Results and Discussion
Bromination of olefins followed by bis-elimination of hydrogen bromide has widely been used to prepare symmetrical dienes from olefins [
1,
3,
4,
5]. Thus, monobromination of 1,5-cyclooctadiene (
1) was carried out in CHCl
3 at -70 °C, according to a previously reported procedure [
1]. Subsequent cyclopropanation with ethyl diazoacetate in the presence of CuSO
4 as catalyst [
6] was then performed. Based on similar reported reactions [
7], we expected to obtain a mixture of two isomers, and indeed column chromatography separation of the reaction products afforded two isomers in a 40:60 ratio, which were identified as the
endo and
exo ethyl 4,5-dibromobicyclo[6.1.0]nonane-9-carboxylates
3a and
3b (
Scheme 1). The stereochemistry of the three-membered ring was established by comparison of coupling constants of H-9 with H-1 and H-8 in the two isomers (8.7 Hz and 4.3 Hz for
3a and
3b, respectively), which confirmed the
cis and
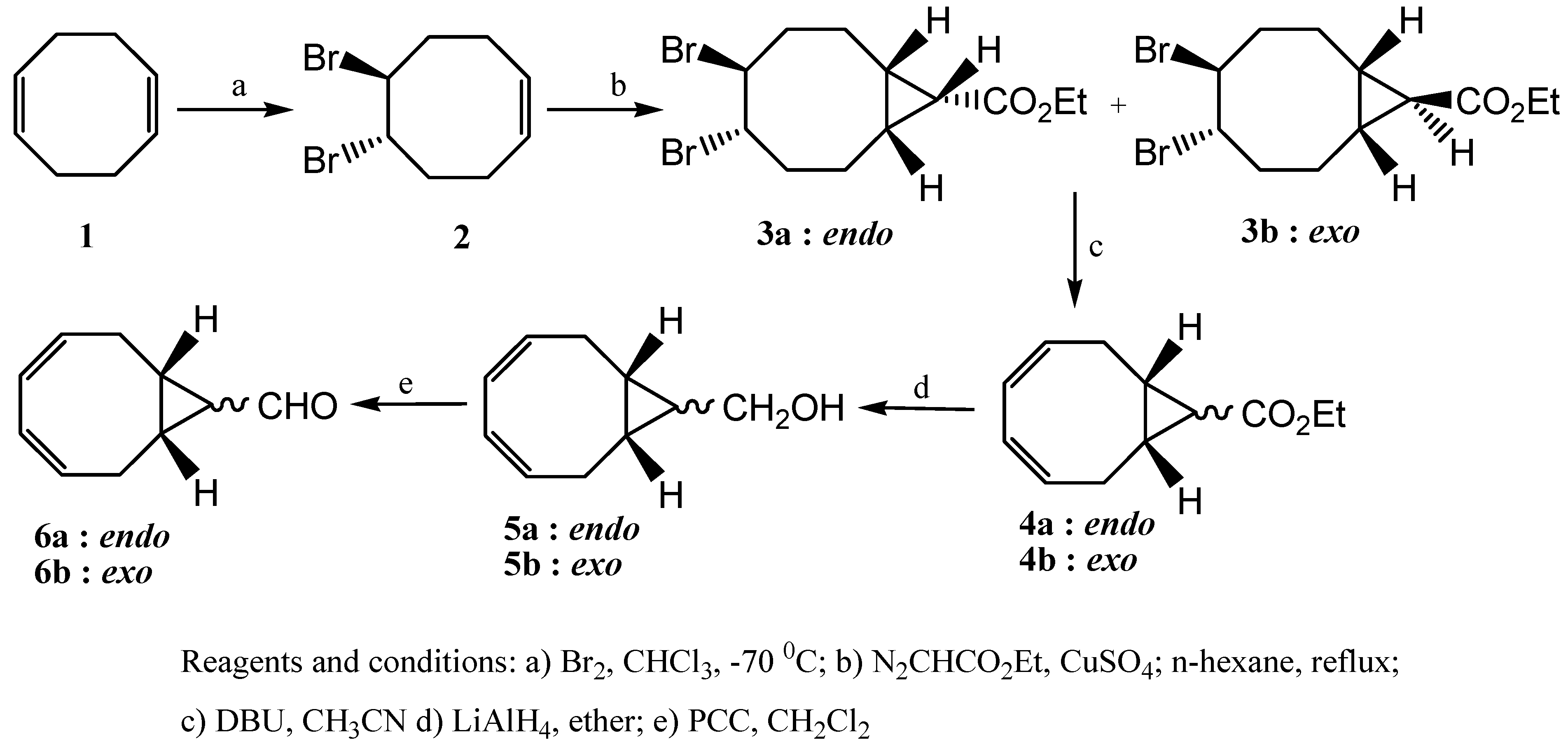
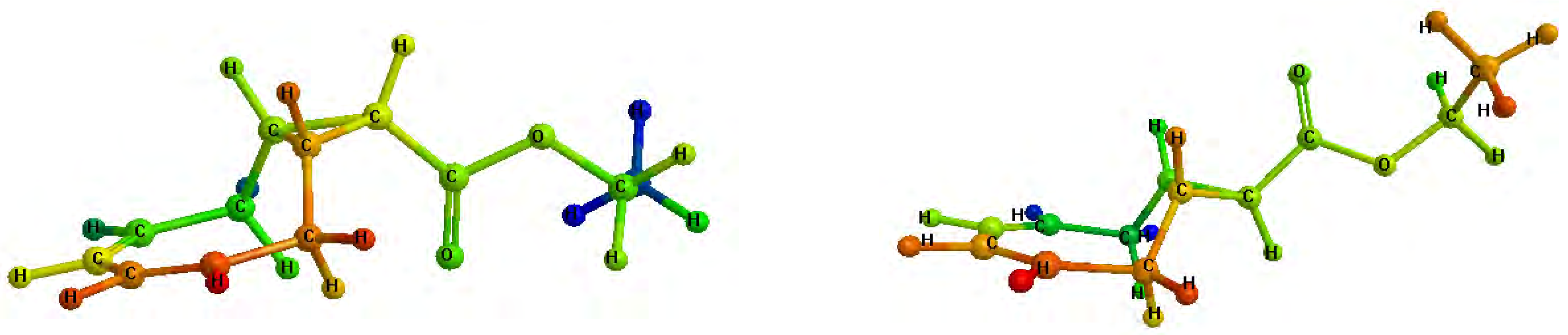
trans configurations with respect to the cyclopropane ring. As a result of their twisted and unsymmetrical conformations, shown in
Figure 1, the isomers
3a and
3b exhibited complicated
1H-NMR spectra (see
Experimental).
Scheme 1.
Synthetic route to bicyclo[6.1.0]nona-3,5-diene-9-carboxaldehyde (6) .
Scheme 1.
Synthetic route to bicyclo[6.1.0]nona-3,5-diene-9-carboxaldehyde (6) .
Bis-elimination of hydrogen bromide from the mixture of 3a and 3b with DBU in acetonitrile resulted in the formation of ethyl bicyclo[6.1.0]nona-3,5-diene-9-carboxylates 4a,b in 75% yield, which gave the corresponding 4a (endo) and 4b (exo) isomers after separation by column chromatography.
Figure 1.
Minimized structures of 3a (left) and 3b (right).
Figure 1.
Minimized structures of 3a (left) and 3b (right).
As shown in
Figure 2, and in contrast to isomers
3a and
3b, compounds
4a and
4b have a symmetry plane which simplifies the corresponding
1H-NMR and
13C-NMR spectra (see
Experimental). Conversion of
4a and
4b to 9-(hydroxymethyl)bicyclo[6.1.0]nona-3,5-dienes
5a (
endo) and
5b (
exo) was carried out by the well known reduction method using lithium aluminum hydride [
8]. Due to the difference between the reduction times needed for
4a and
4b (the former required 8 h, while reduction of the latter was completed after 12 hr),
4a and
4b were reacted separately.
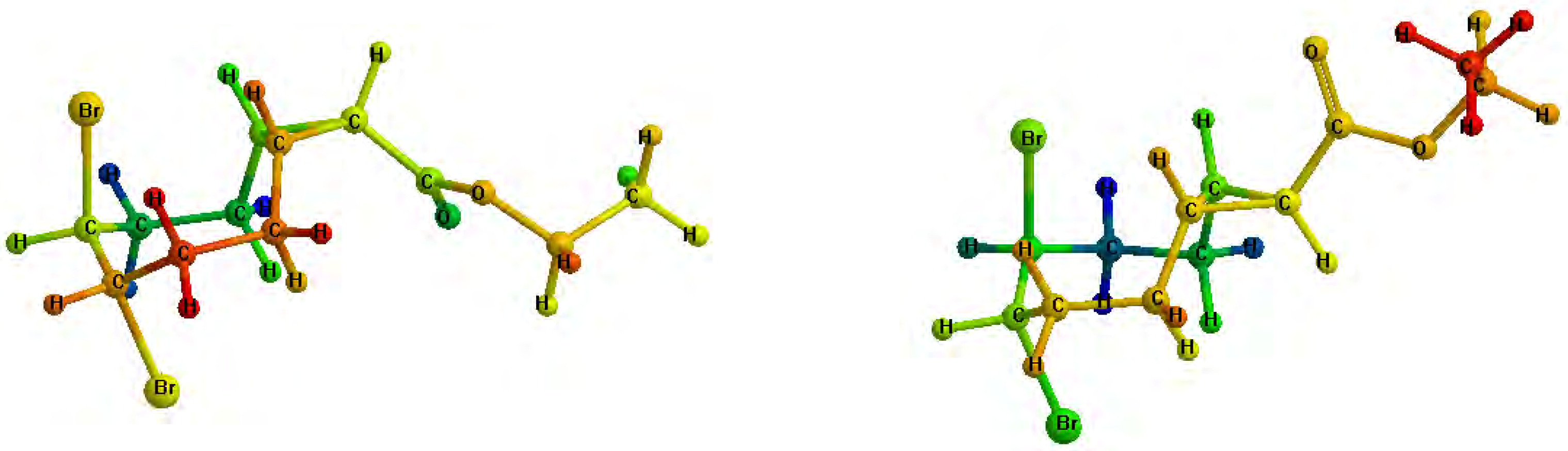
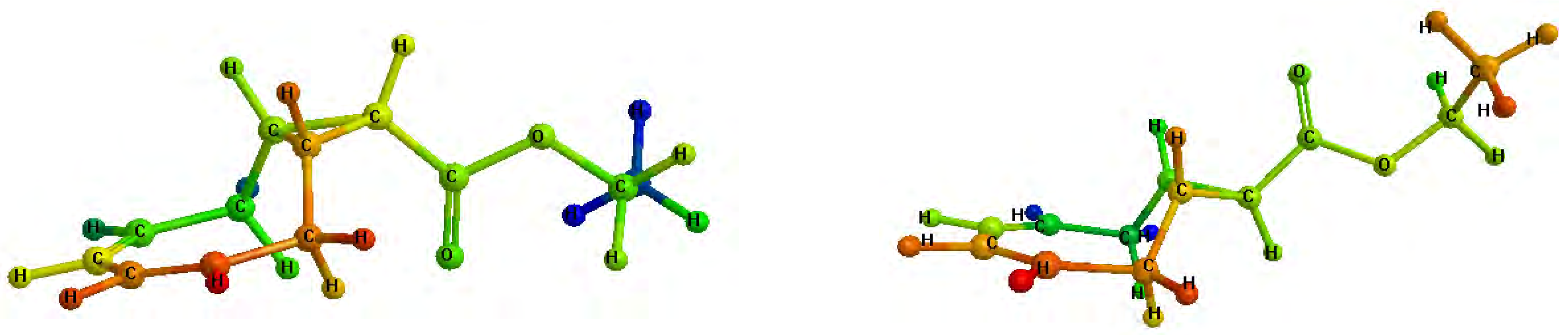
Figure 2.
Minimized structures of 4a (left) and 4b (right).
Figure 2.
Minimized structures of 4a (left) and 4b (right).
PCC is well known as a selective and convenient reagent for converting the primary alcohols to the corresponding aldehydes [
8,
9]. Oxidation of
5a/5b using PCC successfully gave the bicyclo[6,1,0]-nona-3,5-diene-9-carboxaldehydes
6a (
endo) and
6b (
exo) in 71% and 64% yields, respectively.
Experimental
General
All commercially available chemicals and reagents were purchased from the Merck Chemical Company and used without further purification. Melting points were determined on an Electrothermal model 9100 apparatus and are uncorrected. IR spectra were recorded on a Shimadzu 4300 spectrophotometer. The
1H-NMR and
13C-NMR spectra were recorded in CDCl
3 using a DRX-500 AVANCE spectrometer at 298 K. Chemical shifts (δ) are reported in ppm and are referenced to the NMR solvent peak. Mass spectra of the products were obtained with a HP (Agilent Technologies) 5937 Mass Selective Detector with Electron Impact (EI) 70eV, and quadrupole analyzer. Column chromatographies were carried out using silica gel 60 (63-200 mesh). All the reactions were carried out under nitrogen atmosphere and reactions progress was monitored by TLC using aluminium sheets precoated with silica gel Merck 60 FB254B. 5,6-Dibromocyclooct-1-ene (
2) was prepared using 1,5-cyclooctadiene (
1) and bromine, according to the previously reported procedure [
1].
Endo ethyl 4,5-dibromobicyclo[6.1.0]nonane-9-carboxylate (3a) and exo ethyl 4,5-dibromobicyclo-[6.1.0]-nonane-9-carboxylate (3b)
To a stirred refluxing solution of 5,6-dibromocyclooct-1-ene (2, 13.4g, 50 mmol) and anhydrous CuSO4 (2 g) in n-hexane (150 mL) was added a solution of ethyl diazoacetate (6.27 g, 55 mmol) in n-hexane (20 mL) during 30 min. The reaction mixture was then refluxed for one additional hour and filtered while hot in order to remove the CuSO4. The solvent was removed under reduced pressure and the brownish-yellow residue was then recrystallized from methanol to give white crystals (15 g, 85%), consisting of a pair of endo and exo isomers. The two isomers were separated by column chromatography on silica gel 60 using a 90:10 mixture of petroleum ether/ether as eluent to give 3a and 3b. Compound 3a: m.p. 132-133 °C; IR (KBr): 2970, 1724 (C=O), 1469, 1147, 1082, 790 cm-1; 1H-NMR δ: 1.31 (t, 3H, J = 7.1 Hz, CH3), 1.50-1.65 (m, 2H, H-1 and H-8), 1.79 (m, 1H), 1.83 (t, 1H, J = 8.7 Hz, H-9), 1.90 (m, 1H), 2.20 - 2.41(m, 4H), 2.75 (m, 2H), 4.16 (q, 2H, J = 7.1 Hz, OCH2), 4.82 (m, 1H, CHBr), 4.88 (m, 1H, CHBr); 13C-NMR δ: 14.75, 18.64, 19.70, 22.81, 23.04, 25.66, 35.21, 35.30, 54.72, 56.09, 60.24, 172.14 ppm; MS (EI): m/z 354 (M+); Compound 3b: m.p. 123-124 °C; IR (KBr): 2999, 1718 (C=O), 1467, 1174, 981, 783 cm-1; 1H-NMR δ: 1.24 (t, 1H, J = 4.3 Hz, H-9), 1.30 (t, 3H, J = 7.1 Hz, CH3), 1.42 – 1.55 (m, 2H); 1.69, 1.75 (2m, 2H, H-1, H-8), 2.15 (m. 3H), 2.34 (m, 1H), 2.68 (m, 1H), 2.77 (m, 1H), 4.15 (q, 2H, J = 7.1 Hz, OCH2), 4.82 (m, 1H, CHBr), 4.86 (m, 1H, CHBr); 13C-NMR δ: 14.69, 23.45, 24.23, 25.93, 28.18, 28.27, 34.76, 35.21, 52.75, 56.12, 60.64, 173.91 ppm; MS (EI): m/z 354 (M+).
Endo ethyl bicyclo[6.1.0]nona-3,5-diene-9-carboxylates (4a) and exo ethyl bicyclo-[6.1.0]nona-3,5-diene-9-carboxylate (4b)
Ethyl 4,5-dibromobicyclo[6.1.0]nonane-9-carboxylates 3a,b (3.54 g, 10 mmol) were dissolved in acetonitrile (70 mL) and DBU (4.63 g, 30 mmol) was then added in one portion. The solution was refluxed for 24 h and the solvent was then removed under reduced pressure. Water (50 mL) was added to the brown oily residue and the mixture was extracted with diethyl ether (2 × 50 mL). The combined organic extracts was washed with HCl (3M, 2 × 25 mL), then with sodium bicarbonate solution (5 %, 25 mL) and finally with distilled water (25 mL). The solution was dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure. A colorless oily liquid was obtained (1.44 g, 75% yield). The two diene isomers were separated by column chromatography on silica gel 60 using a 90:10 mixture of petroleum ether/ether as eluent to give compounds 4a and 4b as colorless oily liquids. 4a: IR (neat): 2937, 1720, 1398, 1355, 1157 cm-1; 1H-NMR δ: 1.23 ( t, 3H, J = 7.1 Hz, CH3), 1.58 (t, 1H, J = 6.4 Hz, H-9), 1.61 (m, 2H, H-1, H-8), 2.17 (m, 2H, H-2, H-7), 3.03 (m, 2H, H-2, H-7), 4.08 (q, 2H, J = 7.1 Hz, OCH2), 5.61 (m, 2H, H-3, H-6), 5.67 (d, 2H, J = 11.6 Hz, H-4, H-5); 13C-NMR δ: 14.72, 20.31, 23.05, 24.58, 60.31, 126.32, 130.93, 172.48 ppm; MS (EI): m/z 192 (M+); 4b: IR (neat): 2979, 2856, 1722, 1446, 1369, 1163, 995 cm-1; 1H-NMR δ: 1.19 (t, 3H, J = 7.2 Hz, CH3), 1.25 (t, 1H, J = 4.6 Hz, H-9), 1.56 (m, 2H, H-1, H-8), 2.19 (m, 2H, H-2, H-7), 2.50 (m, 2H, H-2, H-7), 4.03 (q, 2H, J = 7.2 Hz, OCH2), 5.60 (m, 2H, H-3, H-6), 5.88 (d, 2H, J = 10.1 Hz, H-4, H-5); 13C-NMR δ: 14.62, 24.63, 25.40, 27.10, 60.60, 129.77, 130.01, 174.45 ppm; MS (EI): m/z 192 (M+).
Endo 9-(hydroxymethyl)bicyclo[6.1.0]nona-3,5-dienes (5a) and exo 9-(hydroxymethyl)bicyclo[6.1.0]-nona-3,5-diene (5b).
Lithium aluminum hydride (270 mg, 7 mmol) and anhydrous diethyl ether (20 mL) were placed in a three-necked round-bottomed flask fitted with a reflux condenser and an addition funnel. Ethyl bicyclo[6.1.0]nona-3,5-diene-9-carboxylate 4a (or 4b) (960 mg, 5 mmol) dissolved in dry ether (5 mL) was added dropwise with magnetic stirring. The addition rate was controlled to maintain gentle reflux. After the addition was complete, the suspension was refluxed for an additional 8 h (in the case of 4a, 12 h for 4b). Ammonium chloride solution (10%, 5 mL) was then added slowly to the reaction mixture precooled to 0 °C in an ice bath. The reaction mixture was stirred for another 10 minutes and then filtered. The precipitate was washed with ether (10 mL). The combined ether filtrates were washed with water (10 mL), dried over anhydrous sodium sulfate and the solvent was then removed under reduced pressure to give 5a (or 5b) as colorless oily liquids. 5a: 640 mg (85%); IR (neat): 3400, 2923, 1440, 1400, 1022, 792, 761, 667 cm-1; 1H-NMR δ: 1.10- 1.18 (m, 3H, H-1, H-8, H-9), 2.36 (m, 4H, 2H-2, 2H-7), 2.91 (s, 1H, OH), 3.74 (d, 2H, J = 7.6, CH2OH), 5.67 (m, 2H, H-3, H-6), 5.74 (d, 2H, J = 11.3, H-4, H-5); 13C-NMR δ: 19.14, 21.18, 24.06, 60.00, 127.20, 131.56 ppm; MS (EI): m/z 150 (M+); 5b: 600 mg (80%); IR (neat): 3379, 3006, 2858, 1442, 1404, 1033, 790 cm-1; 1H-NMR δ: 0.70 (tt, 1H, J = 7.0 Hz, J = 4.8 Hz, H-9), 0.81 (m, 2H, H-1, H-8), 1.60 (s, 1H, OH), 2.15 (m, 2H, H-2, H-7), 2.45 (m, 2H, H-2, H-7), 3.40 (d, 2H, J = 7.0 Hz, CH2OH), 5.60 (d, 2H, H-3, H-6), 5.80 (d, 2H, J = 10.4 Hz, H-4, H-5); 13C-NMR δ: 20.24, 25.95, 28.14, 67.24, 128.85, 131.21 ppm; MS (EI): m/z 150 (M+).
Endo bicyclo[6.1.0]nona-3,5-diene-9-carboxaldehydes (6a) and exo bicyclo[6.1.0]nona-3,5-diene-9-carboxaldehyde (6b)
To a stirring solution of PCC (430 mg, 2 mmol) in dichloromethane (10 mL) was added a solution of 5a (or 5b) (300 mg, 2 mmol) in dichloromethane (2 mL). The mixture was stirred for 22 h at room temperature and was then refluxed for another 30 min. The reaction mixture was then filtered and the brown polymeric residue was washed with dichloromethane (2 × 10 mL). The combined dichloromethane solutions were evaporated under reduced pressure. To the residue was then added ether (30 mL) and the mixture was stirred for 10 min. The solution was filtered and the precipitate was washed with ether (5 mL). The combined ether solution was then washed with portions of 5% sodium bicarbonate solution (5 mL) until the ethereal solution became colorless. The solution was then dried over anhydrous sodium sulfate and the ether was removed under reduced pressure to give 6a (or (6b) as pale yellow liquids. 6a: 210 mg (71%); IR (neat): 3014, 2854, 1693, 1458, 1404, 1145, 993 cm-1; 1H-NMR δ: 1.77 (m, 3H, H-1, H-8, H-9), 2.32 (m, 2H, H-2, H-7), 2.88 (m, 2H, H-2, H-7), 5.60 (m, 2H, H-3, H-6), 5.76 (d, 2H, J = 10.7 Hz, H-4, H-5), 9.59 (d, 1H, J = 4.3 Hz, CHO); 13C-NMR δ: 23.66, 28.07, 30.86, 127.93, 130.69, 202.47 ppm; MS (EI): m/z 148 (M+); 6b: 190 mg (64%); IR (neat): 2954, 2850, 1740, 1470, 1193, 719 cm-1; 1H-NMR (CDCl3) δ: 1.54 (m, 1H, H-9), 1.71 (m, 2H, H-1, H-8), 2.22 (m, 2H, H-2, H-7), 2.50 (m, 2H, H-2, H-7), 5.59 (m, 2H, H-3, H-6), 5.88 (d, 2H, J = 10.1, H-4, H-5), 9.03 (d, 1H, J = 5.2 Hz); 13C-NMR δ: 25.88, 26.95, 30.08, 129.81, 130.15, 201.56 ppm; MS (EI): m/z 148 (M+).

{kind=link}
{kind=link}
{kind=link}