Steroidal Triterpenes: Design of Substrate-Based Inhibitors of Ergosterol and Sitosterol Synthesis

Abstract
:
Introduction
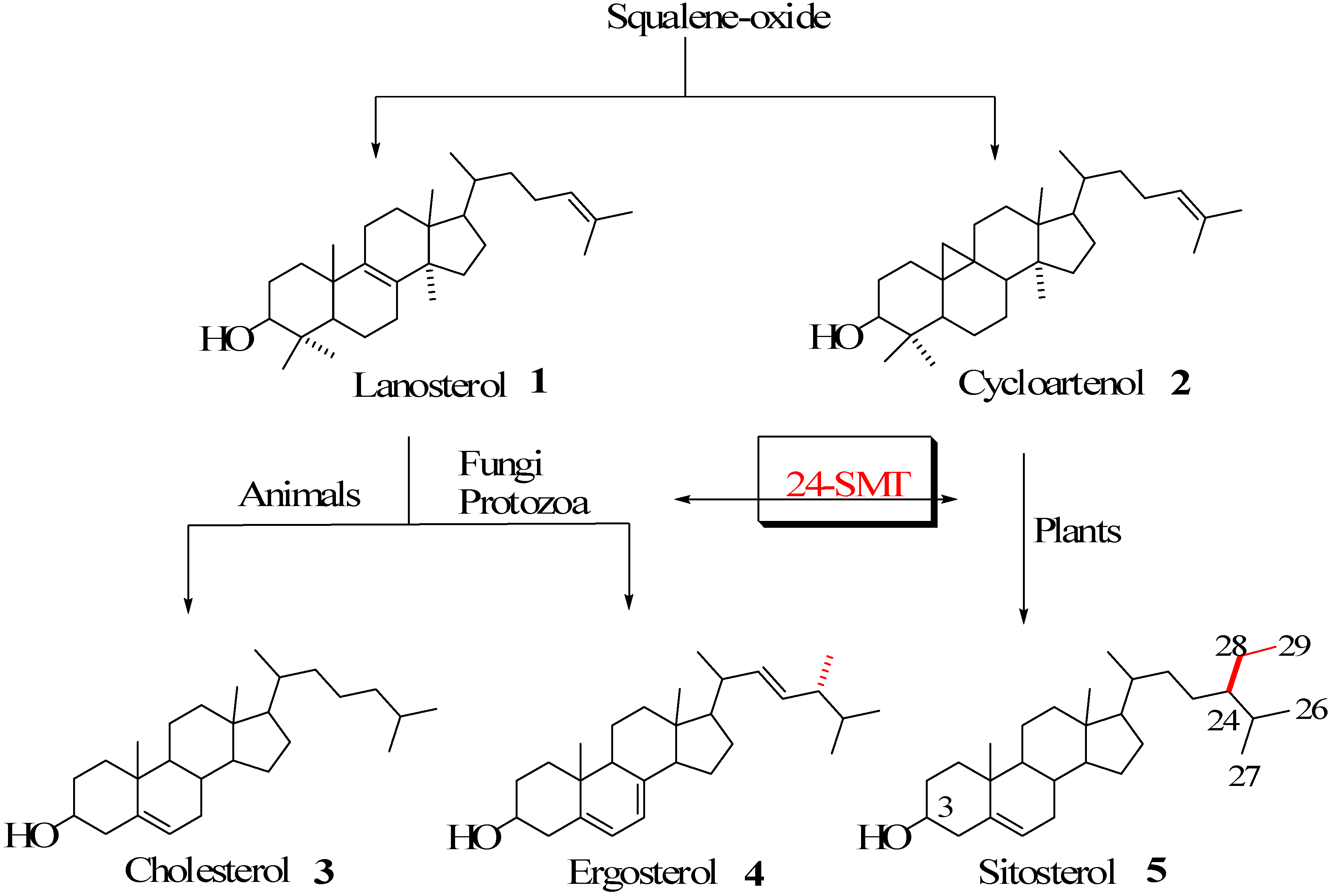
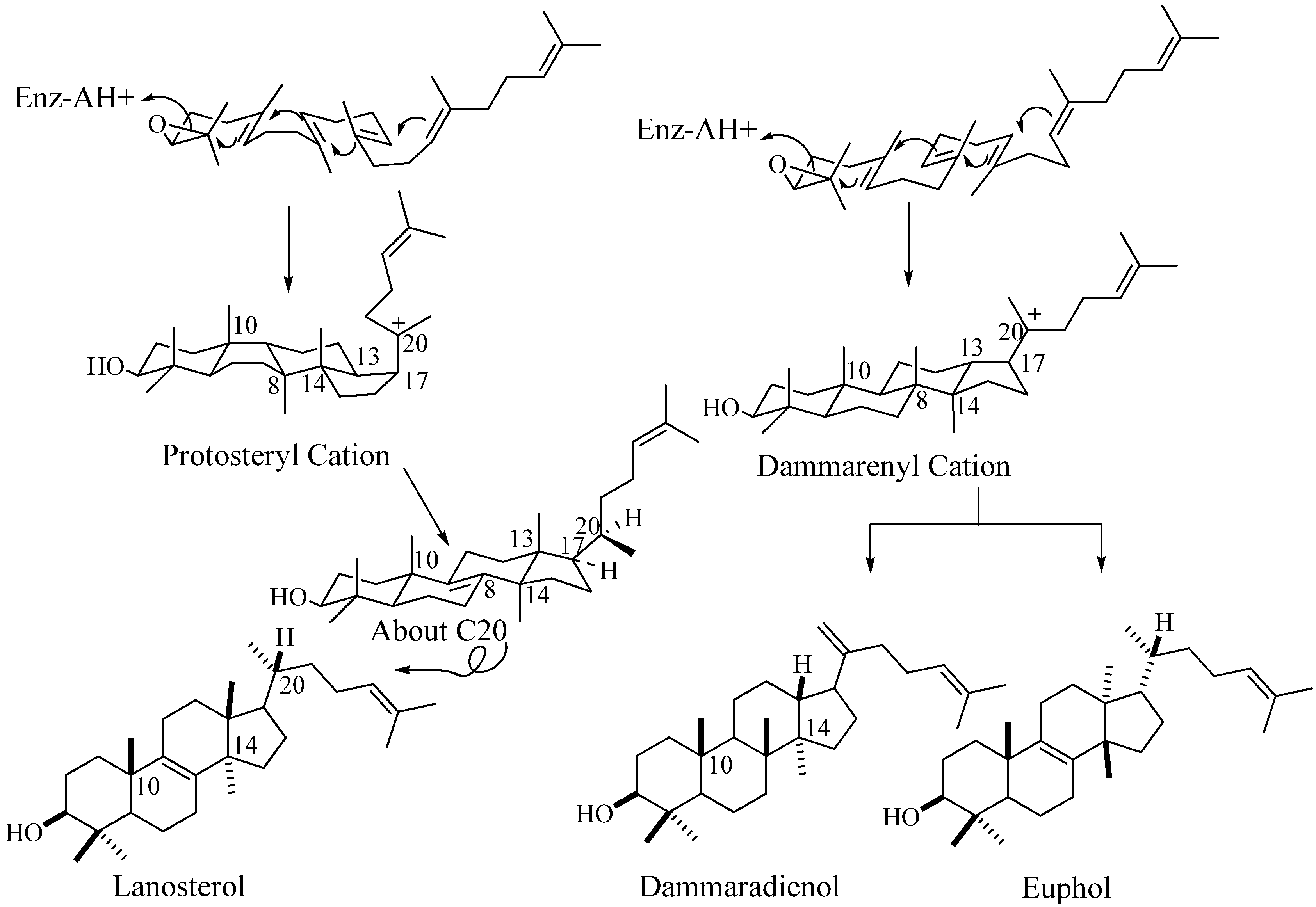
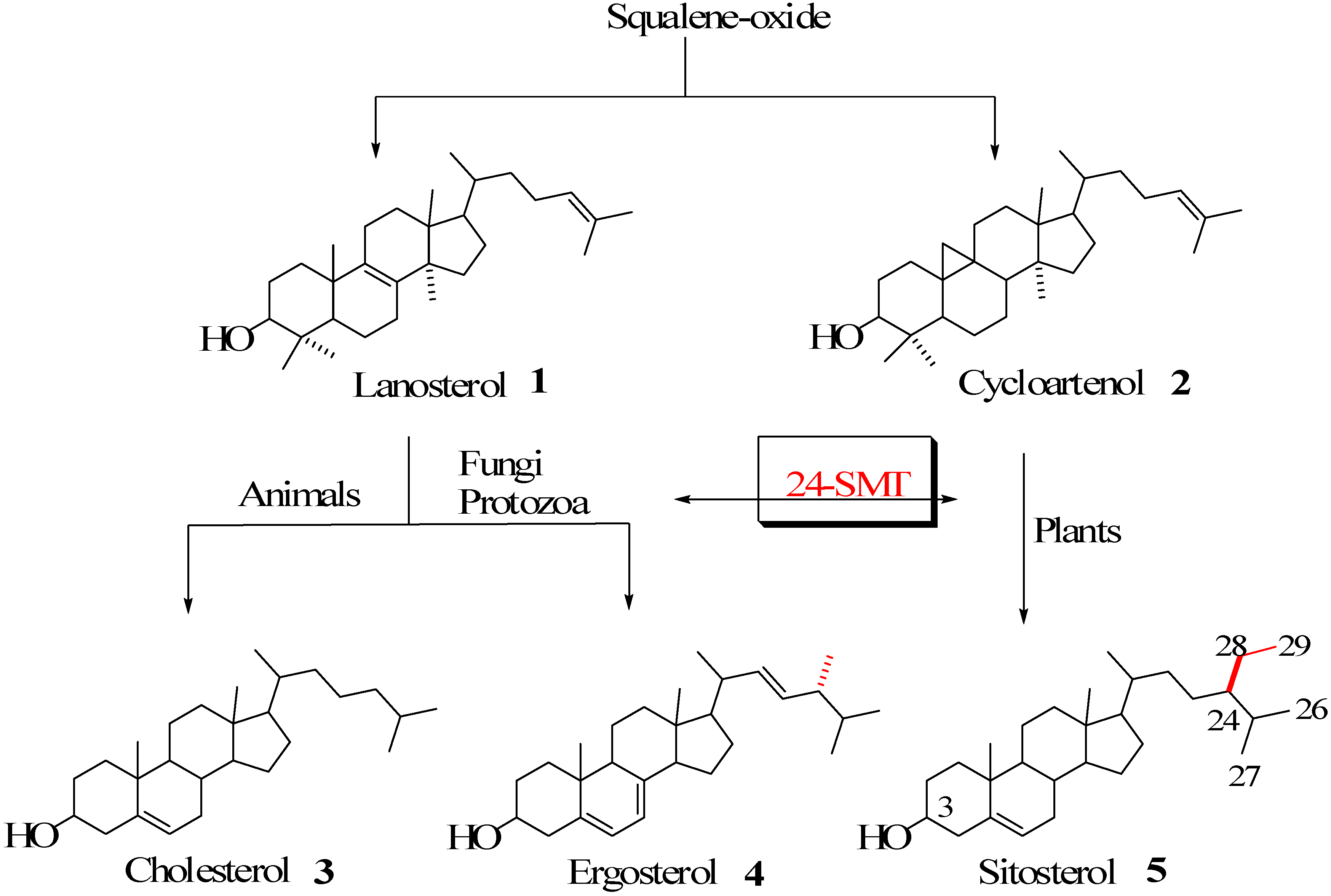
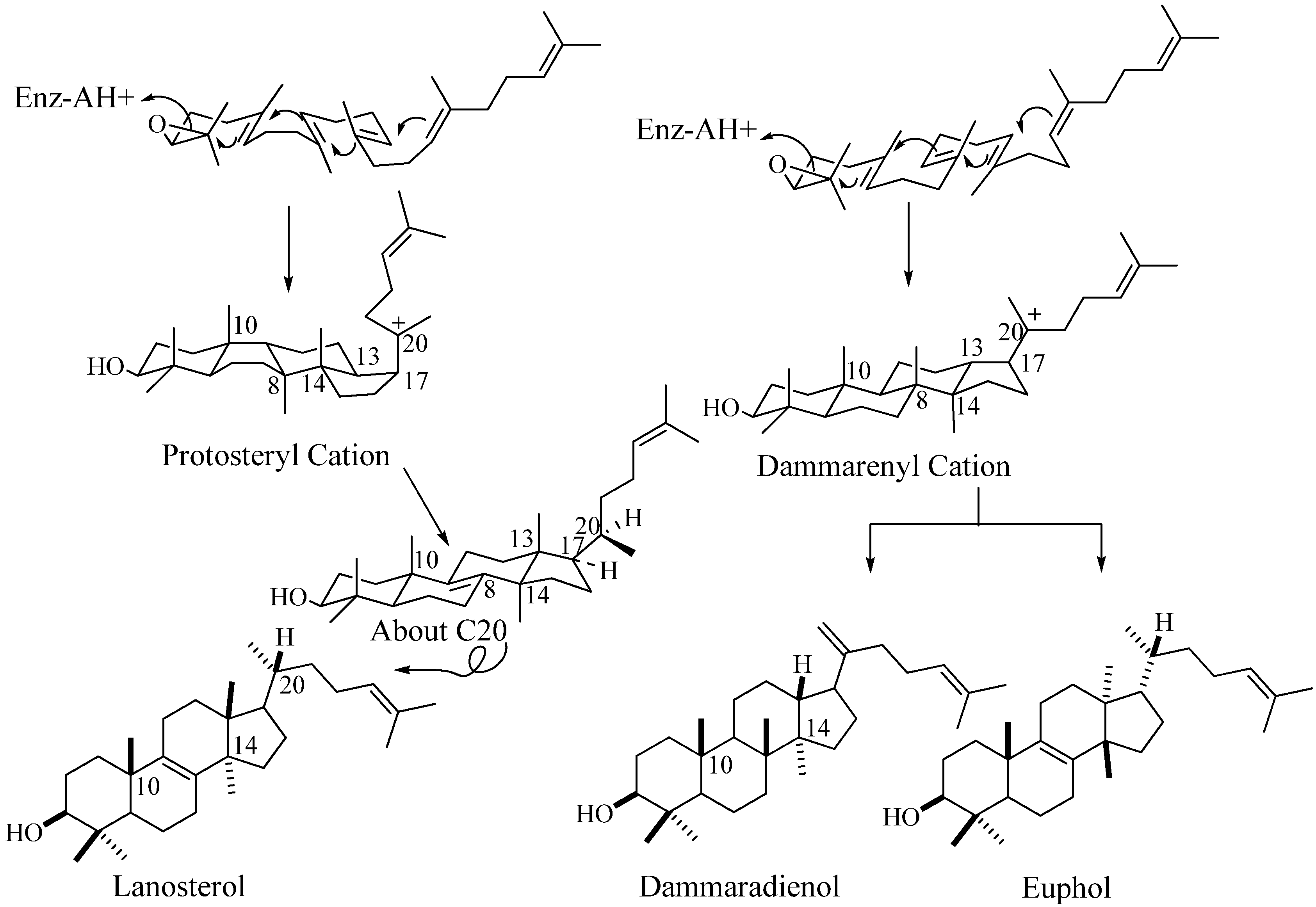
Steroidal Triterpenes
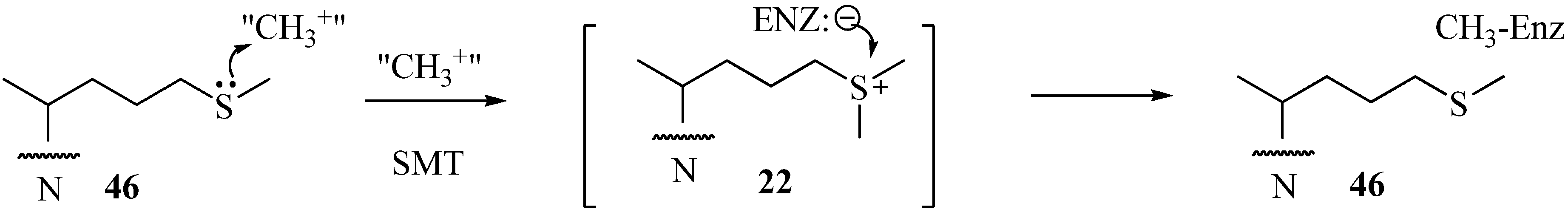
Sterol C24-Methyltranserases
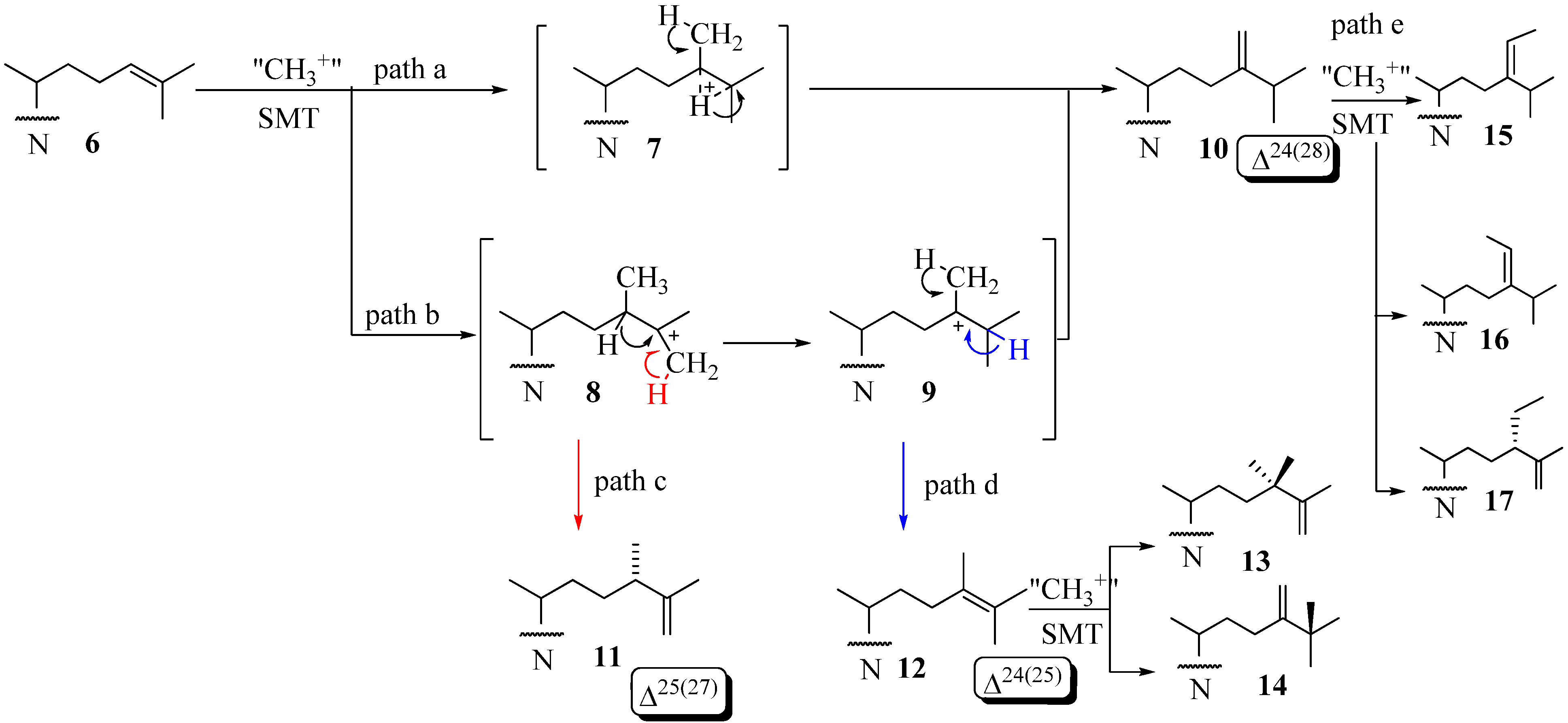
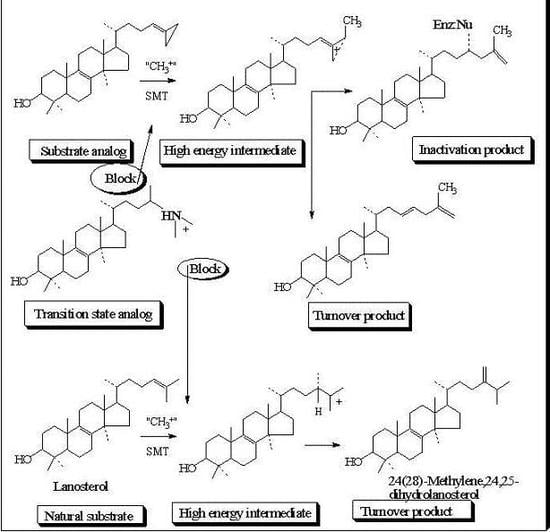
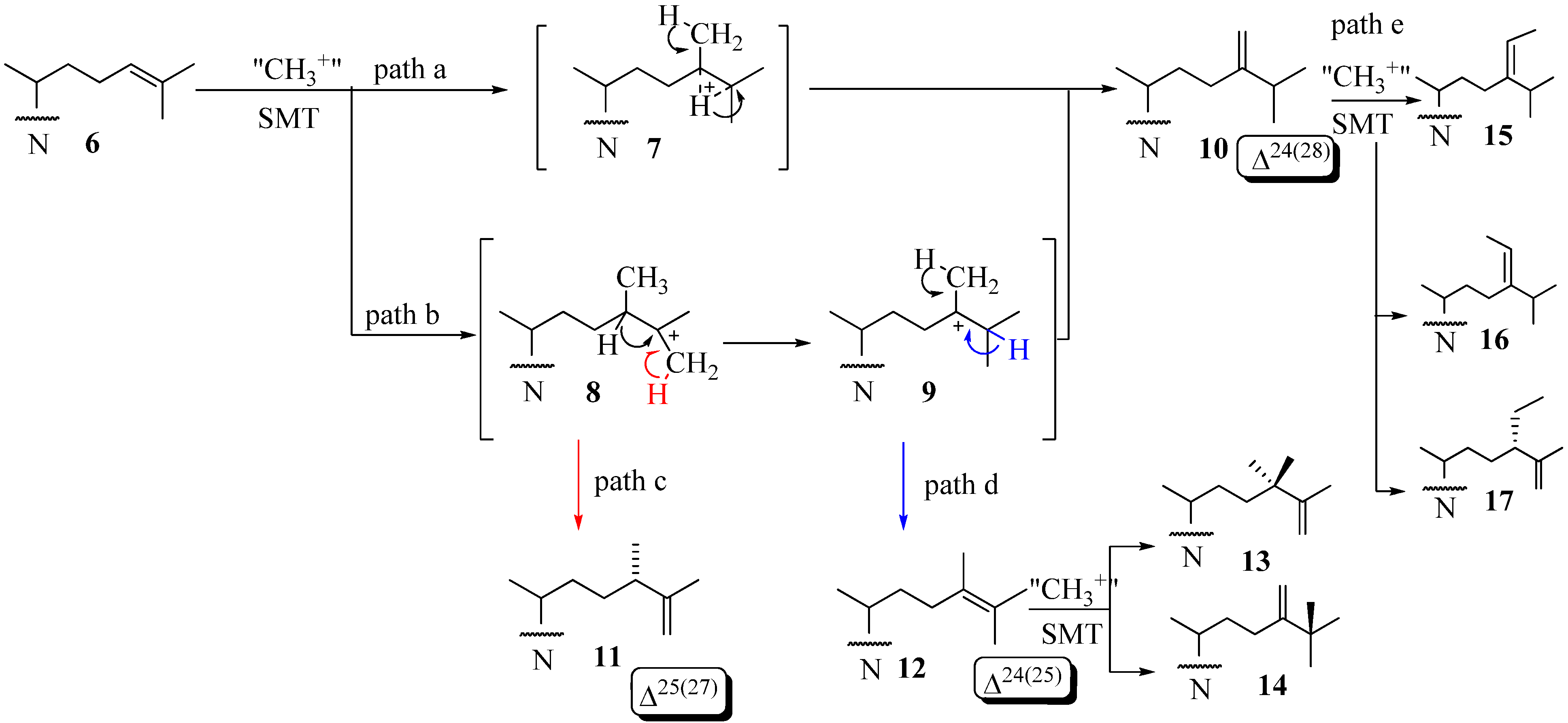
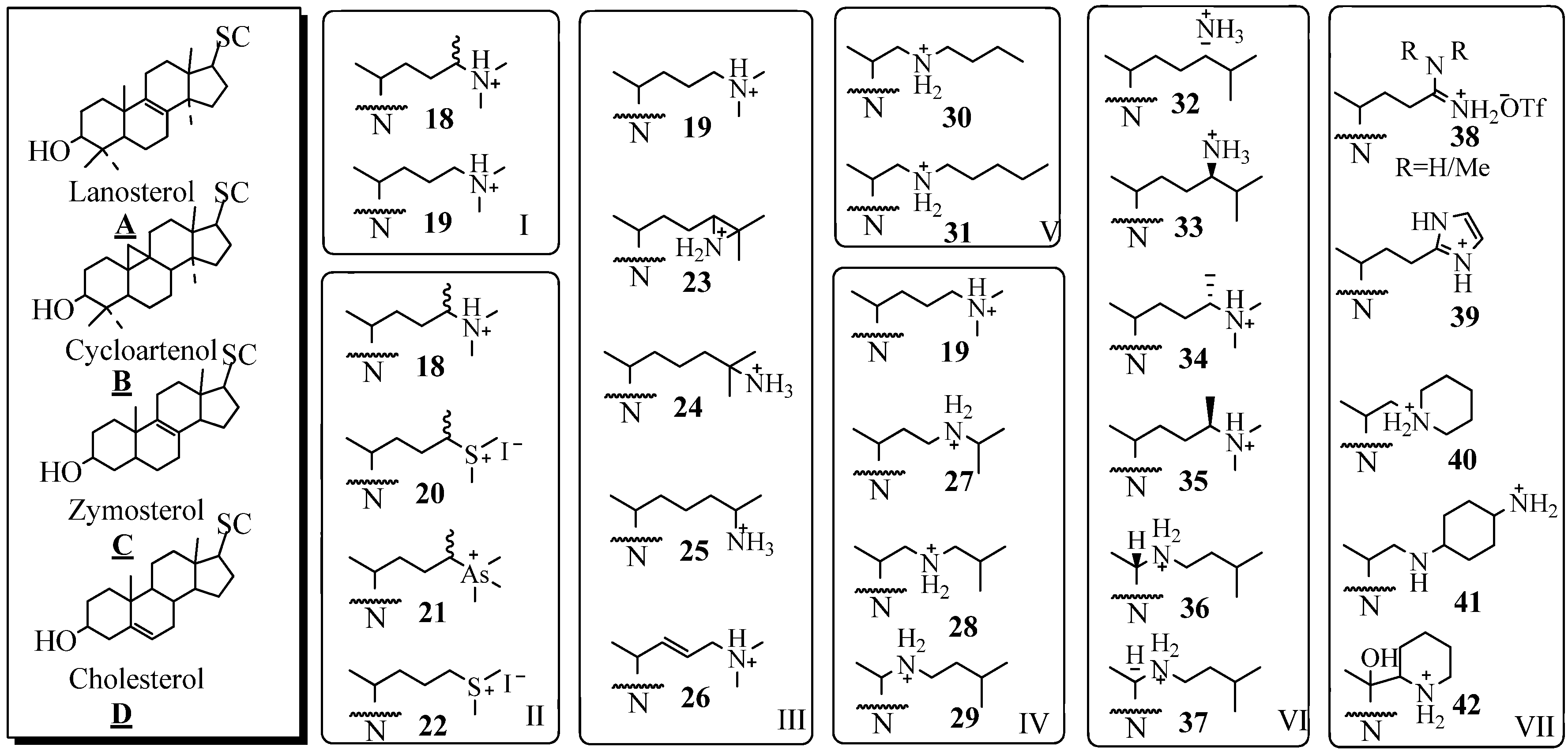
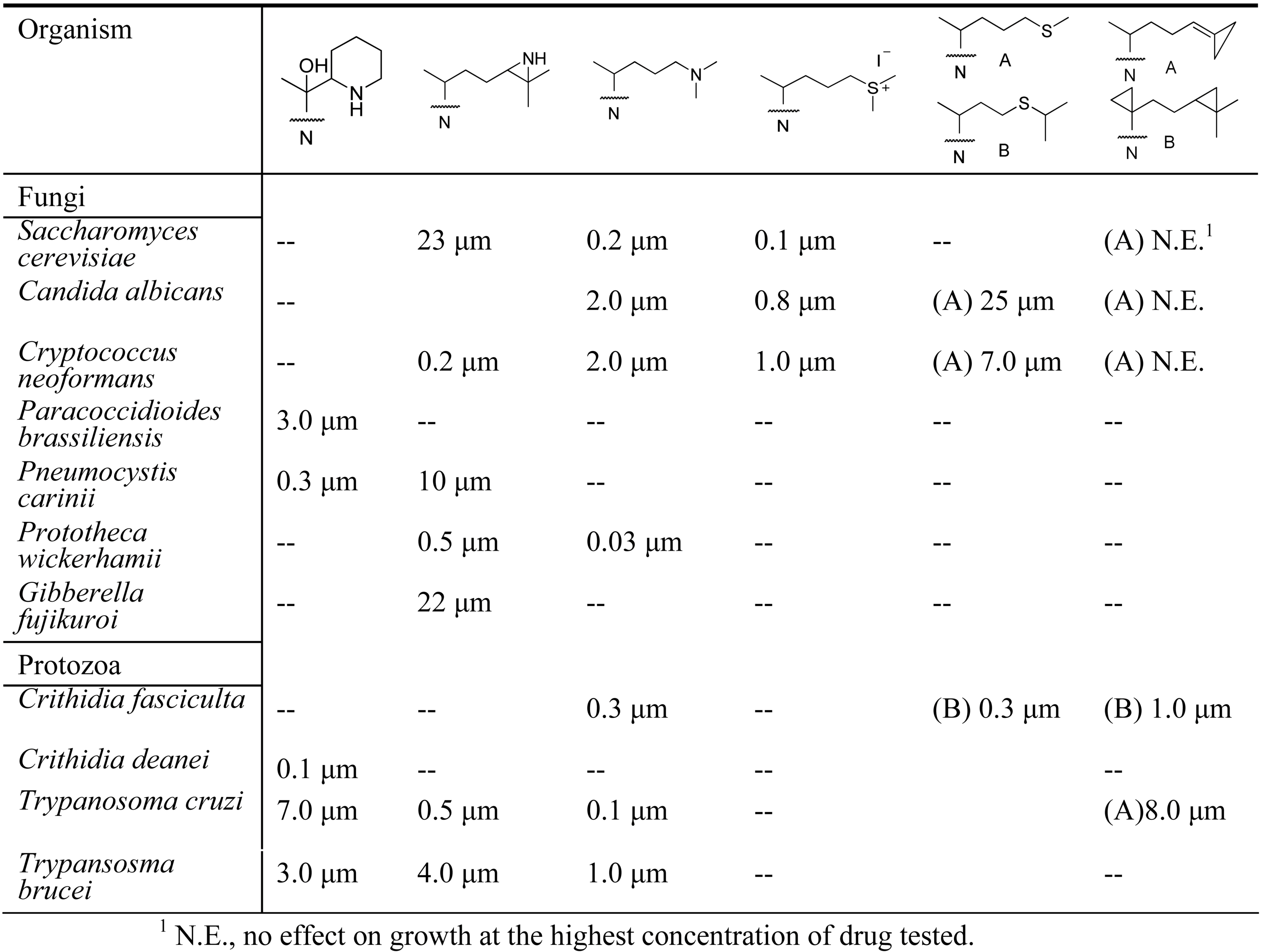
Molecular Parameters of Inhibitors Directed at the 24-SMT
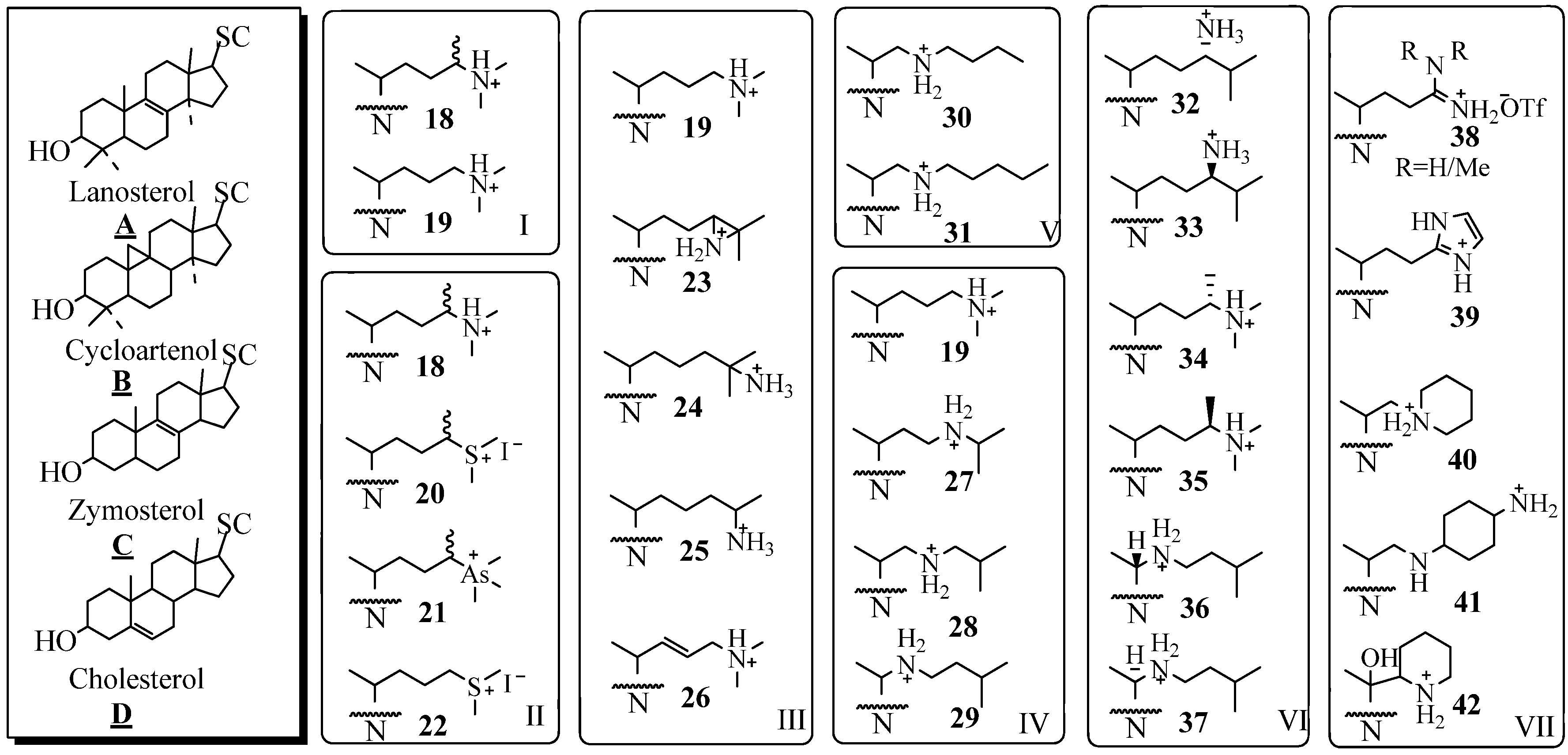
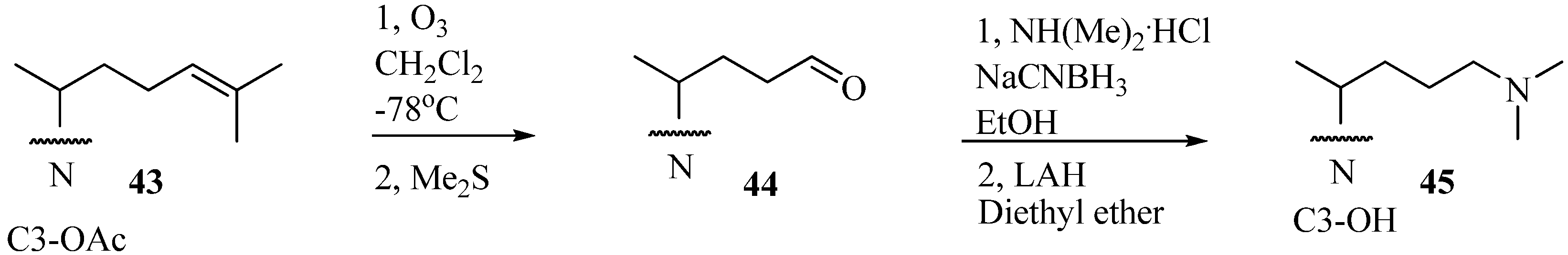
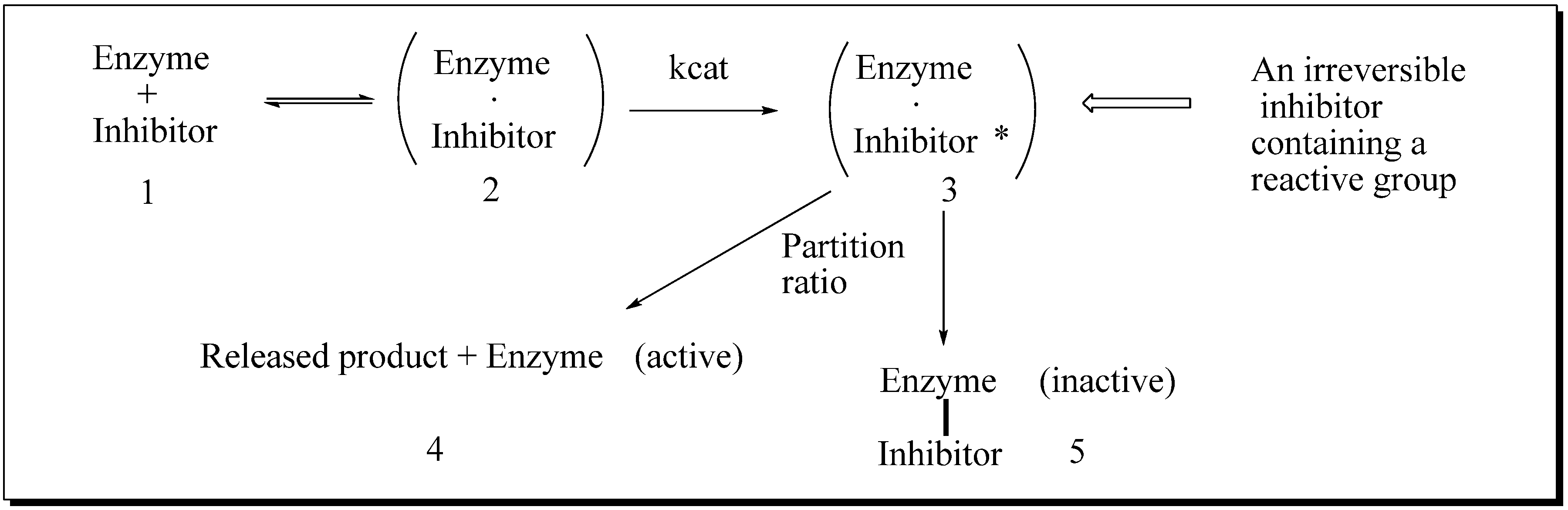
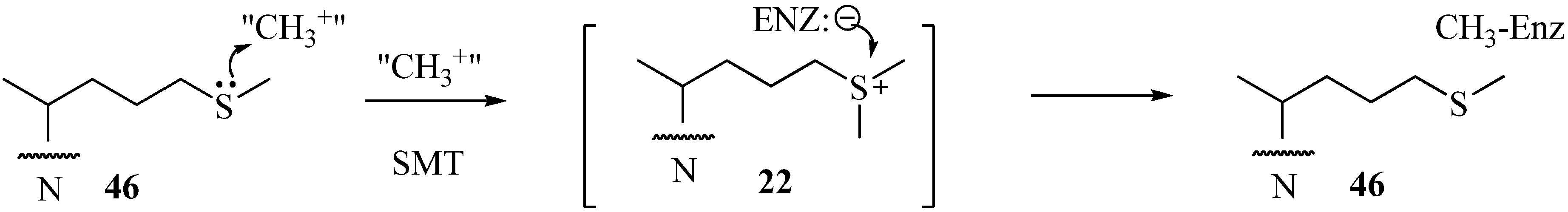
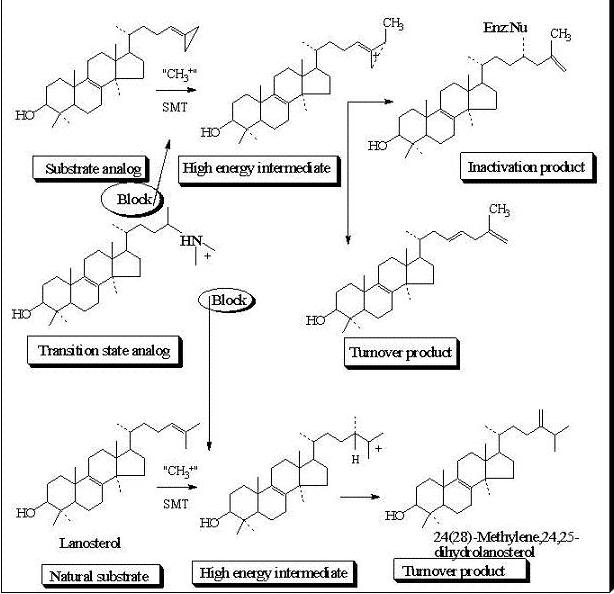
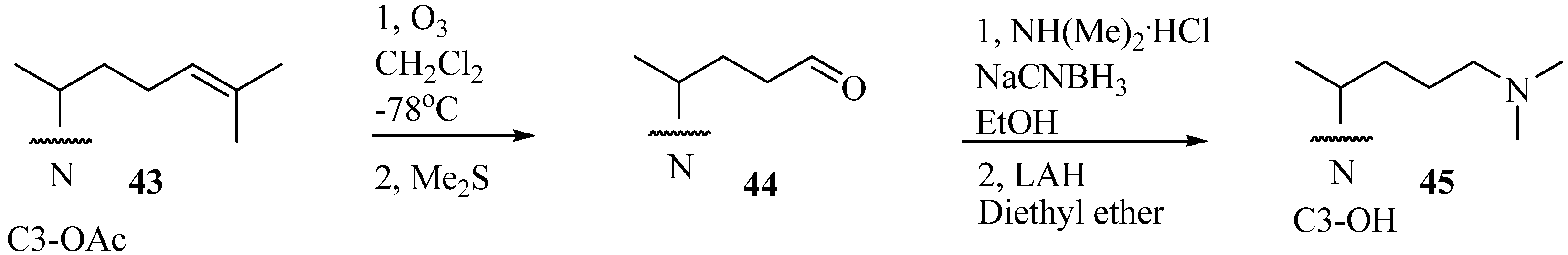
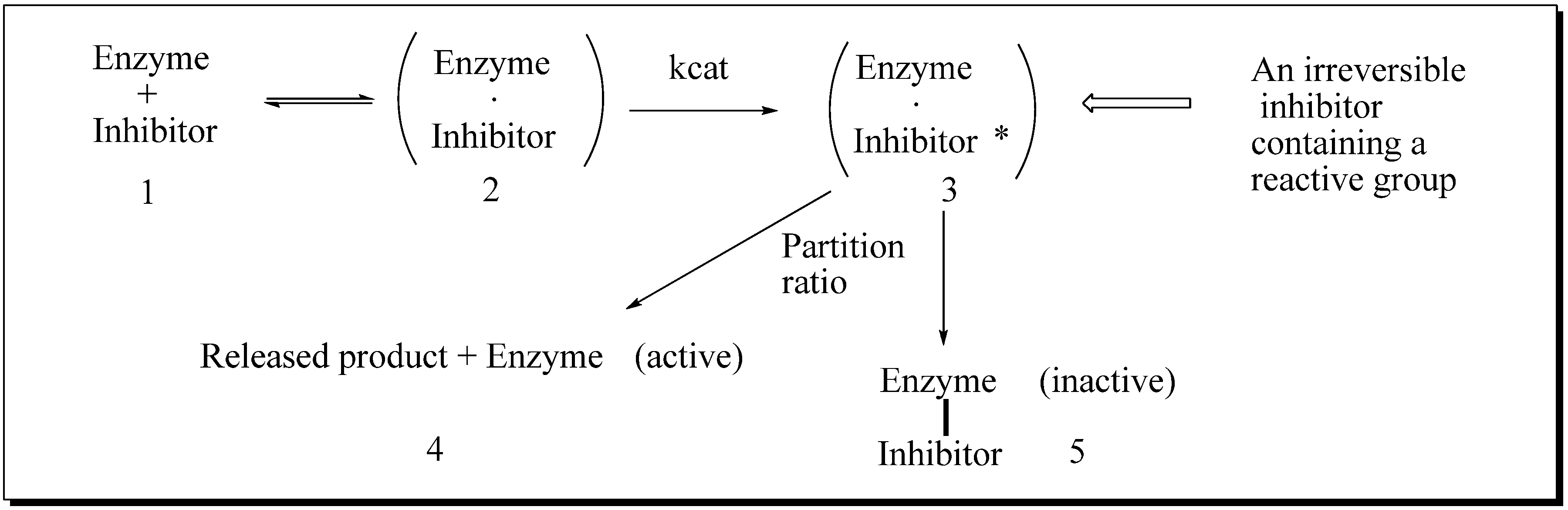
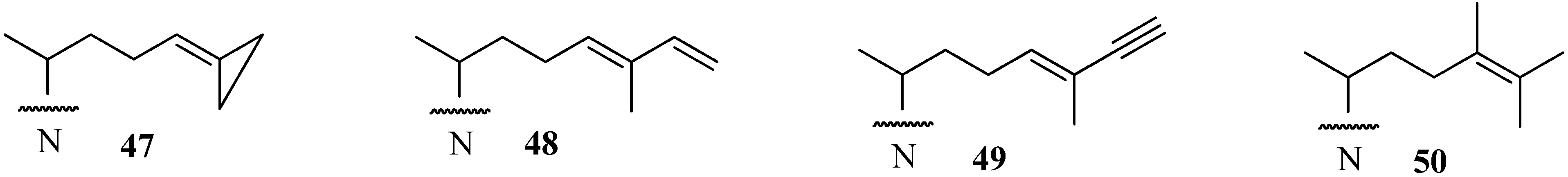
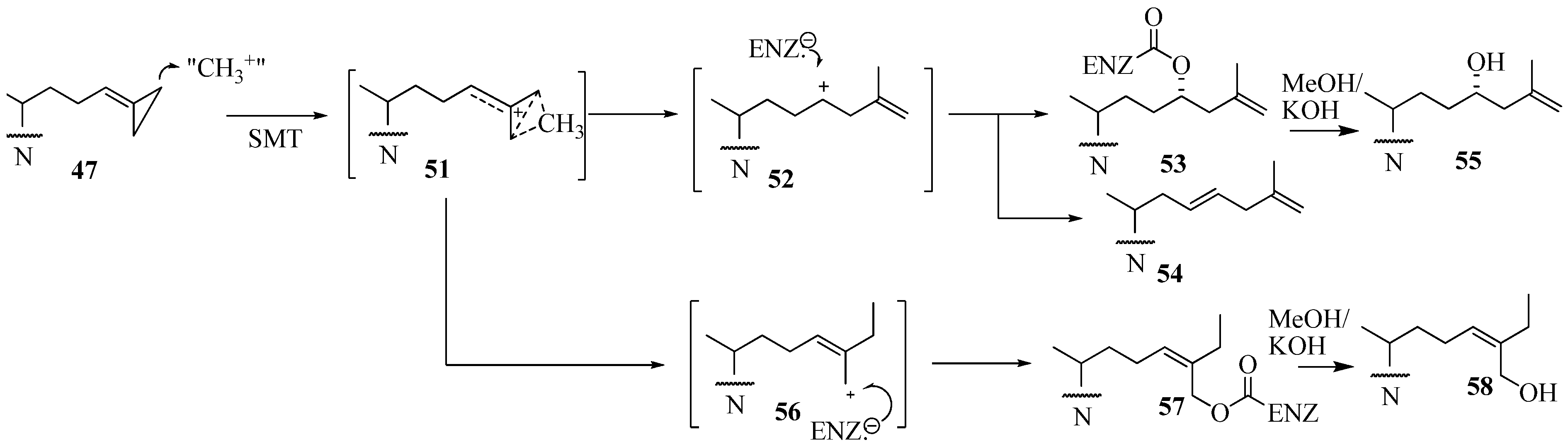
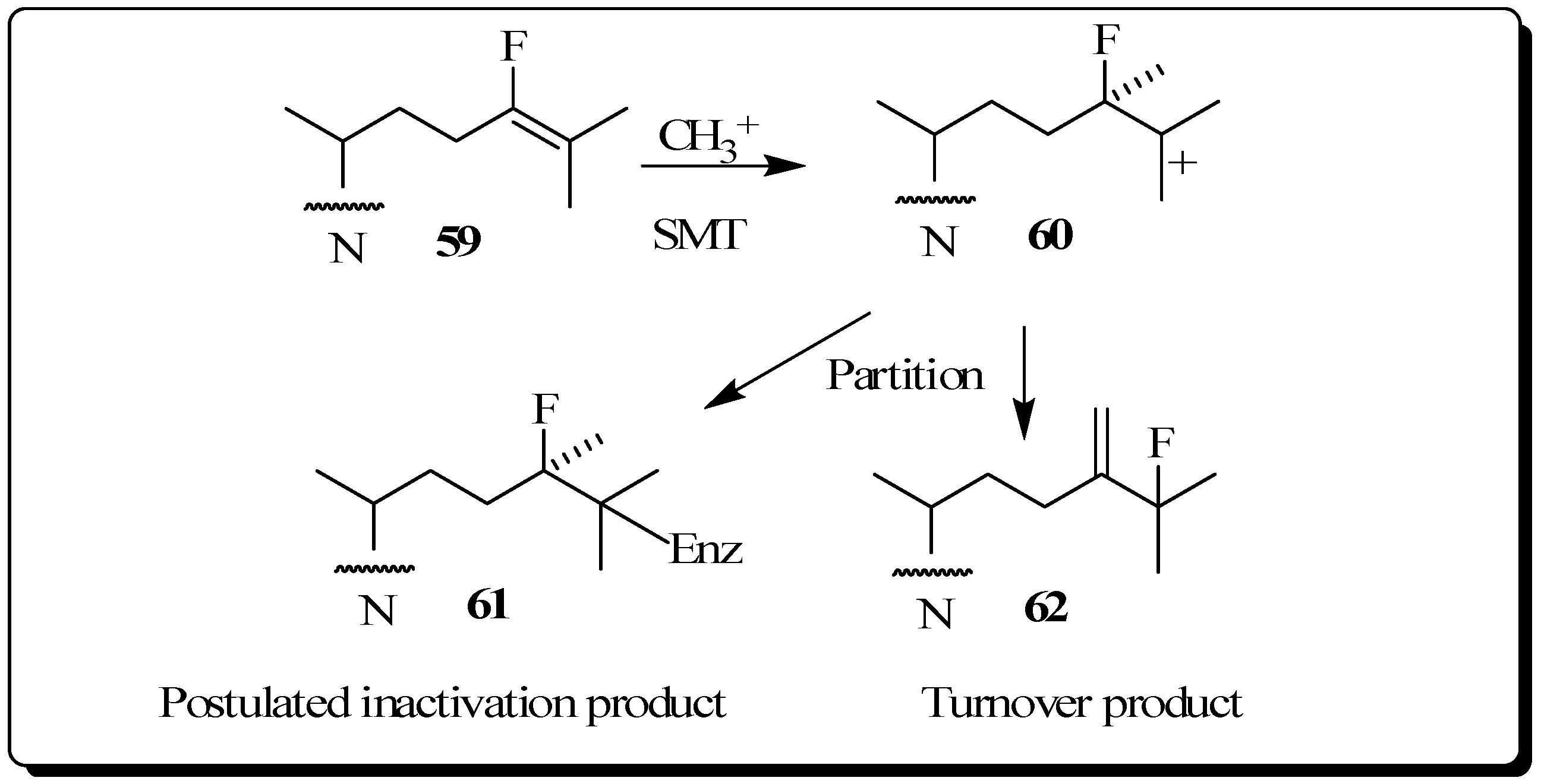
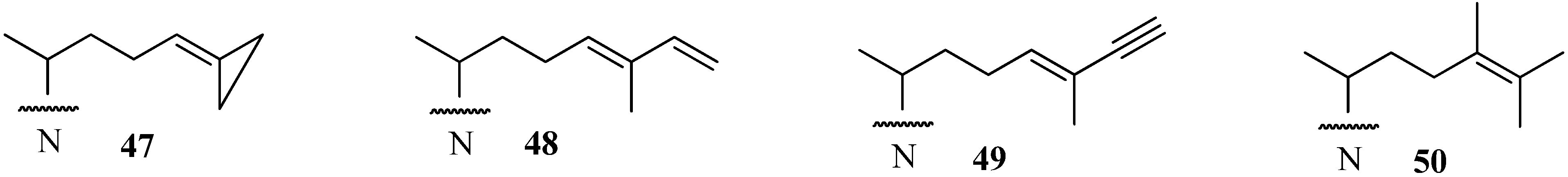
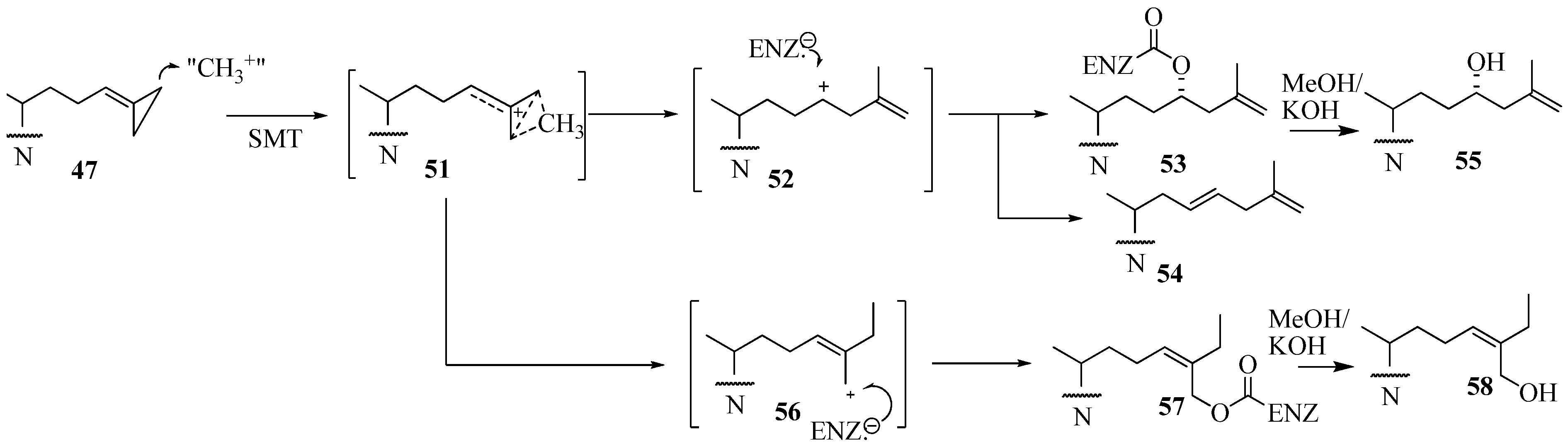
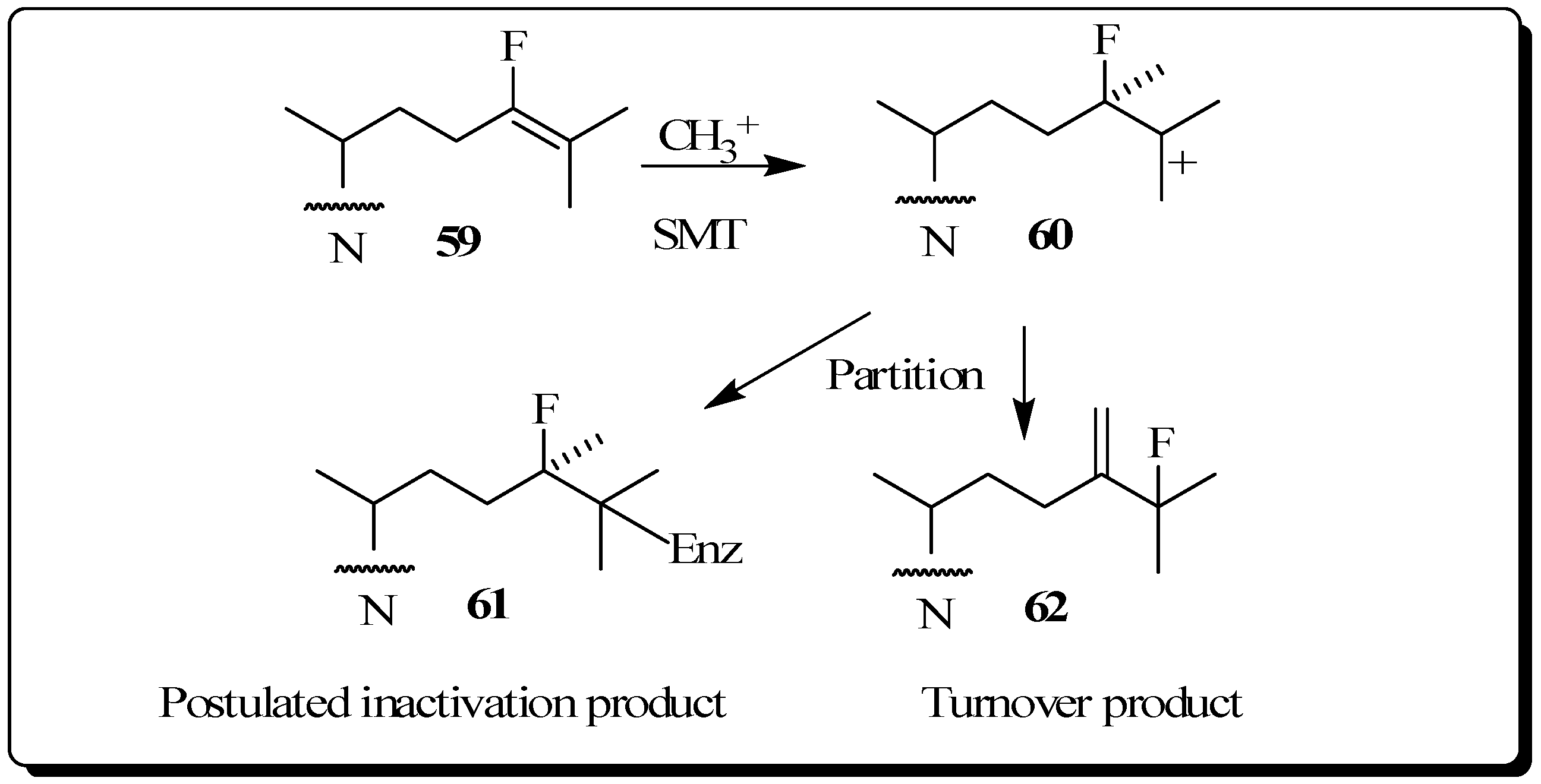
Evaluation of Novel Substrate Analogs as Mechanism-based Inactivators of 24-SMT

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
Conclusions
Acknowledgements
- Sample Availability: Not available.
References and Notes
- Jayasimha, J.; Bowman, C.B.; Pedroza, J.M.; Nes, W.D. Engineering pathway enzymes to understand the function and evolution of sterol structure and activity. Rec. Adv. Phytochem. 2006, 40, 212–251. [Google Scholar]
- Roberts, C.W.; Mcleod, R.; Rice, D.W.; Ginger, M.; Chance, M.L.; Goad, L.J. Fatty acid and sterol metabolism: Potential antimicrobial targets in Apicomplexan and Trypansomatid parasitic protozoa. Mole. Biochem. Parasit. 2003, 126, 129–142. [Google Scholar]
- Benveniste, P. Biosynthesis and accumulation of sterols. Annu. Rev. Plant Biol. 2004, 55, 429–457. [Google Scholar] [CrossRef]
- Goad, L.J.; Akihisa, T. Analysis of Sterols; Blackie Academic & Professional: London, UK, 1997; pp. 1–300. [Google Scholar]
- Varkony, T.H.; Smith, D.H.; Djerassi, C. Computer-assisted structure manipulation: Studies in the biosynthesis of natural products. Tetrahedron 1978, 34, 841–852. [Google Scholar]
- Nes, W.D. Enzyme mechanisms for sterol C-methylations. Phytochemistry 2003, 64, 75–95. [Google Scholar]
- Jimenez-Jimenez, C.; Carrero-Lerida, J.; Sealey-Cardona, M.; Perez, L.M.R.; Urbina, J.A.; Pacanowska, D.G. ∆24(25)-Sterol methyltransferase: Interacellular localization and azazsterol sensitivity in Leishmania major promastigotes overexpressing the enzyme. Int. J.Parasit. 2008, 39, 307–314. [Google Scholar]
- Zinser, E.; Paltauf, F.; Daum, G. Sterol composition of yeast organelle membranes and subcellular distribution of enzymes involved in sterol metabolism. J. Bacteriol. 1993, 175, 2853–2858. [Google Scholar]
- Lederer, E. C-Methylation in biological systems. Israel J. Med. Sci. 1965, 1, 1129–1147. [Google Scholar]
- Nes, W.D.; Zhou, W.; Ganapathy, K.; Liu, J.; Vatsyayan, R.; Chamala, S.; Hernandez, K.; Miranda, M. Sterol 24-C-methyltranferase: An enzymatic target for the disruption of ergosterol biosynthesis and homeostasis in Cryptococcus neoformans. Arch. Biochem. Biophys. 2009, 481, 210–218. [Google Scholar] [CrossRef]
- Kanagasabai, R.; Zhou, W.; Liu, J.; Nguyen, T.T.M.; Veeramachaneni, P.; Nes, W.D. Disruption of ergosterol biosynthesis, growth and the morphological transition in Candida albicans by sterol methyltransferase inhibitors containing sulfur at C25 in the sterol side chain. Lipids 2004, 39, 737–746. [Google Scholar]
- Visbal, G.; Alvarez, A.; Moreno, B.; San-blas, G. S-Adenosyl-L-methionine inhibitors of Δ24-sterol methyltransferase and ∆24(28)-sterol methyltransferase as possible agents against Pareacoccidiodes brasiliensis. Antimicrob. Agents Chem. 2003, 47, 2966–2970. [Google Scholar] [CrossRef]
- Urbina, J.A.; Vivas, J.; Visbal, G.; Contreras, L.M. Modification of the sterol composition of Trypanosoma (Schizotrypanum) cruzi epimastigotes by Δ24(25) -sterol methyltransferase inhibitors and their combinations with ketoconazole. Mol. Biochem. Parasit. 1995, 73, 199–20117. [Google Scholar] [CrossRef]
- Urbina, J.A.; Visbal, G.; Conreras, L.M.; McLaughlin, G.; Docampo, R. Inhibitors of Δ24(25)-sterol methyltransferase block sterol synthesis and cell proliferation in Pneumocystis carinii. Antimicrob. Agents Chemother. 1997, 41, 1428–1432. [Google Scholar]
- Lorente, S.O.; Rodriques, J.C.F.; Jimenez-Jimenez, C.; Joyce-Menekse, M.; Rodriques, C.; Croft, S.L.; Yardley, V.; de Luca-Fradley, K.; Ruiz-Perez, L.M.; Urbina, J.; De Souza, W.; Pacanowska, D.G.; Gilbert, I.H. Novel azasterols as potential agents for treatment of Leishmaniasis and Trypanosomiasis. Antimicrob. Agents Chemother. 2004, 48, 2937–2950. [Google Scholar]
- Wendt, K.U.; Schulz, G.E.; Corey, E.J.; Liu, D.R. Enzyme mechanisms for polycyclic triterpene formation. Angew. Chem. Int. Ed. 2000, 39, 2812–2833. [Google Scholar] [CrossRef]
- Hoshino, T.; Sato, T. Squalene-hopene cyclase: Catalytic mechanism and substrate recognition. Chem. Commun. 2002, 291–301. [Google Scholar] [CrossRef]
- Nes, W.R.; McKean, M.L. Biochemistry of Steroids and Other Isopentenoids; University Park Press: Baltimore, MD, USA, 1977. [Google Scholar]
- De Souza, W.; Rodriques, J.C. Sterol biosynthesis pathway as target for anti-trypanosomatid drugs. Interdiscip. Perspect. Infect. Diseases 2009, 1–19. [Google Scholar] [CrossRef]
- Urbina, J.A. Ergosterol biosynthesis and drug development for Chagas disease. Mem. Inst. Oswaldo Cruz. 2009, 104, 311–318. [Google Scholar] [CrossRef]
- Song, Z.; Nes, W.D. Sterol biosynthesis inhibitors: Potential for transition state analogs and mechanism-based inactivators targeted at sterol methyltransferase. Lipids 2007, 42, 15–33. [Google Scholar] [CrossRef]
- Visba, G.; Sna-blas, G.; Murgich, J.; Franco, H. Paracoccidiodes brasiliensis, Paracoccidiomycosis and antifungal antibiotics. Cur. Drug Targets-Infect. Disorders 2005, 5, 1–16. [Google Scholar] [CrossRef]
- Burbiel, J.; Bracher, F. Azasteroids as antifungals. Steroids 2003, 68, 587–594. [Google Scholar] [CrossRef]
- Kaneshiro, E.S. Sterol biosynthesis in Pneumocystis: Unique steps that define unique targets. Drug Resist. Updates 2002, 5, 259–268. [Google Scholar] [CrossRef]
- Venkatramesh, M.; Guo, D.; Jia, Z.; Nes, W.D. Mechanism and structural requirements for transformation of substrates by the (S)-adenosyl-L methionine: ∆24(25)- sterol methyl transferase from Saccharomyces cerevisiae. Biochim. Biophys. Acta 1996, 1299, 313–324. [Google Scholar] [CrossRef]
- Neelakandan, A.K.; Song, Z.; Wang, J.; Richards, M.H.; Wu, X.; Babu, V.; Nguyen, H.T.; Nes, W.D. Cloning, functional expression, and phylogenetic analysis of plant sterol 24-methyltransferase involved with sitosterol biosynthesis. Phytochemistry 2009, in press. [Google Scholar]
- Nes, W.D.; McCourt, B.S.; Zhou, W.X.; Ma, J.; Marshall, J.A.; Peek, L.A.; Brennan, M. Overexpression, purification, and stereochemical studies of the recombinant (S)-adenosyl-L-methionine: Δ24(25)- to Δ24(28)-sterol methyl transferase enzyme from Saccharomyces cerevisiae. Arch. Biochem. Biophys. 1998, 353, 297–311. [Google Scholar] [CrossRef]
- Nes, W.D.; Jayasimha, P.; Zhou, W.; Ragu, K.; Jin, C.; Jaradat, T.T.; Shaw, R.W.; Bujnicki, J.M. Sterol methyltransferase: Functional analysis of highly conserved residues by site-directed mutagenesis. Biochemistry 2004, 43, 569–576. [Google Scholar]
- Jayasimha, P.; Nes, W.D. Photoaffnity labeling and mutational analysis of 24-C-sterol methyl transferase defines the AdoMet binding site. Lipids 2008, 43, 681–693. [Google Scholar] [CrossRef]
- Ganapathy, K.; Jones, C.W.; Stephens, C.M.; Vatsyayan, R.; Marshall, J.A.; Nes, W.D. Molecular probing of the Saccharomyces cerevisiae sterol 24-C-methyltransferase (SMT) reveals multiple amino acids involved in C2-transfer activity. Biochim. Biophys. Acta 2008, 1781, 344–351. [Google Scholar] [CrossRef]
- Nes, W.D.; Song, Z.; Dennis, A.L.; Zhou, W.; Nam, J.; Miller, M. Biosynthesis of phytosterols: Kinetic mechanism for the enzymatic C-methylation of sterols. J. Biol. Chem. 2003, 278, 34505–34516. [Google Scholar]
- Zhou, W.; Lepesheva, G.I.; Waterman, M.R.; Nes, W.D. Mechanistic analysis of a multiple product sterol methyltransferase from Trypanosoma brucei implicated in ergosterol biosynthesis. J. Biol. Chem. 2006, 281, 6290–6296. [Google Scholar]
- Wang, J.; Nes, W.D. Cyclobranol: A substrate for C25 methyl sterol side chains and potent mechanistic-based inactivator of plant sterol methyltransferase. Bioorg. Med. Chem. Lett. 2008, 18, 3878–3881. [Google Scholar]
- Ganapathy, K.; Liu, J.; Nes, W.D. Site-directed Mutagenesis and Mechanistic Analysis of the Trypanosoma brucei Sterol 24C-methyltransfedrase. In Presented at the 238th Meeting of the American Chemical Society, Washington, D.C., USA, August 19th 2009.
- Nes, W.D.; Sinha, A.; Jayasimha, P.; Zhou, W.; Song, Z.; Dennis, A.L. Probing the sterol binding site of soybean sterol methyltransferase by site-directed mutagenesis: Functional analysis of conserved aromatic amino acids in Region 1. Arch. Biochem. Biophys. 2006, 448, 23–30. [Google Scholar] [CrossRef]
- Nes, W.D.; Marshall, J.A.; Jia, Z.; Jaradat, T.T.; Song, Z.; Jayasimha, P. Active site mapping and substrate channeling in the sterol methyl transferase pathway. J. Biol. Chem. 2002, 277, 42459–42556. [Google Scholar]
- Nes, W.D.; Zhou, W.; Guo, D. Substrate-based inhibitors of the (S)-adenosyl-L-methionine: ∆24(25)-sterol methyl transferase from Saccharomyces cerevisiae. Arch. Biochem. Biophys. 1997, 342, 68–81. [Google Scholar] [CrossRef]
- Mangla, A.T.; Nes, W.D. Sterol C-methyl transferase from Prototheca wickerhamii: Mechanism, sterol specificity and inhibition. Bioorg. Med. Chem. 2000, 8, 925–936. [Google Scholar] [CrossRef]
- Narula, A.S.; Rahier, A.; Benveniste, P.; Schuber, F. 24-Methyl-25-azacycloartenol, an analogue of a carbonium high energy intermediate, is a potent inhibitor of (S)-adenosyl-L-methionine: Sterol C-24-methyltransferase in higher plants. J. Amer. Chem. Soc. 1981, 103, 2408–2409. [Google Scholar]
- Rahier, A.; Genot, J.C.; Schuber, F.; Benveniste, P.; Narula, A.S. Inhibition of S-adenosyl-L-methionine sterol C-24 methyltransfersase by analogues of a carbocationic ion high energy intermediate. J. Biol. Chem. 1984, 259, 15215–15223. [Google Scholar]
- Oehlschlager, A.C.; Anugs, R.H.; Pierce, A.M.; Pierce, H.D., Jr.; Srinivasan, R. Azasterol inhibition of Δ24-sterol methyltransferase in Saccharomyces cerevisiae. Biochemistry 1984, 23, 3582–3589. [Google Scholar]
- Acuna-Johnson, A.P.; Oehlschlager, A.C.; Pierce, A.M.; Pierce, H.D., Jr.; Czyzewska, E.K. Stereochemistry of yeast Δ24-sterol methyltransferase. Bioorg. Med. Chem. 1997, 5, 821–832. [Google Scholar] [CrossRef]
- Janssen, G.G.; Nes, W.D. Structural requirements for transformation of substrates by the (S)-adenosyl-L-methionine: ∆24(25)-sterol methyl transferase. II. Inhibition by analogs of the transition state coordinate. J. Biol. Chem. 1992, 267, 25856–25863. [Google Scholar]
- Ator, M.A.; Schmidt, S.J.; Adams, J.L.; Dolle, R.E.; Kruse, L.I.; Frey, C.L.; Barone, J.M. Synthesis, specificity, and antifungal activity of inhibitors of the Candida Albicans Δ24-sterol methyltransferase. J. Med. Chem. 1992, 35, 100–106. [Google Scholar] [CrossRef]
- Ator, M.A.; Schmidt, S.J.; Adams, J.L.; Dolle, R.E. Mechanism and inhibition of Δ24-sterol methyltransferase from Candida albicans and Candida tropicalis. Biochemistry 1989, 28, 9633–9640. [Google Scholar]
- Rahman, M.D.; Pascal, R.A., Jr. Inhibitors of ergosterol biosynthesis and growth of the Trypanosomatid Protozoan Crithidia fasciculata. J. Biol. Chem. 1990, 265, 4989–4996. [Google Scholar]
- Zhou, W.; Song, S.; Liu, J.; Miller, M.B.; Nes, W.D. 24-Thiacycloartenol, a potent mechanism-based inactivator of plant sterol methyltransferase. Tetrahedron Lett. 2004, 45, 875–878. [Google Scholar]
- Chung, S.K.; Ryoo, C.H.; Yang, H.W.; Shim, J.Y.; Kang, M.G.; Lee, K.W.; Kang, H.I. Synthesis and bioactivities of steroid derivatives as antifungal agents. Tetrahedron 1998, 54, 15899–15914. [Google Scholar]
- Lorente, S.O.; Jimenez-Jimenez, C.; Gros, L.; Yardley, V.; de Luca-Fradley, K.; Croft, S.L.; Urbina, J.A.; Ruiz-Perez, L.M.; Pacanowska, D.G.; Gilbert, I.H. Preparation of transition state analogs of sterol 24-methyltransferase as potential antiparasitics. Bioorg. Med. Chem. 2005, 13, 5435–5453. [Google Scholar]
- Gros, L.; Lorente, S.O.; Jimenez-Jimenez, C.; Yardley, V.; Rattray, L.; Wharton, H.; Little, S.; Croft, S.I.; Ruiz-Perez, L.M.; Gonzalez-Pacanowska, D.; Gilbert, I.H. Evaluation of azasterols as anti-parasitics. J. Med. Chem. 2006, 49, 6094–6103. [Google Scholar]
- Zhou, W.; Cross, G.M.A.; Nes, W.D. Cholesterol import fails to prevent catalyst-based inhibition of ergosterol synthesis and cell proliferation of Trypanosoma brucei. J. Lipid Res. 2007, 48, 665–673. [Google Scholar]
- Chung, S.K.; Lee, K.W.; Kang, H.I.; Yamashita, C.; Kudo, M.; Yoshida, Y. Design and synthesis of potential inhibitors of the ergosterol biosynthesis as antifungal agents. Bioorg. Med. Chem. 2000, 8, 2475–2486. [Google Scholar] [CrossRef]
- Zhou, W.; Song, Z.; Kanagasabai, R.; Liu, J.; Jayasimha, P.; Sinha, A.; Veeramachanemi, P.; Miller, M.B.; Nes, W.D. Mechanism-based enzyme inactivators of phytosterol biosynthesis. Molecules 2004, 9, 185–203. [Google Scholar] [CrossRef]
- Zhou, W.; Song, Z.; Nes, W.D. Comparison of transition state analogs and mechanism-based inhibitors of sterol methyl transferase activity on growth and sterol biosynthesis in human cultured cells. Unpublished data.
- Nes, W.D.; Marshall, J.A.; Zhou, W.; He, L.; Dennis, A.L. Mechanism-based active site modification of sterol methyltransferase by tritium-labeled 26-homocholesta-8,14,24-trien-yn-3β-ol. Tetrahedron Lett. 1998, 39, 8575–8678. [Google Scholar]
- Song, Z.; Nes, W.D. Inactivation of soybean sterol 24-C-methyltransferase by elongated sterol side chains at C26. Bioorg. Med. Chem. Lett. 2007, 17, 5902–5906. [Google Scholar] [CrossRef]
- Nes, W.D.; Jayasimha, P.; Song, Z. Yeast sterol C24-methyltransferase: Role of highly conserved tyrosine in catalytic competence studied by site-directed mutagenesis and thermodynamic analysis. Arch. Biochem. Biophys. 2008, 477, 313–323. [Google Scholar] [CrossRef]
- Nes, C.R.; Liu, J.; Kleschemko, Y.Y.; Villalta, F.; Nes, W.D. The activity of 26,27-dehydrolanosterol and related substrate-analogs of the sterol 24C-methyltransferase against Trypansoma cruzi. In Abstr. at the 238th meeting of the Am. Chem. Soc, Washington, D.C., USA, August 16th, 2009.
- Venkatramesh, M.; Guo, D.; Harman, J.G.; Nes, W.D. Sterol Specificity of the Saccharomyces cerevisiae ERG6 Gene Product Expressed in Escherichia coli. Lipids 1996, 31, 373–377. [Google Scholar] [CrossRef]
- Wang, J.; Liu, J.; Song, Z.; Nes, W.D. Sterol C24-methyltransferase: Mechanistic studies of the C-methylation reaction with 24-fluorocycloartenol. Bioorg. Med. Chem. Lett. 2008, 18, 232–235. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, J.; Nes, W.D. Steroidal Triterpenes: Design of Substrate-Based Inhibitors of Ergosterol and Sitosterol Synthesis. Molecules 2009, 14, 4690-4706. https://doi.org/10.3390/molecules14114690
Liu J, Nes WD. Steroidal Triterpenes: Design of Substrate-Based Inhibitors of Ergosterol and Sitosterol Synthesis. Molecules. 2009; 14(11):4690-4706. https://doi.org/10.3390/molecules14114690
Chicago/Turabian StyleLiu, Jialin, and William David Nes. 2009. "Steroidal Triterpenes: Design of Substrate-Based Inhibitors of Ergosterol and Sitosterol Synthesis" Molecules 14, no. 11: 4690-4706. https://doi.org/10.3390/molecules14114690
APA StyleLiu, J., & Nes, W. D. (2009). Steroidal Triterpenes: Design of Substrate-Based Inhibitors of Ergosterol and Sitosterol Synthesis. Molecules, 14(11), 4690-4706. https://doi.org/10.3390/molecules14114690