Two New Neolignans from Syringa velutina Kom.

 and
and
Abstract
:Introduction
Results and Discussion
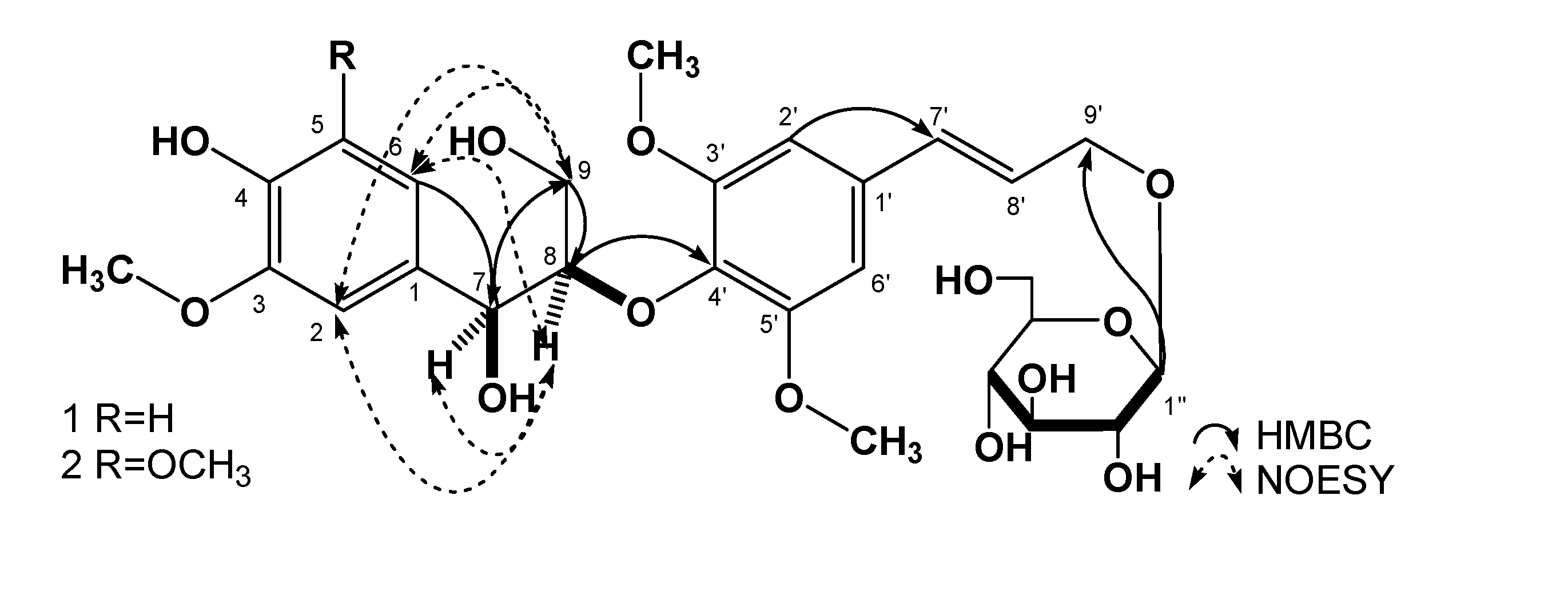

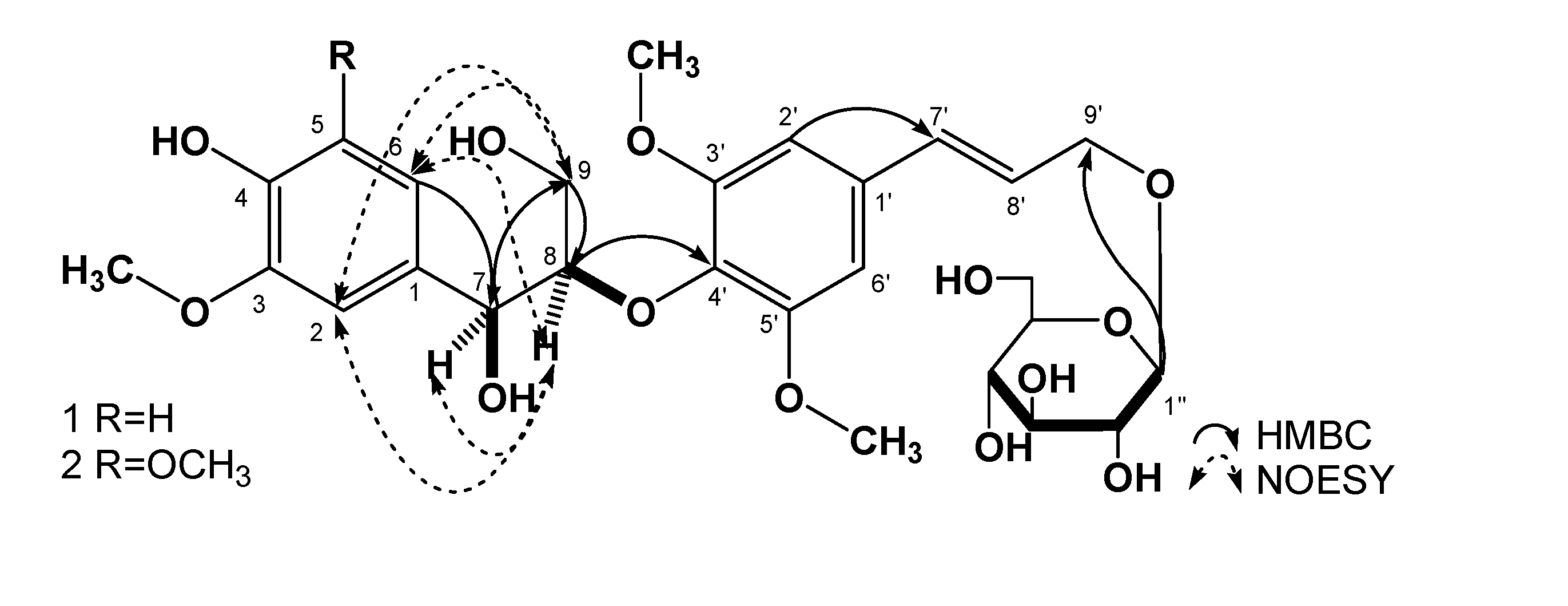{kind=link}
| position | 1 | 2 | |||
|---|---|---|---|---|---|
| δC | δH | HMBC(H→C) | δC | δH | |
| 1 | 133.5 | 132.6 | |||
| 2 | 111.1 | 6.90 (1H, d, J = 1.8 Hz) | C-1, 3, 4, 6, 7 | 104.4 | 6.60 (1H, s) |
| 3 | 147.2 | 147.6 | |||
| 4 | 145.5 | 134.4 | |||
| 5 | 114.9 | 6.68 (1H, d, J = 8.1 Hz) | C-1, 3, 4, 6 | 147.6 | |
| 6 | 119.6 | 6.71 (1H, dd, J =8.1, 1.8 Hz) | C-1, 2, 4, 5, 7 | 104.4 | 6.60 (1H, s) |
| 7 | 72.2 | 4.77 (1H, dd, J =4.8, 4.8 Hz) | C-1, 2, 6, 8, 9 | 72.4 | 4.81 (1H, dd, J =4.8, 4.8 Hz) |
| 8 | 86.4 | 4.11 (1H, m) | C-4′ | 86.4 | 4.15 (1H, m) |
| 9 | 59.9 | 3.39 (1H, m)3.69 (1H, m) | C-7, 8 | 59.9 | 3.40 (1H, m)3.68 (1H, m) |
| 1′ | 132.1 | 132.0 | |||
| 2′, 6′ | 104.0 | 6.73 (2H, s) | C-1′, 4′, 7′ | 103.9 | 6.75 (2H, s) |
| 3′, 5′ | 153.0 | 152.8 | |||
| 4′ | 135.4 | 135.5 | |||
| 7′ | 131.5 | 6.56 (1H, d, J = 15.9 Hz ) | C-1′, 8′, 9′ | 131.4 | 6.57 (1H, d, J = 15.6 Hz ) |
| 8′ | 125.9 | 6.31 (1H, dt, J = 15.9, 5.4 Hz ) | C-1′, 7′, 9′ | 125.8 | 6.34 (1H, dt, J = 15.6, 5.4 Hz ) |
| 9′ | 68.8 | 4.19 (1H, dd, J = 14.4, 5.4 Hz)4.40 (1H, dd, J = 14.4, 5.4 Hz) | C-7′, 8′ | 68.7 | 4.19 (1H, dd, J = 14.4, 5.4 Hz)4.41 (1H, dd, J = 14.4, 5.4 Hz) |
| 4-OH | 8.08 (1H, s) | C-3, 4, 5 | |||
| 7-OH | 5.10 (1H, d, J = 4.8 Hz) | C-1, 7, 8 | |||
| 9-OH | 4.03 (1H, t, J = 6.0 Hz) | C-9 | |||
| Glucose | |||||
| 1′′ | 102.3 | 4.20 (1H, d, J = 7.8 Hz) | C-9′ | 102.2 | 4.21 (1H, d, J = 7.8 Hz) |
| 2′′ | 73.7 | 2.99 (1H, td, J = 7.8, 4.8 Hz) | 73.6 | 3.05(1H,m) | |
| 3″ | 77.1 | 3.08 (1H, m) | 77.0 | 3.09 (1H, m) | |
| 4′′ | 70.3 | 3.04 (1H, m) | 70.2 | 3.07 (1H, m) | |
| 5′′ | 76.9 | 3.13 (1H, td, J = 9.0, 4.8 Hz) | 76.9 | 3.14 (1H, m) | |
| 6′′ | 61.3 | 3.43 (1H, m)3.67 (1H, m) | 61.2 | 3.44 (1H, dd, J = 12.0, 6.0 Hz)3.67 (1H, m) | |
Experimental
General
Plant material
Extraction and Isolation
Acid hydrolysis of 1 and 2
Enzymatic hydrolysis of 1
Acknowledgments
References
- Lu, D.; Li, P.Y. A review of chemical constituents and bioactivities of plants from the genus Syringa. Acad. Period. Changchun College Tradit. Chinese Med. 2001, 4, 58–59. [Google Scholar]
- Zhou, L.G.; Feng, X.S.; Huang, K.Y.; He, L.; Deng, X.M.; Wang, D.C. Studies on chemical constituents of Syringa velutina. Zhong Yao Cai 2008, 31, 976–978. [Google Scholar]
- Zhang, Q.; Sun, L.R. Studies on Chemical Constituents of Serissa serissoides Roots. Zhong Yao Cai 2006, 29, 786–788. [Google Scholar]
- Inoshiri, S.; Sasaki, M.; Kohda, H. Aromatic glycosides from Berchemia racemsoa. Phytochemistry 1987, 26, 2811–2814. [Google Scholar] [CrossRef]
- Nakamura, S.; Qu, Y.; Xu, F.M.; Matsuda, H.; Yoshikawa, M. Structures of new monoterpenes from Thai herbal medicine Curcuma comosa. Chem. Pharm. Bull. 2008, 56, 1604–1606. [Google Scholar] [CrossRef]
- Nattaya, L.; Takeshi, K.; Toshisada, S. Stereochemistry and biosynthesis of 8-O-4 ′ neolignans in Eucommia ulmoides: diastereoselective formation of guaiacylglycerol-8-O-4′-(sinapyl alcohol) ether. Japan Wood Res. Soc. 2005, 51, 370–378. [Google Scholar]
- Morikawa, T.; Matusda, H.; Nishida, N.; Ohgushi, T.; Yoshikawa, M. Medicinal foodstuffs. XXXI. Structures of new aromatic constituents and inhibitors of degranulation in RBL-2H3 cells from a Japanese folk medicine, the stem bark of Acer nikoense. Chem. Pharm. Bull. 2004, 52, 1387–1390. [Google Scholar] [CrossRef]
- Machida, K.; Sakamoto, S.; Kikuchi, M. Structure elucidation and NMR spectral assignments of four neolignan glycosides with enantiometric aglycones from Osmanthus ilicifolius. Magn. Reson. Chem. 2008, 46, 990–994. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compounds 1- 2 are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Feng, X.-S.; Qu, Y.; Xu, L.; Wang, D.-C.; Wu, L.-J.; Meng, F.-H.; Liu, Y.-R. Two New Neolignans from Syringa velutina Kom. Molecules 2009, 14, 953-958. https://doi.org/10.3390/molecules14030953
Feng X-S, Qu Y, Xu L, Wang D-C, Wu L-J, Meng F-H, Liu Y-R. Two New Neolignans from Syringa velutina Kom. Molecules. 2009; 14(3):953-958. https://doi.org/10.3390/molecules14030953
Chicago/Turabian StyleFeng, Xue-Song, Yang Qu, Lei Xu, Da-Cheng Wang, Li-Jun Wu, Fan-Hao Meng, and Ya-Ru Liu. 2009. "Two New Neolignans from Syringa velutina Kom." Molecules 14, no. 3: 953-958. https://doi.org/10.3390/molecules14030953
APA StyleFeng, X.-S., Qu, Y., Xu, L., Wang, D.-C., Wu, L.-J., Meng, F.-H., & Liu, Y.-R. (2009). Two New Neolignans from Syringa velutina Kom. Molecules, 14(3), 953-958. https://doi.org/10.3390/molecules14030953