Synthesis and Cytotoxic Activity of Some 3-Benzyl-5-Arylidenefuran-2(5H)-ones

Abstract
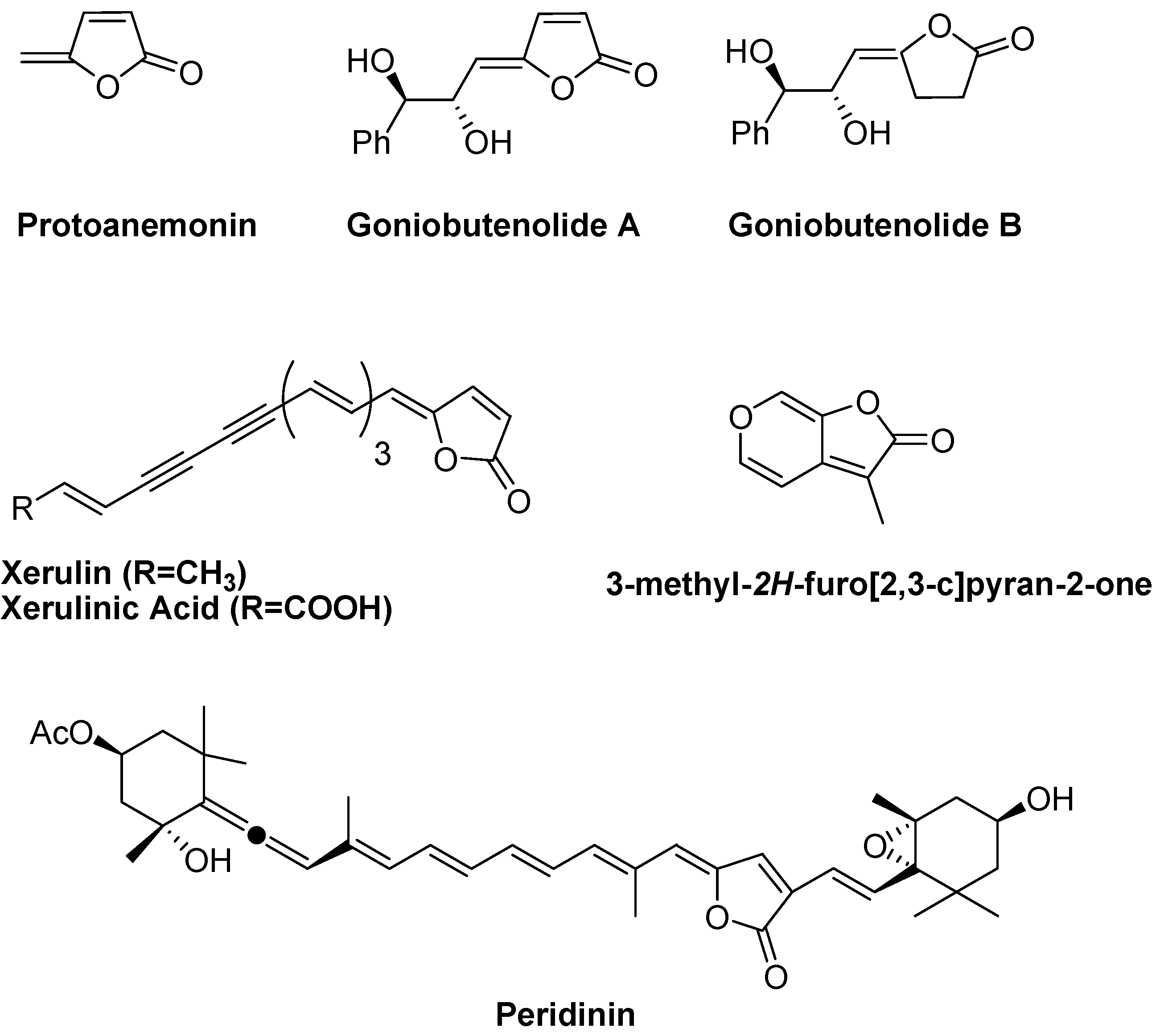
:Introduction
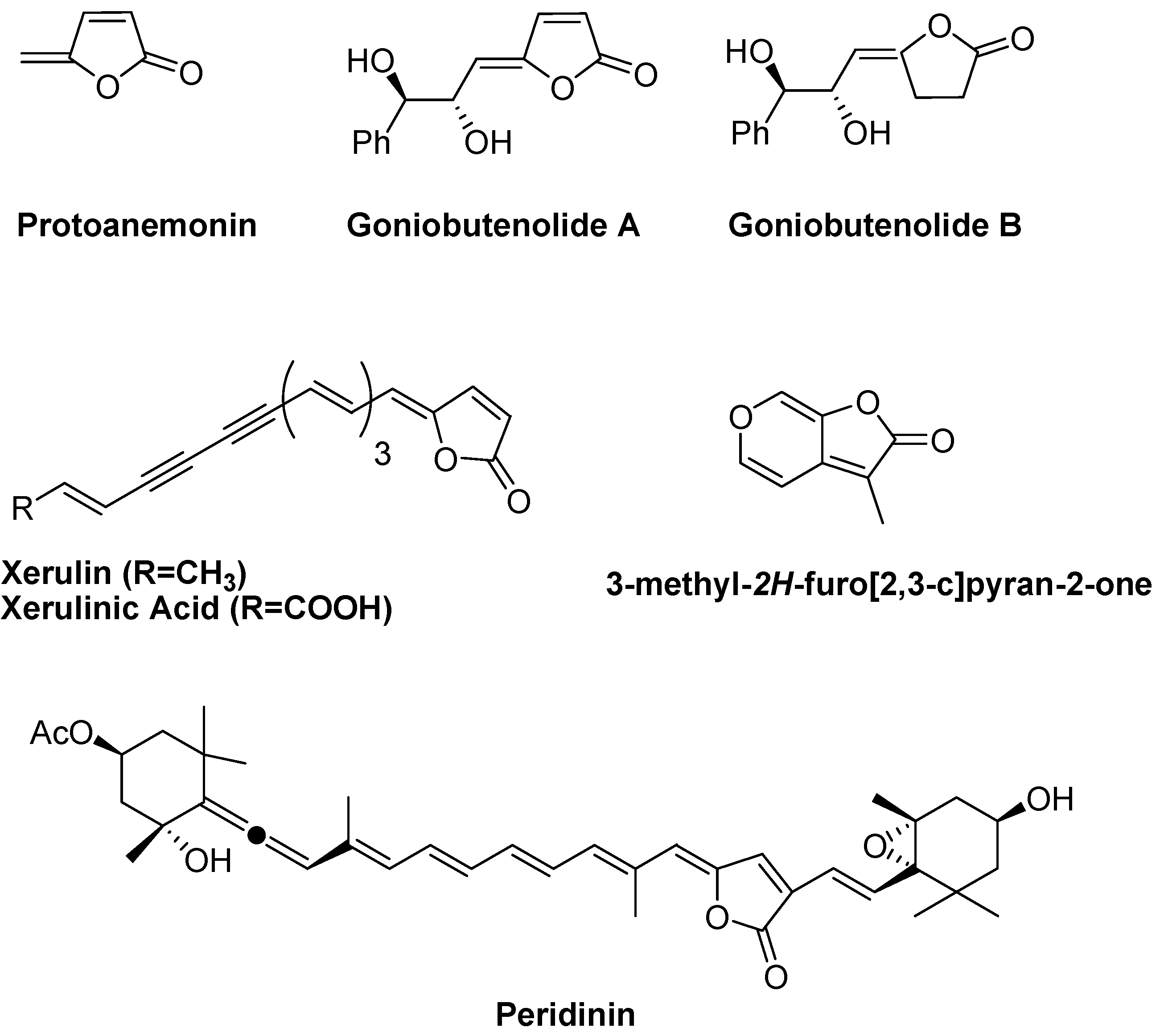
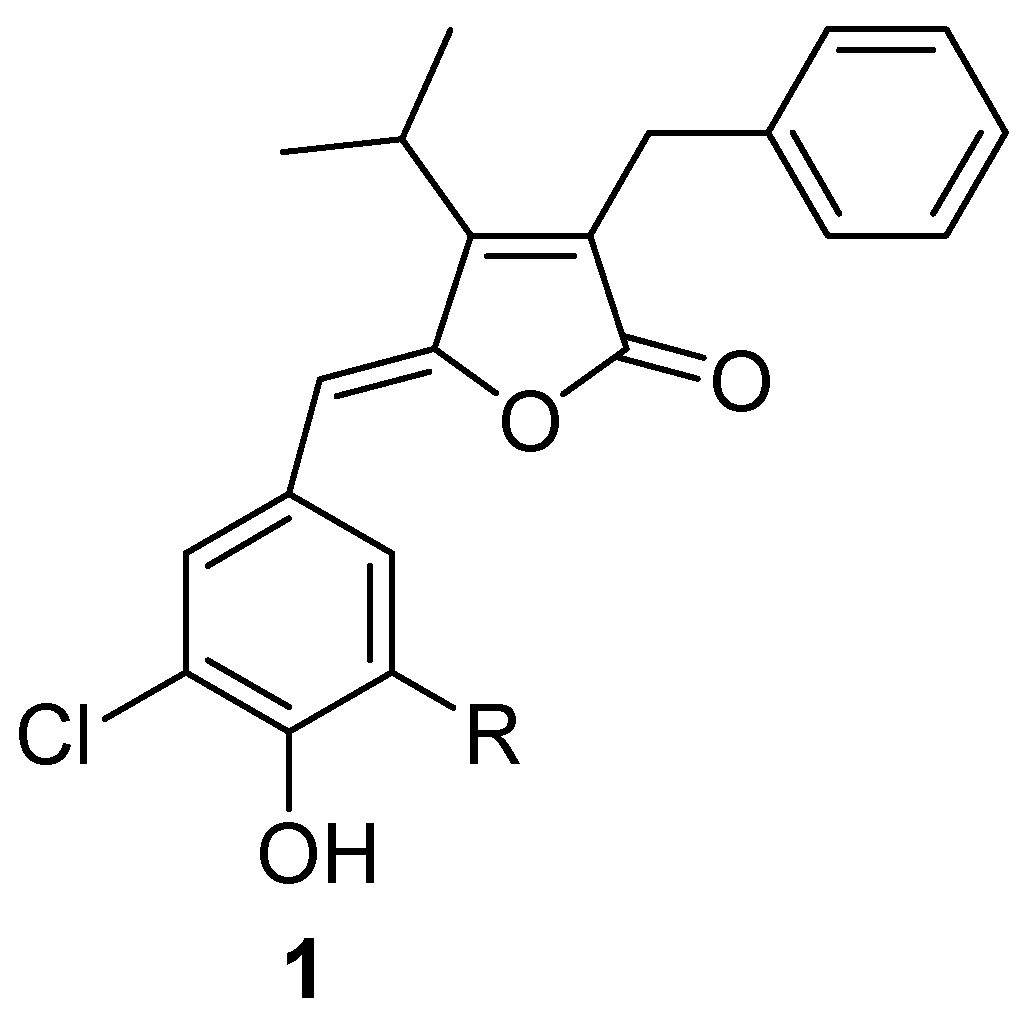
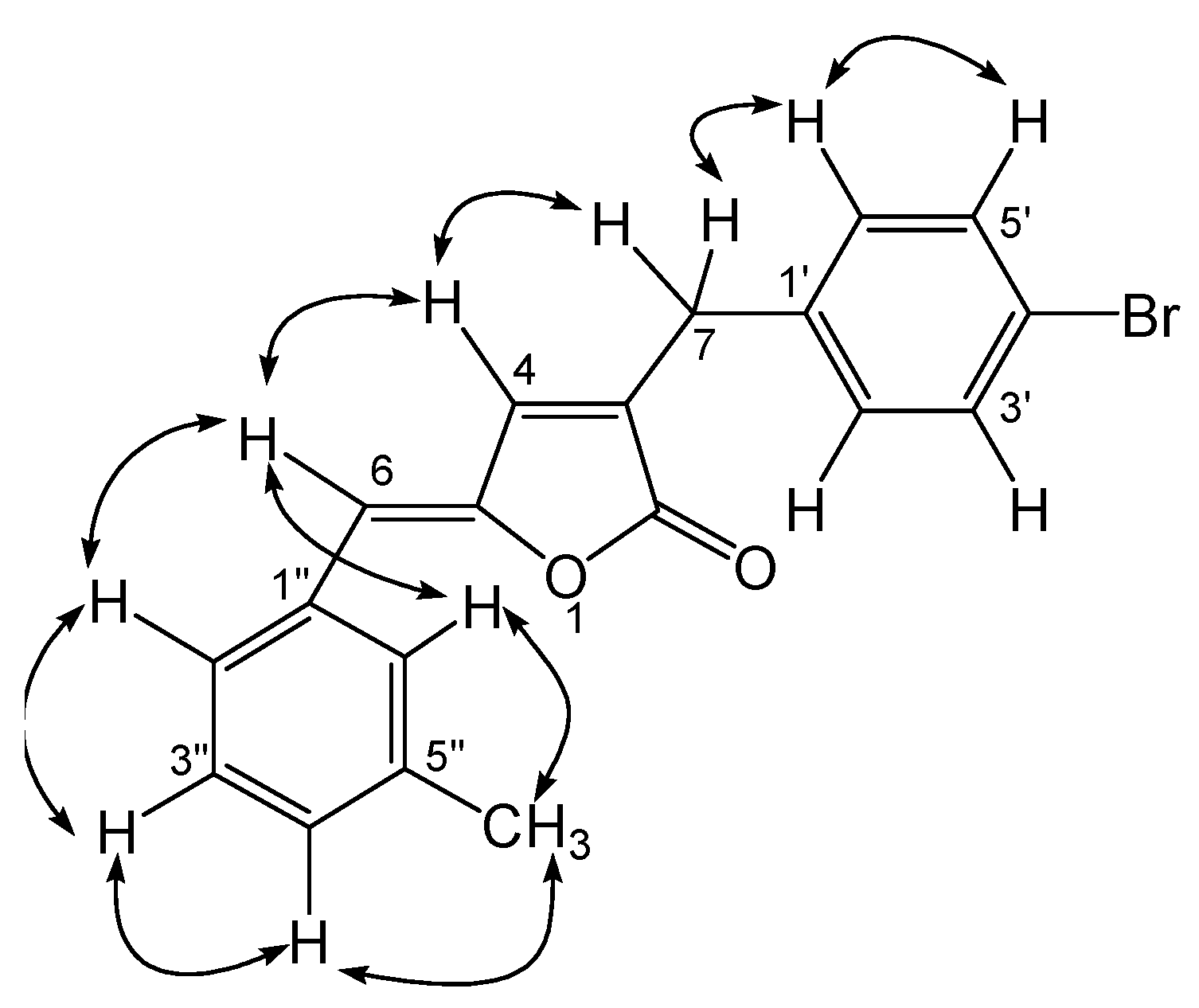
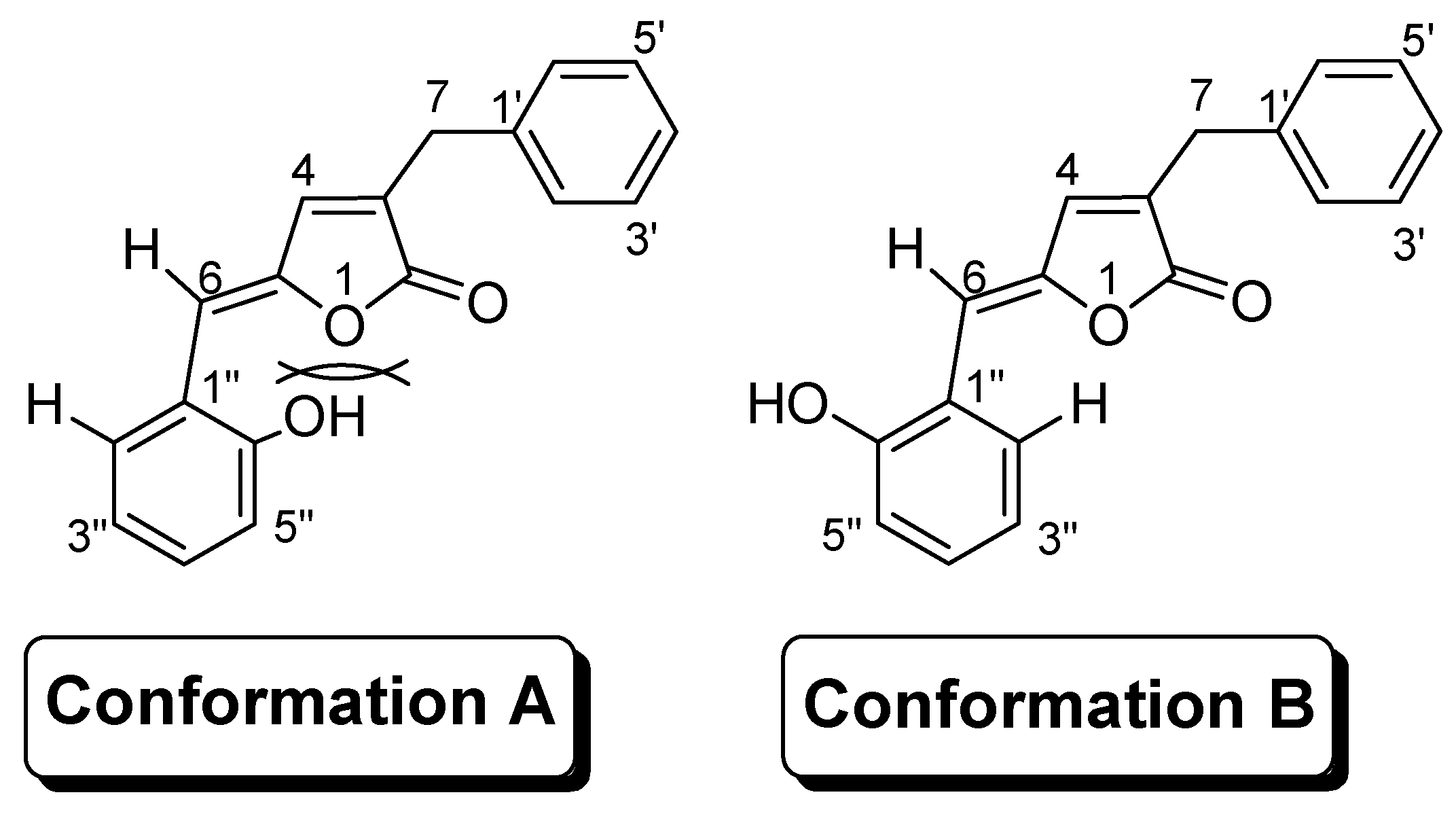
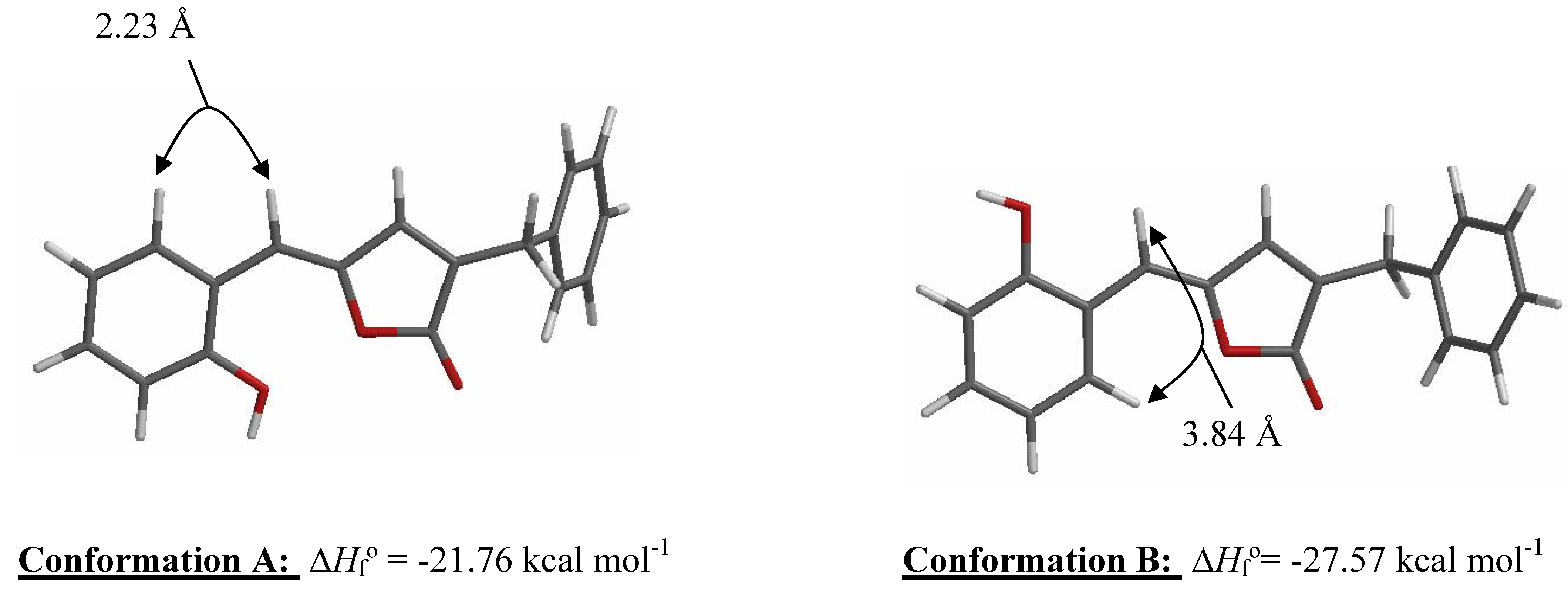
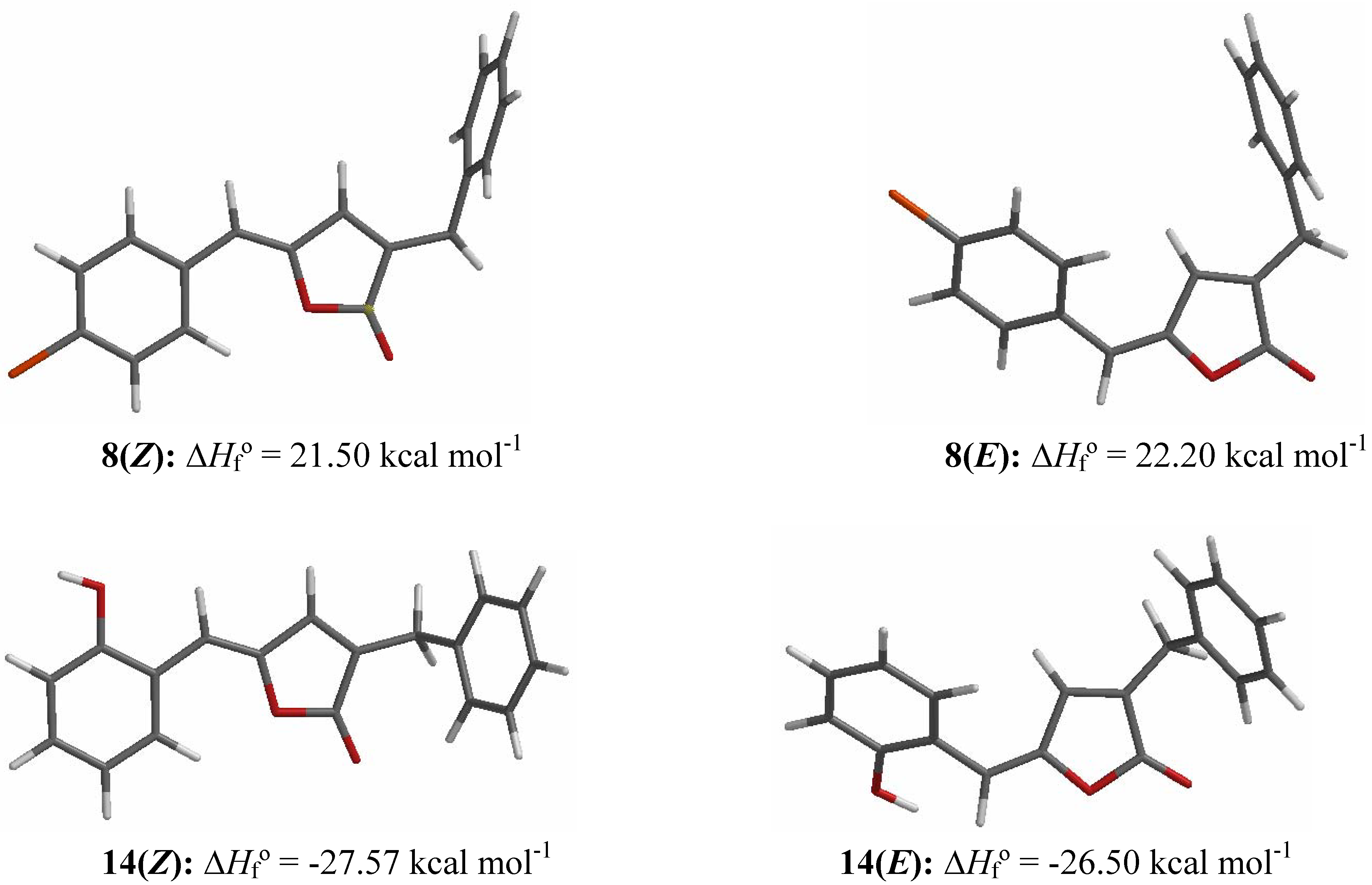
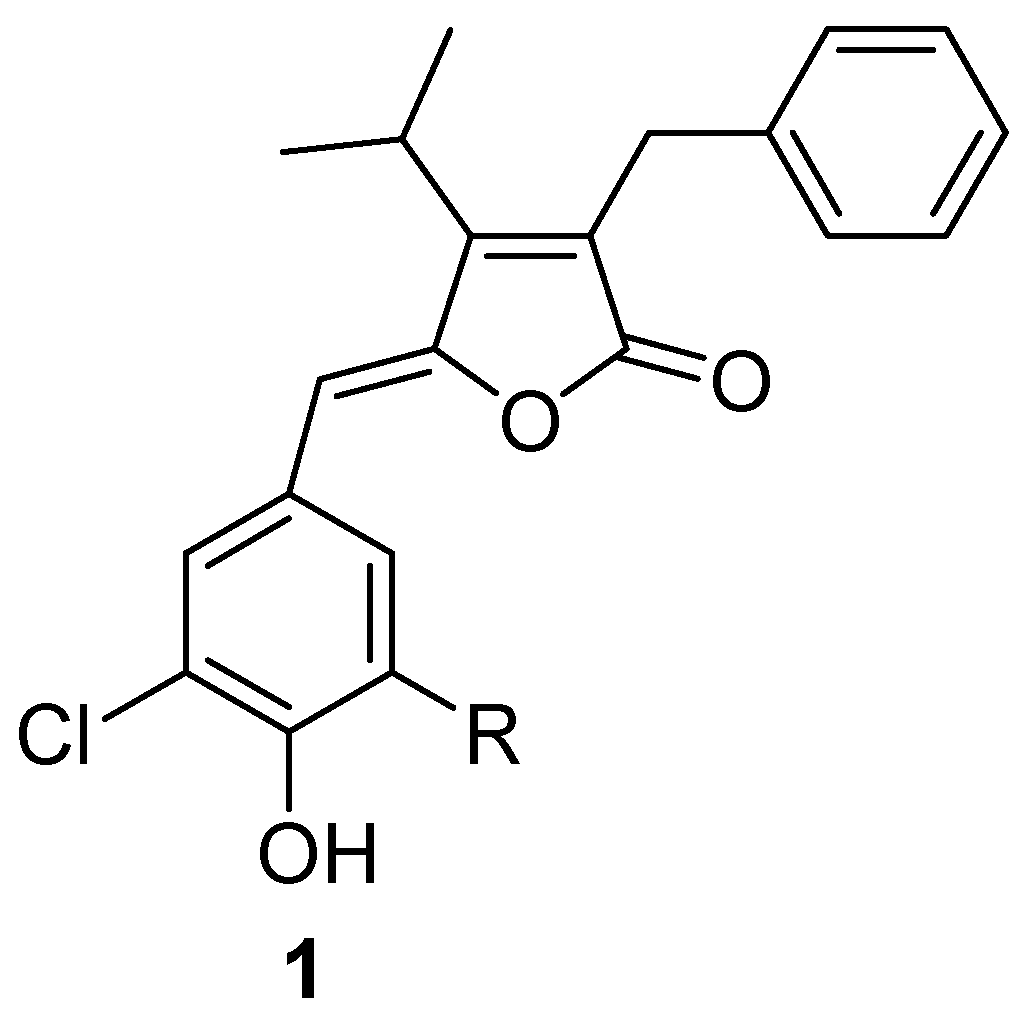
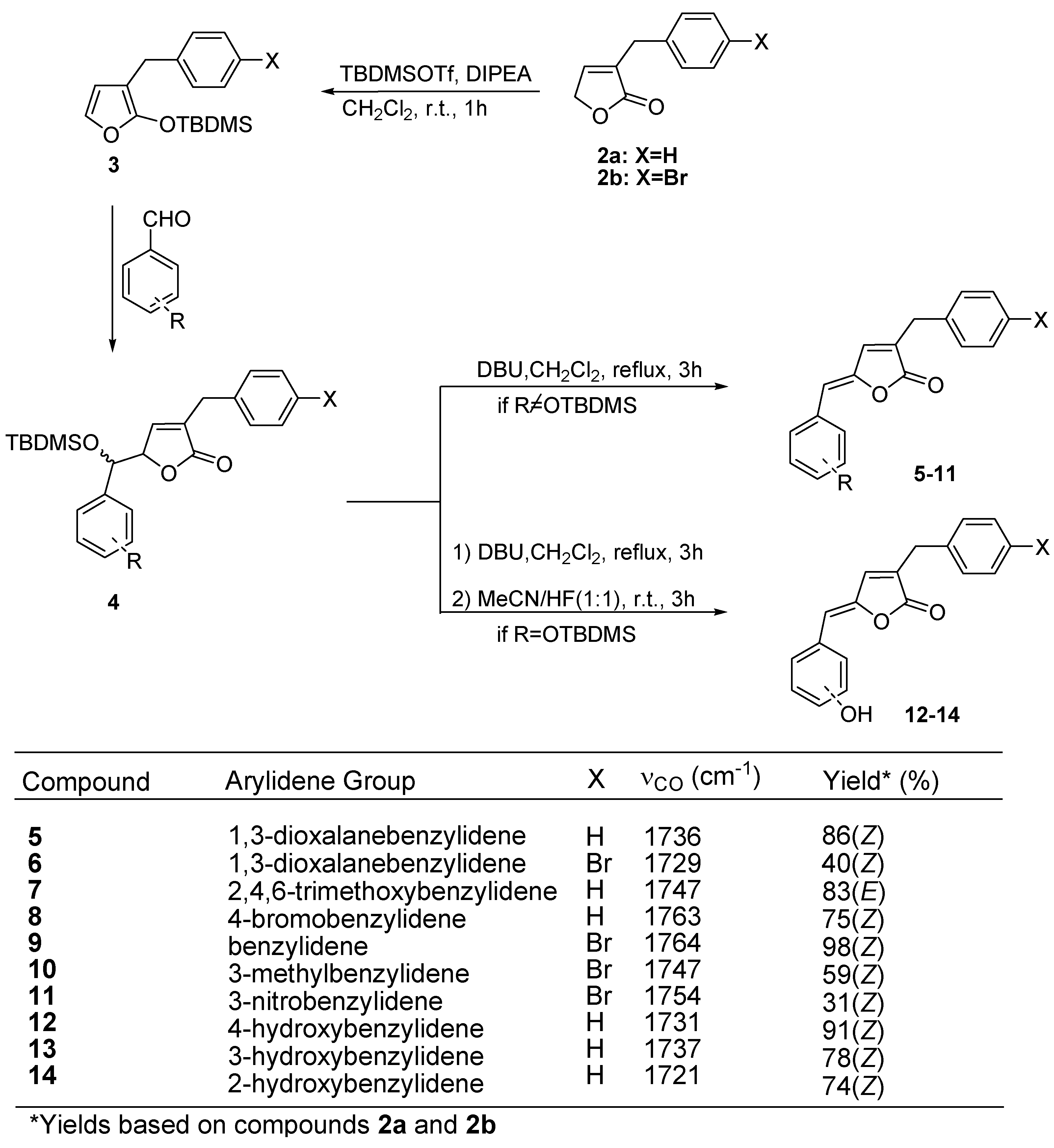
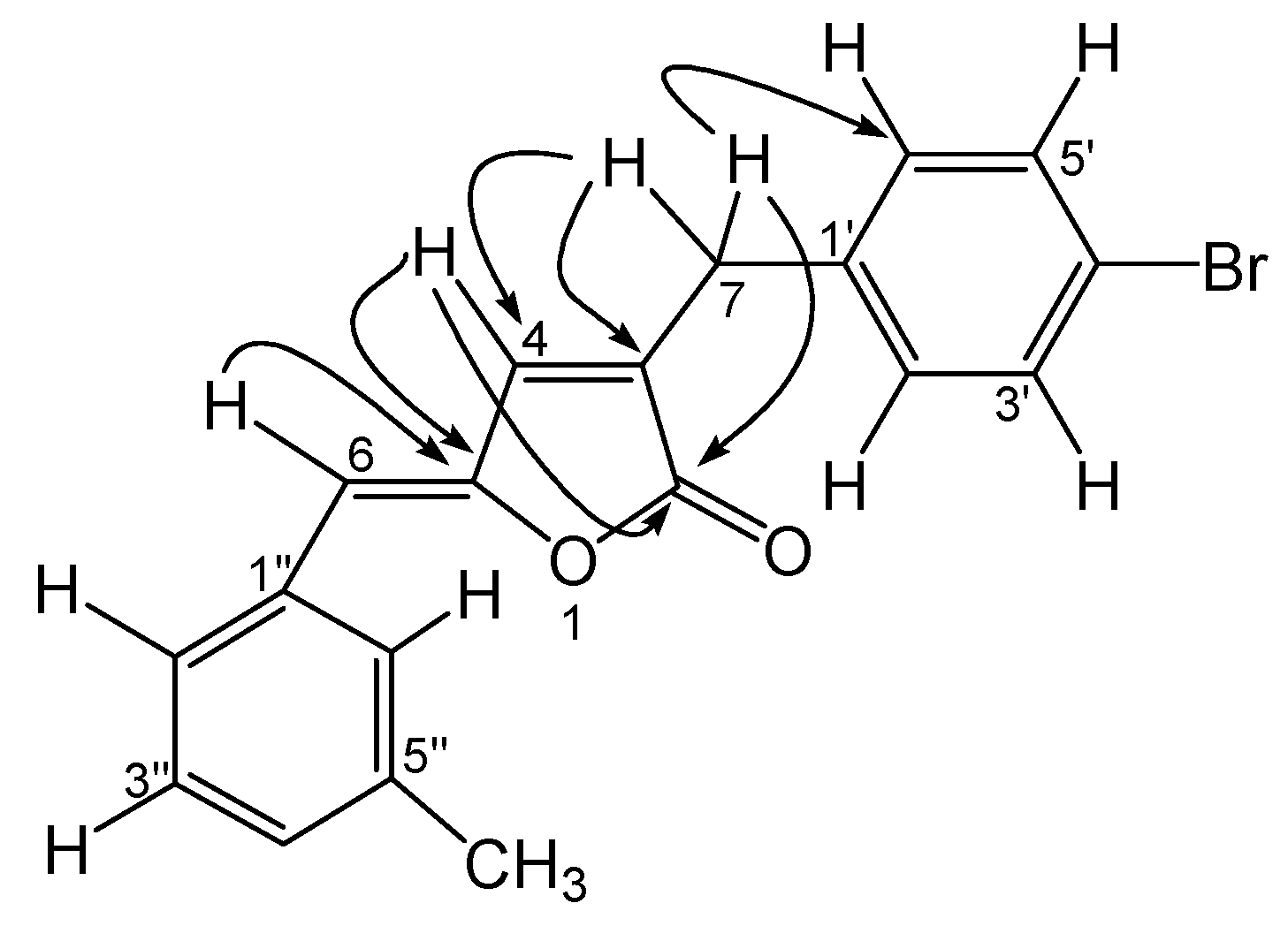
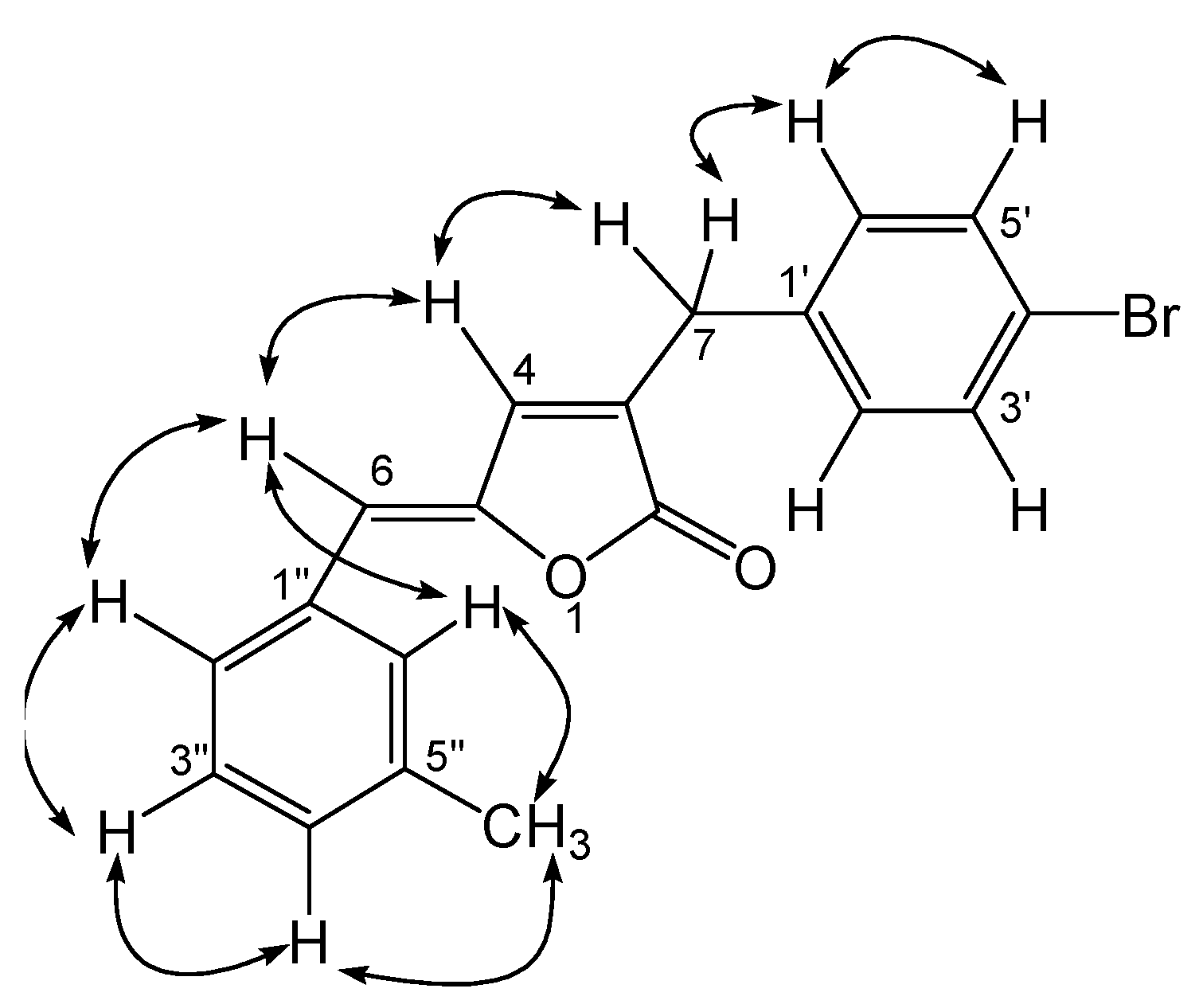
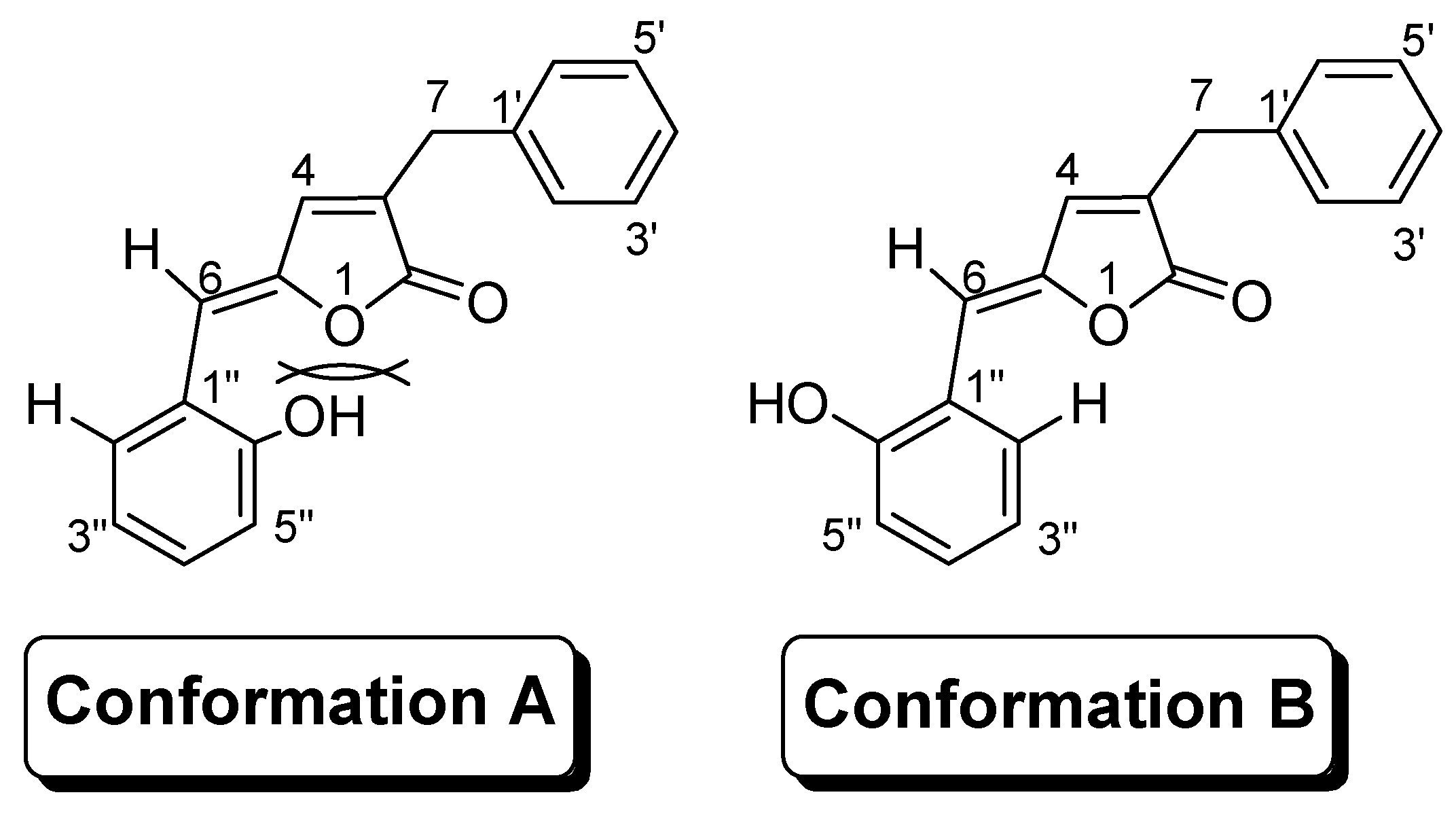
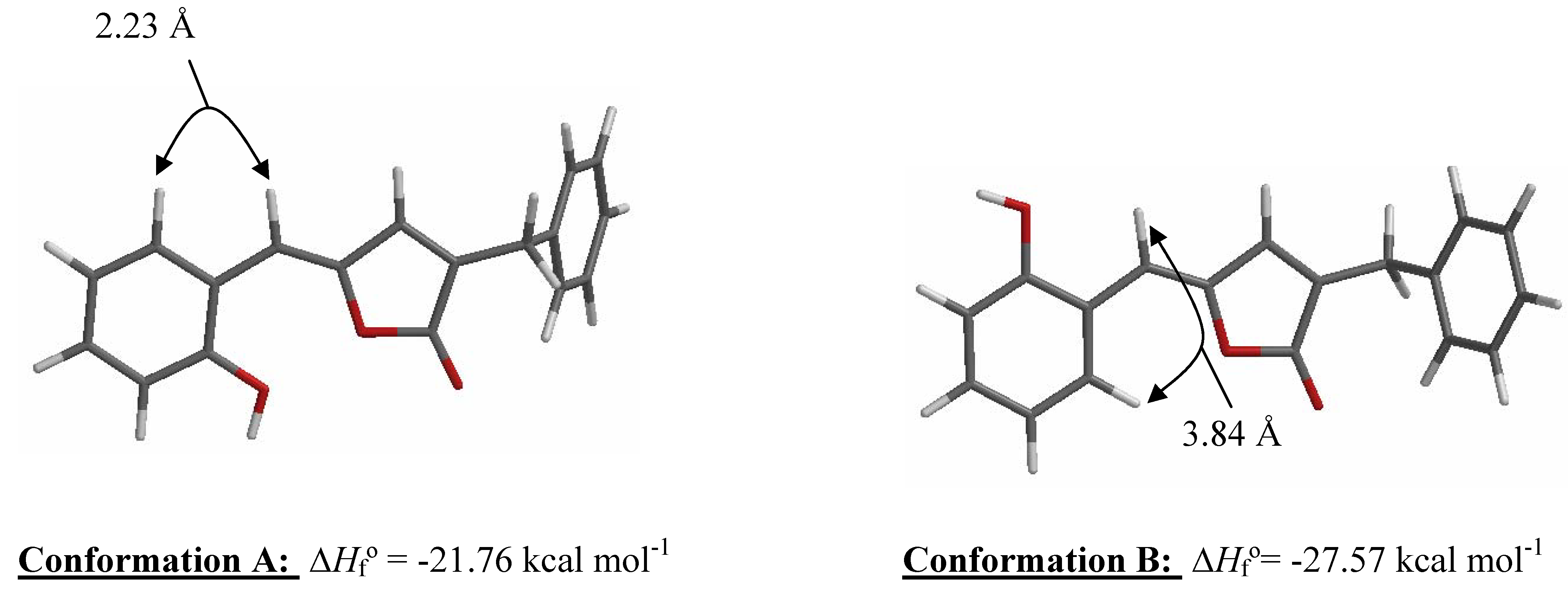
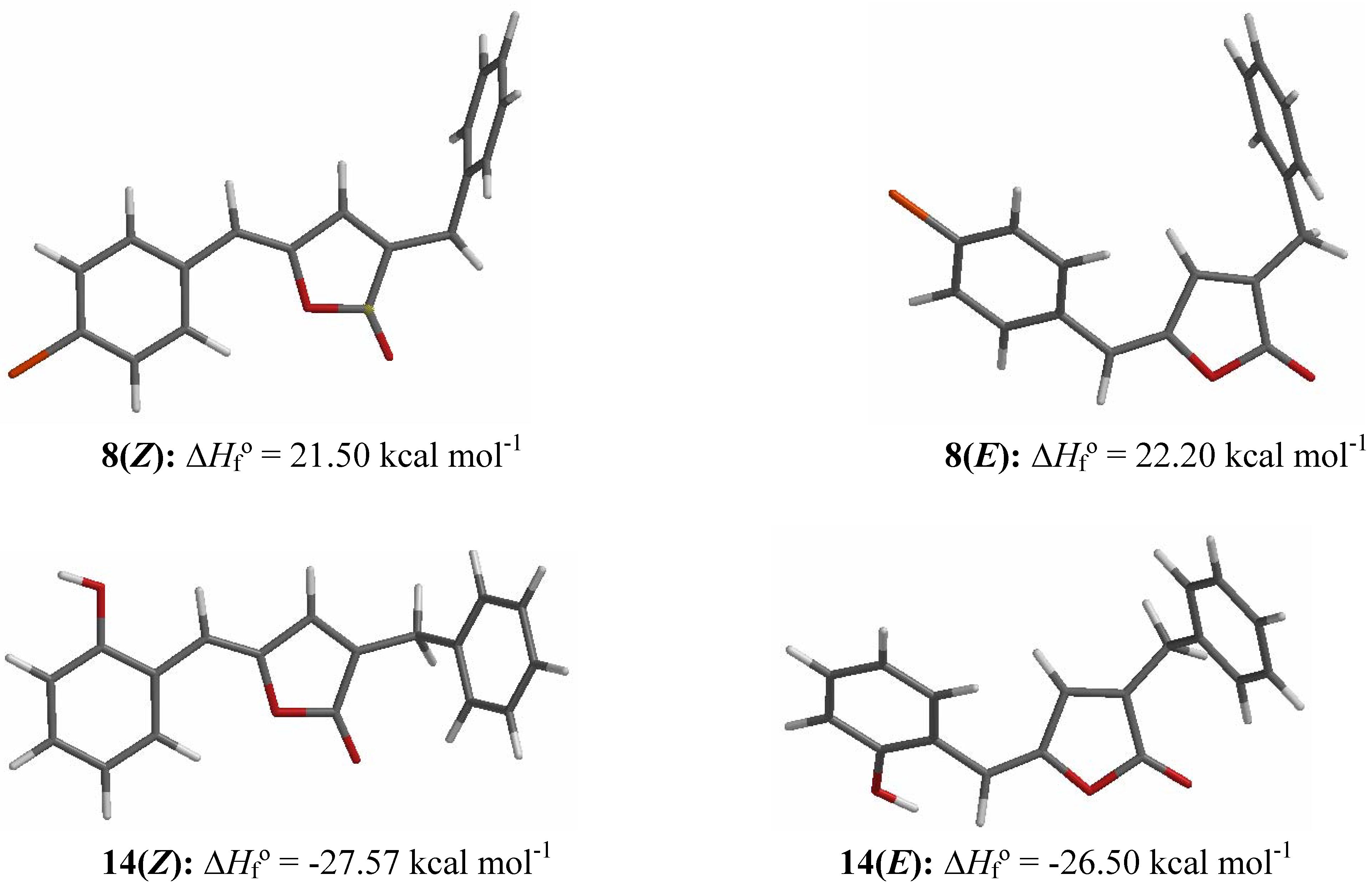
Results and Discussion
Preparation of lactones
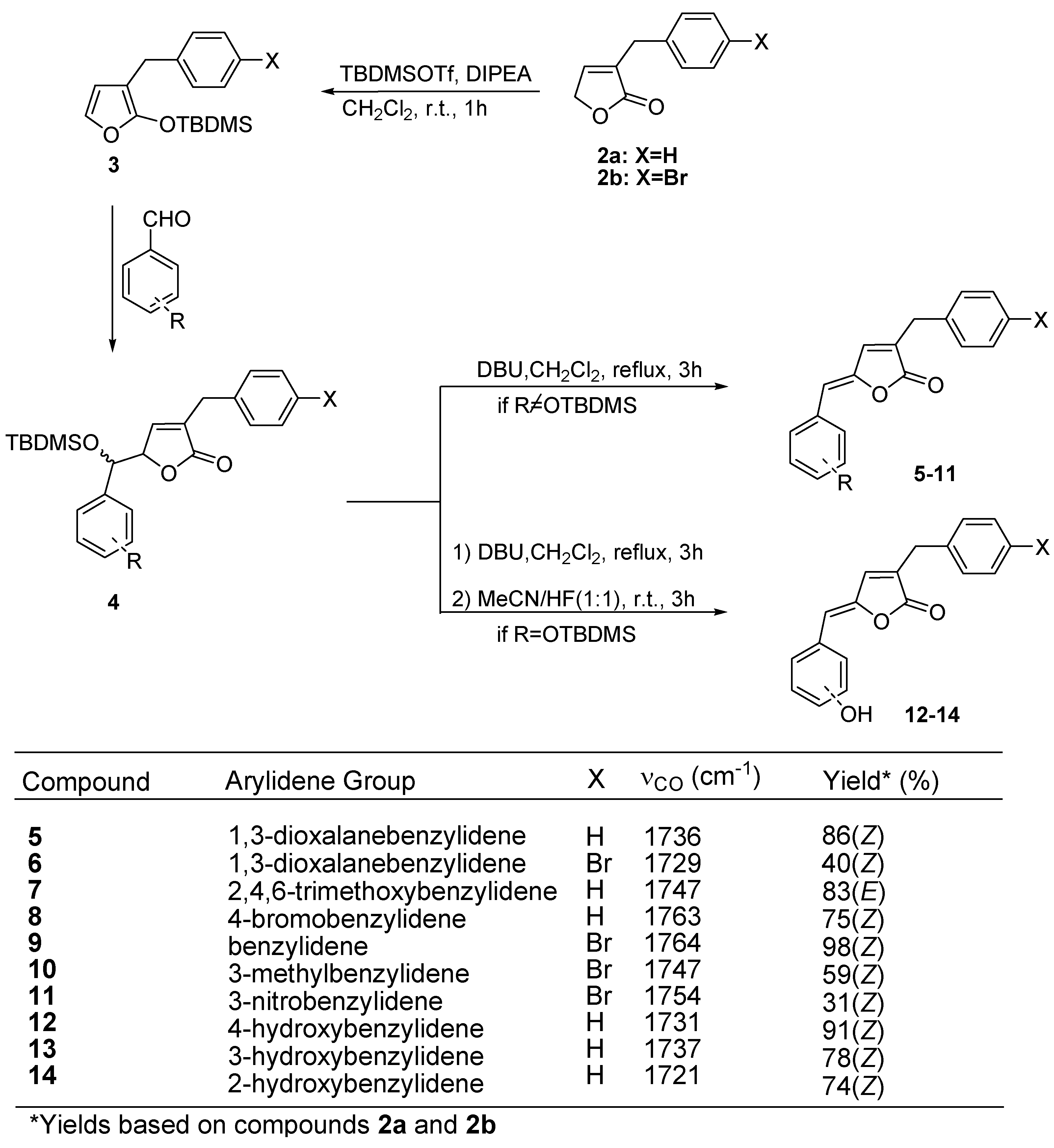
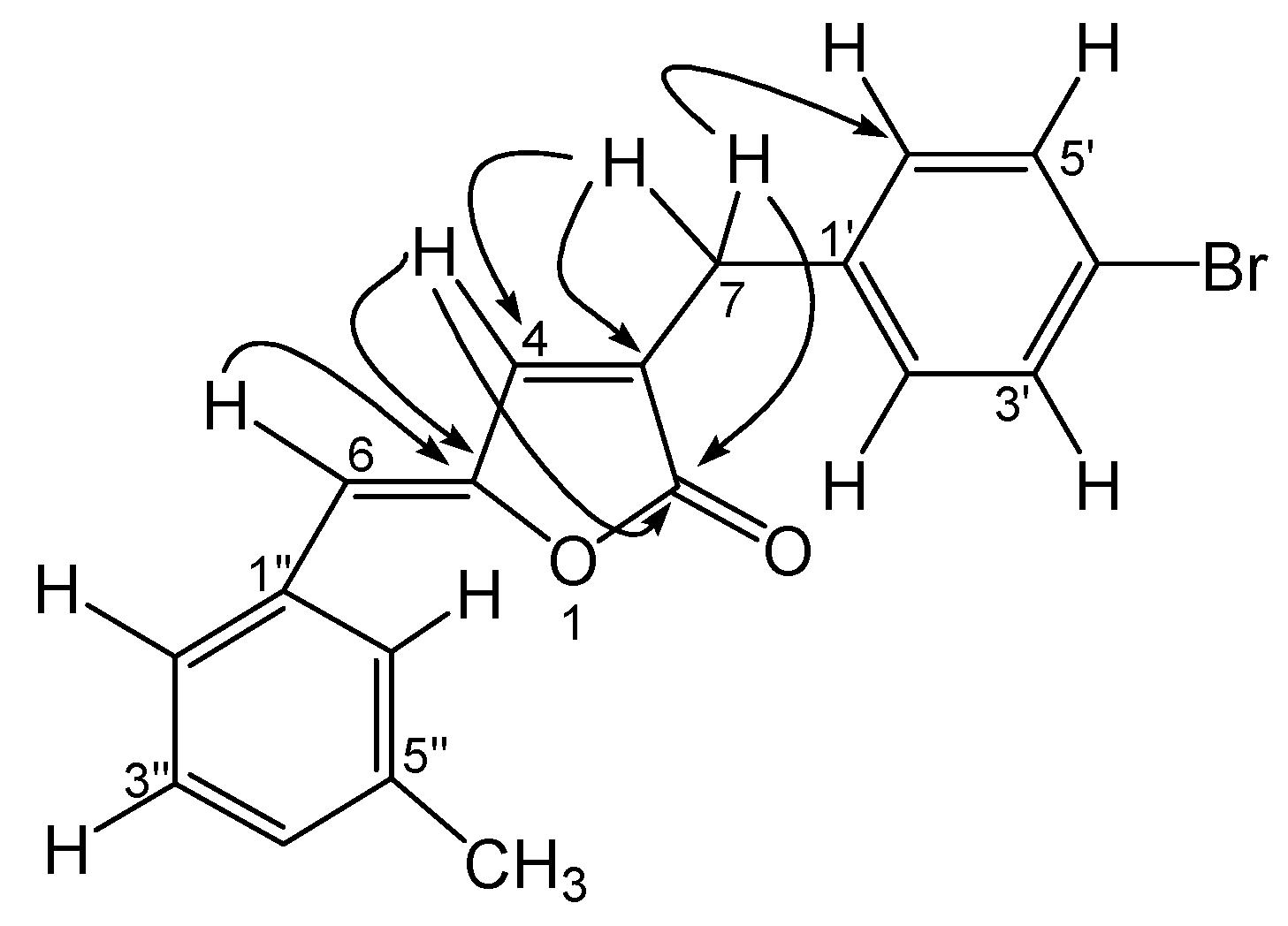
Biological Evaluation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. nº | Cellsa IC50b (μM; confident interval) | |||
|---|---|---|---|---|
| HL-60 | HCT-8 | MDA-MB-435 | SF295 | |
| 5 | >130 | 101.5(95.0-129.6) | >130 | >130 |
| 6 | >130 | >130 | >130 | >130 |
| 7 | >130 | >130 | 119.5(101.3-127.1) | >130 |
| 8 | >130 | 52.8(32.2-80.3) | >130 | 110.2(90.9-128.1) |
| 9 | >130 | 114.4(95.7-128.1) | >130 | >130 |
| 10 | >130 | >130 | 82.8(60.1-90.8) | >130 |
| 11 | >130 | >130 | >130 | >130 |
| 12 | 108.9(88.3-121.4) | >130 | 62.2(44.8-78.0) | >130 |
| 13 | 62.2(48.9-79.8) | 34.9(28.4-42.8) | 20.5(12.2-31.6) | 17.6(12.6-25.9) |
| 14 | 8.9(5.4-15.8) | >130 | >130 | >130 |
| Doxorubicin | 0.04 (0.03-0.05) | 0.02(0.02-0.03) | 0.96(0.68-1.32) | 0.48(0.34-0.72) |
Conclusions
Experimental
General
Synthesis of 5(Z)-3-benzyl-5-(1,3-dioxalenebenzylidene)-furan-2(5H)-one (5)
Biological activity assays
Molecular Modeling and Statistical Analysis
Acknowledgments
References
- Brückner, R. The synthesis of γ-alkylidenebutenolides. Curr. Org. Chem. 2001, 5, 679–718. [Google Scholar]
- Brückner, R. The β-elimination route to setereodefined γ-alkylidenebutenolides. Chem. Commun. 2001, 141–152. [Google Scholar]
- De Souza, M. V. N. The Furan-2(5H)-ones: Recent synthetic methodologies and its application in total synthesis of natural products. Mini-Rev. Org. Chem. 2005, 2, 546–564. [Google Scholar]
- Negishi, E. Regio- and stereoselective synthesis of γ-alkylidenebutenolides and related compounds. Tetrahedron 1997, 53, 6707–6738. [Google Scholar]
- Baer, H.; Holden, M.; Seegal, B.C. The nature of the antibacterial agent from anemone pulsatilla. J. Biol. Chem. 1946, 162, 65–68. [Google Scholar] [PubMed]
- Shaw, E. A synthesis of protoanemonin. The tautomerism of acetylacrylic acid and penicillic acid. J. Am. Chem. Soc. 1946, 68, 2510–2513. [Google Scholar]
- Grundmann, C.; Kober, E. An improved synthesis of protoanemonin. J. Am. Chem. Soc. 1955, 77, 2332–2333. [Google Scholar] [CrossRef]
- Fang, X.; Anderson, J.E.; Chang, C.; McLaughlin, J.L. Three new bioactive styryllactones from Goniothalamus giganteus (Annonaceae). Tetrahedron 1991, 47, 9751–9758. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Tai, V.W.F.; Zhou, Z.H. Enantiospecific synthesis of (+)-goniofufurone, (+)-7-epi-goniofufurone, (+)-goniobutenolide A, (-)-goniobutenolide B, (+)-goniopypyrone, (+)-altho-lactone,(+)-goniotriol, and (+)-7-acetylgoniotriol. J. Org. Chem. 1995, 60, 3121–3130. [Google Scholar] [CrossRef]
- Ko, S.Y.; Lerpiniere, J. Enantioselective synthesis of goniobutenolides A and B. Tetrahedron Lett. 1995, 36, 2101–2104. [Google Scholar] [CrossRef]
- Mukai, C.; Hirai, S.; Kim, I.J.; Kido, M.; Hanaoka, M. Studies on total synthesis of antitumor styryllactones: Stereoselective total synthesis of (+)-goniofufurane, (+)-goniobutenolide A, and (-) goniobutenolide B. Tetrahedron 1996, 52, 6547–6560. [Google Scholar]
- Surivet, J.P.; Vatèle, J.M. Concise total synthesis of (+)-goniofufurone and goniobutenolides A and B. Tetrahedron Lett. 1996, 37, 4373–4376. [Google Scholar] [CrossRef]
- Kuhnt, D.; Anke, T.; Besl, H.; Bross, M.; Herrmann, R.; Mocek, U.; Steffan, B.; Steglich, W. Antibiotics from basidiomycetes. XXXVII. New inhibitors of cholesterol biosynthesis from cultures of Xerula melanotricha Dorfelt. J. Antibiot. 1990, 43, 1413–1420. [Google Scholar]
- Negishi, E.; Hu, Q.; Huang, Z.; Qian, M.; Wang, G. Palladium-catalyzed alkenylation by the Negishi coupling. Aldrichim. Acta 2005, 38, 71–87. [Google Scholar]
- Flematti, G.R.; Ghisalberti, E.L.; Dixon, K.W.; Trengove, R.D. A compound from smoke that promotes seed germination. Science 2004, 305, 977. [Google Scholar] [CrossRef] [PubMed]
- Vaz, B.; Alvarez, R.; Brückner, R.; Lera, A.R. The Stille reaction in the synthesis of carotenoid butenolides: synthesis of 6’-epi-peridinin. Org. Lett. 2005, 7, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Shimizu, Y.; Steiner, J.R.; Clardy, J. Nostoclide I and II, extracellular metabolites from a symbiotic cyanobacterium, Nostoc sp., from the lichen Peltigera Canina. Tetrahedron Lett. 1993, 34, 761–764. [Google Scholar]
- Boukouvalas, J.; Maltais, F.; Lachance, N. Furanolate-based strategy for sequential 2,3,4-trisubstitution of butenolide : Total synthesis of nostoclides I and II. Tetrahedron Lett. 1994, 35, 7897–7900. [Google Scholar] [CrossRef]
- Bellina, F.; Rossi, R. Synthetic applications of 3,4-dihalo-2-(5H)-furanones: A formal total synthesis of nostoclides I and II. Synthesis 2002, 2729–2732. [Google Scholar] [CrossRef]
- Kar, A.; Gogoi, S.; Argade, N.P. Synthesis of naturally occurring bioactive butyrolactones: maculalactones A-C and nostoclide I. Tetrahedron 2005, 61, 5297–5302. [Google Scholar] [CrossRef]
- Barbosa, L.C.A.; Demuner, A.J.; Borges, E.E.L.; Mann, J. Synthesis and evaluation of the plant growth regulatory activity of 8-oxabicyclo[3.2.1] oct-6-en-3-one derivatives. J. Braz. Chem. Soc. 1997, 8, 19–27. [Google Scholar]
- Barbosa, L.C.A.; Costa, A.V.; Piló-Veloso, D.; Lopes, J.L.C.; Terrones, M.G.H.; Diaz, B.K.; Blas, L.H. Phytogrowth-inhibitory lactones derivatives of Glaucolide B. Z.Naturforsch. 2004, 59c, 803–810. [Google Scholar]
- Barbosa, L.C.A.; Maltha, C.R.A.; Demuner, A.J.; Ganen, F.A. Síntese de novas fitotoxinas derivadas do 8-oxabiciclo[3.2.1]oct-6-en-3-ona. Quim. Nova 2005, 28, 444–450. [Google Scholar]
- Chaves, F.C.; Barbosa, L.C.A.; Demuner, A.J.; Silva, A.A. New Helminthosporal analogues with plant-growth regulatory properties synthesized via oxyallyl cation. Z. Naturforsch. 2006, 61b, 1287–1294. [Google Scholar]
- Barbosa, L.C.A.; Alvarenga, E.S.; Demuner, A.J.; Virtuoso, L.S.; Silva, A.A. Synthesis of new phytogrowth-inhibitory substituted aryl-p-benzoquiones. Chem. Biodiv. 2006, 3, 553–567. [Google Scholar] [CrossRef]
- Casiraghi, G.; Zanardi, F.; Appendino, G.; Rasu, G. The vinylogous aldol reaction: a valuable, yet understated carbon-carbon bond-forming maneuver. Chem. Rev. 2000, 100, 1929–1972. [Google Scholar] [CrossRef] [PubMed]
- Boukouvalas, J.; Lachance, N.; Ouellete, M.; Trudeau, M. Facile access to 4-aryl-2(5H)-furanones by Suzuki cross coupling: efficient synthesis of rubrolides C and E. Tetrahedron Lett. 1998, 39, 7665–7668. [Google Scholar] [CrossRef]
- Bellina, F.; Anselmi, C.; Viel, S.; Mannina, L.; Rossi, R. Selective synthesis of (Z)-4-aryl-5-[1-(aryl)methylidene]-3-bromo-2(5H)-furanones. Tetrahedron 2001, 57, 9997–10007. [Google Scholar] [CrossRef]
- Bellina, F.; Anselmi, C.; Rossi, R. Total synthesis of rubrolide M and some of its unnatural congeners. Tetrahedron Lett. 2002, 43, 2023–2027. [Google Scholar] [CrossRef]
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, (3rd edn); John Wiley & Sons, Inc.: New York, 1990. [Google Scholar]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. AM1: A new general purpose quantum chemical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Teixeira, R.R.; Barbosa, L.C.A.; Santana, J.O.; Veloso, D.P.; Ellena, J.; Doriguetto, A.C.; Drew, M.G.B.; Ismail, F.M.D. Synthesis and structural characterization of two nostoclides analogues. J. Mol. Struct. 2007, 837, 197–205. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.A.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bodesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Mosman, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar] [PubMed]
- Berridge, M.V.; Tan, A.S.; McCoy, K.D.; Wang, R. The biochemical and cellular basis of cell proliferation assays that use tetrazolium salts. Biochemica 1996, 4, 15–20. [Google Scholar]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, (3rd edn); Pergamon: Oxford, 1988. [Google Scholar]
- Pettit, G.R.; Grealish, M.P.; Jung, K.; Harmel, E.; Pettit, R.K.; Chaput, J.C.; Schmidt, J.M. Antineoplastic agents.465.Strucutrual modificaction of resveratrol: sodium reverastatin phosphate. J. Med. Chem. 2002, 45, 2534–2542. [Google Scholar]
- Barbosa, L.C.A.; Demuner, A.J.; Alvarenga, E.S.; Oliveira, A.; Diaz, B.K.; Hennsen, B.L. Phytogrowth, and photosynthesis-inhibiting properties of nostoclides analogues. Pest Manag. Sci. 2006, 62, 214–222. [Google Scholar] [CrossRef] [PubMed]
- PC Spartan Pro; Wavefunction, Inc., 1999.
- Sample availability: Small amounts of compounds 5-14 are available from the corresponding author.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Teixeira, R.R.; Barbosa, L.C.; Maltha, C.R.A.; Rocha, M.E.; Bezerra, D.P.; Costa-Lotuf, L.V.; Pessoa, C.; Moraes, M.O. Synthesis and Cytotoxic Activity of Some 3-Benzyl-5-Arylidenefuran-2(5H)-ones. Molecules 2007, 12, 1101-1116. https://doi.org/10.3390/12051101
Teixeira RR, Barbosa LC, Maltha CRA, Rocha ME, Bezerra DP, Costa-Lotuf LV, Pessoa C, Moraes MO. Synthesis and Cytotoxic Activity of Some 3-Benzyl-5-Arylidenefuran-2(5H)-ones. Molecules. 2007; 12(5):1101-1116. https://doi.org/10.3390/12051101
Chicago/Turabian StyleTeixeira, Róbson Ricardo, Luiz Cláudio Barbosa, Célia Regina Alvares Maltha, Marcelo Eça Rocha, Daniel Pereira Bezerra, Letícia Veras Costa-Lotuf, Cláudia Pessoa, and Manoel Odorico Moraes. 2007. "Synthesis and Cytotoxic Activity of Some 3-Benzyl-5-Arylidenefuran-2(5H)-ones" Molecules 12, no. 5: 1101-1116. https://doi.org/10.3390/12051101
APA StyleTeixeira, R. R., Barbosa, L. C., Maltha, C. R. A., Rocha, M. E., Bezerra, D. P., Costa-Lotuf, L. V., Pessoa, C., & Moraes, M. O. (2007). Synthesis and Cytotoxic Activity of Some 3-Benzyl-5-Arylidenefuran-2(5H)-ones. Molecules, 12(5), 1101-1116. https://doi.org/10.3390/12051101