Exploitation of Bile Acid Transport Systems in Prodrug Design

Abstract
:Introduction
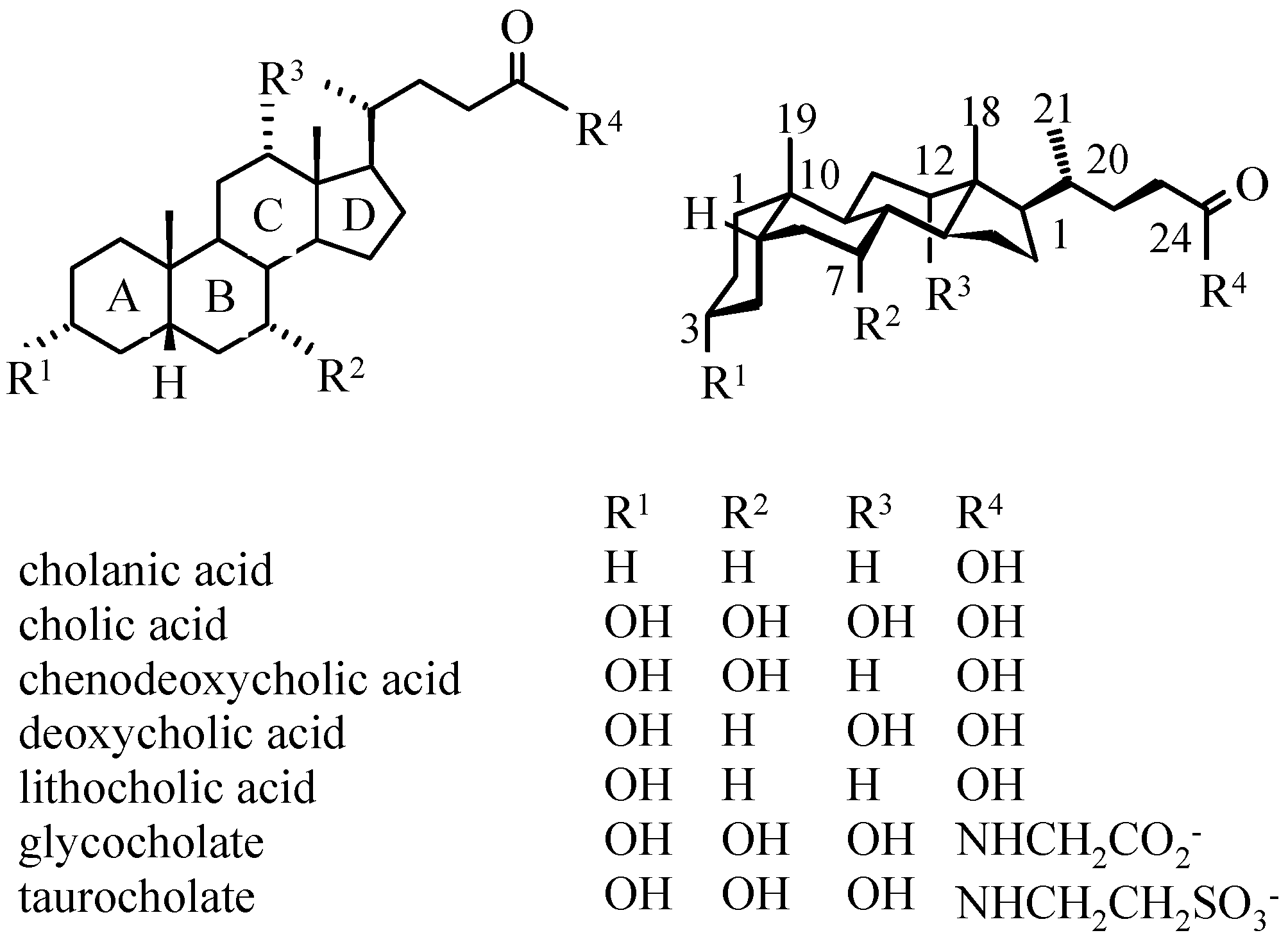
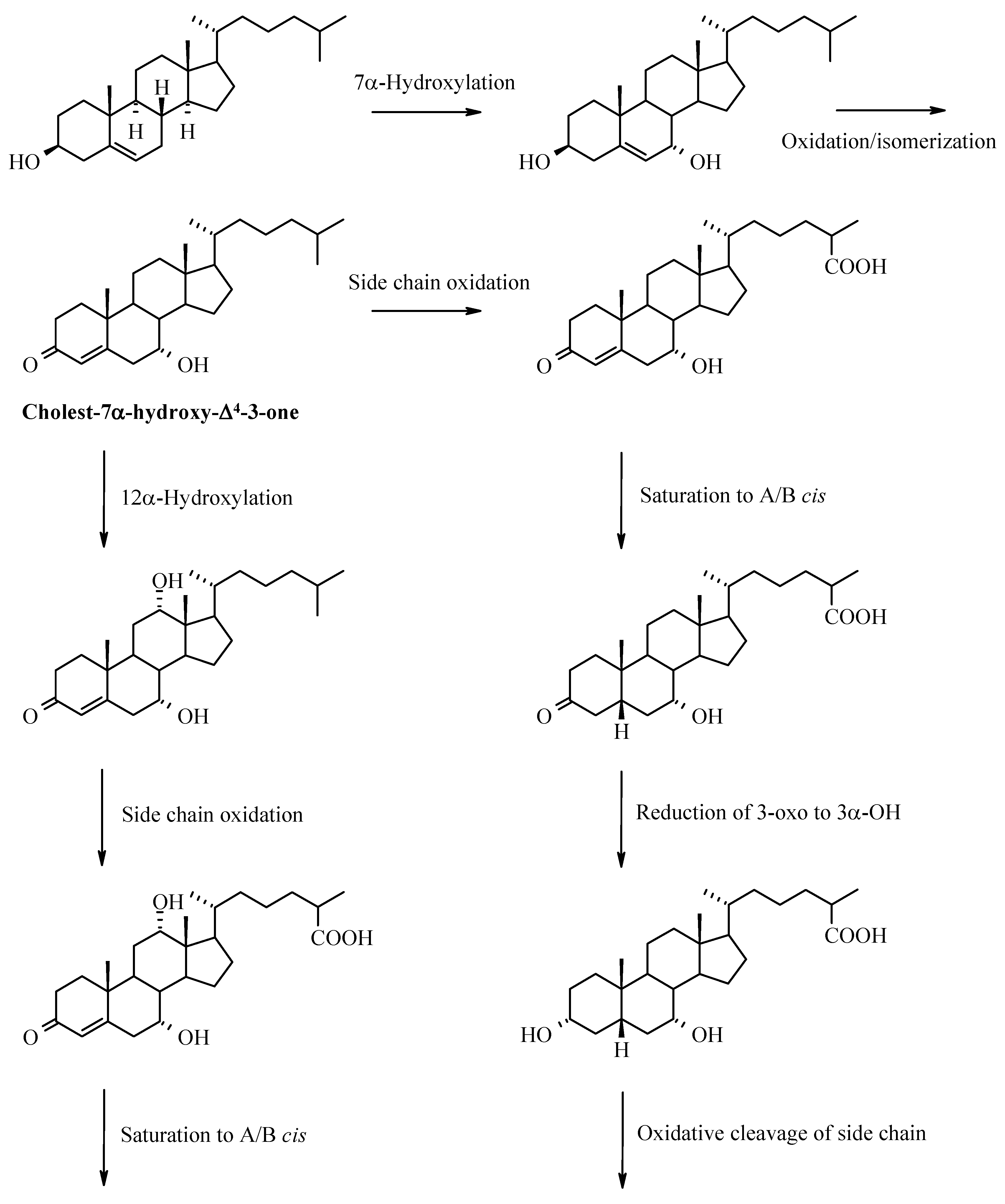
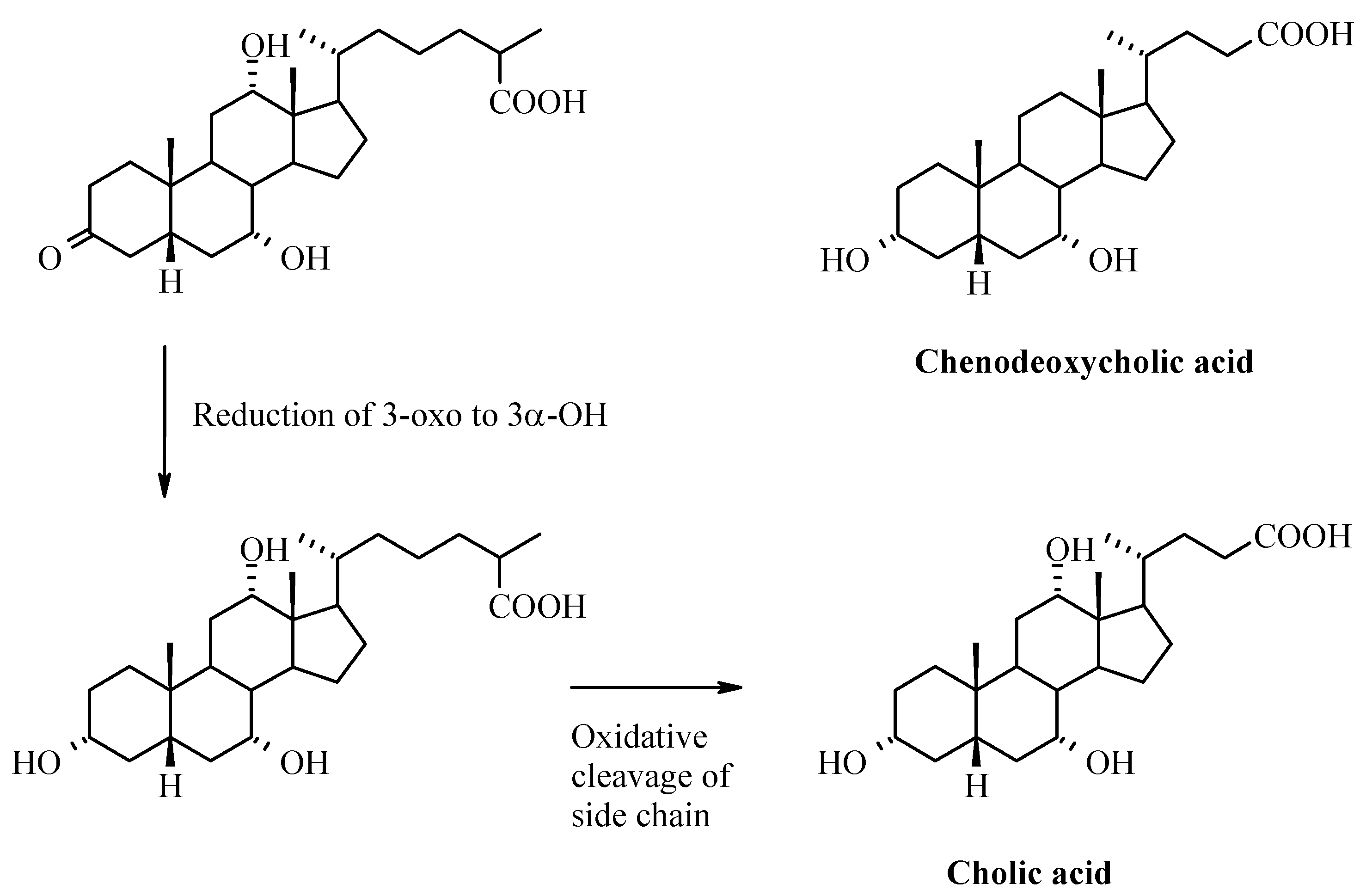
Bile Acids – Chemistry, Biochemistry, and Physiological Functions

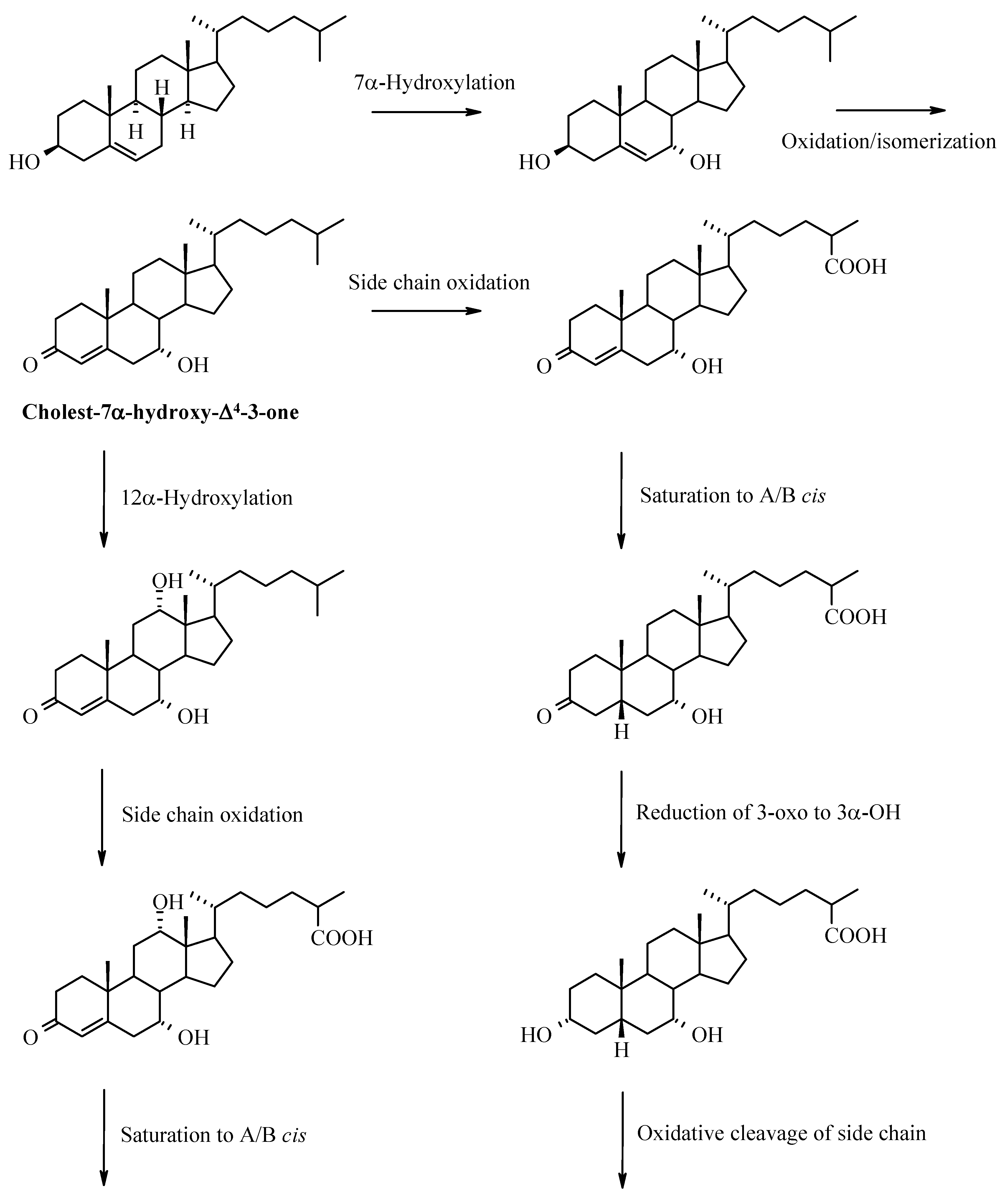
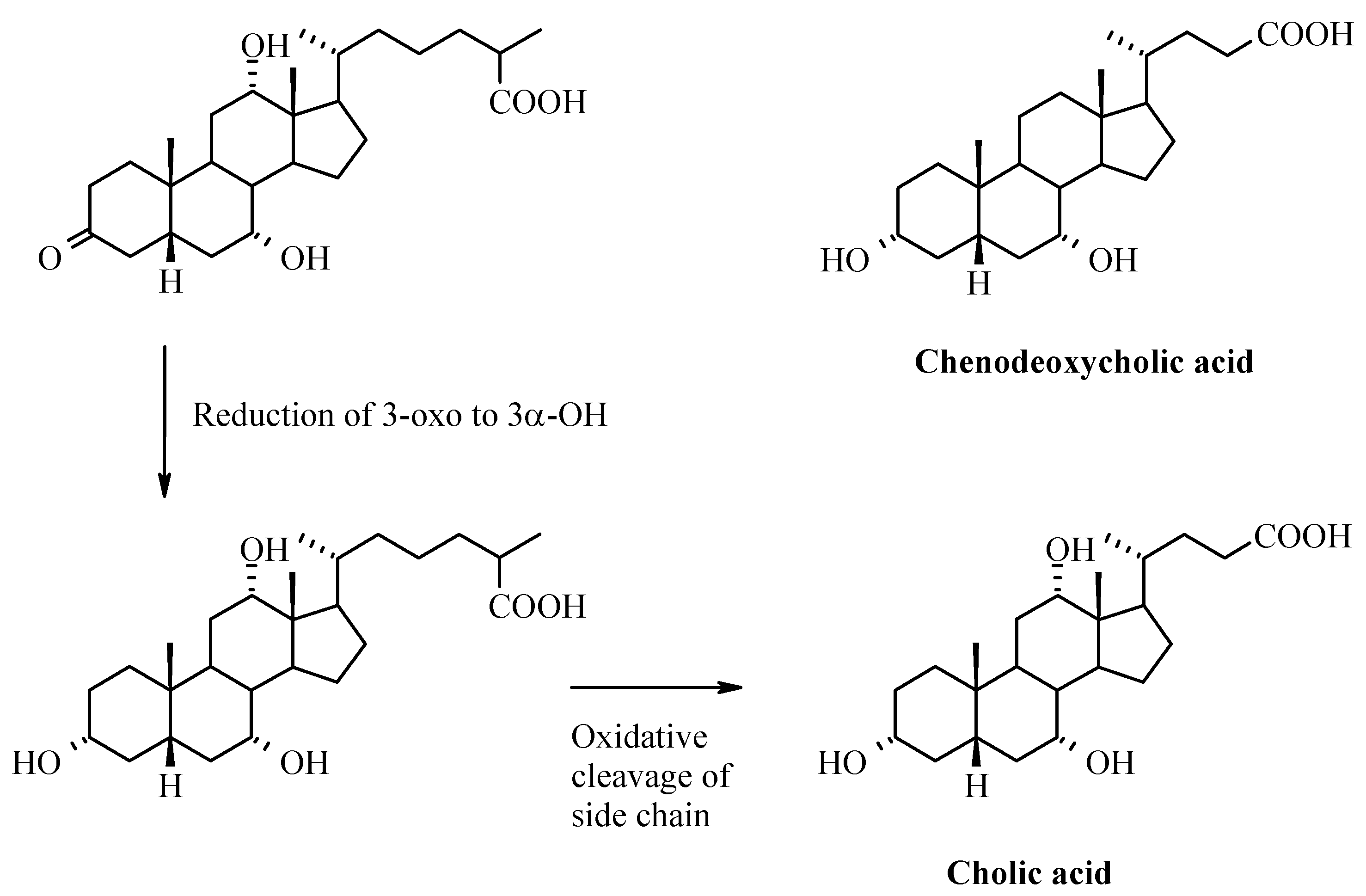
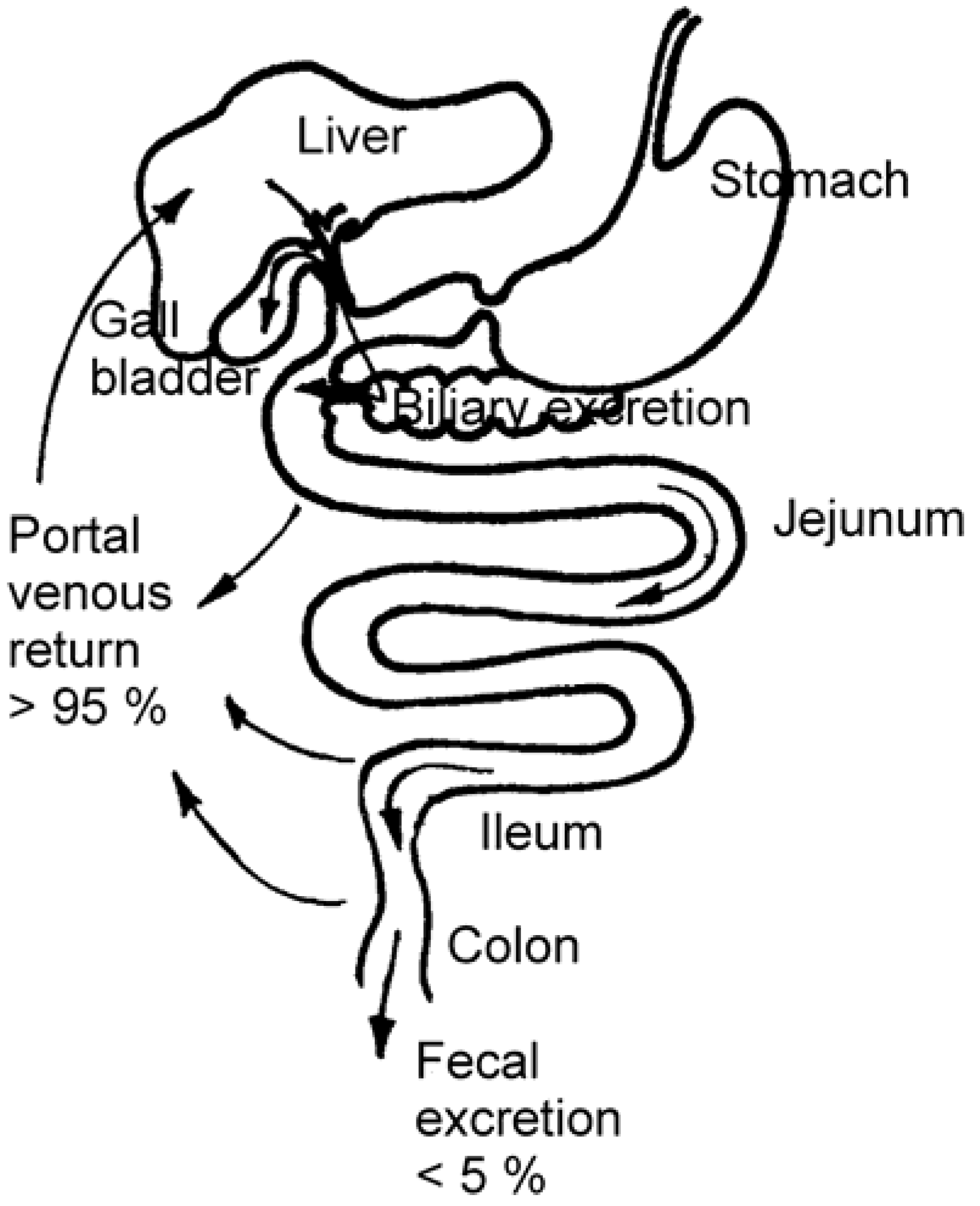
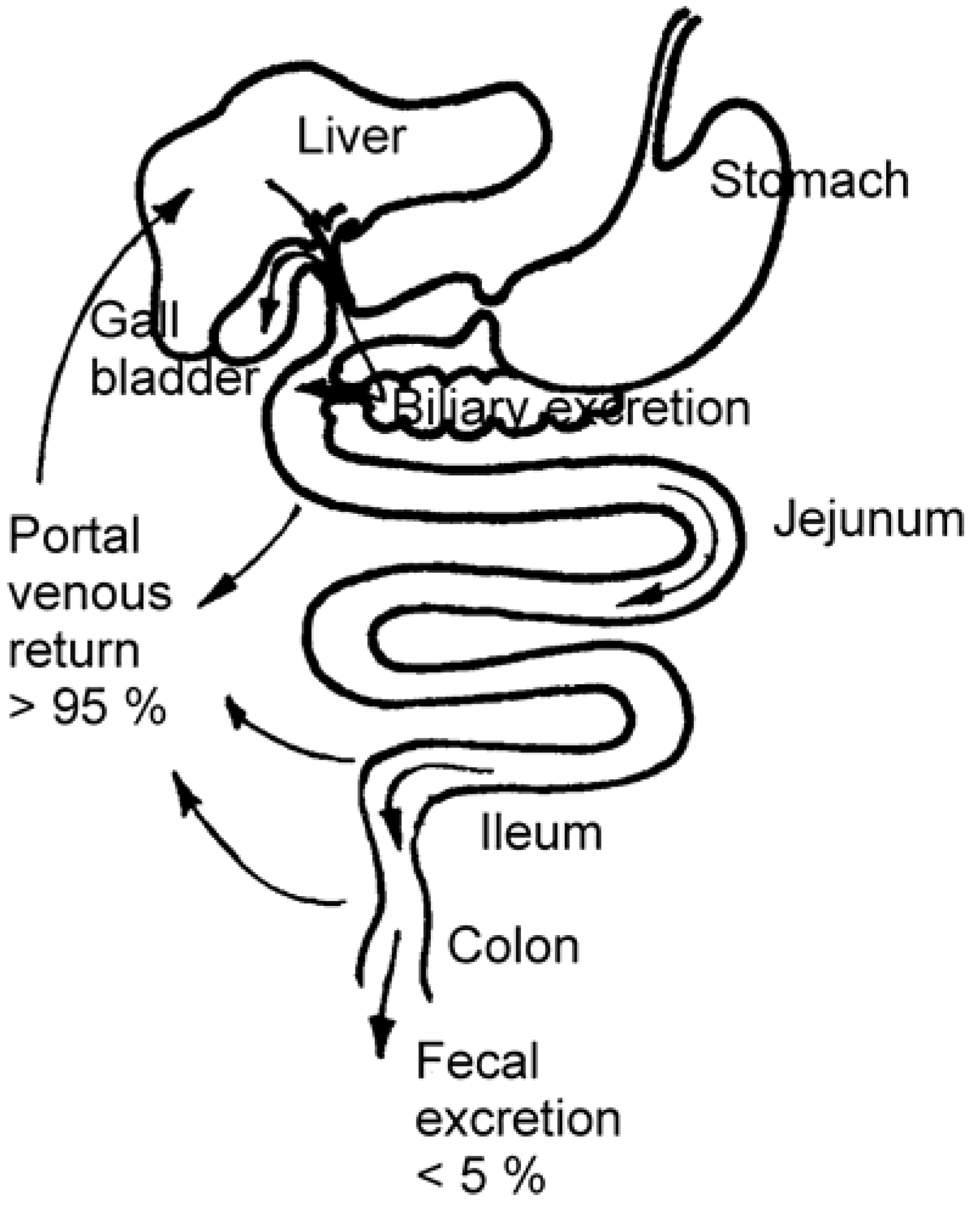
Enterohepatic Circulation
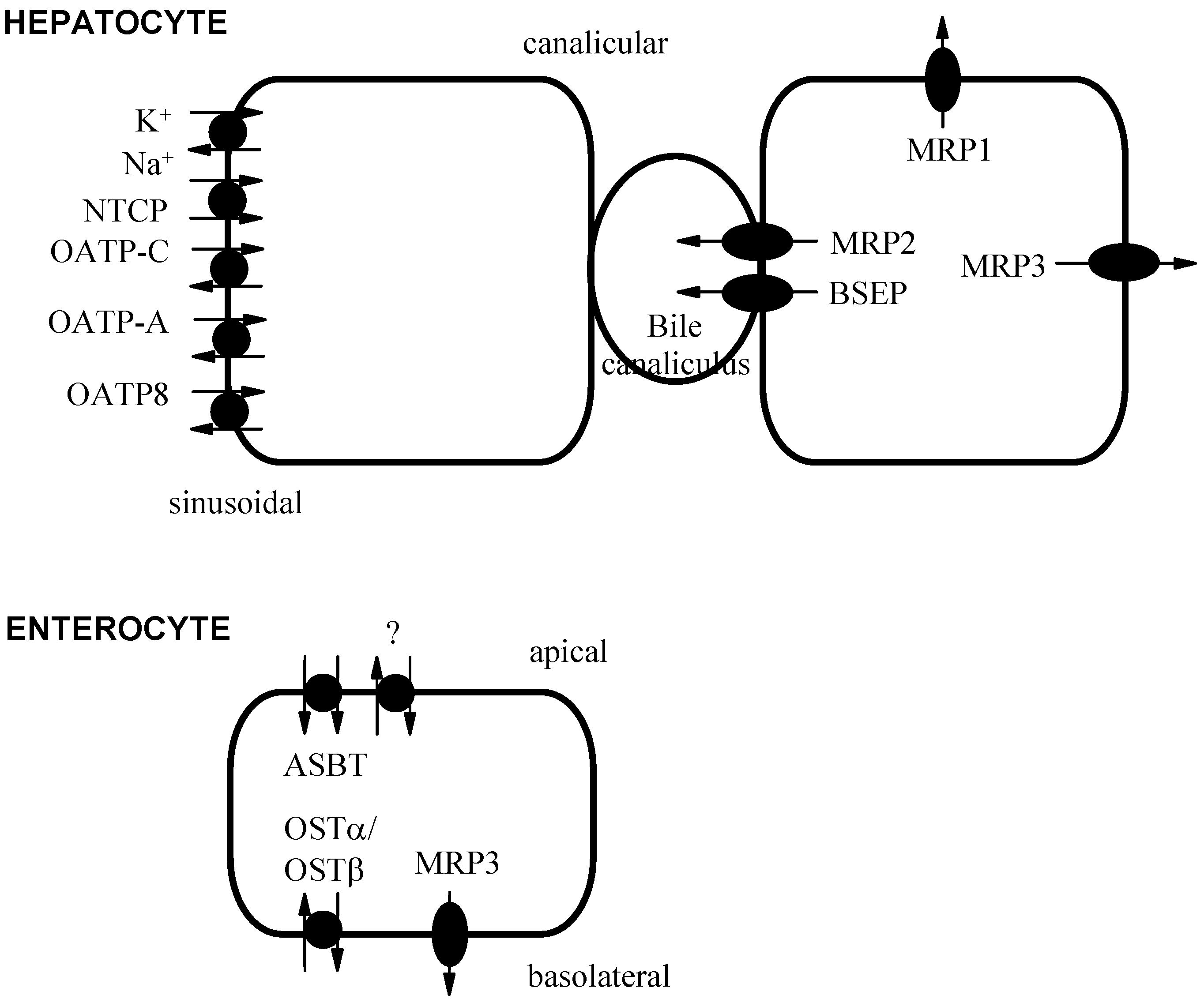
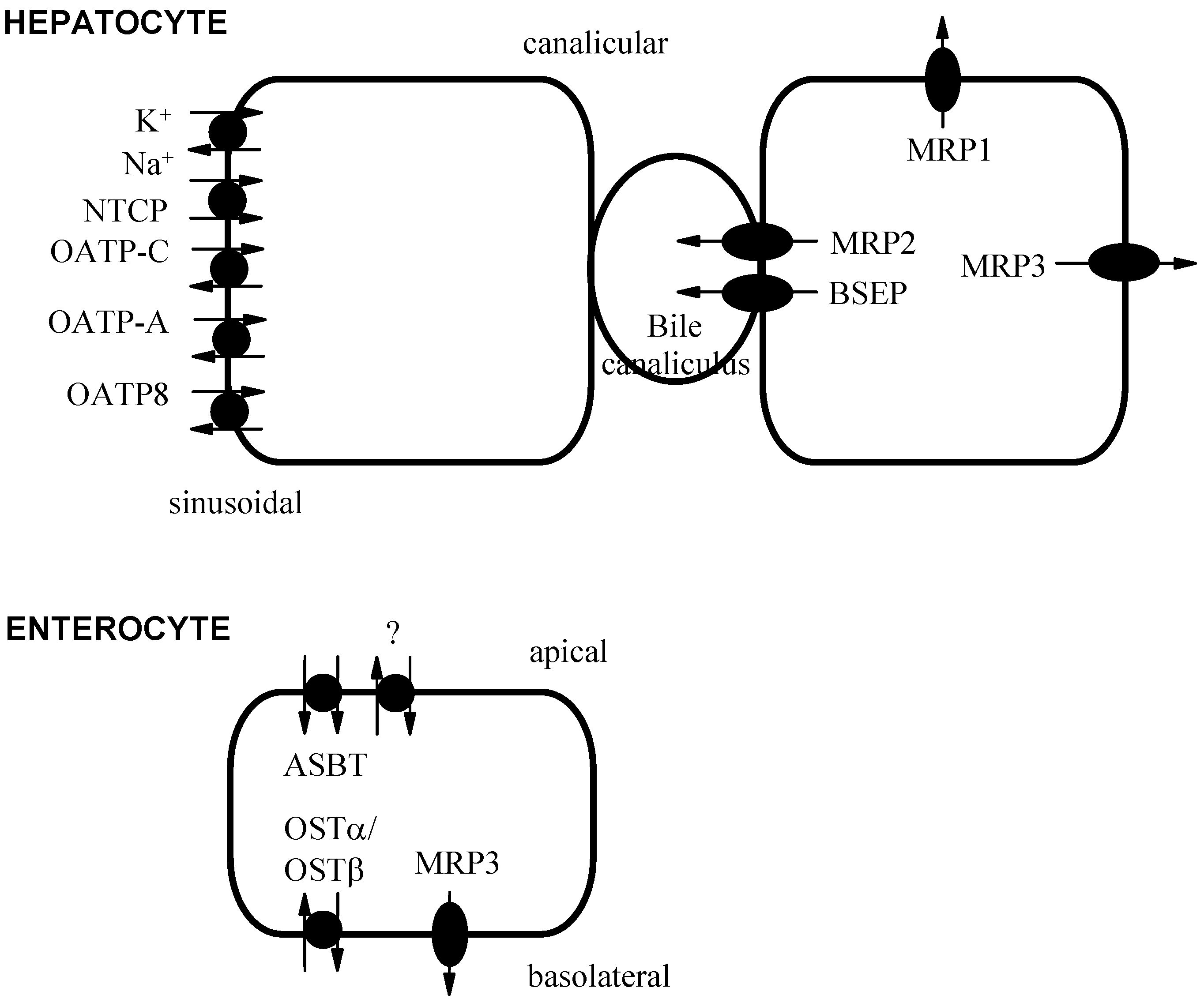
Bile Acid Transport Proteins
Factors Affecting Bile Acid Transport
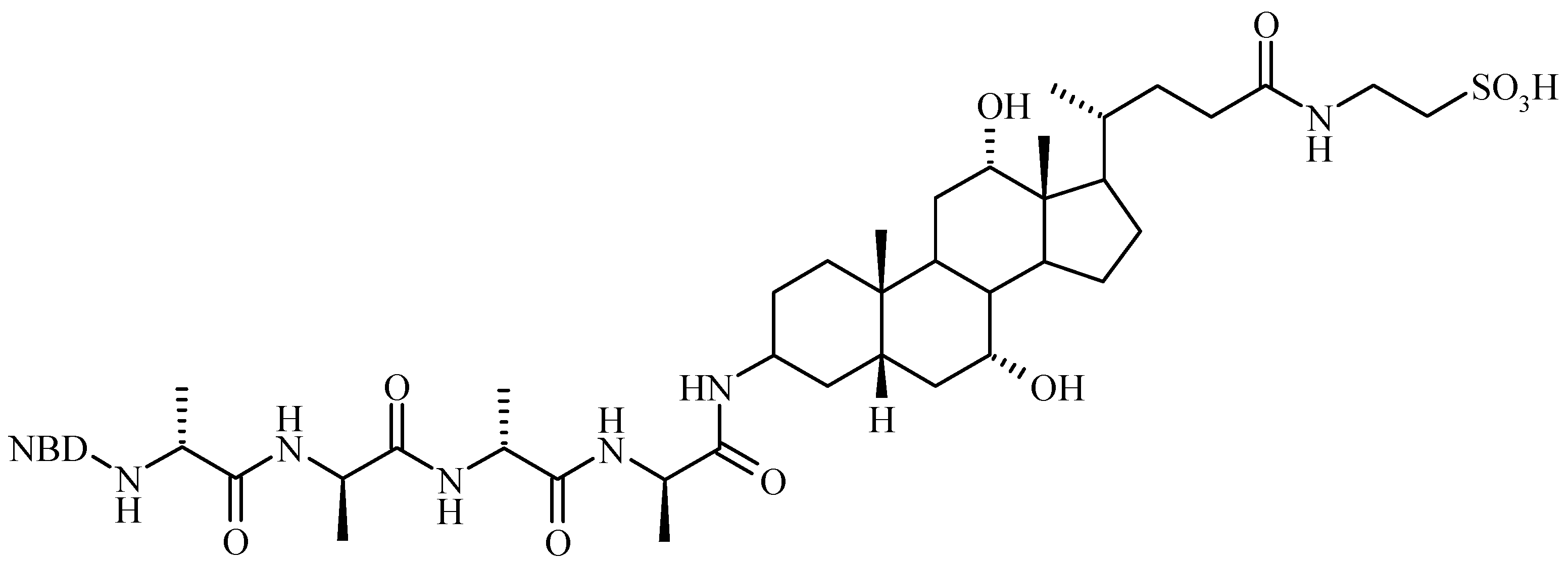
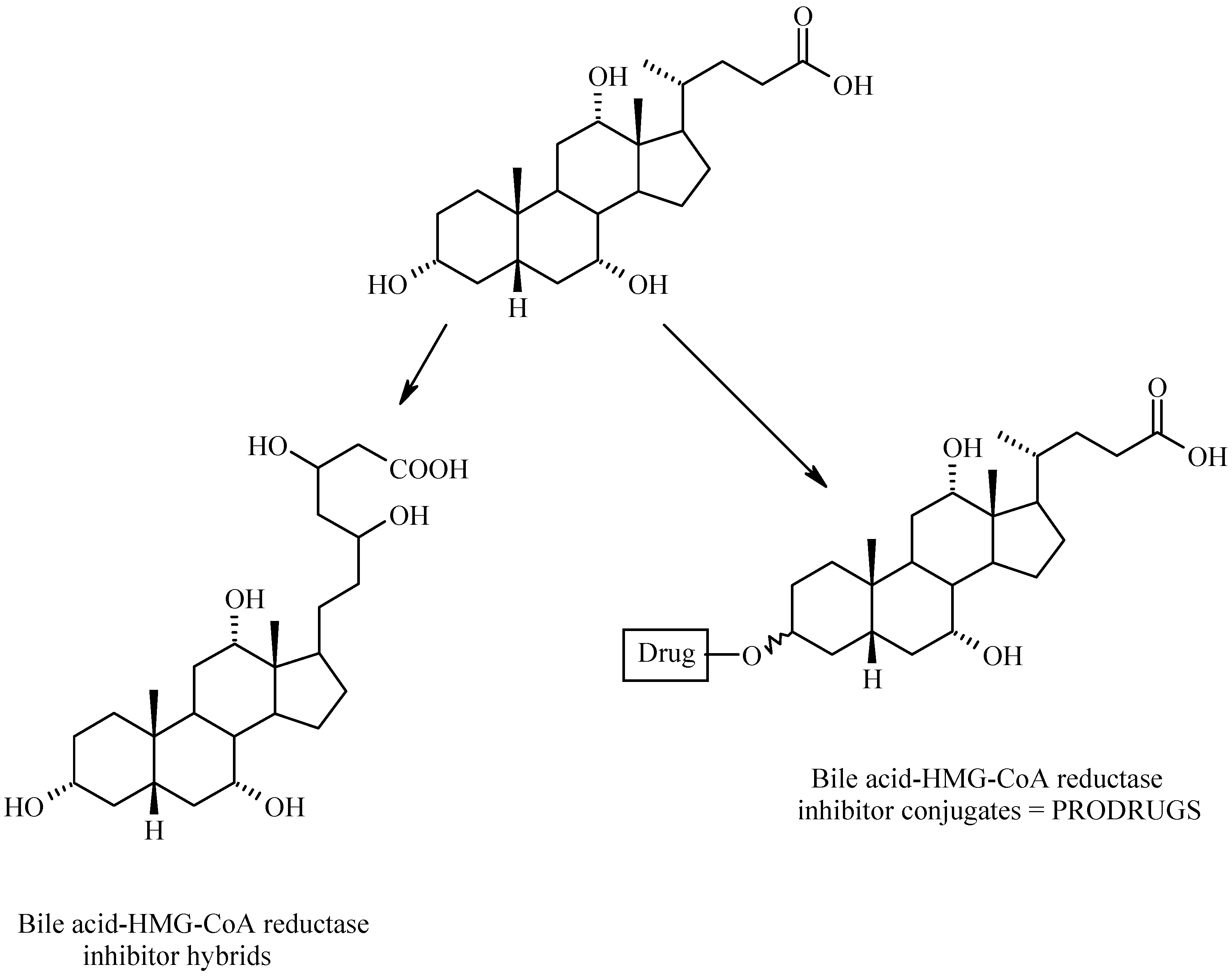
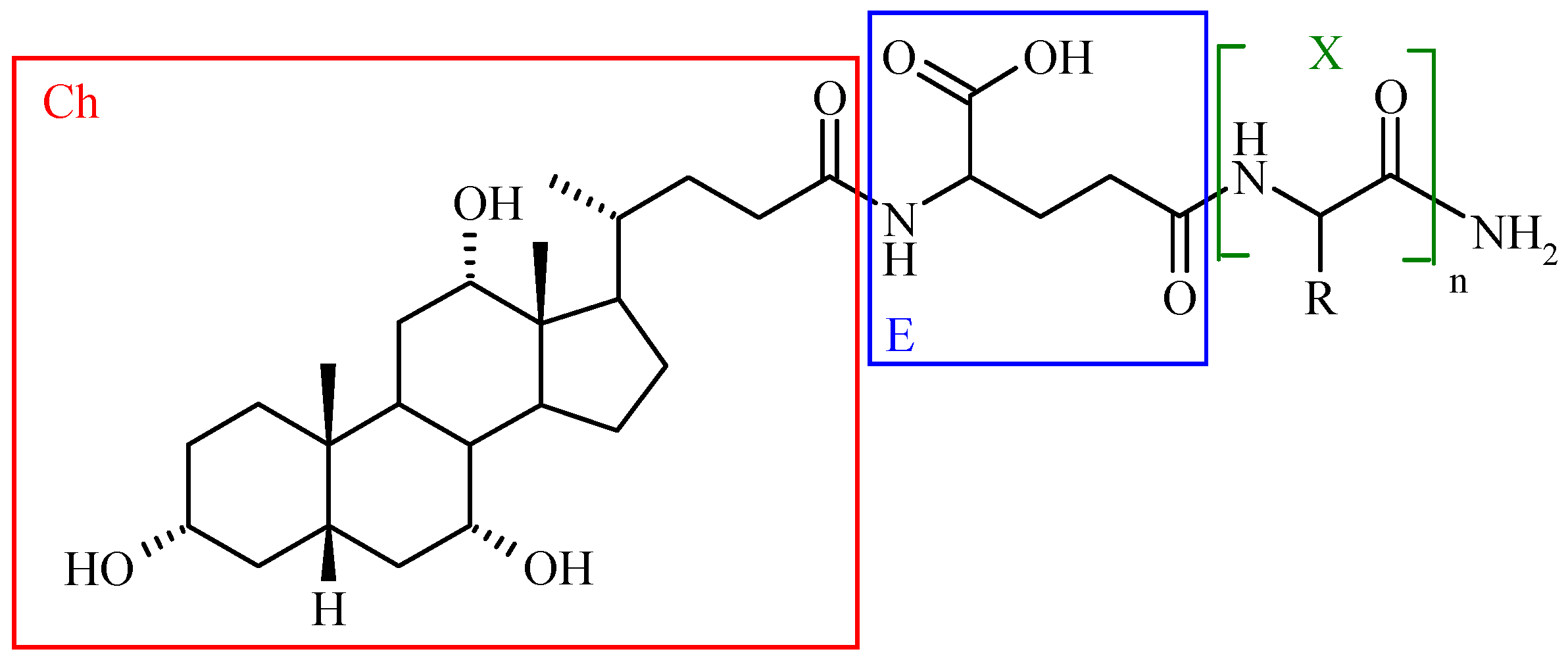
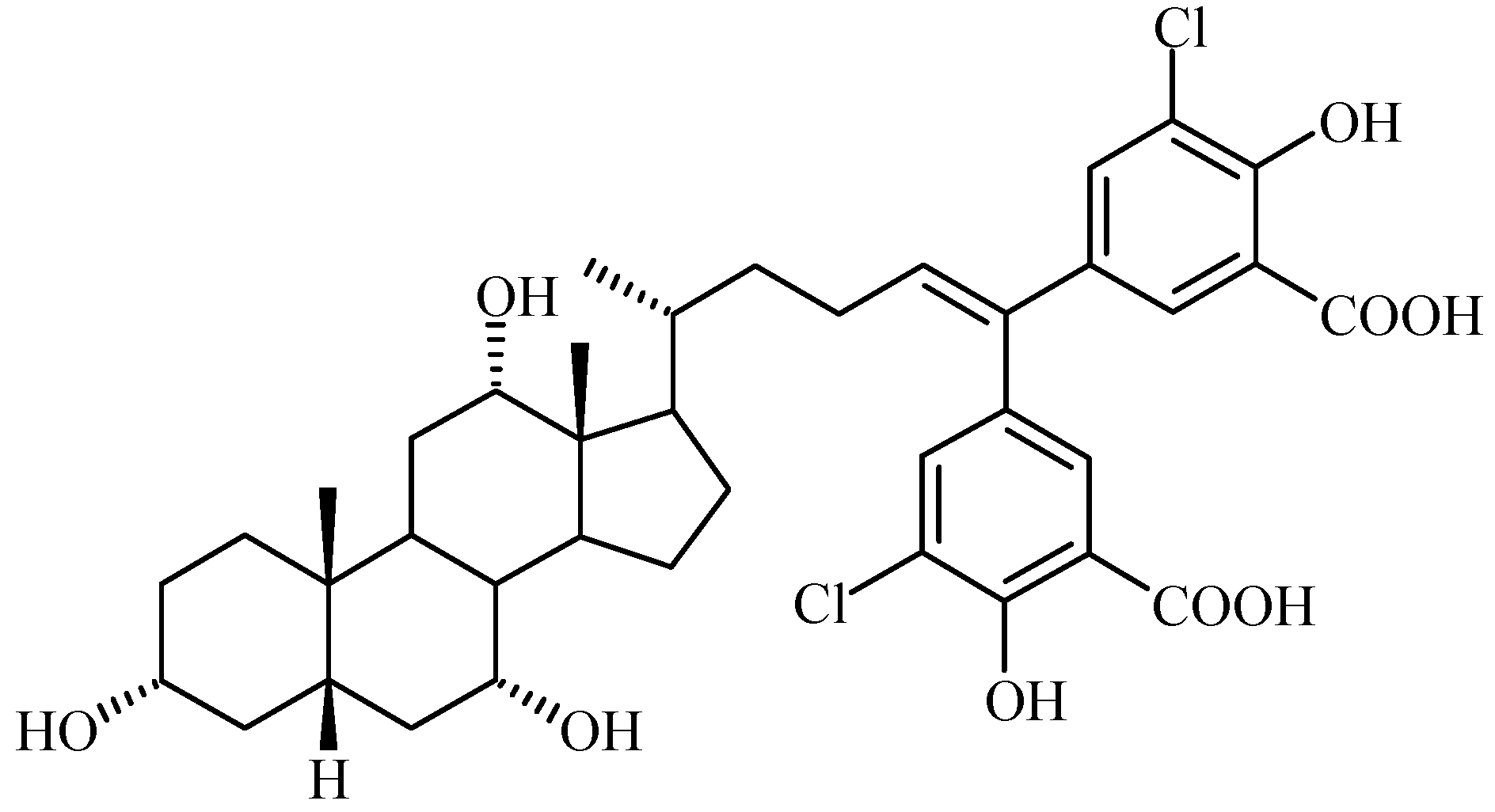
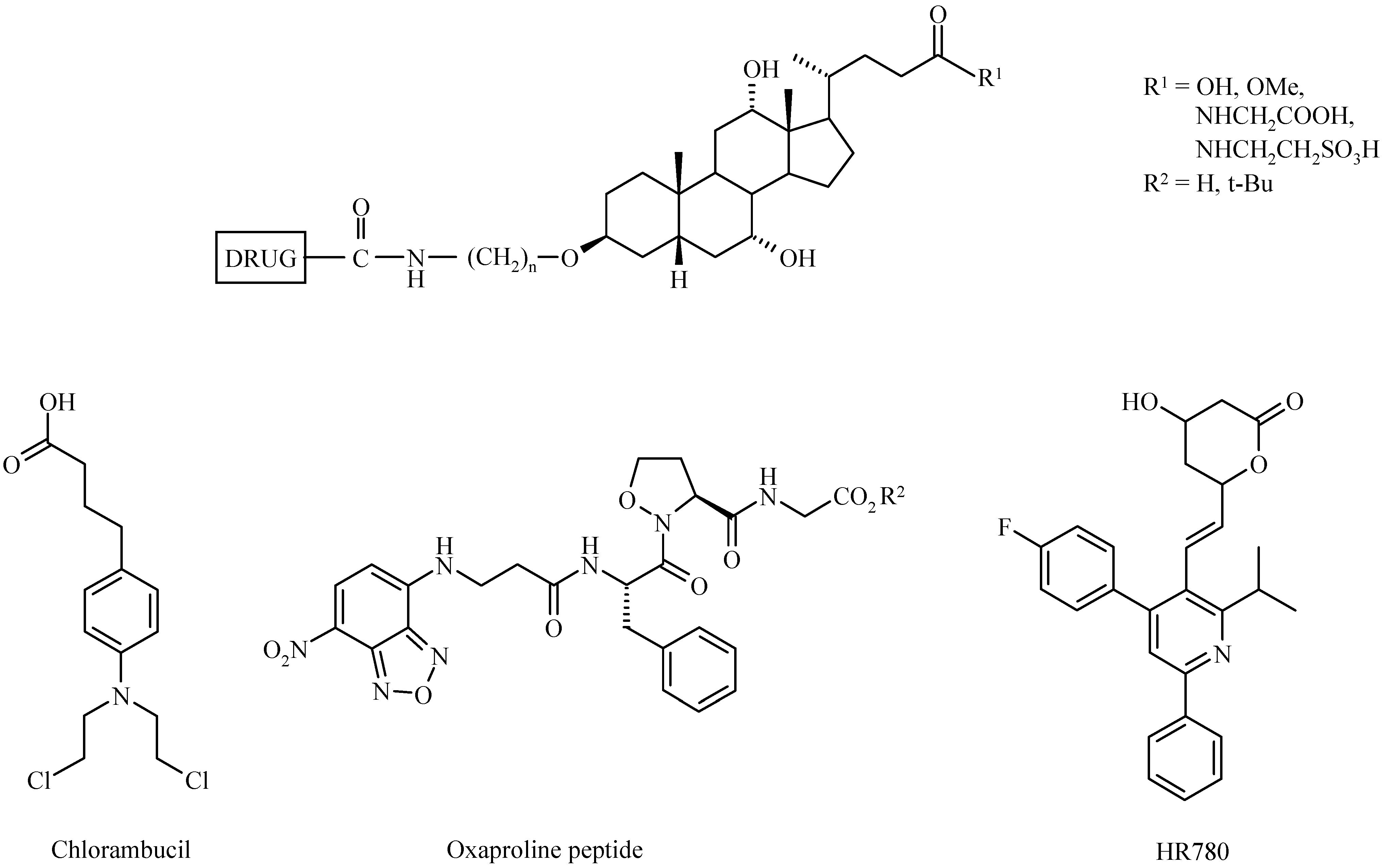
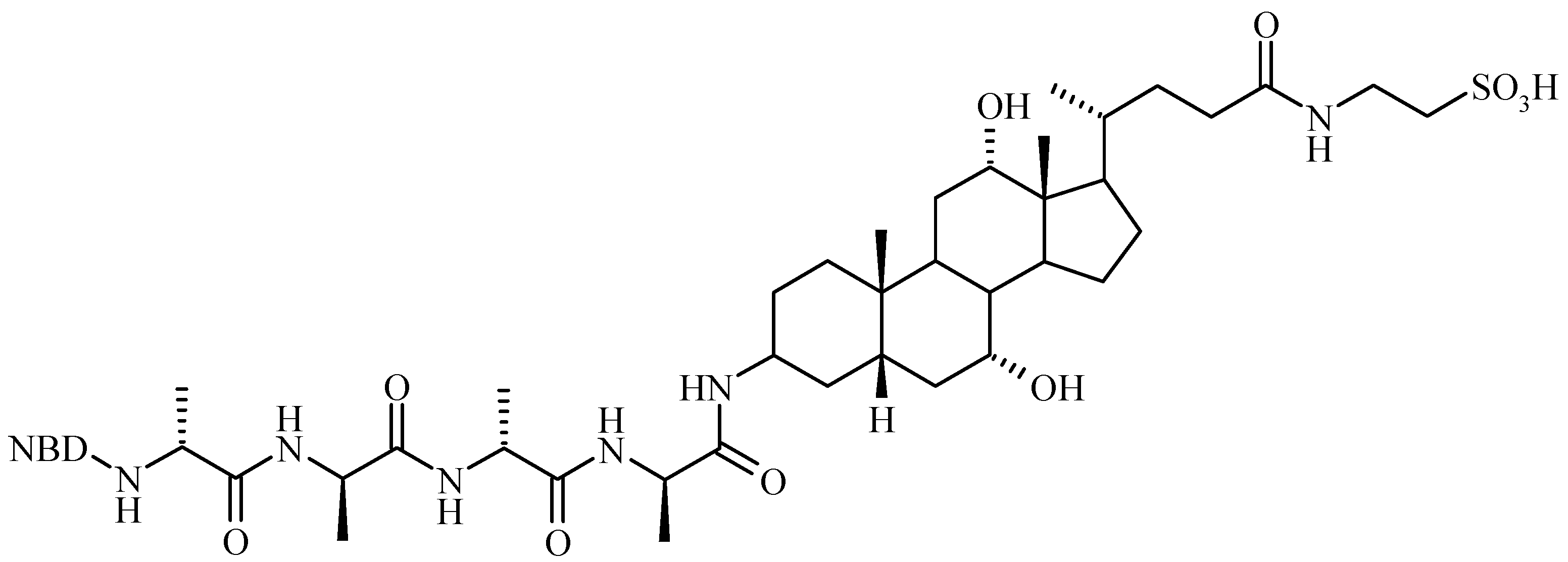
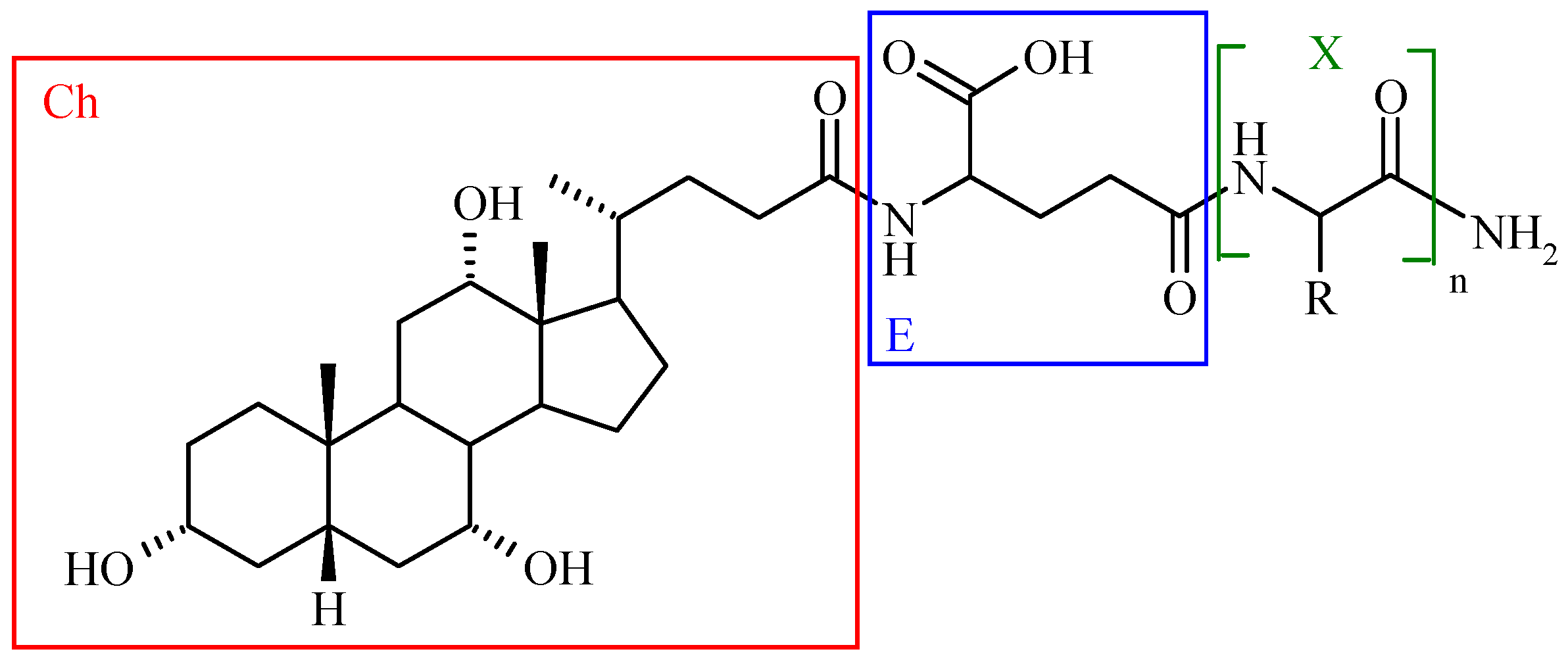
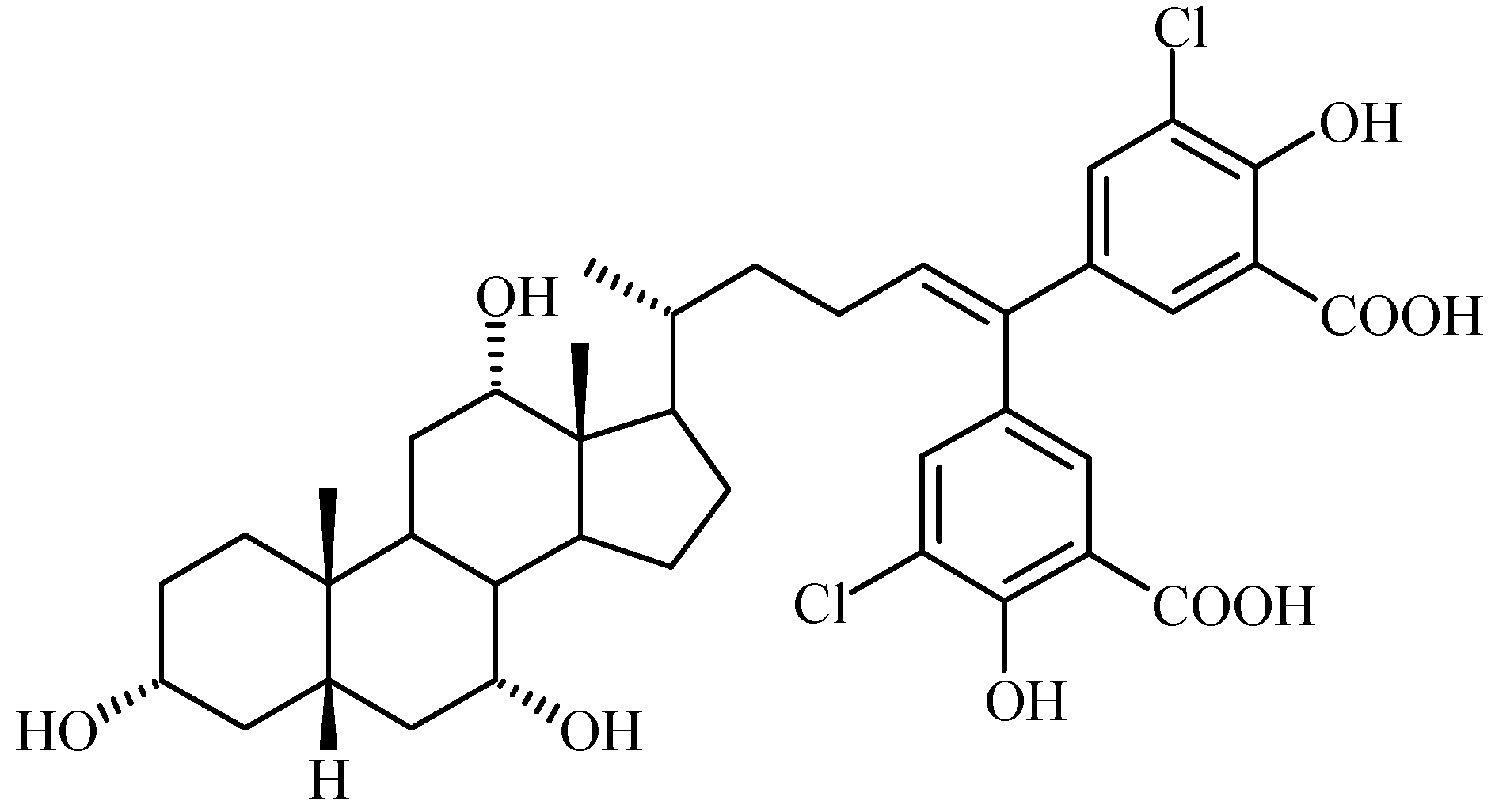
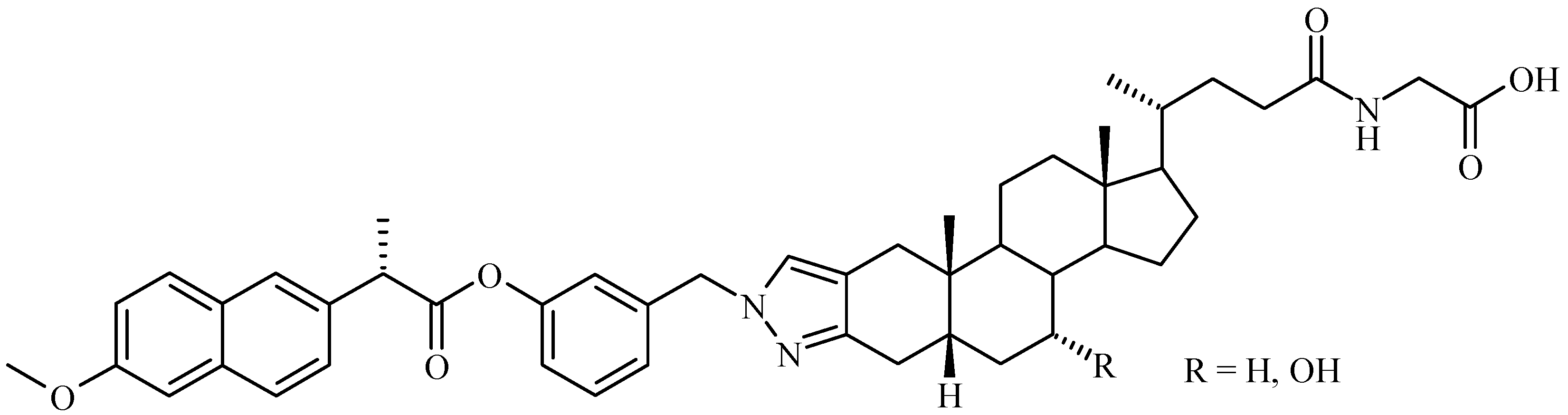
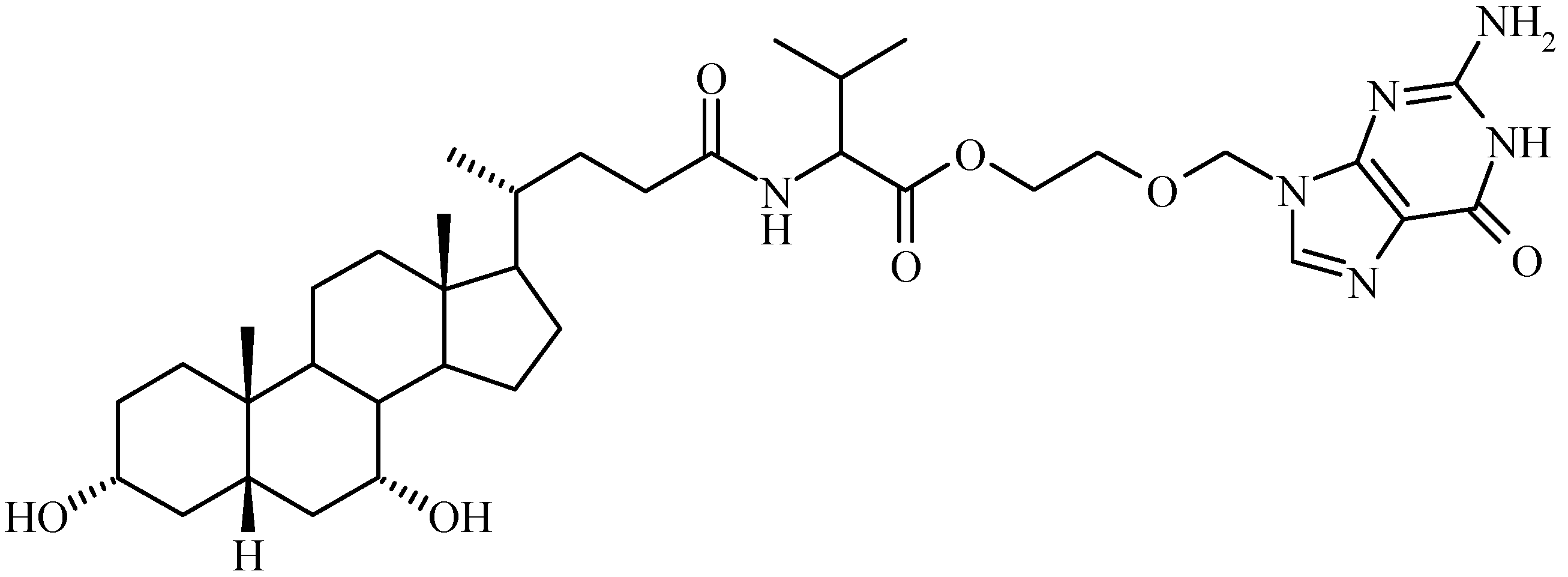
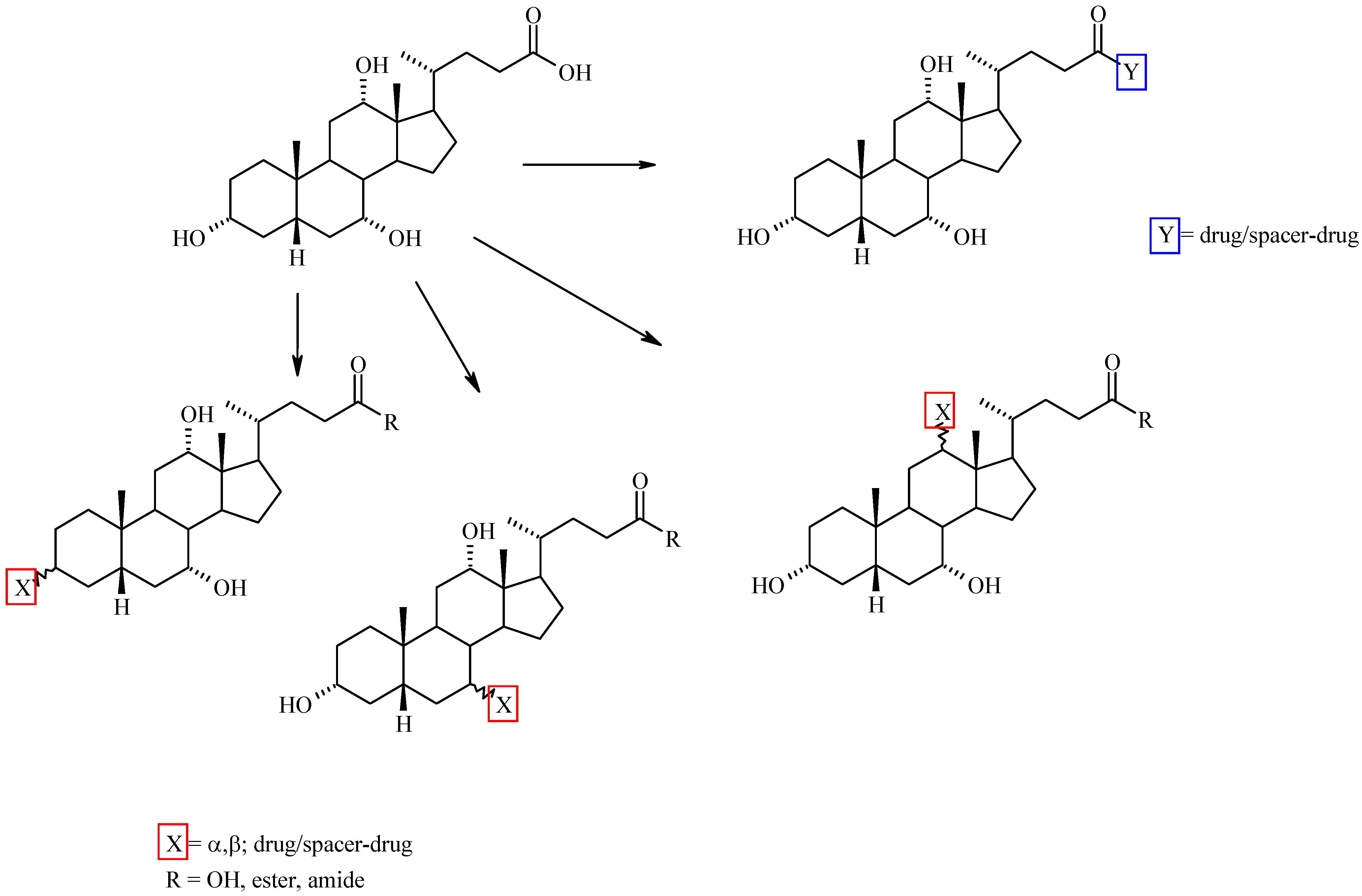
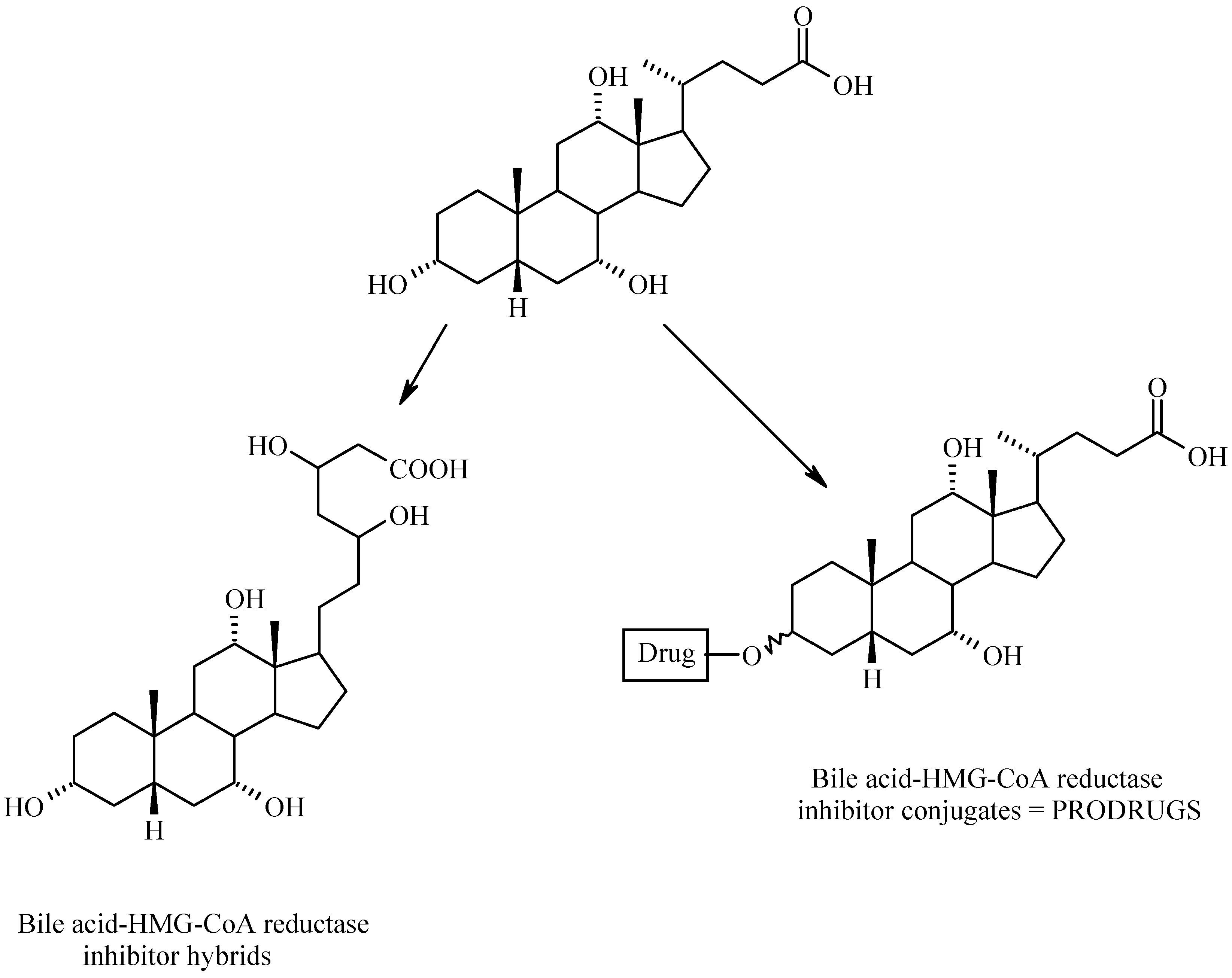
Bile Acid-Containing Prodrugs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Conclusions
Acknowledgements
References
- Yang, C.; Tirucherai, G.S.; Mitra, A.K. Prodrug based optimal drug delivery via membrane transporter/receptor. Exp. Opin. Biol. Ther. 2001, 1, 159–175. [Google Scholar] [CrossRef]
- St-Pierre, M.V.; Kullak-Ublick, G.A.; Hagenbuch, B.; Meier, P.J. Transport of bile acids in hepatic and non-hepatic tissues. J. Exp. Biol. 2001, 204, 1673–1686. [Google Scholar]
- Hofmann, A. The Liver: Biology and Pathobiology; Arias, I.M., Jakoby, W.B., Popper, H., Schachter, D., Shafritz, D.S, Eds.; Raven Press: New York, 1988; pp. 553–572. [Google Scholar]
- Carey, M.C.; Cahalane, M.J. The Liver: Biology and Pathobiology; Arias, I.M., Jakoby, W.B., Popper, H., Schachter, D., Shafritz, D.S., Eds.; Raven Press: New York, 1988; pp. 573–616. [Google Scholar]
- Turley, D.S.; Dietschy, J.M. The Liver: Biology and Pathobiology; Arias, I.M., Jakoby, W.B., Popper, H., Schachter, D., Shafritz, D.S, Eds.; Raven Press: New York, 1988; pp. 617–641. [Google Scholar]
- Petzinger, E. Transport of organic anions in the liver. An update on bile acid, fatty acid, monocarboxylate, anionic amino acid, cholephilic organic anion, and anionic drug transport. Rev. Physiol. Biochem. Pharmacol. 1994, 123, 47–211. [Google Scholar] [CrossRef]
- Stedronsky, E.R. Interaction of bile acids and cholesterol with non-systemic agents having hypocholesterolemic properties. Biochim. Biophys. Acta 1994, 1210, 255–287. [Google Scholar] [CrossRef]
- Enhsen, A.; Kramer, W.; Wess, G. Bile acids in drug discovery. Drug Discov. Today 1998, 3, 409–418. [Google Scholar]
- Soul-Lawton, J.; Seaber, E.; On, N.; Wootton, R.; Rolan, P.; Posner, J. Absolute bioavailability and metabolic disposition of valaciclovir, the l-valyl ester of acyclovir, following oral administration to humans. Antimicrob. Agents Chemother. 1995, 39, 2759–2764. [Google Scholar]
- Han, H.; de Vrueh, R.L.; Rhie, J.K.; Covitz, K.M.; Smith, P.L.; Lee, C.P.; Oh, D.M.; Sadee, W.; Amidon, G.L. 5’-Amino acid esters of antiviral nucleosides, acyclovir and AZT, are absorbed by the intestinal PEPT1 peptide transporter. Pharm. Res. 1998, 15, 1154–1159. [Google Scholar]
- Davis, A.P. Cholaphanes et al.; Steroids as structural components in molecular engineering. Chem. Soc. Rev. 1993, 22, 243–253. [Google Scholar] [CrossRef]
- Li, Y.; Dias, J.R. Dimeric and oligomeric steroids. Chem. Rev. 1997, 97, 283–304. [Google Scholar]
- Wallimann, P.; Marti, T.; Fürer, A.; Diederich, F. Steroids in molecular recognition. Chem. Rev. 1997, 97, 1567–1608. [Google Scholar]
- Davis, A.P.; Bonar-Law, R.P.; Sanders, J.K.M. Comprehensive Supramolecular Chemistry; Atwood, J.L., Davies, J.E.D., MacNicol, D.D., Vögtle, F., Eds.; Elsevier: Oxford, 1996; Vol. 4, pp. 257–286. [Google Scholar]
- Miyata, M.; Sada, K. Comprehensive Supramolecular Chemistry; Atwood, J.L., Davies, J.E.D., MacNicol, D.D., Vögtle, F., Eds.; Elsevier: Oxford, 1996; Vol. 6, pp. 147–176. [Google Scholar]
- Tamminen, J.; Kolehmainen, E. Bile acids as building blocks of supramolecular hosts. Molecules 2001, 6, 21–46. [Google Scholar]
- Virtanen, E.; Kolehmainen, E. Use of bile acids in pharmacological and supramolecular applications. Eur. J. Org. Chem. 2004, 3385–3399. [Google Scholar]
- Mukhopadhyay, S.; Maitra, U. Chemistry and Biology of Bile Acids. Curr. Sci. 2004, 87, 1666–1683. [Google Scholar]
- Kramer, W.; Wess, G. Bile acid transport systems as pharmaceutical targets. Eur. J. Clin. Invest. 1996, 26, 715–732. [Google Scholar]
- Kramer, W.; Glombik, H. Bile acid reabsorption inhibitors (BARI): Novel hypolipidemic drugs. Curr. Med. Chem. 2006, 13, 997–1016. [Google Scholar]
- Kritchevsky, D.; Nair, P.P. The Bile Acids: Chemistry, Physiology, and Metabolism; Nair, P.P., Kritchevsky, D., Eds.; Plenum: New York, 1971; Vol. 1, pp. 3–9. [Google Scholar]
- Jelinek, D.F.; Andersson, S.; Slaughter, C.A.; Russell, D.W. Cloning and regulation of cholesterol 7 alpha-hydroxylase, the rate-limiting enzyme in bile acid biosynthesis. J. Biol. Chem. 1990, 265, 8190–8197. [Google Scholar]
- Falany, C.N.; Johnson, M.R.; Barnes, S.; Diasio, R.B. Glycine and taurine conjugation of bile acids by a single enzyme. Molecular cloning and expression of human liver bile acid CoA:amino acid N-acyltransferase. J. Biol. Chem. 1994, 269, 19375–19379. [Google Scholar]
- Hofmann, A.F.; Mysels, K.J. Bile acid solubility and precipitation in vitro and in vivo: the role of conjugation, pH, and Ca2+ ions. J. Lipid Res. 1992, 33, 617–626. [Google Scholar]
- Balakrishnan, A.; Polli, J.E. Apical sodium dependent transporter (ASBT, SLC10A2): A potential prodrug target. Mol. Pharm. 2006, 3, 223–230. [Google Scholar]
- Chiang, J.Y.L. Regulation of bile acid synthesis. Front. Biosci. 1998, 15, D176–D193. [Google Scholar]
- Pandak, W.M.; Li, Y.C.; Chiang, J.Y.; Studer, E.J.; Gurley, E.C.; Heuman, D.M.; Vlahcevic, Z.R.; Hylemon, P.B. Regulation of cholesterol 7 alpha-hydroxylase mRNA and transcriptional activity by taurocholate and cholesterol in the chronic biliary diverted rat. J. Biol. Chem. 1991, 266, 3416–3421. [Google Scholar]
- Duane, W.C. Effects of lovastatin and dietary cholesterol on sterol homeostasis in healthy human subjects. J. Clin. Invest. 1993, 92, 911–918. [Google Scholar] [CrossRef]
- Nguyen, L.B.; Xu, G.; Shefer, S.; Tint, G.S.; Batta, A.; Salen, G. Comparative regulation of hepatic sterol 27-hydroxylase and cholesterol 7alpha-hydroxylase activities in the rat, guinea pig and rabbit: effects of cholesterol and bile acids. Metabolism 1999, 48, 1542–1548. [Google Scholar]
- Xu, G.; Salen, G.; Shefer, S.; Tint, G.S.; Nguyen, L.B.; Chen, T.S.; Greenblatt, D. Increasing dietary cholesterol induces different regulation of classic and alternative bile acid synthesis. J. Clin. Invest. 1999, 103, 89–95. [Google Scholar]
- Wess, K.; Kramer, W.; Enhsen, A.; Glombik, H.; Baringhaus, K.-H.; Böger, G.; Urmann, M.; Bock, K.; Kleine, H.; Neckermann, G.; Hoffmann, A.; Pittius, C.; Falk, E.; Fehlhaber, H.-W.; Kogler, H.; Friedrich, M. Specific inhibitors of ileal bile acid transport. J. Med. Chem. 1994, 37, 873–875. [Google Scholar]
- Kramer, W.; Müllner, S.; Gutweiler, M.; Kroggel, M. Polymers and oligomers of bile acids, process for their production as use as medicines. EP0549967 1993. [Google Scholar]
- Enhsen, A.; Glombik, H.; Kramer, W.; Wess, G. Bile acid derivatives, a process for their production and their use as medicines. EP0573848 1993. [Google Scholar]
- Wess, G.; Enhsen, A.; Glombik, H.; Kramer, W. Bile acid derivatives, a method for their production and their use as medicines. EP0624593 1994. [Google Scholar]
- Glombik, H.; Enshsen, A.; Kramer, W.; Wess, G. Monomeric bile acid derivatives, a process for their production, and their use as medicines. EP0624594 1994. [Google Scholar]
- Enhsen, A.; Glombik, H.; Kramer, W.; Wess, G. Nor-derivation of bile acids, a process for their production and their use as medicines. EP0624595 1994. [Google Scholar]
- Enhsen, A.; Glombik, H.; Kramer, W.; Wess, G. Tetrazole derivatives of bile acids, a process for their production and their use as medicines. EP0624596 1994. [Google Scholar]
- Kobayashi, K.; Oka, T. Enteroabsorption inhibitor for bile acid. JP6321783 1994. [Google Scholar]
- Kobayashi, K.; Kobayashi, K. Enteroabsorption inhibitor for bile acid. JP6321785 1994. [Google Scholar]
- Vicens, M.; Medarde, M.; Macias, R.I.R.; Larena, M.G.; Villafaina, A.; Serrano, M.A.; Marin, J.J.G. Novel cationic and neutral glycocholic acid and polyamine conjugates able to inhibit transporters involved in hepatic and intestinal bile acid uptake. Bioorg. Med. Chem. 2007, 15, 2359–2367. [Google Scholar]
- Hofmann, A.F. Bile acids as drugs: principles, mechanisms of action, and formulations. Ital. J. Gastroenterol. 1995, 106–113. [Google Scholar]
- Stroup, D.; Crestani, M.; Chiang, J.Y. Identification of a bile acid response element in the cholesterol 7 alpha-hydroxylase gene CYP7A. Am. J. Physiol. 1997, 273, G508–G517. [Google Scholar]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704. [Google Scholar]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; Lehmann, J.M. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Devl. Biol. 2000, 16, 459–481. [Google Scholar]
- Houten, S.M.; Watanabe, M.; Auwerx, J. Endocrine functions of bile acids. EMBO J. 2006, 25, 1419–1425. [Google Scholar]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Constantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6α-Ethyl-chenodeoxycholic acid (6ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar]
- Pellicciari, R.; Constantin, G.; Camaioni, E.; Sadeghpour, B.M.; Entrena, A.; Willson, T.M.; Fiorucci, S.; Clerici, C.; Gioiello, A. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J. Med. Chem. 2004, 47, 4559–4569. [Google Scholar]
- Dawson, P.A.; Hubbert, M.; Haywood, J.; Craddock, A.L.; Zerangue, N.; Christian, W.V.; Ballatori, N. The heteromeric organic solute transporter alpha-beta, Ostα-Ostβ, is an ileal basolateral bile acid transporter. J. Biol. Chem. 2005, 280, 6960–6968. [Google Scholar]
- Ballatori, N.; Christian, W.V.; Lee, J.Y.; Dawson, P.A.; Soroka, C.J.; Boyer, J.L.; Madejczyk, M.S.; Li, N. OSTα-OSTβ: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005, 42, 1270–1279. [Google Scholar]
- Hagenbuch, B.; Meier, P.J. Molecular cloning, chromosomal localization and functional characterization of a human liver Na+/bile acid cotransporter. J. Clin. Invest. 1994, 93, 1326–1331. [Google Scholar]
- Stieger, B.; Hagenbuch, B.; Landmann, L.; Hochli, M.; Schröder, A.; Meier, P.J. In situ localization of the hepatocytic Na+/taurocholate cotransporting polypeptide in rat liver. Gastroenterol. 1994, 107, 1781–1787. [Google Scholar]
- Weinman, S.A. Electrogenicity of Na+-coupled bile acid transporters. Yale J. Biol. Med. 1997, 70, 331–340. [Google Scholar]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am. J. Physiol. 2000, 278, G156–G164. [Google Scholar]
- Kullak-Ublick, G.A.; Ismair, M.G.; Stieger, B.; Landmann, L.; Huber, R.; Pizzagalli, F.; Fattinger, K.; Meier, P.J.; Hagenbuch, B. Organic anion transporting polypeptide B (OATP-B) and its functional comparison with three other OATPS of human liver. Gastroenterology 2001, 120, 525–533. [Google Scholar]
- Gerloff, T.; Stieger, B.; Hagenbuch, B.; Madon, J.; Landmann, L.; Roth, J.; Hofmann, A.F.; Meier, P.J. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J. Biol. Chem. 1998, 273, 10046–10050. [Google Scholar]
- Nishida, T.; Gatmaitan, Z.; Che, M.; Arias, I.M. Rat liver canalicular membrane vesicles contain an ATP-dependent bile acid transport system. Proc. Natl. Acad. Sci. USA 1991, 88, 6590–6594. [Google Scholar]
- Stieger, B.B.O.N.; Meier, P.J. ATP-dependent bile-salt transport in canalicular rat liver plasma-membrane vesicles. Biochem. J. 1992, 284, 67–74. [Google Scholar]
- Strautnieks, S.S.; Bull, L.N.; Knisely, A.S.; Kocoshis, S.A.; Dahl, N.; Arnell, H.; Sokal, E.; Dahan, K.; Childs, S.; Ling, V.; Tanner, M.S.; Kagalwalla, A.F.; Nemeth, A.; Pawlowska, J.; Baker, A.; Mieli-Vergani, G.; Freimer, N.B.; Gardiner, R.M.; Thompson, R.J. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intra-hepatic cholestasis. Nature Genetics 1998, 20, 233–238. [Google Scholar]
- Stieger, B.; Fattinger, K.; Madon, J.; Kullak-Ublick, G.A.; Meier, P.J. Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterol. 2000, 118, 422–430. [Google Scholar]
- Keppler, D.; Leier, I.; Jedlitschky, G. Transport of glutathione conjugates and glucuronides by the multidrug resistance proteins MRP1 and MRP2. Biol. Chem. 1997, 378, 787–791. [Google Scholar]
- Lam, P.; Wang, R.; Ling, V. Bile acid transport in sister of P-glycoprotein (ABCB11) knockout mice. Biocehmistry 2005, 44, 12598–12605. [Google Scholar] [CrossRef]
- Mayer, R.; Kartenbeck, J.; Buchler, M.; Jedlitschky, G.; Leier, I.; Keppler, D. Expression of the MRP gene-encoded conjugate export pump in liver and its selective absence from the canalicular membrane in transport-deficient mutant hepatocytes. J. Cell. Biol. 1995, 131, 137–150. [Google Scholar]
- Kool, M.; van der Linden, M.; de Haas, M.; Scheffer, G.L.; de Vree, J.M.; Smith, A.J.; Jansen, G.; Peters, G.J.; Ponne, N.; Scheper, R.J.; Elferink, R.P.; Baas, F.; Borst, P. MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc. Natl. Acad. Sci. USA 1999, 96, 6914–6919. [Google Scholar]
- König, J.; Rost, D.; Cui, Y.; Keppler, D. Characterization of the human multidrug resistance protein isoform MRP3 localized to the basolateral hepatocyte membrane. Hepatology 1999, 29, 1156–1163. [Google Scholar]
- König, J.; Nies, A.T.; Cui, Y.; Leier, I.; Keppler, D. Conjugate export pumps of the multidrug resistance protein (MRP) family: Localization, substrate specificity and MRP2-mediated drug resistance. Biochim. Biophys. Acta 1999, 1461, 377–394. [Google Scholar] [CrossRef]
- Hirohashi, T.; Suzuki, H.; Takikawa, H.; Sugiyama, Y. ATP-dependent transport of bile salts by rat multidrug resistance-associated protein 3 (Mrp3). J. Biol. Chem. 2000, 275, 2905–2910. [Google Scholar]
- Zeng, H.; Liu, G.; Rea, P.A.; Kruh, G.D. Transport of amphipathic anions by human multidrug resistance protein 3. Cancer Res. 2000, 60, 4779–4784. [Google Scholar]
- Wong, M.H.; Oelkers, P.; Dawson, P.A. Identification of a mutation in the ileal sodium-dependent bile acid transporter gene that abolishes transport activity. J. Biol. Chem. 1995, 270, 27228–27234. [Google Scholar]
- Lazaridis, K.N.; Pham, L.; Tietz, P.; Marinelli, R.A.; deGroen, P.C.; Levine, S.; Dawson, P.A.; LaRusso, N.F. Rat cholangiocytes absorb bile acids at their apical domain via the ileal sodium-dependent bile acid transporter. J. Clin. Invest. 1997, 100, 2714–2721. [Google Scholar]
- Craddock, A.L.; Love, M.W.; Daniel, R.W.; Kirby, L.C.; Walters, H.C.; Wong, M.H.; Dawson, P.A. Expression and transport properties of the human ileal and renal sodium-dependent bile acid transporter. Am. J. Physiol. 1998, 274, G157–G169. [Google Scholar]
- Weinman, S.A.; Carruth, M.W.; Dawson, P.A. Bile acid uptake via the human apical sodium-bile acid cotransporter is electrogenic. J. Biol. Chem. 1998, 273, 34691–34695. [Google Scholar]
- Hallén, S.; Fryklund, J.; Sachs, G. Inhibition of the human sodium/bile acid cotransporters by side-specific methanethiosulfonate sulfhydryl reagents: substrate-controlled accessibility of site of inactivation. Biochemistry 2000, 39, 6743–6750. [Google Scholar]
- Hallén, S.; Björquist, A.; Östlund-Lindqvist, A.M.; Sachs, G. Identification of a region of the ileal-type sodium/bile acid cotransporter interacting with a competitive bile acid transport inhibitor. Biochemistry 2002, 41, 14916–14924. [Google Scholar] [CrossRef]
- Kramer, W.; Girbig, F.; Glombik, H.; Corsiero, D.; Stengelin, S.; Weyland, C. Identification of a ligand-binding site in the Na+/bile acid cotransporting protein from rabbit ileum. J. Biol. Chem. 2001, 276, 36020–36027. [Google Scholar]
- Banerjee, A.; Ray, A; Chang, C.; Swaan, P.W. Site-directed mutagenesis and use of bile acid-MTS conjugates to probe the role of cysteines in the human apical sodium-dependent bile acid transporter (SLC10A2). Biochemistry 2005, 44, 8908–8917. [Google Scholar]
- Ray, A.; Banerjee, A.; Chang, C.; Khantwal, C.M.; Swaan, P.W. Design of novel synthetic MTS conjugates of bile acids for site-directed sulfhydryl labeling of cysteine residues in bile acid binding and transporting proteins. Bioorg. Med. Chem. Lett. 2006, 16, 1473–1476. [Google Scholar]
- Kramer, W.; Stengelin, S.; Baringhaus, K.-H.; Enhsen, A.; Heuer, H.; Becker, W.; Corsiero, D.; Girbig, F.; Noll, R.; Weyland, C. Substrate specificity of the ileal and the hepatic Na(+)/bile acid cotransporters of the rabbit. I. Transport studies with membrane vesicles and cell lines expressing the cloned transporters. J. Lipid. Res. 1999, 40, 1604–1617. [Google Scholar]
- Izzat, N.N.; Deshazer, M.E.; Loose Mitchell, D.S. New molecular targets for cholesterol-lowering therapy. J. Pharmacol. Exp. Ther. 2000, 293, 315–320. [Google Scholar]
- Thompson, G.R.; Naumova, R.P. Novel lipid regulating drugs. Expert. Opin. Invest. Drugs 2000, 9, 2619–2628. [Google Scholar]
- Cattori, V.; Hagenbuch, B.; Stieger, B.; Landmann, L.; Winterhalter, K.H.; Meier, P.J. Characterization of the rat liver organic anion transporting polypeptide 4 (Oatp4) and its comparison with Oatp3. Hepatology 2000, 32, 437A. [Google Scholar]
- Walters, H.C.; Craddock, A.L.; Fusegawa, H.; Willingham, M.C.; Dawson, P.A. Expression, transport properties and chromosomal location of organic anion transporter subtype 3. Am. J. Physiol. 2000, 279, G1188–G1200. [Google Scholar]
- Kramer, W.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Jouvenal, K.; Müller, G.; Tripier, D.; Wess, G. Intestinal bile acid absorption. Sodium-dependent bile acid transport activity in rabbit small intestine correlates with the coexpression of an integral 93-kDa and a peripheral 14-kDa bile acid-binding membrane protein along the duodenum-ileum axis. J. Biol. Chem. 1993, 268, 18305–18046. [Google Scholar]
- Kramer, W.; Girbig, F.; Gutjahr, U.; Kowalewski, S. Radiation-inactivation analysis of the Na+/bile acid co-transport system from rabbit ileum. Biochem. J. 1995, 305, 241–246. [Google Scholar]
- Kramer, W.; Wess, G.; Bewersdorf, U.; Corsiero, D.; Girbig, F.; Weyland, C.; Stengelin, S.; Enhsen, A.; Bock, K.; Kleine, H.; Le Dreau, M.A.; Schäfer, H.F. Topological photoaffinity labeling of the rabbit ileal Na+/bile-salt-cotransport system. Eur. J. Biochem. 1997, 249, 456–464. [Google Scholar]
- Kramer, W.; Corsiero, D.; Friedrich, M.; Girbig, F.; Stengelin, S.; Weyland, C. Intestinal absorption of bile acids: Paradoxical behaviour of the 14 kDa ileal lipid-binding protein in differential photoaffinity labeling. Biochem. J. 1998, 333, 335–341. [Google Scholar]
- Kramer, W.; Sauber, K.; Baringhaus, K.-H.; Kurz, M.; Stengelin, S.; Lange, G.; Corsiero, D.; Girbig, F.; König, W.; Weyland, C. Identification of the bile acid-binding site of the ileal lipid-binding protein by photoaffinity labeling, matrix-assisted laser desorption ionization-mass spectrometry, and NMR structure. J. Biol. Chem. 2001, 276, 7291–7301. [Google Scholar]
- Kurz, M.; Brachvogel, V.; Matter, H.; Stengelin, S.; Thüring, H.; Kramer, W. Insights into the bile acid transportation system: The human ileal lipid-binding protein-cholyltaurine complex and ist comparison with homologous structure. Proteins: Struct., Funct., Gen. 2003, 50, 312–328. [Google Scholar]
- Weinberg, S.L.; Burckhardt, G.; Wilson, F.A. Taurocholate transport by rat intestinal basolateral membrane vesicles. Evidence for the presence of an anion exchange transport system. J. Clin. Invest. 1986, 78, 44–50. [Google Scholar]
- Lazaridis, K.N.; Tietz, P.; Wu, T.; Kip, S.; Dawson, P.A.; LaRusso, N.F. Alternative splicing of the rat sodium/bile acid transporter changes its cellular localization and transport properties. Proc. Natl. Acad. Sci. USA 2000, 97, 11092–11097. [Google Scholar]
- Hirohashi, T.; Suzuki, H.; Chu, X.Y.; Tamai, I.; Tsuji, A.; Sugiyama, Y. Function and expression of multidrug resistance-associated protein family in human adenocarcinoma cells (Caco-2). J.Pharmac. Exp. Ther. 2000, 292, 265–270. [Google Scholar]
- Kiuchi, Y.; Suzuki, H.; Hirohashi, T.; Tyson, C.A.; Sugiyama, Y. cDNA cloning and inducible expression of human multidrug resistance associated protein 3 (Mrp3). FEBS Lett. 1998, 433, 149–152. [Google Scholar]
- Pascual, M.J.; Macias, R.I.R.; Garcia-del-Pozo, J.; Serrano, M.A.; Marin, J.J.G. Enhanced efficiency of the placental barrier to cisplatin through binding to glycocholic acid. Anticancer Res. 2001, 21, 2703–2707. [Google Scholar]
- Lack, L.; Weiner, I.M. Intestinal bile salt transport: structure-activity relationships and other properties. Am. J. Physiol. 1966, 210, 1142–1152. [Google Scholar]
- Baringhaus, K.-H.; Matter, H.; Stengelin, S.; Kramer, W. Substrate specificity of the ileal and the hepatic Na+/bile acid cotransporters of the rabbit. II. A reliable 3D QSAR pharmacophore model for the ileal hepatic Na+/bile acid cotransporter. J. Lipid Res. 1999, 40, 2158–2168. [Google Scholar]
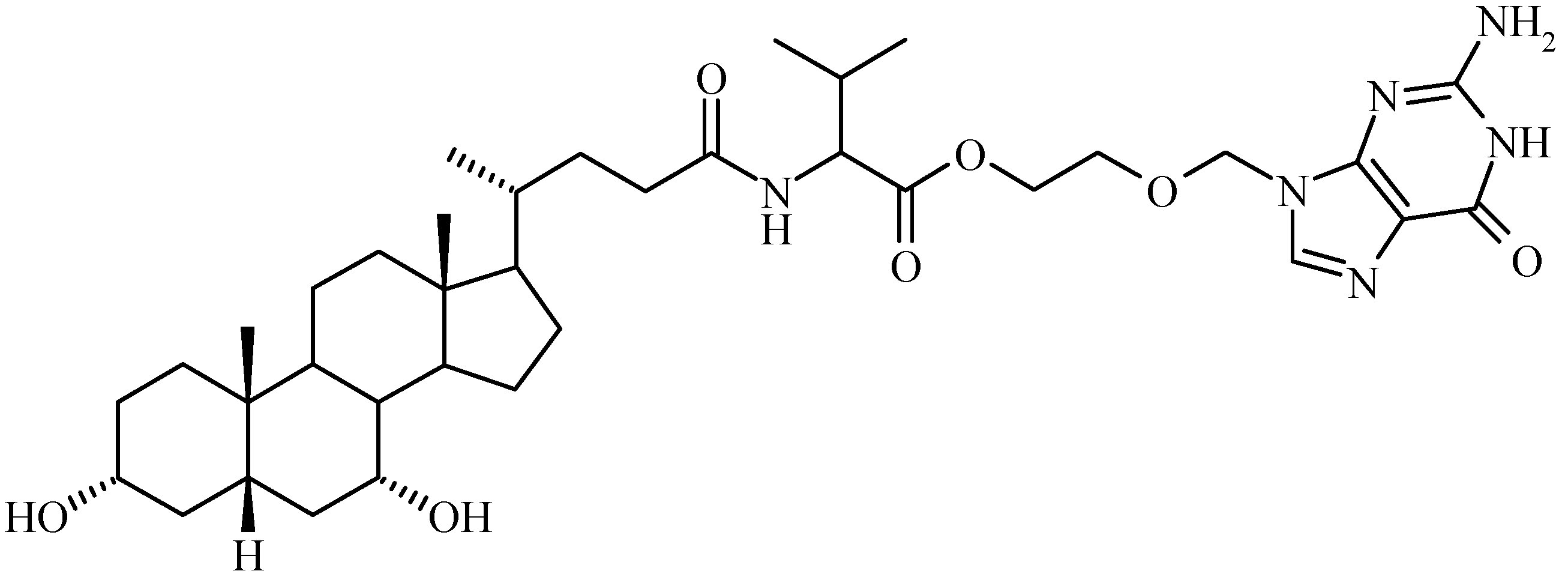
- Tolle-Sander, S.; Lentz, K.A.; Maeda, D.Y.; Coop, A.; Polli, J.E. Increased acyclovir oral bioavailability via a bile acid conjugate. Mol. Pharm. 2004, 1, 40–48. [Google Scholar]
- Balakrishnan, A.; Sussman, D.J.; Polli, J.E. Development of stably transfected monolayer overexpressing the human apical sodium-dependent bile acid transporter (hASBT). Pharm. Res. 2005, 22, 1269–1280. [Google Scholar]
- Balakrishnan, A.; Wring, S.A.; Polli, J.E. Interaction of native bile acids with human apical sodium dependent bile acid transporter (hASBT): Influence of steroidal hydroxylation pattern and C-24 conjugation. Pharm. Res. 2006, 23, 1451–1459. [Google Scholar]
- Swaan, P.W.; Hillgren, K.M.; Szoka, F.C., Jr.; Øie, S. Enhanced transepithelial transport of peptides by conjugation to cholic acid. Bioconj. Chem. 1997, 8, 520–525. [Google Scholar]
- Anwer, M.S.; O’Maille, E.R.; Hofmann, A.F.; DiPietro, R.A.; Michelotti, E. Influence of side-chain charge on hepatic transport of bile acids and bile acid analogues. Am. J. Physiol. Gastrointest. Liver Physiol. 1985, 249, G479–G488. [Google Scholar]
- Asamoto, Y.; Tazuma, S.; Ochi, C.; Chayama, K.; Suzuki, H. Bile-salt hydrophobicity is a key factor regulating rat liver plasma-membrane communication: relation to bilayer structure, fluidity and transporter expression and function. Biochem. J. 2001, 359, 605–610. [Google Scholar]
- Zhang, E.Y.; Knipp, G.T.; Ekins, S.; Swaan, P.W. Structural biology and function of solute transporters: implications for identifying and designing substrates. Drug. Metab. Rev. 2002, 34, 709–750. [Google Scholar]
- Kramer, W.; Wess, G.; Schubert, G.; Bickel, M.; Hoffmann, A.; Baringhaus, K.-H.; Enhsen, A.; Glombik, H.; Müllner, S.; Neckermann, G.; Schulz, S.; Petzinger, E. Bile acids as carriers for drugs. Falk Symp. 1993, 68, 161–176. [Google Scholar]
- Kramer, W.; Wess, G.; Schubert, G.; Bickel, M.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Baringhaus, K.-H.; Enhsen, A.; Glombik, H.; Müllner, S.; Neckermann, G.; Schulz, S.; Petzinger, E. Liver-specific drug targeting by coupling to bile acids. J. Biol. Chem. 1992, 267, 18598–18604. [Google Scholar]
- Wess, G.; Kramer, W.; Schubert, G.; Enhsen, A.; Baringhaus, K.-H.; Glombik, H.; Müllner, S.; Bock, K.; Kleine, H.; John, M.; Neckermann, G.; Hoffmann, A. Synthesis of bile acid-drug conjugates: Potential drug-shuttles for liver specific targeting. Tetrahedron Lett. 1993, 34, 819–822. [Google Scholar]
- Kramer, W.; Wess, G.; Neckermann, G.; Schubert, G.; Fink, J.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Baringhaus, K.-H.; Böger, G.; Enhsen, A.; Falk, E.; Friedrich, M.; Glombik, H.; Hoffmann, A.; Pittius, C.; Urmann, M. Intestinal absorption of peptides by coupling to bile acids. J. Biol. Chem. 1994, 269, 10621–10627. [Google Scholar]
- Wess, G.; Kramer, W.; Bartmann, W.; Enhsen, A.; Glombik, H.; Müllner, S.; Bock, K.; Dries, A.; Kleine, H.; Schmitt, W. Modified bile acids: Preparation of 7α,12α-dihydroxy-3β- and 7α,12α-dihydroxy-3α-(2-hydroxyethoxy)-5β-cholanic acid and their biological activity. Tetrahedron Lett. 1992, 33, 195–198. [Google Scholar]
- Wess, G.; Kramer, W.; Enhsen, A.; Glombik, H.; Baringhaus, K.-H.; Bock, K.; Kleine, H.; Schmitt, W. Preparation of 3α- and 3β-(ω-aminoalkoxy)-7α,12α-dihydroxy-5β-cholanoic acid esters: Versatile shuttles for drug targeting. Tetrahedron Lett. 1993, 34, 817–818. [Google Scholar]
- Kullak-Ublick, G.-A.; Glasa, J.; Böker, C.; Oswald, M.; Grützner, U.; Hagenbuch, B.; Stieger, B.; Meier, P.J.; Beuers, U.; Kramer, W.; Wess, G.; Paumgartner, G. Chlorambucil-taurocholate is transported by bile acid carriers expressed in human hepatocellular carcinomas. Gastroenterology 1997, 113, 1295–1305. [Google Scholar]
- Petzinger, E.; Wickboldt, A.; Pagels, P.; Starke, D.; Kramer, W. Hepatobiliary transport of bile acid amino acid, bile acid peptide, and bile acid oligonucleotide conjugates in rats. Hepatology 1999, 30, 1257–1268. [Google Scholar] [CrossRef]
- Starke, D.; Lischka, K.; Pagels, P.; Uhlmann, E.; Kramer, W.; Wess, G.; Petzinger, E. Bile acid-oligodeoxynucleotide conjugates: Synthesis and liver excretion in rats. Bioorg. Med. Chem. Lett. 2001, 11, 945–949. [Google Scholar]
- Lischka, K.; Starke, D.; Failing, K.; Herling, A.; Kramer, W.; Petzinger, E. Hepatobiliary elimination of bile acid-modified oligodeoxynucleotides in Wistar and TR-rats: Evidence for mrp2 as carrier for oligodeoxynucleotides. Biochem. Pharm. 2003, 66, 565–577. [Google Scholar]
- Lehmann, T.J.; Serwe, M.; Caselmann, W.H.; Engels, J.W. Design and properties of hepatitis C virus antisense oligonucleotides for liver specific drug targeting. Nucleos. Nucleot. Nucl. Acids 2001, 20, 1343–1346. [Google Scholar]
- Lehmann, T.J.; Engels, J.W. Synthesis and properties of bile acid phosphoramidites 5’-tethered to antisense oligodeoxynucleotides against HCV. Bioorg. Med. Chem. 2001, 9, 1827–1835. [Google Scholar]
- Lehninger, A.L.; Nelson, D.L.; Cox, M.M. Principles of BiochemistryWorth Publishers: New York, 1993, 2nd ed; pp. 669–680. [Google Scholar]
- Kramer, W.; Wess, G.; Enhsen, A.; Bock, K.; Falk, E.; Hoffmann, A.; Neckermann, G.; Gantz, D.; Schulz, S.; Nickau, L.; Petzinger, E.; Turley, S.; Dietschy, J.M. Bile acid derived HMG-CoA reductase inhibitors. Biochim. Biophys. Acta 1994, 1227, 137–154. [Google Scholar]
- Wess, G.; Kramer, W.; Han, X.B.; Bock, K.; Enhsen, A.; Glombik, H.; Baringhaus, K.-H.; Böger, G.; Urmann, M.; Hoffmann, A.; Falk, E. Synthesis and biological activity of bile acid-derived HMG-CoA reductase inhibitors. The role of 21-methyl in recognition of HMG-CoA reductase and the ileal bile acid transport system. J. Med. Chem. 1994, 37, 3240–3246. [Google Scholar] [CrossRef]
- Menear, K.A.; Patel, D.; Clay, V.; Howes, C.; Taylor, P.W. A novel approach to the site specific delivery of potential HMG-CoA reductase inhibitors. Bioorg. Med. Chem. Lett. 1992, 2, 285–290. [Google Scholar]
- McCarry, D.G.; Volz, F.A.; Regan, J.R.; Chang, M.N. Compounds having hypocholesterolemic properties. US5216015 1993. [Google Scholar]
- Petzinger, E.; Nickau, L.; Horz, J.A.; Schulz, S.; Wess, G.; Enhsen, A.; Falk, E.; Baringhaus, K.-H.; Glombik, H.; Hoffmann, A.; Müllner, S.; Neckermann, G.; Kramer, W. Hepatobiliary transport of hepatic 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors conjugated with bile acids. Hepatology 1995, 22, 1801–1811. [Google Scholar]
- Kim, D.C.; Harrison, A.W.; Ruwart, M.J.; Wilkinson, K.F.; Fisher, J.F.; Hidalgo, I.J.; Borchardt, R.T. Evaluation of the bile acid transporter in enhancing intestinal permeability to renin-inhibitory peptides. J. Drug Target. 1993, 1, 347–359. [Google Scholar]
- Lee, S.; Lee, J.; Lee, D.Y.; Kim, S.K.; Lee, Y.; Byun, Y. A new drug carrier, Nα-deoxycholyl-l-lysil-methyl-ester, for enhancing insulin absorption in the intestine. Diabetologia 2005, 48, 405–411. [Google Scholar]
- Lee, S.; Kim, K.; Kumar, T.S.; Lee, J.; Kim, S.K.; Lee, D.Y.; Lee, Y.; Byun, Y. Synthesis and biological properties of insulin-deoxycholic acid chemical conjugates. Bioconj. Chem. 2005, 16, 615–620. [Google Scholar]
- Kågedahl, M.; Swaan, P.W.; Redemann, C.T.; Tang, M.; Craik, C.S.; Szoka, F.C., Jr.; Øie, S. Use of the intestinal bile acid transporter for the uptake of cholic acid conjugates with HIV-1 protease inhibitory activity. Pharm. Res. 1997, 14, 176–180. [Google Scholar] [CrossRef]
- Kannan, A.; DeClercq, E.; Pannecouque, C.; Witvrouw, M.; Hartman, T.L.; Turpin, J.A.; Buckheit, R.W., Jr.; Cushman, M. Synthesis and anti-HIV activity of a bile acid analog of cosalane. Tetrahedron 2001, 57, 9385–9391. [Google Scholar]
- Bhat, L.; Gallop, M.A.; Jandeleit, B. Bile acid derived compounds for enhancing oral absorption and systemic bioavailoability of drugs. WO03020214 2003. [Google Scholar]
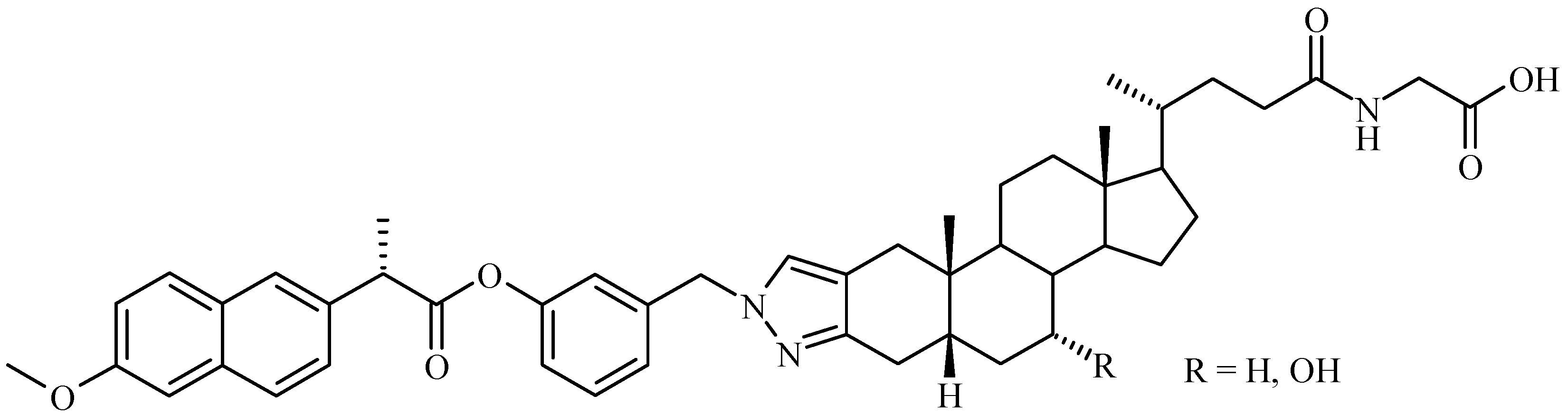
- Bhat, L.; Jandeleit, B.; Dias, T.M.; Moors, T.L.; Gallop, M.A. Synthesis and biological evaluation of novel steroidal pyrazoles as substrates for bile acid transporters. Bioorg. Med. Chem. Lett. 2005, 15, 85–87. [Google Scholar]
- Gallop, M.A.; Cundy, K.C. Compounds for sustained release of orally delivered drugs. WO0228411 2002. [Google Scholar]
- Gallop, M.A.; Cundy, K.C.; Zhou, C.X. Bile acid derived compounds for providing sustained systemic concentrations of drugs after oral administration. WO0228881 2002. [Google Scholar]
- Gallop, M.A.; Cundy, K.C.; Zhou, C.X. Bile acid prodrugs of l-DOPA and their use in the treatment of Parkinsonism. WO0228882 2002. [Google Scholar]
- Gallop, M.A.; Cundy, K.C. Bile acid conjugates for providing sustained systemic concentrations of drugs. WO0228883 2002. [Google Scholar]
- Gallop, M.A.; Cundy, K.C.; Zhou, C.X. Bile acid conjugates for providing sustained systemic concentrations of drugs. WO0232376 2002. [Google Scholar]
- Gallop, M.A.; Cundy, K.C. Bile acid derived compounds for enhancing oral absorption and systemic bioavailoability of drugs. WO0244324 2002. [Google Scholar]
- Marin, G.; Juan, J.; Talavera, C.; Jose, J. Transition metal complexes with cytostatic activity. ES2097085 1997. [Google Scholar]
- Criado, J.J.; Herrera, M.C.; Palomero, M.F.; Medarde, M.; Rodriguez, E.; Marin, J.J.G. Synthesis and characterization of a new bile acid and platinum(II) complex with cytostatic activity. J. Lipid Res. 1997, 38, 1022–1032. [Google Scholar]
- Criado, J.J.; Macias, R.I.R.; Medarde, M.; Monte, M.J.; Serrano, M.A.; Marin, J.J.G. Synthesis and characterization of the new cytostatic complex cis-diammineplatinum(II)-chlorocholylglycinate. Bioconj. Chem. 1997, 8, 453–458. [Google Scholar] [CrossRef]
- Marin, J.J.; Palomero, M.F.; Herrera, M.C.; Macias, R.I.; Criado, J.J.; Serrano, M.A. In vitro cytostatic activity and DNA-interaction of the new liver organotropic complex chloro-bis-cholylglycinate-platinum (II). Anticancer Res. 1998, 18, 1641–1647. [Google Scholar]
- Marin, J.J.G.; Macias, R.I.R.; Criado, J.J.; Bueno, A.; Monte, M.J.; Serrano, M.A. DNA interaction and cytostatic activity of the new liver organotropic complex of cisplatin with glycocholic acid: Bamet-R2. Int. J. Cancer 1998, 78, 346–352. [Google Scholar] [CrossRef]
- Macias, R.I.R.; Monte, M.J.; El-Mir, M.Y.; Villanueva, G.R.; Marin, J.J.G. Transport and biotransformation of the new cytostatic complex cis-diammineplatinum(II)-chlorocholylglycinate (Bamet-R2) by the rat liver. J. Lipid Res. 1998, 39, 1792–1798. [Google Scholar]
- Marin, J.J.G.; Herrera, M.C.; Palomero, M.F.; Macias, R.I.R.; Monte, M.J.; El-Mir, M.Y.; Villanueva, G.R. Rat liver transport and biotransformation of a cytostatic complex of bis-cholylglycinate and platinum (II). J. Hepatol. 1998, 28, 417–425. [Google Scholar]
- Criado, J.J.; Garcia-Moreno, M.C.; Macias, R.R.; Marin, J.J.G.; Medarde, M.; Rodriguez-Fernandez, E. Synthesis and characterization of sodium cis-dichlorochenodeoxycholyl-glycinato(O,N) platinum(II) – Cytostatic activity. BioMetals 1999, 12, 281–288. [Google Scholar]
- Criado, J.J.; Dominguez, M.F.; Medarde, M.; Fernandez, E.R.; Macias, R.I.R.; Marin, J.J.G. Structural characterization, kinetic studies, and in vitro biological activity of new cis-diamminebis-cholylglycinate(O,O’) Pt(II) and cis-diamminebis-ursodeoxycholate(O,O’) Pt(II) complexes. Bioconj. Chem. 2000, 11, 167–174. [Google Scholar]
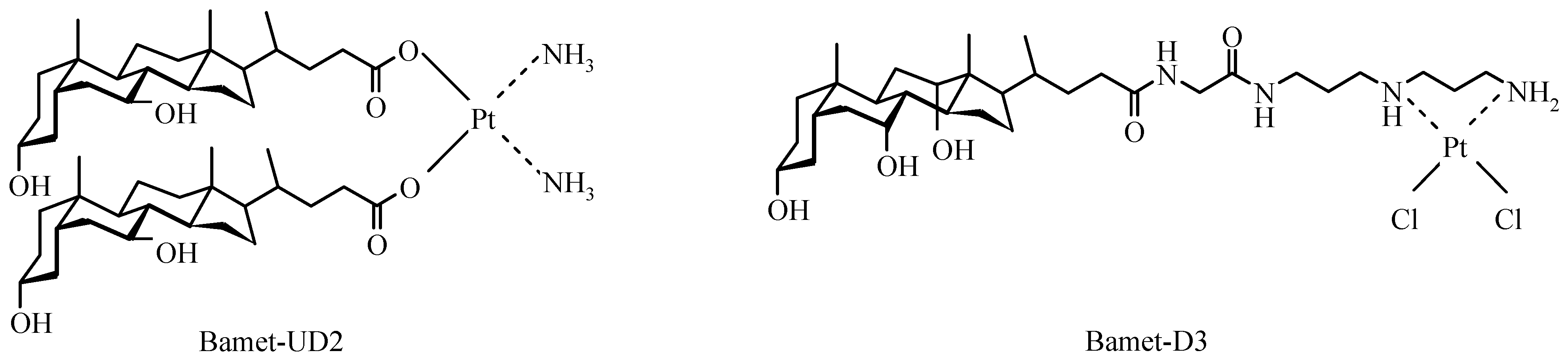
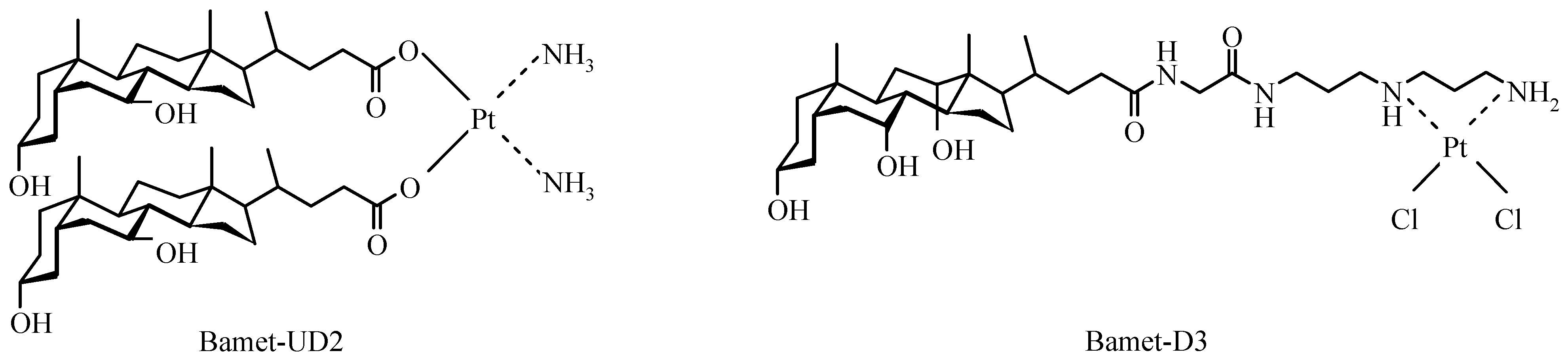
- Martinez-Diez, M.C.; Larena, M.G.; Serrano, M.A.; Macias, R.I.R.; Izco-Basurko, I.; Marin, J.J.G. Relationship between DNA-reactivity and cytostatic effect of two novel bile acid-platinum derivatives, Bamet-UD2 and Bamet-D3. Anticancer Res. 2000, 20, 3315–3322. [Google Scholar]
- Briz, O.; Serrano, M.A.; Macias, R.I.R.; Marin, J.J.G. Overcoming cisplatin resistance in vitro by a free and liposome-encapsulated bile acid derivative: Bamet-R2. Int. J. Cancer 2000, 88, 287–292. [Google Scholar]
- Carrasco, J.; Criado, J.J.; Macias, R.I.; Manzano, J.L.; Marin, J.J.; Medarde, M.; Rodriguez, E. Structural characterization and cytostatic activity of chlorobischolylglycinatogold(III). J. Inorg. Biochem. 2001, 84, 287–292. [Google Scholar]
- Larena, M.G.; Martinez-Diez, M.C.; Monte, M.J.; Dominguez, M.F.; Pascual, M.J.; Marin, J.J. Liver organotropism and biotransformation of a novel platinum-ursodeoxycholate derivative, Bamet-UD2, with enhanced antitumour activity. J. Drug Targ. 2001, 9, 185–200. [Google Scholar]
- Briz, O.; Serrano, M.A.; Rebollo, N.; Hagenbuch, B.; Meier, P.J.; Koepsell, H.; Marin, J.J.G. Carriers involved in targeting the cytostatic bile acid-cisplatin derivatives cis-diammine-chloro-cholylglycinate-platinum(II) and cis-diammine-bisursodeoxycholate-platinum(II) toward liver cells. Mol. Pharmacol. 2002, 61, 853–860. [Google Scholar]
- Criado, J.J.; Manzano, J.L.; Rodriguez-Fernandez, E. New organotropic compounds. Synthesis, characterization and reactivity of Pt(II) and Au(III) complexes with bile acids: DNA interactions and ‘in vitro’ anticancer activity. J. Inorg. Biochem. 2003, 96, 311–320. [Google Scholar] [CrossRef]
- Briz, O.; Macias, R.I.R.; Vallejo, M.; Silva, A.; Serrano, M.A.; Marin, J.J.G. Usefulness of liposomes loaded with cytostatic bile acid derivatives to circumvent chemotherapy resistance of enterohepatic tumors. Mol. Pharmacol. 2003, 63, 742–750. [Google Scholar]
- Paschke, R.; Kalbitz, J.; Paetz, C. Novel spacer linked bile acid-cisplatin compounds as a model for specific drug delivery, synthesis, and characterization. Inorg. Chim. Acta 2000, 304, 241–249. [Google Scholar]
- Campazzi, E.; Cattabriga, M.; Marvelli, L.; Marchi, A.; Rossi, R.; Pieragnoli, M.R.; Fogagnolo, M. Organometallic radiopharmaceuticals: rhenium(I) carbonyl complexes of natural bile acids and derivatives. Inorg. Chim. Acta 1999, 286, 46–54. [Google Scholar]
- Wu, D.; Ji, S.; Wu, Y.; Ju, Y.; Zhao, Y. Design, synthesis, and antitumor activity of bile acid-polyamine-nucleoside conjugates. Bioorg. Med. Chem. Lett. 2007, 17, 2983–2985. [Google Scholar]
- Anelli, P.A.; De Haen, C.; Lattuada, L.; Morosini, P.; Uggeri, F. Bile acid conjugates, derivatives thereof with metal complexes and related uses. WO9532741 1995. [Google Scholar]
- Anelli, P.L.; Brocchetta, M.; De Haen, C.; Gazzotti, O.; Lattuada, L.; Lux, G.; Manfredi, G.; Morosini, P.; Palano, D.; Serleti, M.; Uggeri, F.; Visigalli, M. Blood pool agents for nuclear magnetic resonance diagnostics. WO0038738 2000. [Google Scholar]
- De Haen, C.; Beltrami, A.; Cappelletti, E.; Lattuada, L.; Virtuani, M. Bile acid conjugates with metal ion chelates and the use thereof. WO0164708 2001. [Google Scholar]
- Anelli, P.L.; Lattuada, L.; Lorusso, V.; Lux, G.; Morisetti, A.; Morosini, P.; Serleti, M.; Uggeri, F. Conjugates of gadolinium complexes to bile acids as hepatocyte-directed contrast agents for magnetic resonance imaging. J. Med. Chem. 2004, 47, 3629–3641. [Google Scholar]
- vonDippe, P.; Levy, D. Expression of the bile acid transport protein during liver development and in hepatoma cells. J. Biol. Chem. 1990, 265, 5942–5945. [Google Scholar]
- Polli, J.E.; Coop, A.; Maeda, D.Y.; Lentz, K.A. Bile acid containing prodrugs with enhanced bioavailability. WO0176531 2001. [Google Scholar]
- Jacobson, M.A. Valaciclovir (BW256U87): The l-valyl ester of acyclovir. J. Med. Virol. 1993, Suppl. 1. 150–153. [Google Scholar] [CrossRef]
- Polli, J.E. Formulation approach to enhance the transporter-mediated uptake. WO0025839 2005. [Google Scholar]
- Sample Availability: Contact the author.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Sievänen, E. Exploitation of Bile Acid Transport Systems in Prodrug Design. Molecules 2007, 12, 1859-1889. https://doi.org/10.3390/12081859
Sievänen E. Exploitation of Bile Acid Transport Systems in Prodrug Design. Molecules. 2007; 12(8):1859-1889. https://doi.org/10.3390/12081859
Chicago/Turabian StyleSievänen, Elina. 2007. "Exploitation of Bile Acid Transport Systems in Prodrug Design" Molecules 12, no. 8: 1859-1889. https://doi.org/10.3390/12081859
APA StyleSievänen, E. (2007). Exploitation of Bile Acid Transport Systems in Prodrug Design. Molecules, 12(8), 1859-1889. https://doi.org/10.3390/12081859