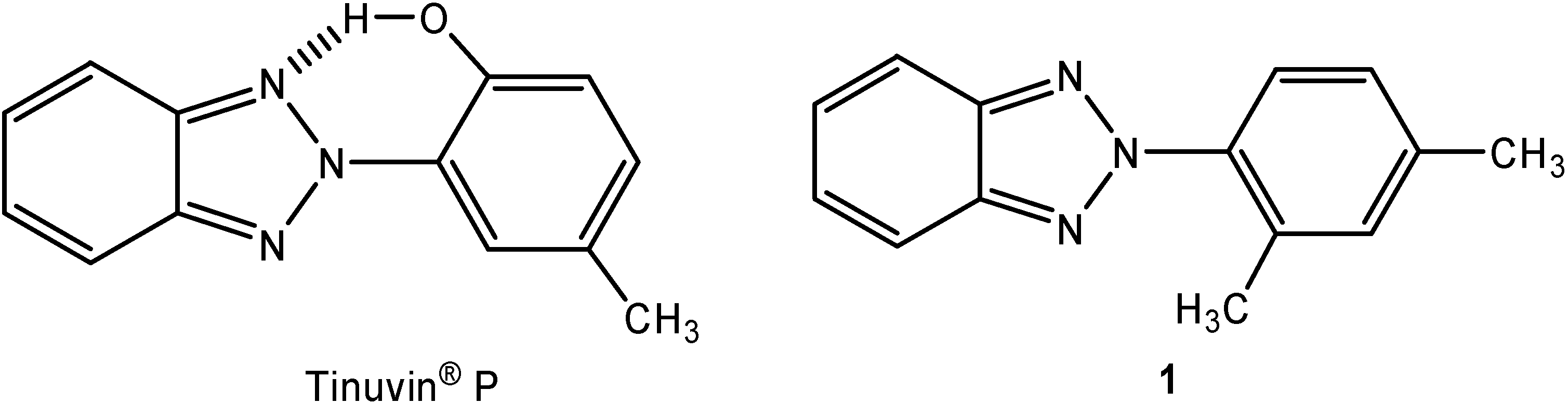
Structural Studies of Two Tinuvin® P Analogs: 2-(2,4-Dimethyl-phenyl)-2H-benzotriazole and 2-Phenyl-2H-benzotriazole

 ,
,
Abstract
:Introduction
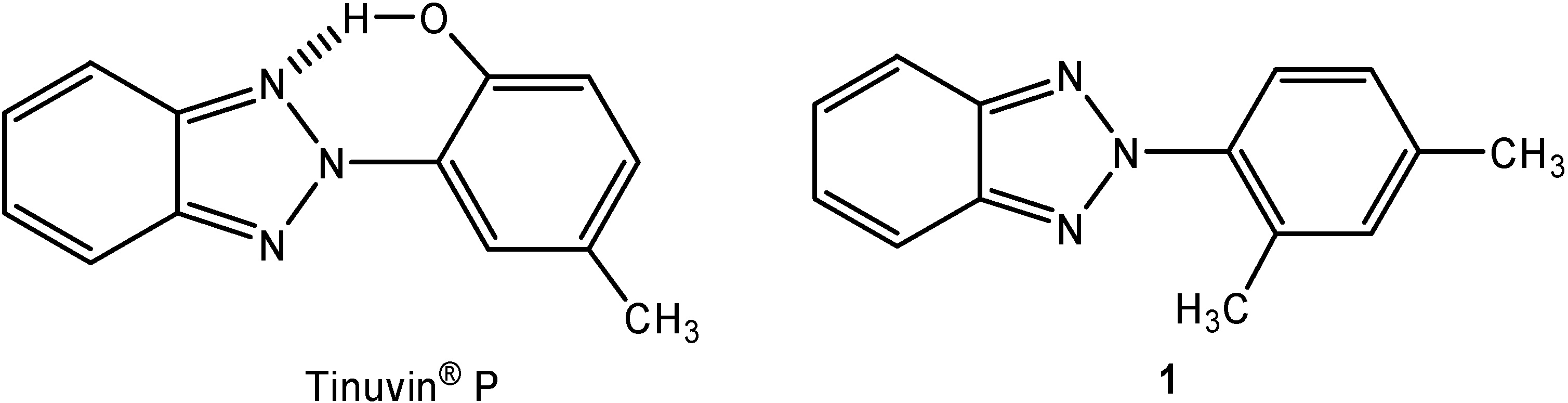
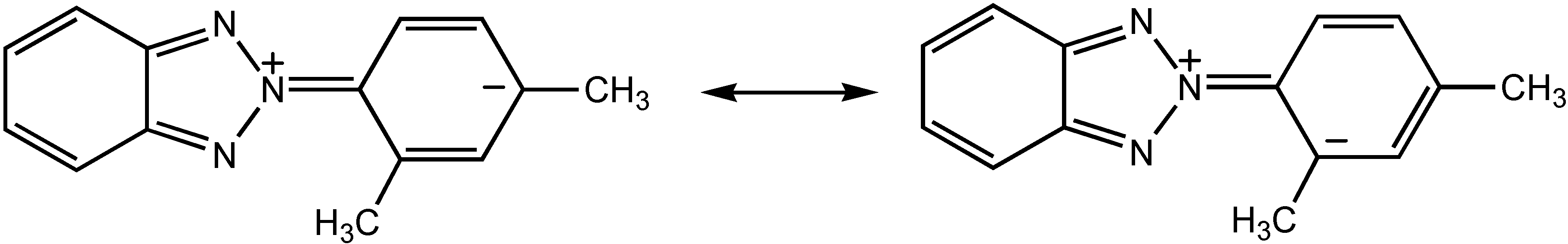
Results and Discussion
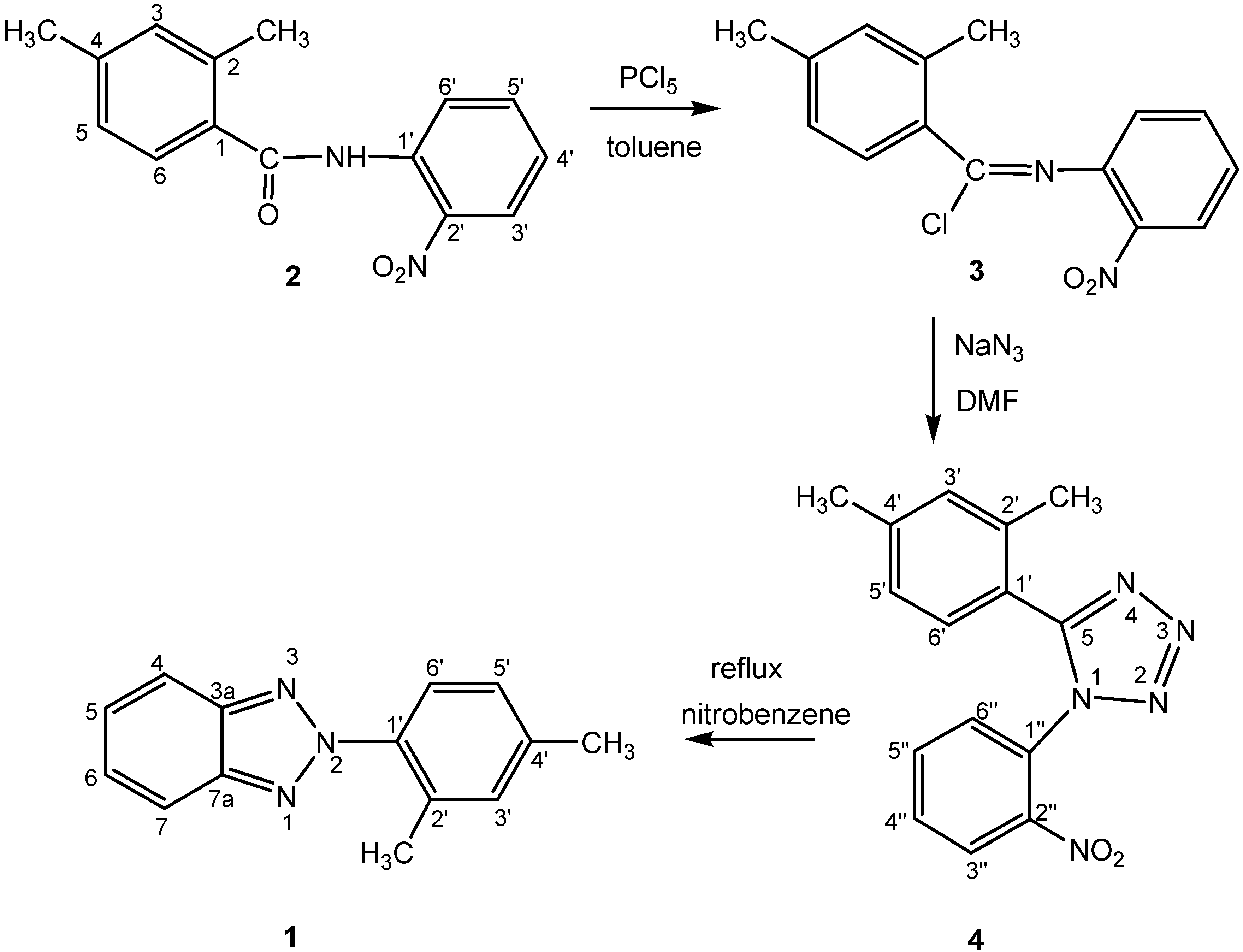
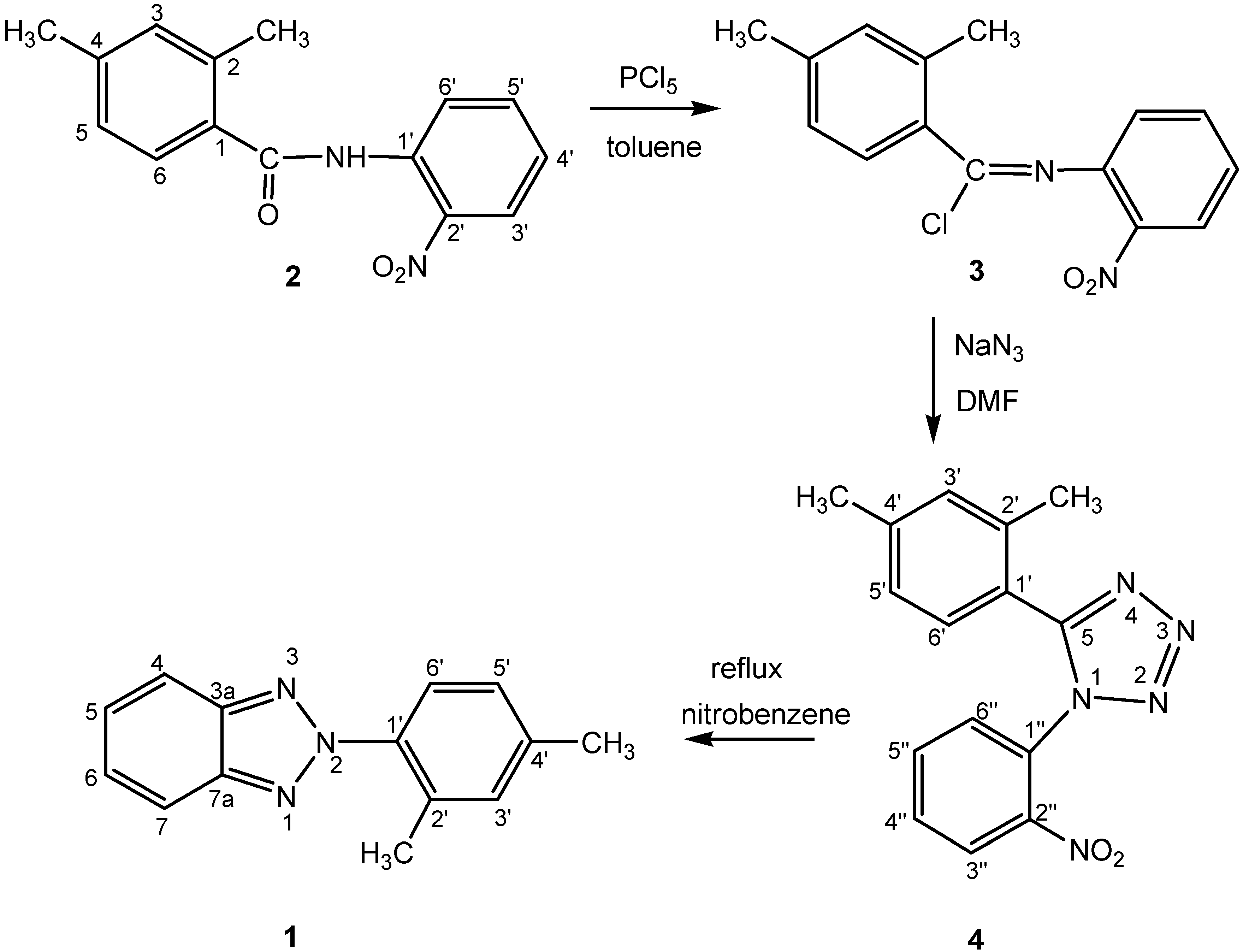
Synthesis
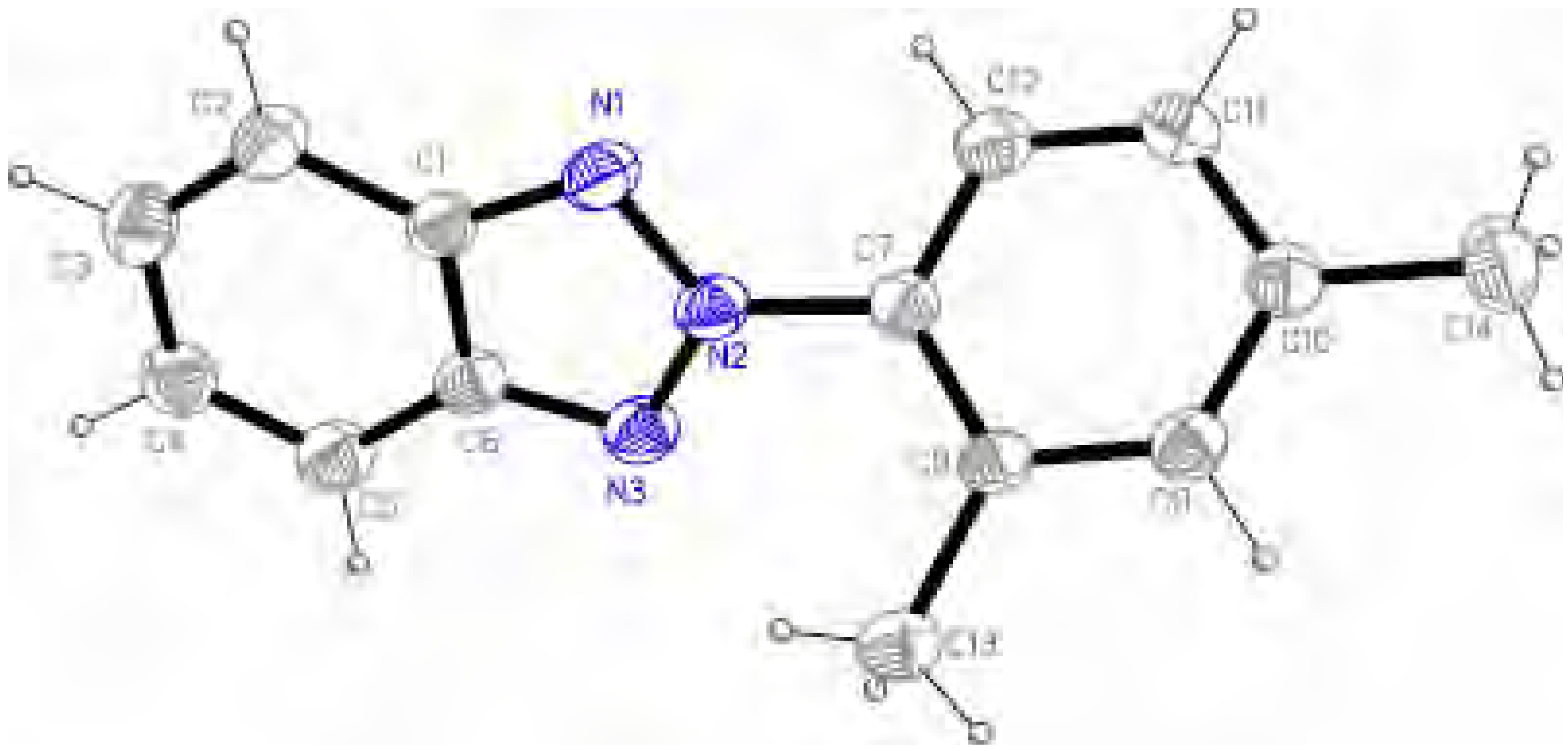
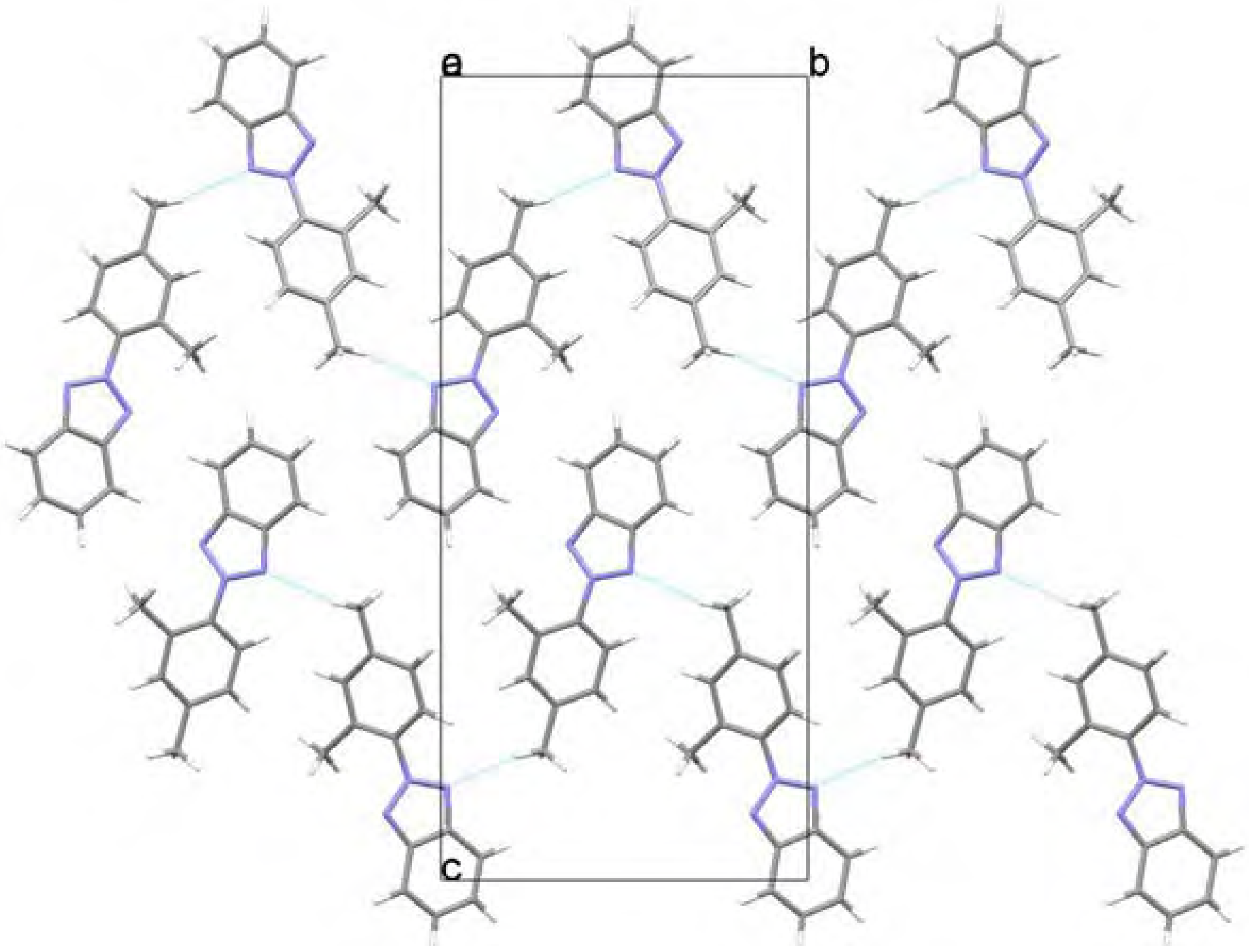
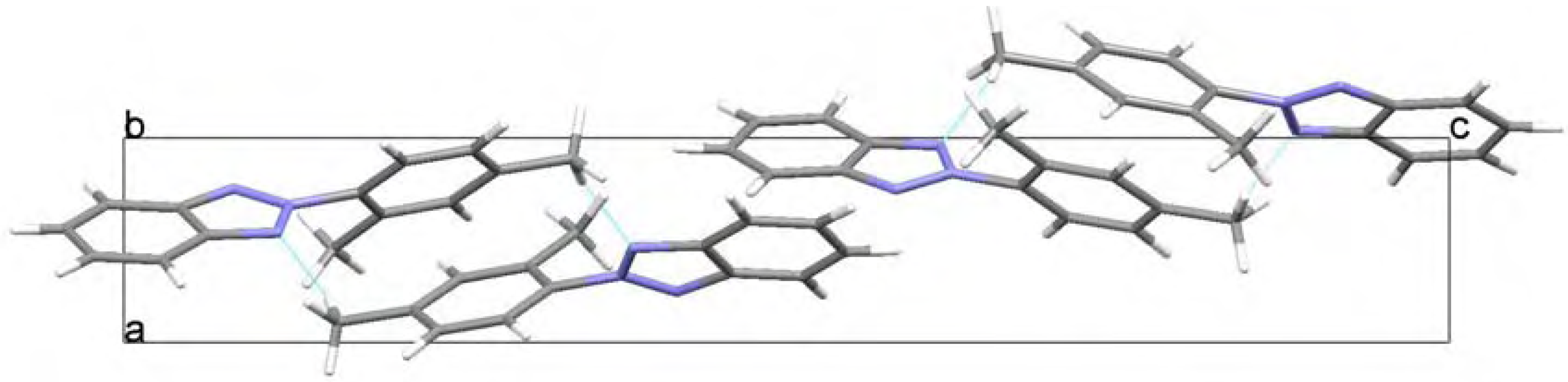
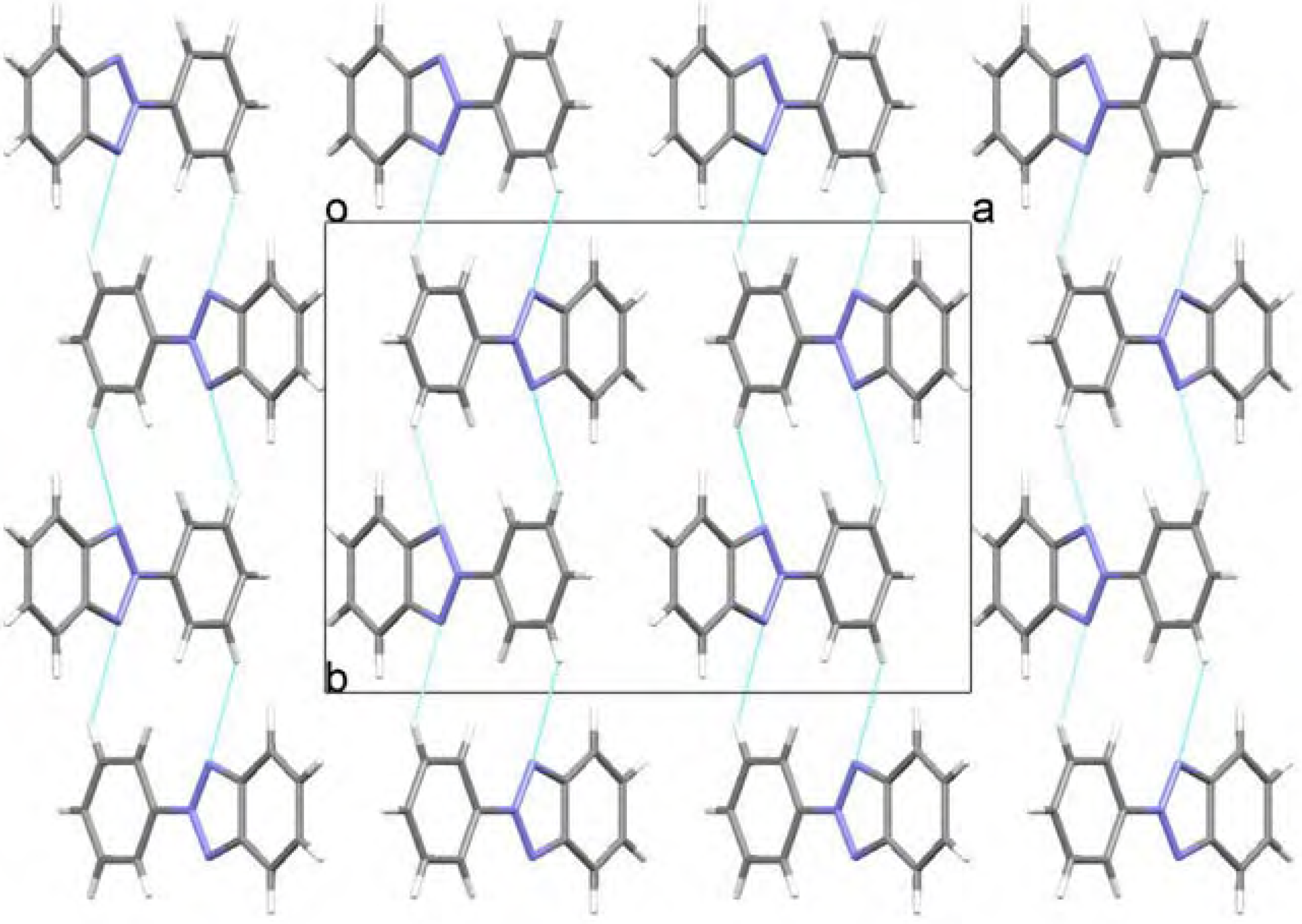
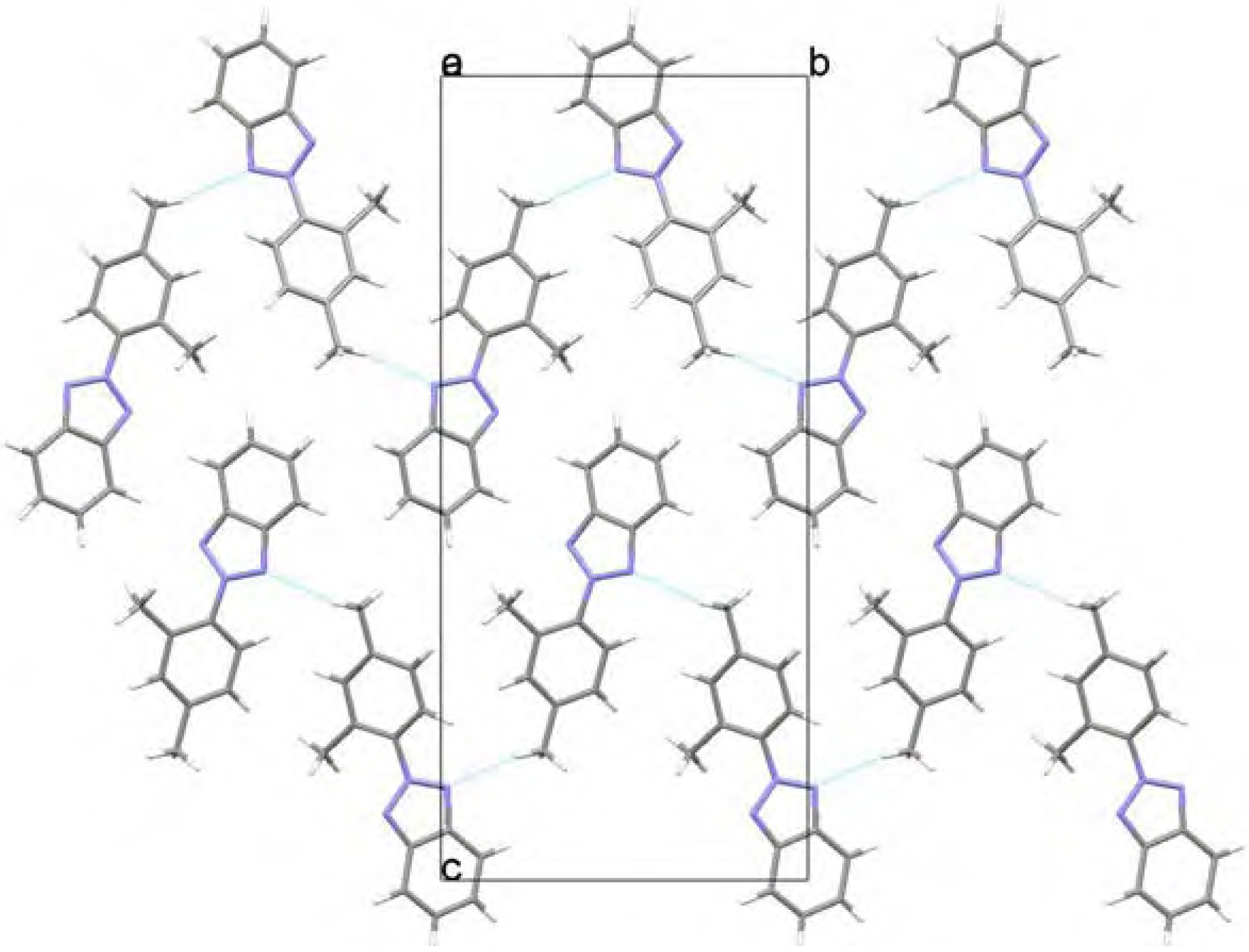
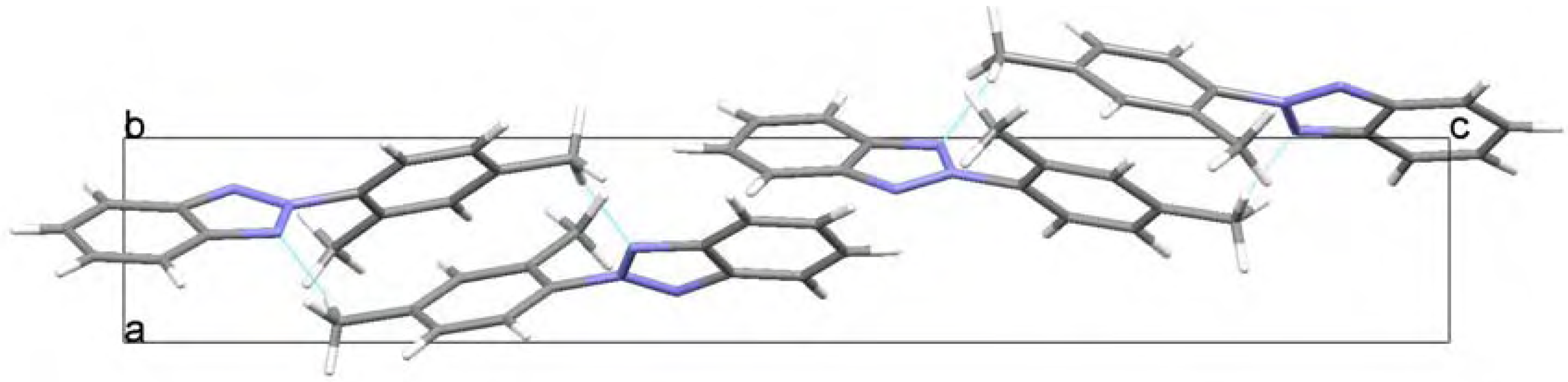
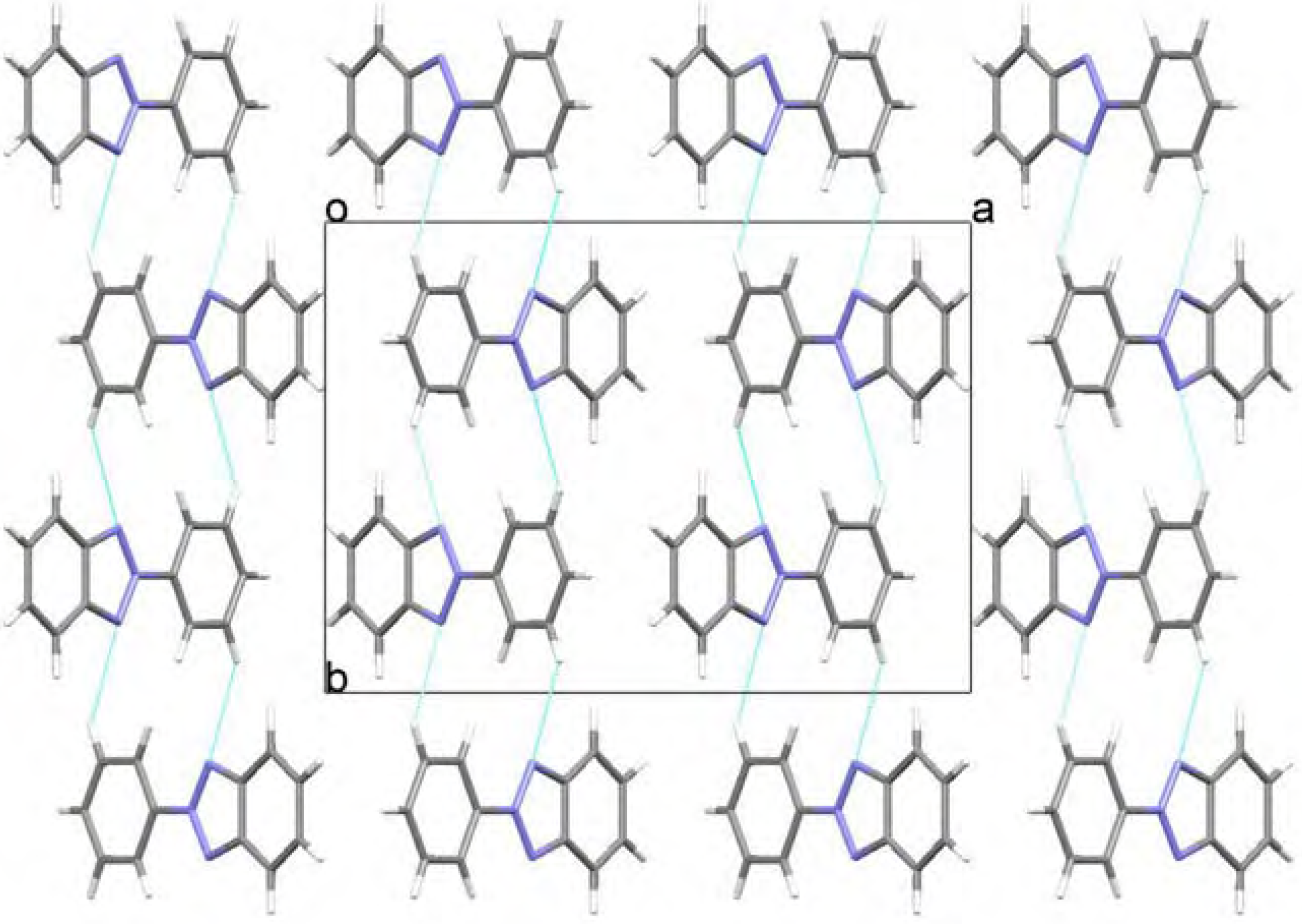
Molecular structure and geometry optimization
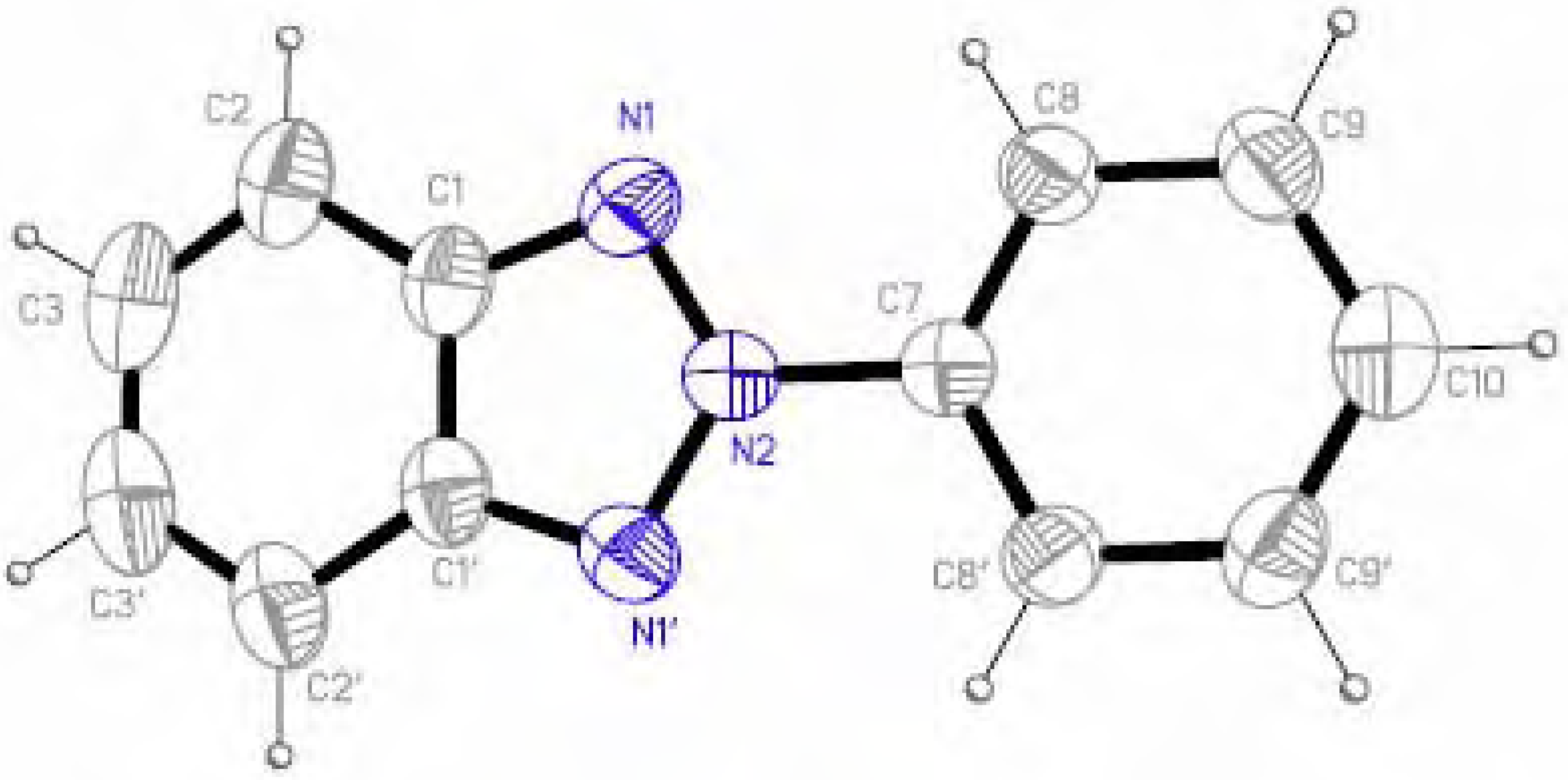

{kind=link}
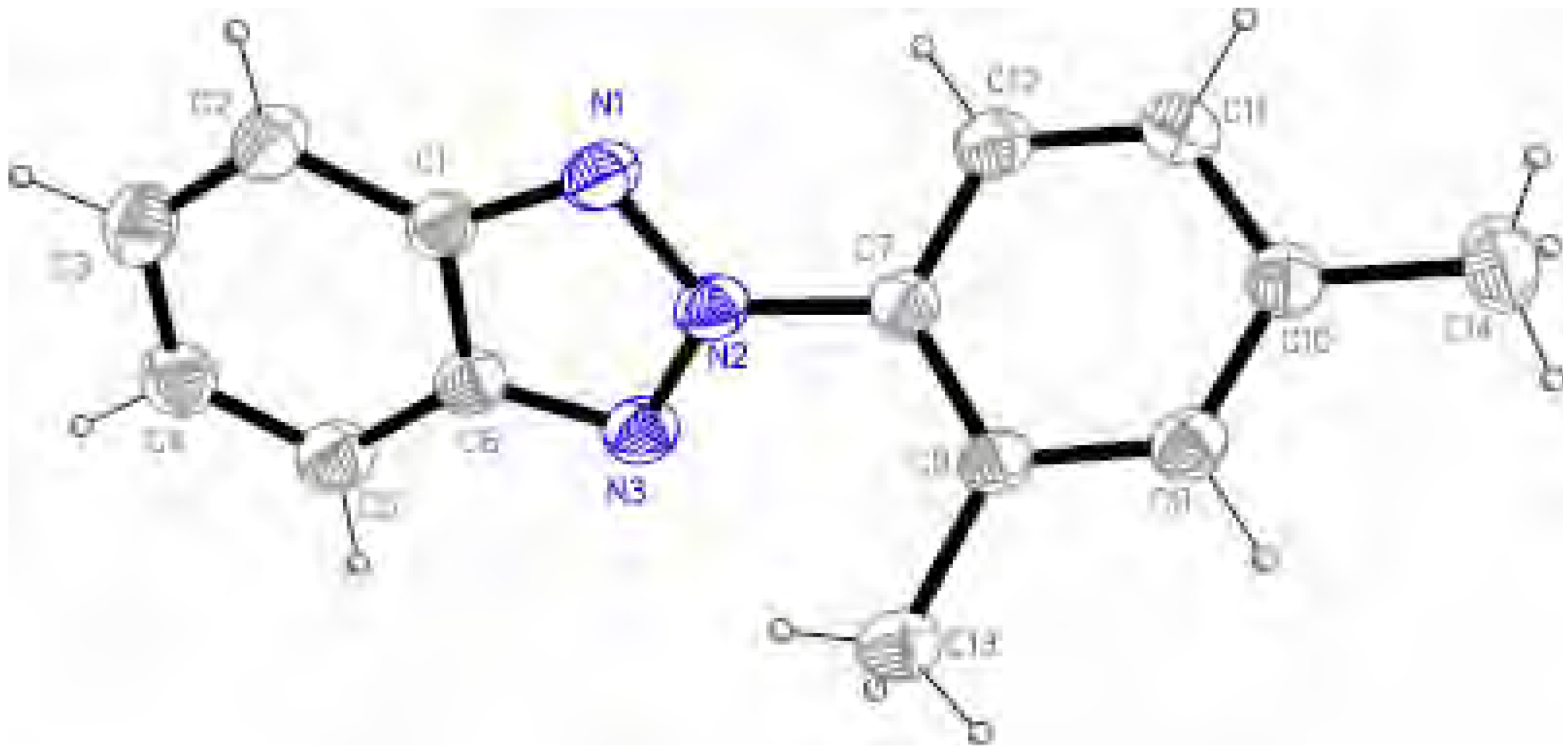{kind=link}
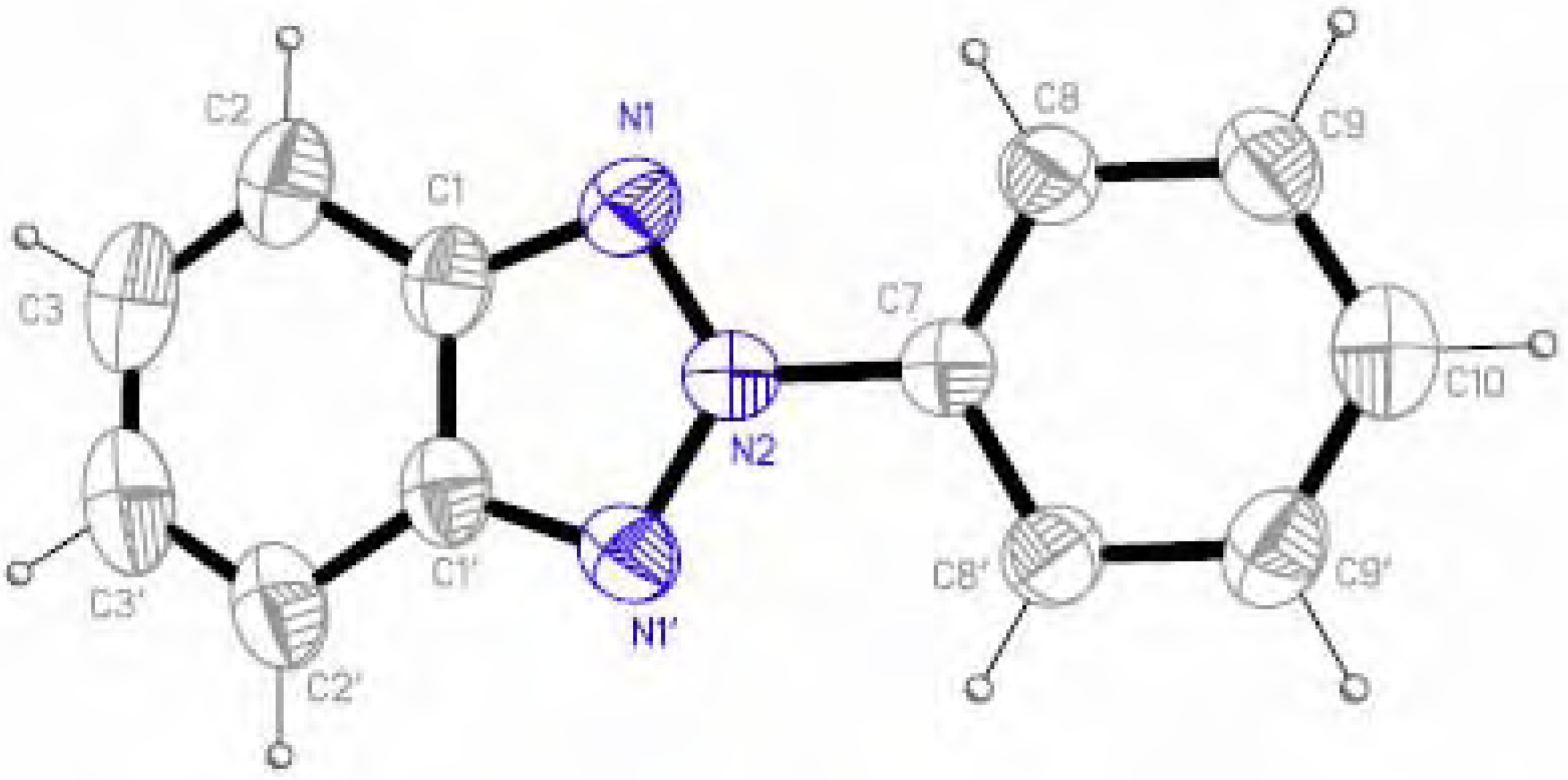{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 5 | ||
|---|---|---|---|
| Bond lengths (Å) | |||
| C1-N1 | 1.353(3) | C1-N1 | 1.348(3) |
| N1-N2 | 1.339(2) | N1-N2 | 1.331(2) |
| N2-N3 | 1.335(2) | N2-N1´ | 1.331(2) |
| N3-C6 | 1.358(3) | N1’-C1’ | 1.348(3) |
| N2-C7 | 1.440(3) | N2-C7 | 1.432(4) |
| C1-C6 | 1.409(3) | C1-C1´ | 1.395(5) |
| C1-C2 | 1.408(3) | C1-C2 | 1.415(4) |
| C2-C3 | 1.385(3) | C2-C3 | 1.361(4) |
| C3-C4 | 1.419(4) | C3-C3´ | 1.395(7) |
| C4-C5 | 1.355(3) | C3´-C2´ | 1.361(4) |
| C5-C6 | 1.409(3) | C2´-C1´ | 1.415(4) |
| C14-H142 | 0.95(4) | C9-H9 | 0.99(3) |
| C14…N1´´ | 3.679(4) | C9···N1´´´ | 3.452(4) |
| N1…H142´´ | 2.73(4) | N1···H9´´´ | 2.76(3) |
| Angles (°) | |||
| N1-N2-C7 | 120.2(2) | N1-N2-C7 | 121.4(1) |
| N1-N2-N3 | 117.2(2) | N1-N2-N1´ | 117.3(3) |
| N3-N2-C7 | 122.6(2) | N1´-N2-C7 | 121.4(1) |
| N2-N1-C1 | 102.5(2) | N2-N1-C1 | 102.4(2) |
| N1-C1-C6 | 109.1(2) | N1-C1-C1´ | 109.0(1) |
| C1-C6-N3 | 108.4(2) | C1-C1´-N1´ | 109.0(1) |
| C6-N3-N2 | 102.8(2) | C1´-N1´-N2 | 102.4(2) |
| C2-C1-C6 | 120.9(2) | C2-C1-C1´ | 121.3(2) |
| C1-C2-C3 | 116.9(2) | C1-C2-C3 | 116.0(3) |
| C3-C4-C5 | 122.4(2) | C3-C3´-C2´ | 122.7(2) |
| C4-C5-C6 | 116.7(2) | C3´-C2´-C1´ | 116.0(3) |
| C5-C6-C1 | 121.2(2) | C2´-C1´-C1 | 121.3(2) |
| C14-H142···N1´´ | 176(3) | C9-H9···N1´´´ | 127(2) |
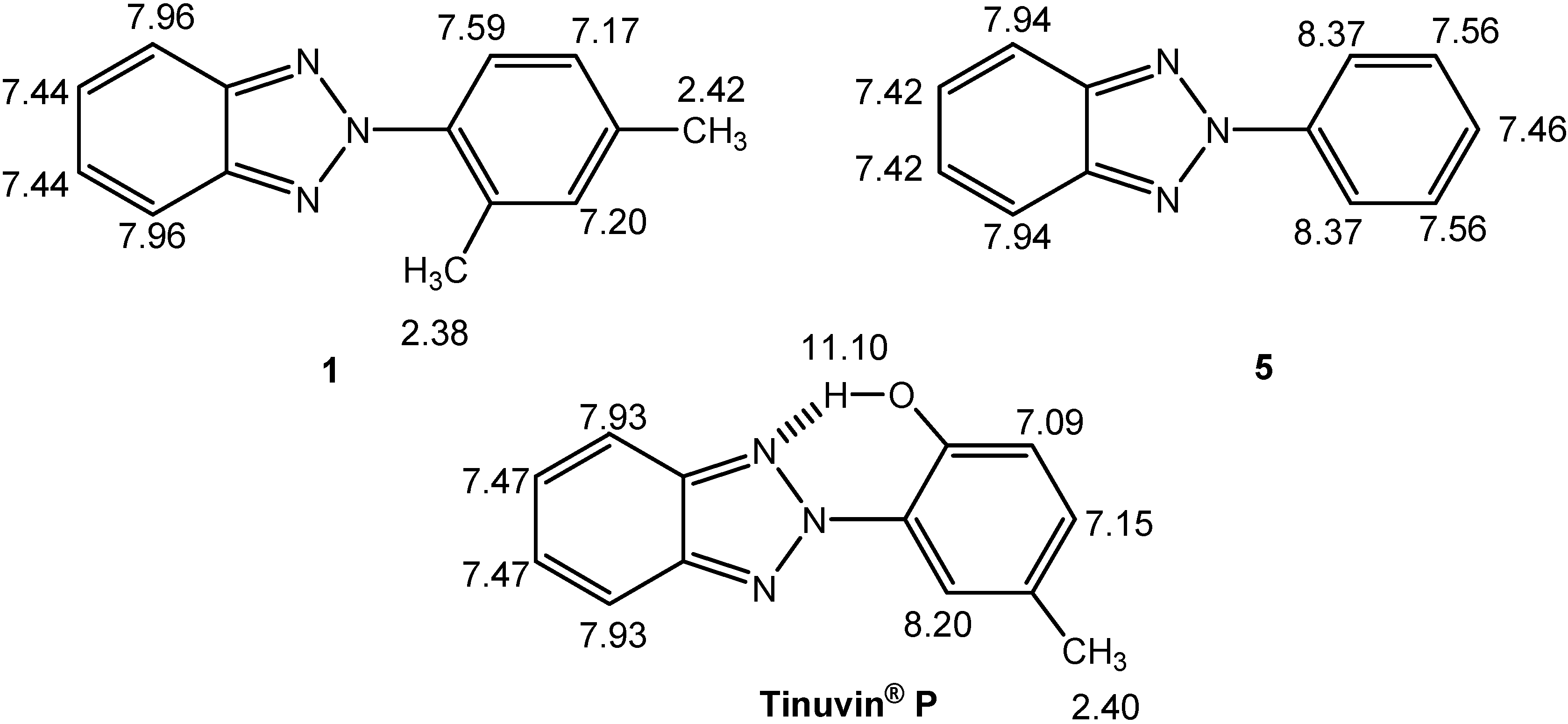
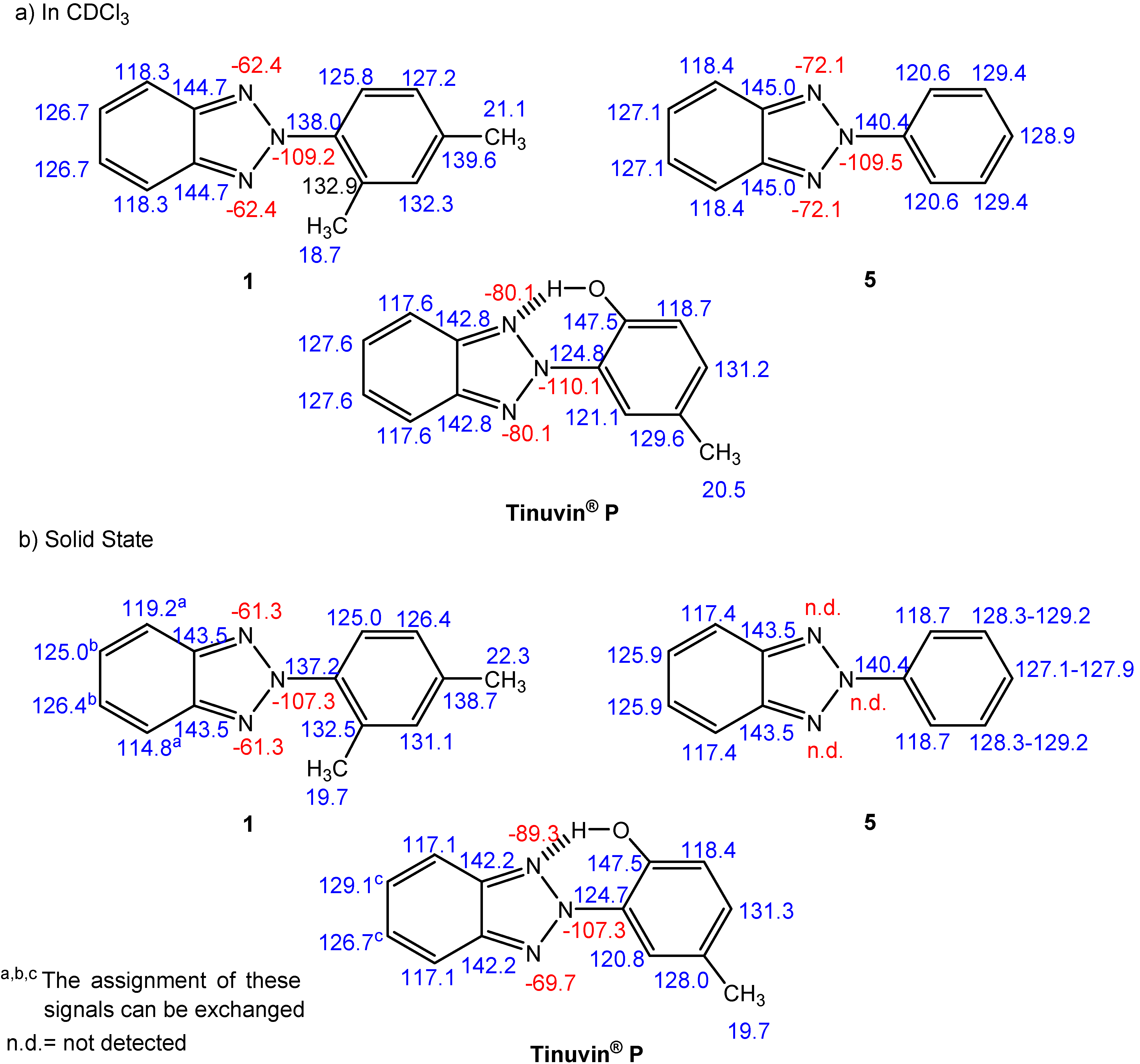
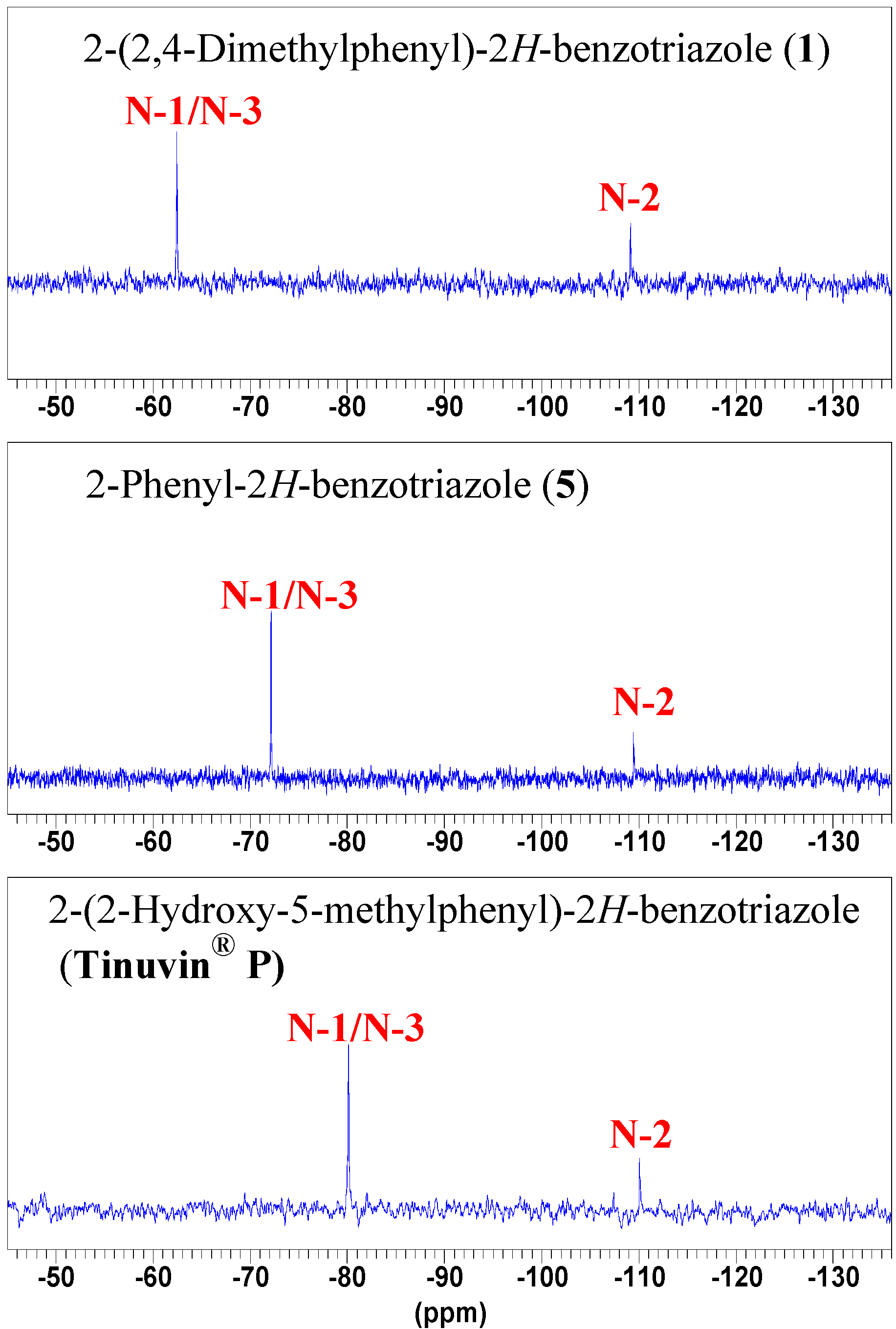
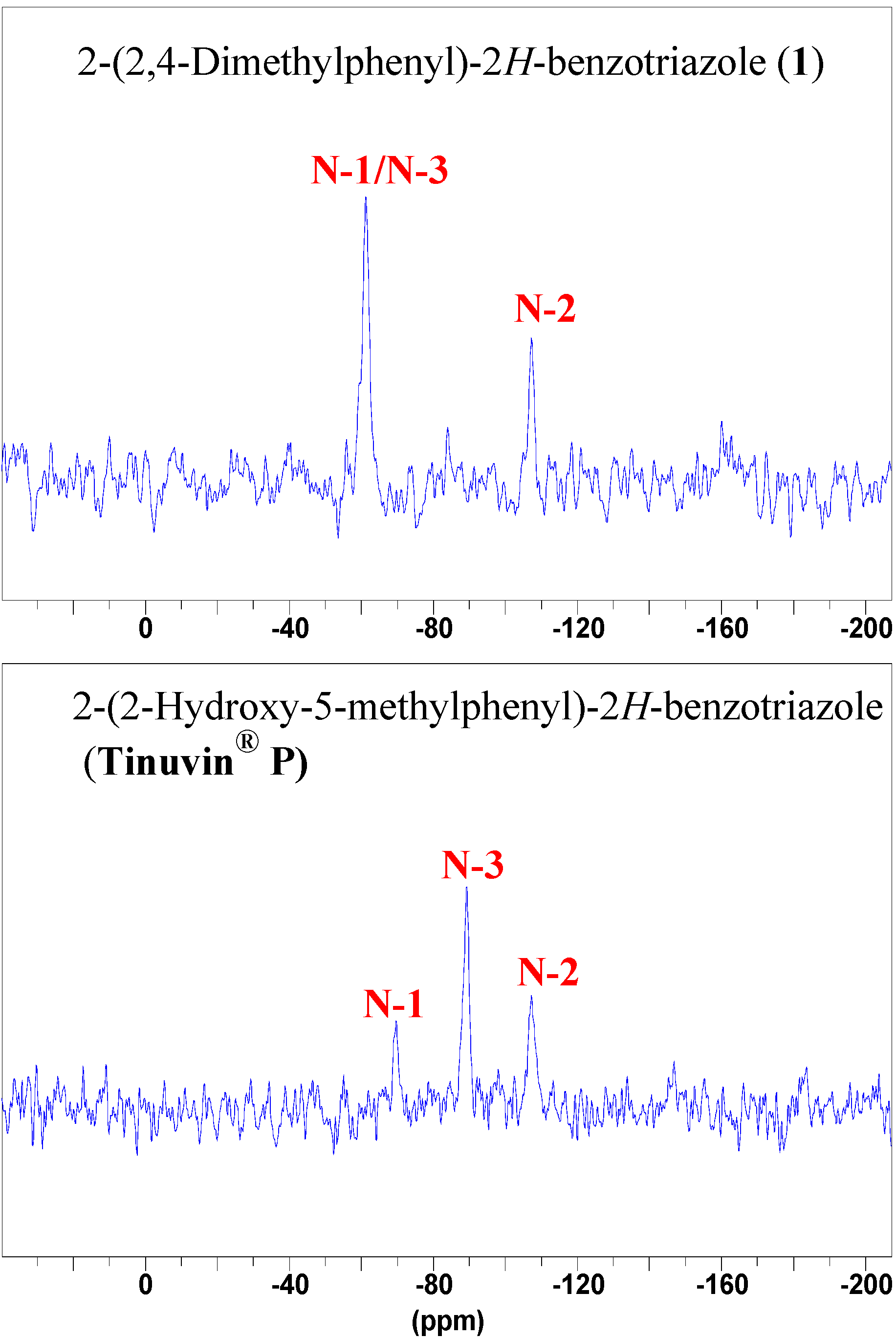
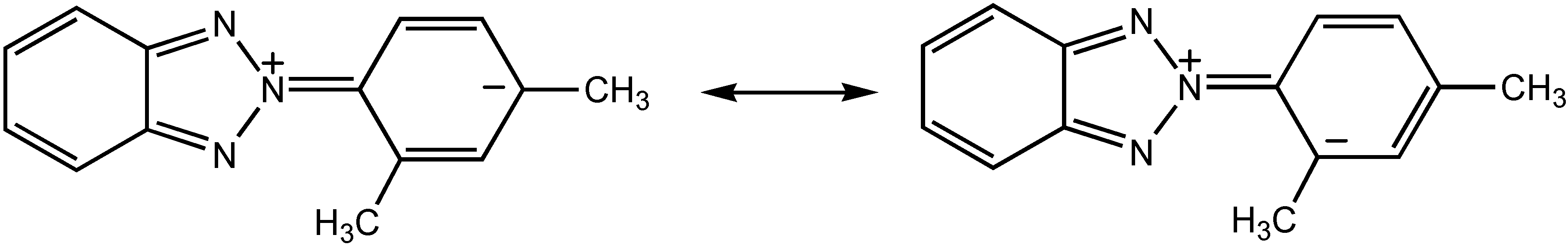
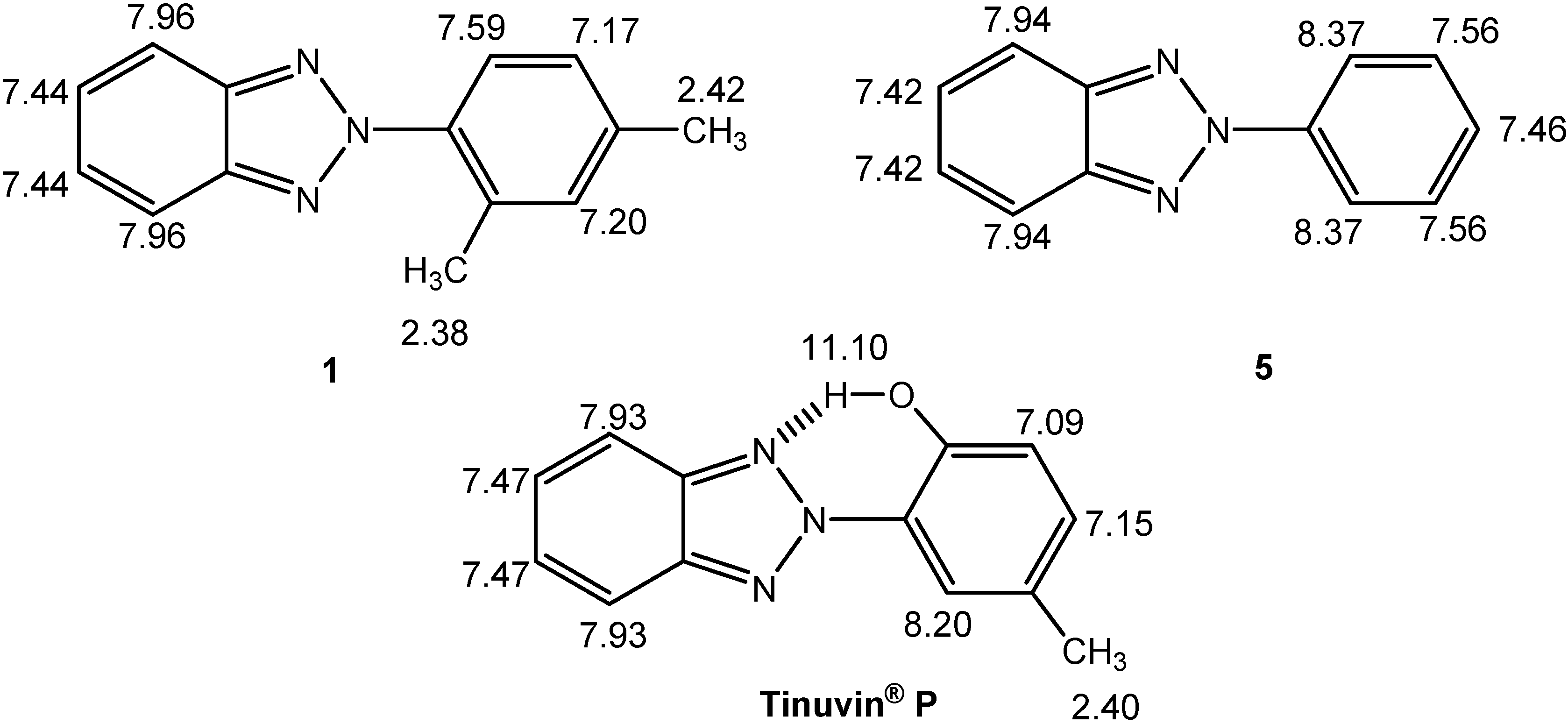
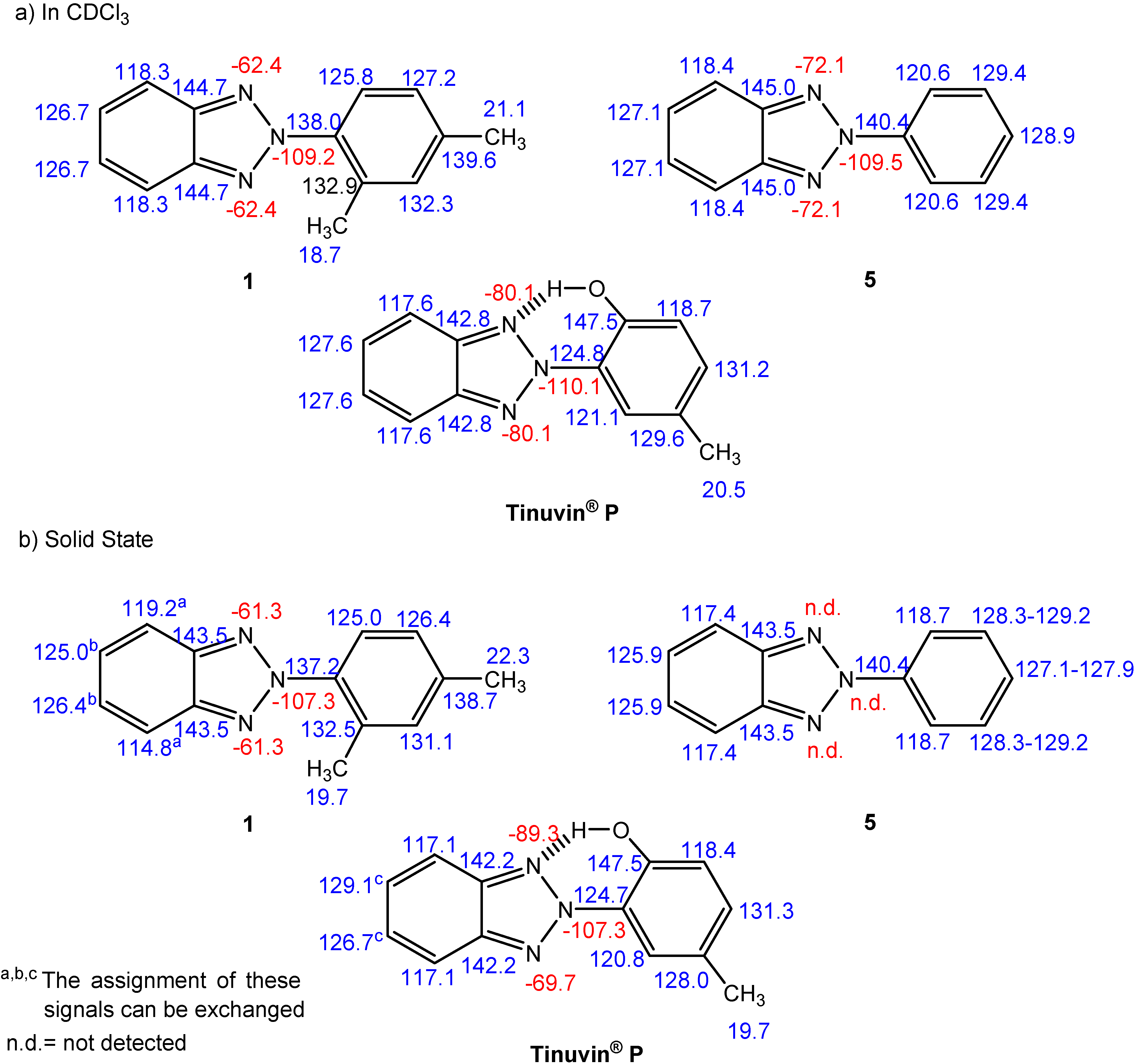
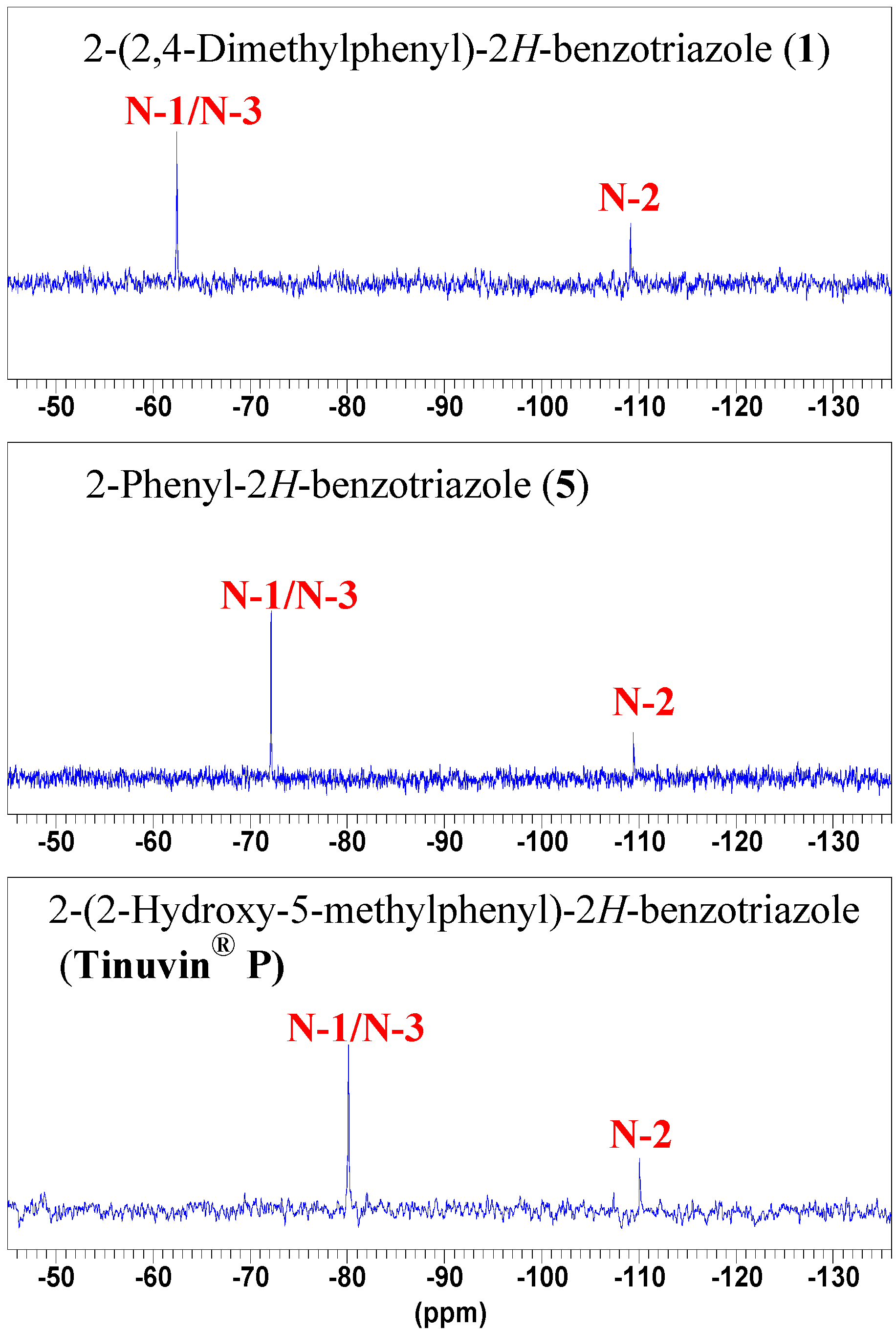
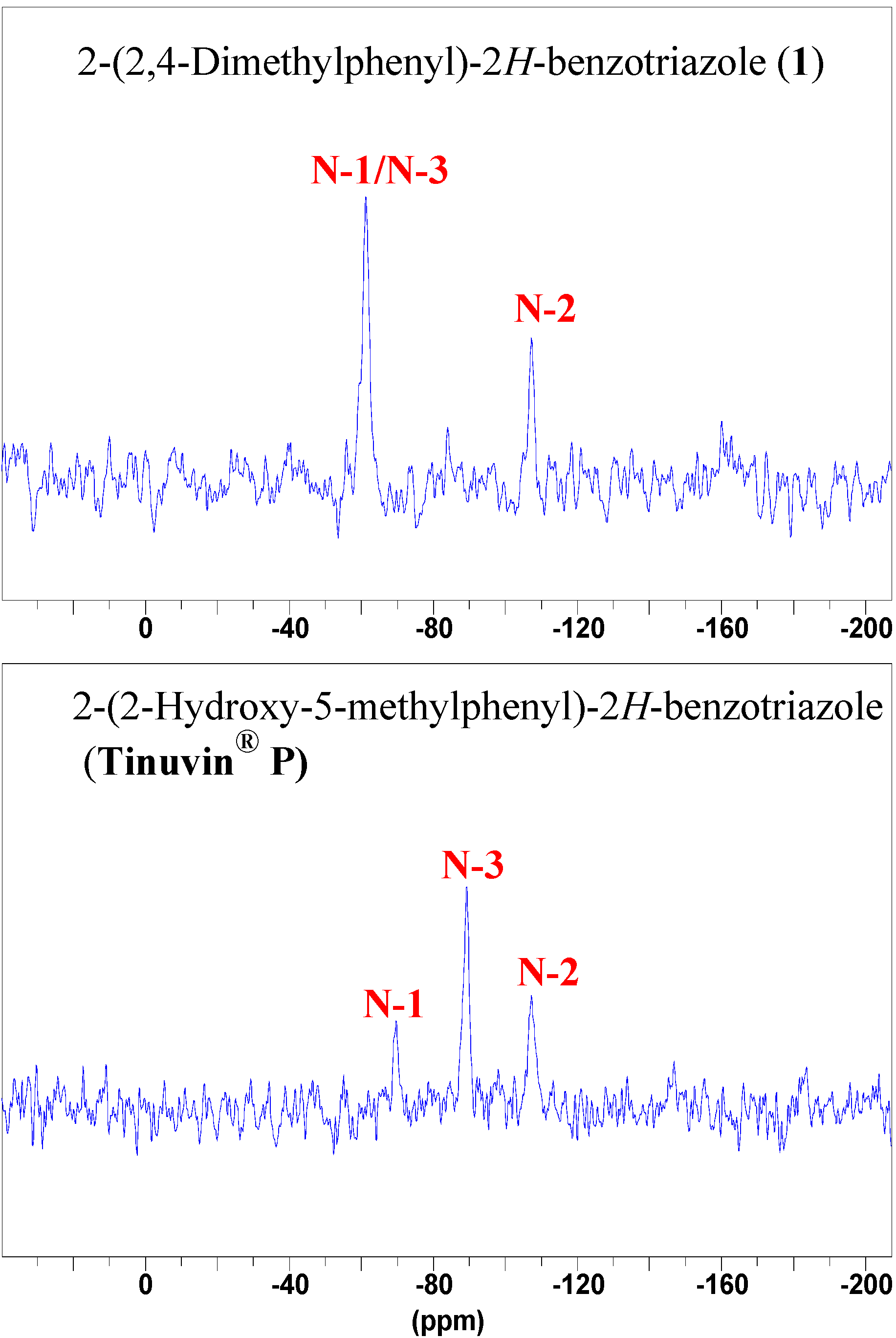
NMR spectroscopy
| Comp. | 1 | 5 | Tinuvin® P |
| δC-ortho | 125.8 + 0.1 + 0.1 = 126.0 | 120.6 | 147.5 –28.8 + 3.0 = 121.7 |
| 132.9 – 9.2 + 0.1 = 123.8 | 121.1 –1.4 –0.7 = 119.0 | ||
| δC-ortho (average) | 124.9 | 120.6 | 120.3 |
| δC-meta | 127.2 + 3.0 – 0.7 = 129.5 | 129.4 | 118.7 +12.8 + 0.1 = 131.6 |
| 132.3 – 0.7 – 0.7 = 130.9 | 129.6 +7.4 – 9.2 = 127.8 | ||
| δC-meta (average) | 130.2 | 124.9 | 129.7 |
| δC-meta-δC-ortho | 5.3 | 8.8 | 9.4 |
| Degree of Interannular Conjugation | Hindered | Extensive | Extensive |
| Comp. | 1 | 5 | Tinuvin® P |
|---|---|---|---|
| δC-ortho | 125.0 + 0.1 + 0.1 = 125.2 | 118.7 | 147.5 –28.8 + 3.0 = 121.7 |
| 132.5 – 9.2 + 0.1 = 123.4 | 120.8 –1.4 –0.7 = 118.7 | ||
| δC-ortho (average) | 124.3 | 118.7 | 120.2 |
| δC-meta | 126.4 + 3.0 – 0.7 = 128.7 | 128.7 | 118.4 +12.8 + 0.1 = 131.3 |
| 131.1 – 0.7 – 0.7 = 129.7 | 128.0 +7.4 – 9.2 = 126.2 | ||
| δC-meta (average) | 129.2 | 128.7 | 128.7 |
| δC-meta-δC-ortho | 4.9 | 10.0 | 8.5 |
| Degree of Interannular Conjugation | Hindered | Extensive | Extensive |
Conclusions
Experimental Section
General
NMR Spectroscopy
2-Phenyl-2H-benzotriazole (5)
Tinuvin® P
Synthesis
X-Ray data collection and structure refinement
| Crystal Data | 1 | 5 |
| Identification code | 651191 | 659842 |
| Empirical formula | C14 H13 N3 | C12H9N3 |
| Formula weight | 223.27 | 195.22 |
| Wavelength (Å) | 0.71073 | 0.71073 |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | P212121 | Pnma |
| Unit cell dimensions | ||
| a (Å) | 3.946(2) | 15.655(2) |
| b (Å) | 11.586(7) | 11.433(2) |
| c (Å) | 25.44(1) | 5.551(1) |
| Volume (Å3) | 1163(1) | 993.6(3) |
| Z | 4 | 4 |
| Density (calculated) (Mg/m3) | 1.275 | 1.305 |
| Absorption coefficient (mm-1) | 0.078 | 0.081 |
| F(000) | 472 | 408 |
| Scan technique | phi and omega | phi and omega |
| Theta range (°) for data collection | 1.60 to 27.00 | 2.60 to 26.00 |
| Index ranges | -4<=h<=4, | -19<=h<=17, |
| -14<=k<=12, | -14<=k<=14, | |
| -32<=l<=32 | -6<=l<=6 | |
| Reflections collected | 10351 | 7629 |
| Independent reflections | 2501 | 1028 |
| [R(int) = 0.0862] | [R(int) = 0.0741] | |
| Completeness to theta max. | 99.4 % | 100% |
| Data / restraints / parameters | 2501 / 0 / 194 | 1028 / 0 / 88 |
| Goodness-of-fit on F2 | 0.938 | 1.079 |
| Final R* indices [F2>2sigma(F2)] | 0.0420 | 0.0457 |
| (1448 reflns. observed) | (569 reflns. observed) | |
| wR** (F2) (all data) | 0.1024 | 0.1573 |
| Largest diff. peak and hole (e.Å–3) | 0.133 and -0.133 | 0.136 and 0.207 |
Acknowledgments
References
- Catalán, J.; Pérez, P.; Fabero, F.; Wilshire, J. F. K.; Claramunt, R. M.; Elguero, J. Photophysical properties of some 2-(2’-hydroxyaryl)benzotriazoles: dramatic effect of an ortho-located bulky tert-butyl group. J. Am. Chem. Soc. 1992, 114, 964–966. [Google Scholar] [CrossRef]
- Catalán, J.; de Paz, J. L. G.; Torres, M. R.; Tornero, J. D. Molecular structure of a unique UV stabilizer: Tinuvin P. J. Chem. Soc. Faraday Trans 1997, 93, 1691–1696, and references therein. [Google Scholar] [CrossRef]
- Houghton, P. G.; Pipe, D. F.; Rees, C. W. Intramolecular reaction between nitro and carbodi-imide groups; a new synthesis of 2-arylbenzotriazoles. J. Chem. Soc. Perkin Trans I 1985, 1471–1479. [Google Scholar] [CrossRef]
- Vickers, S.; Triggle, D. J.; Garrison, D. R. The preparation and diazotisation of some o- and p-aminophenyl benzoates and benzamides. J. Chem. Soc. C 1968, 632–634. [Google Scholar] [CrossRef]
- Windows Titan 1.05, Wavefunction Inc.: Irvine, CA.
- Pretsch, E.; Bühlmann, P.; Affolter, C. Structure Determination of Organic Compounds: Tables of Spectral Data; Springer: New York, 2000. [Google Scholar]
- Catalán, J.; Fabero, F.; Claramunt, R. M.; Santa María, M. D.; Foces-Foces, C.; Cano, F. H.; Martínez-Ripoll, M.; Elguero, J.; Sastre, R. New ultraviolet stabilizers: 3- and 5-(2´-hydroxy-phenyl)pyrazoles. J. Am. Chem. Soc. 1992, 114, 5039–5048. [Google Scholar] [CrossRef]
- Begtrup, M.; Elguero, J.; Faure, R.; Camps, P.; Estopa, C.; Ilavsky, D.; Fruchier, A.; Marzin, C.; de Mendoza, J. Effects of N-substituents on the 13C NMR parameters of azoles. Magn. Reson. Chem. 1988, 26, 134–151. [Google Scholar] [CrossRef]
- Begtrup, M. 13C NMR spectra of phenyl-substituted azoles: a conformational study. Acta Chem. Scand. 1973, 27, 3101–3110. [Google Scholar] [CrossRef]
- Claramunt, R. M.; Sanz, D.; López, C.; Jiménez, J. A.; Jimeno, M. L.; Elguero, J.; Fruchier, A. Susbtituent effects on the 15N NMR parameters of azoles. Magn. Reson. Chem. 1997, 35, 35–75. [Google Scholar] [CrossRef]
- Berger, S.; Braun, S. 200 and More NMR Experiments; Wiley-VCH: Weinheim, 2004. [Google Scholar]
- Sheldrick, G. M. SHELX97, Programs for Crystal Structure Analysis, release 97-2; University of Göttingen: Göttingen, Germany, 1998. [Google Scholar]
- Sample Availability: Samples of the compounds 1 and 5 are available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Claramunt, R.M.; María, D.S.; Pinilla, E.; Torres, M.R.; Elguero, J. Structural Studies of Two Tinuvin® P Analogs: 2-(2,4-Dimethyl-phenyl)-2H-benzotriazole and 2-Phenyl-2H-benzotriazole. Molecules 2007, 12, 2201-2214. https://doi.org/10.3390/12092201
Claramunt RM, María DS, Pinilla E, Torres MR, Elguero J. Structural Studies of Two Tinuvin® P Analogs: 2-(2,4-Dimethyl-phenyl)-2H-benzotriazole and 2-Phenyl-2H-benzotriazole. Molecules. 2007; 12(9):2201-2214. https://doi.org/10.3390/12092201
Chicago/Turabian StyleClaramunt, Rosa María, Dolores Santa María, Elena Pinilla, M. Rosario Torres, and José Elguero. 2007. "Structural Studies of Two Tinuvin® P Analogs: 2-(2,4-Dimethyl-phenyl)-2H-benzotriazole and 2-Phenyl-2H-benzotriazole" Molecules 12, no. 9: 2201-2214. https://doi.org/10.3390/12092201