Synthesis, Structural Studies and Antitumoral Evaluation of C-6 Alkyl and Alkenyl Side Chain Pyrimidine Derivatives S

Abstract
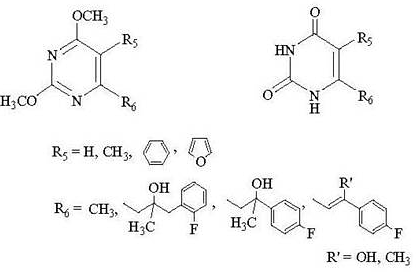
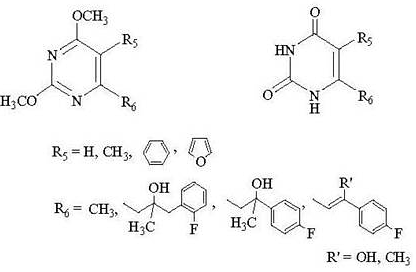
:
Introduction
Results and Discussion
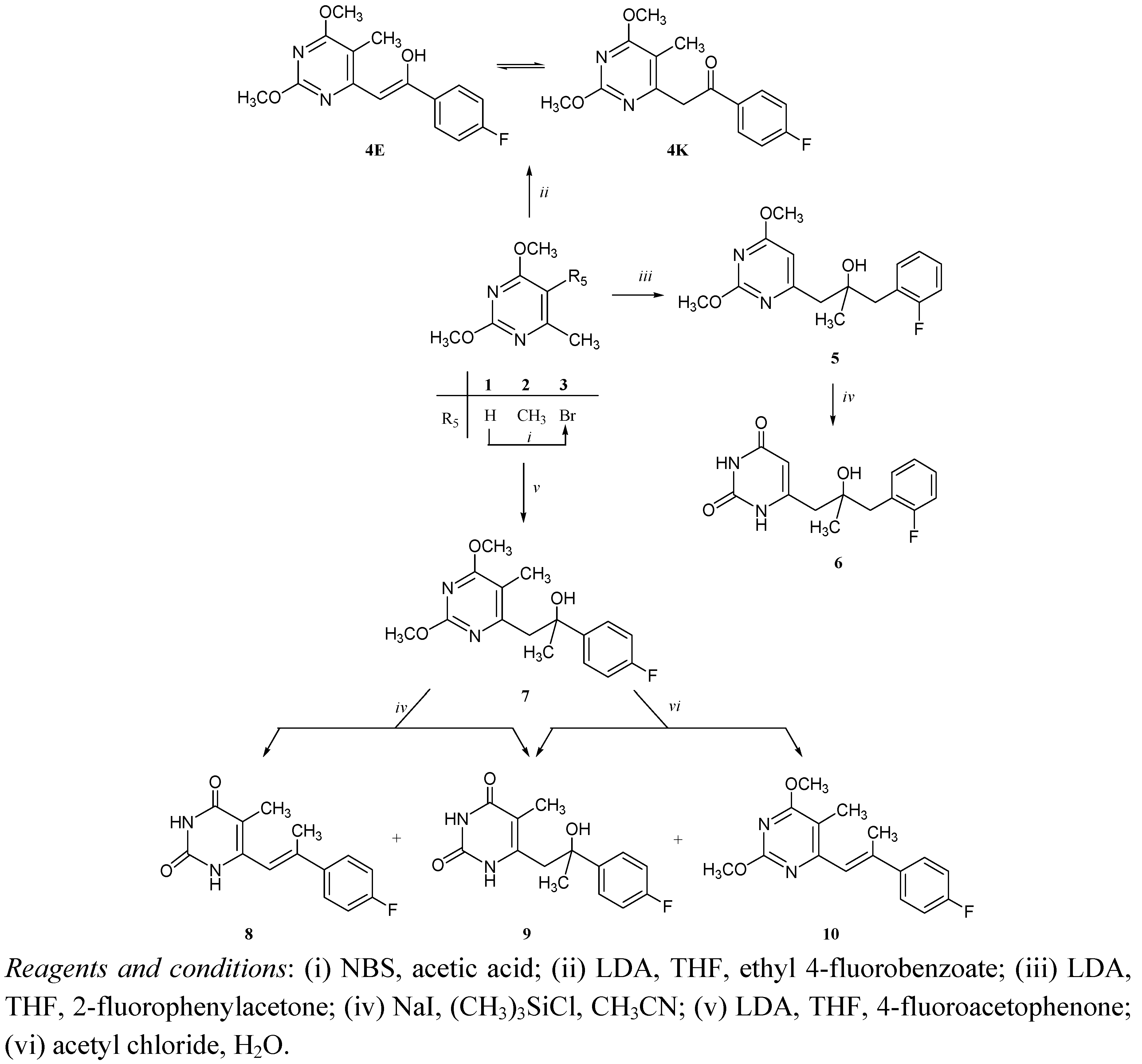
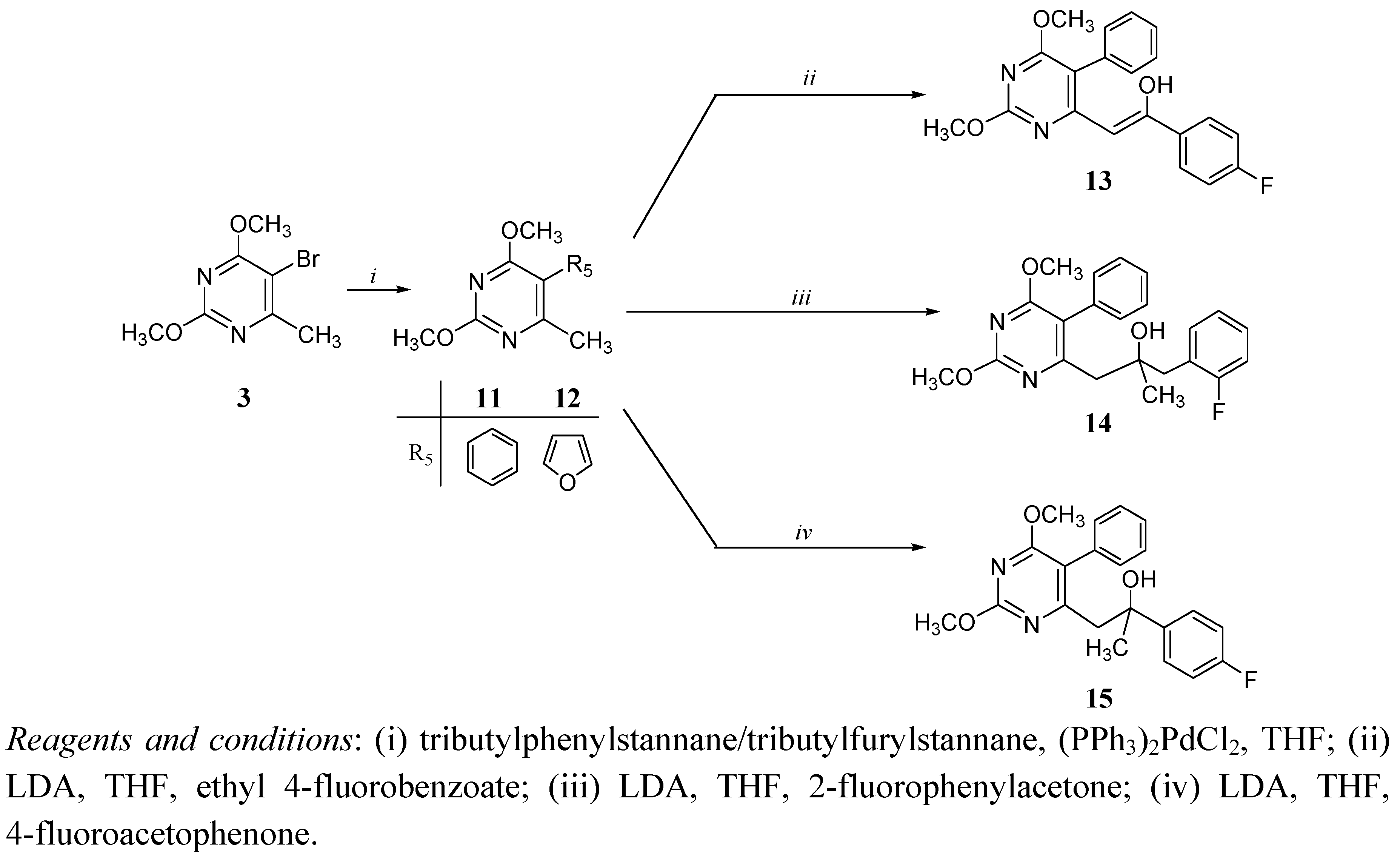
Chemistry
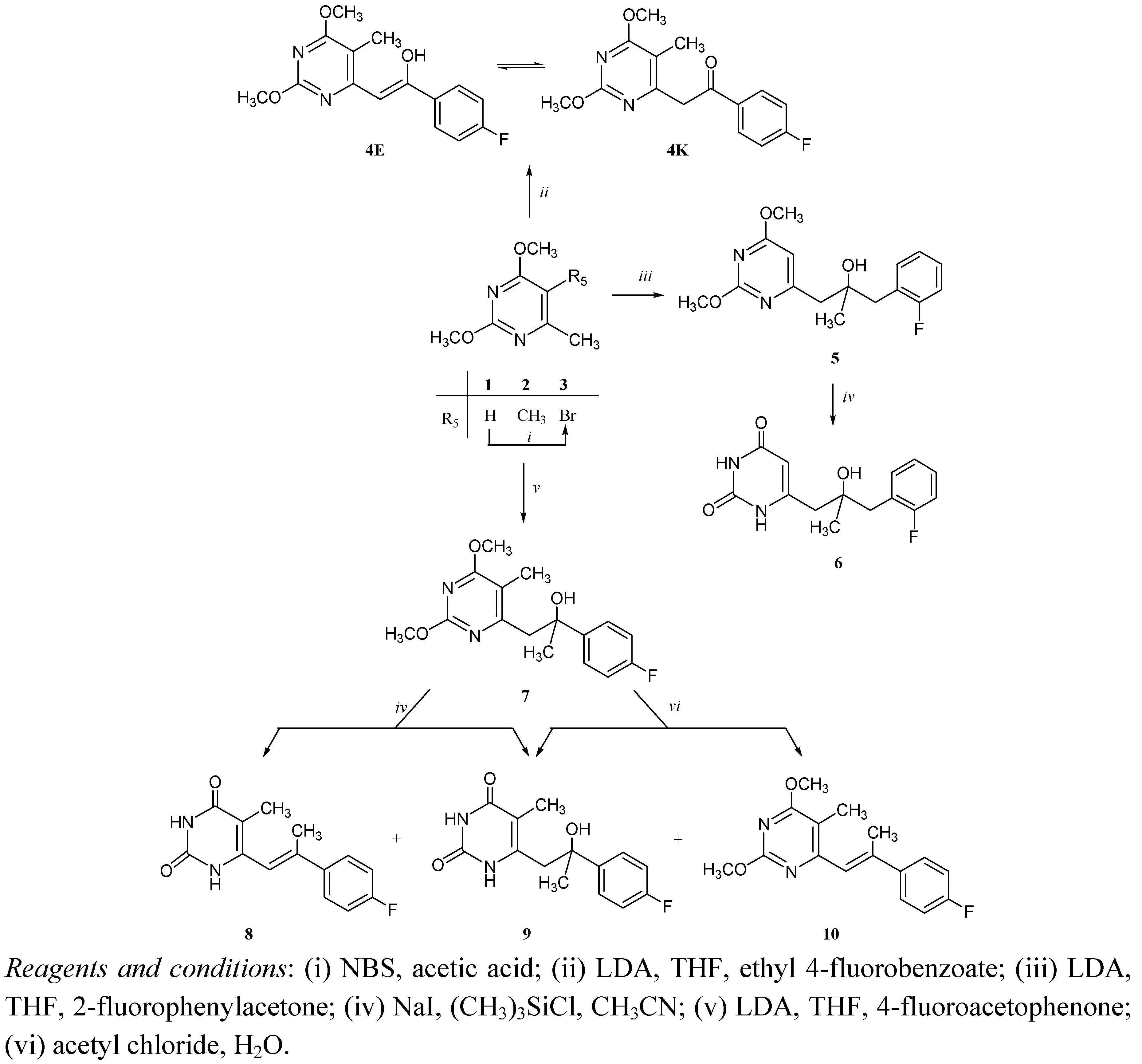
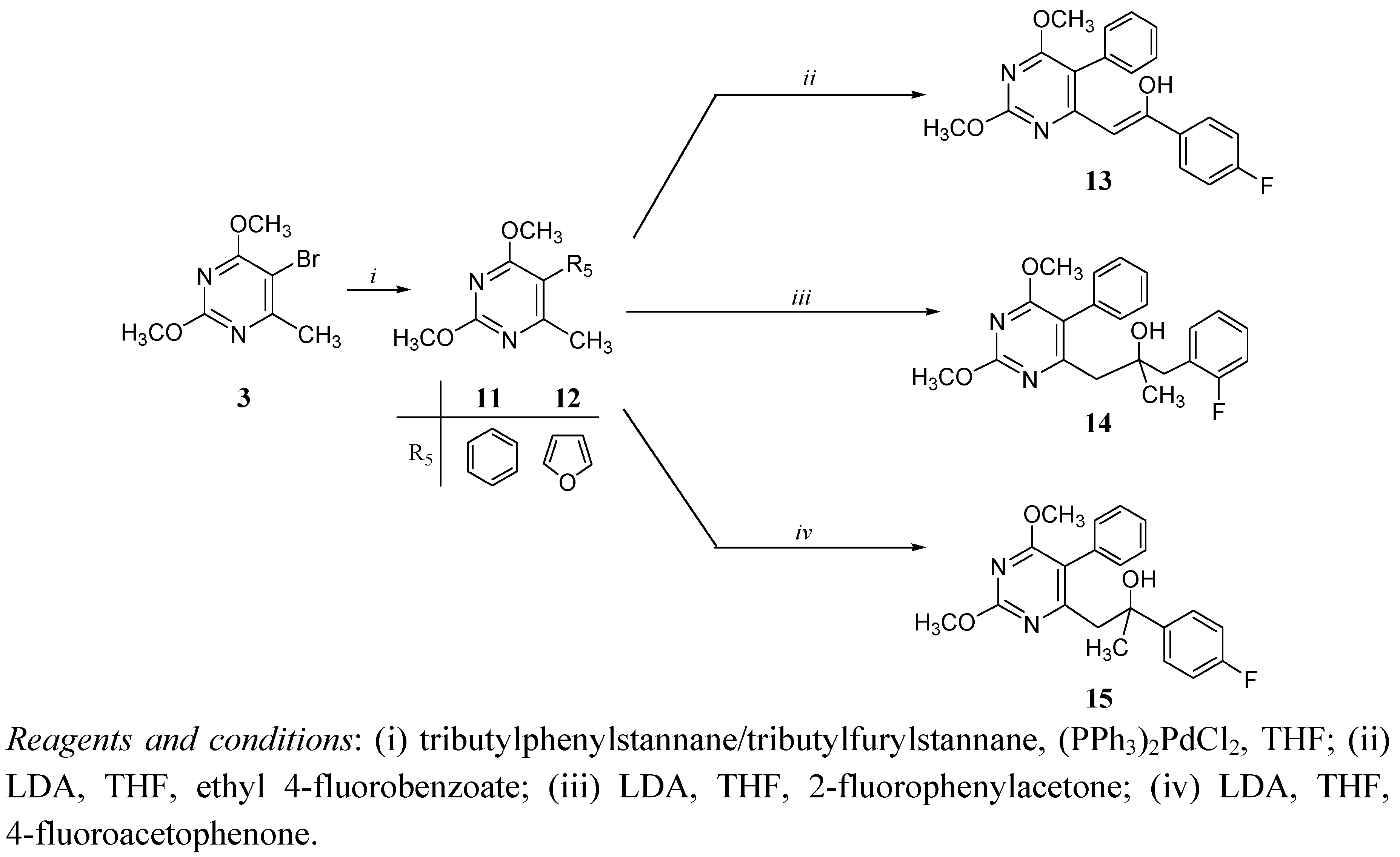
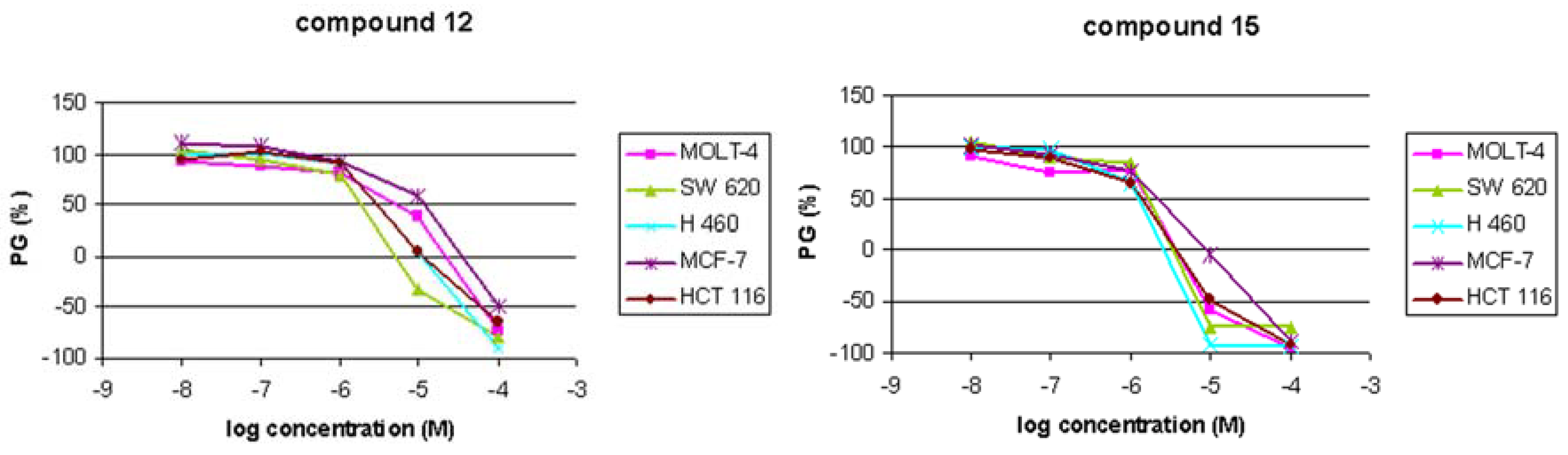
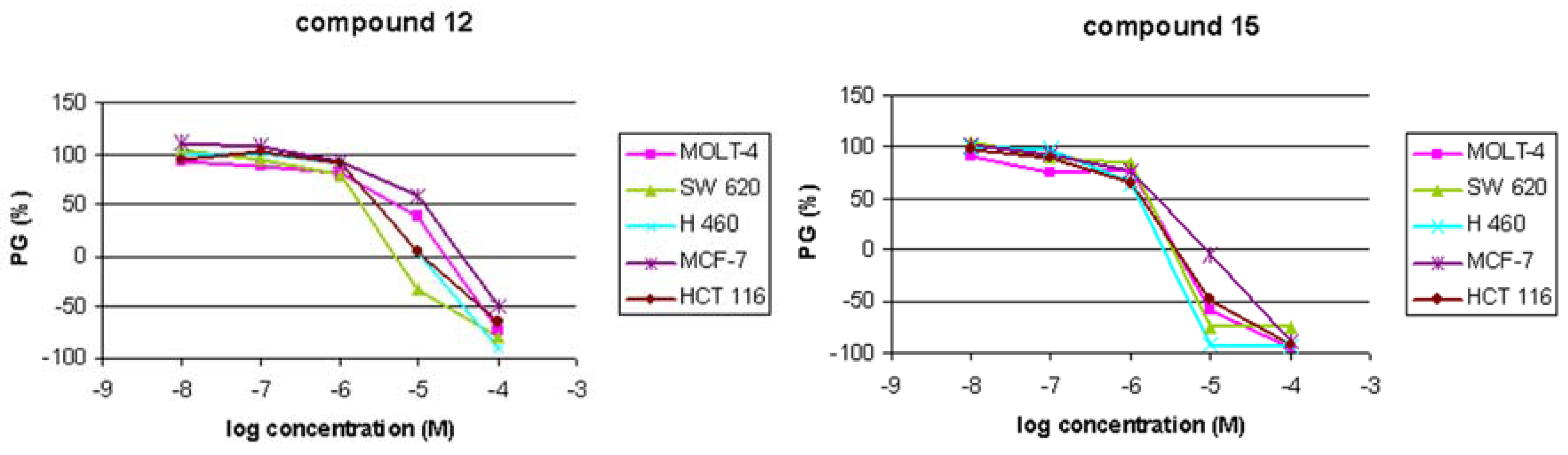
Antitumoral activities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | IC50a (μM) | ||||
|---|---|---|---|---|---|
| Molt-4 | HCT 116 | SW 620 | MCF-7 | H 460 | |
| 4 | 43 ± 41 | 41 ± 10 | 32 ± 19 | 2 ± 1 | 19 ± 5 |
| 6 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 8 | 78 ± 21 | 79 ± 21 | 35 ± 6 | 51 ± 18 | ± 100 |
| 9 | > 100 | ± 100 | > 100 | > 100 | > 100 |
| 10 | 50 ± 45 | 37 ± 0.03 | 32 ± 10 | 42 ± 1 | 75 ± 3 |
| 12 | 6 ± 4 | 3 ± 0.3 | 2 ± 1.6 | 10 ± 6 | 4 ± 3 |
| 13 | N.T. b | 2 ± 0.2 | 2 ± 0.2 | 3 ± 1 | 3 ± 0.1 |
| 14 | N.T. b | 14 ± 0.1 | 17 ± 0.1 | 14 ± 3 | 17 ± 2 |
| 15 | 2 ± 0.03 | 1 ± 0.5 | 2 ± 0.4 | 2 ± 1 | 1 ± 0.4 |
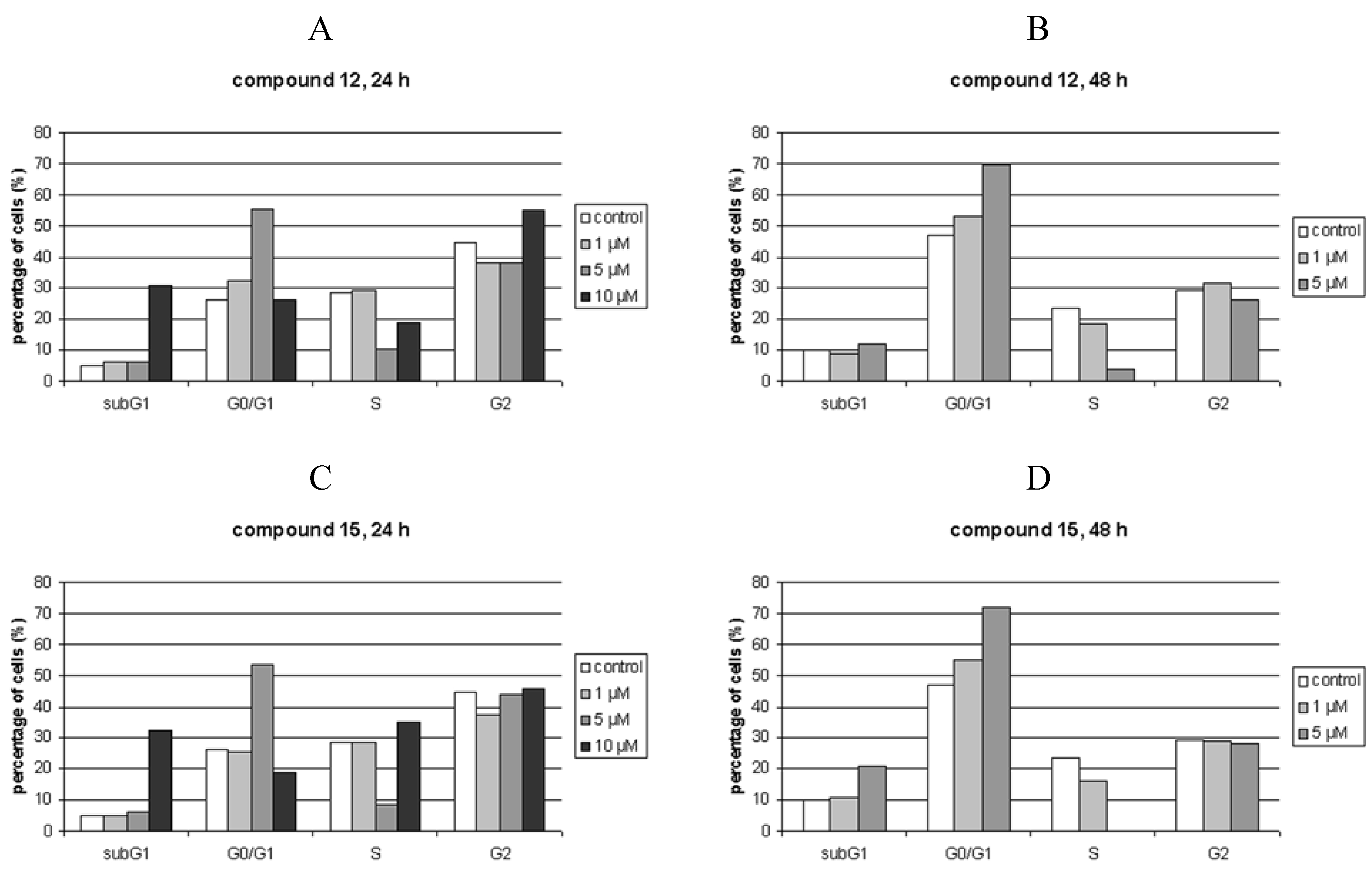
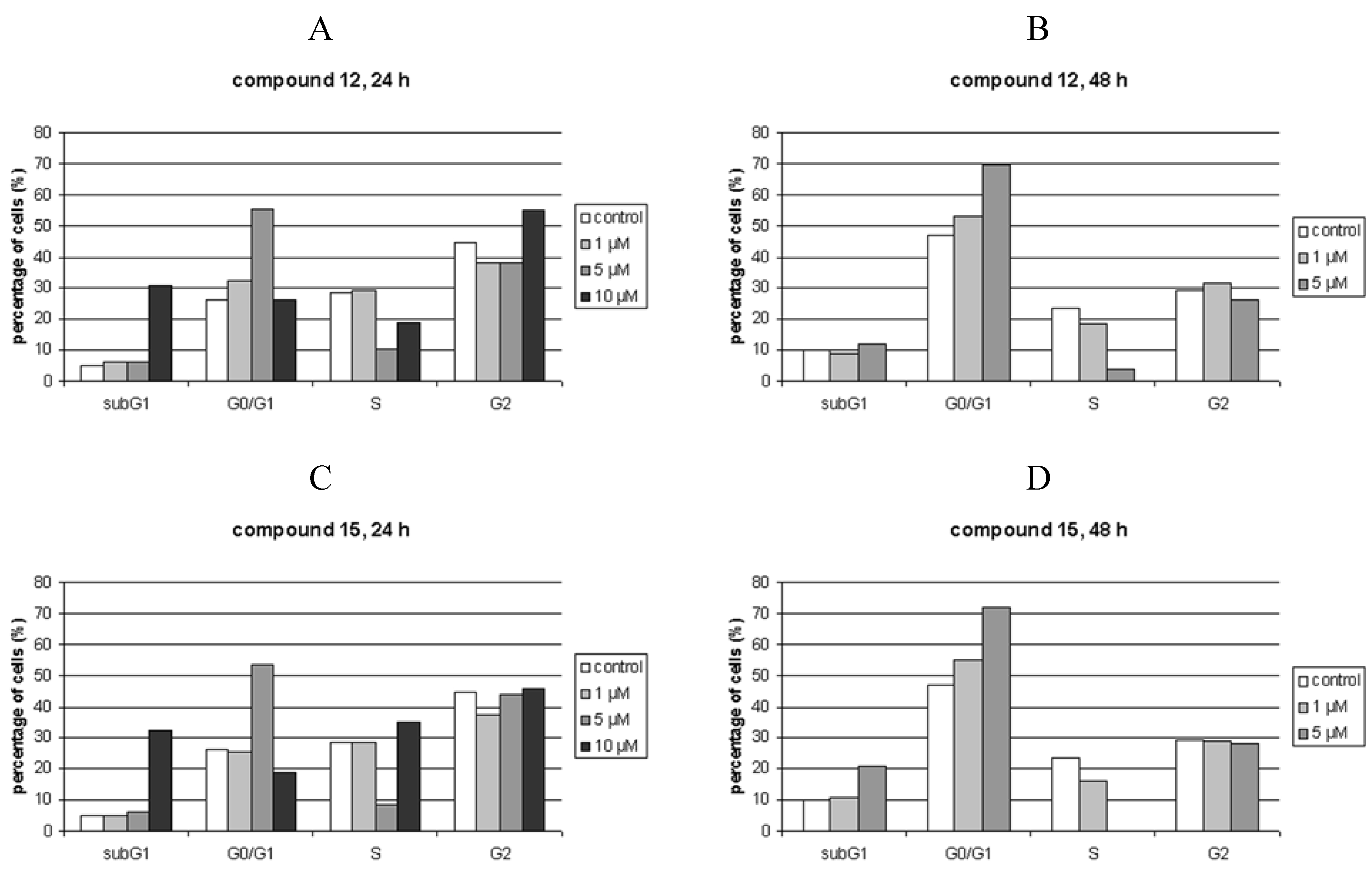
Cell cycle perturbations
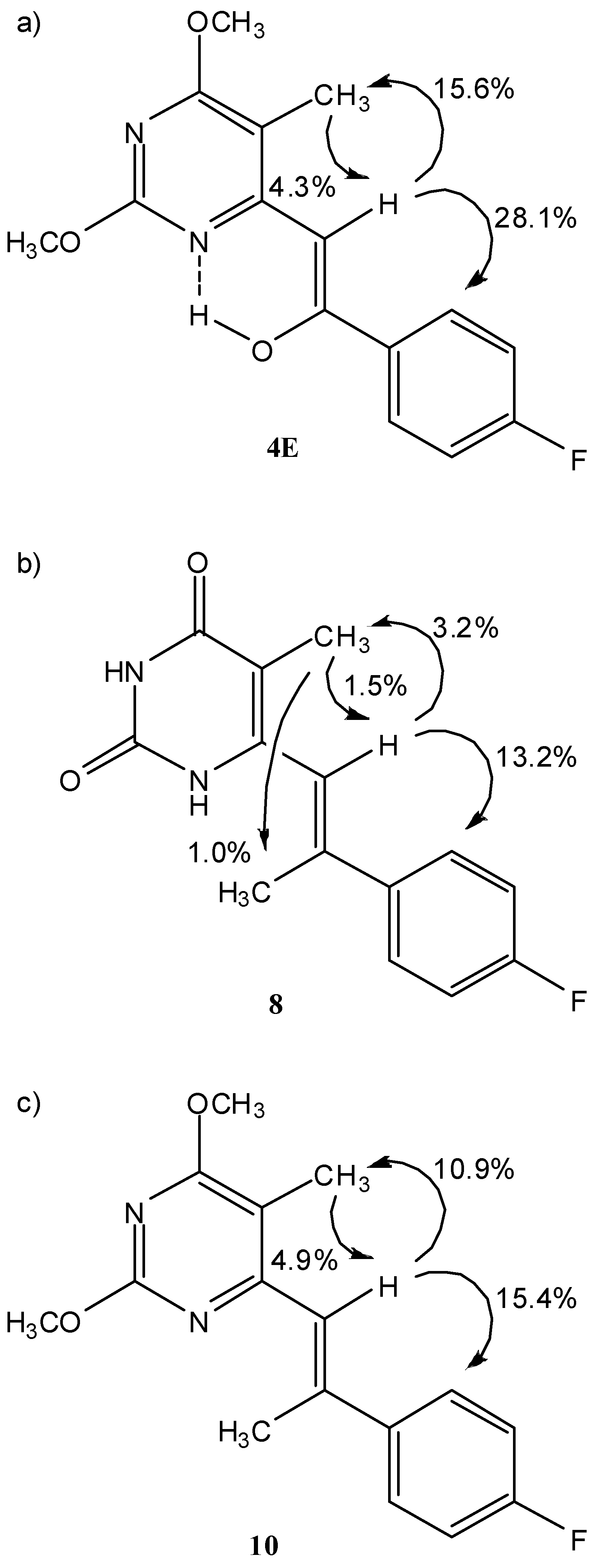
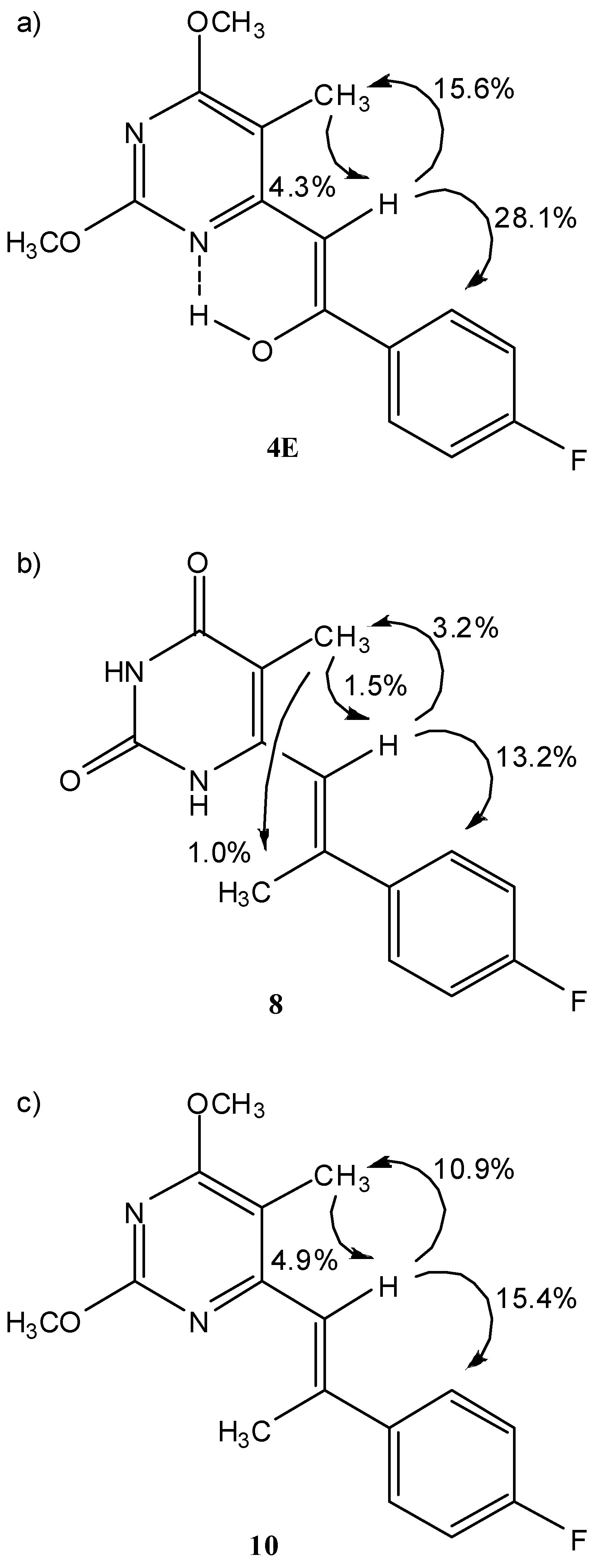
Structural and conformational studies by NMR
| Compd | N1–H | N3–H | H5 | H1' | C2'–OH | C2'–Me | C5–Me | –OCH3 | phenyl | 19F |
|---|---|---|---|---|---|---|---|---|---|---|
| 4Ea | – | – | – | 6.29 (s) | 15.08 (s) | – | 2.09 (s) | 3.95 (s) | 7.97 (m, Hϕ2/Hϕ6) | –110.31 (m) |
| 7.29 (m, Hϕ3/Hϕ5) | ||||||||||
| 4Ka | – | – | – | 4.48 (s) | – | – | 1.97 (s) | 3.75 (s), 3.91 (s) | 8.10 (m, Hϕ2/Hϕ6) | –105.21 (m) |
| 7.37 (m, Hϕ3/Hϕ5) | ||||||||||
| 6b | 10.92 (s) | 10.33 (s) | 5.37 (d) | 2.46 (d) | 4.73 (s) | 1.02 (s) | – | – | 7.14 (m, Hϕ3) | –115.83 (m) |
| 7.27 (m, Hϕ4) | ||||||||||
| 7.14 (m, Hϕ5) | ||||||||||
| 7.33 (m, Hϕ6) | ||||||||||
| 8c | 11.06 (s) | 10.59 (s) | – | 6.39 (m) | – | 2.02 (d) | 1.70 (d) | – | 7.64 (m, Hϕ2/Hϕ6) | –113.36 (m) |
| 7.24 (m, Hϕ3/Hϕ5) | ||||||||||
| 9d | 10.93 (s) | 10.33 (s) | – | 2.73 (d) 2.82 (d) | 5.52 (b) | 1.52 (s) | 1.50 (s) | – | 7.51 (m, Hϕ2/Hϕ6) | – |
| 7.13 (m, Hϕ3/Hϕ5) | ||||||||||
| 10e | – | – | – | 6.78 (q) | – | 2.43 (d) | 2.06 (s) | 3.89 (s), 3.92 (s) | 7.66 (m, Hϕ2/Hϕ6) | –113.79 (m) |
| 7.24 (m, Hϕ3/Hϕ5) | ||||||||||
| 13 | – | – | – | 6.93 (s) | 11.67 (s) | – | – | 3.79 (s) | 7.40 (m, Hϕ2/Hϕ6) | – |
| 7.33 (m, Hϕ3/Hϕ5) | ||||||||||
| 3.90 (s) | ||||||||||
| 7.22 (m, 5Hϕ) | ||||||||||
| 14f | – | – | – | 2.61 (s) | 5.19 (s) | 0.95 (s) | – | 3.80 (s) | 7.17 (m, Hϕ3) | – |
| 7.31 (m, Hϕ4) | ||||||||||
| 7.19 (m, Hϕ5) | ||||||||||
| 3.92 (s) | ||||||||||
| 7.38 (m, Hϕ6) | ||||||||||
| 7.02 (m, 5Hϕ) |
Experimental
General
Procedures for the preparation of compounds
6-[2-(4’-Fluorophenyl)-2-hydroxyethenyl]-2,4-dimethoxy-5-methylpyrimidine (4)
6-[2-(2’-Fluorobenzyl)-2-hydroxypropyl]pyrimidin-2,4-dione (6)
6-[2-(4’-Fluorophenyl)-1-propenyl]-5-methylpyrimidin-2,4-dione (8) and 6-[2-(4’-fluorophenyl)-2-hydroxypropyl]-5-methylpyrimidin-2,4-dione (9)
6-[2-(4’-Fluorophenyl)-2-hydroxypropyl]-5-methylpyrimidin-2,4-dione (9) and 6-[2-(4’-fluorophenyl)-1-propenyl]-2,4-dimethoxy-5-methylpyrimidine (10)
6-[2-(4’-Fluorophenyl)-2-hydroxy-1-ethenyl]-2,4-dimethoxy-5-phenylpyrimidine (13)
6-[2-(2’-Fluorobenzyl)-2-hydroxypropyl]-2,4-dimethoxy-5-phenylpyrimidine (14)
Antitumor activity assays
Cell cycle analysis
Conclusions
Acknowledgments
- Sample Availability: Samples of compounds 1-15 are available from the authors.
References
- Böhm, H.J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr. Fluorine chemistry at the millennium. J. Fluorine Chem. 2005, 126, 157–163. [Google Scholar] [CrossRef]
- Isanbor, C.; O'Hagan, D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Howard, J.A.K.; Hoy, V.J.; O'Hagan, D.; Smith, G.T. How good is fluorine as a hydrogen bond acceptor? Tetrahedron 1996, 52, 12613–12622. [Google Scholar] [CrossRef]
- Bergstrom, D.E.; Swartling, D.J. Structure, Reactivity, Synthesis and Applications. In Fluorine-Containing Molecules; Liebman, J.F., Greenberg, A., Dolbier, W.R., Jr., Eds.; VCH: New York, NY, USA, 1988; pp. 259–308. [Google Scholar]
- Öğretir, C.; Yaman, M. AM1, PM3 and MNDO study of the tautomeric equilibria of 2-, 4- or 5-hydroxypyrimidin derivatives and their azo- and thio- analogs. J. Mol. Struct. (Theochem) 1999, 458, 217–226. [Google Scholar] [CrossRef]
- Williams, M.; Kowaluk, E.A.; Arneric, S.P. Emerging molecular approaches to pain therapy. J. Med. Chem. 1999, 42, 1481–1500. [Google Scholar] [CrossRef]
- Bradshaw, T.K.; Hutchinson, D.N. 5-Substituted pyrimidine nucleosides and nucleotides. Chem. Soc. Rev. 1977, 6, 43–62. [Google Scholar]
- Prekupec, S.; Makuc, D.; Plavec, J.; Kraljević, S.; Kralj, M.; Pavelić, K.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E.; Raić-Malić, S.; Mintas, M. Antiviral and cytostatic evaluation of the novel 6-acyclic chain substituted thymine derivatives. Antiviral Chem. Chemother. 2005, 16, 327–338. [Google Scholar]
- Prekupec, S.; Makuc, D.; Plavec, J.; Šuman, L.; Kralj, M.; Pavelić, K.; Balzarini, J.; De Clercq, E.; Mintas, M.; Raić-Malić, S. Antiviral and cytostatic evaluation of the novel 6-acyclic chain substituted thymine derivatives. J. Med. Chem. 2007, 50, 3037–3045. [Google Scholar] [CrossRef]
- Shields, A.F.; Lim, K.; Grierson, J.; Link, J.; Krohn, K.A. Utilization of Labeled Thymidine in DNA Synthesis: Studies for PET. J. Nucl. Med. 1990, 31, 337–342. [Google Scholar]
- Schlenker, J. Ueber 4.5-Dimethylpyrimidin. Berichte 1901, 34, 2812–2829. [Google Scholar]
- Raić-Malić, S.; Johayem, A.; Ametamey, S.; Batinac, S.; De Clercq, E.; Folkers, G.; Scapozza, L. Synthesis, 18F-radiolabelling and biological evaluations of C-6 alkylated pyrimidine nucleoside analogues. Nucleos. Nucleot. Nucleic. Acids 2004, 23, 1707–1721. [Google Scholar] [CrossRef]
- Middleton, W.J. New fluorinating reagents. Dialkylaminosulfur fluorides. J. Org. Chem. 1975, 40, 574–578. [Google Scholar] [CrossRef]
- Agrofoglio, L.A.; Gillaizeau, I.; Saito, Y. Palladium-Assisted routes to nucleosides. Chem. Rev. 2003, 103, 1875–1916. [Google Scholar]
- Stille, J.K. The Palladium-catalyzed Cross-coupling reactions of organotin reagents with organic electrophiles [New Synthetic Methods (58)]. Angew. Chem. Int. Ed. Engl. 1986, 25, 508–524. [Google Scholar] [CrossRef]
- Stille, J.K. Palladium catalyzed coupling of organotin reagents with organic electrophiles. Pure Appl. Chem. 1985, 57, 1771–1780. [Google Scholar] [CrossRef]
- Gazivoda, T.; Krištafor, S.; Cetina, M.; Nagl, A.; Raić-Malić, S. Synthesis and structural studies of C-5 aryl and C-6 alkyl substituted pyrimidine derivatives. Struct. Chem. 2008, 19, 441–449. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krištafor, S.; Gazivoda Kraljević, T.; Makuc, D.; Plavec, J.; Šuman, L.; Kralj, M.; Raić-Malić, S. Synthesis, Structural Studies and Antitumoral Evaluation of C-6 Alkyl and Alkenyl Side Chain Pyrimidine Derivatives S. Molecules 2009, 14, 4866-4879. https://doi.org/10.3390/molecules14124866
Krištafor S, Gazivoda Kraljević T, Makuc D, Plavec J, Šuman L, Kralj M, Raić-Malić S. Synthesis, Structural Studies and Antitumoral Evaluation of C-6 Alkyl and Alkenyl Side Chain Pyrimidine Derivatives S. Molecules. 2009; 14(12):4866-4879. https://doi.org/10.3390/molecules14124866
Chicago/Turabian StyleKrištafor, Svjetlana, Tatjana Gazivoda Kraljević, Damjan Makuc, Janez Plavec, Lidija Šuman, Marijeta Kralj, and Silvana Raić-Malić. 2009. "Synthesis, Structural Studies and Antitumoral Evaluation of C-6 Alkyl and Alkenyl Side Chain Pyrimidine Derivatives S" Molecules 14, no. 12: 4866-4879. https://doi.org/10.3390/molecules14124866
APA StyleKrištafor, S., Gazivoda Kraljević, T., Makuc, D., Plavec, J., Šuman, L., Kralj, M., & Raić-Malić, S. (2009). Synthesis, Structural Studies and Antitumoral Evaluation of C-6 Alkyl and Alkenyl Side Chain Pyrimidine Derivatives S. Molecules, 14(12), 4866-4879. https://doi.org/10.3390/molecules14124866