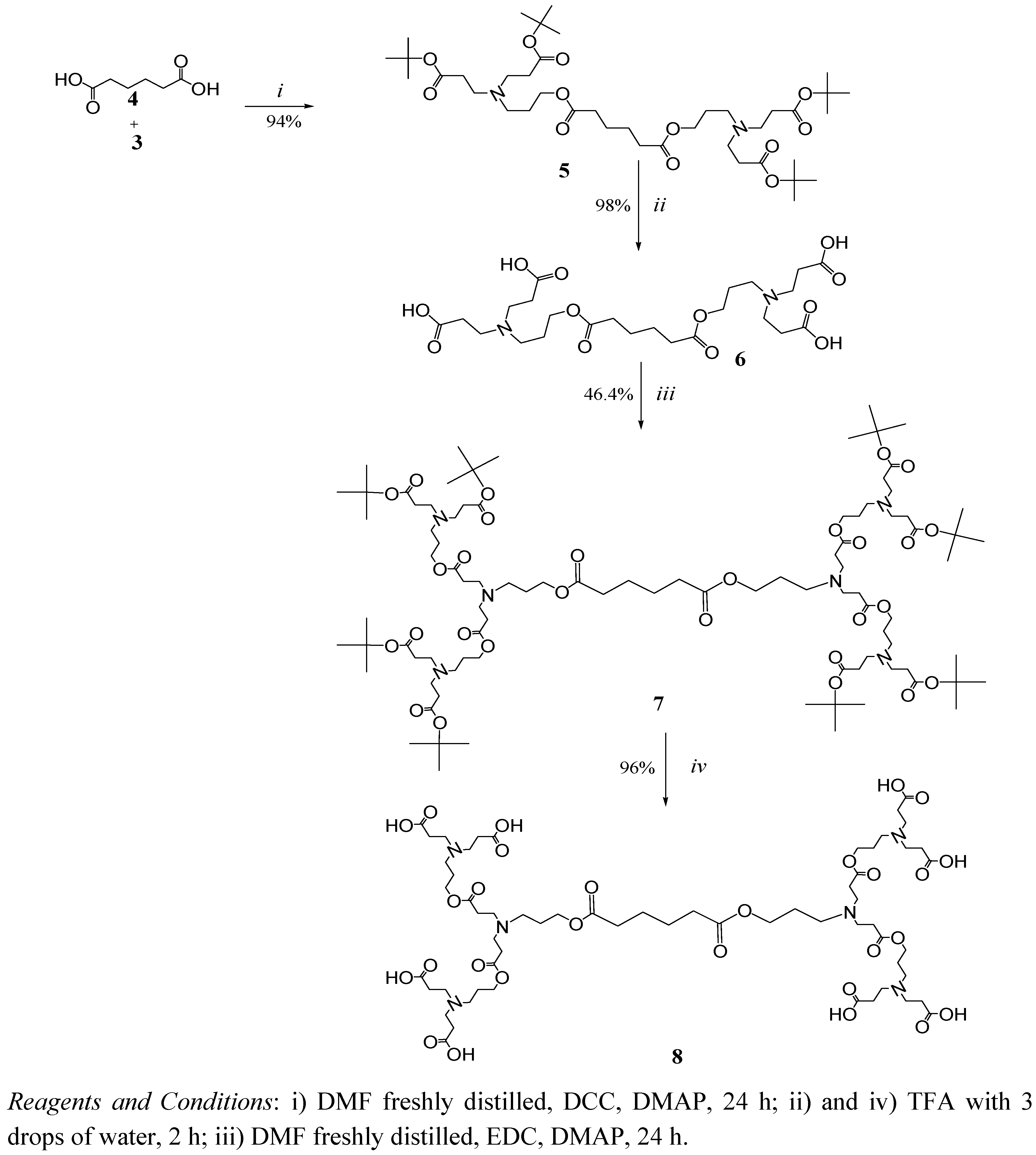
2. Results and Discussion

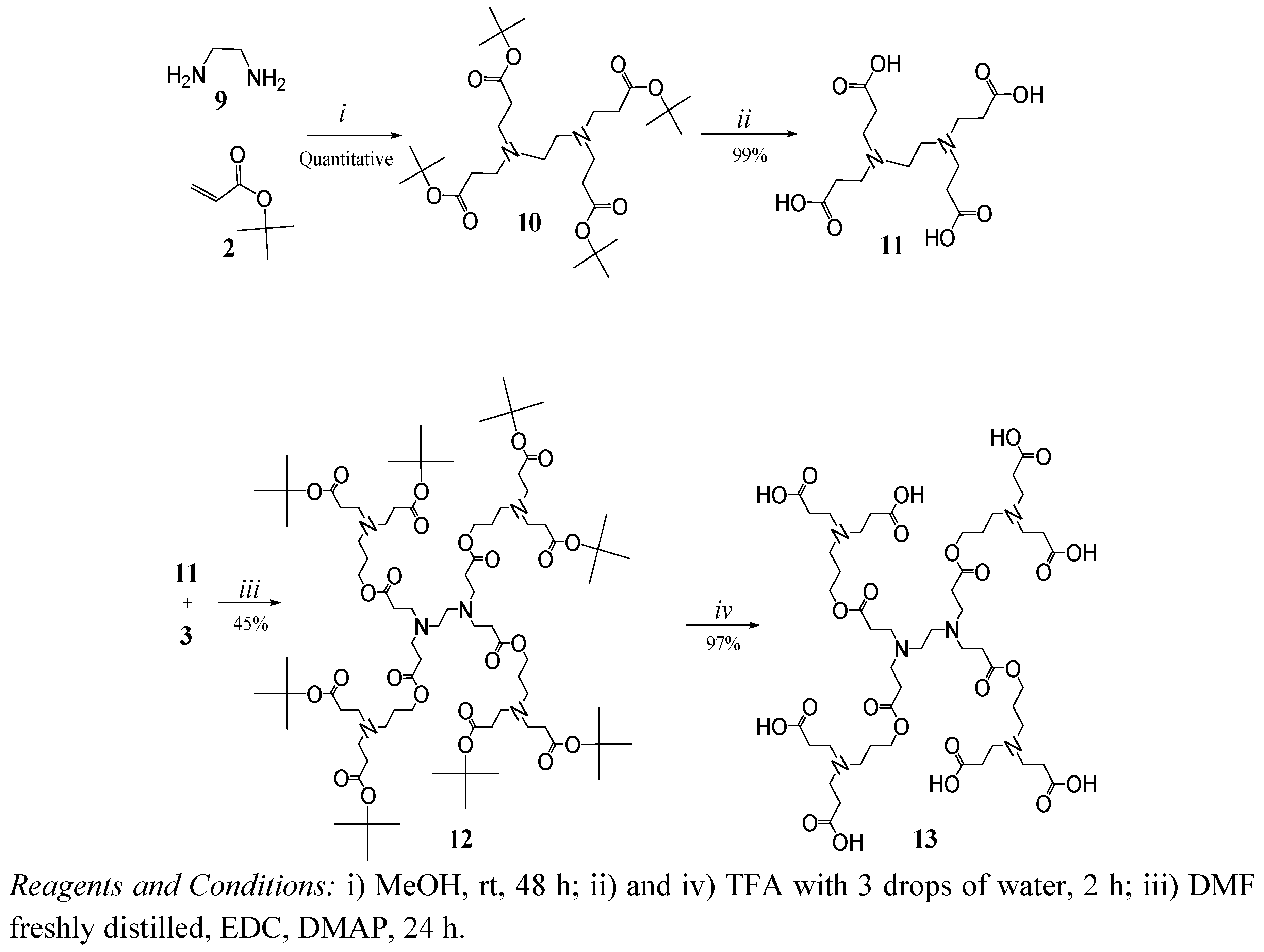
3. Biological Section

{kind=link}
{kind=link}
{kind=link}
{kind=link}
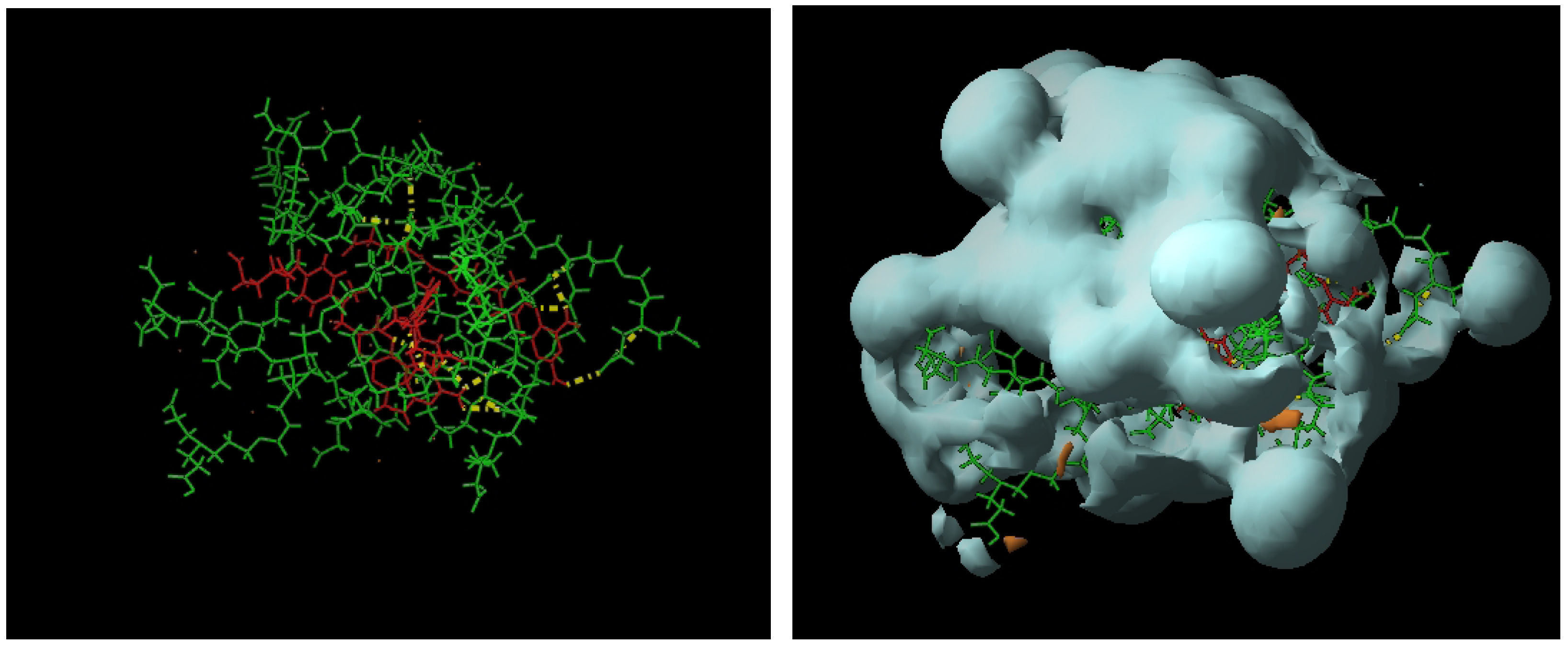{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MT2 | U251 | PC-3 | K-563 | HCT-15 | MCF7 | SKLU-1 |
|---|---|---|---|---|---|---|---|
| 3 (hydrolyzed) | +3.4±2.0 | 10.6±1.2 | +6.6±5.0 | 6.5±1.2 | 7.8±1.4 | 24.0±1.2 | 17.9±1.3 |
| 4 | +3.2±1.5 | +13.7±2.9 | +3.7±0.8 | +12.0±1.3 | +6.5±1.0 | +11.1±6.0 | +5.1±0.5 |
| 6 | +3.0±1.5 | 1.6±1.0 | +5.2±0.2 | 15.3±5.8 | +3.5±3.3 | +21.4±5.4 | +2.3±0.8 |
| 8 | +6.1±1.0 | 9.8±0.4 | 12.6±3.6 | 7.7±0.0 | +5.9±2.0 | +10.2±2.2 | 2.4±0.4 |
| 11 | 6.7±2.2 | 19.7±5.0 | +1.7±1.0 | 11.8±2.4 | 9.9±3.3 | +9.8±4.2 | +5.4±1.3 |
| 13 | 1.3±0.6 | 11.2±0.5 | +9.6±2.1 | 1.3±0.3 | +4.4±4.0 | +6.6±1.2 | 2.1±0.1 |
| Polyglycidolb | +5.8±0.6 | 1.6±0.1 | +5.4±1.8 | 6.4±0.08 | 4.8±1.5 | 5.3±0.2 | 5.5±1.5 |
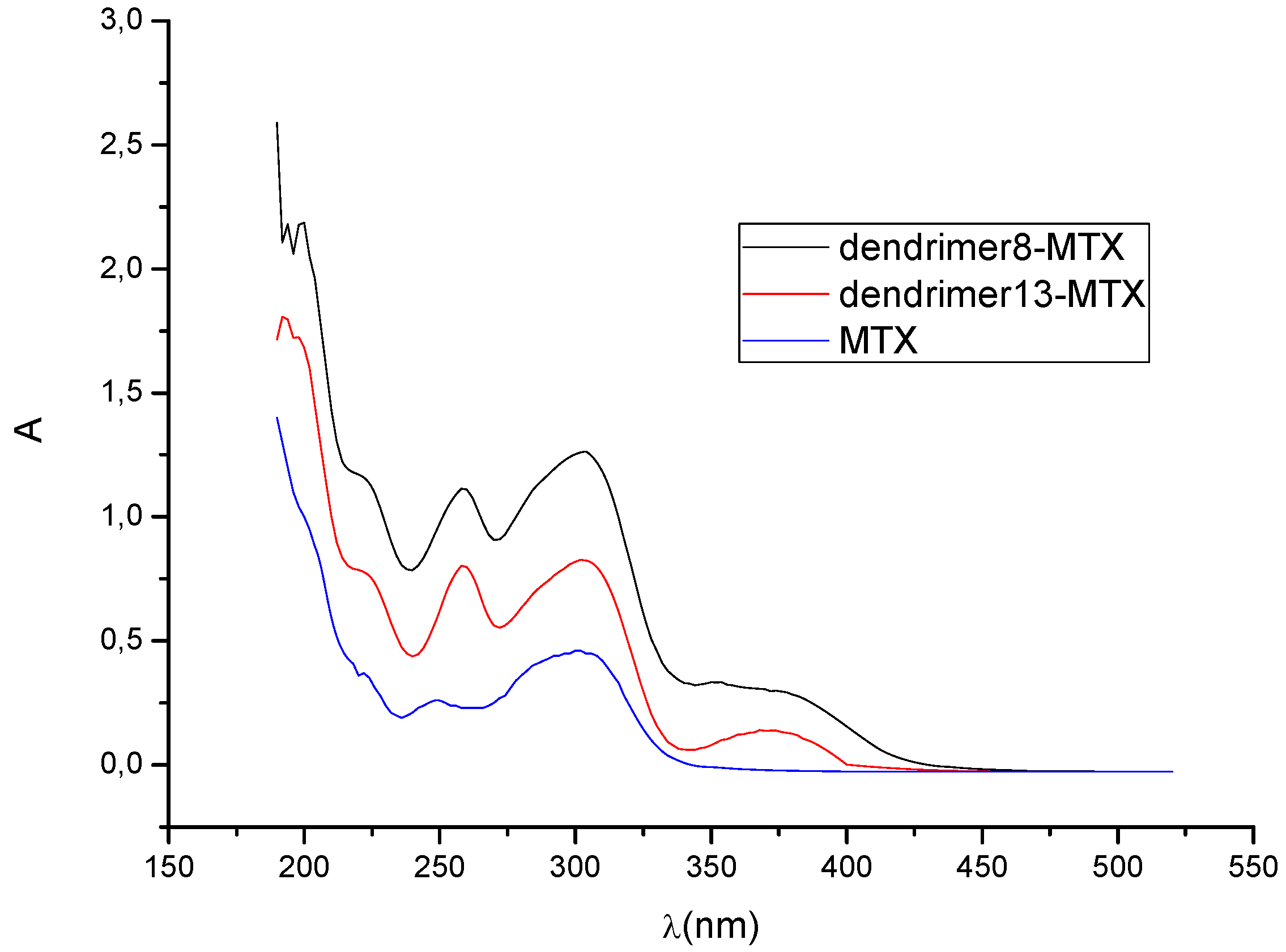
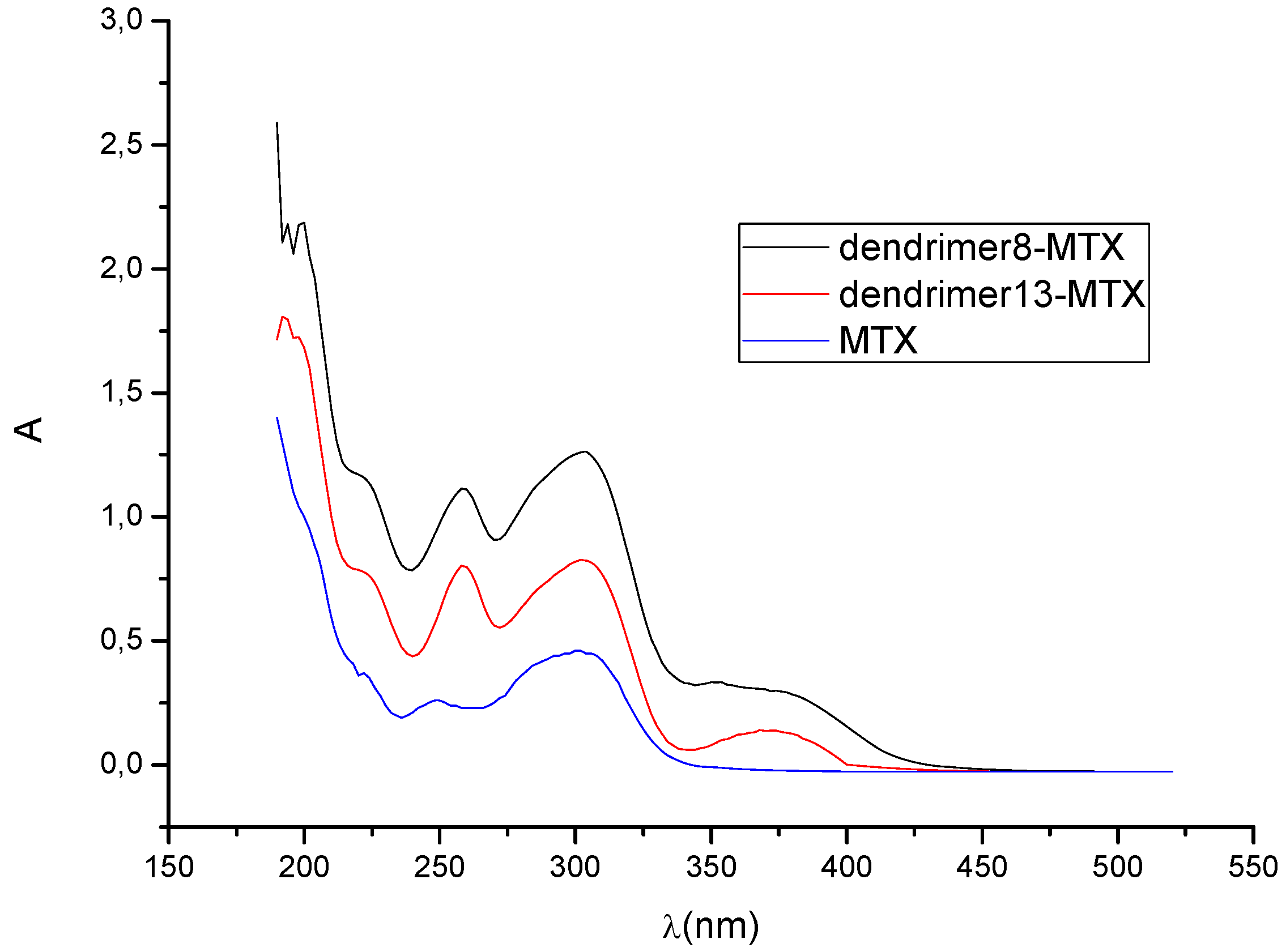
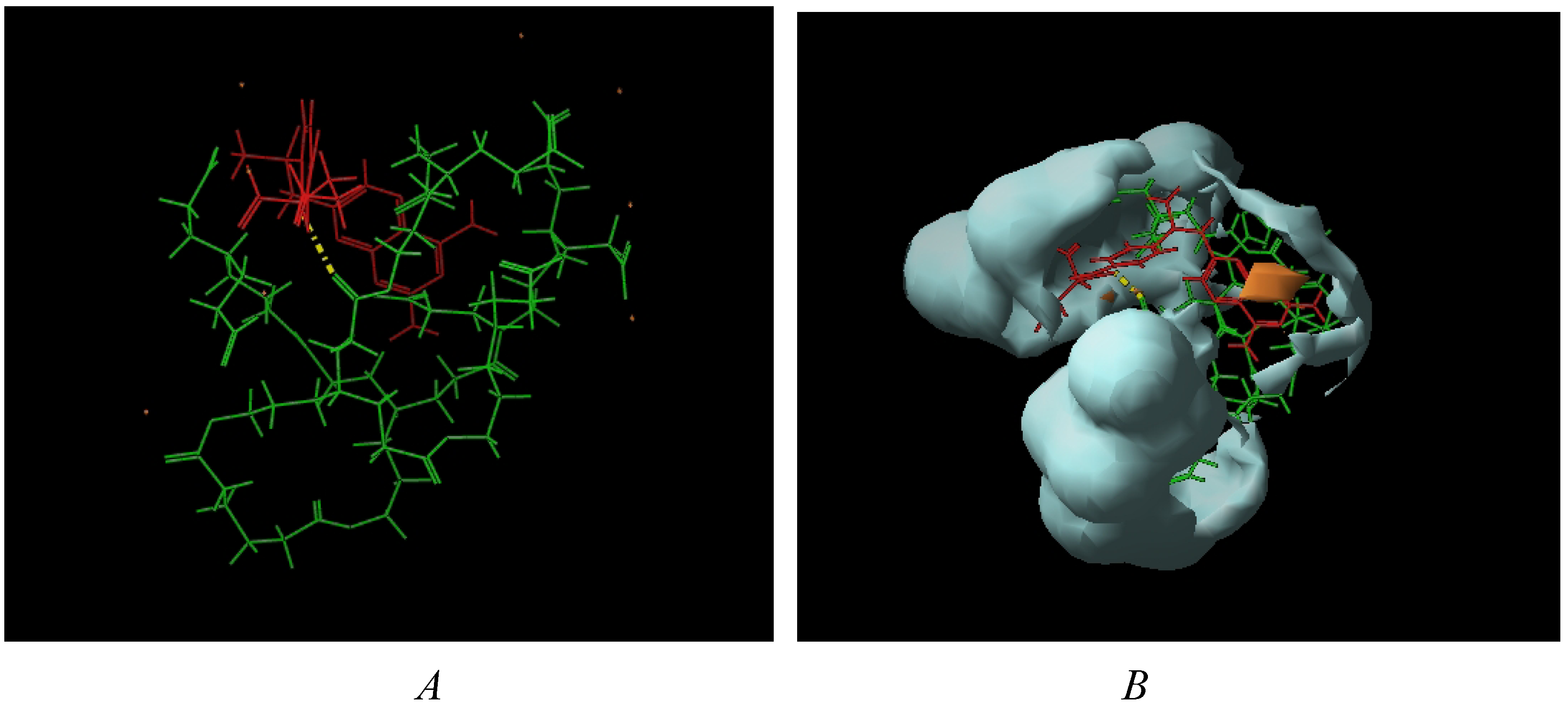
4. Dendrimers as Enhancers of Solubility: Preliminary Studies with MTX

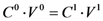
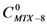 ) by the concentration of MTX in water alone (
) by the concentration of MTX in water alone ( 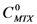 )), can be expressed as follows:
)), can be expressed as follows:

5. Conclusions
6. Experimental
6.1. General
6.1.1. General Procedure for the Synthesis of tert-Butyl Ester Terminated Dendrimer
6.1.2. General Procedure for the Hydrolysis of tert-Butyl Esters
6.1.3. Polyglycidol (Reference Compound for Biological Assays).
6.2. Cell Lines Culture and Culture Medium
6.3. Cytotoxicity Assay
6.4. Formation of Dendrimer-MTX Complexes
6.4.1. Dendrimer 8-MTX Complex
6.4.2. Dendrimer 13-MTX Complex
6.4.3. Calculation of Complex Stoichiometry
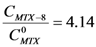 ; isolating CMTX-8 and dividing by CD, we have:
; isolating CMTX-8 and dividing by CD, we have: 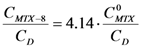 . If
. If 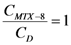 and CD = 1.7 mM,
and CD = 1.7 mM, 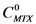 will be:
will be: 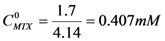 . With this result and evoking the concentration ratio for dendrimer 13, we have:
. With this result and evoking the concentration ratio for dendrimer 13, we have:
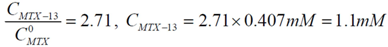
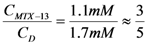
Supplementary Materials
Acknowledgments
References
- Astruc, D.; Chardac, F. Dendritic catalysts and dendrimers in catalysis. Chem. Rev. 2001, 101, 2991–3031. [Google Scholar] [CrossRef]
- Segura, J.L.; Giacalone, F.; Gómez, R.; Martín, N.; Guldi, D.M.; Luo, C.; Swartz, A.; Riedel, I.; Chirvase, D.; Parisi, J.; Dyakonov, V.; Sariciftci, N.S.; Padinger, F. Design, synthesis and photovoltaic properties of [60]fullerene based molecular materials. Mater. Sci. Eng. C 2005, 25, 835–842. [Google Scholar] [CrossRef]
- Ramos, E.; Guadarrama, P.; Terán, G.; Fomine, S. Push–pull hyperbranched molecules. A theoretical study. J. Phys. Org. Chem. 2009, 22, 9–16. [Google Scholar]
- Stiriba, S.E.; Frey, H.; Haag, R. Dendritic polymers in biomedical applications: from potential to clinical use in diagnostics and therapy. Angew. Chem. Int. Ed. 2002, 41, 1329–1334. [Google Scholar] [CrossRef]
- Lee, C.C.; MacKay, J.A.; Fréchet, J.M.J.; Szoka, F.C. Designing dendrimers for biological applications. Nature Biotech. 2005, 23, 1517–1526. [Google Scholar] [CrossRef]
- Halkes, S.B.A.; Vrasidas, I.; Rooijer, G.R.; Van den Berg, A.J.J.; Liskamp, R.M.J.; Pieters, R.J. Synthesis and biological activity of polygalloyl-dendrimers as stable tannic acid mimics. Bioorg. Med. Chem. Lett. 2002, 12, 1567–1570. [Google Scholar] [CrossRef]
- Aulenta, F.; Hayes, W.; Rannard, S. Dendrimers: a new class of nanoscopic containers and delivery devices. Eur. Polym. J. 2003, 39, 1741–1771. [Google Scholar] [CrossRef]
- Boas, U.; Heegaard, P.M.H. Dendrimers in drug research. Chem. Soc. Rev. 2004, 33, 43–63. [Google Scholar]
- Esfand, R.; Tomalia, D.A. Poly(amidoamine) (PAMAM) dendrimers: from biomimicry to drug delivery and biomedical applications. Drug Discov. Today 2001, 6, 427–436. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hal, M.; Kallos, G.; Martin, S. A New Class of Polymers: Starburst-Dendritic Macromolecules. Polym. J. (Tokyo) 1985, 17, 117–132. [Google Scholar] [CrossRef]
- Bertino, J.R.; Cronstein, B.N. Methotrexate (Milestones in Drug Therapy); Birkhäuser Verlag AG: Basel, Switzerland, 2000. [Google Scholar]
- Fernandez, L.; Calderón, M.; Martinelli, M.; Strumia, M.; Cerecetto, H.; González, M.; Silber, J.J.; Santo, M.J. Evaluation of a new dendrimeric structure as prospective drugs carrier for intravenous administration of antichagasic active compounds. Phys. Org. Chem. 2008, 21, 1079–1085. [Google Scholar] [CrossRef]
- Morgan, M.T.; Carnahan, M.A.; Immoos, C.E.; Ribeiro, A.A.; Finkelstein, S.; Lee, S.J.; Grinstaff, M.W. Dendritic molecular capsules for hydrophobic compounds. J. Am. Chem. Soc. 2003, 125, 15485–15489. [Google Scholar]
- Padilla De Jesús, O.L.; Ihre, H.R.; Gagne, L.; Fréchet, J.M.; Szoka Jr, F.C. Polyester dendritic systems for drug delivery applications: In vitro and in vivo evaluation. Bioconjugate Chem. 2002, 13, 453–461. [Google Scholar] [CrossRef]
- Malik, N.; Wiwattanapatapee, R.; Klopsch, R.; Lorenz, K.; Frey, H.; Weenerc, J.W.; Meijerc, E.W.; Paulusd, W.; Duncan, R. Dendrimers: Relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125i-labelled polyamidoamine dendrimers in vivo. J. Control. Rel. 2000, 65, 133–148. [Google Scholar] [CrossRef]
- Fuchs, S.; Kapp, T.; Otto, H.; Schoneberg, T.; Franke, P.; Gust, R.; Schluter, A.D. A set of surface-modified dendrimers with potential application as drug delivery vehicles: Synthesis, in vitro cytotoxicity, and intracellular localization. Chem. Eur. J. 2004, 10, 1167–1192. [Google Scholar] [CrossRef]
- Krishna, T.R.; Jain, S.; Tatu, U.S.; Jayaraman, N. Synthesis of water soluble and non-toxic poly(ether imine) dendrimers. Tetrahedron 2005, 61, 4281–4288. [Google Scholar] [CrossRef]
- Griffin, C.; Sharda, N.; Sood, D.; Nair, J.; McNulty, J.; Pandey, S. Selective cytotoxicity of pancratistatin-related natural Amaryllidaceae alkaloids: Evaluation of the activity of two new compounds. Canc. Cell Int. 2007, 7, 10. [Google Scholar] [CrossRef]
- Kainthan, R.K.; Hester, S.R.; Levin, E.; Devine, D.V.; Brooks, D.E. In vitro Biological Evaluation of High Molecular Weight Hyperbranched Polyglycerols. Biomaterials 2007, 28, 4581–4590. [Google Scholar]
- Kainthan, R.K.; Brooks, D.E. In vivo biological evaluation of high molecular weight hyperbranched polyglycerols. Biomaterials 2007, 28, 4779–4787. [Google Scholar] [CrossRef]
- Barkin, R.L. Acetaminophen, aspirin, or ibuprofen in combination analgesic products. Am. J. Ther. 2001, 8, 433–442. [Google Scholar] [CrossRef]
- Saha, P.; Kim, K.J.; Yamahara, H.; Crandall, E.D.; Lee, V.H.L. Influence of lipophilicity on beta-blocker permeation across rat alveolar epithelial cell monolayers. J. Control. Release 1994, 32, 191–200. [Google Scholar] [CrossRef]
- Alvarez-Figueroa, M.J.; Delgado-Charro, M.B.; Blanco-Méndez, J. Passive and iontophoretic transdermal penetration of methotrexate. Int. J. Pharm. 2001, 212, 101–107. [Google Scholar] [CrossRef]
- Chang, G.; Guida, W.C.; Still, W.C. An internal coordinate monte carlo method for searching conformational space. J. Am. Chem. Soc. 1989, 111, 4379–4386. [Google Scholar] [CrossRef]
- Saunders, M.; Houk, K.N.; Wu, Y.D.; Still, W.C.; Lipton, M.; Chang, G.; Guida, W.C. Conformations of cycloheptadecane. a comparison of methods for conformational searching. J. Am. Chem. Soc. 1990, 112, 1419–1427. [Google Scholar]
- Goodford, P. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the opls all-atom force field on conformational energetics and properties of organic liquid. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kolossvary, I.; Guida, W.C. Low Mode Search. An efficient, automated computational method for conformational analisys: Application to cyclic and acyclic alkanes and cyclic peptides. J. Am. Chem. Soc. 1996, 118, 5011–5019. [Google Scholar] [CrossRef]
- Maestro, version 6.5.007, Schrödinger Inc.: New York, NY, USA, 2004.
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paul, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; Gray-Goodrich, M.; Campbell, H.; Mayo, J.; Boyd, M. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. JNCI 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Soto-Castro, D.; Cruz-Morales, J.A.; Apan, M.T.R.; Guadarrama, P. Synthesis of Non-Cytotoxic Poly(Ester-Amine) Dendrimers as Potential Solubility Enhancers for Drugs: Methotrexate as a Case Study. Molecules 2010, 15, 8082-8097. https://doi.org/10.3390/molecules15118082
Soto-Castro D, Cruz-Morales JA, Apan MTR, Guadarrama P. Synthesis of Non-Cytotoxic Poly(Ester-Amine) Dendrimers as Potential Solubility Enhancers for Drugs: Methotrexate as a Case Study. Molecules. 2010; 15(11):8082-8097. https://doi.org/10.3390/molecules15118082
Chicago/Turabian StyleSoto-Castro, Delia, Jorge A. Cruz-Morales, María Teresa Ramírez Apan, and Patricia Guadarrama. 2010. "Synthesis of Non-Cytotoxic Poly(Ester-Amine) Dendrimers as Potential Solubility Enhancers for Drugs: Methotrexate as a Case Study" Molecules 15, no. 11: 8082-8097. https://doi.org/10.3390/molecules15118082
APA StyleSoto-Castro, D., Cruz-Morales, J. A., Apan, M. T. R., & Guadarrama, P. (2010). Synthesis of Non-Cytotoxic Poly(Ester-Amine) Dendrimers as Potential Solubility Enhancers for Drugs: Methotrexate as a Case Study. Molecules, 15(11), 8082-8097. https://doi.org/10.3390/molecules15118082