The Morita-Baylis-Hillman Reaction: Insights into Asymmetry and Reaction Mechanisms by Electrospray Ionization Mass Spectrometry


Abstract
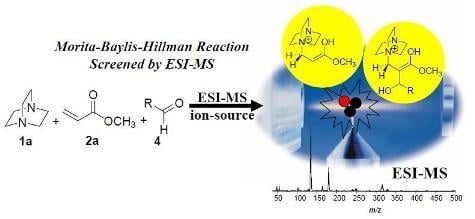
:
1. Introduction
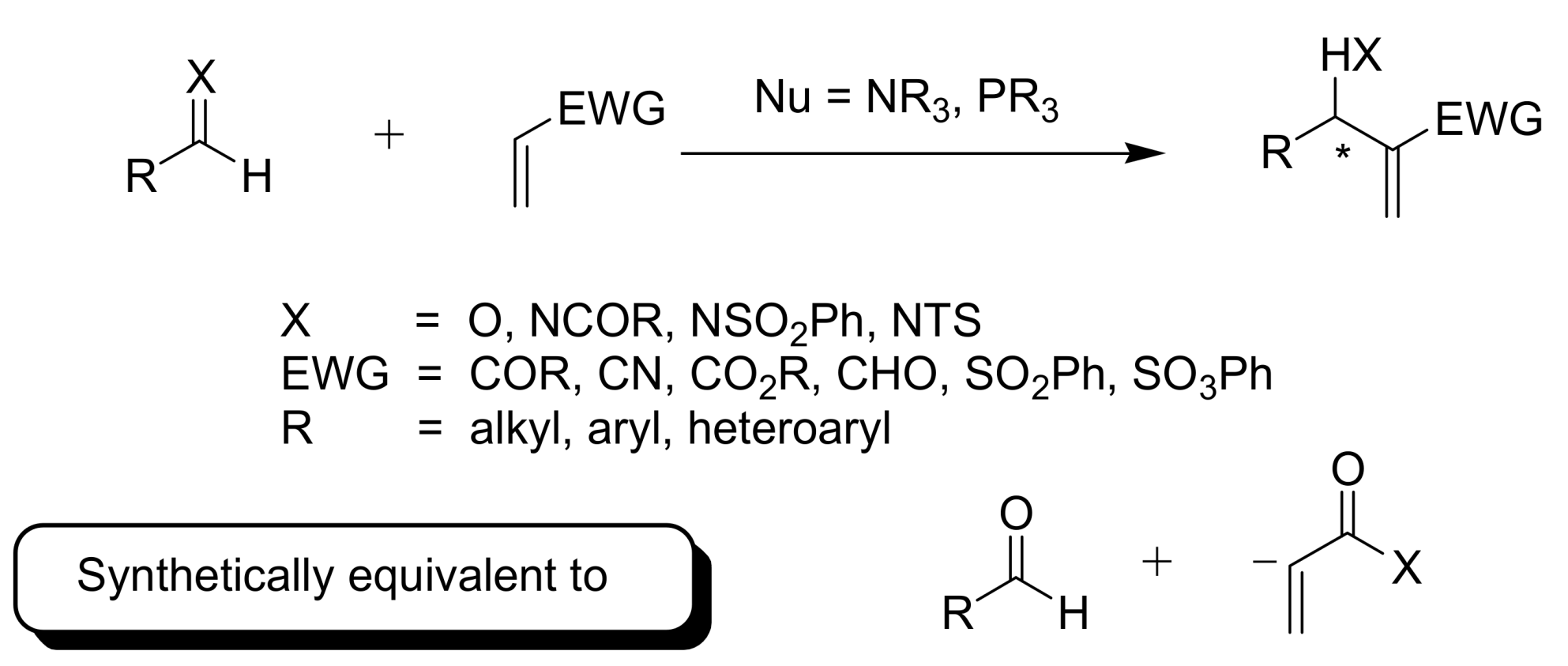
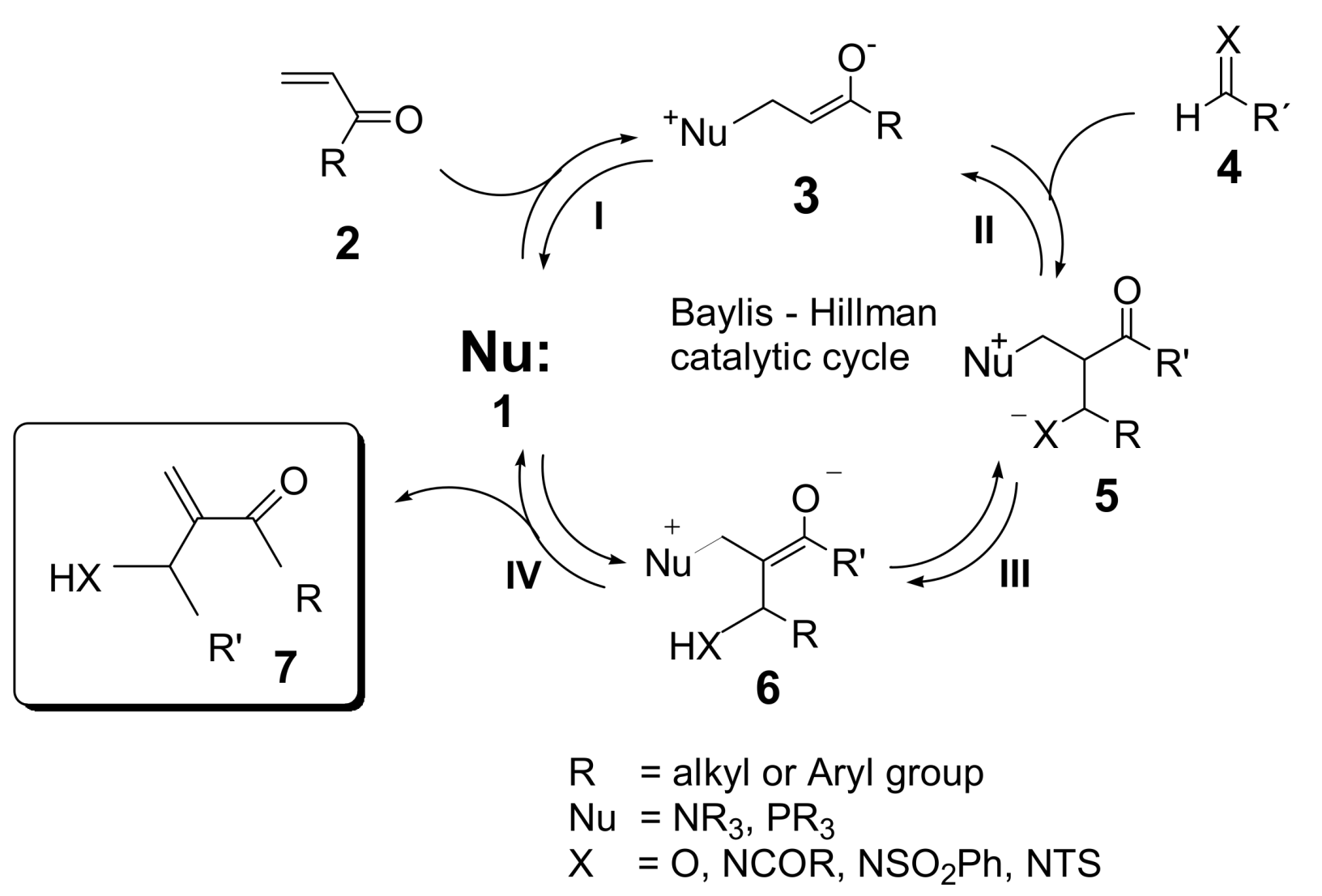
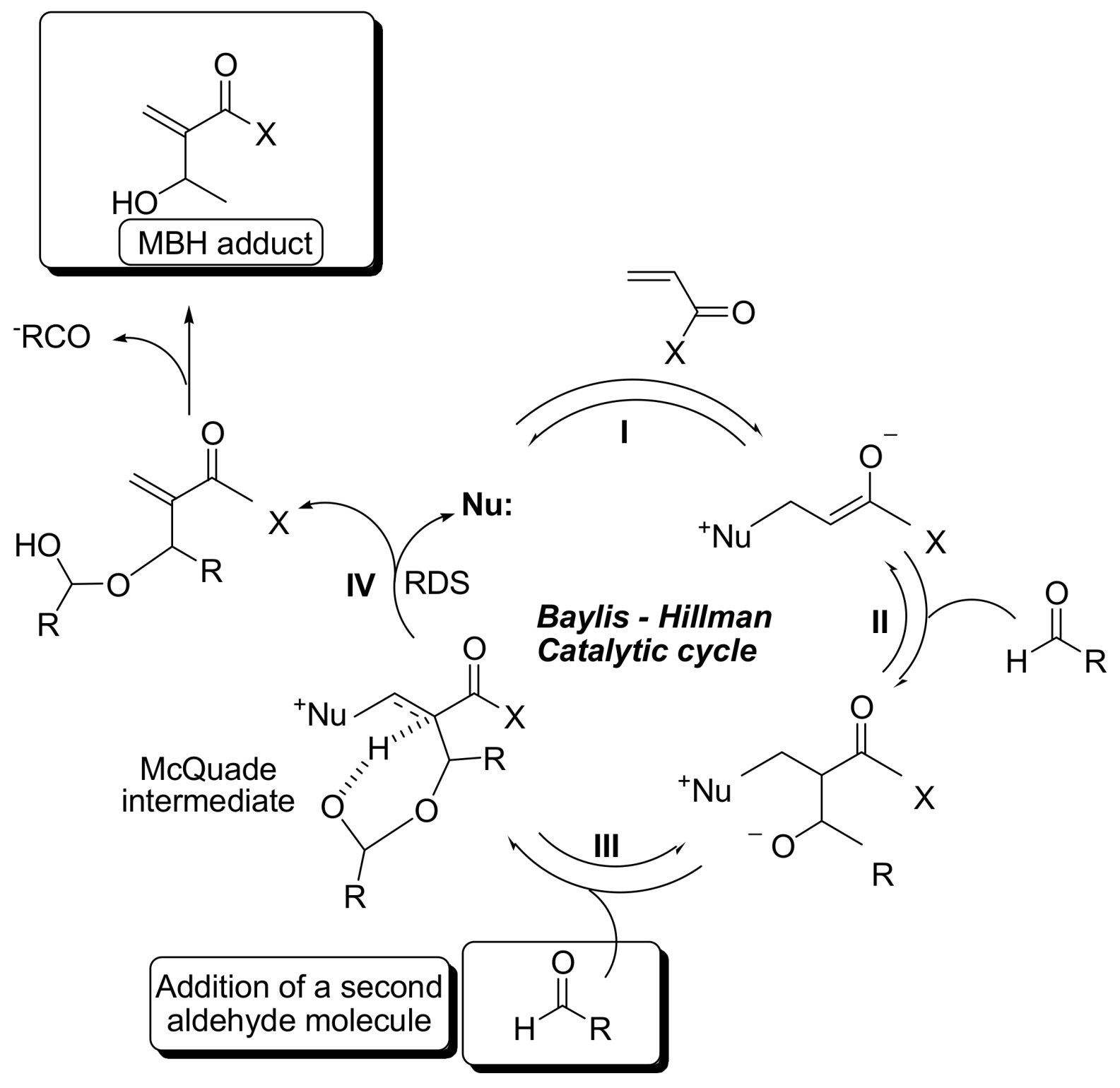
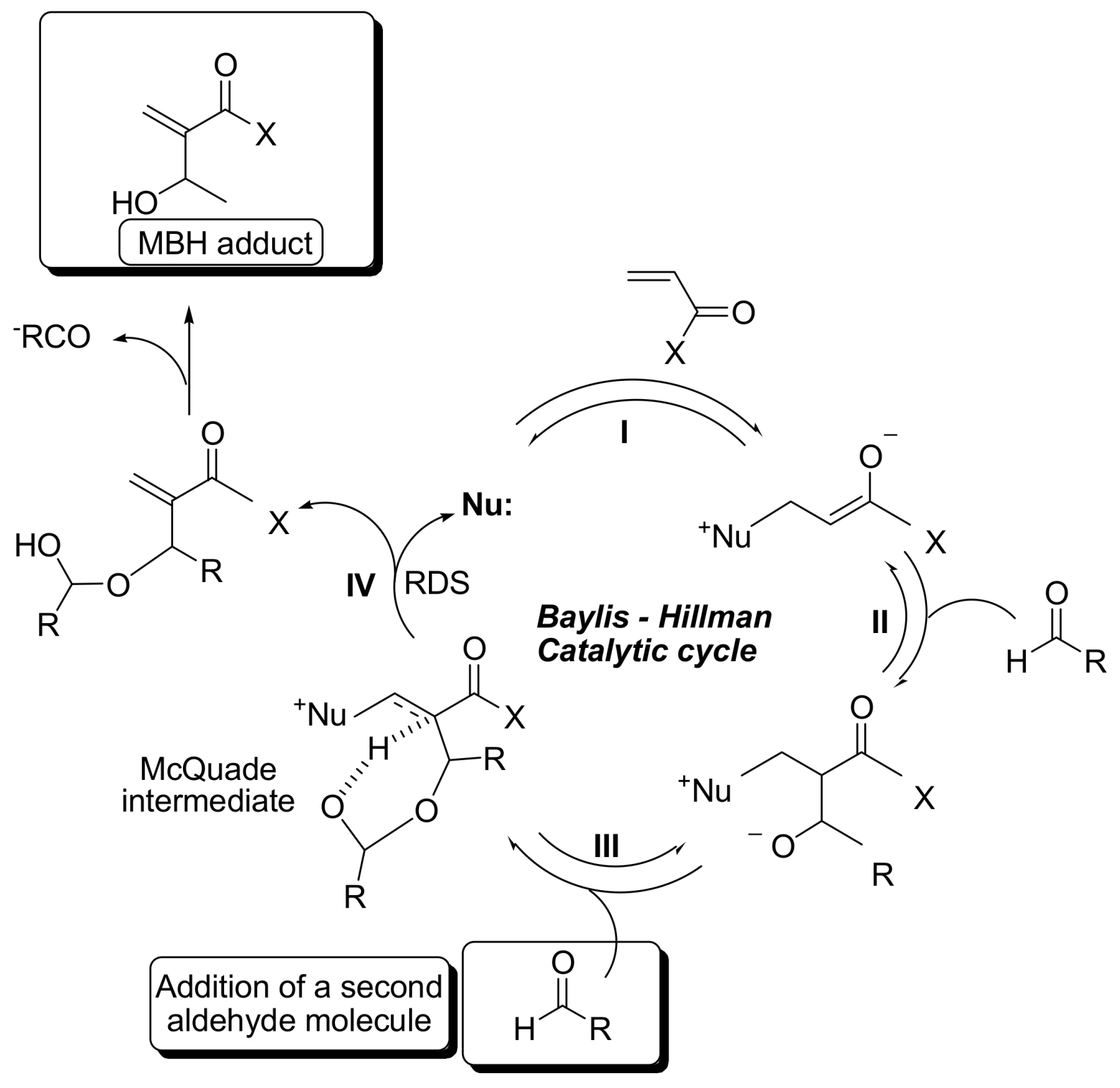
2. Mechanism. Earlier Work
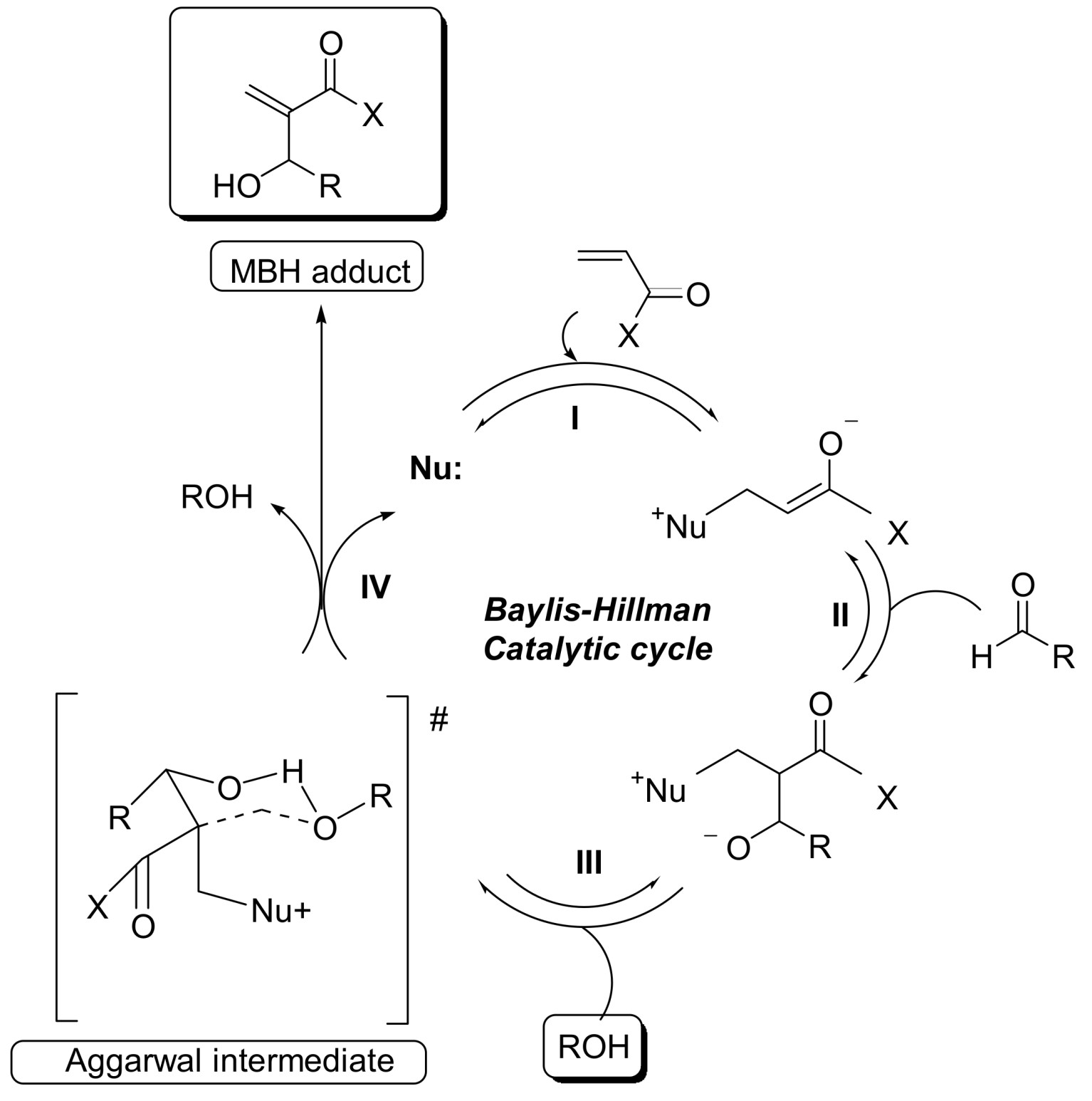
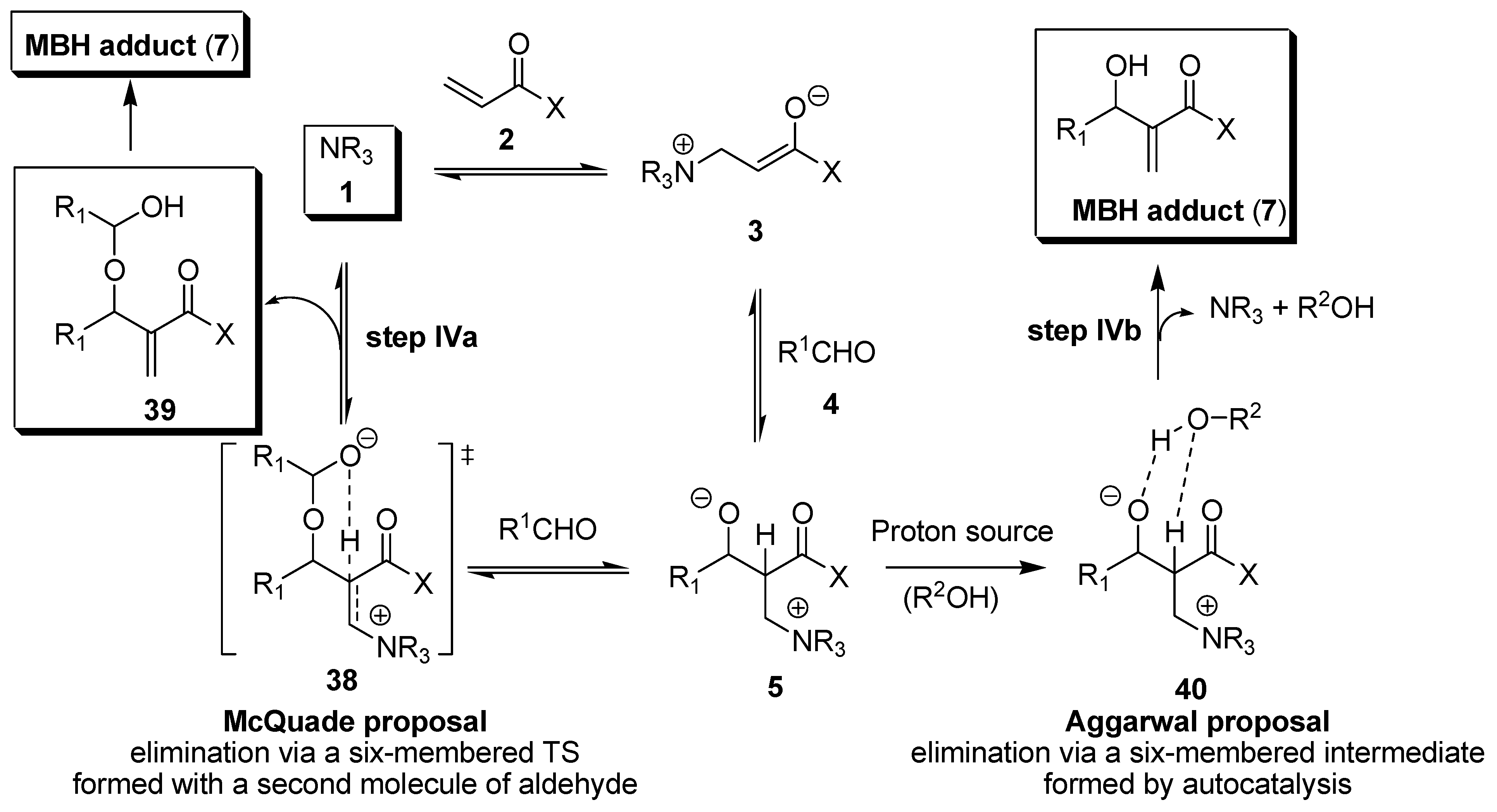
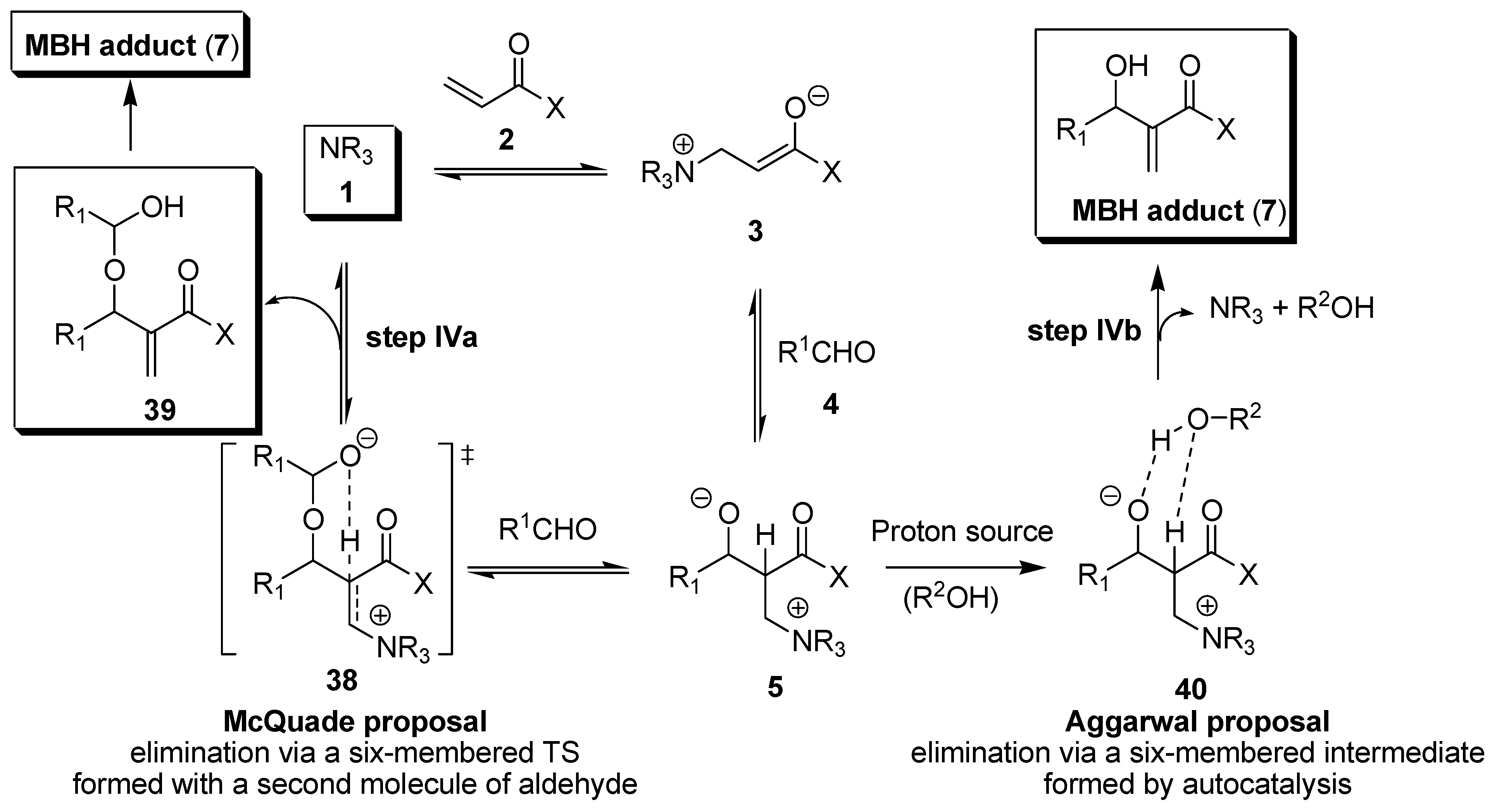
3. Recent Analysis of MBH Reaction Mechanism
4. Reaction Times, Yield and Stereochemistry of MBH Reactions
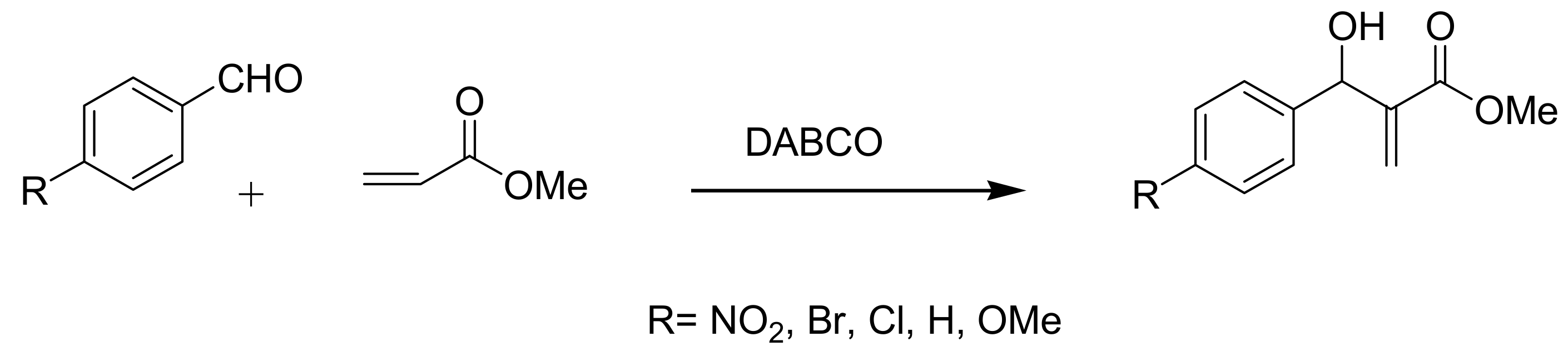
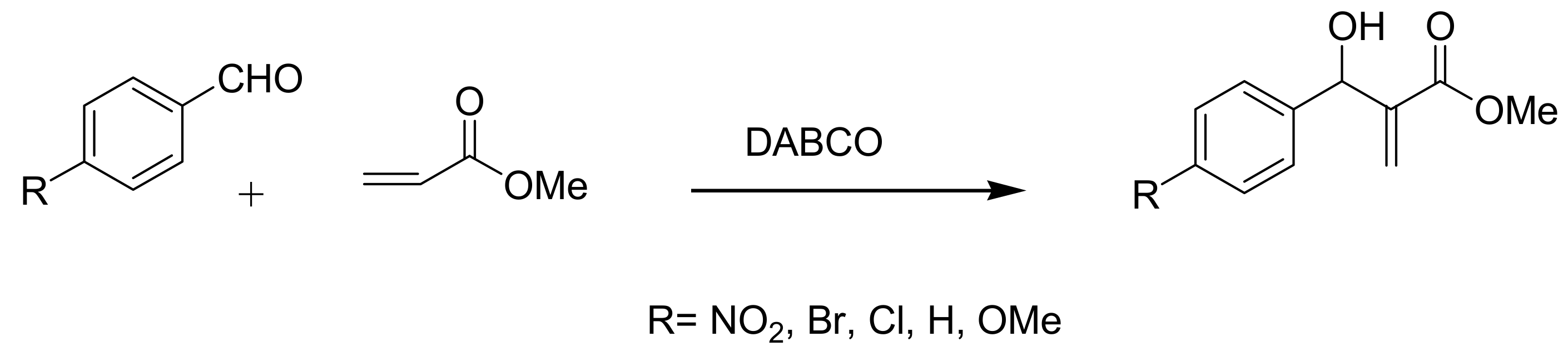
4.1. Stereochemistry of Michael acceptor
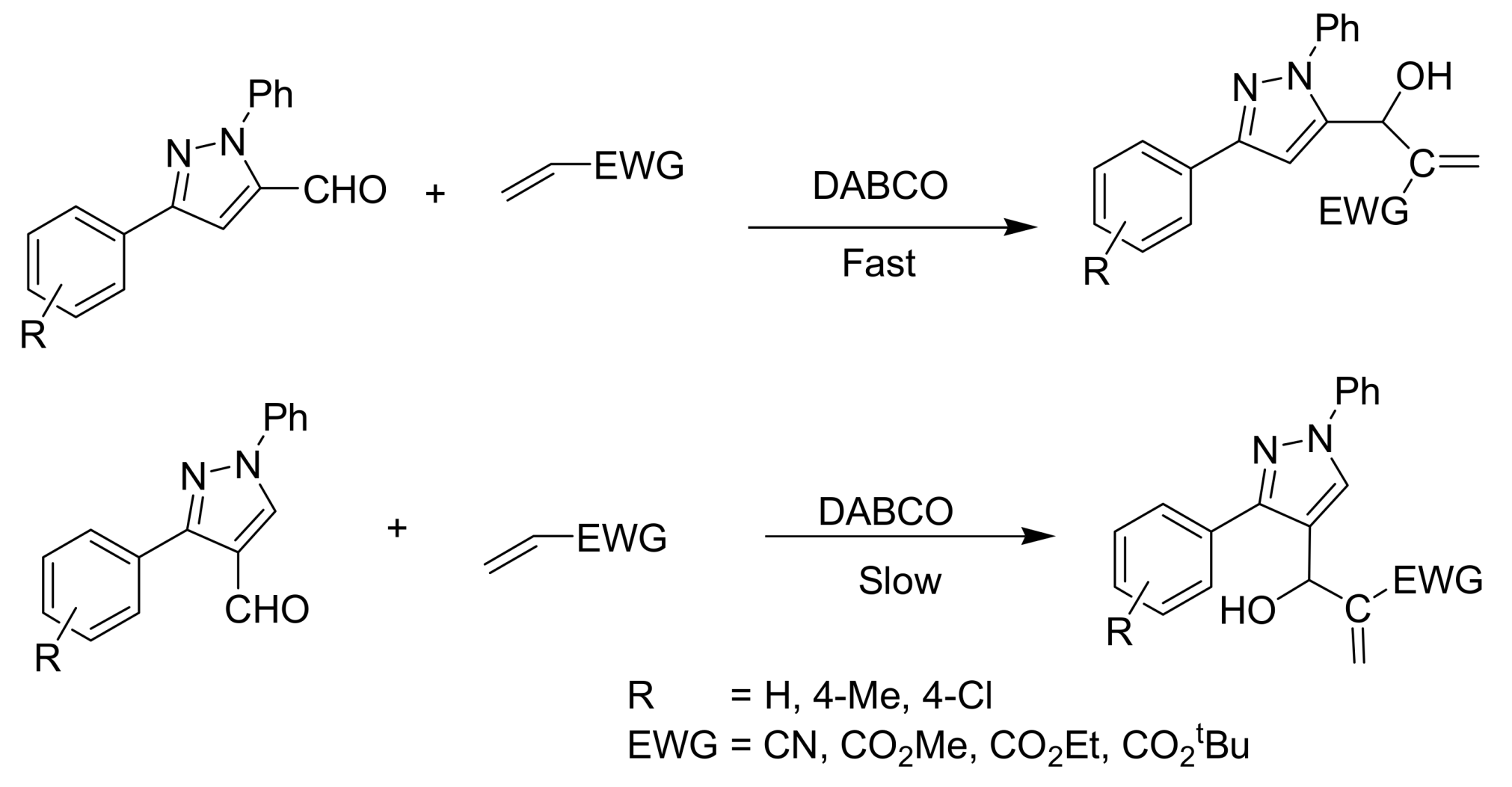
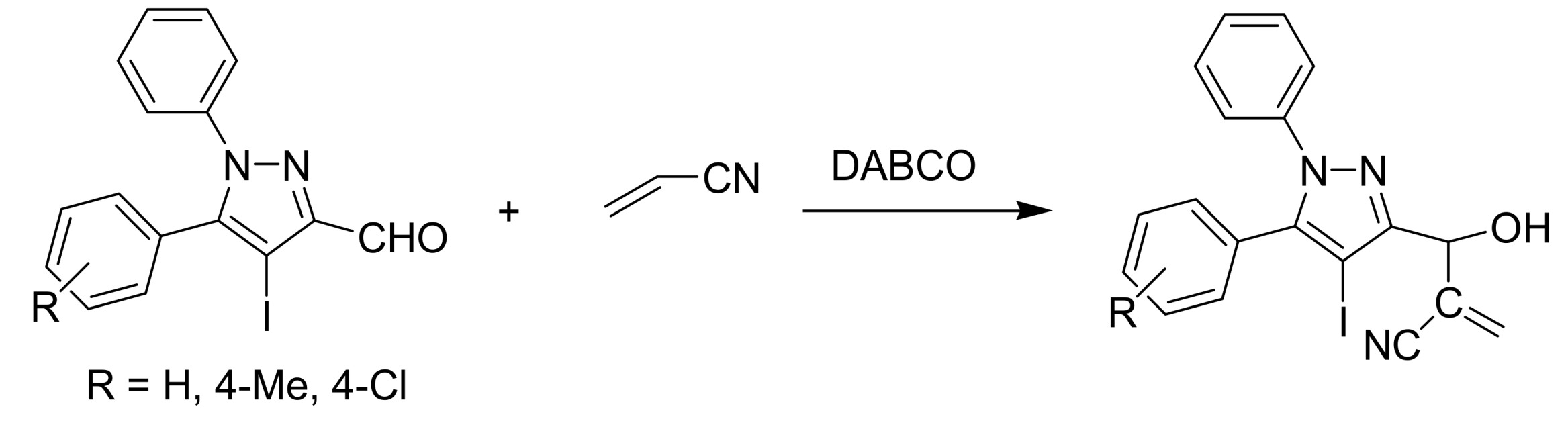
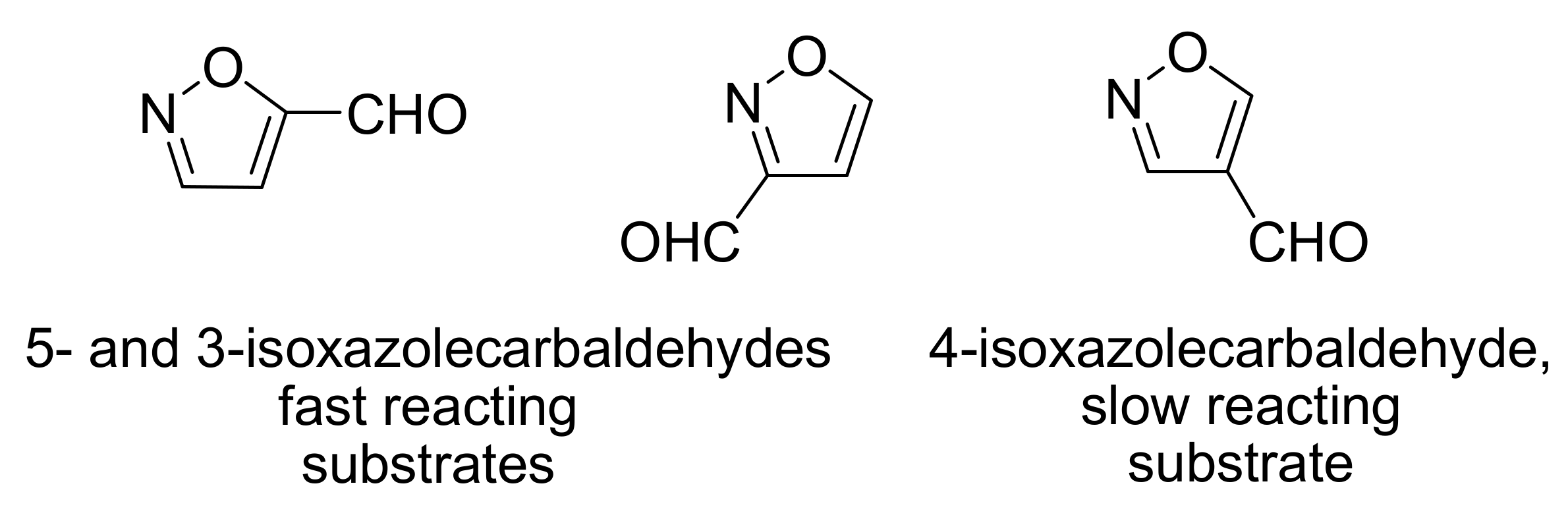
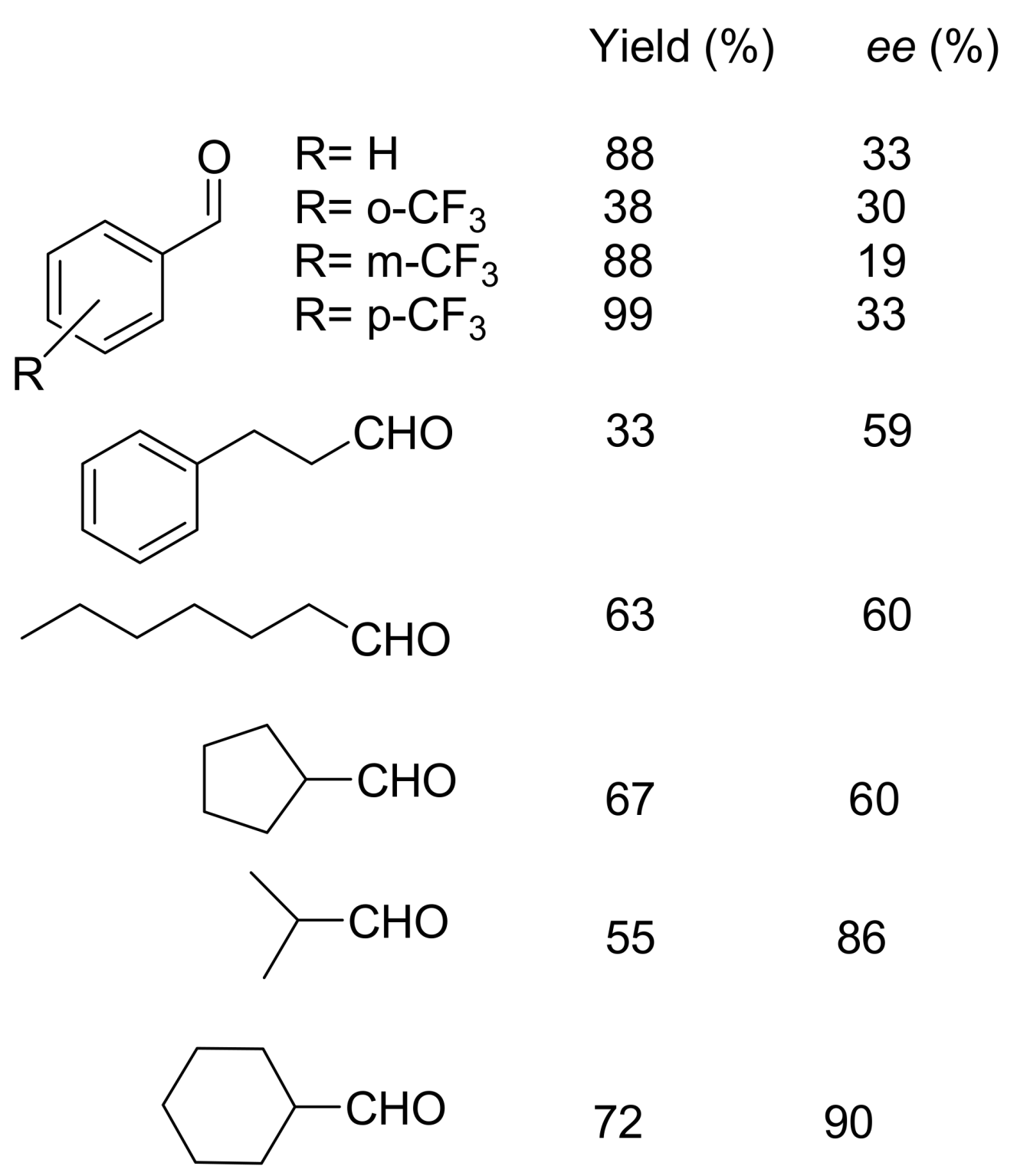
4.2. Studies on the reactivity of the formyl group
4.3. Three-component and one-pot MBH reactions
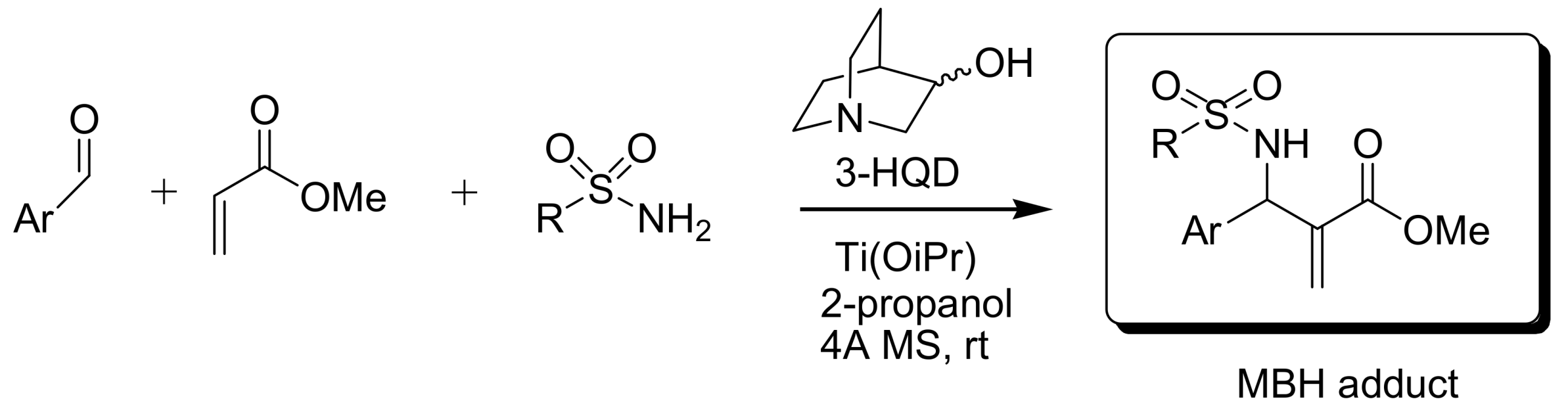
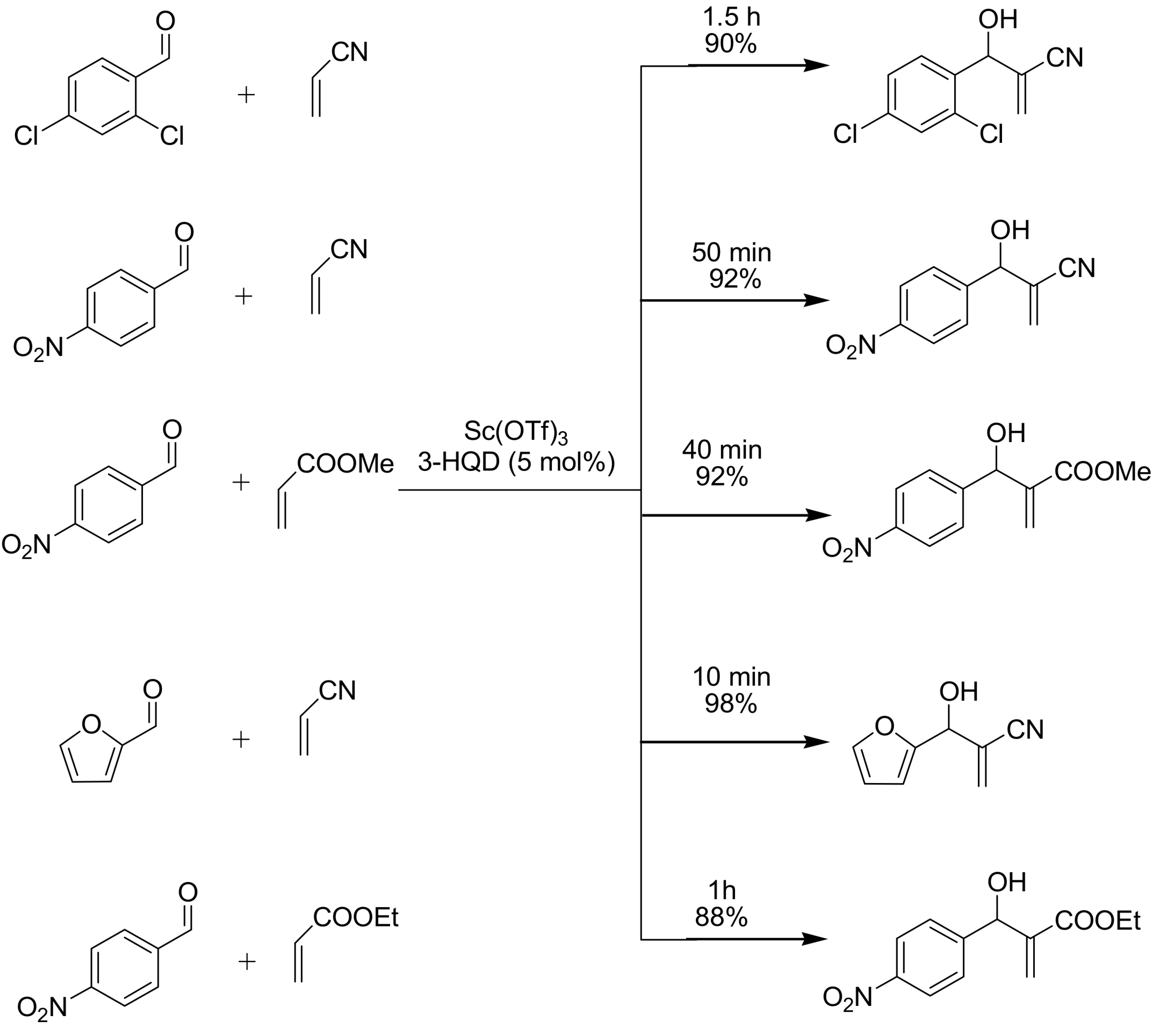
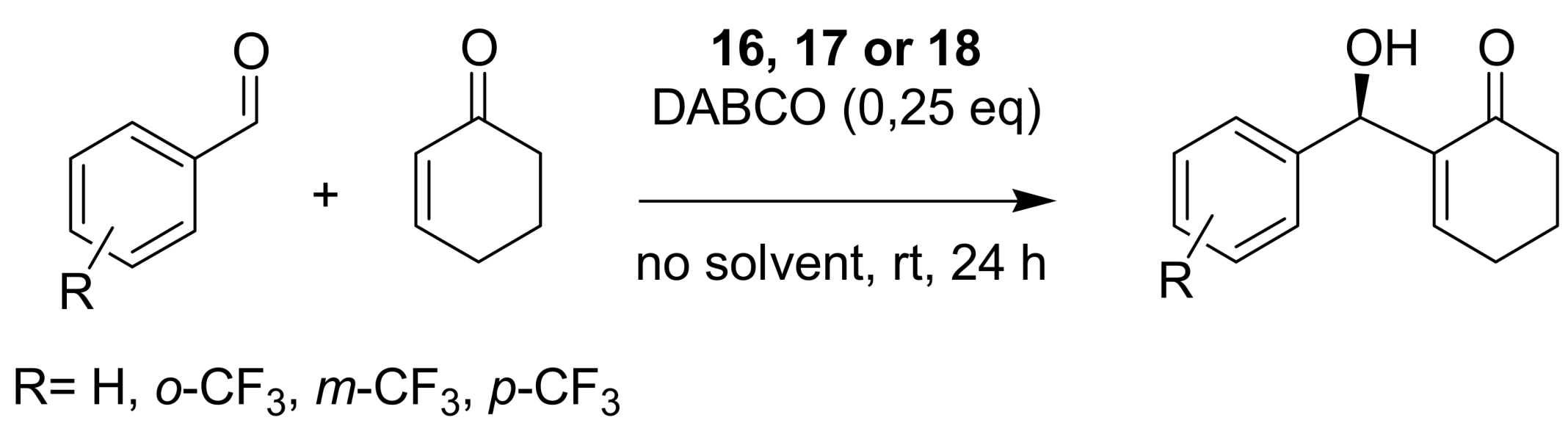
4.4. Catalytic asymmetric induction for MBH reactions
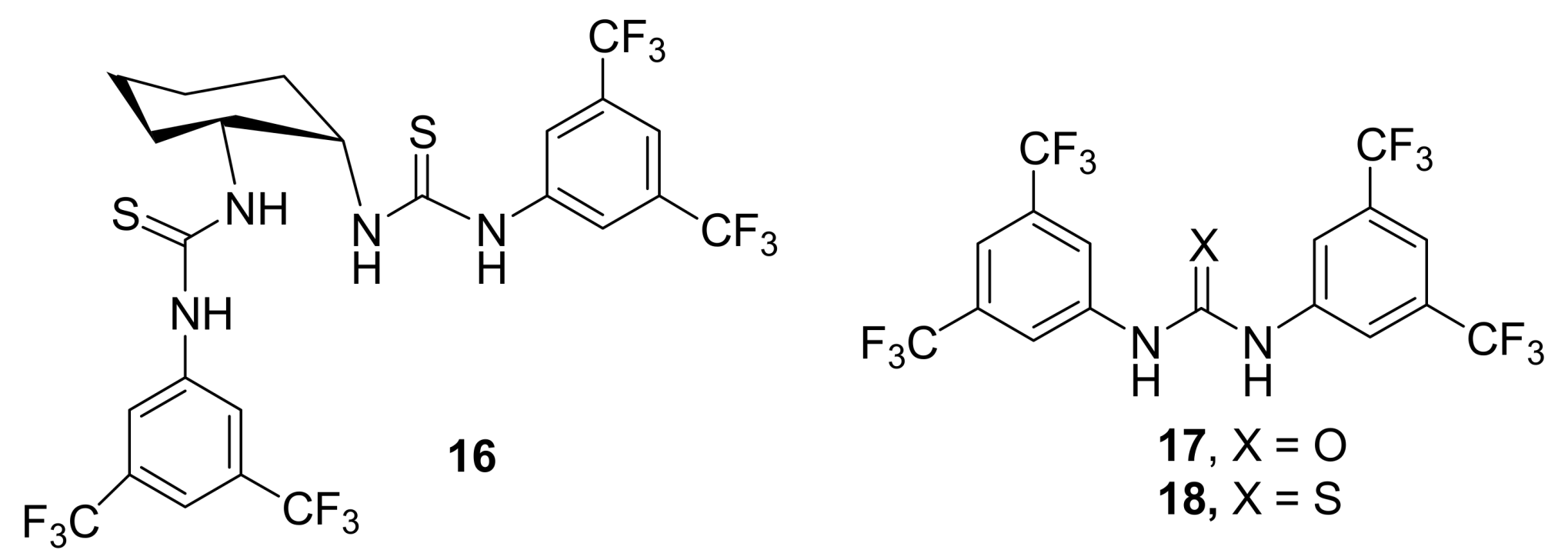
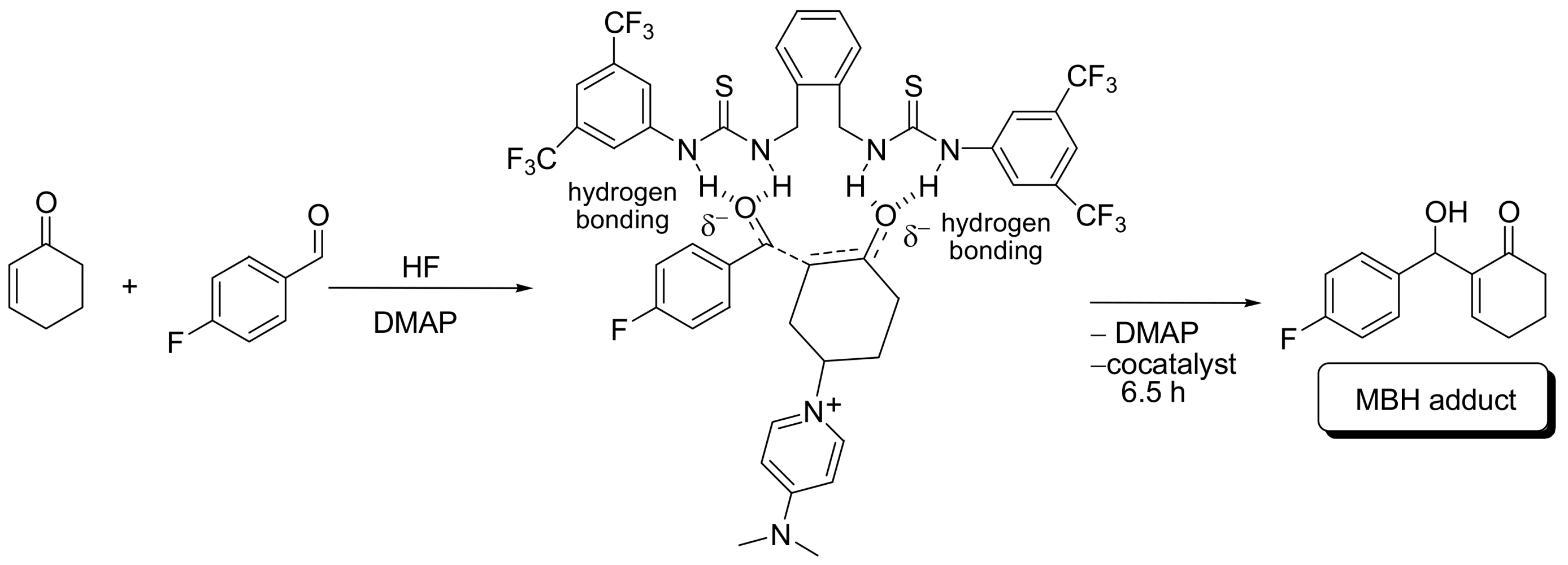
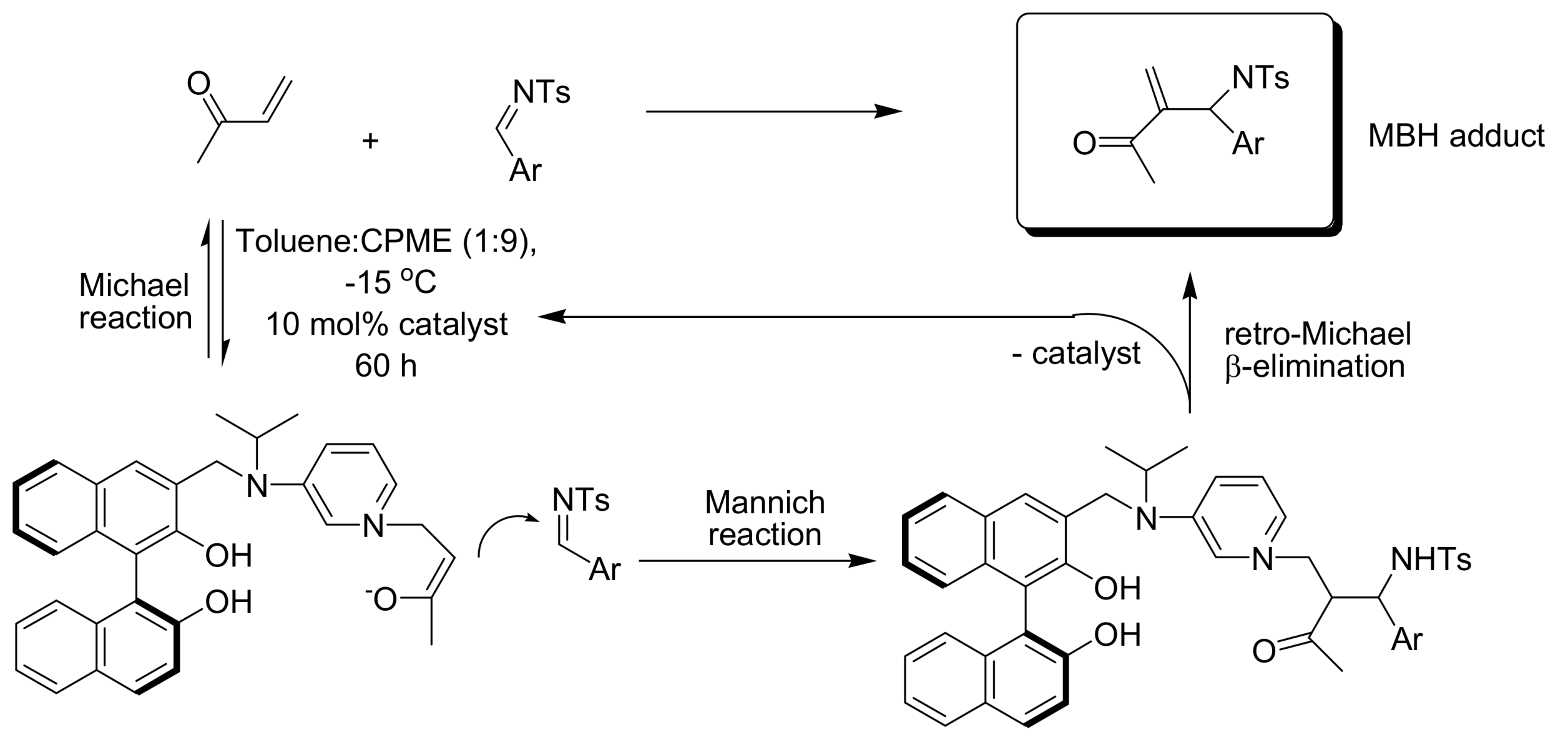
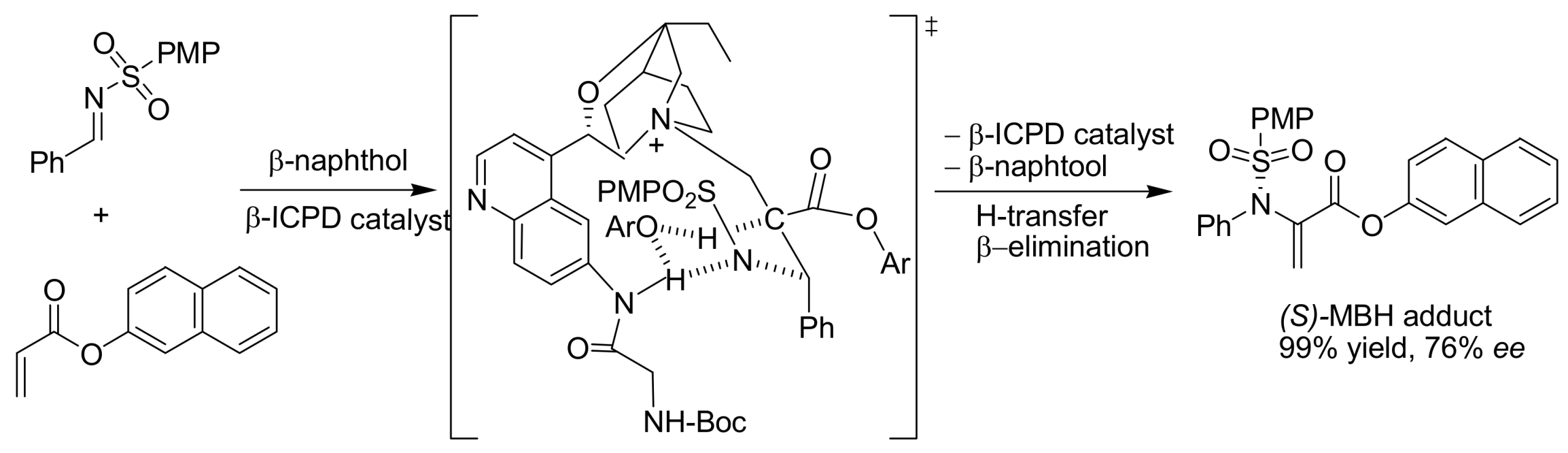
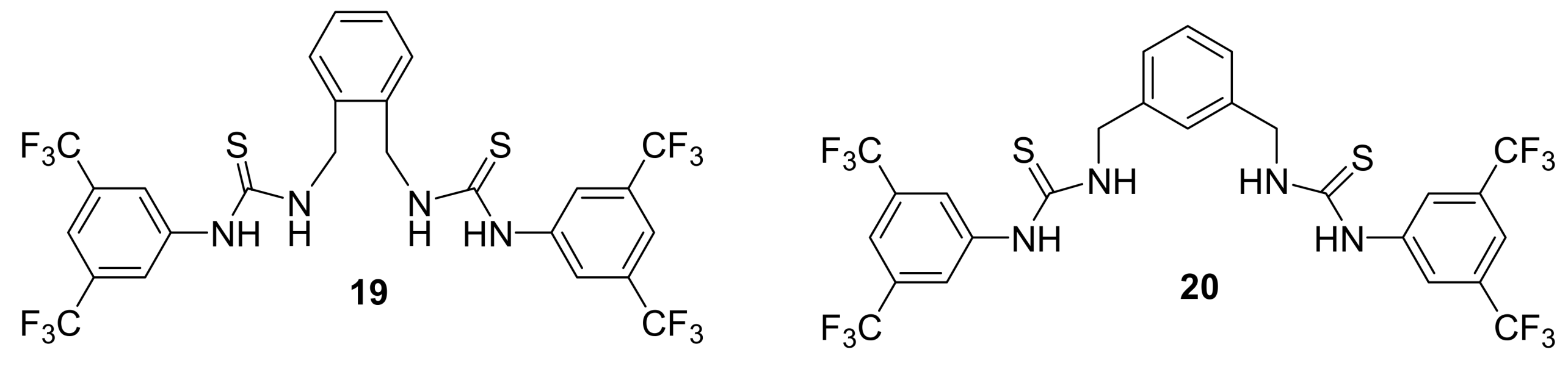
4.4.1. Asymmetric induction using chiral (thio)ureas
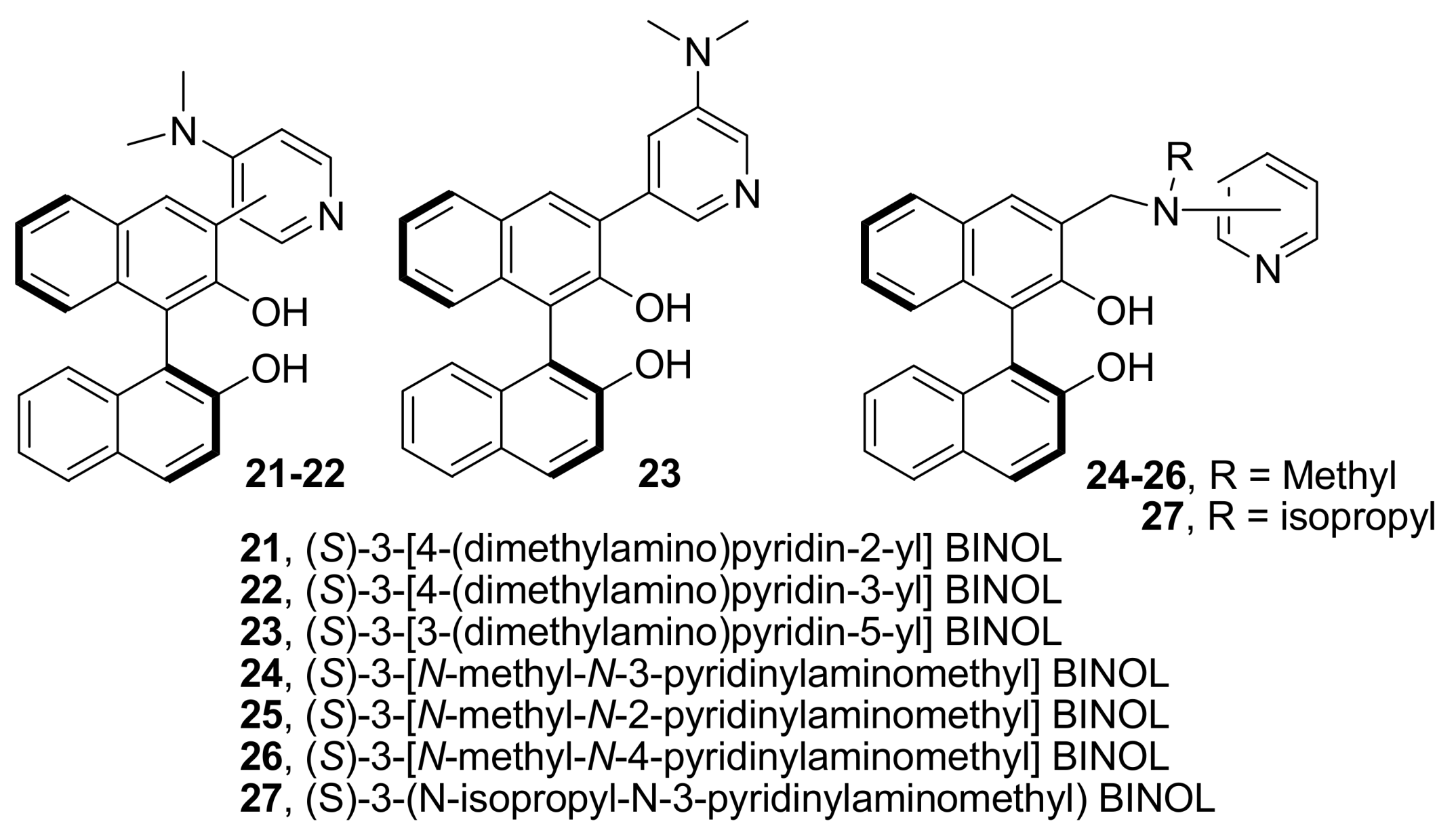
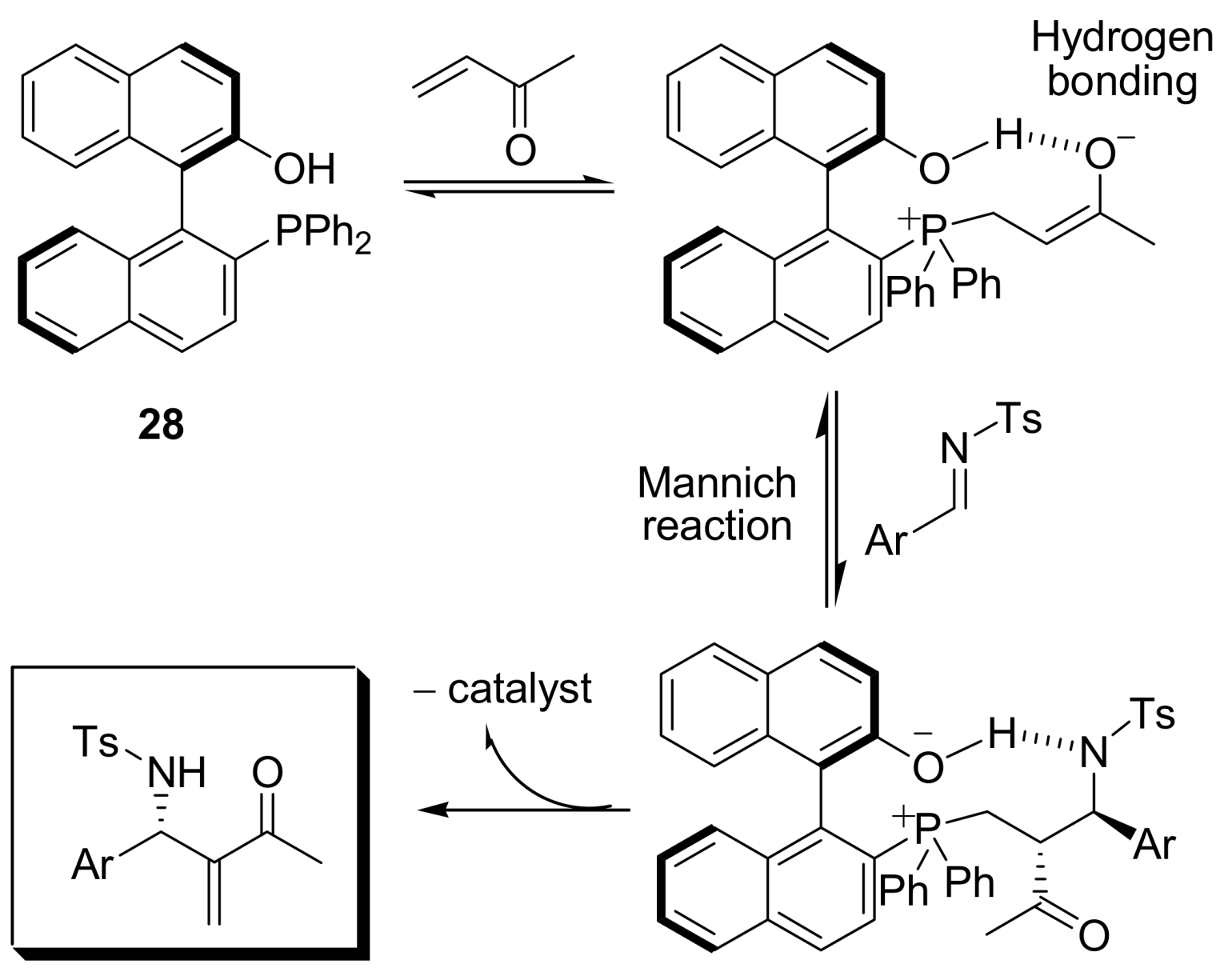
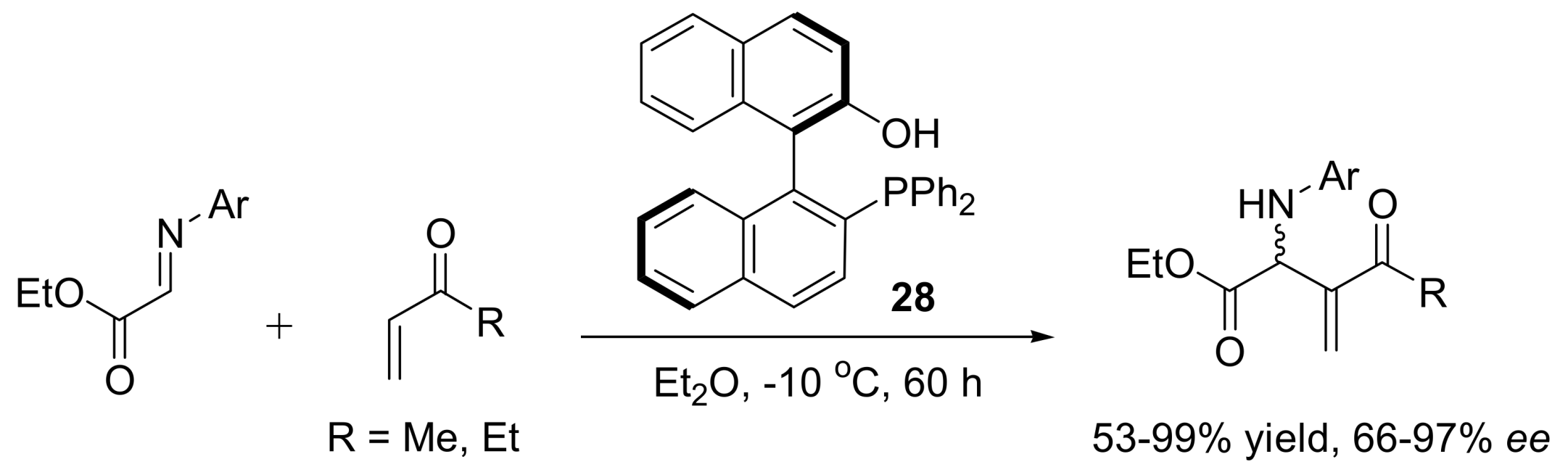
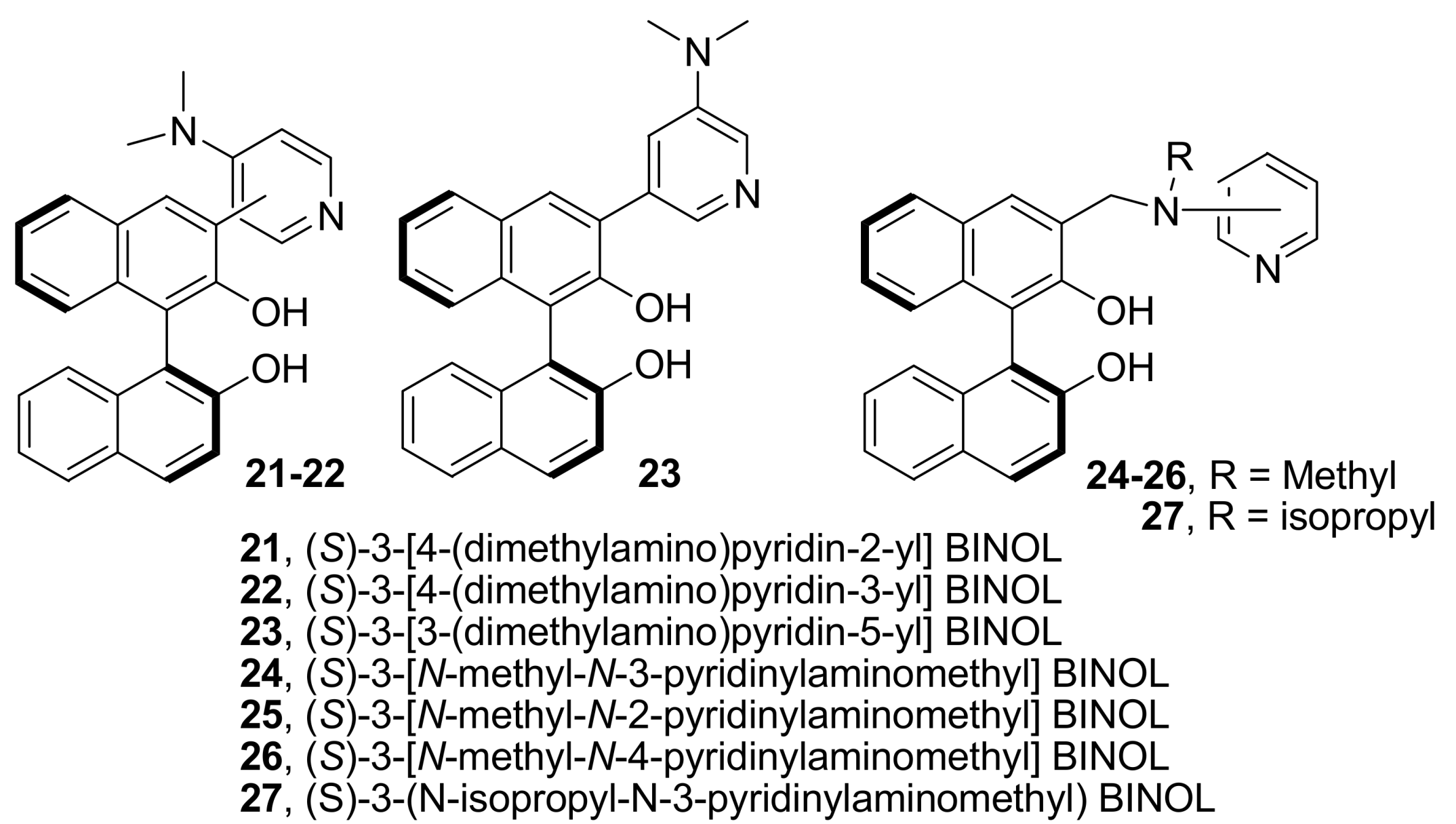
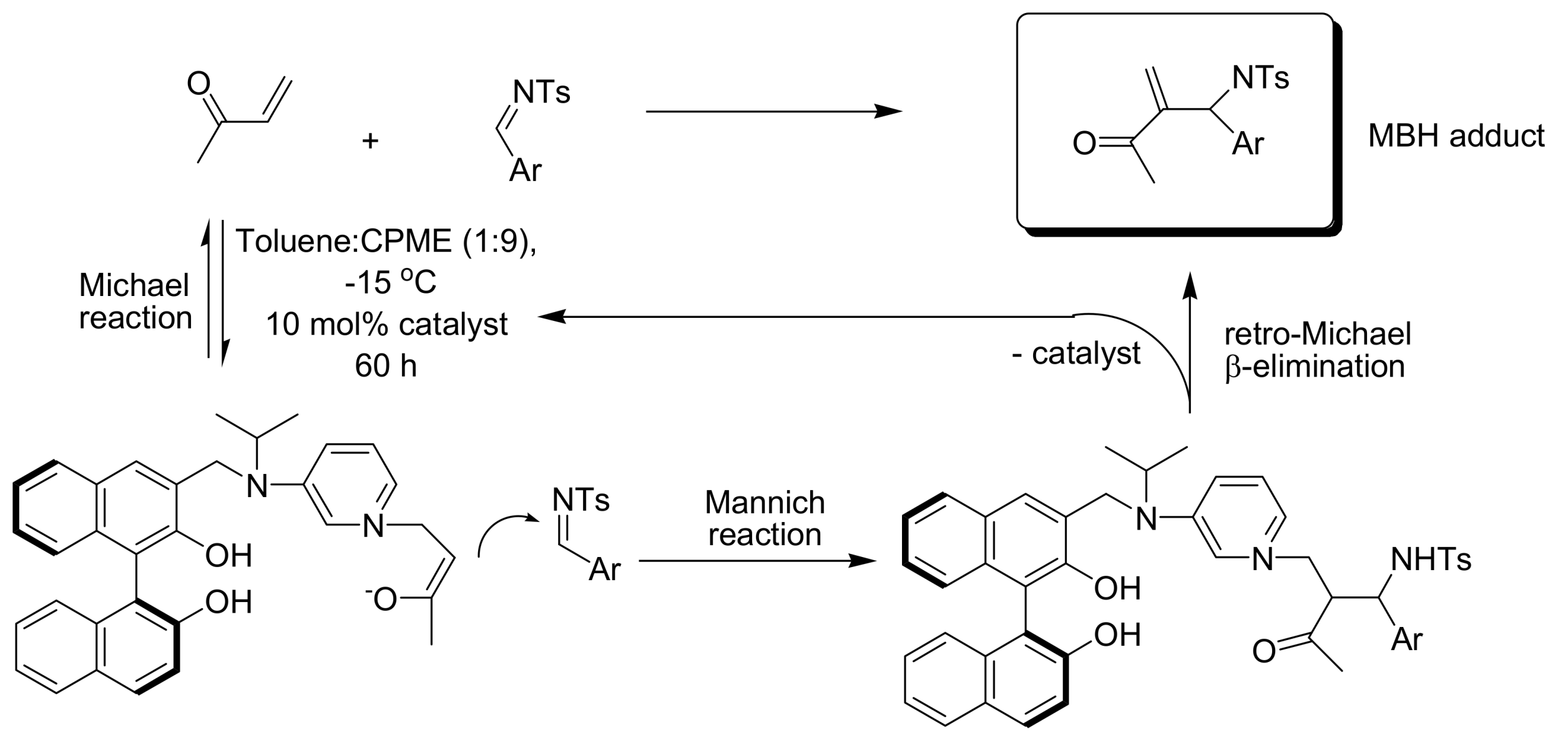
4.4.2. Asymmetric induction using biphenols
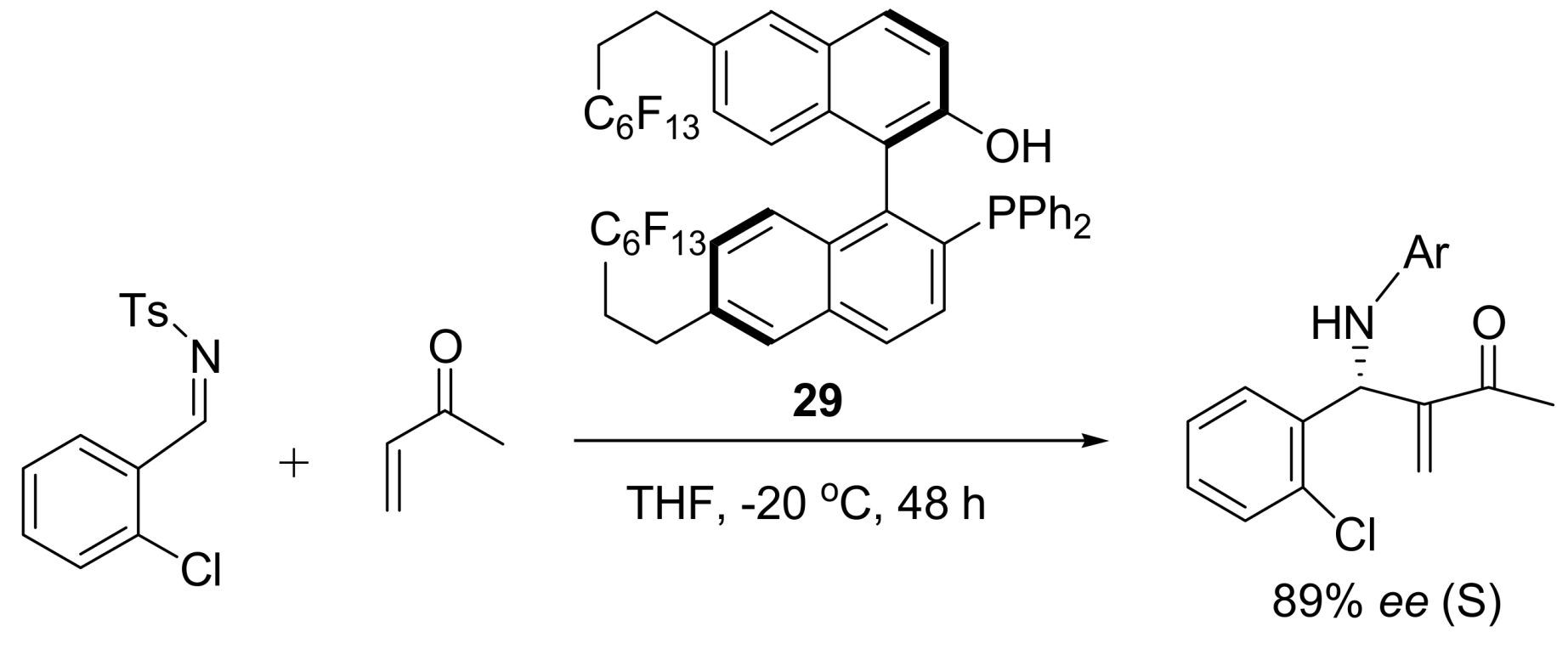
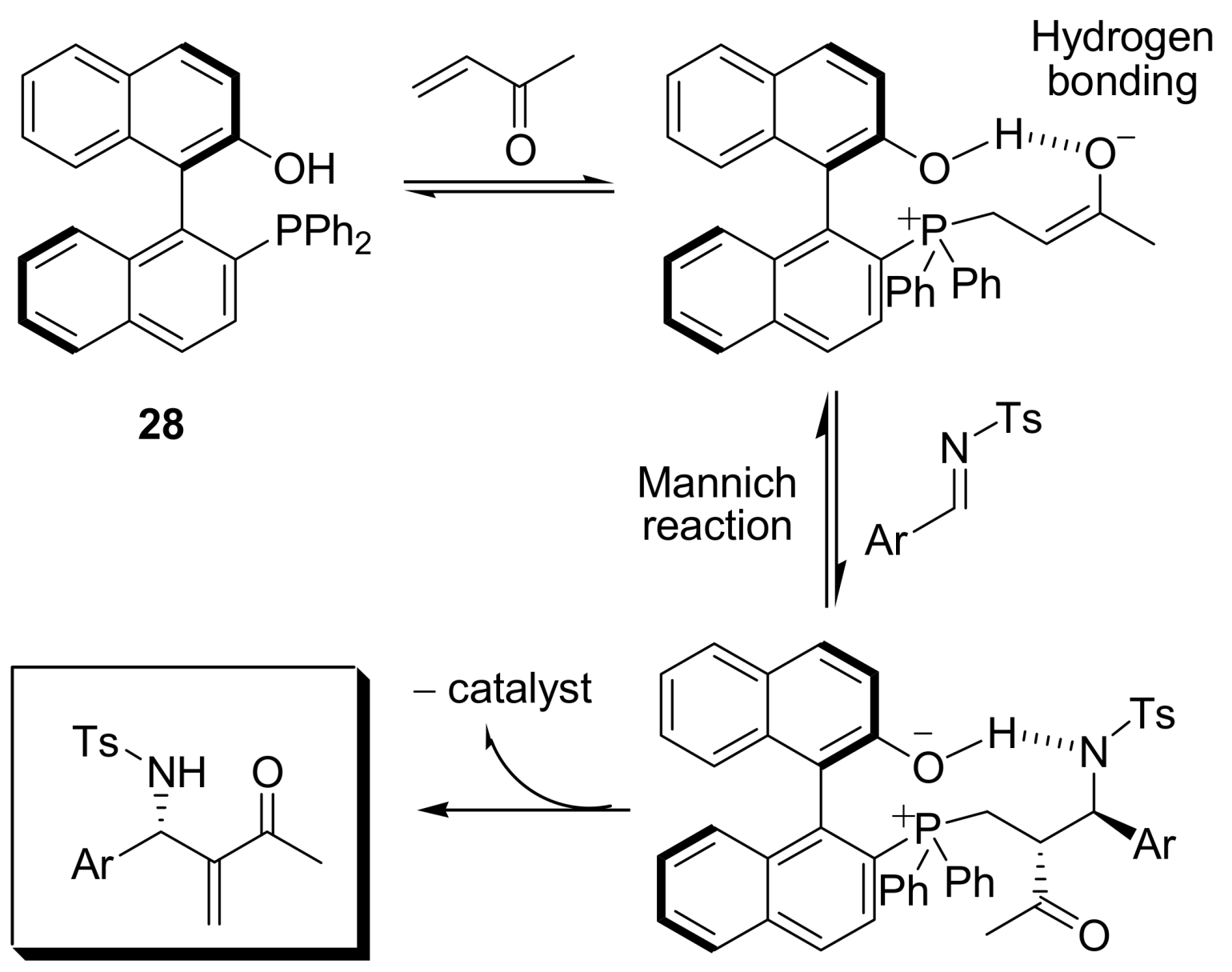
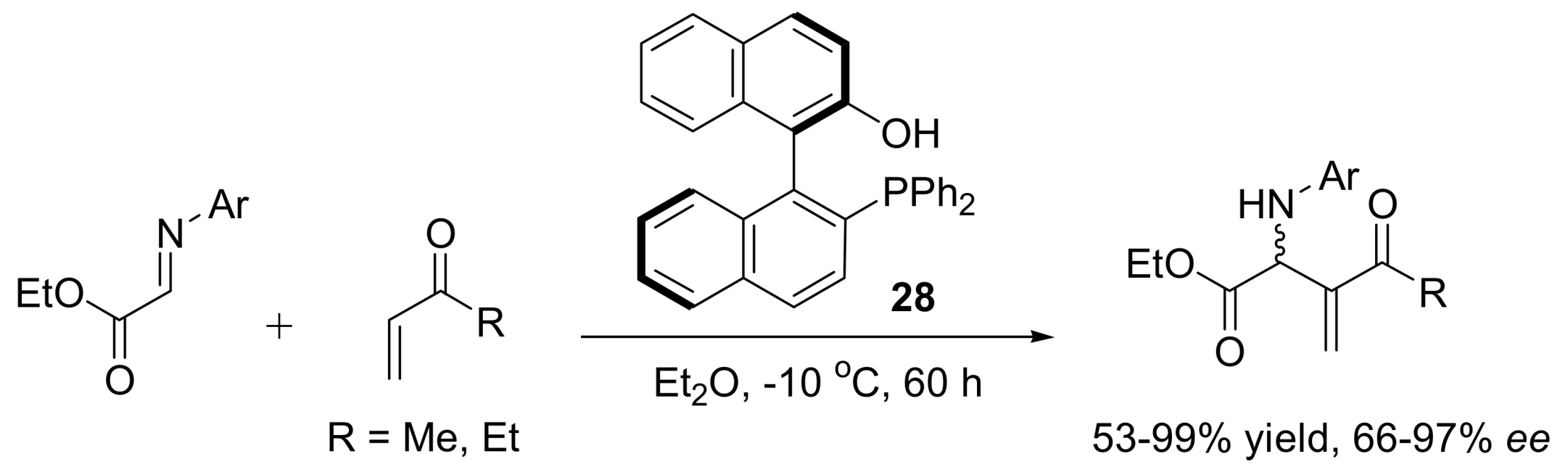
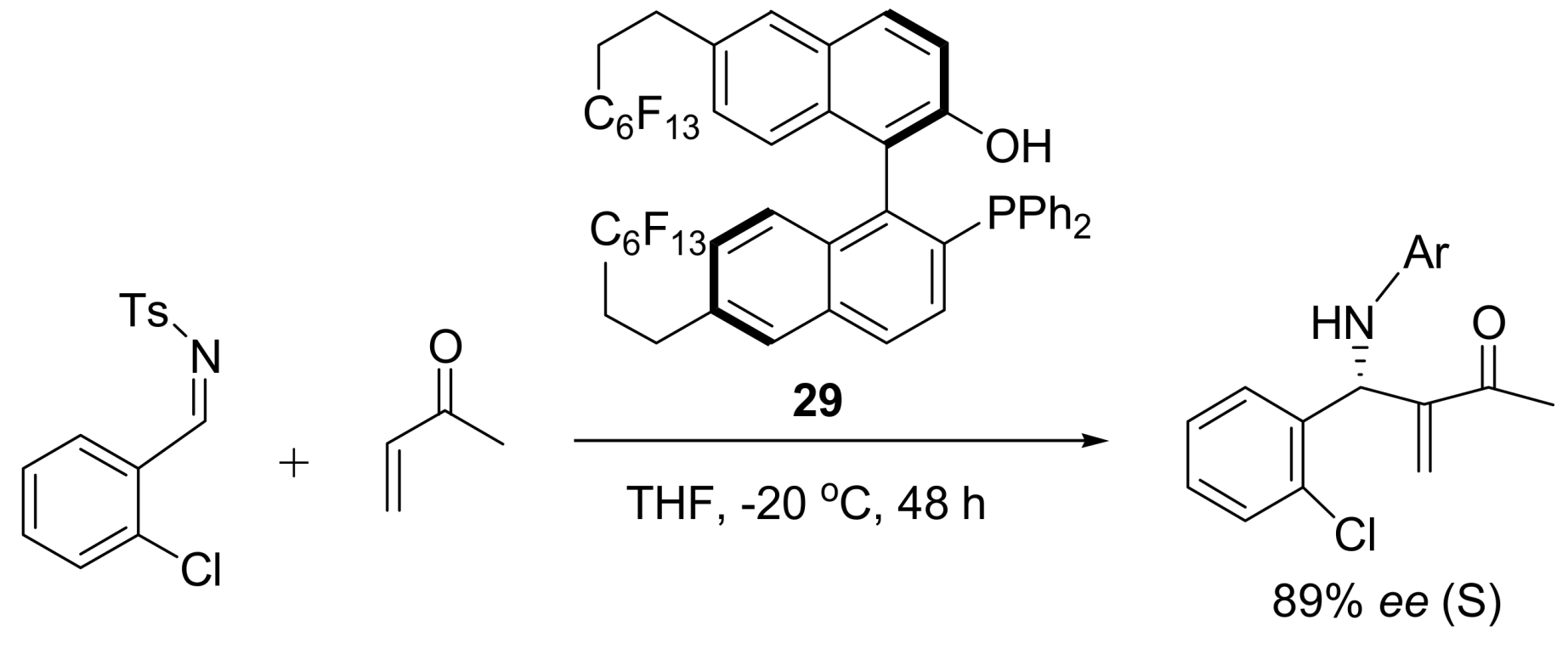
4.4.3. Asymmetric induction using phosphines
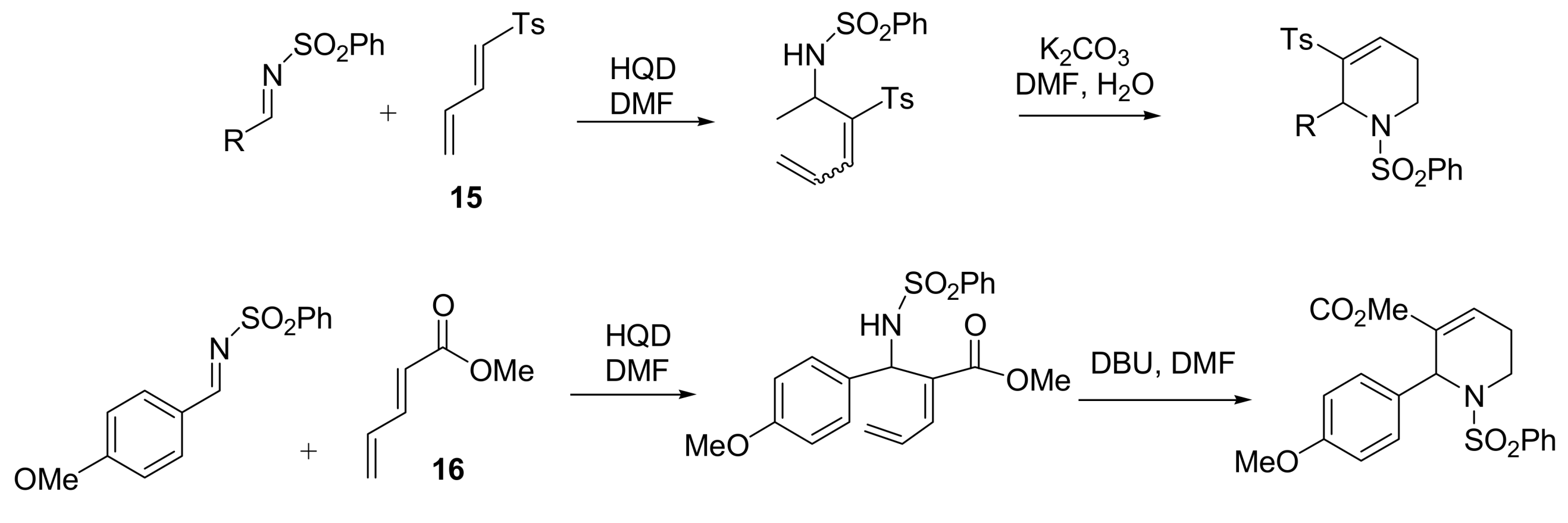
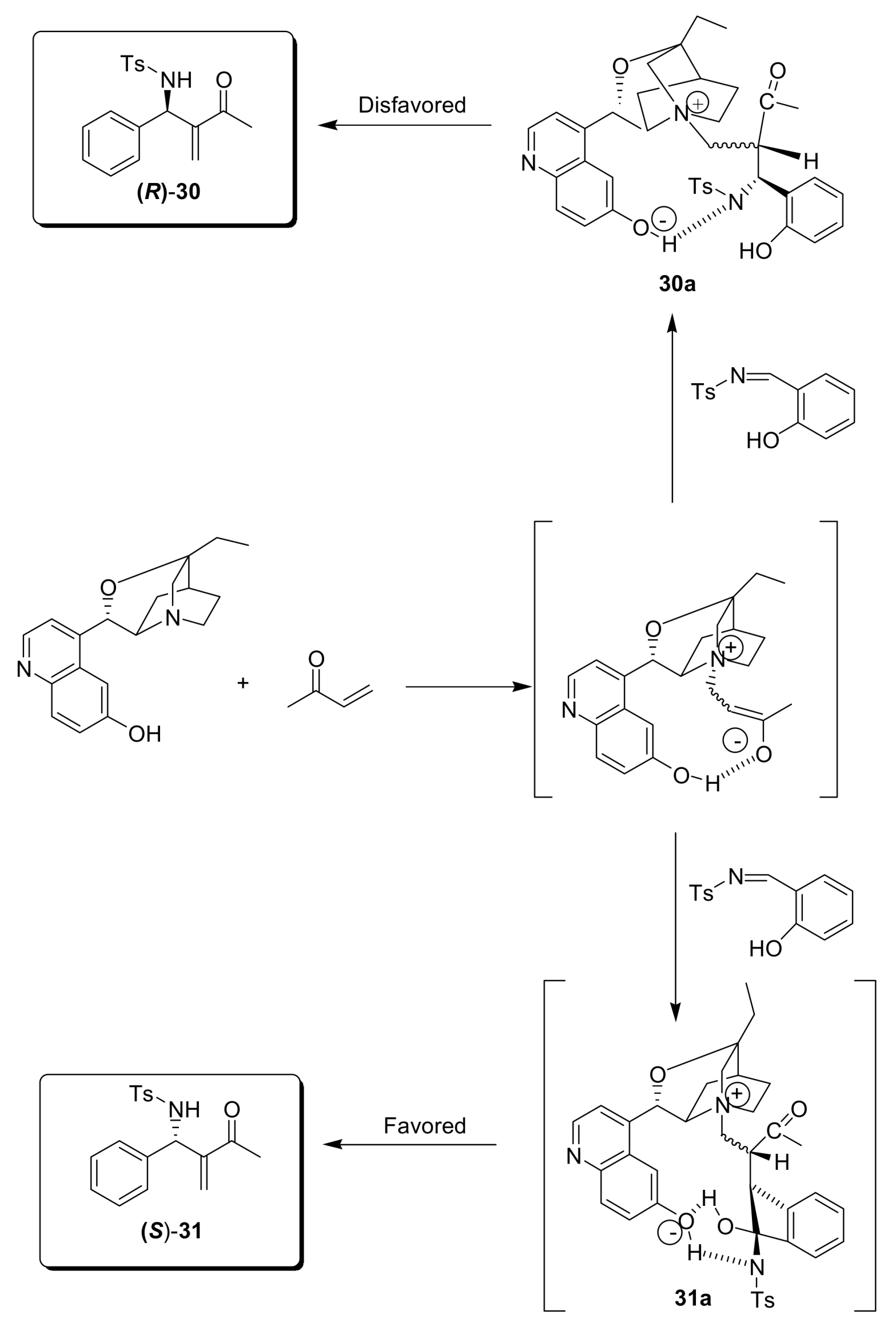
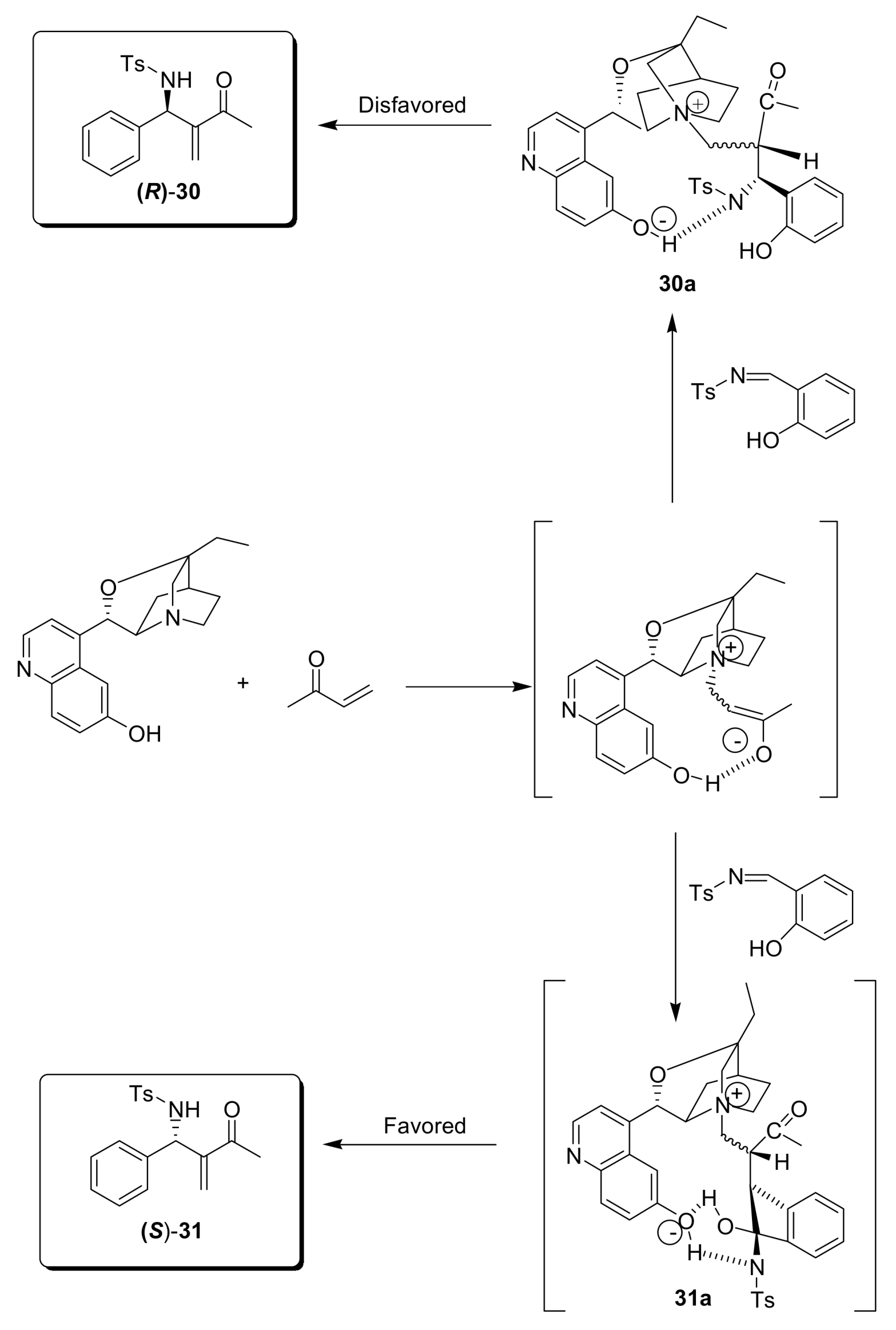
4.4.4. Asymmetric induction using chiral amines
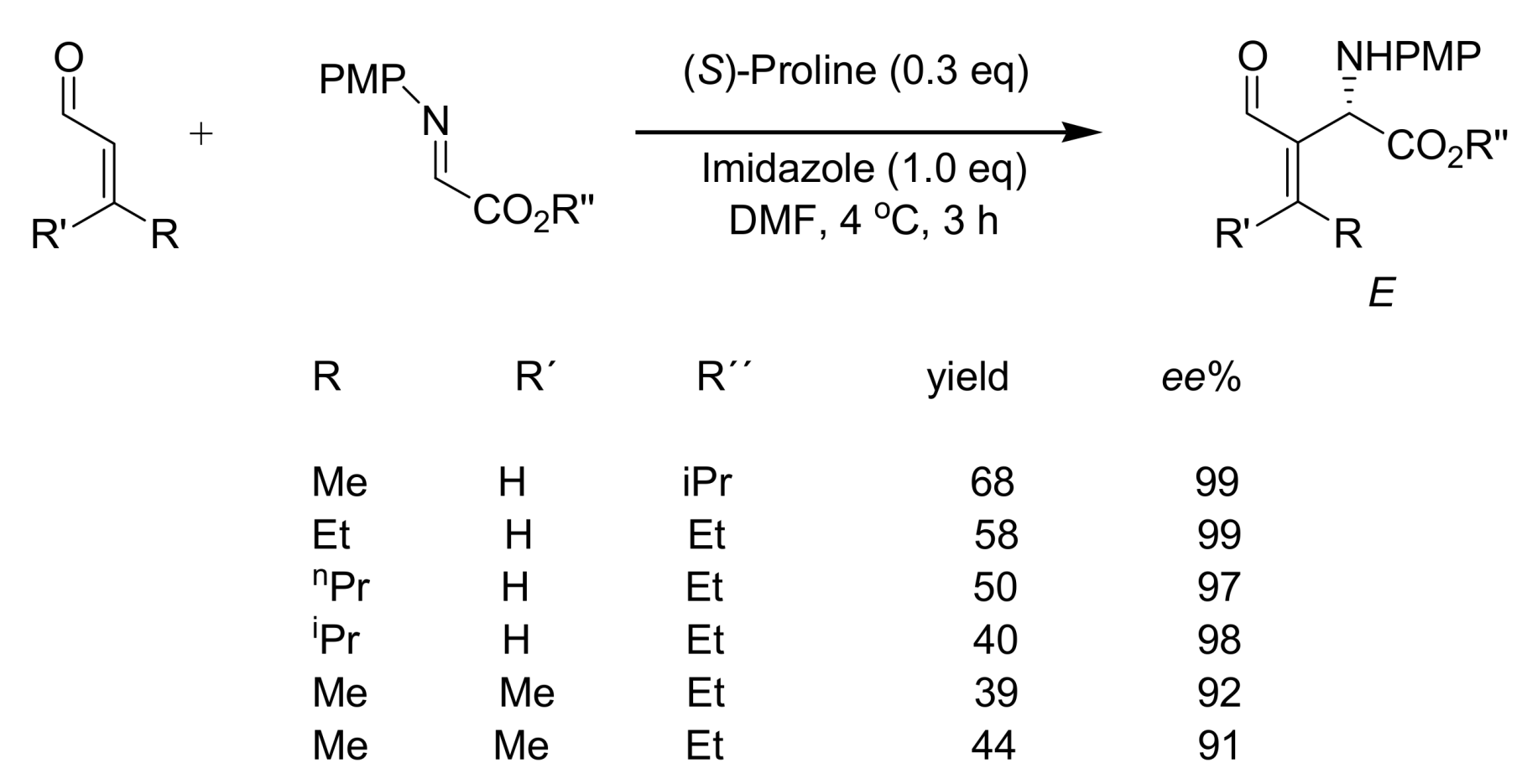
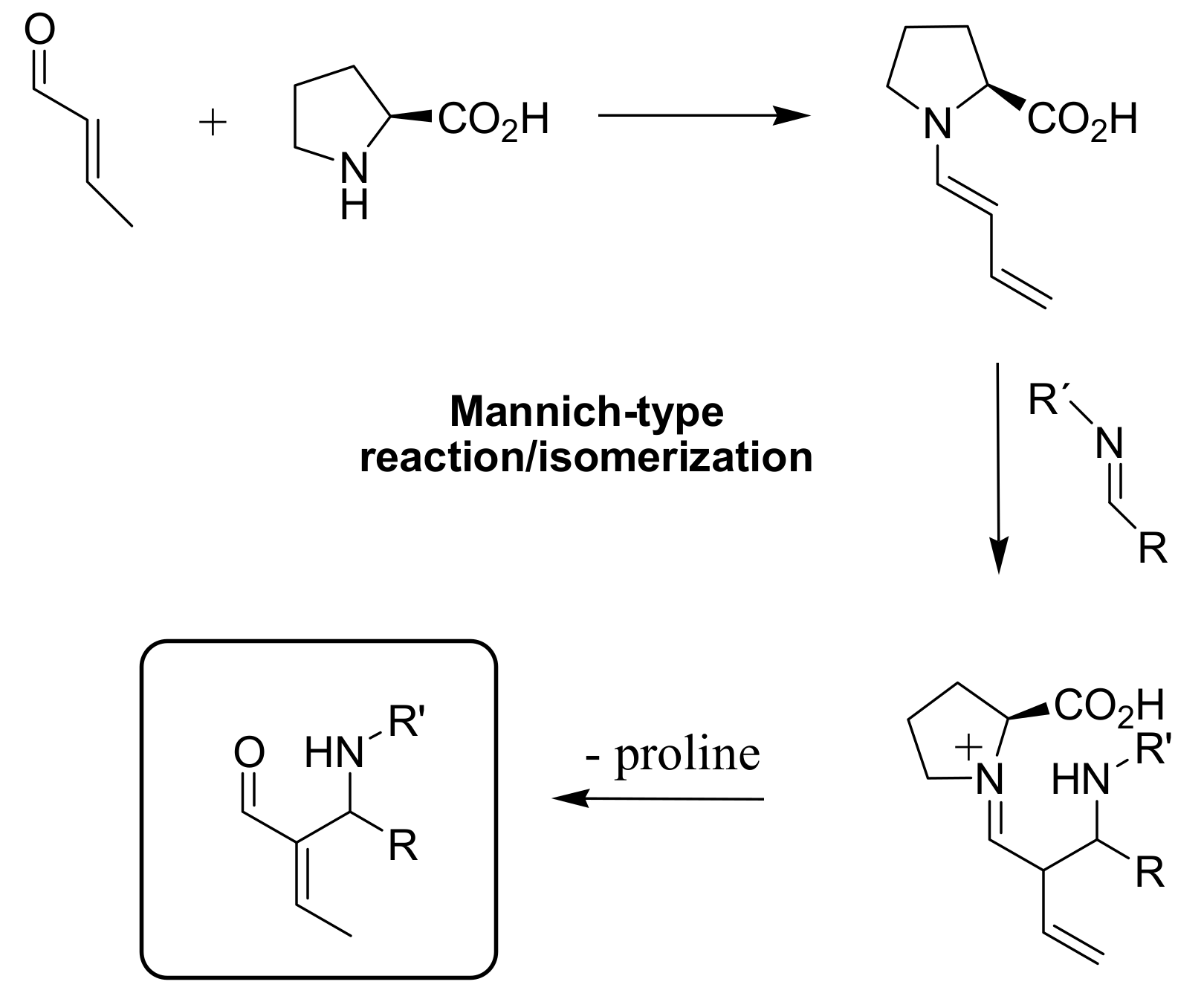
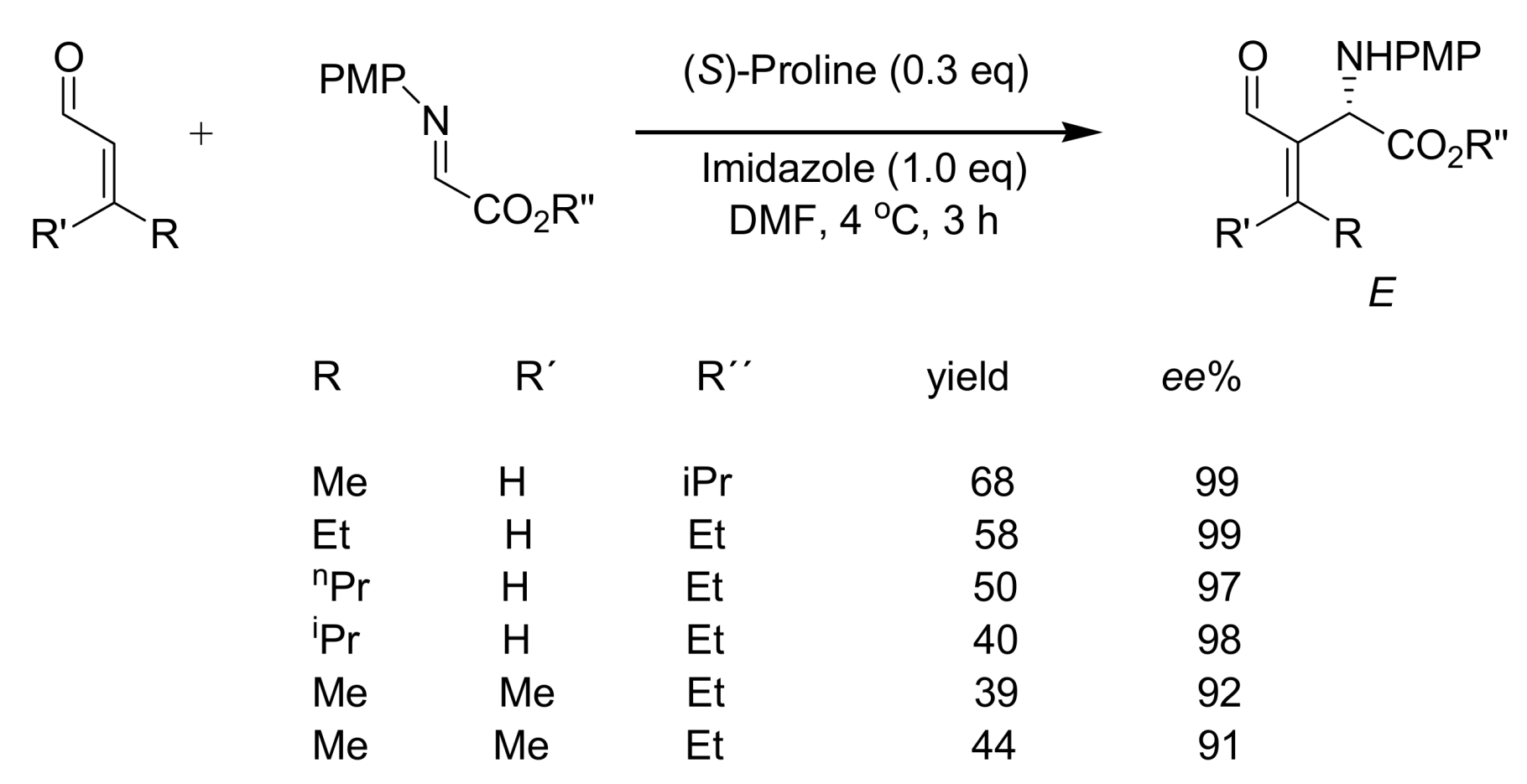
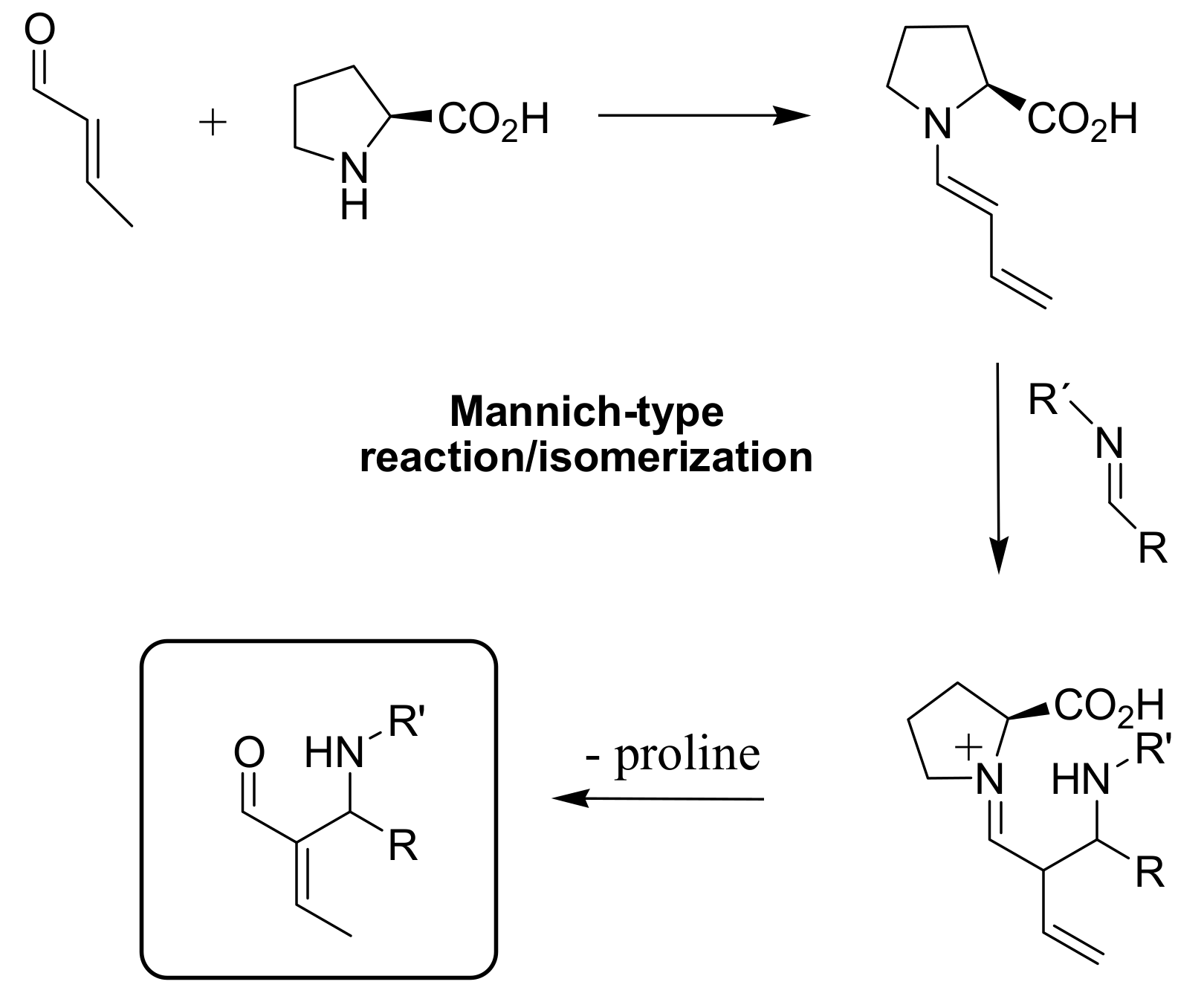
4.5. Amino acids used as catalysts for MBH reaction
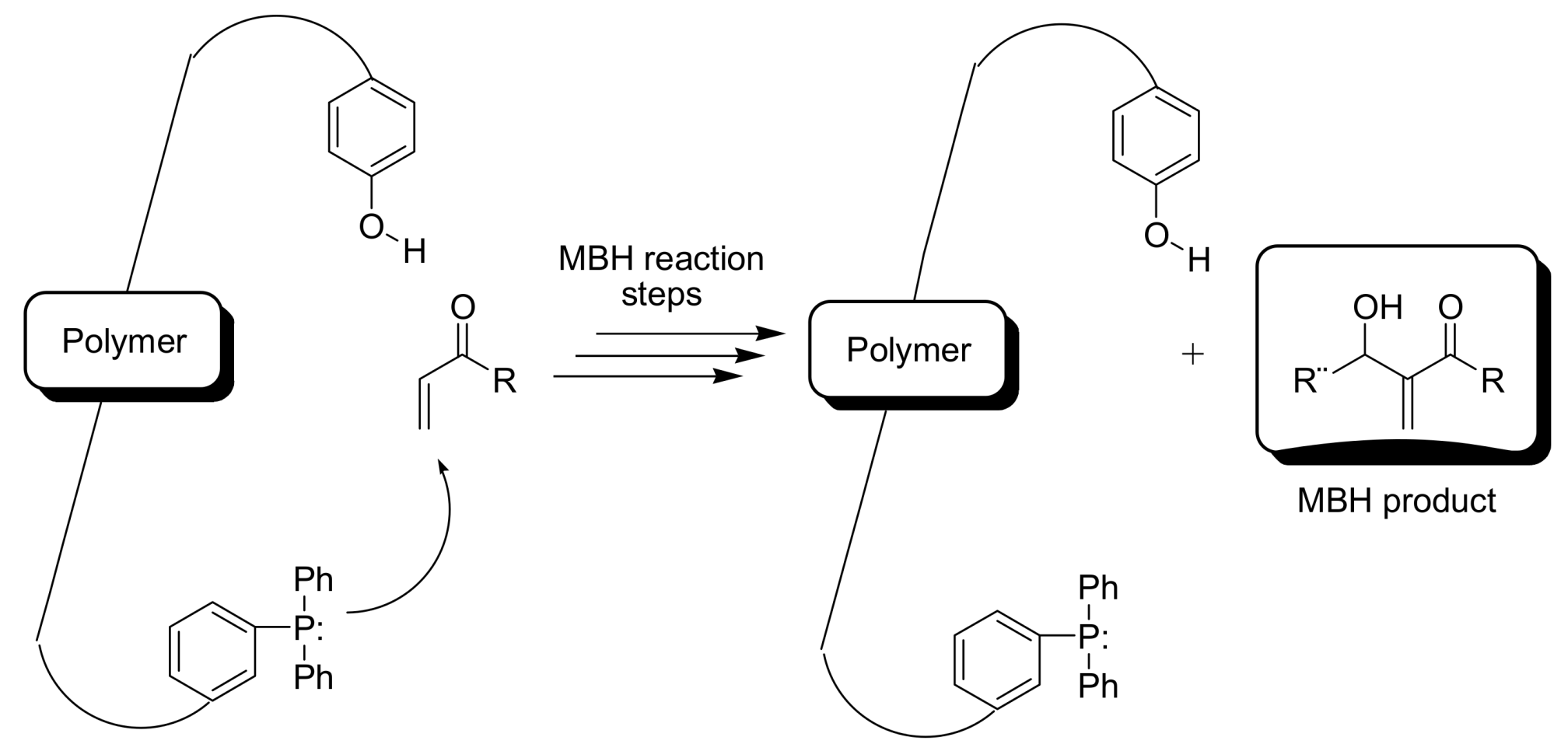
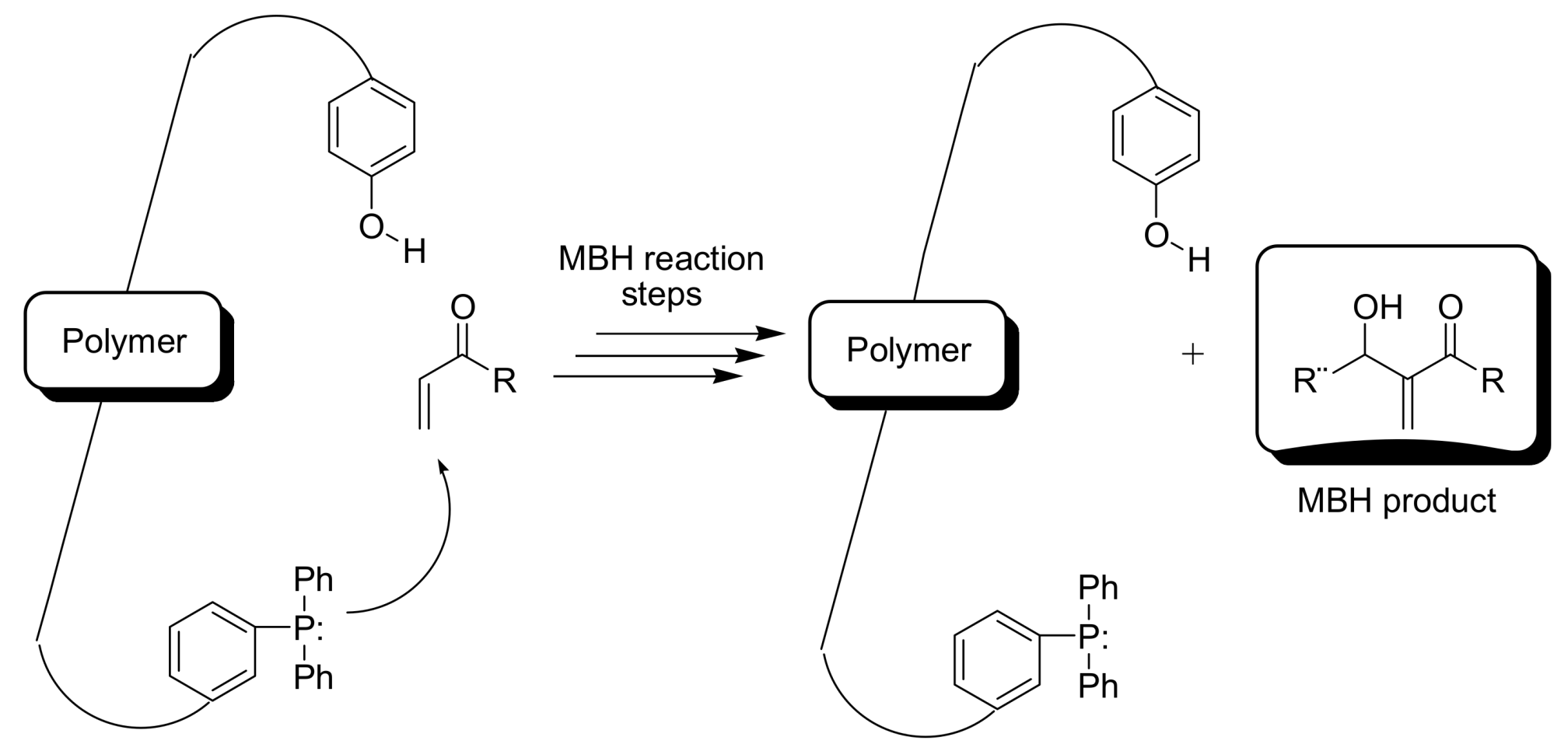
4.5.1. Polymers as organocatalysts
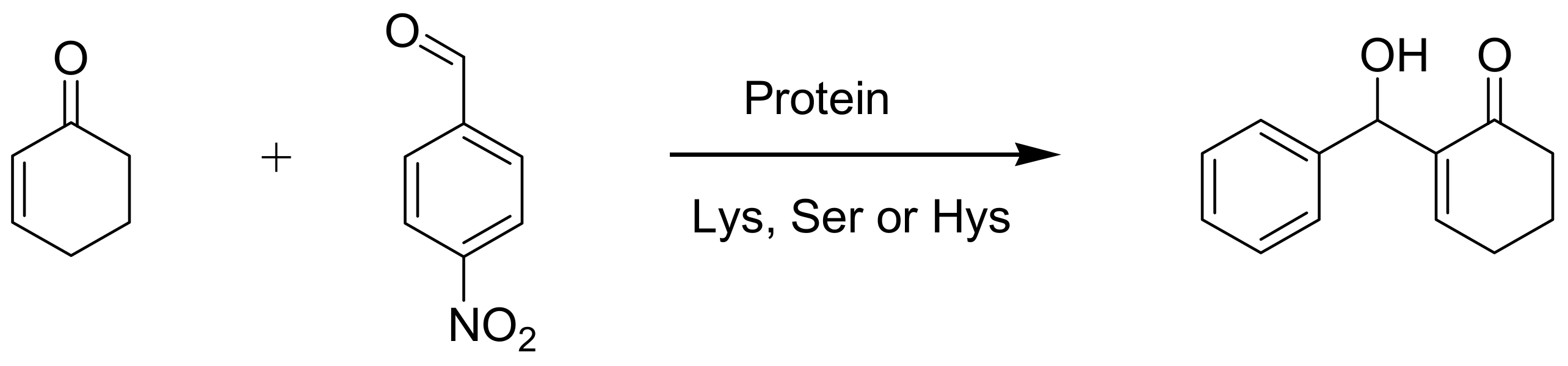
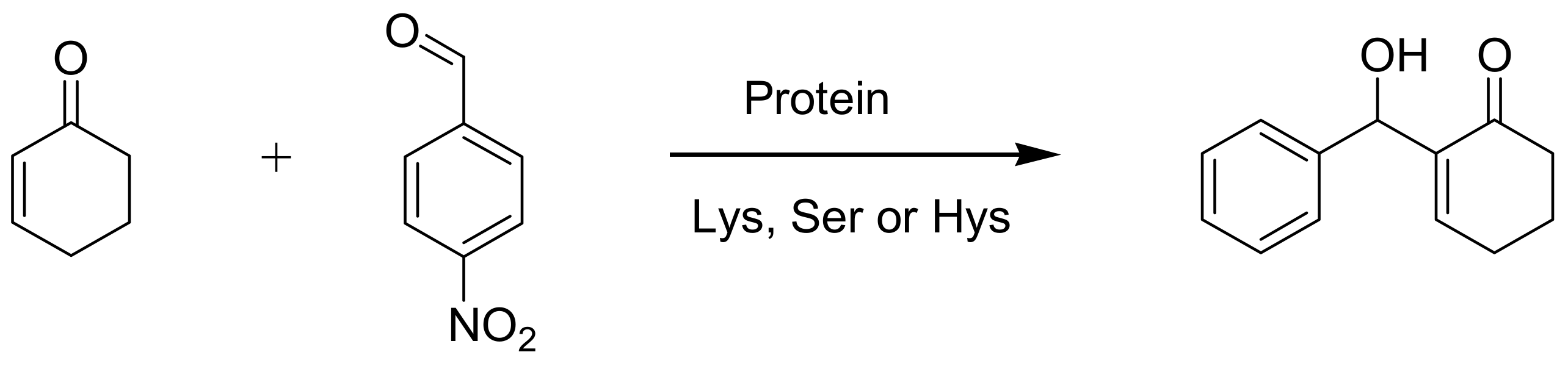
4.6. Enzymes as catalysts
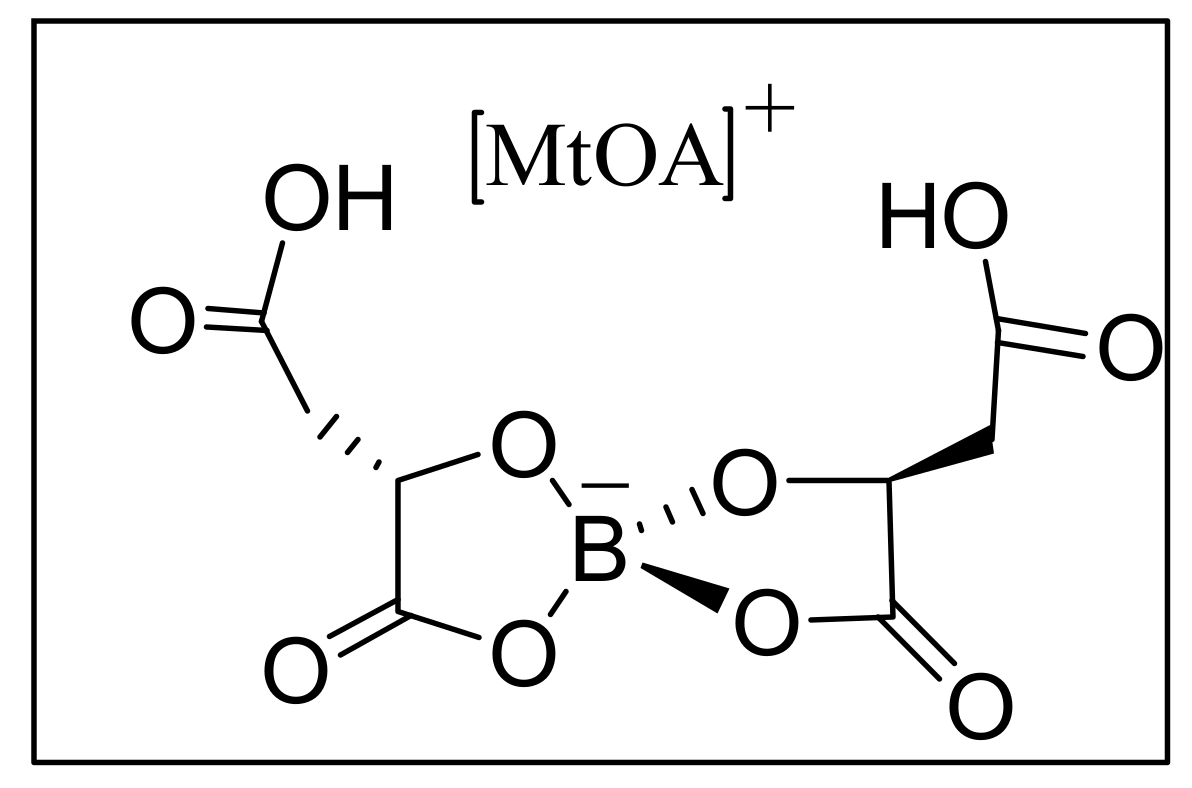
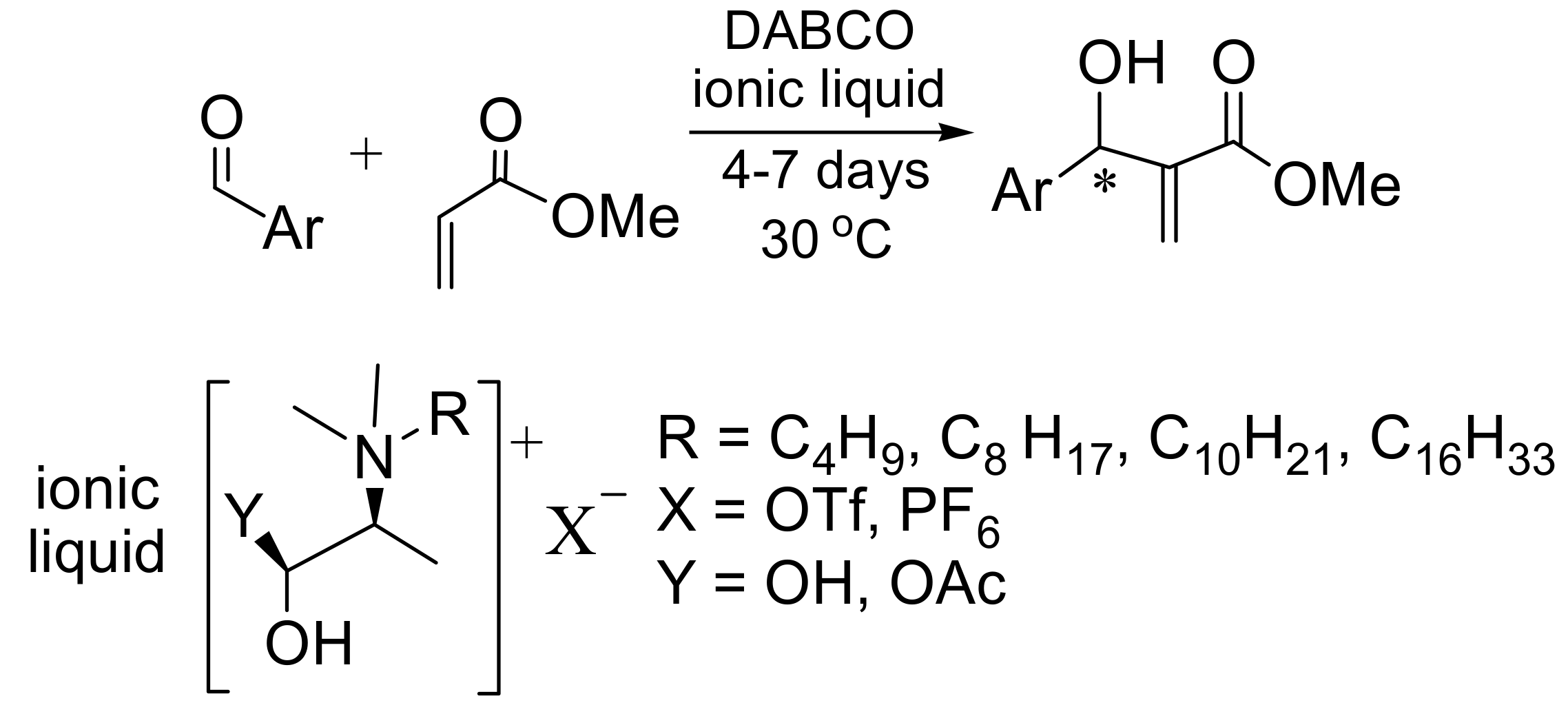
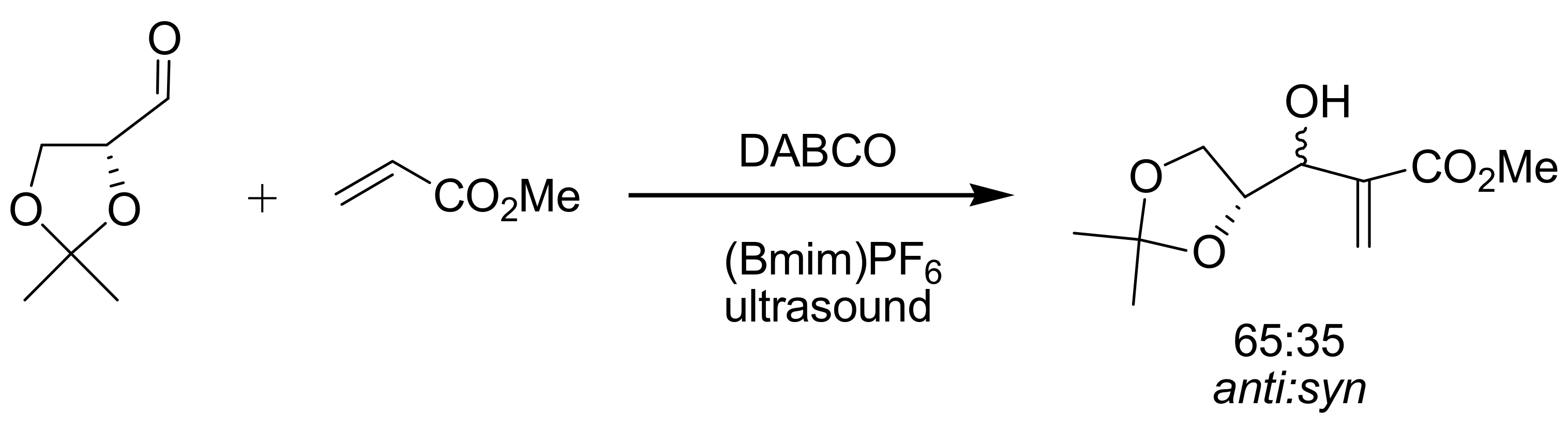
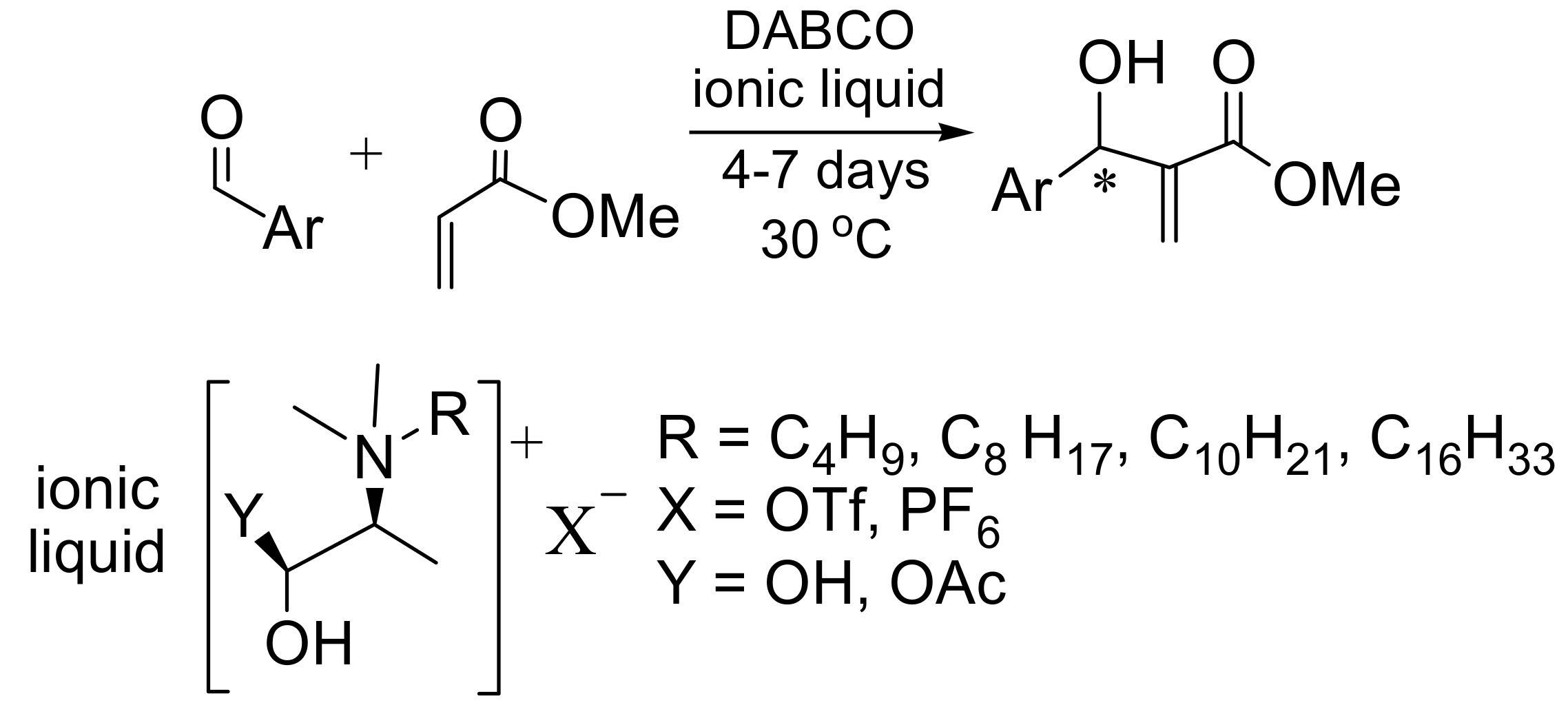
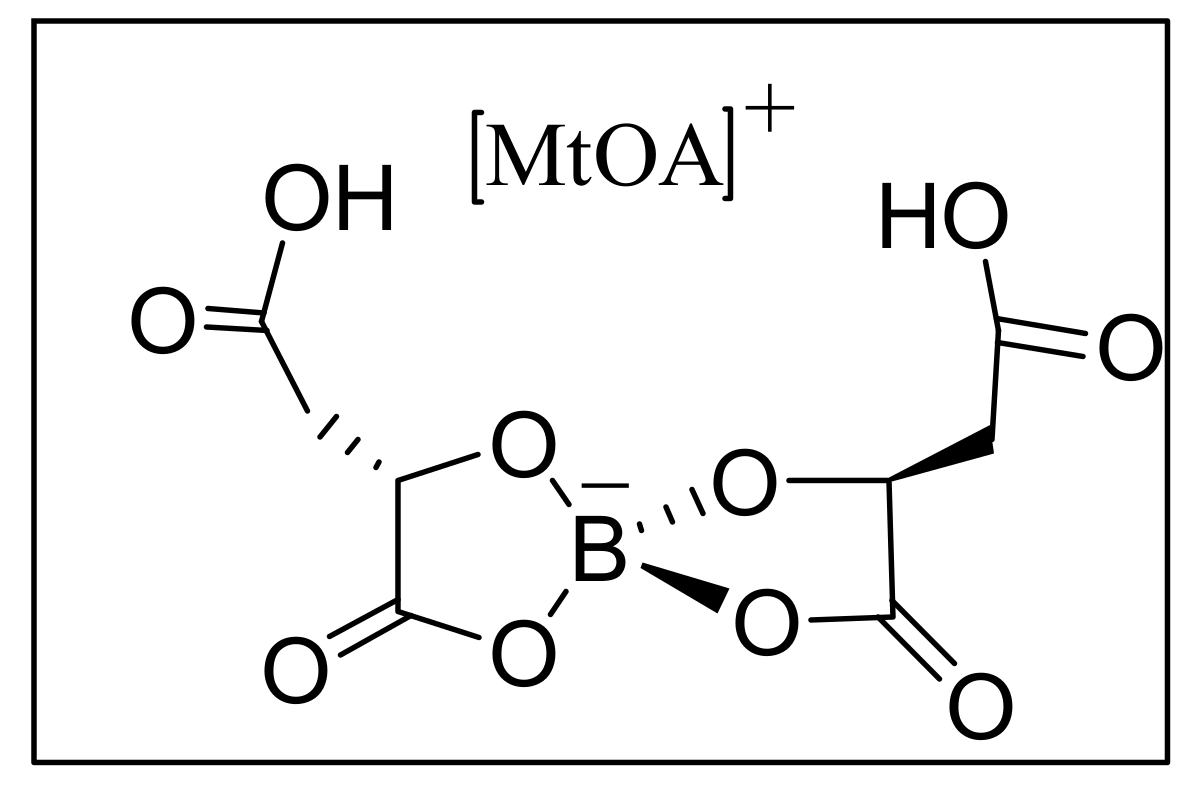
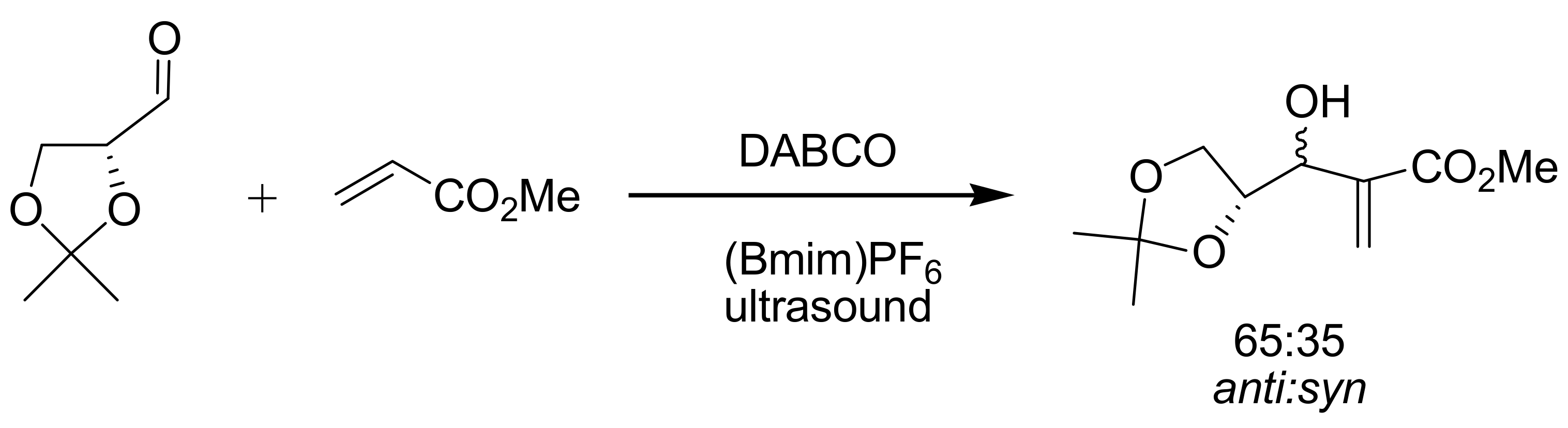
4.7. Ionic liquids as additives for MBH reaction
4.8. Acceleration of MBH reaction through mechanochemistry
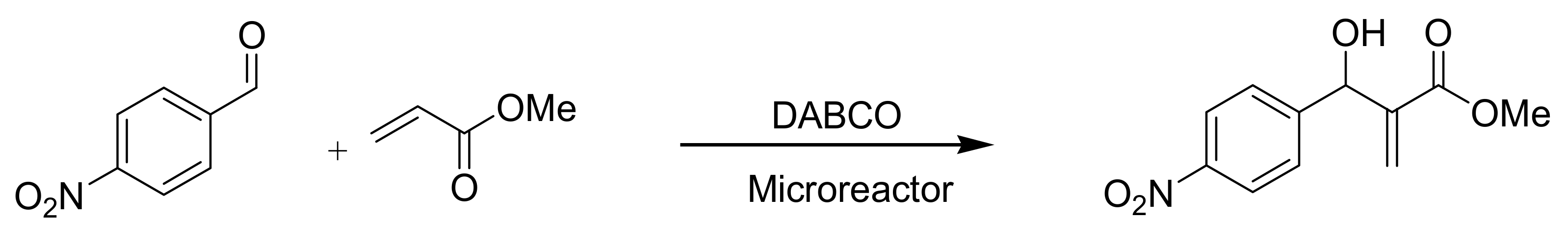
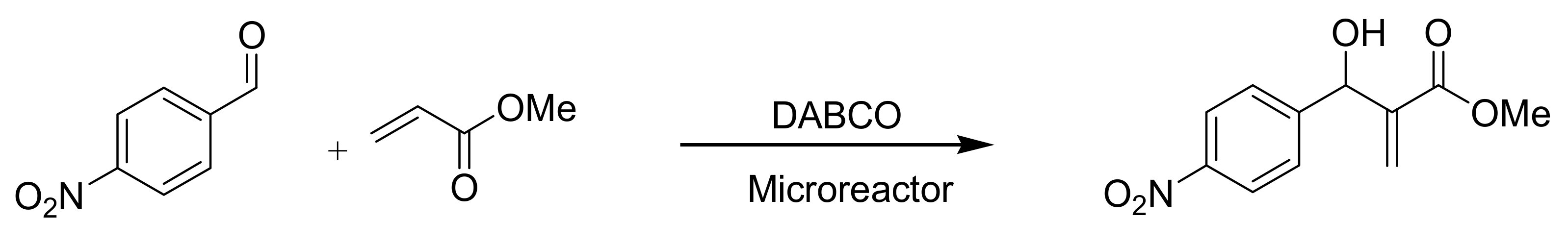
4.9. Online MBH reaction
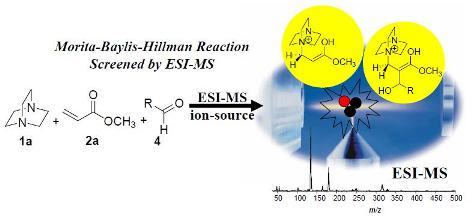
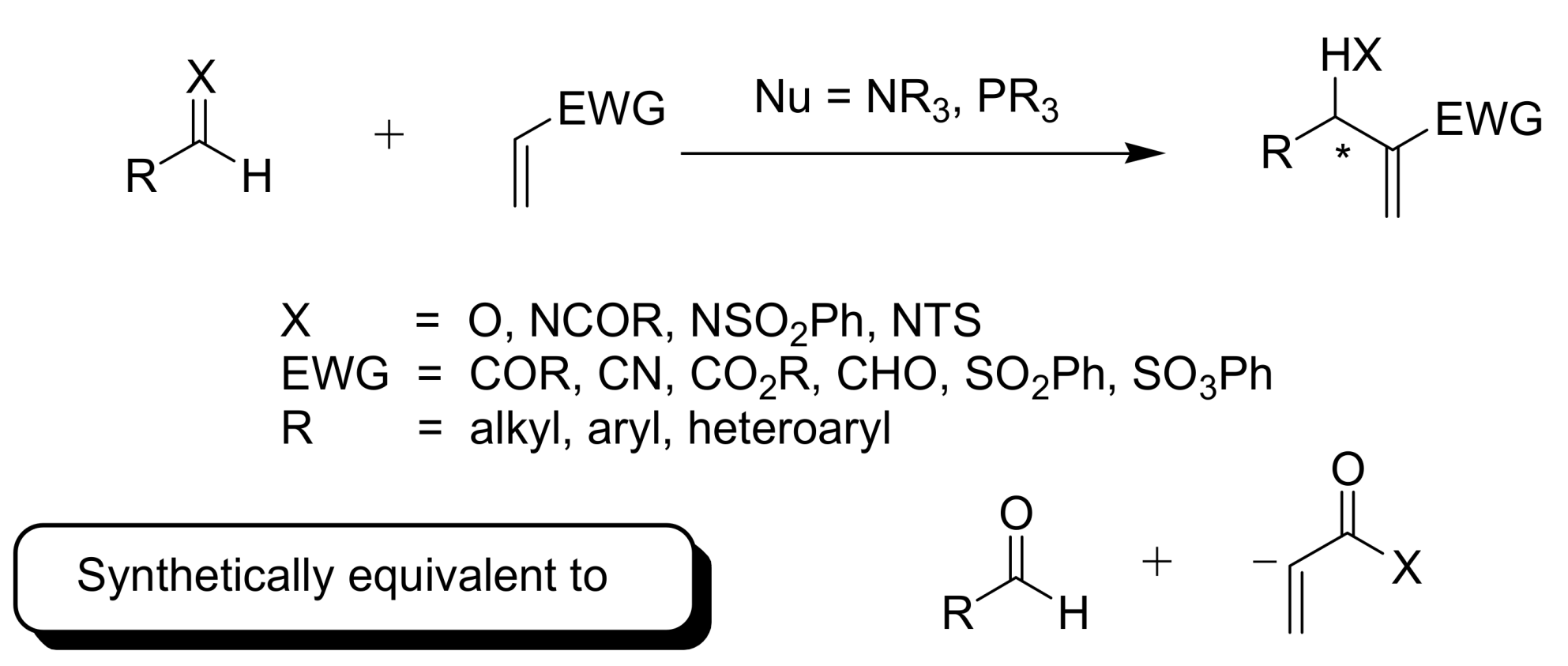
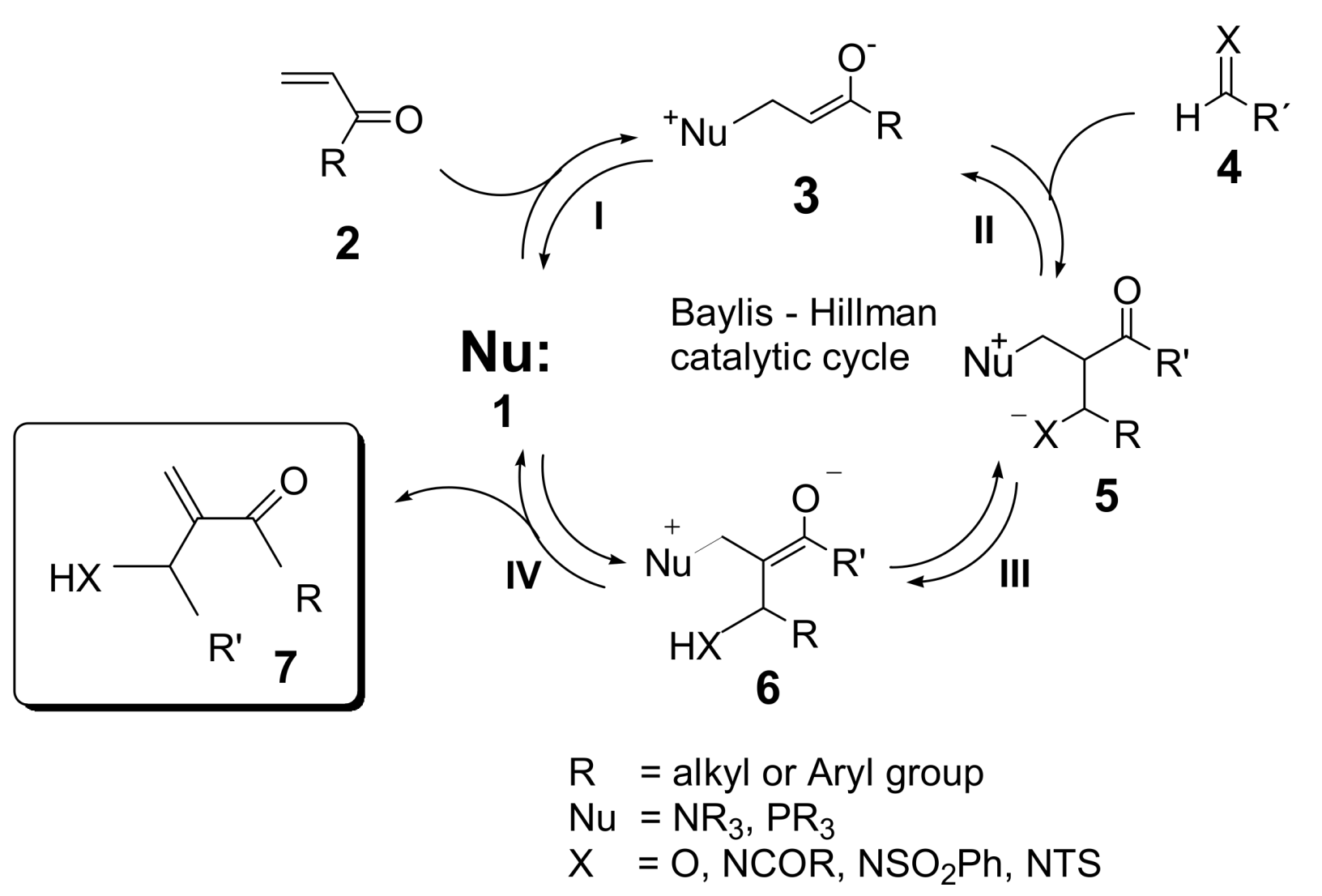
5. Probing the MBH Mechanism by Online ESI–MS(/MS)
5.1. Preservation of the charge in the transit of ions from solution to the gas phase using the ESI technique
5.1.1. Developing methods to study reaction mechanisms
5.1.1.1. Monitoring methods
5.1.1.2. Off-line monitoring
5.1.1.3. Online monitoring
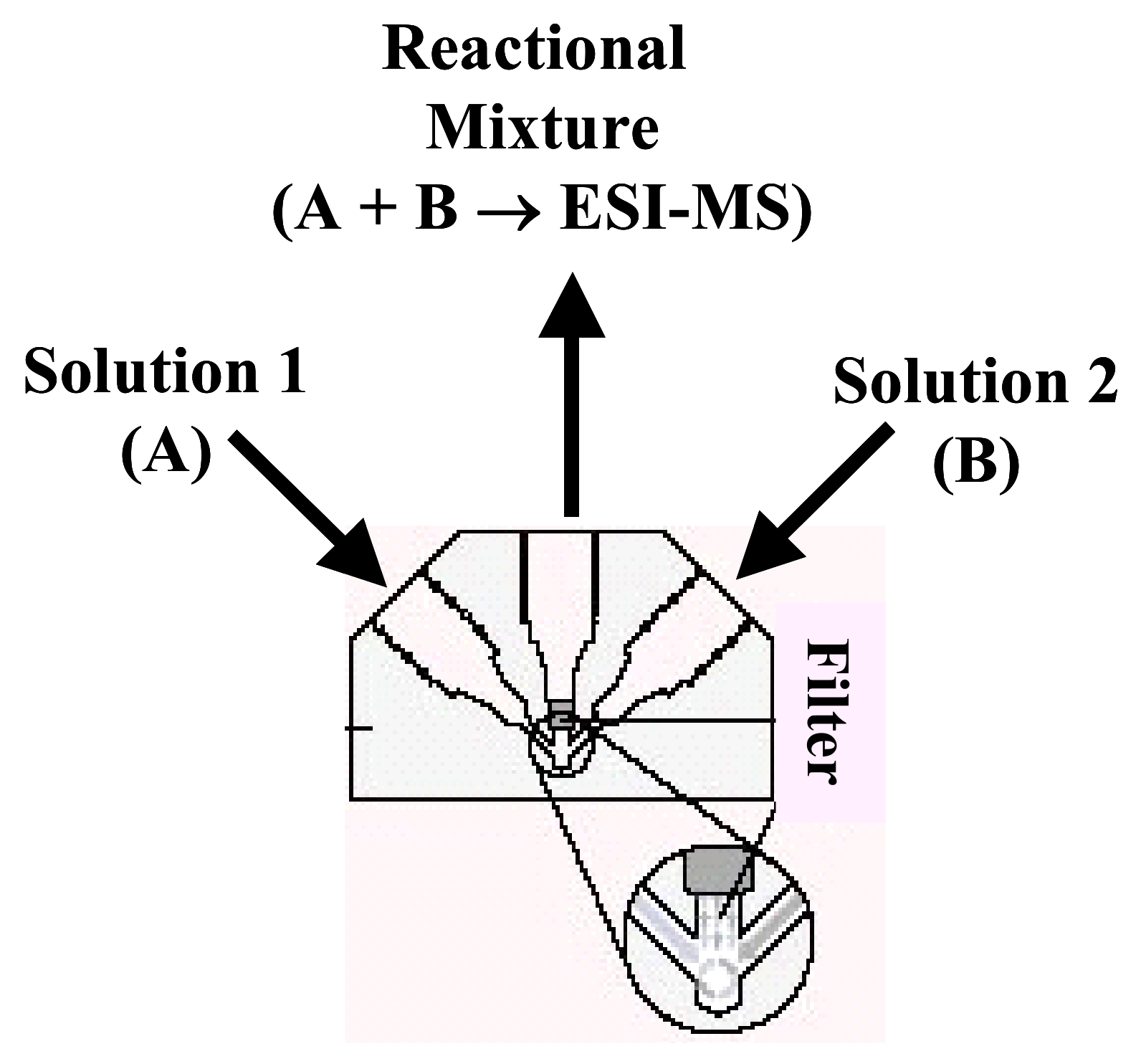
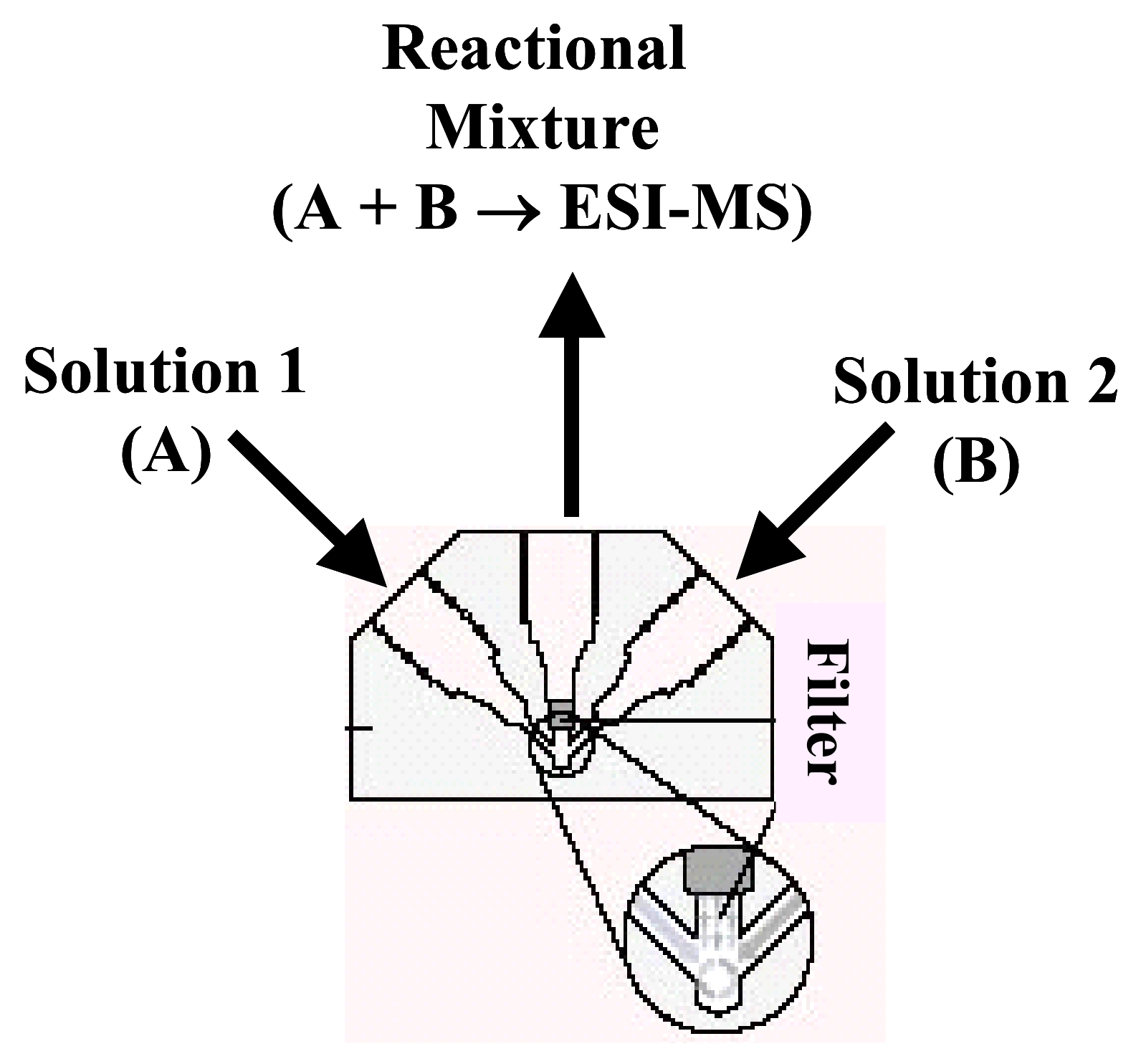
5.1.2. Microreactor
5.1.2.1. Peek mixing tee as microreactor
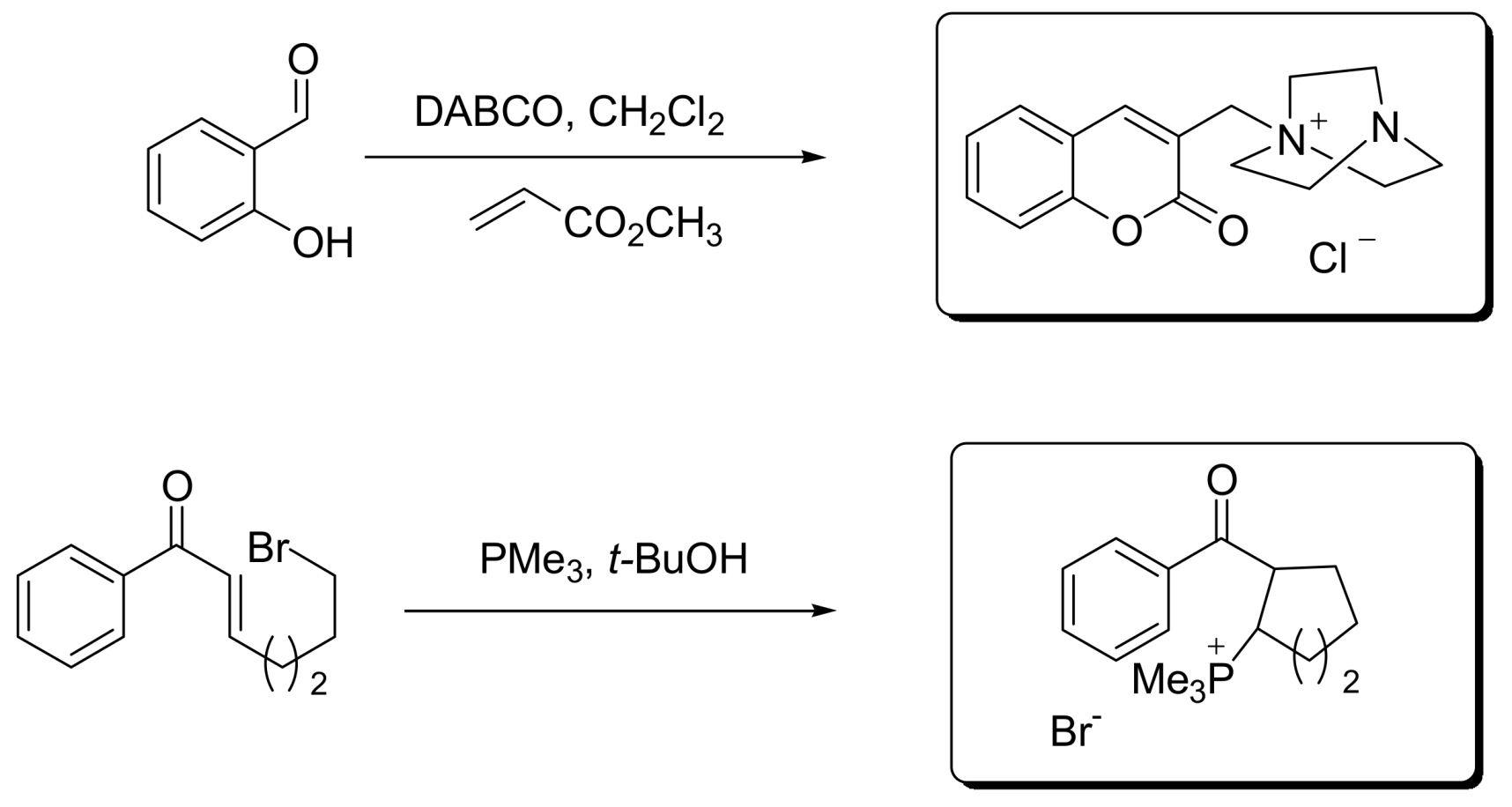
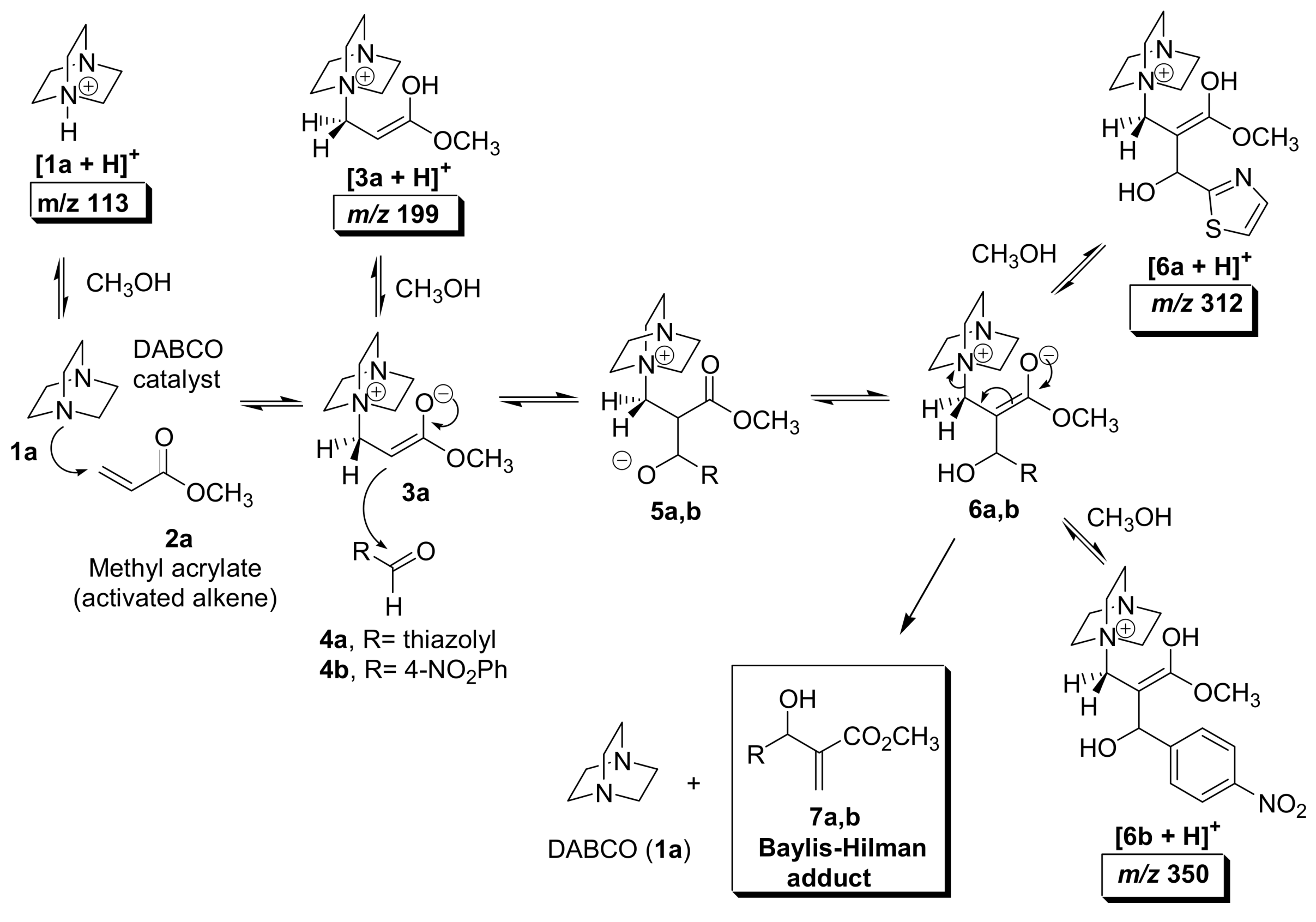
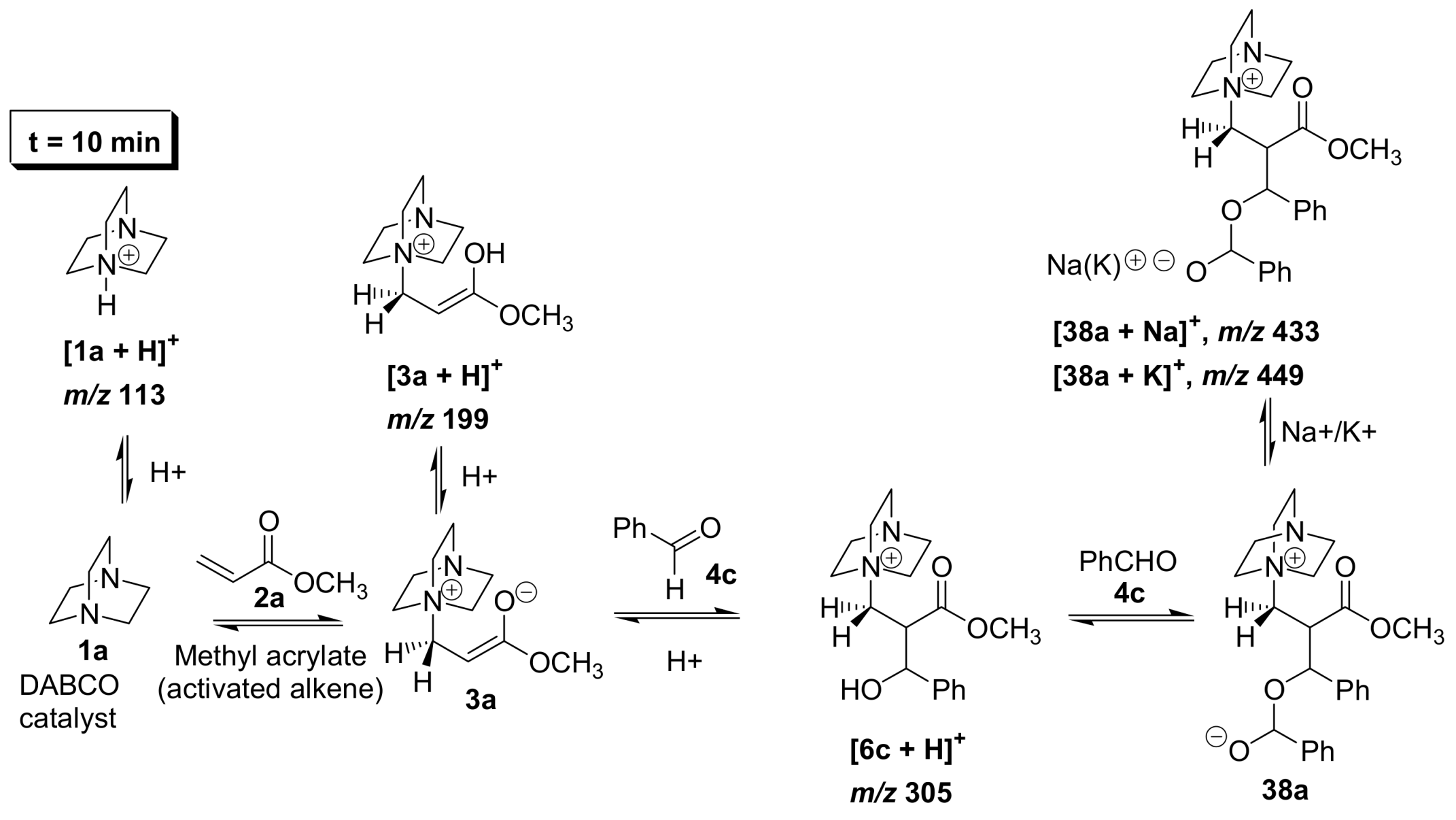
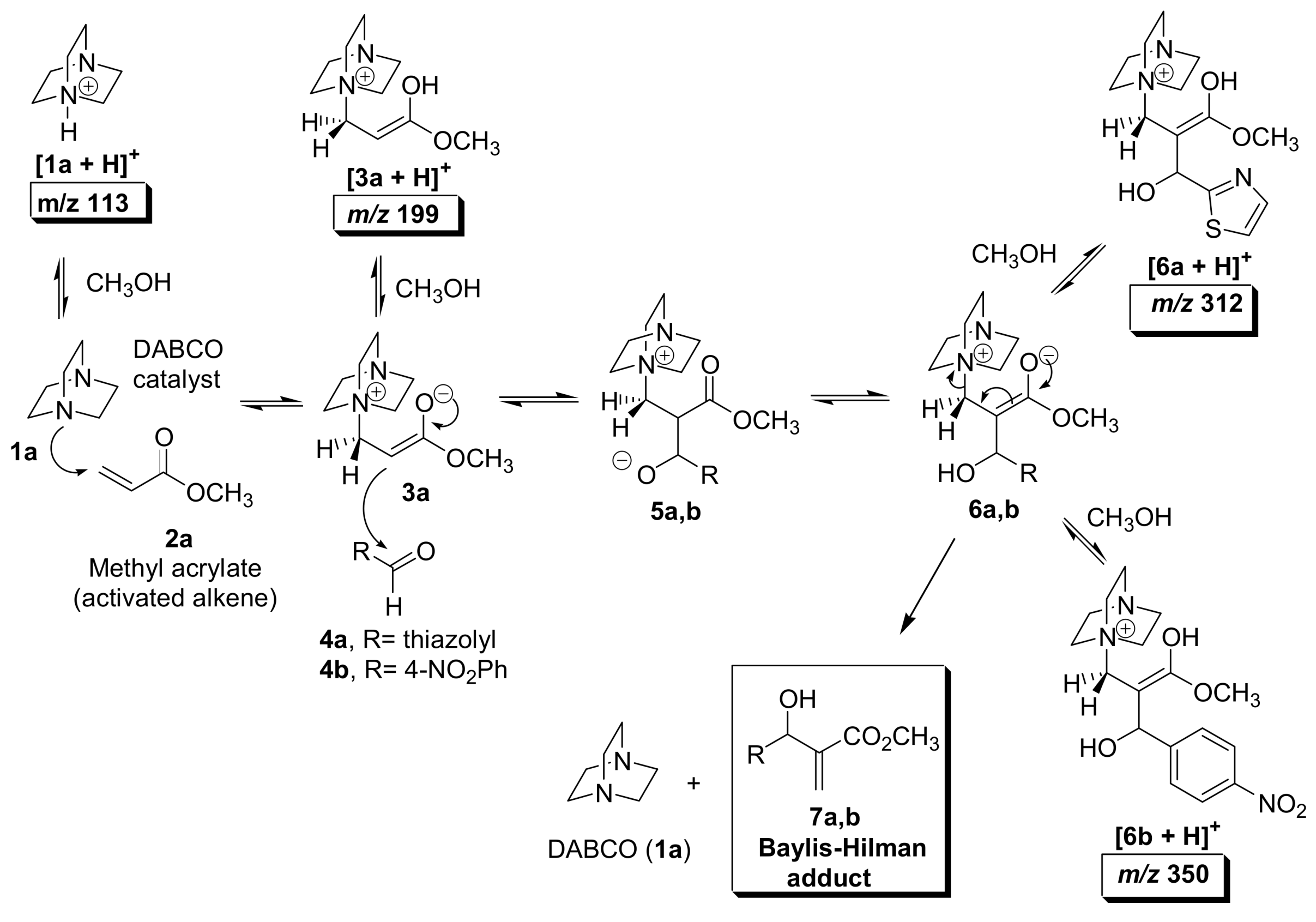
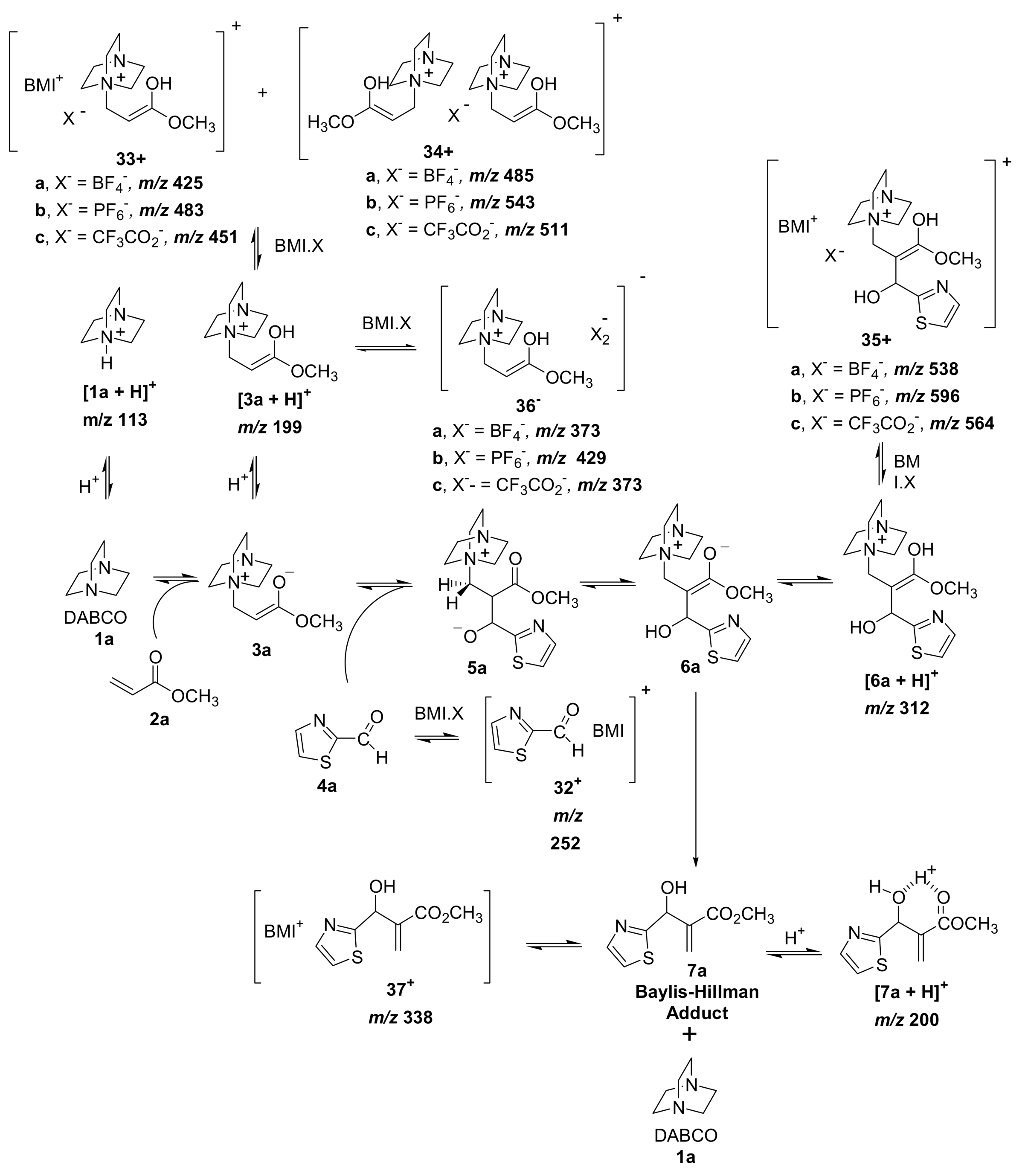
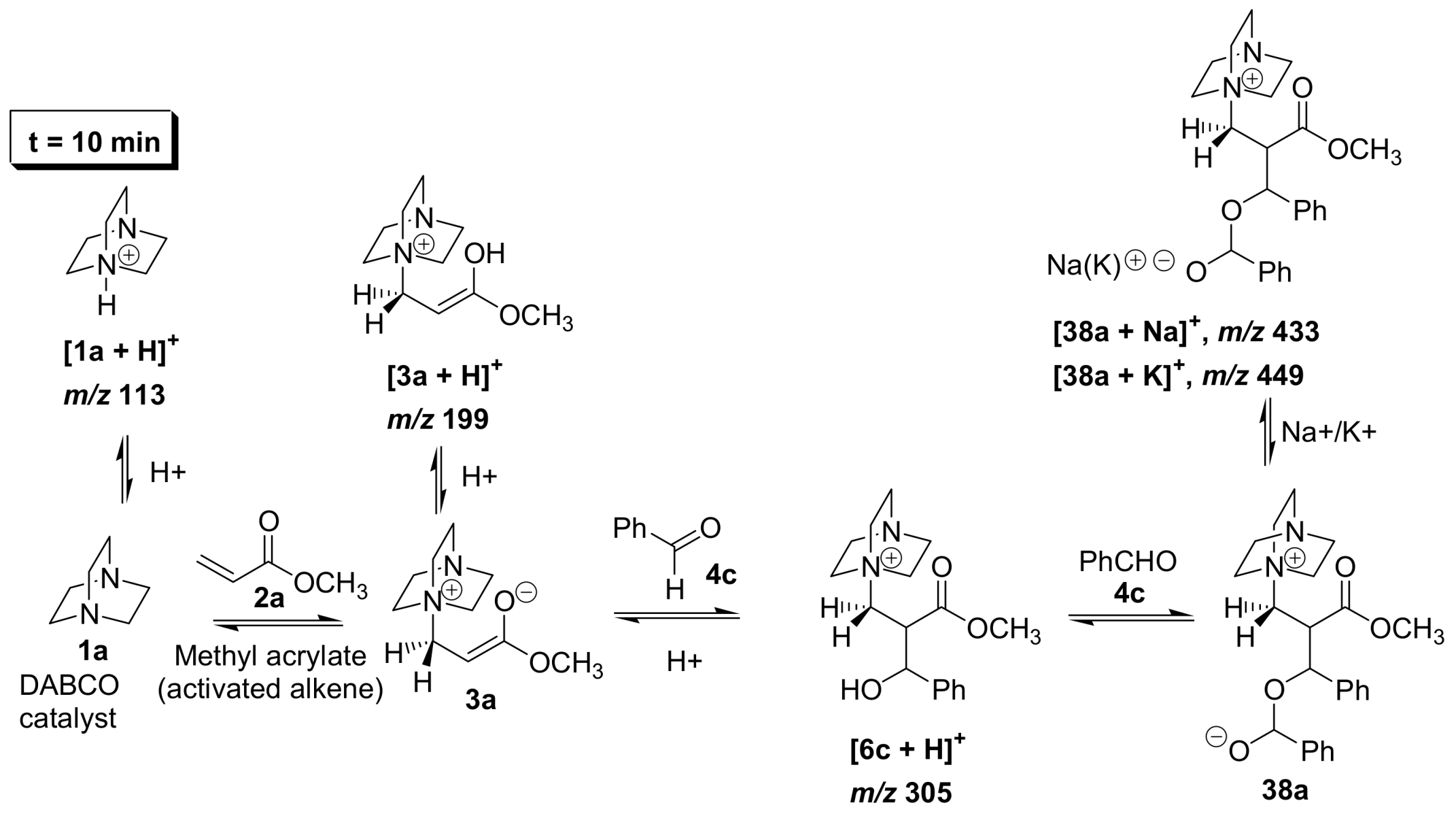
5.2. Detection of MBH cycle intermediates
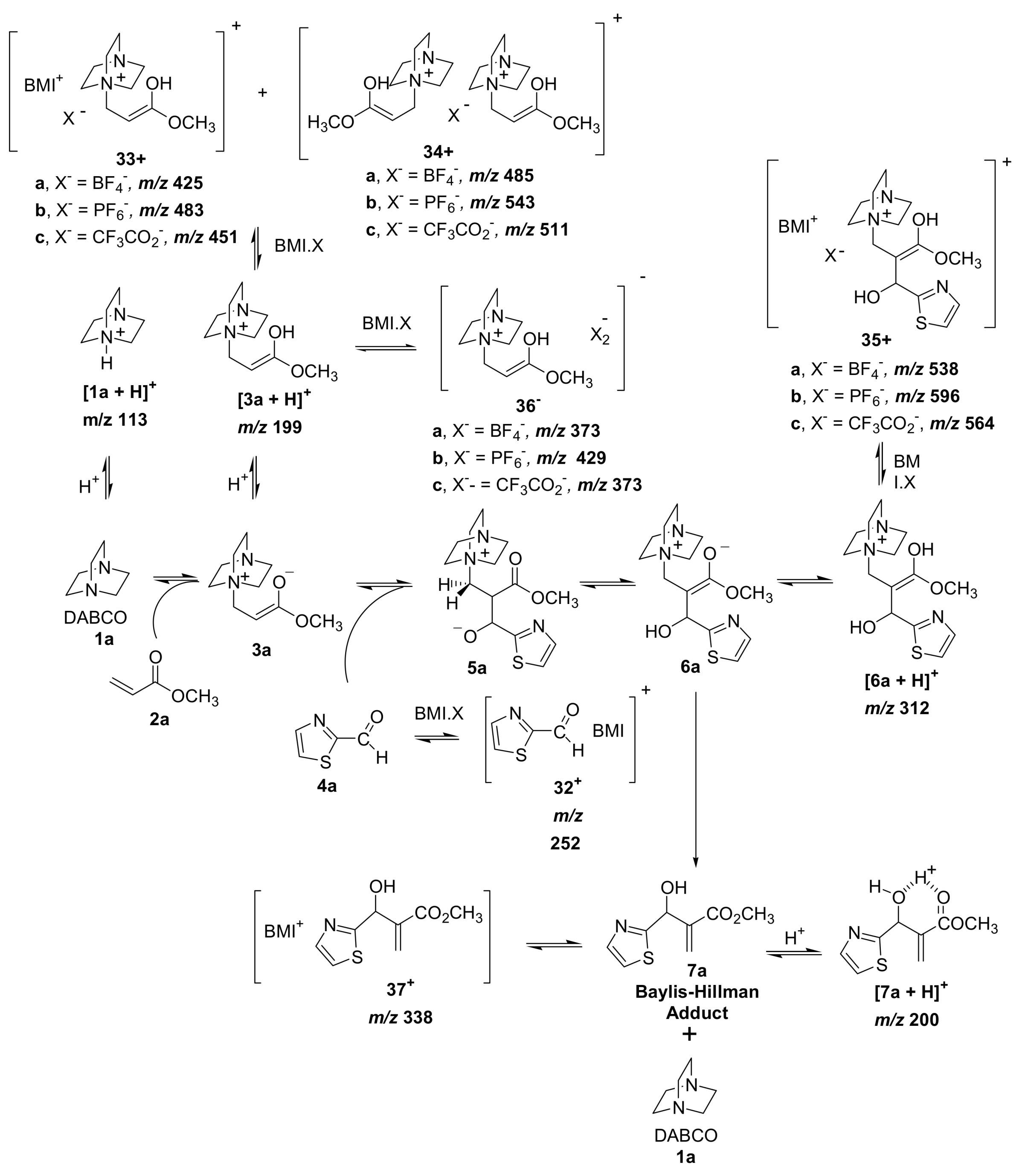
5.3. Morita–Baylis–Hillman reaction co-catalyzed by ionic liquids
5.4. Dualistic nature of Morita–Baylis–Hillman reaction probed by ESI–MS
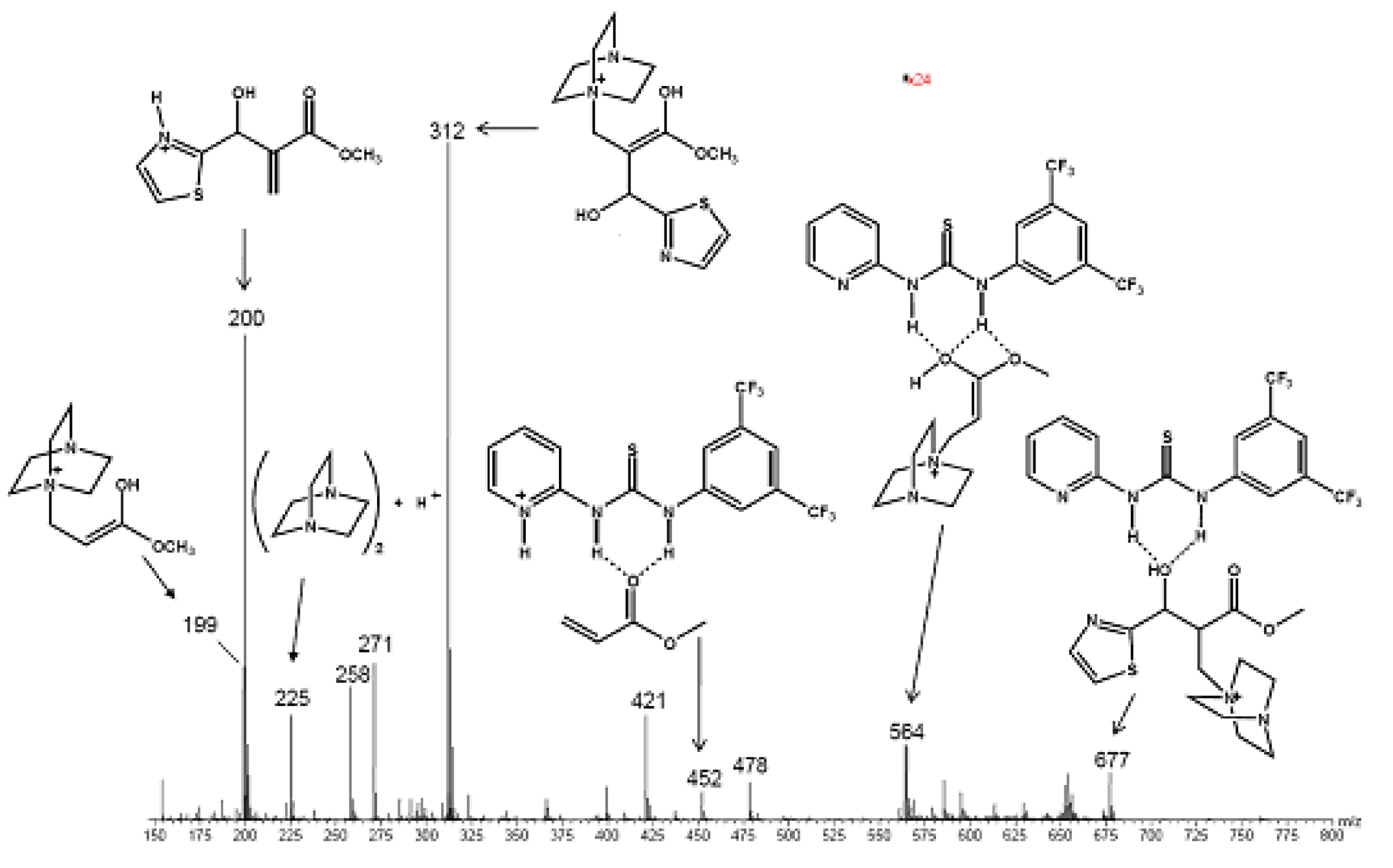
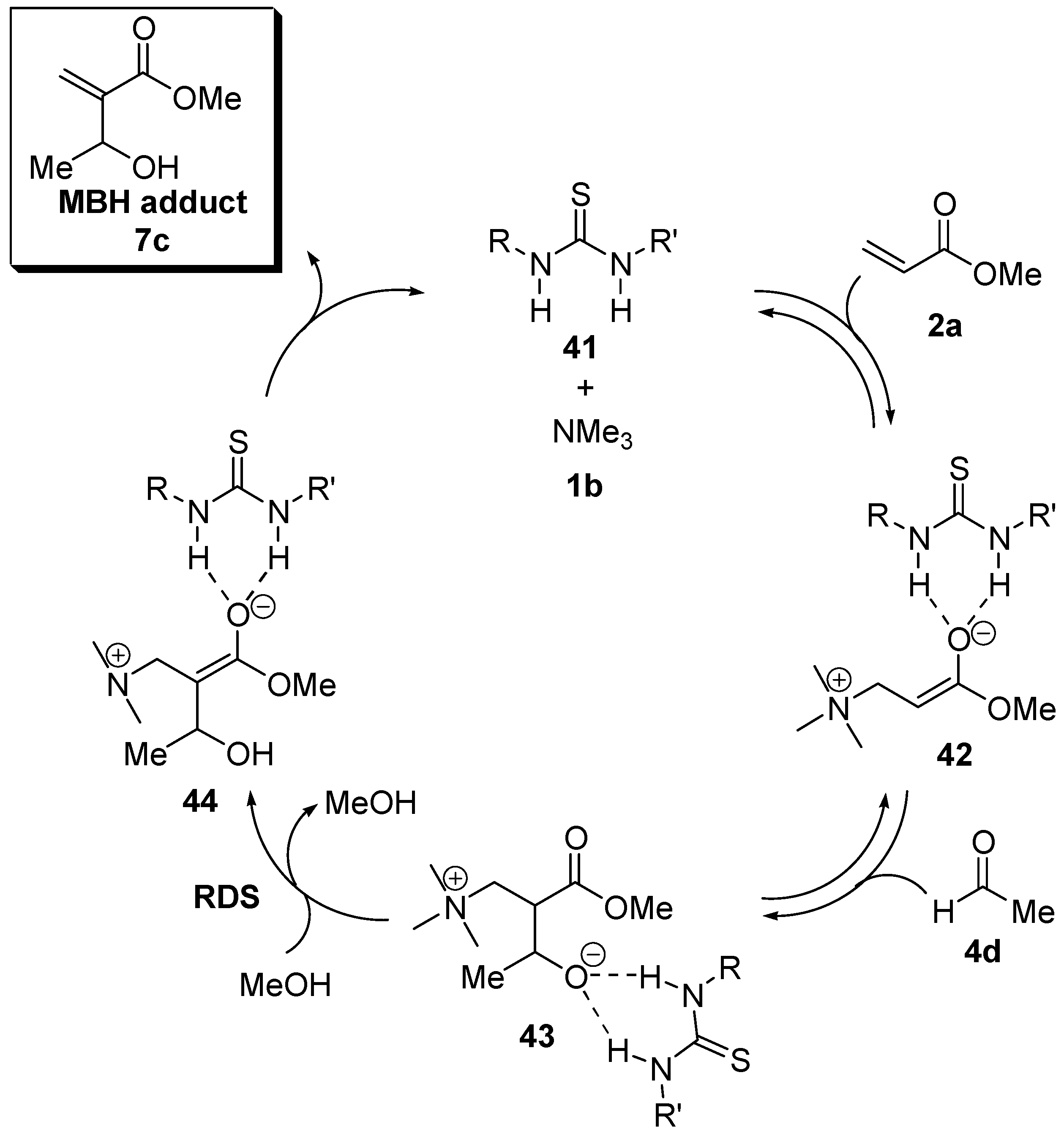
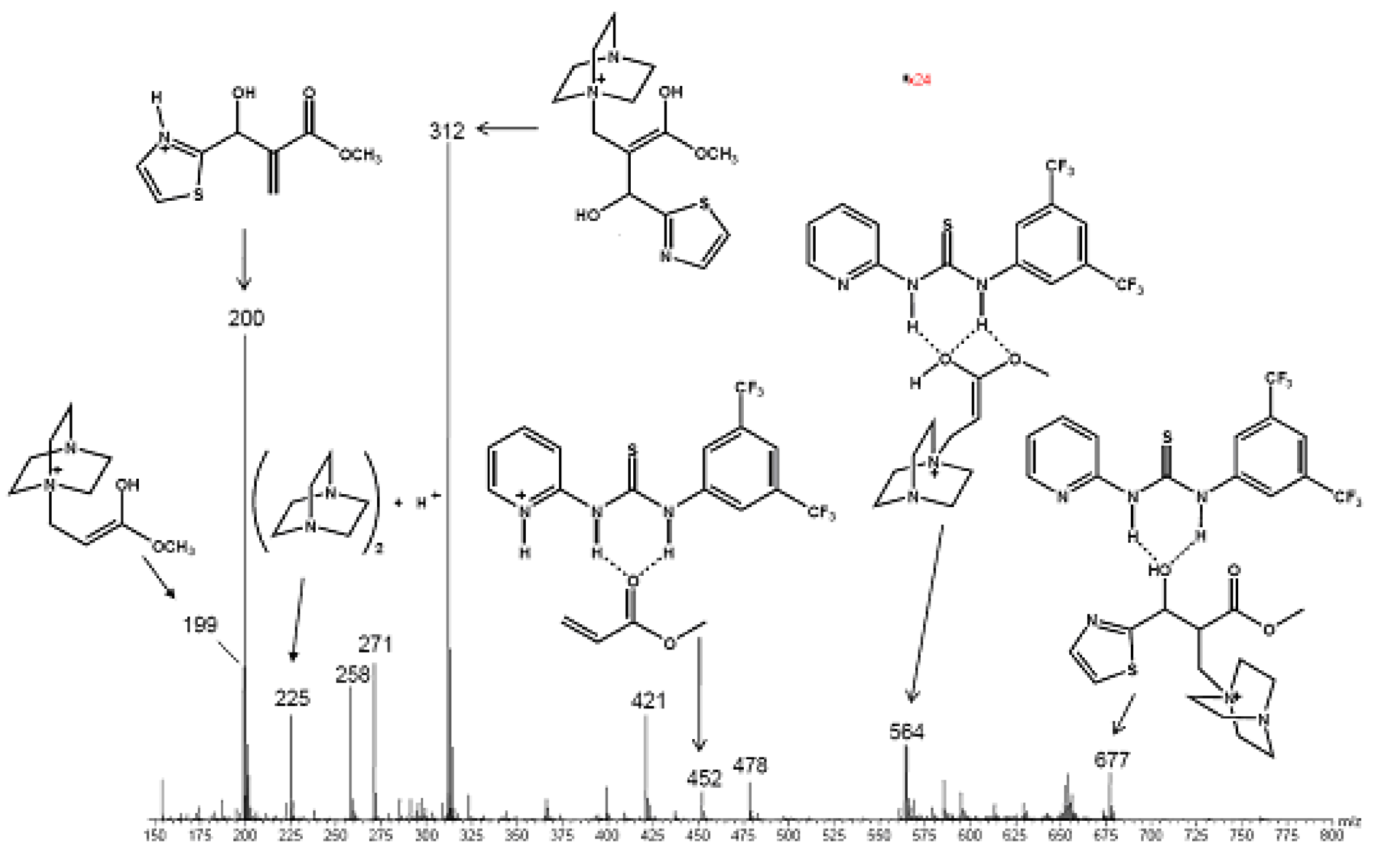
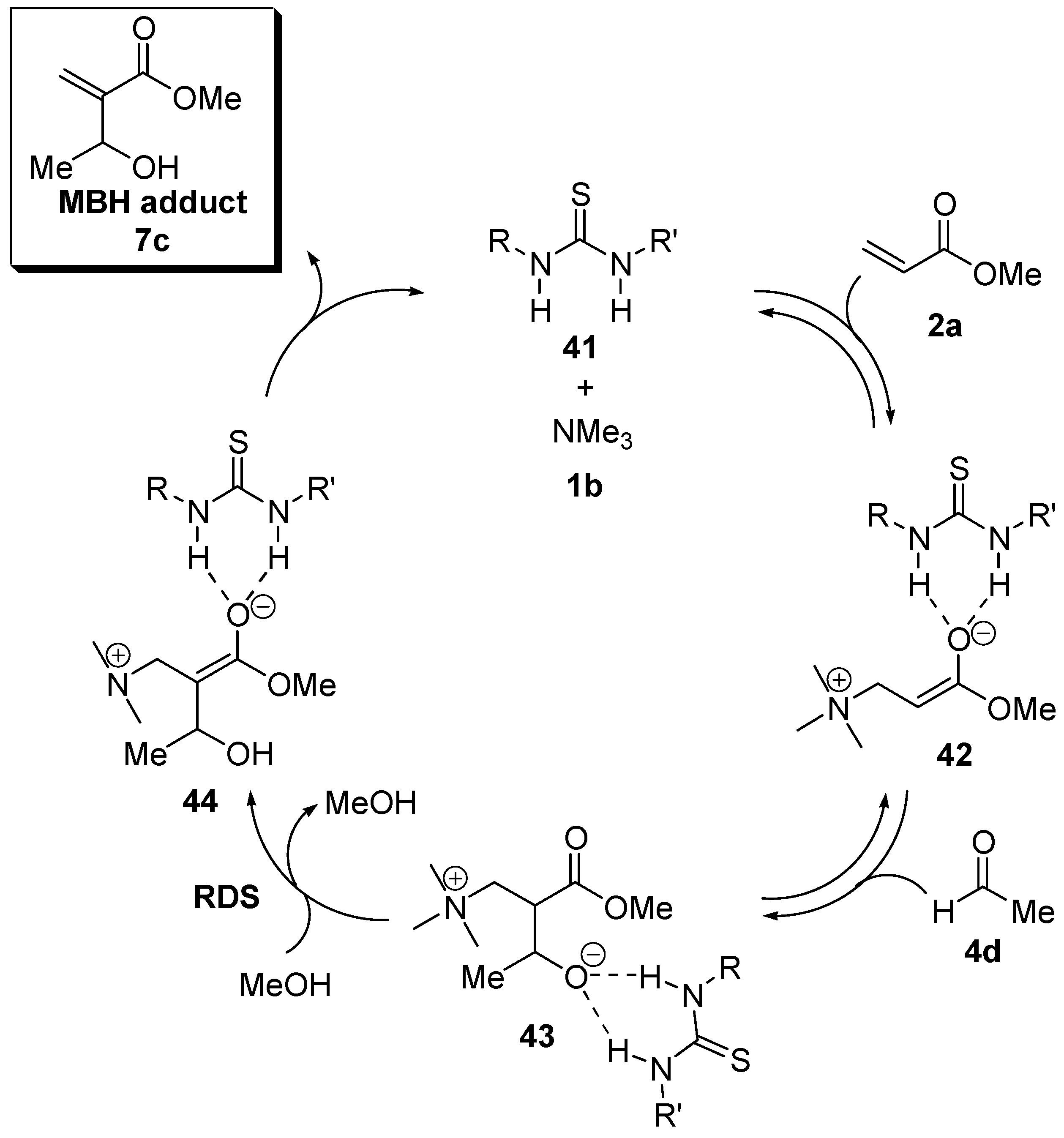
5.5. (Thio)ureas catalyzed Morita–Baylis–Hillman reaction analyzed by ESI–MS
6. Conclusions
Acknowledgements
References and Notes
- Das, B.; Chowdhury, N.; Banerjee, J.; Anjoy, M. A facile one-pot stereoselective synthesis of trisubstituted (E)-2-methylalk-2-enoic acids from unactivated Baylis−Hillman adducts and a simple access to some important insect pheromones. Tetrahedron Lett. 2006, 47, 6615–6618. [Google Scholar] [CrossRef]
- Amarante, G.W.; Rezende, P.; Cavallaro, M.; Coelho, F. Acyloins from Morita−Baylis−Hillman adducts: an alternative approach to the racemic total synthesis of bupropion. Tetrahedron Lett. 2008, 49, 3744–3748. [Google Scholar] [CrossRef]
- Wasnaire, P.; Wiaux, M.; Touillaux, R.; Markó, I.E. Reductive cyclisation of Morita−Baylis−Hillman adducts. A simple approach towards substituted hydrindanones and decalones. Tetrahedron Lett. 2006, 47, 985–989. [Google Scholar] [CrossRef]
- Masson, G.; Housseman, C.; Zhu, J. The enantioselective Morita−Baylis−Hillman reaction and its aza counterpart. Angew. Chem. Int. Ed. 2007, 46, 4614–4628. [Google Scholar] [CrossRef] [PubMed]
- Basavaiah, D.; Rao, A.J.; Satyanarayana, T. Recent advances in the Baylis−Hillman reaction and applications. Chem. Rev. 2003, 103, 811–892. [Google Scholar] [CrossRef] [PubMed]
- Basavaiah, D.; Rao, D.P.; Hyma, R.S. The Baylis−Hillman reaction: a novel carbon-carbon forming reaction. Tetrahedron 1996, 52, 8001–8062. [Google Scholar] [CrossRef]
- Declerck, V.; Martinez, J.; Lamaty, F. Aza-Baylis−Hillman Reaction. Chem. Rev. 2009, 109, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.S.; Isaacs, N.S. Mechanism of substitution reactions of acrylic derivatives. J. Phys. Org. Chem. 1990, 3, 285–288. [Google Scholar] [CrossRef]
- Denmark, S.E.; Gregory, L.B. Lewis base catalysis in organic synthesis. Angew. Chem. Int. Ed. 2008, 47, 1560–1638. [Google Scholar] [CrossRef] [PubMed]
- Marko, I.E.; Giles, P.R.; Hindley, N.J. Catalytic enantioselective Baylis−Hillman reactions. Correlation between pressure and enantiomeric excess. Tetrahedron 1997, 53, 1015–1024. [Google Scholar] [CrossRef]
- Drewes, S.E.; Njamela, O.L.; Emslie, N.D.; Ramesar, N.; Field, J.S. Intramolecular Baylis−Hillman reaction: A pathway to substituted coumarins. Synth. Commun. 1993, 23, 2807–2815. [Google Scholar] [CrossRef]
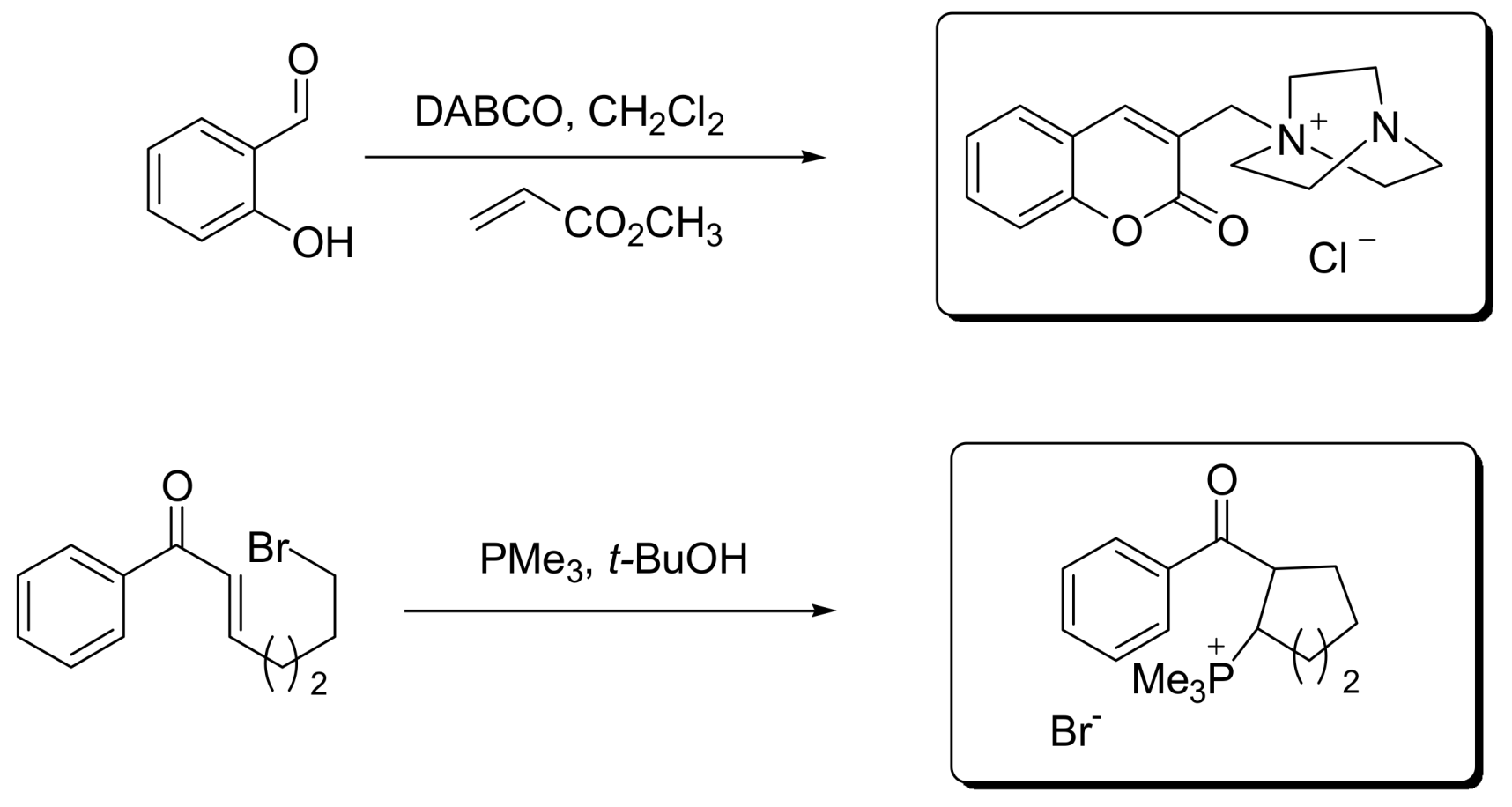
- Krafft, M.E.; Haxell, T.F.N.; Seibert, K.A.; Abboud, K.A. Mechanistic Implications in the Morita−Baylis−Hillman alkylation: Isolation and characterization of an Intermediate. J. Am. Chem. Soc. 2006, 128, 4174–4175. [Google Scholar] [CrossRef] [PubMed]
- Price, K.E.; Broadwater, S.J.; Jung, H.M.; McQuade, D.T. Baylis−Hillman mechanism: A new Interpretation in aprotic solvents. Org. Lett. 2005, 7, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Price, K.E.; Broadwater, S.J.; Walker, B.J.; McQuade, D.T. A new interpretation of the Baylis−Hillman mechanism. J. Org. Chem. 2005, 70, 3980–3987. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K.; Fulford, S.Y.; Lloyd-Jones, G.C. Re-evaluation of the mechanism of the Baylis−Hillman reaction - implications for asymmetric catalysis. Angew. Chem. Int. Ed. 2005, 44, 1706–1708. [Google Scholar] [CrossRef] [PubMed]
- Robiette, R.; Aggarwal, V.K.; Harvey, J.N. Mechanism of the Morita−Baylis−Hillman reaction: A computational investigation. J. Am. Chem. Soc. 2007, 129, 15513–15525. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Sunoj, R.B. Ab Initio and density functional theory evidence on the rate-limiting step in the Morita−Baylis−Hillman reaction. Org. Lett. 2007, 9, 4873–4876. [Google Scholar] [CrossRef] [PubMed]
- Teng, W.D.; Huang, R.; Kwong, C.K.W.; Shi, M.; Toy, P.H. Influence of Michael acceptor stereochemistry on intramolecular Morita−Baylis−Hillman reactions. J. Org. Chem. 2006, 71, 368–371. [Google Scholar] [CrossRef] [PubMed]
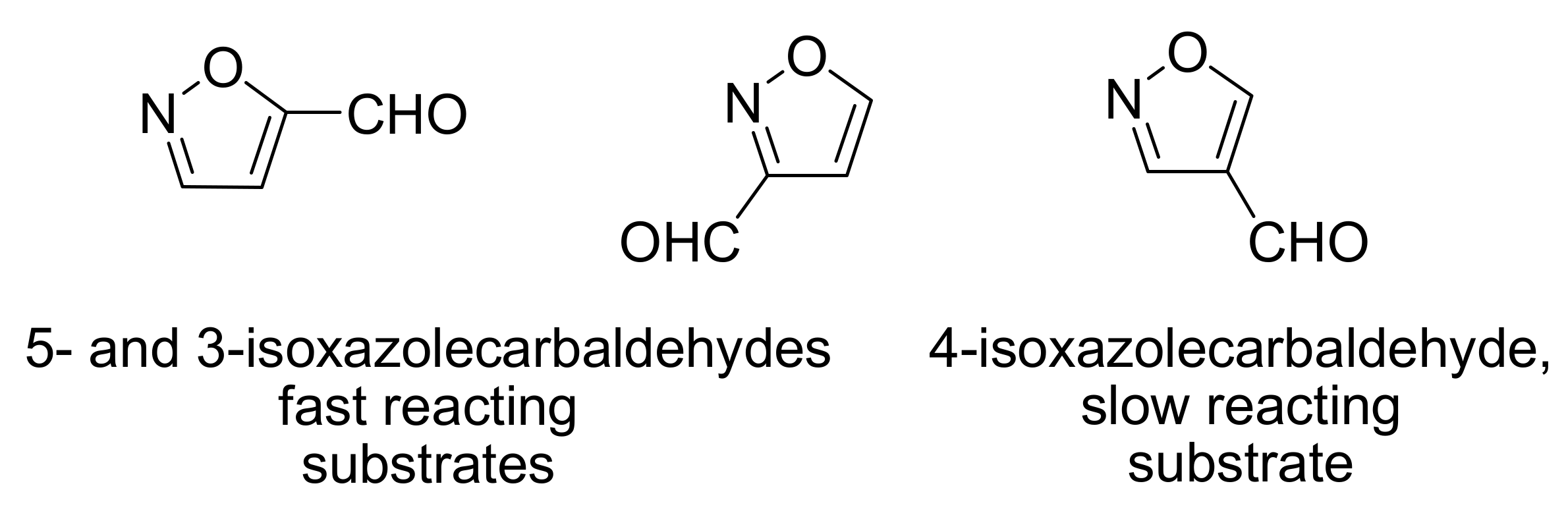
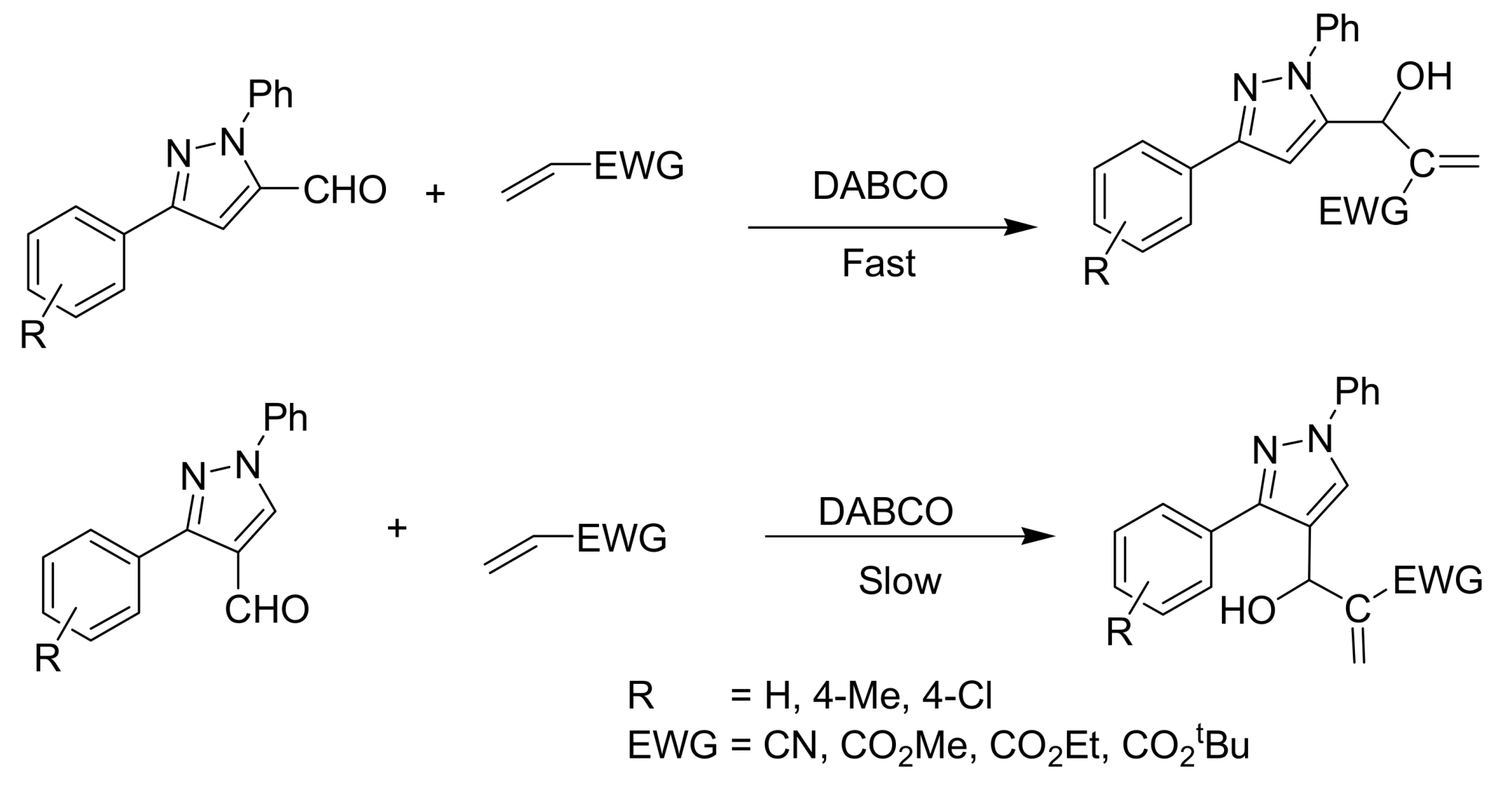
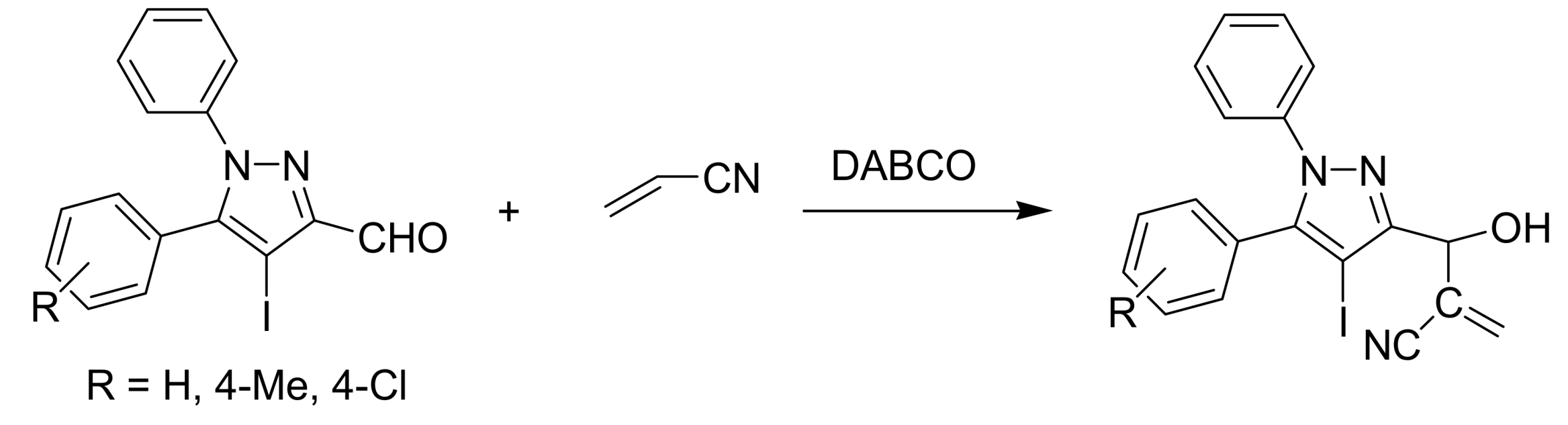
- Nag, S.; Singh, V.; Batra, S. Studies on the Baylis−Hillman reaction of pyrazolecarbaldehydes under the influence of DABCO: positional effect on the reactivity of the formyl group. ARKIVOC 2007, 14, 185–203. [Google Scholar]
- Balan, D.; Adolfsson, H. Titanium Isopropoxide as efficient catalyst for the Aza-Baylis−Hillman Reaction. Selective formation of b-methylene-a-amino acid derivatives. J. Org. Chem. 2002, 67, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Sorbetti, J.M.; Clary, K.N.; Rankic, D.A.; Wulff, J.E.; Parvez, M.; Back, T.G. Aza-Morita−Baylis−Hillman reactions and cyclizations of conjugated dienes activated by sulfone, ester, and keto groups. J. Org. Chem. 2006, 71, 368–371. [Google Scholar]
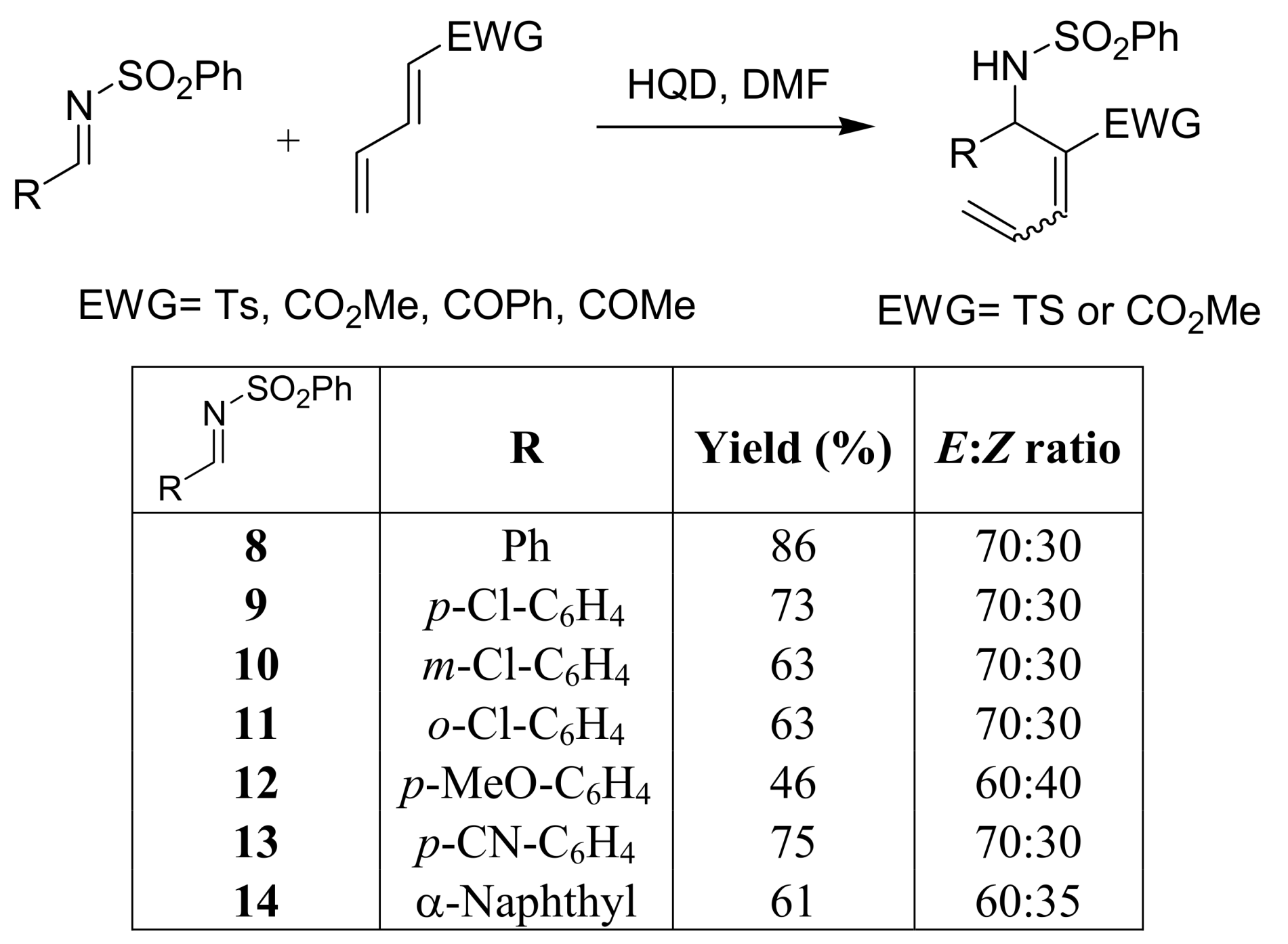
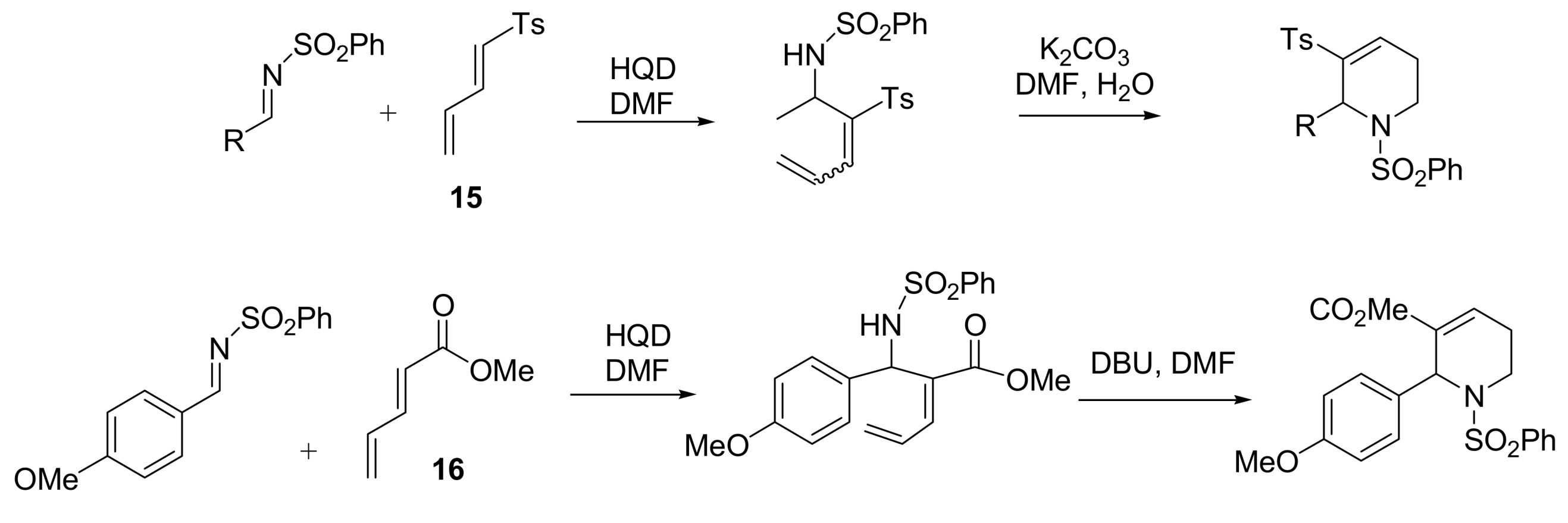
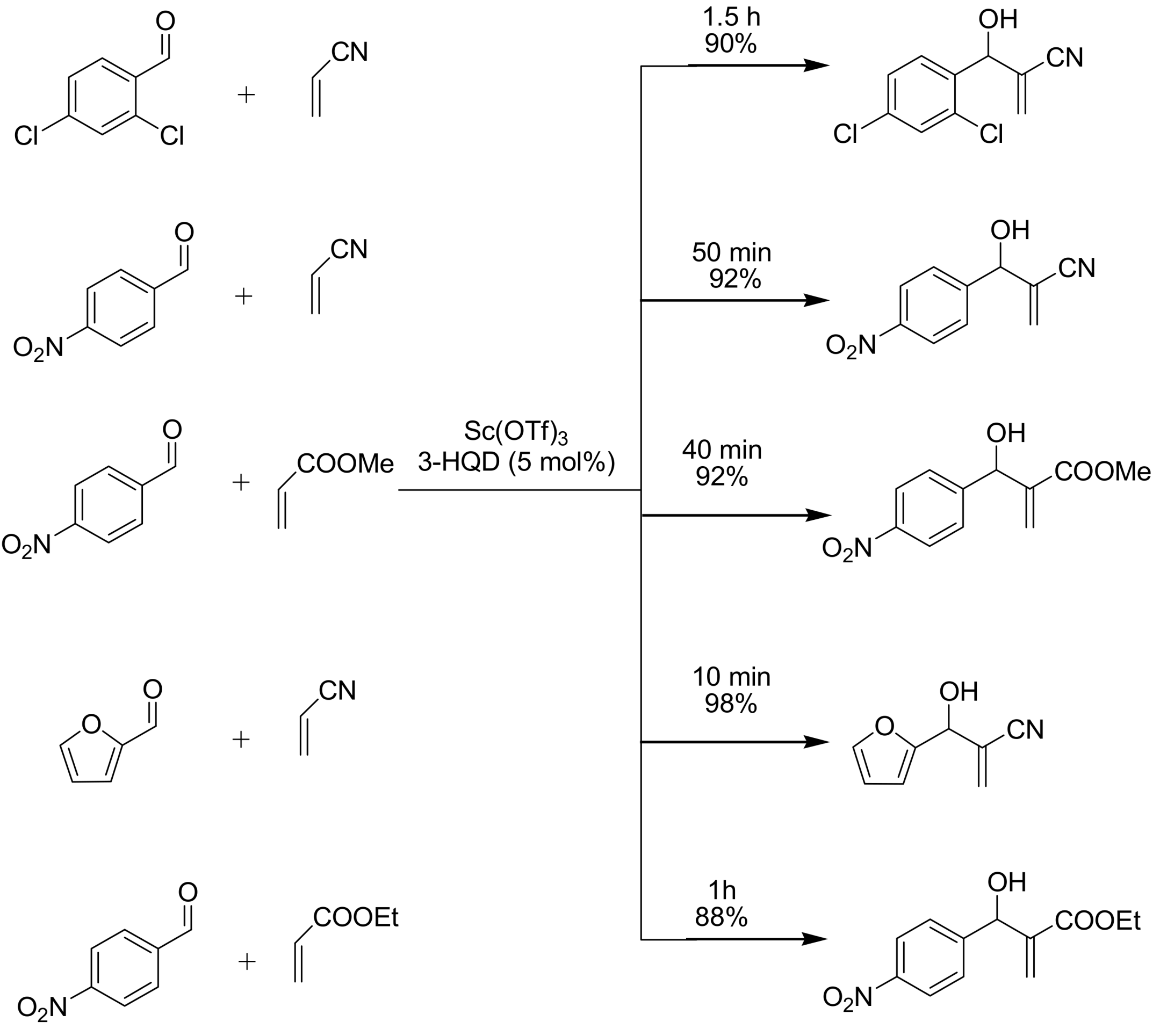
- Shang, Y.; Wang, D.; Wu, J. Novel Sc(OTf)3/3-HQD catalyst for Morita–Baylis–Hillman reaction. Synth. Commun. 2009, 1, 1035–1045. [Google Scholar] [CrossRef]
- Shi, M.; Chen, L.H.; Li, C.Q. Chiral phosphine Lewis bases catalyzed asymmetric aza-Baylis-Hillman reaction of N-sulfonated imines with activated olefins. J. Am. Chem. Soc. 2005, 127, 3790–3800. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Tanaka, K.; Horiia, A.; Takizawa, S.; Sasai, H. Conformational lock in a Brønsted acid–Lewis base organocatalyst for the aza-Morita–Baylis–Hillman reaction. Tetrahedron Asymmetry 2006, 17, 578–583. [Google Scholar] [CrossRef]
- Iwabuchi, Y.; Nakatani, M.; Yokoyama, N.; Hatakeyama, S. Chiral amine-catalyzed asymmetric Baylis−Hillman reaction: A reliable route to highly enantiomerically enriched (alpha-Methylene-beta-hydroxy)esters. J. Am. Chem. Soc. 1999, 121, 10219–10220. [Google Scholar] [CrossRef]
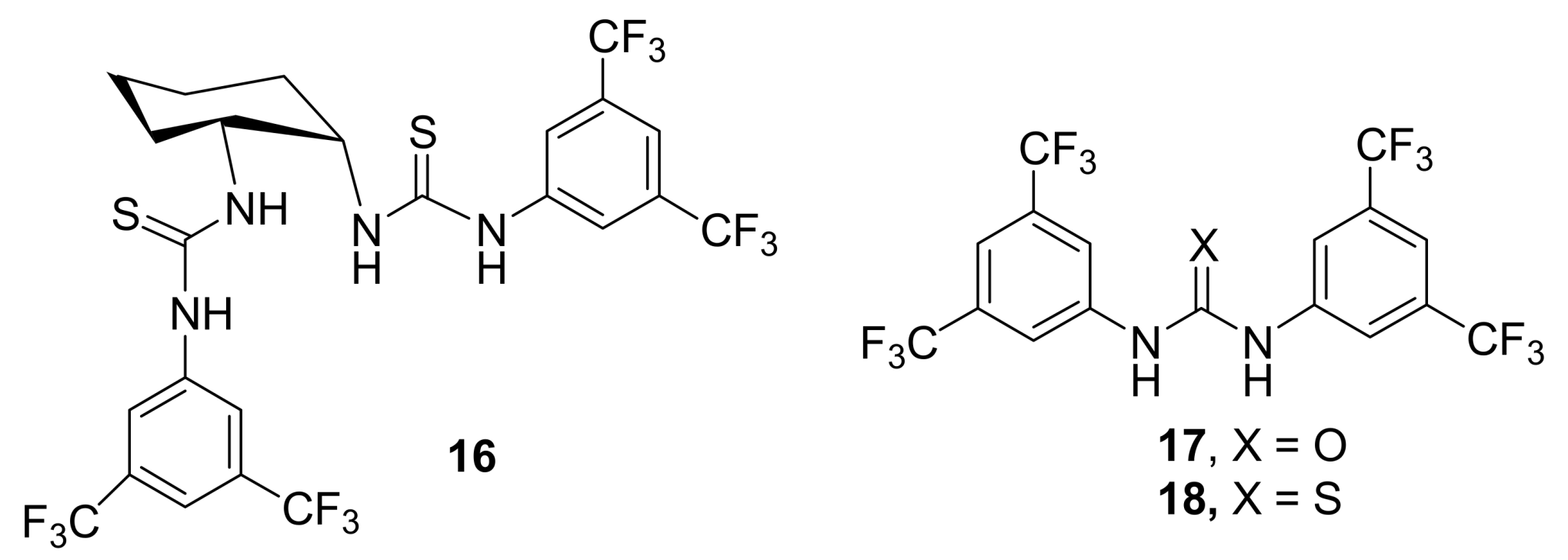
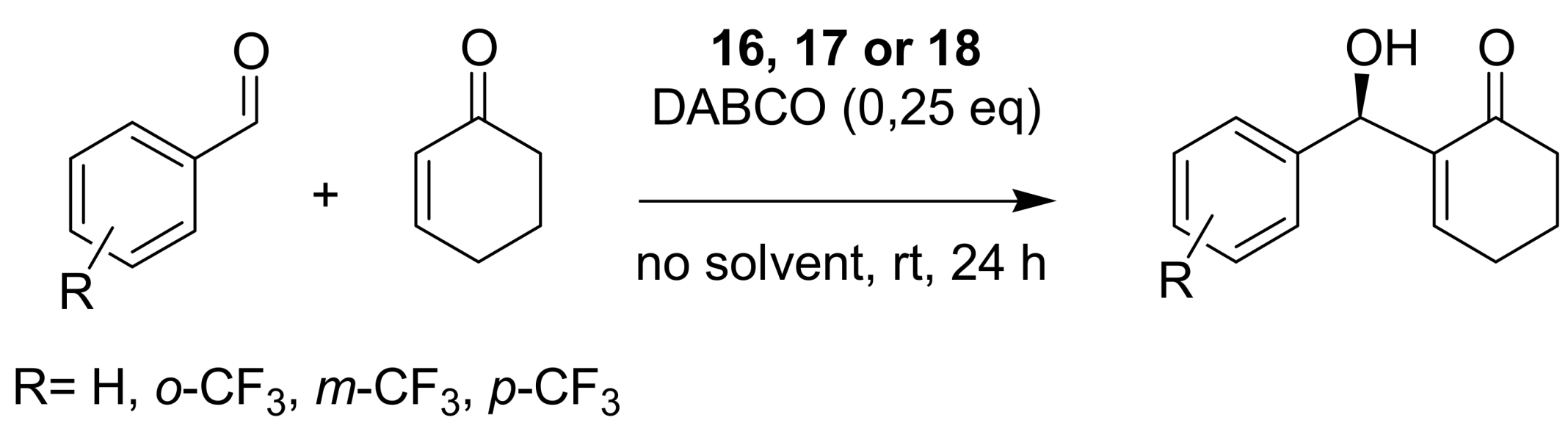
- Sohtome, Y.; Tanatani, A.; Hashimoto, Y.; Kazuo, N. Development of bis-thiourea-type organocatalyst for asymmetric Baylis−Hillman reaction. Tetrahedron Lett. 2004, 45, 5589–5592. [Google Scholar] [CrossRef]
- Connon, S.J. Organocatalysis mediated by (thio)urea derivatives. Chem. Eur. J. 2006, 12, 5418–5427. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.E.S.; Turega, S.M.; Clarke, M.L.; Philp, D. A rationally designed cocatalyst for the Morita−Baylis−Hillman reaction. Tetrahedron Lett. 2008, 49, 4666–4669. [Google Scholar] [CrossRef]
- Perlmutter, P.; Teo, C.C. A simple synthesis of 2-methylidene-3-aminopropanoates. Tetrahedron Lett. 1984, 25, 5951–5952. [Google Scholar] [CrossRef]
- Shi, M.; Ma, G.N.; Gao, J. Chiral bifunctional organocatalyst in asymmetric Aza-Morita−Baylis−Hillman reactions of ethyl (Arylimino)acetates with methyl vinyl ketone and ethyl vinyl ketone. J. Org. Chem. 2007, 72, 9779–9781. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Chen, L.H.; Teng, W.D. Asymmetric aza-Morita−Baylis−Hillman reaction of N-sulfonated imines with methyl vinyl ketone catalyzed by chiral phosphine lewis bases bearing perfluoroalkanes as "Pony tails". Adv. Synth. Catal. 2005, 347, 1781–1789. [Google Scholar] [CrossRef]
- Shi, M.; Qi, M.J.; Liu, X.G. Asymmetric catalytic aza-Morita−Baylis−Hillman reaction (aza-MBH): an interesting functional group-caused reversal of asymmetric induction. Chem. Commun. 2008, 6025–6027. [Google Scholar] [CrossRef] [PubMed]
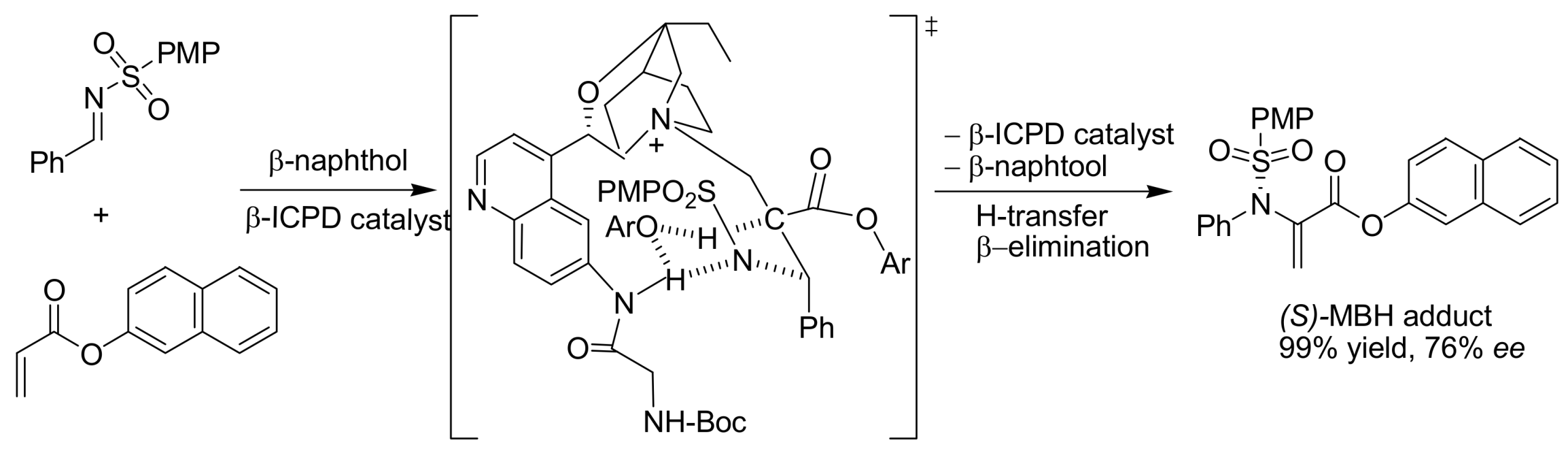
- Abermil, N.; Masson, G.; Zhu, J. Highly enantioselective aza Morita−Baylis−Hillman reaction catalyzed by bifunctional β-isocupreidine derivatives. J. Am. Chem. Soc. 2008, 130, 12596–12597. [Google Scholar] [CrossRef] [PubMed]
- Utsumi, N.; Zhang, H.; Tanaka, F.; Barbas, C.F. A way to highly enantiomerically enriched aza-Morita−Baylis−Hillman-type products. Angew. Chem. Int. Ed. 2007, 46, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Kwong, C.K.W.; Huang, R.; Zhang, M.; Shi, M.; Toy, P.H. Bifunctional polymeric organocatalysts and their application in the cooperative catalysis of Morita−Baylis−Hillman reactions. Chem. Eur. J. 2007, 13, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T.; Mondiere, R.; Carballeira, J.D. Enzyme promiscuity: first protein-catalyzed Morita−Baylis−Hillman reaction. Tetrahedron Lett. 2007, 48, 1679–1681. [Google Scholar] [CrossRef]
- Pegot, B.; Vo-Thanh, G.; Gori, D.; Loupy, A. First application of chiral ionic liquids in asymmetric Baylis−Hillman reaction. Tetrahedron Lett. 2004, 45, 6425–6428. [Google Scholar] [CrossRef]
- Gausepohl, R.; Buskens, P.; Kleinen, J.; Bruckmann, A.; Lehmann, C.W.; Klankermayer, J.; Leitner, W. Highly enantioselective Aza-Baylis−Hillman reaction in a chiral reaction medium. Angew. Chem. Int. Ed. 2006, 45, 3689–3692. [Google Scholar] [CrossRef] [PubMed]
- Porto, R.S.; Amarante, G.W.; Cavallaro, M.; Poppi, R.; Coelho, F. Improved catalysis of Morita−Baylis−Hillman reaction.The strong synergic effect using both an imidazolic ionic liquid and a temperature. Tetrahedron Lett. 2009, 50, 1184–1187. [Google Scholar] [CrossRef]
- Suryanarayana, C. Materials and process design through mechanochemical routes. Prog. Mat. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Mack, J.; Shumba, M. Rate enhancement of the Morita−Baylis−Hillman reaction through mechanochemistry. Green Chem. 2007, 9, 328–330. [Google Scholar] [CrossRef]
- Acke, D.R.J.; Stevens, C.V. Study of the Baylis−Hillman reaction in a microreactor environment: first continuous production of Baylis−Hillman adducts. Org. Process Res. Dev. 2006, 10, 417–422. [Google Scholar] [CrossRef]
- Santos, L.S. Online mechanistic Investigations of catalyzed reactions by electrospray ionization mass spectrometry: A tool to intercept transient species in solution. Eur. J. Org. Chem. 2008, 235–253. [Google Scholar] [CrossRef]
- Santos, L.S.; Rosso, G.B.; Pilli, R.A.; Eberlin, M.N. The mechanism of the Stille reaction Investigated by electrospray ionization mass spectrometry. J. Org. Chem. 2007, 72, 5809–5812. [Google Scholar] [CrossRef] [PubMed]
- Abella, C.A.M.; Benassi, M.; Santos, L.S.; Eberlin, M.N.; Coelho, F. The mechanism of Troger’s base formation probed by electrospray ionization mass spectrometry. J. Org. Chem. 2007, 72, 4048–4054. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S.; Knaack, L.; Metzger, J.O. Investigation of chemical reactions in solution using API-MS. Int. J. Mass Spectrom. 2005, 246, 84–104. [Google Scholar] [CrossRef]
- Chen, P. Electrospray ionization tandem mass spectrometry in high-throughput screening of homogeneous catalysts. Angew. Chem. Int. Ed. 2003, 42, 2832–2847. [Google Scholar] [CrossRef] [PubMed]
- Eberlin, M.N. Electrospray ionization mass spectrometry: a major tool to investigate reaction mechanisms in both solution and the gas phase. Eur. J. Mass Spectrom. 2007, 13, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization-principles and practice. Mass Spectrom. Rev. 1990, 9, 37–70. [Google Scholar] [CrossRef]
- Fenn, J.B. Electrospray wings for molecular elephants. Angew. Chem. Int. Ed. 2003, 42, 3871–3894. [Google Scholar] [CrossRef] [PubMed]
- Kebarle, P.; Tang, L. From ions in solution to ions in the gas phase. Anal. Chem. 1993, 65, 972–986. [Google Scholar] [CrossRef]
- Gaskell, S.J. Electrospray: principles and practice. J. Mass Spectrom. 1997, 32, 677–688. [Google Scholar] [CrossRef]
- Jones, J.L.; Dongre, A.R.; Somogyi, A.; Wysocki, V.H. Sequence dependence of peptide fragmentation efficiency curves determined by electrospray-ionization surface-induced dissociation mass-spectrometry. J. Am. Chem. Soc. 1994, 116, 8368–8369. [Google Scholar] [CrossRef]
- De la Mora, J.F.; Van Berckel, G.J.; Enke, C.G.; Cole, R.B.; Martinez-Sanchez, M.B.; Fenn, J.B. Electrochemical processes in electrospray ionization mass spectrometry - Discussion. J. Mass Spectrom. 2000, 35, 939–952. [Google Scholar] [CrossRef]
- Van Berkel, G.J. The Electrolytic Nature of Electrospray. In Electrospray Ionization Mass Spectrometry, 2nd Edition; Cole, R.B, Ed.; Wiley: New York, NY, USA, 1997; pp. 65–105. [Google Scholar]
- KaneMaguire, L.A.P.; Kanitz, R.; Sheil, M.M. Electrospray mass spectrometry of neutral pi-hydrocarbon organometallic complexes. Inorg. Chim. Acta 1996, 245, 209–214. [Google Scholar] [CrossRef]
- Vandell, V.E.; Limbach, P.A. Electrospray ionization mass spectrometry of metalloporphyrins. J. Mass Spectrom. 1998, 33, 212–220. [Google Scholar] [CrossRef]
- Lavanant, H.; Hecquet, E.; Hoppilliard, Y. Complexes of L-histidine with Fe2+, Co2+, Ni2+, Cu2+, Zn2+ studied by electrospray ionization mass spectrometry. Int. J. Mass Spectrom. 1999, 187, 11–23. [Google Scholar] [CrossRef]
- Van Berkel, G.J.; Giles, G.E.; Bullock, J.S.; Gray, L.J. Computational simulation of redox reactions within a metal electrospray emitter. Anal. Chem. 1999, 71, 5288–5296. [Google Scholar] [CrossRef] [PubMed]
- Van Berkel, G.J.; Asano, K.G.; Schnier, P.D. Electrochemical processes in a wire-in-a-capillary bulk-loaded, nano-electrospray emitter. J. Am. Soc. Mass Spectrom. 2001, 12, 853–862. [Google Scholar] [CrossRef]
- Van Berkel, G.J.; Asano, K.G.; Kertesz, V. Enhanced study and control of analyte oxidation in electrospray using a thin-channel, planar electrode emitter. Anal. Chem. 2002, 74, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Van Berkel, G.J.; Asano, K.G.; Gragner, M.C. Controlling analyte electrochemistry in an electrospray ion source with a three-electrode emitter cell. Anal. Chem. 2004, 76, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zimmerman, D.M.; Chung-Phillips, A.; Cassady, C.J. Experimental and ab initio studies of the gas-phase basicities of polyglycines. J. Am. Chem. Soc. 1993, 115, 10812–10822. [Google Scholar] [CrossRef]
- Blades, A.T.; Jayaweera, P.; Ikonomou, M.G.; Kebarle, P. Studies of alkaline-earth and transition-metal M+2 gas-phase ion chemistry. J. Chem. Phys. 1990, 92, 5900–5906. [Google Scholar] [CrossRef]
- Gianelli, L.; Amendola, V.; Fabbrizzi, L.; Pallavicini, P.; Mellerio, G.G. Investigation of reduction of Cu(II) complexes in positive-ion mode electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 2001, 15, 2347–2353. [Google Scholar] [CrossRef]
- Vachet, R.W.; Hartman, J.A.R.; Callahan, J.H. Ion-molecule reactions in a quadrupole ion trap as a probe of the gas-phase structure of metal complexes. J. Mass Spectrom. 1998, 33, 1209–1225. [Google Scholar] [CrossRef]
- Vachet, R.W.; Callahan, J.H. Gas, solution, and solid state coordination environments for the nickel(II) complexes of a series of aminopyridine ligands of varying coordination number. Inorg. Chim. Acta 2000, 297, 79–87. [Google Scholar]
- Vachet, R.W.; Hartman, J.R.; Gertner, J.W.; Callahan, J.H. Investigation of metal complex coordination structure using collision-induced dissociation and ion-molecule reactions in a quadrupole ion trap mass spectrometer. Int. J. Mass Spectrom. 2001, 204, 101–112. [Google Scholar] [CrossRef]
- Hartman, J.R.; Vachet, R.W.; Pearson, W.; Wheat, R.J.; Callahan, J.H. A comparison of the gas, solution, and solid state coordination environments for the copper(II) complexes of a series of aminopyridine ligands of varying coordination number. Inorg. Chim. Acta 2003, 343, 119–132. [Google Scholar] [CrossRef]
- Schäfer, A.; Fischer, B.; Paul, H.; Bosshard, R.; Hesse, M.; Viscontini, M. Pterin Chemistry.94. electrospray-ionization mass-spectrometry - Detection of a radical cation present in solution - New results on the chemistry of (tetrahydropteridinone)-metal complexes. Helv. Chim. Acta 1992, 75, 1955–1964. [Google Scholar] [CrossRef]
- Schäfer, A.; Paul, H.; Fischer, B.; Hesse, M.; Viscontini, M. Reaction of 5,6,7,8-tetrahydropterin with Iron(III) acetylacetonate - Detection of radical Cations by electrospray-ionization mass-spectrometry. Helv. Chim. Acta 1995, 78, 1763–1776. [Google Scholar] [CrossRef]
- Aliprantis, A.O.; Canary, J.W. Observation of catalytic Intermediates in the Suzuki reaction by electrospray mass-spectrometry. J. Am. Chem. Soc. 1994, 116, 6985–6986. [Google Scholar] [CrossRef]
- Moreno-Manas, M.; Perez, M.; Pleixats, R. Palladium-catalyzed Suzuki-type self-coupling of arylboronic acids.A mechanistic study. J. Org. Chem. 1996, 61, 2346–2351. [Google Scholar] [CrossRef]
- Hinderling, C.; Chen, P. Rapid screening of olefin polymerization catalyst libraries by electrospray ionization tandem mass spectrometry. Angew. Chem. Int. Ed. 1999, 38, 2253–2556. [Google Scholar] [CrossRef]
- Hinderling, C.; Chen, P. Mass spectrometric assay of polymerization catalysts for combinational screening. Int. J. Mass Spectrom. 2000, 195, 377–383. [Google Scholar] [CrossRef]
- Hambitzer, G.; Heitbaum, J. Electrochemical thermospray mass spectrometry. Anal. Chem. 1986, 58, 1067–1070. [Google Scholar] [CrossRef]
- Sam, J.W.; Tang, X.J.; Magliozzo, R.S.; Peisach, J. Electrospray mass-spectrometry of iron bleomycin-II - Investigation of the reaction of Fe(III) bleomycin with iodosylbenzene. J. Am. Chem. Soc. 1995, 117, 1012–1218. [Google Scholar] [CrossRef]
- Griep-Raming, J.; Meyer, S.; Bruhn, T.; Metzger, J.O. Investigation of reactive intermediates of chemical reactions in solution by electrospray ionization mass spectrometry: radical chain reactions. Angew. Chem. Int. Ed. 2002, 41, 2738–2742. [Google Scholar] [CrossRef]
- Meyer, S.; Metzger, J.O. Use of electrospray ionization mass spectrometry for the investigation of radical cation chain reactions in solution: detection of transient radical cations. Anal. Bioanal. Chem. 2003, 377, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S.; Pavam, C.H.; Almeida, W.P.; Coelho, F.; Eberlin, M.N. Probing the mechanism of the Baylis-Hillman reaction by electrospray ionization mass and tandem mass spectrometry. Angew. Chem. Int. Ed. 2004, 43, 4330–4333. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.M.R.; Rabe, J. A new, efficient and stereocontrolled synthesis of trisubstituted alkenes via functionalized acrylic esters. Angew. Chem. Int. Ed. 1983, 22, 796–797. [Google Scholar]
- Bode, M.L.; Kaye, P.T. A kinetic and mechanistic study of the Baylis−Hillman reaction. Tetrahedron Lett. 1991, 32, 5611–5614. [Google Scholar] [CrossRef]
- Fort, Y.; Berthe, M.C.; Caubere, P. The Baylis−Hillman reaction-Mechanism and applications revisited. Tetrahedron Lett. 1992, 48, 6371–6384. [Google Scholar] [CrossRef]
- Santos, L.S.; DaSilveira Neto, B.A.; Consorti, C.S.; Pavam, C.H.; Almeida, W.P.; Coelho, F.; Dupont, J.; Eberlin, M.N. The role of ionic liquids in co-catalysis of Baylis−Hillman reaction: Interception of supramolecular species via electrospray ionization mass spectrometry. J. Phys. Org. Chem. 2006, 19, 731–736. [Google Scholar] [CrossRef]
- Rosa, J.N.; Afonso, C.A.M.; Santos, A.G. Ionic liquids as a recyclable reaction medium for the Baylis-Hillman reaction. Tetrahedron Lett. 2001, 57, 4189–4193. [Google Scholar] [CrossRef]
- Amarante, G.W; Milagre, H.M; Vaz, B.G; Vilacha Ferreira, B.R.; Eberlin, M.N.; Coelho, F. Dualistic nature of the mechanism of the Morita-Baylis-Hillman reaction probed by electrospray ionization mass spectrometry. J. Org. Chem. 2009, 74, 3031–3037. [Google Scholar] [CrossRef] [PubMed]
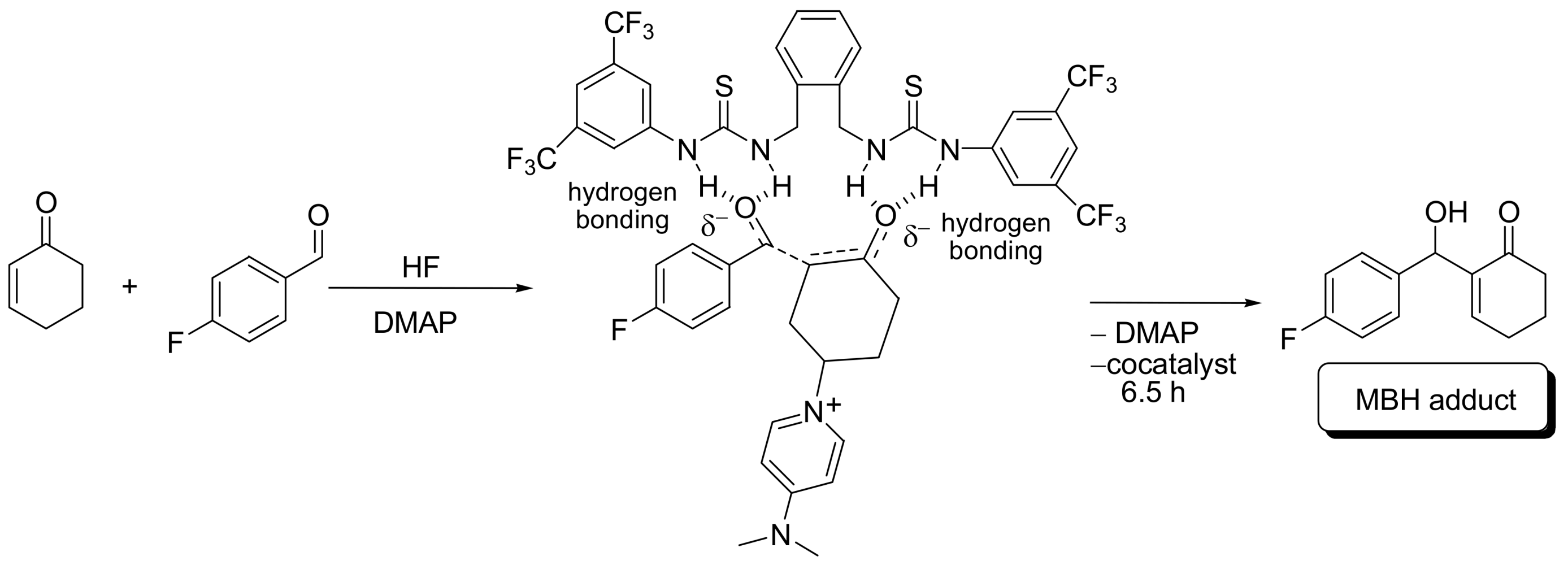
- Amarante, G.W.; Benassi, M.; Milagre, H.M.S.; Braga, A.A.C.; Maseras, F.; Eberlin, M.N.; Coelho, F. Brønsted Acid-catalyzed Morita-Baylis-Hillman reaction: A new mechanistic view for (Thio)ureas revealed by ESI-MS monitoring and DFT calculations. Chem. Eur. J. 2009. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carrasco-Sanchez, V.; Simirgiotis, M.J.; Santos, L.S. The Morita-Baylis-Hillman Reaction: Insights into Asymmetry and Reaction Mechanisms by Electrospray Ionization Mass Spectrometry. Molecules 2009, 14, 3989-4021. https://doi.org/10.3390/molecules14103989
Carrasco-Sanchez V, Simirgiotis MJ, Santos LS. The Morita-Baylis-Hillman Reaction: Insights into Asymmetry and Reaction Mechanisms by Electrospray Ionization Mass Spectrometry. Molecules. 2009; 14(10):3989-4021. https://doi.org/10.3390/molecules14103989
Chicago/Turabian StyleCarrasco-Sanchez, Verónica, Mario J. Simirgiotis, and Leonardo S. Santos. 2009. "The Morita-Baylis-Hillman Reaction: Insights into Asymmetry and Reaction Mechanisms by Electrospray Ionization Mass Spectrometry" Molecules 14, no. 10: 3989-4021. https://doi.org/10.3390/molecules14103989
APA StyleCarrasco-Sanchez, V., Simirgiotis, M. J., & Santos, L. S. (2009). The Morita-Baylis-Hillman Reaction: Insights into Asymmetry and Reaction Mechanisms by Electrospray Ionization Mass Spectrometry. Molecules, 14(10), 3989-4021. https://doi.org/10.3390/molecules14103989