Imidazole-based Potential Bi- and Tridentate Nitrogen Ligands: Synthesis, Characterization and Application in Asymmetric Catalysis

Abstract
:Introduction
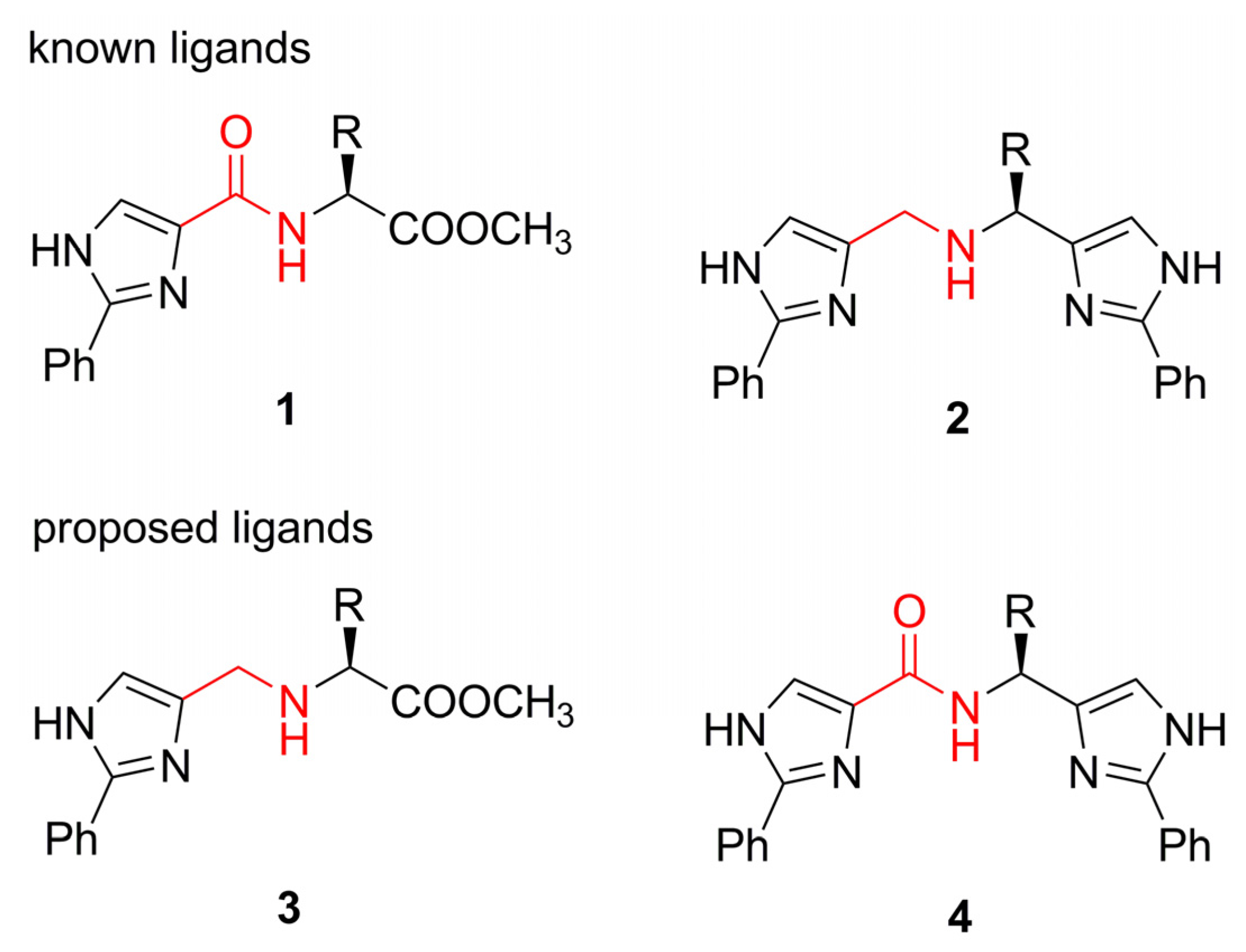
Results and Discussion
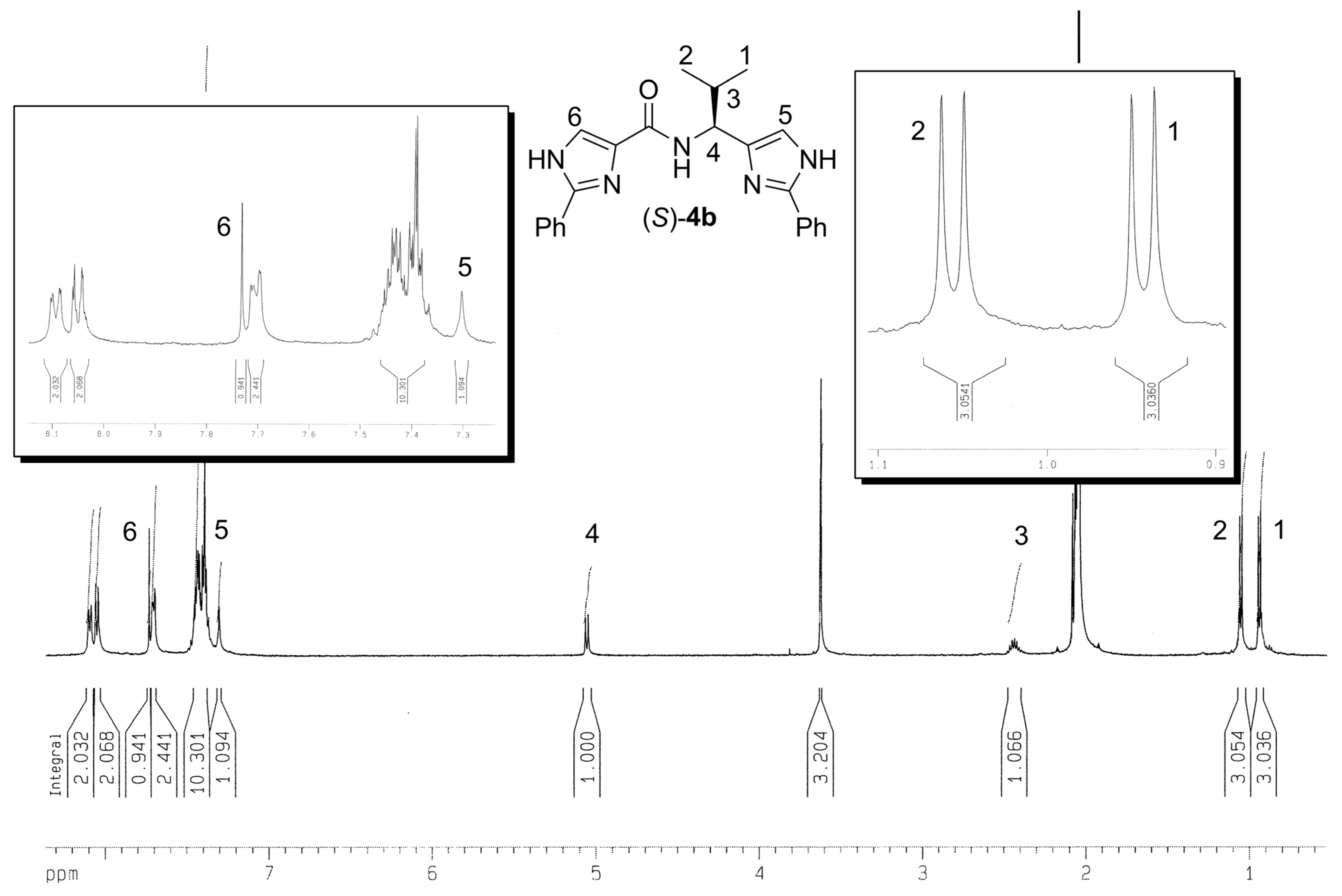
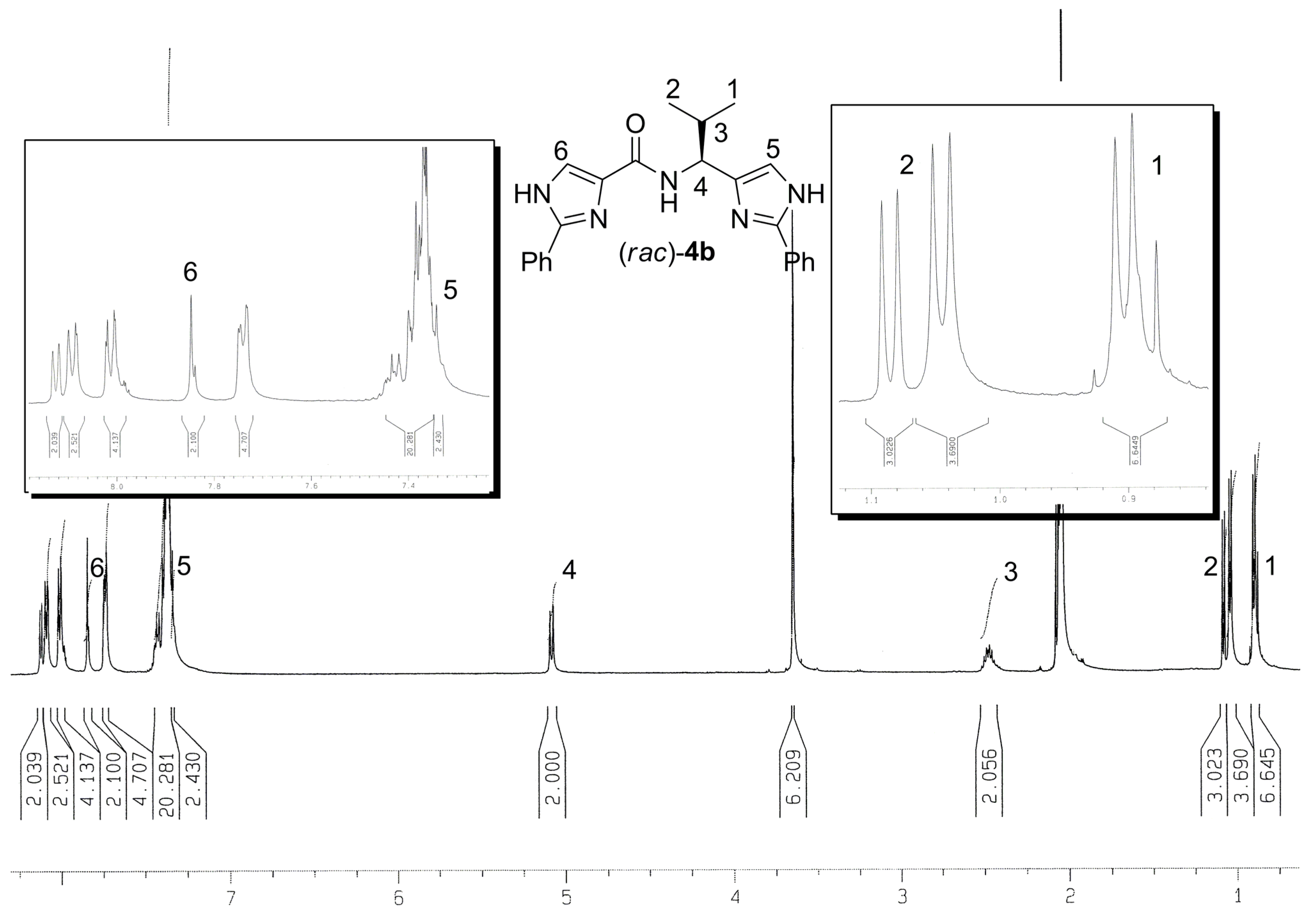
Ligand synthesis
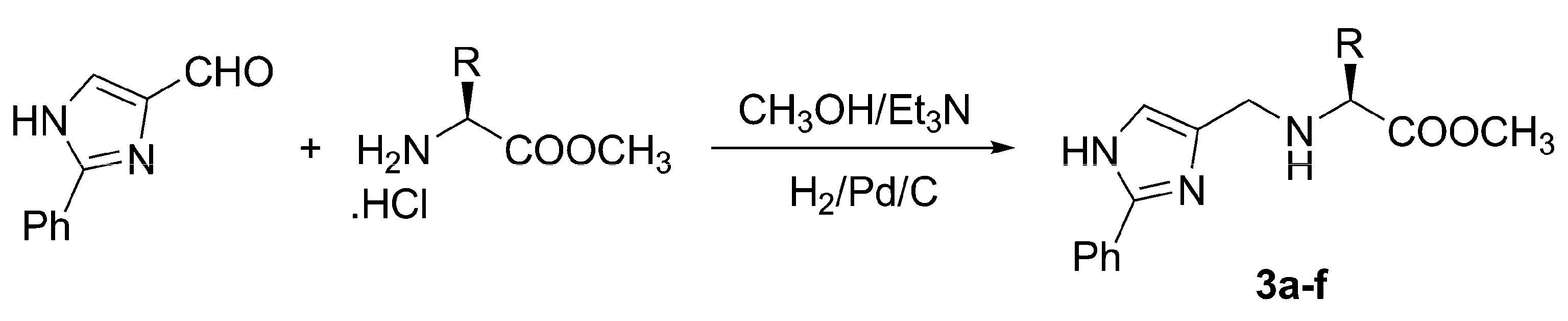

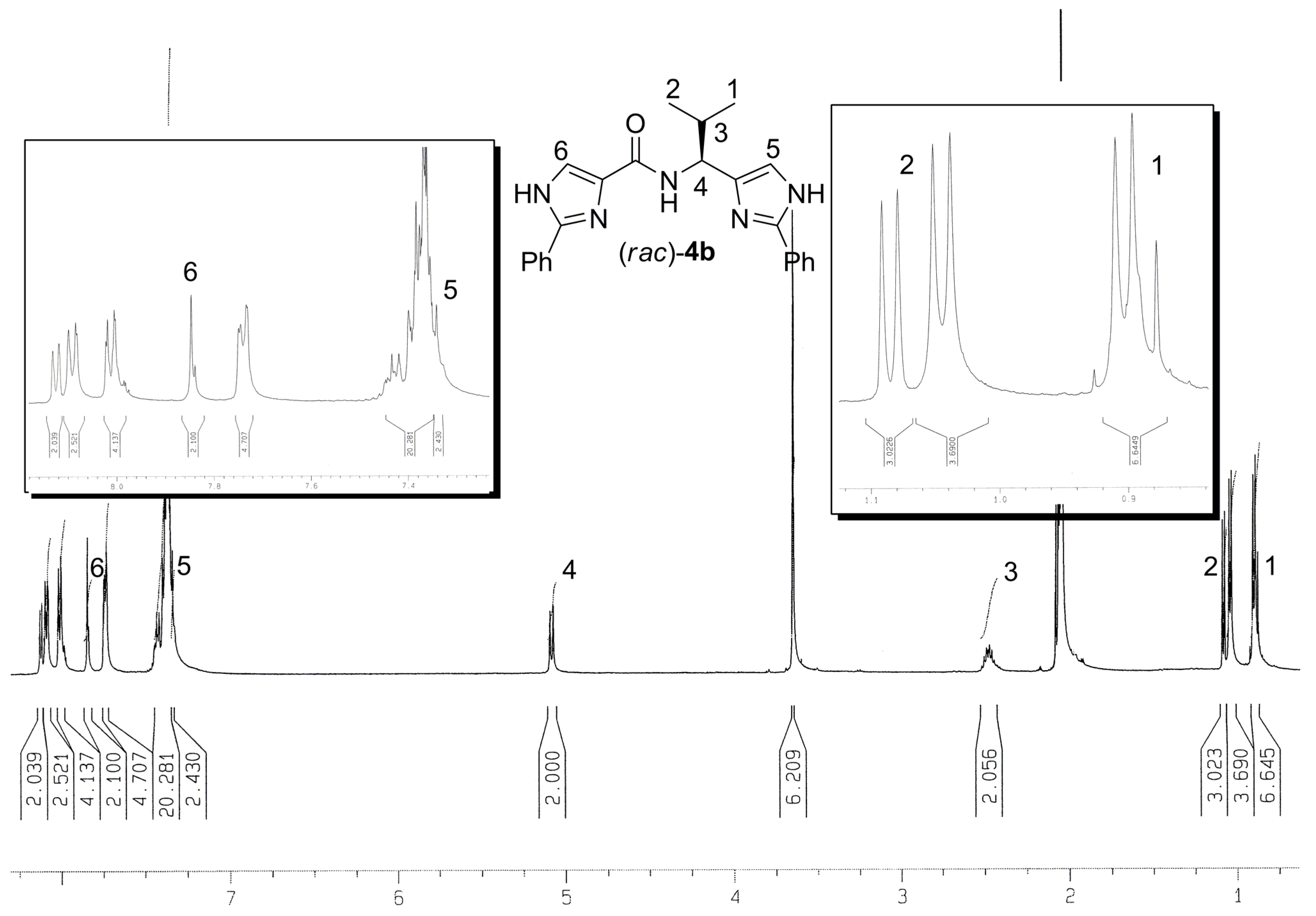
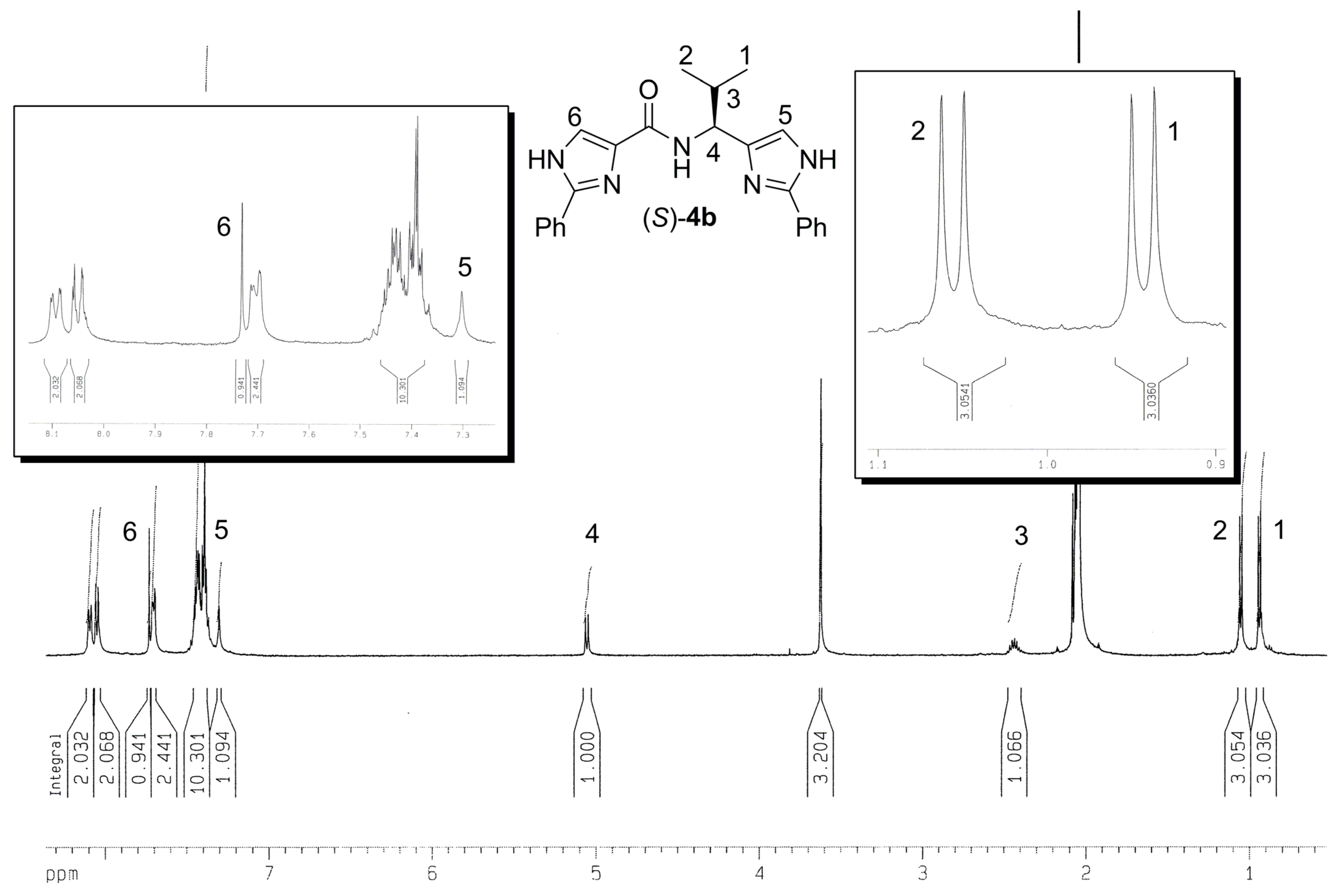{kind=link}
{kind=link}
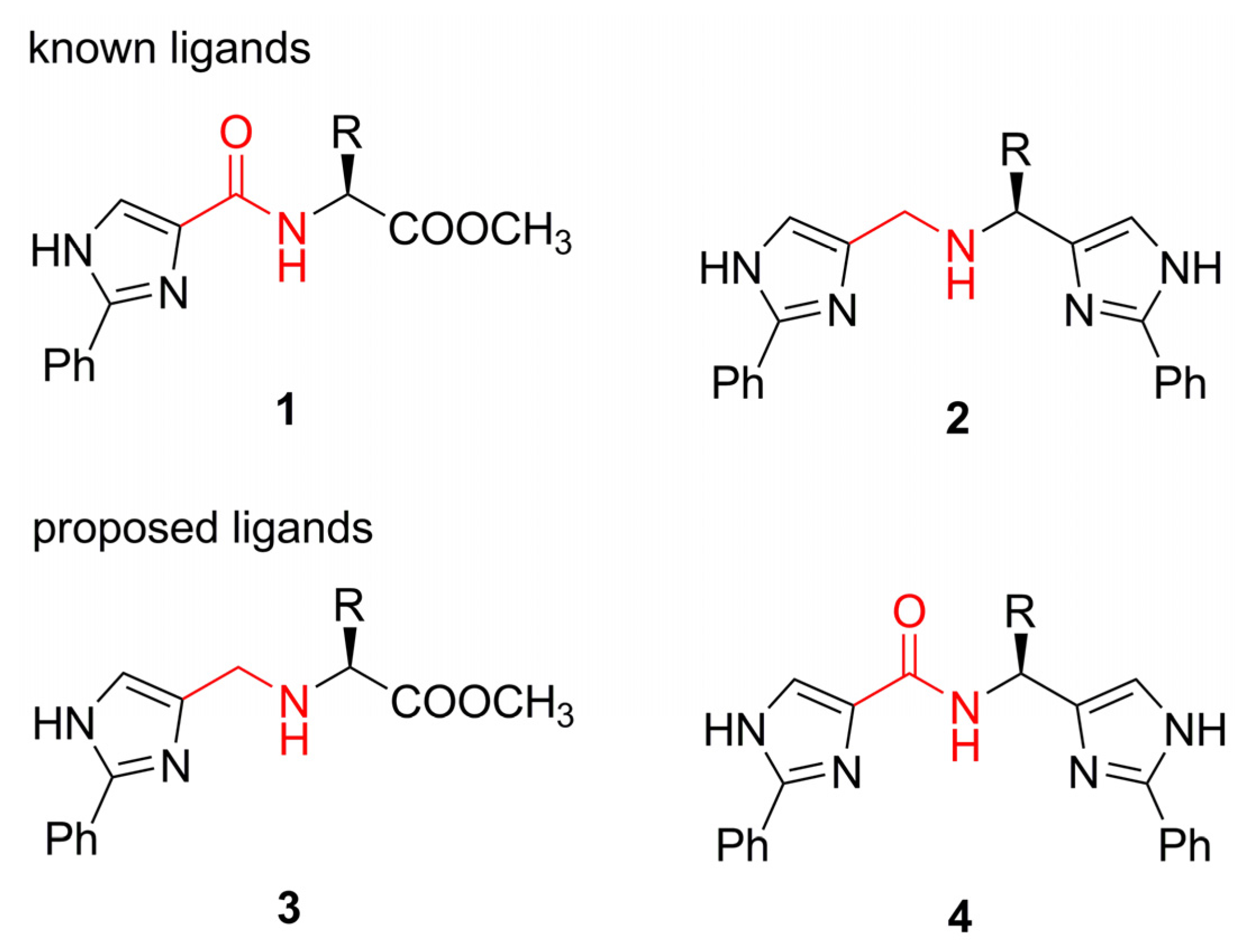{kind=link}
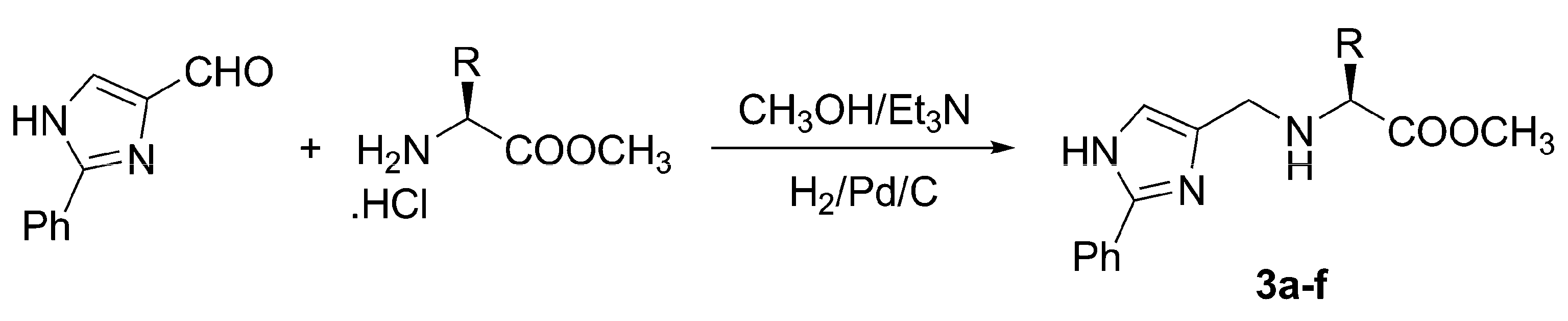{kind=link}
{kind=link}
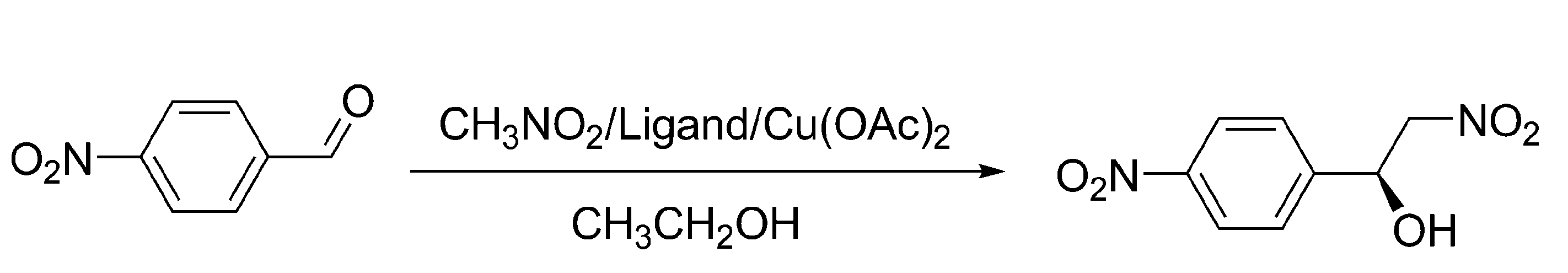{kind=link}
| Comp. | R / Source of chirality | Yield [%] | e.e. [%] | [α]D20 (c 0.05, CH3OH) |
|---|---|---|---|---|
| 3a | CH3 / (S)-Alanine | 56 | > 95 | -8.9 |
| 3b | CH(CH3)2 / (S)-Valine | 73 | > 95 | -22.8 |
| 3c | CH2CH(CH3)2 / (S)-Leucine | 34 | > 95 | -22.0 |
| 3d | CH(CH3)CH2CH3 / (S)-Isoleucine | 38 | > 95 | -9.2 |
| 3e | CH2Ph / (S)-Phenylalanine | 66 | > 95 | -13.4 |
| 3f | Ph / (R)-Phenylglycine | 23 | > 95 | -16.7 |
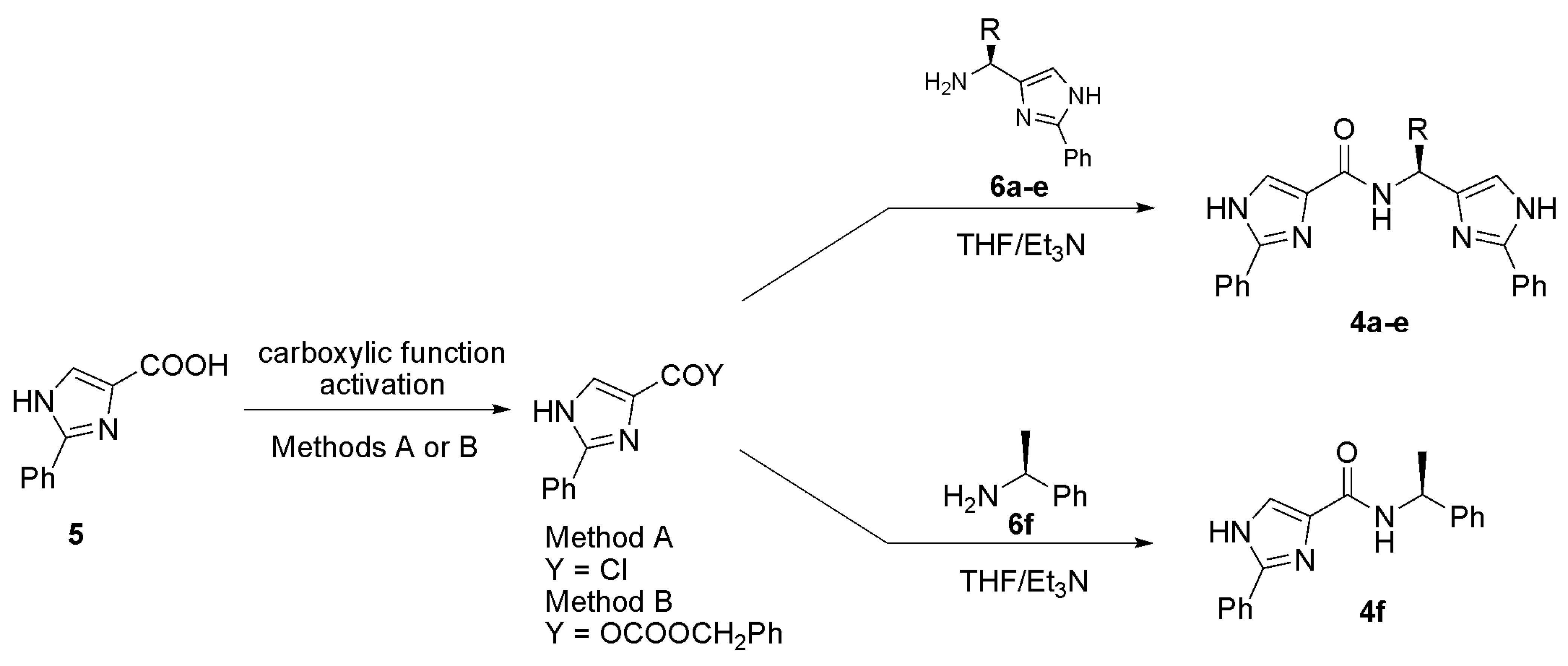
| Comp. | R / Source of chirality | Yield[a] [%] | e.e. [%] | [α]D20 (c 0.05, CH3OH) |
|---|---|---|---|---|
| 4a | CH3 / (S)-Alanine | 23/24 | > 95 | +95.6 |
| 4b | CH(CH3)2 / (S)-Valine | 30/35 | > 95 | +48.0 |
| 4c | CH2CH(CH3)2 / (S)-Leucine | 16/25 | > 95 | +48.8 |
| 4d | CH(CH3)CH2CH3 / (S)-Isoleucine | 13/34 | > 95 | +36.0 |
| 4e | CH2Ph / (S)-Phenylalanine | 17/22 | > 95 | +33.0 |
| 4f | CH3 / (S)-1-Phenylethanamine | 44/42 | > 95 | +142.0 |
Asymmetric catalysis
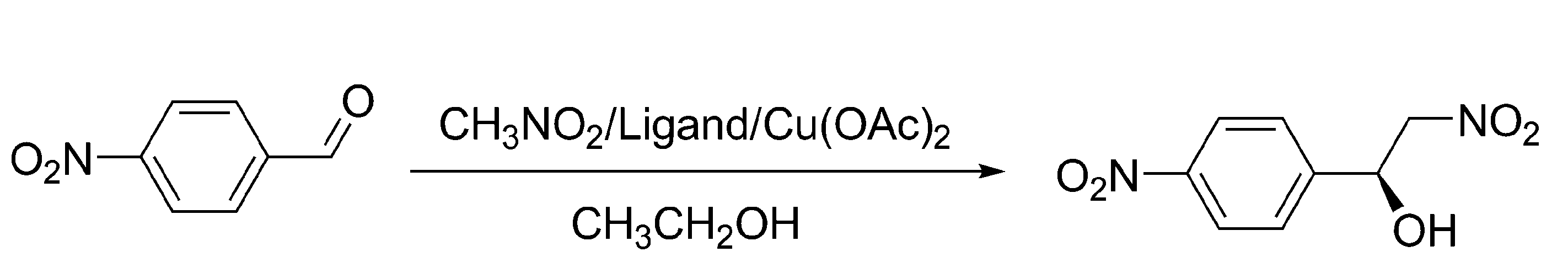
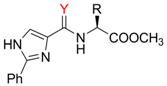 | 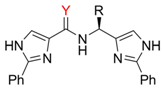 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Lig. | R |  | Yield [%] | ee [%] | Lig. | R | 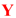 | Yield [%] | ee [%] |
| 3a | CH3 | H, H | 98 | 10 | 4a | CH3 | O | 94 | 10 |
| 3b | CH(CH3)2 | H, H | 96 | 7 | 4b | CH(CH3)2 | O | 91 | 6 |
| 3c | CH2CH(CH3)2 | H, H | 94 | 15 | 4c | CH2CH(CH3)2 | O | 94 | 14 |
| 3d | CH(CH3)CH2CH3 | H, H | 97 | 10 | 4d | CH(CH3)CH2CH3 | O | 89 | 8 |
| 3e | CH2Ph | H, H | 95 | 14 | 4e | CH2Ph | O | 99 | 15 |
| 3f | Ph | H, H | 93 | 9 | 4f | see Scheme 3 | O | 91 | 5 |
| 1a[a] | CH3 | O | 79 | 1 | 2a[b] | CH3 | H, H | 94 | 13 |
| 1b[a] | CH(CH3)2 | O | 84 | 3 | 2b[b] | CH(CH3)2 | H, H | 95 | 13 |
| 1c[a] | CH2CH(CH3)2 | O | 85 | 8 | 2c[b] | CH2CH(CH3)2 | H, H | 96 | 15 |
| 1d[a] | CH(CH3)CH2CH3 | O | 91 | 4 | 2d[c] | CH(CH3)CH2CH3 | H, H | - | - |
| 1e[a] | CH2Ph | O | 90 | 3 | 2e[b] | CH2Ph | H, H | 96 | 19 |
| 1f[a] | Ph | O | 70 | 4 | |||||
Conclusions
Experimental
General
General method for reductive amination. Preparation of 3a-f
General procedure for the preparation of 4a-f
Method A
Method B
Acknowledgements
References
- Corelli, F.; Summa, V.; Brogi, A.; Monteagudo, E.; Botta, M. Chiral azole derivatives. 2.1 Synthesis of enantiomerically pure 1-alkylimidazoles. J. Org. Chem. 1995, 60, 2008–2015. [Google Scholar] [CrossRef]
- Groarke, M.; McKervey, M. A.; Nieuwenhuyzen, M. Synthesis of amino acid-derived imidazoles from enantiopure N-protected α-amino glyoxals. Tetrahedron Lett. 2000, 41, 1275–1278. [Google Scholar]
- Rűther, T.; Done, M.C.; Cavell, K.J.; Peacock, E.J.; Skelton, B.W.; White, A.H. Novel methylpalladium(II) complexes bearing tridentate imidazole-based chelate ligands: synthesis, structural characterization, and reactivity. Organometallics 2001, 20, 5522–5531. [Google Scholar]
- Bureš, F.; Kulhánek, J. Chiral imidazole derivatives synthesis from enantiopure N-protected α-amino acids. Tetrahedron Asymmetry 2005, 16, 1347–1354. [Google Scholar]
- Jiang, H.-Y.; Zhou, C.-H.; Luo, K.; Chen, H.; Lan, J.-B.; Xie, R.-G. Chiral imidazole metalloenzyme models: synthesis and enantioselective hydrolysis for α-amino acid esters. J. Mol. Catal. A: Chem. 2006, 260, 288–294. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Ishida, Y.; Sasaki, D.; Saigo, K. Synthesis of enantiopure 1-substituted, 1,2‒disubstituted, and 1,4,5-trisubstituted imidazoles from 1,2-amino alcohols. Tetrahedron 2006, 62, 8199–8206. [Google Scholar] [CrossRef]
- Marek, A.; Kulhánek, J.; Ludwig, M.; Bureš, F. Facile synthesis of optically active imidazole derivatives. Molecules 2007, 12, 1183–1190. [Google Scholar] [CrossRef]
- Thomas, P.J.; Axtell, A.T.; Klosin, J.; Peng, W.; Rand, C.L.; Clark, T.P.; Landis, C.R.; Abboud, K.A. Asymmetric hydroformylation of vinyl acetate: Application in the synthesis of optically active isoxazolines and imidazoles. Org. Lett. 2007, 9, 2665–2668. [Google Scholar] [CrossRef]
- Gulevich, A.V.; Balenkova, E.S.; Nenajdenko, V.G. The first example a diastereoselective Thio-Ugi reaction: A new synthetic approach to chiral imidazole derivatives. J. Org. Chem. 2007, 72, 7878–7885. [Google Scholar] [CrossRef]
- Mlostoń, G.; Mucha, P.; Urbaniak, K.; Broda, K.; Heimgartner, H. Synthesis of optically active 1−(1-phenylethyl)-1H-imidazoles derived from 1-phenylethylamine. Helv. Chim. Acta 2008, 91, 232–236. [Google Scholar] [CrossRef]
- Gerstenberg, B.S.; Lin, J.; Mimieux, Y.S.; Brown, L.E.; Oliver, A.G.; Konopelski, J.P. Structural characterization of an enantiomerically pure amino acid imidazolide and direct formation of the β-lactam nucleus from an α-amino acid. Org. Lett. 2008, 10, 369–372. [Google Scholar] [CrossRef]
- De Luca, L. Naturally occurring and synthetic imidazoles: Their chemistry and their biological activities. Curr. Med. Chem. 2006, 13, 1–23. [Google Scholar]
- Boiani, M.; González, M. Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini-Rev. Med. Chem. 2005, 5, 409–424. [Google Scholar] [CrossRef]
- Suwiński, J.; Szczepankeiwicz, W.; Świerczek, K.; Walczak, K. Synthesis of chiral imidazole derivatives as purine precursors. Eur. J. Org. Chem. 2003, 1080–1084. [Google Scholar]
- Katsuki, I.; Motoda, Y.; Sunatsuki, Y.; Matsumoto, N.; Nakashima, T.; Kojima, M. Spontaneous resolution induced by self-organization of chiral self-complementary cobalt(III) complexes with achiral tripod-type ligands containing three imidazole groups. J. Am. Chem. Soc. 2002, 124, 629–640. [Google Scholar]
- Nakamura, H.; Fujii, M.; Sunatsuki, Y.; Kojima, M.; Matsumoto, N. Cobalt(III) complexes of a tripodal ligand containing three imidazole groups: Properties and structures of racemic and optically active species. Eur. J. Inorg. Chem. 2008, 1258–1267. [Google Scholar]
- Bureš, F.; Szotkowski, T.; Kulhánek, J.; Pytela, O.; Ludwig, M.; Holčapek, M. Novel nitrogen ligands based on imidazole derivatives and their application in asymmetric catalysis. Tetrahedron Asymmetry 2006, 17, 900–907. [Google Scholar] [CrossRef]
- Hojabri, L.; Hartikka, A.; Moghaddam, F.M.; Arvidsson, P.I. A new imidazole-containing imidazolidinone catalyst for organocatalyzed asymmetric conjugate addition of nitroalkanes to aldehydes. Adv. Synth. Catal. 2007, 349, 740–748. [Google Scholar] [CrossRef]
- Kotsuki, H.; Hayakawa, H.; Wakao, M.; Shimanouchi, T.; Ochi, M. Synthesis of novel chiral diazole ligands for enantioselective addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 1995, 6, 2665–2668. [Google Scholar] [CrossRef]
- Perl, N.R.; Leighton, J.L. Enantioselective Imidazole-Directed Allylation of Aldimines and Ketimines. Org. Lett. 2007, 9, 3699–3701. [Google Scholar]
- Shitama, H.; Katsuki, T. Synthesis of metal-(pentadentate-salen) complexes: Asymmetric epoxidation with aqueous hydrogen peroxide and asymmetric cyclopropanation (salenH2: N,N’-bis(salicylidene)ethylene-1,2-diamine. Chem. Eur. J. 2007, 13, 4849–4858. [Google Scholar]
- Gullotti, M.; Santagostini, L.; Pagliarin, R.; Granata, A.; Casella, L. Synthesis and characterization of new chiral octadentate nitrogen ligands and related copper(II) complexes as catalysts for stereoselective oxidation of catechols. J. Mol. Catal. A: Chem. 2005, 235, 271–284. [Google Scholar] [CrossRef]
- Hodgson, R.; Douthwaite, R.E. Synthesis and asymmetric catalytic application of chiral imidazollium-phosphines derived from (1R,2R)-trans-diaminocyclohexane. J. Organomet. Chem. 2005, 690, 5822–5831. [Google Scholar] [CrossRef]
- Sívek, R.; Pytela, O.; Bureš, F. Design and synthesis of optically active 2−phenylimdiazolecarboxamides featuring amino acid motive. J. Heterocyclic Chem. 2008, in press. [Google Scholar]
- Paul, R.; Brockman, J.A.; Hallett, W.A.; Hanifin, J.W.; Tarrant, M.E.; Torley, L.W.; Callahan, F.M.; Fabio, P.F.; Johnson, B.D.; Lenhard, R. H.; Schaub, R.E.; Wissner, A. Imidazo[1,5-d][1,2,4]triazines as potential antiasthma agents. J. Med. Chem. 1985, 28, 1704–1716. [Google Scholar] [CrossRef]
- Bull, S.D.; Davies, S.G.; Jones, S.; Sanganee, H.J. Asymmetric alkylation using SuperQuat auxiliaries – an investigation into the synthesis and stability of enolates derived from 5,5-disubstituted oxazolidin-2-ones. J. Chem. Soc., Perkin Trans. 1 1999, 387–398. [Google Scholar]
- Luzzio, F.A. The Henry reaction: recent examples. Tetrahedron 2001, 57, 915–945. [Google Scholar] [CrossRef]
- Evans, D.A.; Seidel, D.; Rueping, M.; Hon, Wai Lam; Shaw, J.T.; Downey, C.W. A new copper acetate-bis(oxazoline)-catalyzed, enantioselective Henry reaction. J. Am. Chem. Soc. 2003, 125, 12692–12693. [Google Scholar]
- Gan, C.; Lai, G.; Zhang, Z.; Wang, Z.; Zhou, M.-M. Efficient and enantioselective nitroaldol reaction catalyzed by copper Schiff-base complexes. Tetrahedron Asymmetry 2006, 17, 725–728. [Google Scholar] [CrossRef]
- Trost, B.M.; Yeh, V.S.C. A dinuclear Zn catalyst for the asymmetric nitroaldol (Henry) reaction. Angew. Chem. Int. Ed. Engl. 2002, 41, 861–863. [Google Scholar] [CrossRef]
- Sasai, H.; Suzuki, T.; Arai, S.; Arai, T.; Shibasaki, M. Basic character of rare earth metal alkoxides. Utilization in catalytic C-C bond-forming reactions and catalytic asymmetric nitroaldol reactions. J. Am. Chem. Soc. 1992, 114, 4418–4420. [Google Scholar]
- Sample Availability: Samples of compounds 3a-f and 4a-f are available from the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sívek, R.; Bureš, F.; Pytela, O.; Kulhánek, J. Imidazole-based Potential Bi- and Tridentate Nitrogen Ligands: Synthesis, Characterization and Application in Asymmetric Catalysis. Molecules 2008, 13, 2326-2339. https://doi.org/10.3390/molecules13092326
Sívek R, Bureš F, Pytela O, Kulhánek J. Imidazole-based Potential Bi- and Tridentate Nitrogen Ligands: Synthesis, Characterization and Application in Asymmetric Catalysis. Molecules. 2008; 13(9):2326-2339. https://doi.org/10.3390/molecules13092326
Chicago/Turabian StyleSívek, Roman, Filip Bureš, Oldřich Pytela, and Jiří Kulhánek. 2008. "Imidazole-based Potential Bi- and Tridentate Nitrogen Ligands: Synthesis, Characterization and Application in Asymmetric Catalysis" Molecules 13, no. 9: 2326-2339. https://doi.org/10.3390/molecules13092326
APA StyleSívek, R., Bureš, F., Pytela, O., & Kulhánek, J. (2008). Imidazole-based Potential Bi- and Tridentate Nitrogen Ligands: Synthesis, Characterization and Application in Asymmetric Catalysis. Molecules, 13(9), 2326-2339. https://doi.org/10.3390/molecules13092326