Experimental
General
All chemicals were purchased from Aldrich Chemical Co. Nitrobenzene was freshly distilled from 4Å molecular sieve; Benzene, HMPA, and 1,1,2,-tetrachloroethane were dried over 4Å molecular sieve; THF was freshly distilled from sodium granules. Other reagents were used as supplied. Melting points were determined on an Electrothermal melting point apparatus. Both 1H-NMR and 13C-NMR were acquired on Varian VXG-400 S equipment. Mass spectra was recorded on either VG-MM-1212 or MS 902/ZAB instruments. Optical rotations were measured on a Perkin Elmer Polarimeter 343 (PerkinElmer INC., USA). HPLC was carried on a Varian LC Star system (Varian Associates Inc., USA); The column used for reversed-phase analysis was purchase from Vydac (Hesperia, CA 92345, USA), size: 0.46×25 cm packed with 5 μm C18 modified silica gel; the chiral column was available from Phenomenex® (Torrance, CA, USA), model: Chirex (R)-NGLY and DNB; size: 0.46×25 cm. Method used for RP-HPLC analysis: isocratic using pH 2.5 aqueous phosphate buffer/acetonitrile (8/2, V/V); flow rate: 0.5 mL/min; wavelength: 280 nm; Method used for chiral HPLC: isocratic using 0.03 mol/L NH4Ac in methanol; flow rate: 0.5 ml/min; wavelength: 280 nm.
Fluorescence determinations
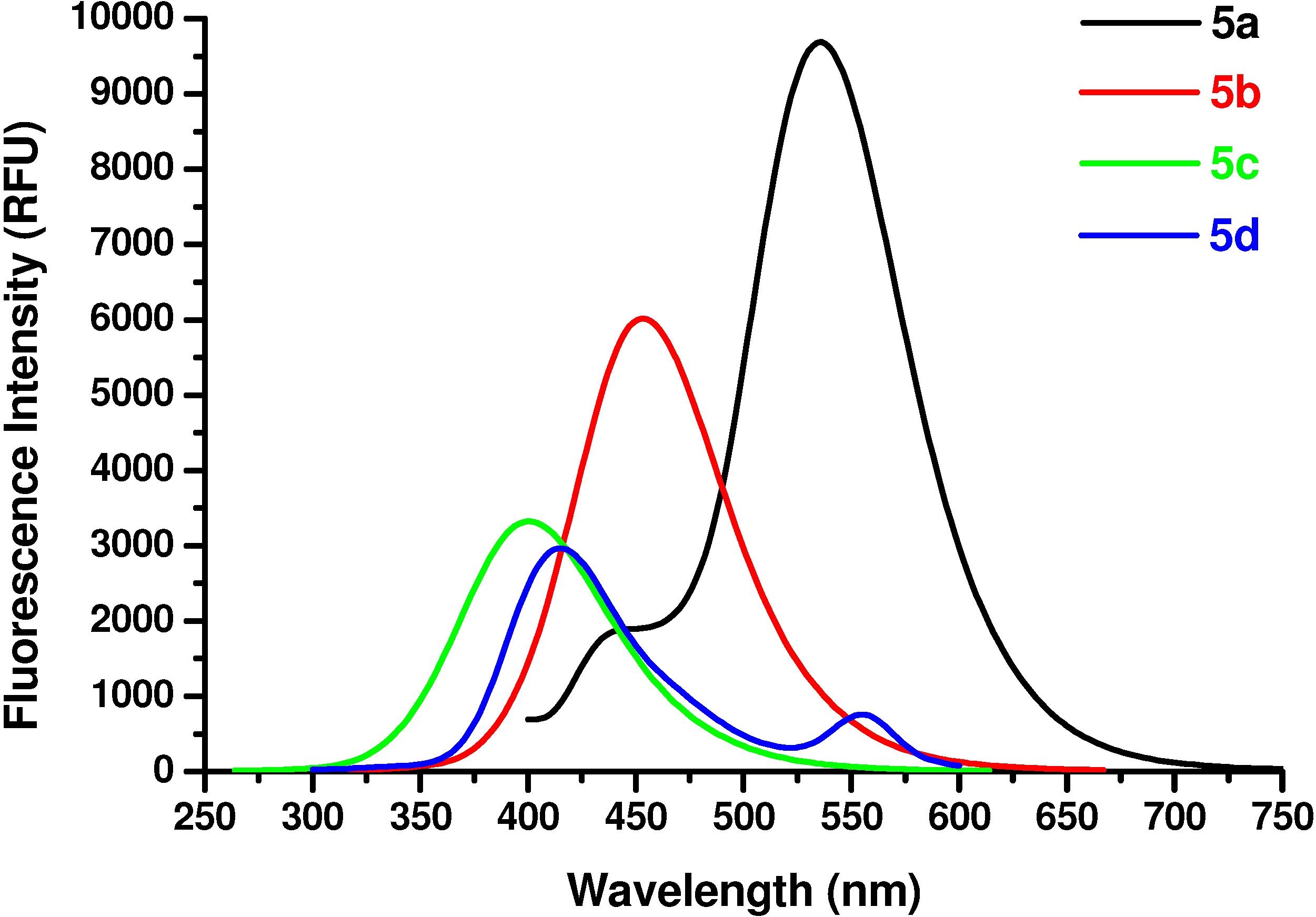
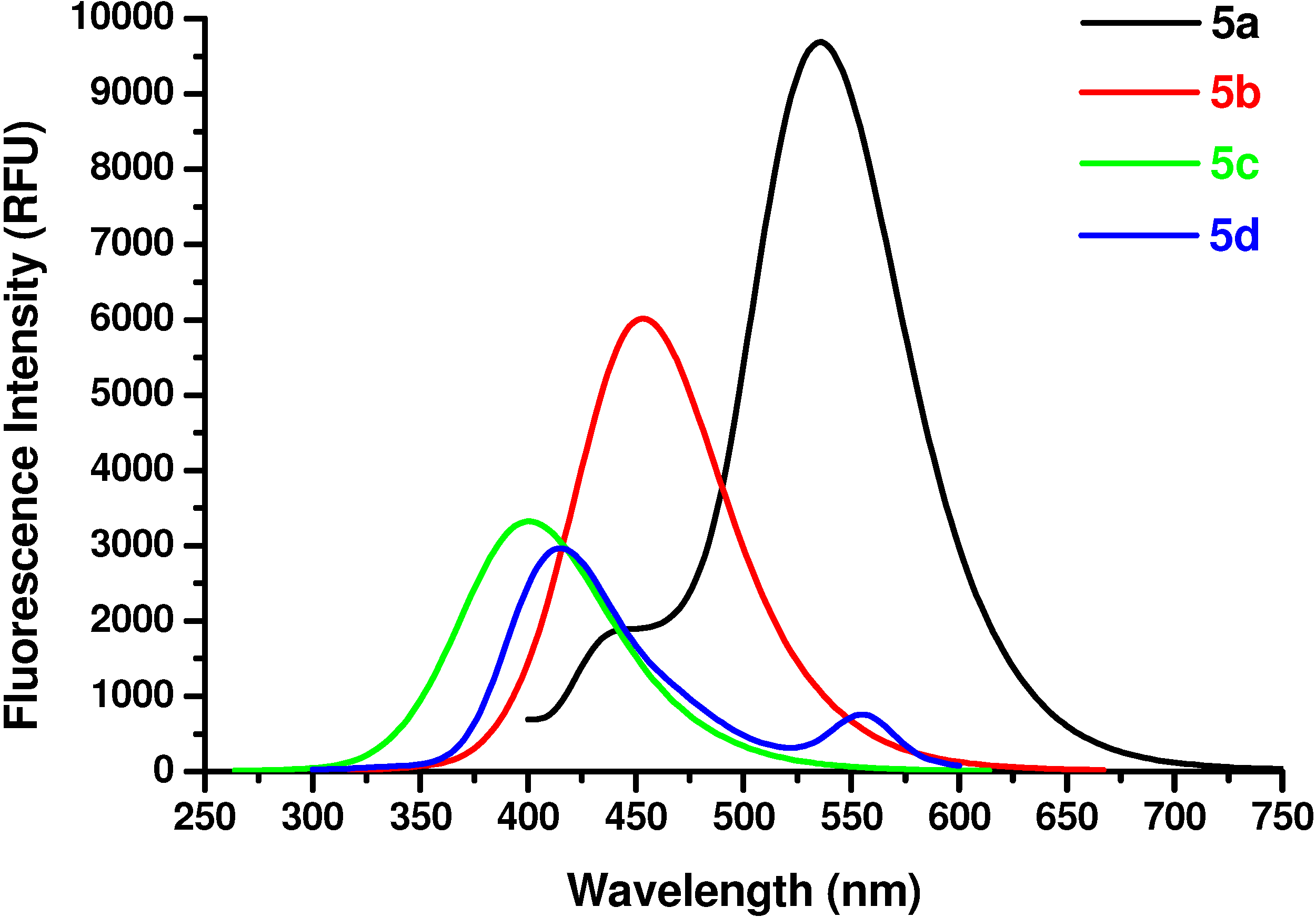
The determinations were carried out on a Gemini EM instrument (Molecular Devices Corporation, Sunnyvale, USA). The excitation wavelength was 350 nm, the cutoff was 420 nm, and the step was 2 nm. The spectral resolution was 2 nm for excitation and for emission. Solutions of sample compounds and reference amino acid in Tris/HCl buffer (pH 6.6) at a concentration of 2×10
-5 mol/L were used. Fluorescence quantum yields (
ϕ) were determined with
N-acetyl-
L-tryptophanamide (NATA) (
ϕNATA = 0.14) as reference. The quantum yield was calculated based on the following equation:
where the subscripts
S and
R refer to the sample and reference compound (NATA), respectively.
E is the integrated area under the corrected emission spectrum.
A is the absorbance of the solution at the excitation wavelength (
A < 0.05) and (
nS/
nR)
2 is the correction for the refractive index.
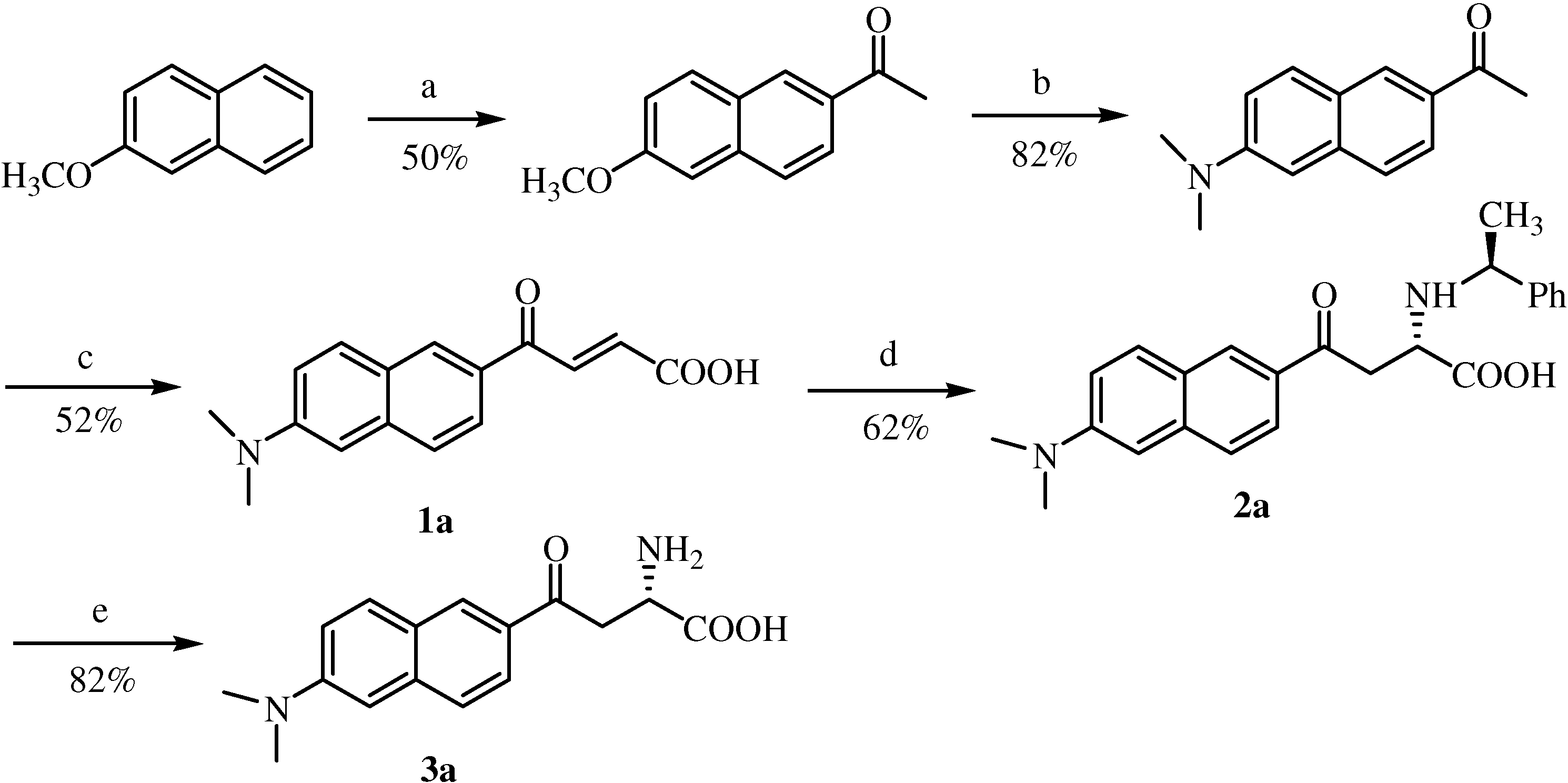
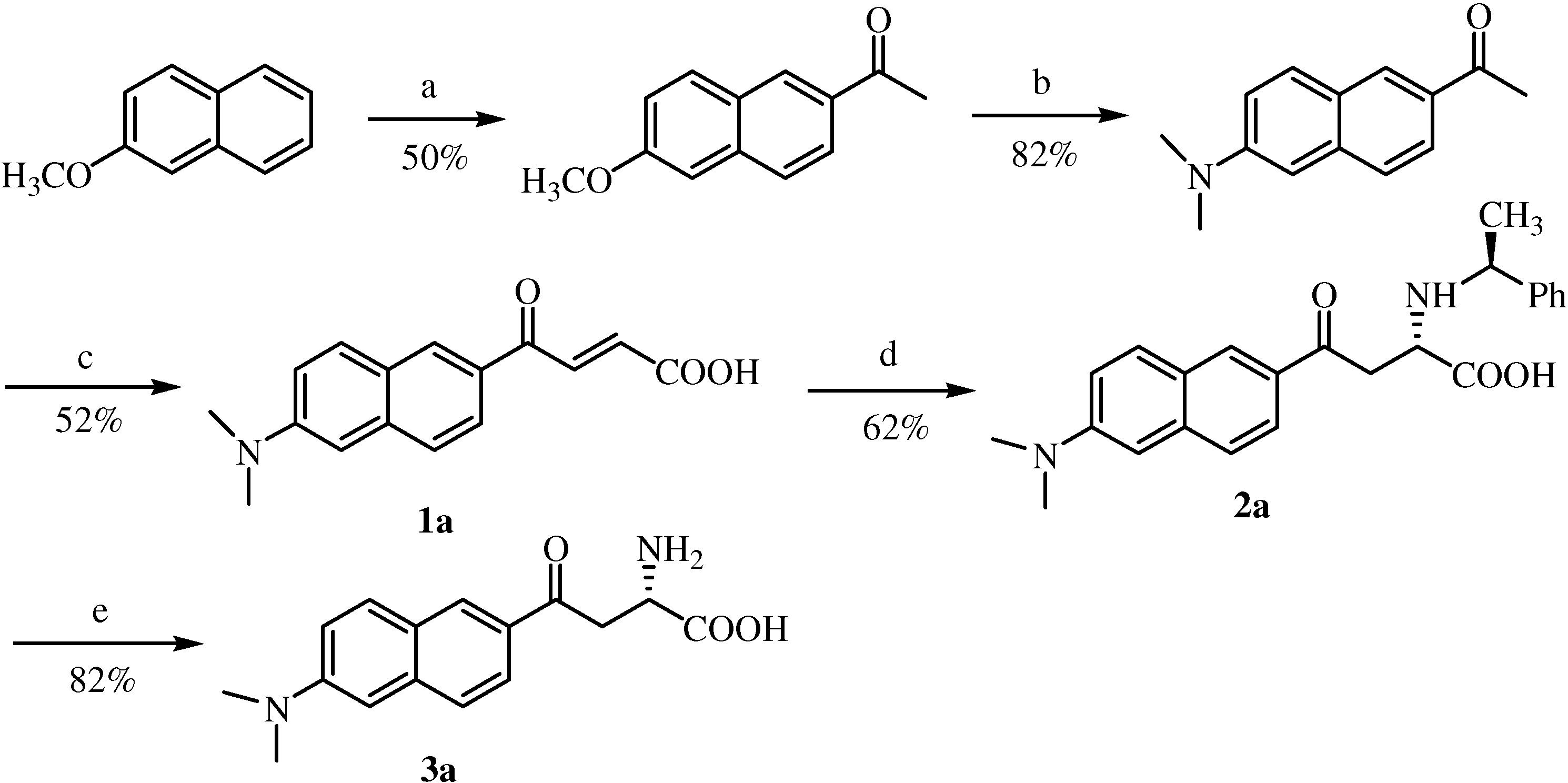
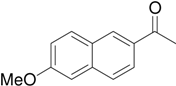
Synthesis of 6-methoxy-2-acetylnaphthalene
After following a procedure described in literature [
6] the title compound was separated and purified by flash liquid chromatography (ethyl acetate/
n-hexane, 1:4,
v/v). The fraction corresponding to a
Rf value of 0.42 was collected and further purified by crystallization (
n-hexane) to give 12.6 g (50%) of the title compound as colorless plates. Mp 106-108°C (lit. [
11] 104-105°C);
1H-NMR (CDCl
3, 400 MHz) δ 8.39 (d, 1H,
J=1.5 Hz), 8.0 (dd, 1H,
J=8.6, 1.8 Hz), 7.85 (d, 1H,
J=8.8 Hz), 7.76 (d, 1H,
J=8.6 Hz), 7.2 (dd, 1H,
J=8.9, 2.5 Hz), 7.15 (d, 1H,
J=2.4 Hz), 3.95 (s, 3H), 2.7 (s, 6H);
13C-NMR (100.6 MHz, CDCl3) δ 198.1, 160.0, 137.5, 132.8, 131.3, 130.3, 128.0, 127.3, 124.8, 119.9, 105.9, 55.6, 26.8.
Synthesis of 6-N,N-dimethyamino-2-acetylnaphthalene (acedan)
A literature procedure was followed [
6]. The title compound was separated and purified by flash liquid chromatography (ethyl acetate/
n-hexane, 3:7,
v/v). The fraction with a
Rf value of 0.49 was then further purified by crystallization (
n-hexane/chloroform) to give 4.4 g (82%) of the title compound as needle-like crystals; mp 154-156°C;
1H-NMR (400 MHz, CDCl
3) δ 8.3 (s, 1H), 7.92 (dd, 1H,
J=8.7, 1.8 Hz), 7.79 (d, 1H,
J=9.2 Hz), 7.63 (d, 1H,
J=8.6 Hz), 7.17 (d, 1H,
J=6.2 Hz), 6.9 (s, 1H), 3.1 (s, 6H), 2.66 (s, 3H);
13C-NMR (106 MHz, CDCl
3) δ 197.9, 150.0, 137.8, 131.2, 131.0, 130.6, 126.5, 125.2, 124.8, 116.5,105.8, 40.8, 26.7.
Synthesis of 4’-methoxyacetophenone
Anhydrous powdered AlCl
3 (20 g, 0.15 mol) and acetyl chloride (10.7 mL, 0.15 mol) were suspended in dried 1,1,2,2-tetrachloroethane (100 mL). The suspension was cooled down to 0 °C in an ice bath, then anisole (10.8 mL, 0.1 mol) was added via syringe. The mixture was stirred at 0 °C for another 4 hours and then warmed up to room temperature and stirred overnight. The reaction was terminated by pouring the mixture into a solution of 4 M hydrochloric acid (100 mL) mixed with cracked ice. The suspension was then extracted with ethyl acetate (3×50 mL). All the extracts were combined and washed with water (3×20 mL) and brine (2×20 mL), respectively. The organic layer was then dried over anhydrous MgSO
4. After filtration and removal of ethyl acetate, the residue was purified by flash liquid chromatography (ethyl acetate/
n-hexane, 3:7,
v/v). The fraction with an
Rf value of 0.51 was collected to give 13.6 g (91%) of the target compound as oil, which gradually solidified. Mp 37-39 °C (lit. [
12] 36-38 °C);
1H-NMR (400 MHz, CDCl
3) δ 7.91 (dd, 2H,
J = 1.5, 9.0 Hz), 6.90 (dd, 2H,
J = 1.5, 9.0 Hz), 3.84 (s, 3H), 2.53 (s, 3H);
13C-NMR (106 MHz, CDCl
3) δ 197.0, 163.7, 130.8, 130.5, 113.9, 44.8, 25.6.
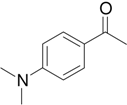
Synthesis of 4’-(N, N-dimethyl)aminoacetophenone
The same procedure described for the synthesis of acedan was used employing 4’-methoxy-acetophenone (3.42 g, 22.8 mmol). The crude residue was separated and purified by flash liquid chromatography (ethyl acetate/n-hexane, 3:7, v/v). The fraction with Rf 0.45 was collected to give 1.213 g (33%) of the title compound as a light yellow solid. Mp 103-105 °C; 1H-NMR (400 MHz, CDCl3) δ 7.89 (d, 2H, J = 8.6 Hz), 6.68 (d, 2H, J = 8.8 Hz), 3.06 (s, 6H), 2.51 (s, 3H); 13C-NMR (106 MHz, CDCl3) δ 196.6, 153.4, 130.7, 111.2, 40.5, 26.2.
Typical experimental procedure for the synthesis of E-acryl acids derivatives via an aldol reaction
Aryl 2-acetyl acetone (10 mmol) and glyoxylic acid monohydrate (0.92 g, 10 mmol) were stirred in dried methanol and 20 wt% tetramethylammonium hydroxide in methanol (5.8 mL, 11 mmol) was added via syringe. The temperature gradually rose to room temperature and the mixture was stirred for 24 h. The reaction mixture was then poured into 2 M aqueous H3PO4 solution (20 mL) mixed with cracked ice. The mixture was concentrated to a volume of about 20 mL under reduced pressure at 39 °C. The residues were then extracted with chloroform (3×30 mL). All the extracts were combined and washed with water (3×10 mL) and brine (2×10 mL). The organic layer was then dried over anhydrous MgSO4. Removal of chloroform was followed by reduced pressure evaporation at room temperature. To the residue p-toluenesulfonic acid (0.2 g, 1 mmol) and benzene (50 mL) were added, the flask was equipped with a Dean-Stark trap and the solution was refluxed for 4 h, then the benzene was removed by rotatory evaporation under reduced pressure at r.t. The residues was dissolved in chloroform (50 mL) and washed with water (3×10 mL) and brine (2×10 mL). The chloroform layer was then dried over anhydrous MgSO4. After removal of chloroform, the residues were purified by liquid chromatography (EtOAc/n-hexane/HOAc, 60/38/2, v/v/v).
6’-(N,N-Dimethylamino)-2’-naphthoylacryl acid (1a). Rf = 0.63; deep red solid; yield: 52%; Mp. 144-146 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.60 (d, 1H, J = 1.5 Hz), 8.08 (d, 1H, J = 15.6 Hz), 7.97 (d, 1H, J = 9.2 Hz), 7.88 (m, 1H), 7.71 (m, 1H), 7.28 (m, 1H), 6.95 (m, 1H), 6.69 (d, 1H, J = 15.6 Hz), 3.06 (s, 6H); 13C-NMR (106 MHz, DMSO-d6) δ 188.2, 167.3, 151.3, 138.4, 137.1, 132.5, 131.9, 130.0, 127.2, 126.6, 125.0, 124.8, 117.1, 106.9, 40.7; FAB-MS m/z: calcd. for C16H14NO3 for [M-H]+ 268.11; found 268.1; daughter peak: 224, 209, 196, 181.
6’-Methoxy-2’-naphthoylacryl acid (1b). Rf = 0.64; yellow solid; yield: 70%; Mp. 135-136 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.75 (d, 1H, J = 1.5 Hz), 7.88-8.11 (m, 4H), 7.38-7.42 (m, 1H), 7.22-7.28 (m, 1H), 6.72 (d, 1H, J = 15.6 Hz), 3.9 (s, 3H); 13C-NMR (106 MHz, DMSO-d6) δ 198.3, 189.2, 176.4, 167.1, 160.6, 160.1, 136.9, 133.2, 132.4, 128.3, 128.1, 125.0, 120.4, 106.9, 56.2; FAB-MS m/z: calcd. for C15H13O4 ([M+H]+) 257.07, found 257.1; daughter peak: 211.07, 183.07, 159.07, 144.05.
p-Methoxybenzoylacrylic acid (1c). Rf = 0.5; white solid; yield: 66%; Mp. 126-128 °C; 1H-NMR (400 MHz, CDCl3) δ 8.03 (m, 3H), 7.01 (m, 2H), 6.86 (d, 1H, J = 15.6 Hz), 3.90 (s, 3H); 13C-NMR (106 MHz, CDCl3) δ 187.6, 171.0, 164.6, 138.8, 131.7, 131.0, 129.7, 114.5, 55.9; FAB-MS m/z: calcd. for C11H10O4 ([M+H]+) 207.19, found 207.17.
4’-(N, N-Dimethyl)benzoylacryl acid (1d). Rf = 0.43; golden solid; yield: 63%; Mp. 146-148 °C; 1H-NMR (400 MHz, DMSO-d6) δ 7.89 (m, 2H), 7.68 (m, 1H), 6.75 (m, 2H), 6.60 (d, 1H, J = 15.4 Hz), 3.03 (s, 6H); 13C-NMR (106 MHz, DMSO-d6) δ 186.1, 167.4, 154.5, 137.3, 131.7, 131.6, 130.8, 124.4,111.7, 40.5; FAB-MS m/z: calcd. for C12H13NO3 ([M+H]+) 219.24, found 219.21; daughter peak: 174.1, 146.1.
Typical experimental procedure for the synthesis of compounds 2a~2d by asymmetric Michael addition.
To a vigorously stirred solution of trans-acrylic acid 1a~1d (2.0 mmol) in ethanol (60 mL) at 50 to 60 °C, S-1-phenylethylamine (0.28 mL, 2.2 mmol) was added dropwise via syringe. After the addition was complete, the mixture was stirred at the same temperature for 24 h. Precipitation was first observed ca. 1 h into the reaction and increased as time went on. The mixture was then cooled down to room temperature, and the precipitates were collected by filtration and washed with cold ethanol. The precipitates were quite pure, judged from both RP-HPLC and chiral HPLC analytical results. Further purification by re-crystallization from acetonitrile/aqueous phosphate buffer (pH 2.5) was also performed.
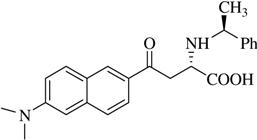
(αS,1S)-α-[N-(1-Methyl)benzyl]-β-[(6’-dimethylamino)-2’-naphthoyl]alanine (
2a).
1H-NMR (400 MHz, DMSO-d
6/TFA) δ 8.43 (d, 1H,
J=1.5 Hz,), 7.90 (d, J = 9.3 Hz, 1H), 7.80 (dd, 1H,
J=8.8, 1.8 Hz) 7.68 (d, 1H,
J=8.8 Hz), 7.40-7.54 (m, 5H), 7.28 (dd, 1H,
J=9.2, 2.5 Hz), 6.97 (d, 1H,
J=2.4 Hz), 4.58 (m, 1H), 3.93 (m, 1H), 3.72 (m, 2H), 3.06 (s, 6H), 1.61 (d, 3H,
J=6.8 Hz);
13C-NMR (100.6 MHz, DMSO-d
6/TFA) δ 194.6, 170.5, 150.8, 138.2, 137.0, 131.5, 131.3, 129.9, 129.7, 128.6, 126.9, 125.2, 124.4, 117.3, 114.4, 105.8, 58.7, 53.5, 40.5, 20.6; FAB-MS
m/z: calcd. for C
24H
27N
2O
3 ([M+H]
+) 391.19, found 391.21; daughter peaks: 287, 214, 198, 172, 105;
![Molecules 12 01170 i013]()
= +94.7 ° (c = 0.21, methanol/0.1 mol/L H
2SO
4, 3/1,
v/v).
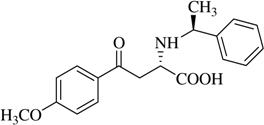
(αS,1S)-α-[N-(1-Methyl)benzyl]-β-[(6’-methoxy)-2’-naphthoyl]alanine (
2b).
1H-NMR (400 MHz, DMSO-d
6/TFA) δ 8.57 (s, 1H), 8.01 (d, 1H,
J=9.2 Hz), 7.90 (m, 2H), 7.52 (d, 1H,
J=8.0 Hz), 7.42 (m, 5H), 7.24 (m, 1H), 4.58 (m, 1H), 3.89 (s, 1H), 3.88 (s, 3H), 3.68 (m, 2H), 1.62 (d, 3H,
J=6.8 Hz);
13C- NMR (100.6 MHz, DMSO-d
6/TFA) δ 195.2, 170.4, 160.4.9, 137.9, 136.9, 131.9, 131.1, 129.9, 129.7, 128.6, 128.0, 127.8, 124.6, 120.4, 120.1, 117.2, 114.3, 111.4, 106.6, 58.7, 55.9, 53.4, 20.5; FAB-MS
m/z: calcd. for C
23H
24NO
4 ([M+H]
+) 378.16, found 378.2; daughter peaks: 274, 201, 185, 159, 105;
![Molecules 12 01170 i013]()
= +85.5 ° (c = 0.47, methanol/0.1 mol/L H
2SO
4, 3/1,
v/v).
(αS,1S)-α-[N-(1-Methyl)benzyl]-β-[(4’-methoxy)benzoyl]alanine (
2c).
1H-NMR (400 MHz, DMSO-d
6/TFA) δ 7.92 (dd, 2H,
J=1.6, 8.8 Hz), 7.40-7.51 (m, 5H), 7.03 (dd, 2H,
J=1.6, 8.8 Hz), 4.59 (m, 1H), 3.89 (m, 1H), 3.82 (s, 3H), 3.61-3.46 (m, 2H), 1.60 (d, 3H, J = 6.6 Hz);
13C-NMR (100.6 MHz, DMSO-d
6/TFA) δ 194.0, 170.4, 164.4, 136.9, 131.2, 129.9, 129.7, 128.8, 128.6, 120.1, 117.2, 114.6, 114.3, 111.4, 58.7, 56.1, 53.4, 20.5; FAB-MS
m/z: calcd. for C
19H
22NO
4 ([M+H]
+) 328.15; found 328.2;
![Molecules 12 01170 i013]()
= +63.9 ° (c = 0.49, methanol/0.1 mol/L H
2SO
4, 3/1,
v/v)
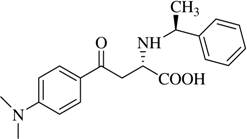
(αS,1S)-α-[N-(1-Methyl)benzyl]-β-[(4’-dimethylamino)benzoyl]alanine (
2d).
1H-NMR (400 MHz, DMSO-d
6/TFA) δ 7.75 (d, 2H,
J=9.0 Hz), 7.38-7.50 (m, 5H), 6.68 (d, 2H,
J=9.0 Hz), 4.59 (m, 1H), 3.82 (s, 1H), 3.41 (m, 2H), 2.98 (s, 6H), 1.59 (d, 3H,
J=6.9 Hz);
13C-NMR (100.6 MHz, DMSO-d
6/TFA) δ 192.7, 170.5, 154.3, 136.9, 130.8, 129.9, 129.7, 128.5, 123.2, 120.0, 117.1, 114.3, 111.4, 58.7, 53.6, 40.4, 20.5;
m/z (FAB-MS): calcd. for C
20H
25N
2O
3 ([M+H]
+) 341.18; found 341.15; daughter peak: 237, 220, 164, 148, 122, 105, 88, 74;
![Molecules 12 01170 i013]()
= +113.2° (c=0.28, methanol/0.1 mol/L H
2SO
4, 3/1, V/V)
Typical deprotection procedure
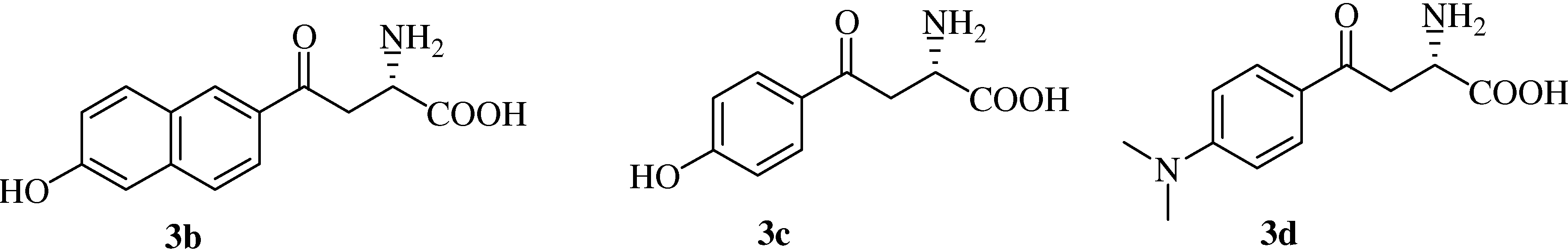
To α-[N-(1-S-methyl)benzyl]alanine derivatives 2a~2d (2 mmol) in a 100 mL flask, 6 N aqueous HBr solution (30 mL) was added. The flask was wrapped with aluminum foil. Under stirring, the mixture was kept refluxing for 12 h. After cooling down, the mixture was concentrated to sticky oil under reduced pressure at 45 °C and cold water (50 mL) was added. The precipitates were collected by filtration and washed carefully with cold water, acetone and ether and then further purified by crystallization using hot water.
(αS)-β-[6’-(N,N-Dimethyl)amino-2’-naphthoyl]alanine (
3a). Yield: 0.61 g (82%);
1H-NMR (400 MHz, D
2O+DCl) δ 8.68 (s, 2H), 8.19 (d, 1H,
J=8.1 Hz), 7.96 (s, 2H), 7.78 (d, 1H,
J=6.8 Hz), 4.35 (m, 1H), 3.84 (m, 2H), 3.16 (s, 6H); FAB-MS
m/z: calcd. for C
16H
19N
2O
3 ([M+H]
+) 287.33; found 287.1.
![Molecules 12 01170 i013]()
= -42.3° (c=0.23, 1 mol/L HCl).
(αS)-β-(6’-Hydroxy-2’-naphthoyl)alanine (
3b). Yield: 0.59 g (87%);
1H-NMR (400 MHz, D
2O+DCl) δ 8.68 (s, 1H), 8.13 (d, 1H,
J= 9.2 Hz), 8.02 (m, 2H), 7.63 (d, 1H,
J=8.0 Hz,), 7.35 (m, 1H), 4.35 (m, 1H), 3.73 (m, 2H); FAB-MS
m/z: calcd. for C
14H
14NO
4 ([M+H]
+) 260.08; found 260.10.
![Molecules 12 01170 i013]()
= -35.3° (c=0.45, 1 mol/L HCl).
(αS)-β-[(4’-Hydroxy)benzoyl]alanine (
3c). Yield: 0.52 g (91%);
1H-NMR (400 MHz, D
2O+DCl) δ 8.02 (dd, 2H,
J=1.6, 8.8 Hz), 7.13 (dd, 2H,
J=1.6, 8.8 Hz), 4.36 (m, 1H), 3.71 (m, 2H); FAB-MS
m/z: calcd. for C
10H
11NO
4 ([M+H]
+) 210.71; found 210.65.
![Molecules 12 01170 i013]()
= -17.6° (c=0.5, 1 mol/L HCl).
(αS)-β-[(4’-Dimethylamino)benzoyl]alanine (
3d). Yield: 0.53 g (83%);
1H-NMR (400 MHz, D
2O+DCl) δ 7.86 (d, 2H,
J=9.0 Hz), 6.89 (d, 2H,
J=9.0 Hz), 4.36 (m, 1H), 3.52 (m, 2H), 3.11 (s, 6H); FAB-MS
m/z: calcd. for C
10H
12NO
4 ([M+H]
+) 237.12; found 237.05.
![Molecules 12 01170 i013]()
= -25.3° (c=0.52, 1 mol/L HCl).
Typical experimental Fmoc-proctection procedure
To the above free amino acid HBr salt (2.0 mmol) dissolved in DMF (20 mL), aqueous Na2CO3 solution (10 mL, 0.53 g, 5.0 mmol) was added. The mixture was cooled down to 0 °C with an ice-water bath. 9-Fluorenylmethyl chloroformate (0.62 g, 2.4 mmol) was added in small portions under stirring in the ice-water bath for 4 hr. The solution was then warmed up to room temperature and stirred for another 8 hr. After the end of reaction, the mixture was poured into solution of crushed ice and water (100 mL) and concentrated hydrochloric acid was added slowly until the pH value was around 1~2. The mixture was extracted with chloroform (150 mL). The organics was washed with water (2×30 mL) and brine, respectively, and dried over anhydrous MgSO4. After removal of solvent by rotary evaporation, the residues were separated and purified by flash liquid chromatography with ethyl acetate in chloroform (3:7, v/v).
α-N-Fmoc-(αS)-β-[6’-(N,N-Dimethyl)amino-2’-naphthoyl]alanine (4a). Rf = 0.62. Yield: 0.84 g (83%); 1H-NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 7.84 (br, 1H), 7.68 (m, 3H), 7.55 (m, 3H), 7.52-7.12 (m, 5H), 6.84 (s, 1H), 6.10 (s, 1H), 4.84 (br, 1H), 4.35-4.16 (m, 3H), 3.88 (m, 1H), 3.62 (m, 1H), 3.09 (s, 6H); 13C-NMR (106 MHz, CDCl3) δ 163.3, 156.5, 150.6, 144.2, 144.0, 141.4, 138.2, 131.2, 130.9, 129.6, 127.8, 127.3, 126.5, 125.5, 125.1, 124.5, 120.0, 116.4, 105.4, 67.4, 55.2, 52.3, 47.3, 36.9; FAB-MS: calcd. C31H28N2O5 508.55, found 508.2.
α-N-Fmoc-(αS)-β-(6’-Hydroxy-2’-naphthoyl)alanine (4b). Rf= 0.57; Yield: 0.86 g (89%); 1H-NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 8.02 (s, 1H), 7.91-7.72 (m, 4H), 7.55 (m, 2H), 7.42-7.23 (m, 6H), 7.09 (m, 1H), 7.06 (s, 1H), 4.94 (br, 1H), 4.65-4.46 (m, 3H), 3.28 (m, 1H), 3.12 (m, 1H); 13C-NMR (106 MHz, CDCl3) δ 165.6, 158.2, 156.5, 150.6, 145.1, 142.3, 138.2, 131.8, 130.6, 128.9, 127.0, 126.8, 126.5, 125.5, 118.4, 109.4, 66.8, 56.2, 50.9, 46.1; FAB-MS: calcd. C29H23NO6 481.5, found 482.3.
α-N-Fmoc-(αS)-β-[(4’-Hydroxy)benzoyl]alanine (4c). Rf = 0.55; Yield: 0.79 g (92%); 1H-NMR (400 MHz, CDCl3) δ 7.91-7.8 (m, 4H), 7.53 (s, 2H), 7.41-7.23 (m, 5H), 6.86 (m, 2H), 5.04 (br, 1H), 4.71-4.36 (m, 3H), 3.08 (m, 1H), 2.93 (m, 1H); 13C-NMR (106 MHz, CDCl3) δ 175.6, 168.2, 166.5, 158.6, 144.2, 142.7, 130.8, 128.9, 128.3, 126.8, 116.4, 66.8, 55.2, 51.4, 44.9; FAB-MS: calcd. C25H21NO6 431.44, found 432.5.
α-N-Fmoc-(αS)-β-[(4’-Dimethylamino)benzoyl]alanine (4d). Rf = 0.60; Yield: 0.81 g (88%); 1H-NMR (400 MHz, CDCl3) δ 7.88-7.81 (m, 4H), 7.57 (m, 2H), 7.45-7.12 (m, 5H), 6.94 (s, 2H), 4.96 (br, 1H), 4.42-4.08 (m, 3H), 3.18 (m, 1H), 2.89 (m, 1H), 3.02 (s, 6H); 13C-NMR (106 MHz, CDCl3) δ 173.3, 168.5, 162.6, 154.2, 142.8, 141.4, 129.7, 128.6, 126.5, 126.2, 114.45, 68.2, 55.6, 50.7, 46.3, 38.9; FAB-MS: calcd. C27H26N2O5 458.51, found 458.6.
Typical experimental procedure for the synthesis of N-acetylated aminoamides
N-acetylated and carboxamidated Ald, Alb and other two amino acids were synthesized on a TentaGel S RAM resin (Sigma-Aldrich) respectively. After removal of the Fmoc group on the resins, the N-Fmoc-protected amino acids were attached to the resin respectively by using PyBOP/HOBt as coupling reagents. Fmoc deprotection, acetylation and cleavage from the resin by TFA/anisole treatment yielded the title compounds. Crude residues were purified by preparative reversed-phase HPLC using a linear gradient of 20-65% MeOH in 0.1% TFA over 25 min, respectively.
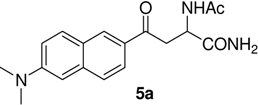
N-Ac-(αS)-β-[6’-(N, N-Dimethyl)amino-2’-naphthoyl]alanyl amide (5a). 1H-NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H), 8.06 (s, 1H), 7.9 (d, 9.2, 1H), 7.8 (d, 7.1, 1H), 7.66 (d, 8.8, 1H), 7.23 (s, 2H), 7.06 (d, 7.9, 1H), 6.94 (s, 1H), 4.70 (m, 1H), 3.25 (m, 1H), 2.93 (m, 1H), 3.05 (s, 6H), 1.79 (s, 3H); 13C-NMR (100.6 MHz, DMSO-d6) δ 196.6, 174.0, 169.9, 150.9, 137.9, 131.4, 130.3, 129.5, 126.6, 125.2, 124.5, 117.1, 105.4, 49.7, 40.8, 39.5, 23.2; FAB-MS m/z: calcd. for C18H22N3O3 ([M+H]+) 328.16; found 328.13.
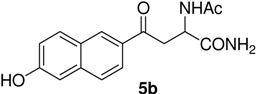
N-Ac-(αS)-β-(6’-Hydroxy-2’-naphthoyl)alanyl amide (5b). 1H-NMR (400 MHz, DMSO-d6) δ 9.85 (s, 1H), 8.53 (s, 1H), 8.24 (s, 1H), 8.09 (d, 8.5, 1H), 7.92 (d, 6.9, 1H), 7.83 (d, 8.5, 1H), 7.26 (s, 2H), 7.09 (s, 1H), 7.06 (s, 1H), 5.11 (m, 1H), 3.25 (m, 1H), 2.96 (m, 1H), 1.86 (s, 3H); 13C-NMR (100.6 MHz, DMSO-d6) δ 196.6, 174.0, 169.9, 159.9, 134.5, 131.6, 131.4, 130.7, 127.7, 126.2, 125.5, 118.6, 108.4, 50.7, 45.8, 23.2; FAB-MS m/z: calcd. for C16H17N2O4 ([M+H]+) 301.31; found 301.33.
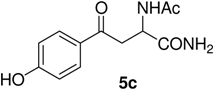
N-Ac-(αS)-β-[(4’-Hydroxy)benzoyl]alanyl amide (5c). 1H-NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 8.42 (s, 1H), 7.91 (d, 9.0, 2H), 7.24 (s, 2H), 6.78 (d, 9.0, 2H), 4.86 (m, 1H), 3.62 (m, 2H), 1.96 (s, 3H); 13C- NMR (100.6 MHz, DMSO-d6) δ 192.8, 174.2, 170.1, 162.6, 130.4, 129.5, 117.3, 51.5, 45.6, 22.8; FAB-MS m/z: calcd. for C12H14N2O4 ([M+H]+) 251.25; found 251.23.
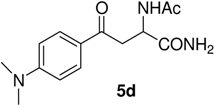
N-Ac-(αS)-β-[(4’-dimethylamino)benzoyl]alanyl amide (5d). 1H-NMR (400 MHz, DMSO-d6) δ 8.33 (s, 1H), 7.86 (d, 9.0, 2H), 7.21 (s, 2H), 6.89 (d, 9.0, 2H), 4.36 (m, 1H), 3.52 (m, 2H), 3.11 (s, 6H), 1.86 (s, 3H); 13C-NMR (100.6 MHz, DMSO-d6) δ 192.8, 174.2, 170.1, 154.3, 129.9, 125.2, 114.3, 51.2, 44.6, 39.7, 22.8; FAB-MS m/z: calcd. for C14H20N3O3 ([M+H]+) 278.14; found 278.11.


{kind=link}
{kind=link}
{kind=link}


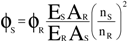
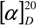 = +94.7 ° (c = 0.21, methanol/0.1 mol/L H2SO4, 3/1, v/v).
= +94.7 ° (c = 0.21, methanol/0.1 mol/L H2SO4, 3/1, v/v).